

Abstract
Circular forms of RNA were first discovered in plant viroids and later found in a variety of animal viruses. These circular RNAs lack free 5’ and 3’ ends, granting protection from exonucleases. This review is focused on the methods that are used to investigate virus-encoded circular RNAs. Using DNA viruses that are prevalent among human as examples, we begin with features of circular RNAs and the unique methods to enrich for circular RNAs. Next, we discuss the computational methods for RNA-sequencing analysis to discover new virus-encoded circular RNAs. Many strategies are similar to analyzing cellular RNAs, but some unique aspects of virus-encoded circular RNAs that are likely due to highly packed viral genomes and non-canonical use of splicing machinery, are described herein. We illustrate the various methods of validating expression of specific virus-encoded circular RNAs. Finally, we discuss methods to study functions of circular RNAs and the current technical challenges that remain for investigating virus-encoded circular RNAs.
Keywords: Circular RNA, Virus, Epstein-Barr virus, Kaposi sarcoma herpesvirus, human papilloma virus, viral non-coding RNA
1. Introduction
Circular RNA (circRNA) has long history in virology. In 1976, Sanger et al. showed that viroids, pathogenic and infectious agent mainly in plants, are non-coding circRNAs [1] Plant viroids can interact with the RNA-induced silencing complex (RISC), with endogenous siRNAs, and RNA-binding proteins (RBPs), which may be important for viroid replication [2]. In the animal world, Hepatitis delta virus (HDV) was found to have a 1.7 kilobase negative-strand closed circRNA genome in 1986. HDV, unlike plant viroids, has the coding capacity of HDAg proteins. Both viroids and HDV genomic circRNAs depend on eukaryotic RNA polymerases for their replication [3]. As such, these circRNAs of infectious pathogens have features akin to human circRNAs. Human circRNAs are a novel class of ncRNAs (non-coding RNAs) with gene regulatory functions, which are formed by a back-splicing process (Fig. 1A) after transcription by RNA polymerase II [4]. One of the proposed regulatory mechanisms of circRNAs is to interact with microRNAs (miRNAs) or RBPs, thus sequestering them (called the “sponge” function) [5–7]. Protein-coding capacity of certain human circRNAs have been proposed as well [4]There are differences between human circRNAs and viroids/HDV in origin and biogenesis with human circRNAs being generated through transcription and back-splicing by the spliceosome. They also do not contain entire genomes of the species. On the other hand, Viroid and HDV represent RNA genomes, but certain similarity exists at the level of interactions with other molecules like RNAs and proteins.
Figure 1.
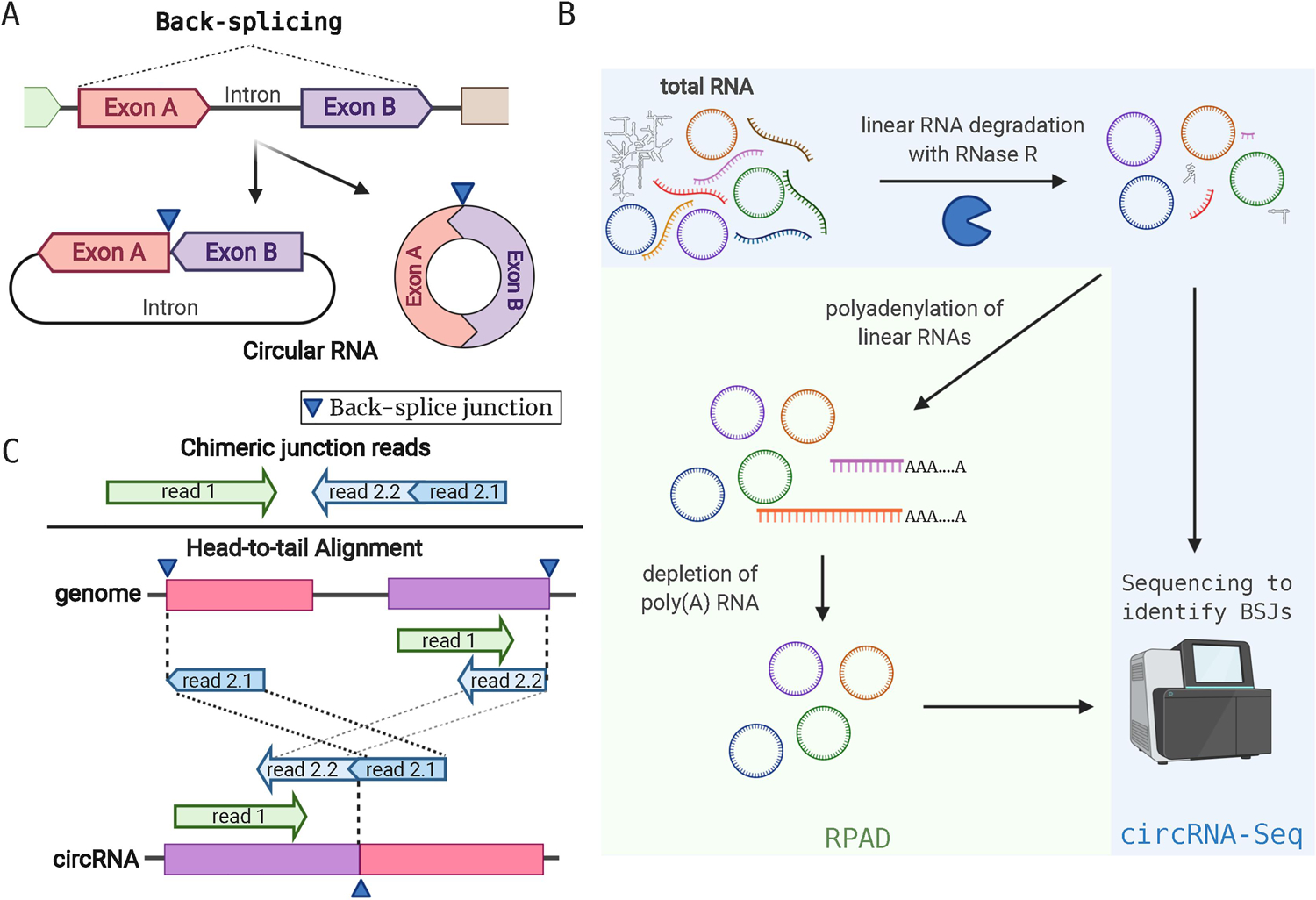
Features of circRNAs and circRNA Detection by RNA-sequencing. A. CircRNAs from back-splicing events that fuse the 5’ end of an exon (exon A) with the 3’ end of a separate exon (exon B). In this example, two forms of circRNAs can arise depending on the intron between exon A and B: it can be exonic (right) or exon-intron circRNAs. B. Total RNA purifications contain a mixture of linear RNAs and circRNAs. RNase R degrades linear RNAs and enriches samples for circRNAs. This enriched material can be used for RNA-sequencing called circRNA-Seq. Alternatively, after RNase R digestion, contaminating linear RNAs can be polyadenylated and removed to further enrich for circRNAs (RPAD). C. Detection of circRNAs from short-read sequencing is performed by identifying “head-to-tail” alignment on the genome sequence. Top, typical paired-end reads with chimeric junction are shown. Read 2 is mapped to two separate locations (2.1 and 2.2). Bottom, paired reads (read 1 and 2) originate from a circRNA, but on the genome read2 is chimerically mapped in head-to-tail manner (read 2.1 maps ahead of read 2.2). In this example, read1 and 2.2 are partially overlapping.
With the exceptions of viroids and HDV, most known viruses do not have circRNA genomes. What are the motivations to investigate viral circRNAs then? Viral ncRNAs have been found to be critical components for successful infection by various viruses. Oncogenic herpesviruses are particularly well-known examples; Epstein-Barr virus and Kaposi Sarcoma herpesvirus have relatively large DNA genomes. Both evolved to encode their own miRNAs and long non-coding RNAs (lncRNAs) to regulate cell proliferation, avoid apoptosis, or evade immune surveillance. These functions by viral miRNAs are crucial particularly during the latent phase, when viruses are dormant and most viral genes are turned off, but ncRNAs are abundant [8]As such, identifying and characterizing viral ncRNAs has been of interest in virology. Recently, circRNAs encoded in sub-viral genome loci were identified from various DNA viruses. In this review, we refer to the novel, back-spliced circRNAs transcribed by RNA polymerases from viral genome as virus-encoded circRNAs. EBV and KSHV were among the first to be found expressing multiple circRNAs, and Human papilloma virus (HPV) was also reported to encode circRNAs [9–13]. The functions of viral circRNAs remain largely unexplored.
In this review, we discuss current studies of virus-encoded circRNAs focusing on the methods to screen, validate, and quantitate virus-encoded circRNAs. Unlike viroids or HDV, these recently identified viral circRNAs are similar to human circRNAs regarding formation of back-splice junctions (BSJs), but have peculiar features like inter-ORF, intra-exon back-splicing, and variability at the junctions of BSJs. These features may be due to the fact that genomes of viruses have high gene density and the splicing machinery is often a controlling point of oncogenic viruses [14]. Upon screening and validation of virus-encoded circRNAs, these peculiarities should be considered to have different characteristics compared to human circRNAs.
2. Methods to study virus-encoded circRNAs
Small non-coding RNAs like miRNAs were missed by traditional RNA purification methods because these small RNAs were thought to be only degraded RNAs. Similarly, circRNAs were missed in previous RNA-sequencing experiments because normal genome and alignment algorithm would not allow mapping of reads containing back-splice junctions as discussed further in this section. Further, mRNA purification was used to enrich for protein-encoding mRNAs and to remove contaminating ribosomal RNAs. However, this poly-A pull-down method also removed circRNAs that lack poly-A tails. The use of total RNA sequencing with ribosomal RNA depletion has allowed multiple discoveries of cellular and viral circRNAs. As of now, only DNA viruses, but no RNA viruses have been found to encode circRNAs from back-spliced RNA pol II products [9–13]. These viral circRNAs are believed to form in a similar fashion as cellular circRNAs.
2.1. Comprehensive methods to identify viral circRNAs
Before discussing more specific aspects of circRNA discovery (section 3), we want to provide a simplified overview of the methods involved in the process. The most sensitive methods of de novo discovery of viral circRNAs rely on RNA-Seq of ribosomal RNA-depleted total RNA followed by circRNA enrichment. However, we also acknowledge that circRNAs can be found in RNA-seq assays without an enrichment step for circRNAs. Methods to enrich for circRNAs are shown in Fig. 1B. Typically the circRNA-Seq method relies upon the use of RNase R, an exonuclease that degrades linear RNA, but not circRNA. This material after RNase R treatment can be used for making RNA sequencing libraries. However, longer circRNAs may be more prone to be accidentally linearized via hydrolysis or omnipresent ribonucleases during RNA purification compared to shorter circRNAs, leading to the degradation by RNase R and to loss of detection. Recently, it was found that RNase R-mediated digestions can stall at G-rich regions in mRNAs and could lead to contamination of linear RNAs in samples enriched for circRNAs. Replacing Li+ with K+ in the conventional RNase R reaction buffer solves the issue and allows more efficient circRNA enrichment [15] Alternatively, the RPAD (RNase R treatment followed by Polyadenylation and poly(A)+ RNA Depletion) method can be used to further enrich for circRNAs before library generation [16]. RPAD polyadenylates residual linear RNAs after RNase R treatment and removes them by a poly(A)-pull down step to further purify circRNAs. Longer read sequencing (typically 100–150 base pair-end reads) with deeper sequencing than traditional transcriptome assays (more than 60 million reads) is recommended for circRNA detection. This is because identification of BSJs is done by detecting “head-to-tail” alignments (Fig. 1C) on viral genomes; usable reads have to (i) contain a BSJ and (ii) the BSJ has to be flanked at least by 15–20 nucleotides for reliable mapping i.e. many genuine reads from circRNAs would be discarded if they do not have optimal sequencing reads in order to detect BSJ sequences. For circRNA detection, we use CIRCexplorer2 [17] that takes an output from STAR aligner [18], or CIRI2 [19] that requires BWA aligner [20]. CIRCexplorer2 and CIRI2 are algorithms to detect head-to-tail alignments (Fig. 1B) from corresponding aligners. Both are comparable among other similar software and are reliable and reproducible for detecting new BSJs, particularly when combined [21]. Note that for STAR has a two-pass alignment mode. In this mode, splice junctions detected in first alignment are used as additional annotated junctions and allows for better chimeric read detection [22].
2.2. Methods to validate expression of circRNAs
Once BSJs have been detected after the RNA-sequencing, alignment, and circRNA detection programs, it is important to validate these potential circRNAs. One common method is to use divergent PCR primers (Fig. 2A). These divergent primers are designed with 5’ and 3’ directionality to extend in opposite directions on templates from linear transcripts, but extend towards each other when bound to templates generated from back-spliced junctions. PCR products from divergent primers can be used to quantitate circRNAs using real-time PCR. Additionally, these PCR products can be subjected to DNA sequencing to validate expected BSJs and validate newly discovered BSJs. RNA samples can be treated with RNase R prior to divergent PCR to degrade linear RNAs. PCR products generated with divergent PCR primers that are also resistant to RNase R provides compelling evidence of a BSJ from a circRNA.
Figure 2.
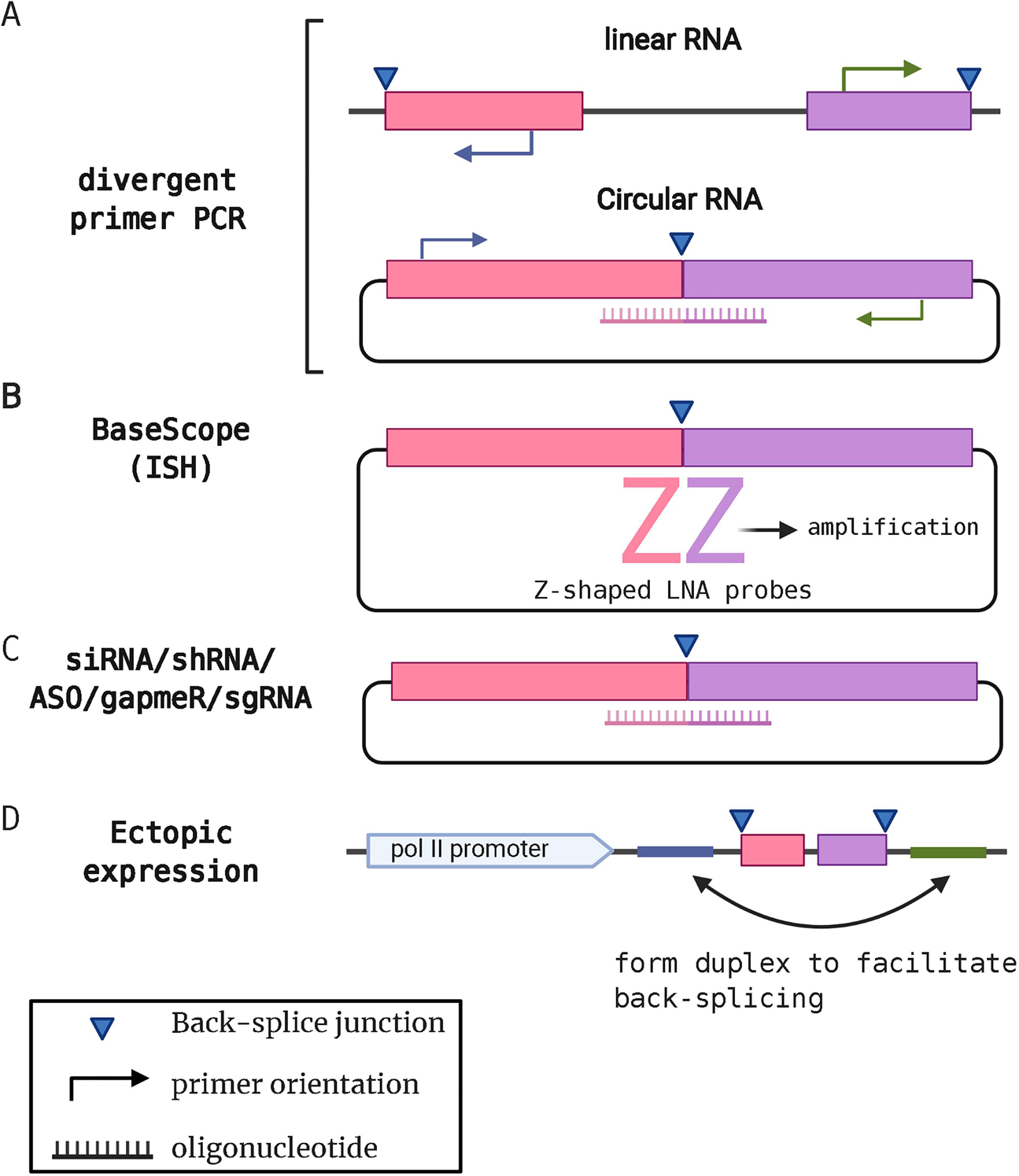
Validation and Functional Analysis of circRNAs. A. Validation of potential circRNAs identified by circRNA-Seq is performed by using divergent PCR primer pairs. These divergent primers only produce a product from circRNAs in combination with a Taqman probe. B. In situ hybridization using BaseScope or similar methods can detect the unique BSJ in cells. A double-Z probe pair are locked nucleic acid oligos and each Z binds to each side of 18–25 nt flanking region of the BSJ. Only when both Z probes are bound in close proximity, then the detection signal can be amplified and chromogenically or fluorescently detected. C. Small nucleotide-based suppressors like siRNAs (small interfering RNA), shRNA (short hairpin RNA), gapmeRs, antisense oligos (ASO), or sgRNA (guide RNA) can be designed to target BSJs in circRNAs for loss-of-function studies. D. circRNA expression vectors that contain flanking sequences that form hairpin structure to promote back-splicing can be used for gain-of-function studies. These flanking regions are often derived from genes that are known to generate circRNAs such as ZKSCAN1.
Circularity can be further proven by linearizing circRNAs. Chemical hydrolysis or targeted degradation with RNase H, an endonuclease targeting DNA:RNA hybrids, can be used in this purpose [23]. For RNase H, antisense oligo (ASO) DNAs can be designed to target specific circRNAs so that ASO can bind to the circRNA such that RNase H degrades the hybrid. Linearized circRNAs will not be amplified with divergent primer RT-PCR. Northern blotting can be also used to distinguish linear and circRNAs since they will show a different migration pattern on polyacrylamide gels [10,23].
Since the BSJs are unique to the circRNAs these regions can be targeted with special probes and amplification to detect the corresponding circRNAs in cell culture and in tissues using fluorescence in situ hybridization (FISH), aptamers, or BaseScope technology (Reviewed in [24]). BaseScope is widely used to visualize viral circRNAs [10,13,25]. utilizes a tandem of Z-shaped locked nucleic acids (LNA) as probes (Fig. 2B). They can be designed to specifically bind to 18–25 nt of each side of flanking regions of BSJs with high specificity [26] Signal is amplified only when both Z probes are in proximity to be detectable as chromogenic signal in cell lines or human tissues. Compared to traditional methods utilizing tiling of probes or MS2 tagging, BaseScope allows for detection of natural circRNAs without background detection of linear RNA products.
2.3. Methods to study functions of viral circRNAs
Many of the methods employed to study the functions of cellular circRNAs can be used for viral circRNAs, but the study of viral circRNAs benefits from the fact that uninfected cells represent a great negative control since they do not possess any viral circRNAs. Gain-of-function assays can use ectopic expression of a specific viral circRNA in uninfected cells. Ectopic expression of circRNAs using plasmids was developed by the Wilusz group [27]. Introns before exon 2 and after exon 3 of the ZKSCAN1 gene have reverse-complementary sequences that form a double-stranded structure to facilitate back-splicing and thus promote production of circRNAs (Fig. 2D). This system utilizes an existing splicing mechanism of host cells. One potential downside is that this plasmid does produce a certain level of linear RNAs in addition to desired circRNAs. One method to avoid this is to use synthesized circRNAs. Products of in vitro transcription can be ligated with T4 RNA ligase or chemically with cyanogen bromide [28] or with self-cleaving intron of Anabaena [29]. There are, however, discussion on the immunogenicity of synthesized circRNAs; T4 based circRNAs are reported to be immunogenic [30] whereas Anabaena intron-based circRNAs with stringent HPLS purification diminished the immunogenicity [31]. In contrast, ZKSCAN1-based circRNAs do not cause immune responses [30,32]. The interactions between circRNAs and the innate immune system are still inconclusive, and researchers should choose an appropriate system depending on the purpose of their specific study.
Specific viral circRNAs can be targeted for loss-of-function assays using genetic disruption and targeting with RNAi using guides to BSJs (Fig. 2C). Since BSJs are unique to circRNAs, siRNAs/shRNAs, or gapmers (LNA/DNA based ASO to induce RNase H-mediated RNA degradation) designed to target BSJs should not cleave linear RNAs directly. Still, multiple siRNAs should be tested since circRNA-specific siRNAs bind to both sides of flanking regions of BSJ and 5’ends of the siRNA may work as miRNAs thus causing off-target effects leading to suppression of linear RNAs. Recently, a circRNA knock-down system with CRISPRi based on Cas13 was established [33]. CRISPR-Cas13 system was reported to be more efficient than shRNAs in knock-down circRNAs and thus a promising platform for future loss-of-function studies.
To study mechanistic aspects of circRNAs, coding capacity and interactions with other molecules should be investigated. Polysome profiling can determine whether circRNAs are interacting with ribosomes and thus likely to be translated. This technique has been also used for viral circRNAs [10,12,25]. Other types of molecules to potentially interact with circRNAs are miRNAs and RBPs. CircRNAs can work as sponges of miRNAs or RBPs to suppress their functions. A specific viral circRNA from HPV was examined for its sponging ability to miRNAs. The circRNA sequence was conjugated to luciferase reporter gene to test their ability to be directly bound by miRNAs of interest [34]. More direct method is to pull-down RNA-RNA complex. For example, Liu et al used a circRNA-specific probe to capture viral circRNA-human miRNA complexes [34]. A comprehensive way is to pull-down RISC (RNA-induced silencing complex), a protein-miRNA complex to regulate target RNA. Antibodies specific to Argonaute, the core protein of RISC, allows immunoprecipitation of RISC-circRNA complex [4]. A similar approach could be used to identify other RBPs interacting with circRNAs. Combining gain- and loss-of-functions methods as well as direct screening of interacting partners will lead to deeper understanding of functions of circRNAs. In turn, showing functions also will be a potent evidence of viral circRNAs to be more than “junk” RNAs, as some lncRNAs have been perceived. We discuss these approaches and specific findings on virus-encoded circRNAs using herpesviruses and human papilloma virus (HPV) as examples in section 3.
3. Virus-encoded circRNAs
The approach to identify virus-encoded circRNAs has been analogous to human circRNAs. Starting from circRNA-enriched RNA-Seq, these predicted circRNAs are validated by sanger sequencing, divergent primer PCR, and in situ hybridization. Several virus-encoded circRNAs have, however, peculiar features such as very complicated back-splicing variations that complicate the analysis and need novel techniques to study them further. Viral genomes are very different compared to human genome in various ways: they are densely packed with ORFs; often, both + and − strand encode for proteins; they encode for many polycistronic transcripts; and transcription/splicing machineries are co-regulated by viral gene products. These features likely contribute to the way virus-encoded circRNAs are formed. In this section, we discuss current advancement in identifying and characterizing viral circRNAs.
3.1. Identification of viral circRNAs
Human herpesviruses, namely Epstein-Barr virus (EBV/Human herpesvirus 4) and Kaposi Sarcoma herpesvirus (KSHV/Human herpesvirus 8) were the first viruses to be found encoding circRNAs in their genomes. EBV and KSHV are oncogenic virus with large DNA genomes (172kb and 165 kb, respectively) harboring more than 80 ORFs, miRNAs, and long non-coding RNAs. Upon infection, EBV and KSHV’s circular genomes reside in the nucleus without integrating into human genomes (in contrast to retroviruses). Rather, viral genomes are tethered to host chromosomes via viral DNA-binding proteins [35]. Since these viral plasmids, called episomes, are methylated, chromatinized, and distributed to daughter cells, they can be treated as extra chromosomes when analyzing viral transcriptomes, including viral non-coding RNAs. One thing to note, however, is that unlike human chromosomes, episome copy numbers per cell are dynamic and heterogenous, depending on the conditions; EBV and KSHV life cycles largely consist of the latent phase, when most of viral genes are silent and without any virus replication, and the lytic phase, during which viral genomes are highly active and replicated for virion production [35]. Screening efforts to identify EBV and KSHV-encoded circRNAs have been conducted in variable infection conditions and cell types.
RNA-Seq of circRNA-enriched total RNA (circRNA-Seq, Fig. 1B) was employed to identify circRNAs encoded in EBV/KSHV genome. Ungerleider et al. and our group employed RNase R to enrich for circRNAs in EBV/KSHV-positive cell lines as well as de novo infected human primary cell cultures [9,11], whereas Toptan et al. utilized RPAD [10]. Regardless of enrichment methods and cell conditions, existence of virus-encoded circRNAs were confirmed, though functional studies are limited in these initial reports.
In one study, the authors found back-spliced junctions from more than 30 new EBV circRNAs from nine EBV positive cell lines at latent or lytic phases [11]. Ungerleider et al. treated total RNA with RNase R and utilized python script “find_circ” [7] developed by the Rajewsky group, which detects head-to-tail alignment with BSJs flanked by GU/AG canonical splicing sites, and developed a tool named circleVis to visualize back-splicing with box (exons) and curves (splicing sites). These EBV-encoded circRNAs came from eight different genes. The majority of these eight viral genes encode several circRNA back-splicing isoforms. In particular, in one EBV locus called BART, they found an RPMS1-derived circRNA, that was confirmed in three studies [11,13,34] and this locus can express more than 10 circRNA isoforms in at least one cell line for each type of EBV latency [11]. Similarly, the EBV LMP2 locus was found to encode multiple circRNAs. It should be noted that splicing is rare in most DNA virus transcripts. However, both the RPMS1 and LMP2 ORFs harbor multiple exons, and have many well-defined splicing donor/acceptor sites of back-spliced junctions. In a simultaneous effort, Toptan et al. found EBV-encoded circRNAs including four variants of circRPMS1 from patient-derived PTLD (posttransplant lymphoproliferative disease) samples, signifying the expression and importance in human disease [10]. The authors performed RPAD for circRNA enrichment and used CIRI2 for detection of viral circRNAs. Existence of circRNAs has been explored with RT-PCR with divergent primers as well as BaseScope detection method [10,26]. Multiple assays were thus used to validate expression of these newly discovered virus-encoded circRNAs.
In KSHV, at least 10 different viral genes have been found to express circRNAs in a combination of KSHV-infected lymphocytes, epithelial and endothelial cells [9,10,36]. In particular, circRNAs encoded in the KSHV v-IRF4 gene were confirmed in all three independent studies that investigated KSHV latent infections [9,10,37] regardless of detection method (find_circ by Ungereleder et al., CIRI2 by Toptan et al., or CircExplorer2 by our group). Similar to EBV, KSHV-encoded circRNAs were confirmed in KSHV-positive cell lines as well as samples from patients with Kaposi sarcoma (KS), primary effusion lymphoma (PEL), and multicentric Castleman’s disease (MCD) [9,10]. BaseScope assays were also applied to detect KSHV-encoded circRNAs in KSHV-positive cell lines [25] Abere et al also found that KSHV-encoded circRNAs can be integrated within virions. Extra-cellular circRNAs have another potential function for viruses to manipulate the microenvironment [38] to a more pro-viral one, as what was similarly proposed for EBV miRNAs.
Zhao et al. searched NCBI Sequence Read Archive (SRA) for HPV total RNA sequencing data and found 154 RNA-Seq samples from 12 projects [12]. HPV, similar to EBV and KSHV, is an oncogenic DNA virus with circular DNA genomes, though the HPV genome is relatively small (8 kb for HPV16). Zhao et al. developed an HPV-encoded circRNA discovery tool called vircircRNA. Similar to previously mentioned BSJ detection tools, this pipeline maps reads from RNA-Seq to a viral genome and identifies head-to-tail alignment at canonical GU-AG splice donor-acceptor motifs [12]. The predicted splicing results are visualized as box (exon) and curve (splicing) diagrams like circleVis [11]. The authors particularly focused on circE7, a circRNA in the HPV16 strain, and performed further validations, including amplification with divergent-PCR followed by sanger sequencing for circRNA validation and Northern blots of HPV16-positive cell lines [12]. A similar approach utilizing total RNA-Seq (without circRNA enrichment) was carried out for EBV circRNAs. Huang et al. performed RNA-Seq of rRNA-depleted EBV-positive cell lines and found one variant of circRPMS1 (back-spliced at exon 4 to exon 2, E4-E2), which was validated by divergent PCR and detected by BaseScope using a BSJ-specific probe [13].
Thus, it is possible to discover legitimate and functional virus-encoded circRNAs from total RNA-Seq. However, we would argue that circRNA-enriched RNA-Seq is still the more-sensitive way to identify virus-encoded circRNAs. According to Ungerleider et al. (RNase R treated), the E4-E2 variant found in Huang et al (total RNA) is only one of more than 10 variants of circRPMS1. Also, the E4-E2 variant is not the most abundant EBV circRNA. Since head-to-tail reads of viral origin typically account for less than 1% of cellular BSJs, unless the virus is in the lytic phase of infection (Table 1), it has been more challenging to discover virus-encoded circRNAs compared to human circRNAs. Enrichment of circRNAs is, therefore, crucial for comprehensive understanding of viral circRNAs.
Table 1.
De novo prediction of virus-encoded circRNAs from EBV and KSHV genomes byCircExplorer2 in publicly available data [10,11,37] on EBV (NC_007605.1), KSHV (NC_009333.1), and human genome (GRCh38). Certain samples were chemically induced for lytic cycle. PTLD, posttransplant lymphoproliferative disorder.
| GEO ID | sample ID | sample name | circRNA enrichment method | Sample type | KSHV BSJ counts | EBV BSJ counts | Hs BSJ counts | Ratio (viral/Hs) | Reference |
|---|---|---|---|---|---|---|---|---|---|
| GSE124711 | GSM3543750 | BCBL1 (induced) | RNase R | B cell line | 101 | 0 | 283921 | 0.04% | [37] |
|
| |||||||||
| GSM3543751 | BCBL1 (uninduced) | RNase R | B cell line | 2994 | 0 | 325402 | 0.92% | [37] | |
|
| |||||||||
| GSE116675 | GSM3258253 | Akata (uninduced) | RNase R | B cell line | 0 | 151 | 441337 | 0.03% | [11] |
|
| |||||||||
| GSM3258256 | Akata (induced) | RNase R | B cell line | 0 | 7378 | 495653 | 1.49% | [11] | |
|
| |||||||||
| GSM3258259 | Mutu I (uninduced) | RNase R | B cell line | 0 | 184 | 575870 | 0.03% | [11] | |
|
| |||||||||
| GSM3258260 | Mutu I (induced) | RNase R | B cell line | 0 | 1458 | 1406572 | 0.10% | [11] | |
|
| |||||||||
| GSM3258262 | Mutu III | RNase R | B cell line | 0 | 134 | 186673 | 0.07% | [11] | |
|
| |||||||||
| GSM3258264 | Jijoye | RNase R | B cell line | 2 | 561 | 532847 | 0.11% | [11] | |
|
| |||||||||
| GSM3258265 | JY | RNase R | B cell line | 0 | 486 | 357609 | 0.14% | [11] | |
|
| |||||||||
| GSM3258268 | SNU719 | RNase R | Gastric Carcinoma Cell line | 0 | 116 | 396343 | 0.03% | [11] | |
|
| |||||||||
| GSM3258269 | YCCEL1 | RNase R | Gastric Carcinoma cell line | 0 | 1151 | 309750 | 0.37% | [11] | |
|
| |||||||||
| GSE117798 | GSM3309421 | PTLD4 | RPAD | Non-infected clinical sample | 0 | 0 | 36080 | 0.00% | [10] |
|
| |||||||||
| GSM3309422 | PTLD5 | RPAD | non-infected clinical sample | 0 | 0 | 35022 | 0.00% | [10] | |
|
| |||||||||
| GSM3309423 | PTLD6 | RPAD | Infected clinical sample | 0 | 23 | 39136 | 0.06% | [10] | |
|
| |||||||||
| GSM3309424 | PTLD9 | RPAD | Infected clinical sample | 100 | 75 | 84214 | 0.21% | [10] | |
|
| |||||||||
| GSM3309425 | BC1 (uninduced) | RPAD | B cell line | 1003 | 339 | 277773 | 0.48% | [10] | |
|
| |||||||||
| GSM3309426 | BC1 (induced) | RPAD | B cell line | 365584 | 1040 | 266652 | 137.49% | [10] | |
|
| |||||||||
| GSM3309427 | BCBL1 (uninduced) | RPAD | B cell line | 1270 | 3 | 122805 | 1.04% | [10] | |
|
| |||||||||
| GSM3309428 | BCBL1 (induced) | RPAD | B cell line | 208489 | 0 | 121801 | 171.17% | [10] | |
|
| |||||||||
When viral miRNAs were first discovered, it was a surprising discovery that limited conservation of viral miRNAs existed between similar viruses. Likewise, the only viral circRNA conservation reported to date is an isoform of circRPMS1 between EBV and rLCV [37], which has 65% conservation at the primary sequence level. It is possible that viral circRNAs are conserved in their RNA and protein interactions, structures, or functions rather than their primary sequences.
3.2. Functional studies of virus-encoded circRNAs.
Virus-encoded circRNAs are expressed in various viral life stages and cell types, as we have discussed, but are they intentionally encoded by viruses? One approach to evaluate the importance of virus-encoded circRNAs is to determine their functions.
Abundance and potential functions of HPV circE7 were further explored, suggesting its importance in HPV virology. Expression of circE7 is as abundant as the linear E7 transcript and most of the circRNA is enriched in the cytoplasm. The circE7 contains the full-length of E7 coding sequence. When ectopically expressed in 293T cells, circE7 has N6-methyladenosine (m6A) modifications, which allowed translation into protein in cap-dependent manner [12]. Indeed, associations with polysomes and circE7 were shown, in contrast to the herpesviral circRNAs that were not associated with polysomes [10].
In order to seek potential functions of KSHV-encoded circRNAs, we cloned KSHV-encoded circRNAs into ZKSCAN1-based expression vectors to exogenously express circRNAs of interest in KSHV-infected cells (Fig. 2). Though the mechanism is unclear, certain virus-encoded circRNAs regulated cell proliferation, suggesting potential functions of KSHV circRNAs during infection [9].
Ectopic expression of a variant of EBV’s circRPMS1 was also performed using a pCMV-driven circRNA cassette similar to the ZKSCAN1 system in EBV-negative AGS cells (Fig. 2D). Ectopic circRPMS1 reduced expression of human miRNAs that are predicted to bind circRPMS1[13]. This suggests that circRPMS1 can function as a sponge for miRNAs. In an independent knock-down study, circRPMS1 was suppressed via transfecting plasmids encoding an shRNA targeted against circRPMS1 (Fig. 2C). With this approach, they found that a different group of human miRNAs, namely miR-31, 203, and 451, were found to be affected by circRPMS1 expression levels [34]. They further performed luciferase reporter assays and circRNA-miRNA pull-down to confirm the interaction [34](see section 2.3 for more details). Interestingly, one would not expect miRNA levels to be reduced since, in the sponge model, miRNAs are merely bound by circRNAs. It is possible, therefore, that circRPMS1 may utilize other mechanisms like target-directed miRNA degradation (TDMD) in which RNAs cause degradation of bound miRNAs. HCMV’s UL144–145, for example, was shown to degrade hsa-miR-17 and hsa-miR-20 [39].
Functional studies of viral circRNAs are thus under way for various viruses and methods are largely derived from human circRNA research. There are, however, multiple feature unique to viral circRNAs that may pose difficulties to utilize conventional functional assays.
3.3. Unconventional features of virus-encoded circRNA
3.3.1. circRNA-specific BSJs within viral exons
Human circRNAs are typically back-spliced at known intron-exon junctions of host unspliced transcripts (i.e. like alternative splicing isoforms) [4]. Certain known virus-encoded circRNAs do not follow traditional splicing patterns. For example, KSHV vIRF4 mRNA transcript contains two coding exons and an intron in between, but back-splicing does not occur at the splicing donor and acceptor of these exons for these virus-encoded circRNAs. Rather, back-splicing happens within previously annotated exons (exon 1 and exon 2) to create circvIRF4 with and without the intron [10,36]. This circRNA-specific splicing donor/acceptor patterns exist in other viruses. For example, EBV circBHLF1 is made from back-splicing within the single exon viral gene BHLF1 [11] (Fig. 3A). HPV expresses a polycistronic transcript coding for E6, E7, and E1 in this order. Meanwhile, back-splicing of circE7 occurs between donor/acceptor within E6 and E1 such that circE7 contains the 3’ end of E6, all of E7, and the 5’ end of E1 exon [12] (Fig. 3B). We have to note that circRNAs derived from multi-exon viral genes like EBV LMP2 or RPMS1 are indeed back-spliced at previously known canonical acceptor/donor at coding exon-intron junctions. However, one has to acknowledge that even single exon genes can produce circRNAs using unique splice sites.
Figure 3.
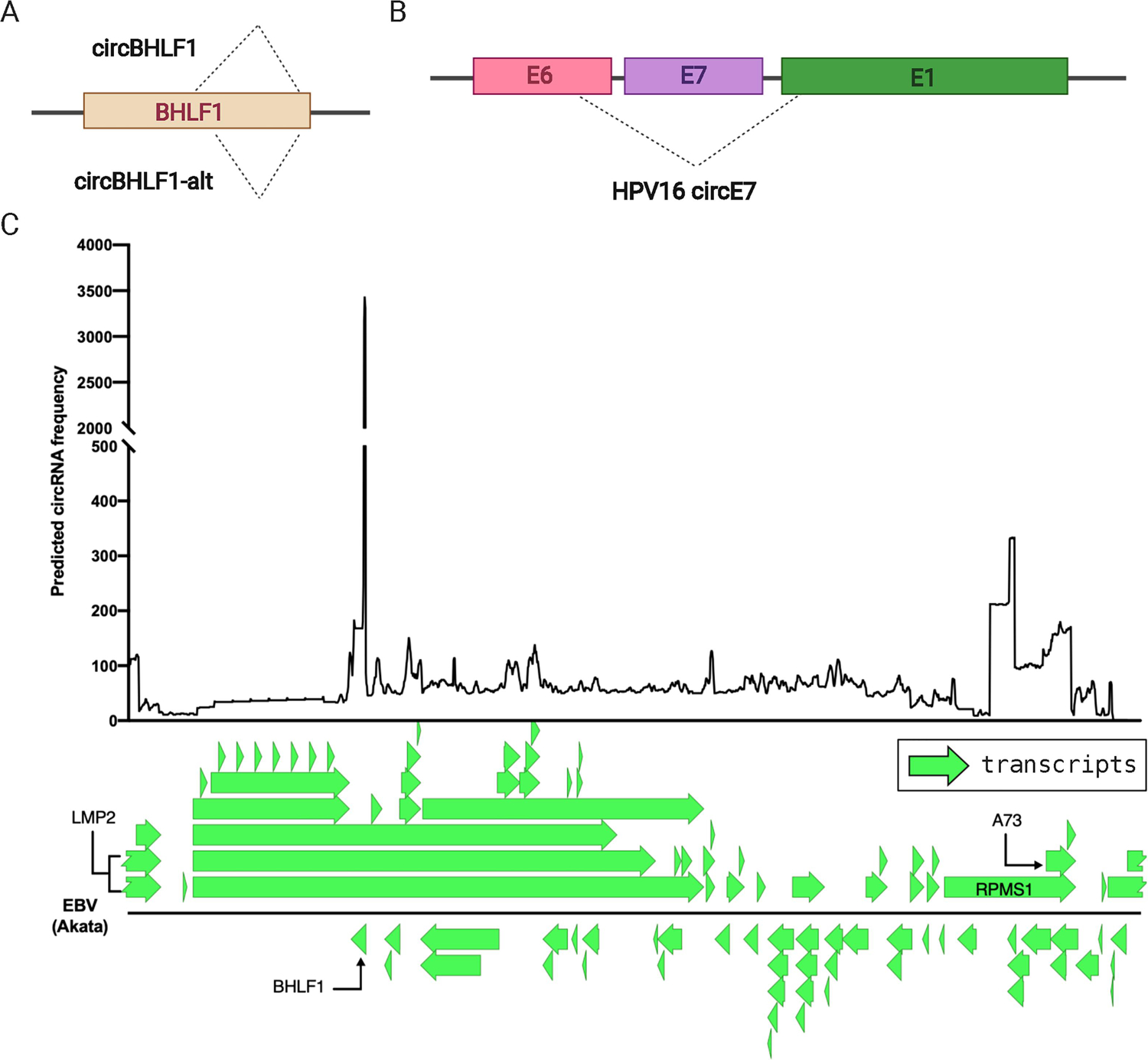
Unique Features of Virus-Encoded circRNAs. A. Back-splicing from the EBV BHLF1 gene display variability in the BSJs. circBHLF1 uses canonical splice sites whereas circBHLF1-alt uses alternative splice donor 9 bp upstream of circBHLF1 [11]. B. From HPV, one circRNA contains fragments of three protein-encoding genes. C. Histogram of virus-encoded circRNAs predicted by CircExplorer2 across the EBV genome (NC_007605.1). BSJ-containing reads were collected and used to predict locations and abundance of EBV circRNA in reactivated EBV-positive Akata cells from GSM3258253 [11] (top). BHLF1 and RPMS1 region hosts highly expressed circRNAs and indicated in the transcript map of EBV (NC_007605.1).
3.3.2. ORF-spanning circRNAs
Other unusual viral circRNAs are the ones spanning beyond a single gene as seen in HPV circE7. EBV has many such examples: A73, RPMS1, and LMP2 are located at close proximity and some circRNAs are back-spliced at intron-exon junctions of RPMS1 and LMP2, or LMP2 and A73. Similarly, other EBV-encoded circRNAs span the aforementioned BHLF1 and BGLF1, BDLF1, or BILF2 [11]. Additionally, we have reported that a KSHV-encoded circRNA harbors parts of ORF35 and ORF36 genes [9]. Unlike the human host genome, all these viral genomes are packed with ORFs and many of them are transcribed in polycistronic manner. In theory, host splicing machinery can also work on those transcripts to cause back-splicing. This is fascinating because many viral genes like BHLF1 or ORF35 contain a single exon and do not need splicing before translation even though they host circRNAs. It is, therefore, interesting to explore whether host splicing machinery is somehow recruited to viral transcripts to specifically produce circRNAs. The circRNAs that span multiple genes do not seem unusual, in DNA viruses at least, and thus these circRNAs should be taken into account when screening for virus-encoded circRNAs, particularly at or around packed ORFs or regions coding for polycistronic transcripts.
3.3.3. Use of alternative splicing sites and variable ends of BSJs of virus-encoded circRNAs
The typical circRNA identification computational pipeline includes the use of canonical splice site of GU/AG as a criterion [11–13,37]. There are, however, notable exceptions in virus-encoded circRNAs. EBV’s circBHLF1 was originally detected as the BSJ was flanked by GU/AG, but through divergent PCR, the authors found a circBHLF1 isoform (termed circBHLF1-alt) that uses a non-canonical splice donor nine nucleotides upstream of the original circBHLF1 [11] (Fig. 3A). During lytic reactivation of the EBV-positive cell line Akata, circBHLF1-alt-originated reads were three-fold more than from circBHLF1. Given that circBHLF1 has the highest expression among EBV-encoded circRNAs (Fig. 3C), these results suggest that the circa that uses alternative splice sites can be the dominant form of virus-encoded circRNAs in certain conditions.
Possibly by similar mechanism or a lack of precise control in back-splicing, some KSHV-encoded circRNAs are reported to have variable BSJs [9,10]. Toptan et al and our group found head-to-tail BSJ reads at KSHV PAN/K7.3 region. There are more than 30 variants of circRNAs ranging from 200 ~900 nucleotides in this ~1kb PAN/K7.3 region. Though each variant has low read counts, PAN/K7.3 is the most abundant region in virus-encoded circRNAs in aggregate. Back-splicing junctions of circPAN/K7.3 are highly variable, lacking obvious sequence motifs like canonical splicing donor/acceptor bases. PAN is a viral lncRNA transcribed by pol II and can be the most abundant transcript during lytic reactivation, but was not previously known to be spliced [40]. Importantly, expression of certain circPAN isoforms was confirmed by BaseScope in situ hybridization in KSHV-positive cell lines. Therefore, it is unlikely to be mere artifact during library constructions [25]. Similar variabilities were observed in other KSHV circRNAs located at ORF35, 36, or ORF61, all of which are part of polycistronic transcripts. BSJs were cloned and Sanger sequencing was performed to evaluate the variability of the BSJs [9]. We do not know whether these virus-encoded circRNAs produced by these cryptic back-splicing events are intentional viral factors or mere byproducts of gene expression from dense viral genomes. Functional studies are needed to answer the question. However, this BSJ variability suggests that specific BSJs are not important for the potential functions of these virus-encoded circRNAs, but rather that circRNA sequences outside of the BSJs are the more functionally relevant sequences.
4. Current challenges
Virus-encoded circRNAs are thus identified and validated in various systems and viruses, but the field is still in early stages. Unique features of viral genomes require modified and optimized ways to comprehensively screen virus-encoded circRNAs. Functional analysis is missing for most circRNAs. In this section, we discuss specific challenges and potential solutions.
4.1. Comprehensive cataloguing of virus-encoded circRNAs
Multiple back-splice isoforms of circRNAs have been detected from human transcripts with multiple exons, but for virus-encoded circRNAs, one cannot ignore ORF-spanning back-spliced reads. Also, apparent back-splice sites within known viral ORFs should not be discarded as observed in numerous examples discussed in Section 3. This causes a challenge to select genuine circRNA-originated sequencing reads from noise. Too strict of a selection method based on canonical splicing sites (based on previous poly-A RNA sequencing), may lead to the loss of relevant isoforms of virus-encoded circRNAs as seen in circBHLF1 and circBHLF1-alt that could be detected only after RT-PCR with divergent primers [11]. Thus, validation assays with Sanger sequencing and BaseScope are crucial.
One potential problem that creates uncertainty in circRNA detection is the reliance on short-read sequencing technologies. Library preparation requires ligation steps that might introduce artifacts and circRNA-Seq gives only distinct information of the flanking BSJs. Recent third-generation long-read sequencing techniques like Oxford Nanopore [41] may serve as more reliable identification tools by sequencing circRNAs without reverse transcription. Recently, a technique to sequence full-length circRNAs, termed isoCirc, utilizing reverse-transcription, rolling-circle amplification methods, and Nanopore sequencing [42]. Though this method is promising, long read, direct sequencing also has merit, raising the chance to discover previously unknown splicing isoforms in herpesvirus transcripts [43] and can detect RNA modifications such as m6A. Direct long read RNA sequencing may be applied to circRNAs by nicking circRNAs with hydrolysis [23], adding poly-A tails to nicked ends, and then continuing with 3’ capture and Nanopore sequencing.
4.2. Targeting circRNAs, but not linear RNAs
To detect (Taqman probe or BaseScope) or target circRNAs (siRNAs, LNA gapmers, or CRISPR-mediated mutation of back-splice sites) specifically, BSJs have to be well defined since BSJs are the only unique sequence compared to linear counterparts in general. Variabilities at BSJs and use of non-canonical back-splice sites (Sections 3.1 and 3.2) pose challenges to target specific virus-encoded circRNAs. For example, circBHLF1 and circBHLF1-alt vary only 9 nucleotides and may be redundant in function [11]. Detection or knock-down/out of both isoforms may be necessary for proper loss-of-function study. This complication would be further problematic for hypervariable circRNAs such as circPAN/K7.3 where more than 30 isoforms are detected. One possible approach would be to first identify more abundant isoforms and then multiplex knock-down reagents to target multiple forms simultaneously. Such dominant isoforms can be assessed by circRNA-Seq or, potentially by deep-sequence the amplicons from divergent RT-PCR (Section 2). Simultaneous suppression of multiple isoforms could be accomplished by using a pool of siRNAs or guide RNAs for the CRISPR-Cas13 method [33]; each siRNA or guide RNA would be designed to target one of the major isoforms. These pools could potentially degrade the most abundant isoforms. Finally, making genetic mutations to disrupt a specific BSJ will likely leave related circRNA isoforms unaltered, complicating genetic loss-of-function experimental designs to assess virus-encoded circRNA functions.
5. Conclusions
Viral non-coding RNAs are of keen interest in virology, particularly for DNA viruses. Some lncRNAs like PAN in KSHV are essential for viral life cycle and viral miRNAs play crucial roles to maintain latency and survival of infected cells in vitro and in vivo. Virus-encoded circRNAs are a potential new addition to the group: their gene-regulatory potentials are already intriguing as observed in EBV, KSHV, and HPV, even at this early stage of the research. Virus-encoded circRNAs have longer half-life due to its closed-circular nature and resistance to exonucleases [25], but the lack of poly(A) might have further implications during KSHV infection. For example, KSHV’s SOX protein causes general degradation of human mRNAs depending on poly(A) and poly(A) binding protein. Therefore, circRNAs might evade this host shut-off mechanism. Thus, the relatively higher stability of viral circRNAs also make them attractive targets to follow.
The effort to identify virus-encoded circRNAs started as analogy to human circRNAs, but we have seen many peculiarities that necessitate new techniques optimized for viruses. Because of this problem, at least partially, functional evidence for virus-encoded circRNAs is still limited in vitro, let alone in vivo. Exploring methods to identify circRNAs comprehensively and accurately, to quantify them, and to determine their functions in infected conditions are needed to fully understand the potential of virus-encoded circRNAs.
Highlights:
Multiple DNA viruses were found to encode circular RNAs.
Virus-encoded circRNAs have distinct features compared to human circRNAs.
More functional studies are needed to understand the potential of viral circRNAs.
Acknowledgements:
This work was supported by the Intramural Research Program of the Center for Cancer Research, NCI, NIH (1ZIABC011176). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We would like to acknowledge Anna Serquiña for critical reading of this manuscript and Guruswamy Mahesh for support in data collection. Figures were created with BioRender.com.
Abbreviations:
- circRNA
circular RNA
- EBV
Epstein-Barr virus
- KSHV
Kaposi sarcoma herpesvirus
- HPV
human papilloma virus
- rLCV
Rhesus lymphocryptovirus
- HDV
hepatitis delta virus
- RBP
RNA-binding protein
- ncRNA
non-coding RNA
- miRNA
microRNA
- lncRNA
long non-coding RNA
- ORF
open-reading frame
- BSJ
back-splice junction
- RPAD
RNase R treatment, polyadenylation, and poly(A)+ RNA depletion
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- [1].Sanger HL, Klotz G, Riesner D, Gross HJ, Kleinschmidt AK, Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures, Proc. Natl. Acad. Sci. U.S.a 73 (1976) 3852–3856. 10.1073/pnas.73.11.3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tsagris EM, Martínez de Alba AE, Gozmanova M, Kalantidis K, Viroids, Cell Microbiol. 10 (2008) 2168–2179. 10.1111/j.1462-5822.2008.01231.x. [DOI] [PubMed] [Google Scholar]
- [3].Kos A, Dijkema R, Arnberg AC, van der Meide PH, Schellekens H, The hepatitis delta (delta) virus possesses a circular RNA, Nature. 323 (1986) 558–560. 10.1038/323558a0. [DOI] [PubMed] [Google Scholar]
- [4].Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, Kjems J, The biogenesis, biology and characterization of circular RNAs, Nat Rev Genet. 20 (2019) 675–691. 10.1038/s41576-019-0158-7. [DOI] [PubMed] [Google Scholar]
- [5].Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, et al. , Natural RNA circles function as efficient microRNA sponges, 495 (2013) 384–388. 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- [6].Hansen TB, Wiklund ED, Bramsen JB, Villadsen SB, Statham AL, Clark SJ, et al. , miRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA, Embo J. 30 (2011) 4414–4422. 10.1038/emboj.2011.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, et al. , Circular RNAs are a large class of animal RNAs with regulatory potency, Nature. 495 (2013) 333–338. 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- [8].Tagawa T, Serquiña A, Kook I, Ziegelbauer J, Viral non-coding RNAs: Stealth strategies in the tug-of-war between humans and herpesviruses, Semin Cell Dev Biol. 13 (2020) 607. 10.1016/j.semcdb.2020.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Tagawa T, Gao S, Koparde VN, Gonzalez M, Spouge JL, Serquiña AP, et al. , Discovery of Kaposi’s sarcoma herpesvirus-encoded circular RNAs and a human antiviral circular RNA, Proc Natl Acad Sci USA. 115 (2018) 12805–12810. 10.1073/pnas.1816183115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Toptan T, Abere B, Nalesnik MA, Swerdlow SH, Ranganathan S, Lee N, et al. , Circular DNA tumor viruses make circular RNAs, Proc Natl Acad Sci USA. 115 (2018) E8737–E8745. 10.1073/pnas.1811728115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ungerleider N, Concha M, Lin Z, Roberts C, Wang X, Cao S, et al. , The Epstein Barr virus circRNAome, PLoS Pathog. 14 (2018) e1007206. 10.1371/journal.ppat.1007206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhao J, Lee EE, Kim J, Yang R, Chamseddin B, Ni C, et al. , Transforming activity of an oncoprotein-encoding circular RNA from human papillomavirus, Nat Commun. 10 (2019) 18. 10.1038/s41467-019-10246-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Huang J-T, Chen J-N, Gong L-P, Bi Y-H, Liang J, Zhou L, et al. , Identification of virus-encoded circular RNA, Virology. 529 (2019) 144–151. 10.1016/j.virol.2019.01.014. [DOI] [PubMed] [Google Scholar]
- [14].Ajiro M, Zheng Z-M, Oncogenes and RNA splicing of human tumor viruses, Emerg Microbes Infect. 3 (2014) e63–16. 10.1038/emi.2014.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Xiao M-S, Wilusz JE, An improved method for circular RNA purification using RNase R that efficiently removes linear RNAs containing G-quadruplexes or structured 3’ ends, Nucleic Acids Res. 47 (2019) 8755–8769. 10.1093/nar/gkz576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pandey PR, Rout PK, Das A, Gorospe M, Panda AC, RPAD (RNase R treatment, polyadenylation, and poly(A)+ RNA depletion) method to isolate highly pure circular RNA, Methods. (2018). 10.1016/j.ymeth.2018.10.022. [DOI] [PMC free article] [PubMed]
- [17].Zhang X-O, Dong R, Zhang Y, Zhang J-L, Luo Z, Zhang J, et al. , Diverse alternative back-splicing and alternative splicing landscape of circular RNAs, Genome Research. 26 (2016) 1277–1287. 10.1101/gr.202895.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. , STAR: ultrafast universal RNA-seq aligner, Bioinformatics. 29 (2012) bts635–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gao Y, Zhang J, Zhao F, Circular RNA identification based on multiple seed matching, Briefings in Bioinformatics. 19 (2018) 803–810. 10.1093/bib/bbx014. [DOI] [PubMed] [Google Scholar]
- [20].Li H, Durbin R, Fast and accurate short read alignment with Burrows-Wheeler transform, Bioinformatics (Oxford, England). 25 (2009) 1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hansen TB, Improved circRNA Identification by Combining Prediction Algorithms, Front Cell Dev Biol. 6 (2018) 20. 10.3389/fcell.2018.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Veeneman BA, Shukla S, Dhanasekaran SM, Chinnaiyan AM, Nesvizhskii AI, Two-pass alignment improves novel splice junction quantification, Bioinformatics (Oxford, England). 32 (2016) 43–49. 10.1093/bioinformatics/btv642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Capel B, Swain A, Nicolis S, Hacker A, Walter M, Koopman P, et al. , Circular transcripts of the testis-determining gene Sry in adult mouse testis, Cell. 73 (1993) 1019–1030. 10.1016/0092-8674(93)90279-y. [DOI] [PubMed] [Google Scholar]
- [24].Bejugam PR, Das A, Panda AC, Seeing Is Believing: Visualizing Circular RNAs, ncRNA. 6 (2020) 45. 10.3390/ncrna6040045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Abere B, Li J, Zhou H, Toptan T, Moore PS, Chang Y, Kaposi’s Sarcoma-Associated Herpesvirus-Encoded circRNAs Are Expressed in Infected Tumor Tissues and Are Incorporated into Virions, mBio. 11 (2020) 1865. 10.1128/mBio.03027-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Baker A-M, Huang W, Wang X-MM, Jansen M, Ma X-J, Kim J, et al. , Robust RNA-based in situ mutation detection delineates colorectal cancer subclonal evolution, Nat Commun. 8 (2017) 1998–8. 10.1038/s41467-017-02295-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kramer MC, Liang D, Tatomer DC, Gold B, March ZM, Cherry S, et al. , Combinatorial control of Drosophila circular RNA expression by intronic repeats, hnRNPs, and SR proteins, Genes Dev. 29 (2015) 2168–2182. 10.1101/gad.270421.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Petkovic S, Müller S, RNA circularization strategies in vivo and in vitro, Nucleic Acids Res. 43 (2015) 2454–2465. 10.1093/nar/gkv045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wesselhoeft RA, Kowalski PS, Anderson DG, Engineering circular RNA for potent and stable translation in eukaryotic cells, Nat Commun. 9 (2018) 2629–10. 10.1038/s41467-018-05096-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chen YG, Chen R, Ahmad S, Verma R, Kasturi SP, Amaya L, et al. , N6-Methyladenosine Modification Controls Circular RNA Immunity, Molecular Cell. 76 (2019) 96–109. 10.1016/j.molcel.2019.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wesselhoeft RA, Kowalski PS, Parker-Hale FC, Huang Y, Bisaria N, Anderson DG, RNA Circularization Diminishes Immunogenicity and Can Extend Translation Duration In Vivo, Molecular Cell. 74 (2019) 508–520. 10.1016/j.molcel.2019.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chen YG, Kim MV, Chen X, Batista PJ, Aoyama S, Wilusz JE, et al. , Sensing Self and Foreign Circular RNAs by Intron Identity, Molecular Cell. 67 (2017) 228–238. 10.1016/j.molcel.2017.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Li S, Li X, Xue W, Zhang L, Yang L-Z, Cao S-M, et al. , Screening for functional circular RNAs using the CRISPR-Cas13 system, Nat Meth. 18 (2021) 51–59. 10.1038/s41592-020-01011-4. [DOI] [PubMed] [Google Scholar]
- [34].Liu Q, Shuai M, Xia Y, Knockdown of EBV-encoded circRNA circRPMS1 suppresses nasopharyngeal carcinoma cell proliferation and metastasis through sponging multiple miRNAs, Cmar. 11 (2019) 8023–8031. 10.2147/CMAR.S218967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lieberman PM, Epigenetics and Genetics of Viral Latency, Cell Host Microbe. 19 (2016) 619–628. 10.1016/j.chom.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ungerleider NA, Jain V, Wang Y, Maness NJ, Blair RV, Alvarez X, et al. , Comparative Analysis of Gammaherpesvirus Circular RNA Repertoires: Conserved and Unique Viral Circular RNAs, J. Virol. 93 (2019). 10.1128/JVI.01952-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ungerleider NA, Jain V, Wang Y, Maness NJ, Blair RV, Alvarez X, et al. , Comparative Analysis of Gammaherpesvirus Circular RNA Repertoires: Conserved and Unique Viral Circular RNAs, J. Virol. 93 (2019) 10110–15. 10.1128/JVI.01952-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Shi X, Wang B, Feng X, Xu Y, Lu K, Sun M, circRNAs and Exosomes: A Mysterious Frontier for Human Cancer, Molecular Therapy: Nucleic Acid. 19 (2020) 384–392. 10.1016/j.omtn.2019.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lee S, Song J, Kim S, Kim J, Hong Y, Kim Y, et al. , Selective Degradation of Host MicroRNAs by an Intergenic HCMV Noncoding RNA Accelerates Virus Production, Cell Host Microbe. 13 (2013) 678–690. 10.1016/j.chom.2013.05.007. [DOI] [PubMed] [Google Scholar]
- [40].Rossetto C, Pari G, PAN’s Labyrinth: Molecular Biology of Kaposi’s Sarcoma-Associated Herpesvirus (KSHV) PAN RNA, a Multifunctional Long Noncoding RNA, Viruses. 6 (2014) 4212–4226. 10.3390/v6114212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Jain M, Olsen HE, Paten B, Akeson M, The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community, Genome Biol. 17 (2016) 239–11. 10.1186/s13059-016-1103-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Xin R, Gao Y, Gao Y, Wang R, Kadash-Edmondson KE, Liu B, et al. , isoCirc catalogs full-length circular RNA isoforms in human transcriptomes, Nat Commun. 12 (2021) 266–11. 10.1038/s41467-020-20459-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Depledge DP, Srinivas KP, Sadaoka T, Bready D, Mori Y, Placantonakis DG, et al. , Direct RNA sequencing on nanopore arrays redefines the transcriptional complexity of a viral pathogen, Nat Commun. 10 (2019) 754–13. 10.1038/s41467-019-08734-9. [DOI] [PMC free article] [PubMed] [Google Scholar]