

Abstract
Progressive supranuclear palsy causes diverse clinical presentations, including classical Richardson’s syndrome and several variant phenotypes. Clinical trials of disease-modifying therapies have recently been completed, with more planned for the next 2 years. However, many people with progressive supranuclear palsy do not meet eligibility criteria for these clinical trials. Understanding clinical progression with different phenotypes would improve trial design and enhance the accuracy of risk–benefit and cost–benefit assessments of new treatments for progressive supranuclear palsy. We set out to determine rates of motor and cognitive progression of possible, probable and definite progressive supranuclear palsy, with different phenotypes, from a representative cohort in a regional UK healthcare service. Longitudinal clinical data from people with Richardson’s syndrome and variant phenotypes were analysed using linear mixed-modelling, using both the full and modified versions of the Progressive Supranuclear Palsy Rating Scale, Mini-Mental State Examination and the revised Addenbrooke’s Cognitive Examination. Subgroup analyses considered patients meeting recent Phase II trial entry criteria and patients with neuropathological confirmation. Two hundred and twenty-seven patients [male = 59%, mean age (±standard deviation), 71.8 (±7.0) years] were followed for a mean 21.6 (±15.6) months. One hundred and seventy-four (77%) had Richardson’s syndrome at the outset, 25 had cortical variant presentations (13%, frontal, corticobasal, speech and language variants) and 28 had subcortical variant presentations (14%, parkinsonism, postural instability and gait freezing variants). Across all participants, annual progression in Richardson’s syndrome was faster than variant phenotypes on the Mini-Mental State Examination (−1.8 versus −0.9/year, P = 0.005) and revised Addenbrooke’s Cognitive Examination (−5.3 versus −3.0/year, P = 0.01) but not the Progressive Supranuclear Palsy Rating Scale (9.0 versus 7.1/year, P = 0.2) nor the modified Progressive Supranuclear Palsy Rating Scale (2.7 versus 2.3/year, P = 0.4). However, for those with more than 1 years’ follow-up, a significant difference was observed between Richardson’s syndrome and variant phenotypes in Progressive Supranuclear Palsy Rating Scale (8.7 versus 6.3/year, P = 0.04). Survival was longer in variant phenotypes than Richardson’s syndrome [7.3 (±3.9) versus 5.6 (±2.0) years, P = 0.02]. Pathologically confirmed cases (n = 49) supported these findings. Patients meeting basic trial-eligibility criteria (n = 129) progressed faster on the Progressive Supranuclear Palsy Rating Scale than trial-not-eligible patients (10.1 versus 6.1/year, P = 0.001). In conclusion, phenotypes other than Richardson’s syndrome show slower progression and longer survival. Trial criteria do not select representative progressive supranuclear palsy cases. This has implications for trial design, and application of trial results to clinically more diverse patient populations.
Keywords: progressive supranuclear palsy, heterogeneity, selection bias, prognosis, Richardson’s syndrome
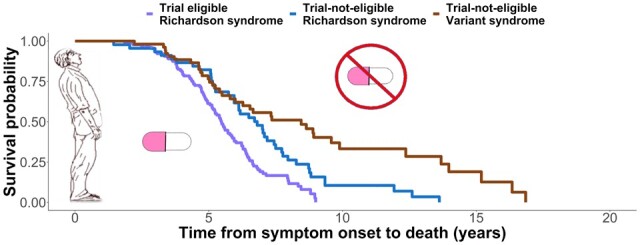
Street et al. show that progression and survival with progressive supranuclear palsy vary according to people’s symptoms and clinical trial eligibility. Common clinical trial criteria exclude many patients. Their exclusion may limit the relevance of clinical trials and affect the relative risks and benefits of future treatments for many people.
Graphical Abstract
Graphical Abstract.
Introduction
When the International Movement Disorders Society endorsed study group for progressive supranuclear palsy (PSP) revised the clinical diagnostic criteria, a major aim was to improve patient access to clinical trials.1 The revision also succeeded in increasing diagnostic sensitivity to clinical phenotypes other than the classical ‘Richardson’s syndrome’.2,3 The ‘variant’ presentations of PSP include those with predominant gait freezing, parkinsonism, behavioural/dysexecutive or frontal, oculomotor, speech and language, corticobasal or postural instability features, without the combination of classical symptoms to meet Richardson’s syndrome criteria. Approximately half of people with PSP pathology may present with a variant phenotype,4,5 which can be divided broadly into ‘cortical’ (e.g. frontal, corticobasal, speech and language) and ‘subcortical’ groups (e.g. parkinsonism, predominant gait freezing, postural instability, oculomotor).4–6 Clinical progression and the distribution of neuropathology differs between Richardson’s syndrome, cortical and subcortical phenotypes.5,7 The prognosis of Richardson’s syndrome is poor, with typical survival 5–7 years from onset, or 3–4 years from diagnosis,8–13 but less is known about prognosis in phenotypic variants.
Several large-scale multicentre clinical trials of disease-modifying therapies have been undertaken in PSP,14,15 with several more Phase II studies planned (e.g. NCT04008355, NCT04184063, NCT03446807, NCT04253132). However, these trials have been constrained by (i) potential ascertainment biases, (ii) restricted phenotypes and (iii) reliance on clinical diagnostic criteria with long delays to pathological confirmation. Together, these factors may impair the generalizability of clinical trial outcomes even when a future compound proves effective to slow the course of disease. Our study therefore considers the rate of progression over a range of PSP phenotypes identified in a regional healthcare service, using common measures of disease severity.
We consider the participants in three groups: (i) a full natural history cohort, clinically representative of the regional UK healthcare service, (ii) a subset meeting principal inclusion/exclusion criteria to a recent multicentre clinical trial (NCT03068468) and (iii) a subset with neuropathological confirmation. Recent clinical trials have combined inclusion and exclusion criteria that effectively exclude a large proportion of patients with symptomatic PSP principally due to restrictive diagnostic (e.g. Richardson’s syndrome only) and disease course (e.g. presence of symptoms for <5 years) specifications. Notwithstanding the negative results of recent Phase II/III trials, they raise the issue of generalizability of successful outcomes to a broader range of phenotypes and stages. Specifically, if the rate of progression of different phenotypes differs, then the risk–benefit ratio and cost–benefit ratio of a treatment may differ.
For each of the three groups, we evaluate progression in terms of widely used measures of motor, cognitive and global function. The PSP Rating Scale (PSPRS)16 is widely used in clinical practice, observational cohorts and clinical trials. Annual progression of the total score (0–100) is commonly reported at 9–12 points in Richardson’s syndrome,17 with higher progression scores accurately predicting poorer survival in meta-analysis of multiple studies.8 However, the standard 28-item PSPRS has also been criticized for inclusion of items that are not patient-centred. A modified version (mPSPRS) uses a subset of 14 items focused on more clinically meaningful and patient-relevant outcomes although missing several cognitive and behavioural items.18 Cognitive and behavioural changes are common in PSP, with apathy, impulsivity and language or executive dysfunction preceding or accompanying classical motor features.19–24 Such cognitive features of PSP predict survival.8,9,25–27 In contrast to subcortical variants of PSP phenotypes, the rate of progression of cortical variant phenotypes inferred from cross-sectional data is similar to Richardson’s syndrome5 although survival may be longer.28 For this study we used Mini-Mental State Examination (MMSE) and revised Addenbrooke’s Cognitive Examination (ACE-R) scores as markers of cognitive function.
Here, we assess the impact of cohort selection and phenotypic variance on the rate of disease progression. We predicted that those eligible for clinical trials progress differently to the wider spectrum of people with PSP, but that the clinicopathological correlations are high enough that the progression of non-trial participants is not a result of diagnostic error.
Materials and methods
Participants
The study is set in the National Health Service (NHS) specialist PSP clinic in Cambridge, UK. This clinic receives referrals from primary care sources as well as movement disorders, cognitive disorders and gerontological secondary care services. Between June 2007 and November 2019, 227 patients were assessed with consent for their data to be used for observational research; and retrospectively identified as fitting 2017 Movement Disorders Society Criteria1 for a diagnosis of ‘probable’ or ‘possible’ PSP. Data from clinical care records (with consent) were augmented by data from the observational studies including the epidemiological Pick’s Disease and Progressive Supranuclear Palsy: Prevalence and Incidence protocol (PIPPIN)10 and the ‘Diagnosis and prognosis in PSP and CBD’ protocol.17 The Cambridge University Hospitals NHS Trust uses EPIC Healthcare records since 2014, with paper records and alternative electronic medical records before 2014. Cases of multiple PSP diagnoses at presentation were resolved under the hierarchical multiple allocations extinction criteria.29 The census date was set as 9 February 2021.
Neuropathological confirmation was available for 49 patients who donated their brain to the Cambridge Brain Bank. ‘Variant PSP’ cases include all those with a PSP-phenotype other than Richardson’s syndrome at first clinic attendance, acknowledging that the majority of variant cases progress to Richardson’s syndrome during the course of disease.2
We repeated the principal analyses after additional application of the general inclusion/exclusion criteria from a recent Phase II clinical trial (NCT03068468) using medical records including known medical conditions, prior or concomitant drug therapies and laboratory test findings. These general criteria are listed in Supplementary Table 1 and are similar to other recent trials of disease-modifying therapies in PSP (NCT04253132, NCT02985879, NCT04185415). Independent ambulation was defined according to a score on the Gait section of the first presentation PSPRS of ≤3.
The study was conducted in accordance with the 1964 Helsinki declaration and fulfilled the criteria of the STROBE Statement. Participants gave written, informed consent. Local ethical approval was granted for the ‘Pick’s Disease and Progressive Supranuclear Palsy Prevalence and Incidence’ protocol (12/EE/0475) by the East of England Cambridge Central Research Ethics Committee in October 2015 and the ‘Diagnosis and prognosis in Progressive Supranuclear Palsy and Corticobasal Degeneration’ protocol (07/Q0102/3) in March 2007 by the East of England Essex Research Ethics Committee. The neuropathological data were obtained with ethical approval from the Health Research Authority, NHS England (IRAS—202 802, ‘Neurodegeneration Research in Dementia’). Where mental capacity was lost later in the course of the illness, or after death, data were retained in accordance with the original consent.
Statistical analysis
Electronic data capture tools were hosted at the University of Cambridge for curation and management of data.30,31 R studio (version 4.0.3, R Core Team, 2020) was used for the analysis of demographic and clinical data. Missing observations in individual subscores constituted <2% of the dataset and were imputed using the Multiple Imputation via Chained Equations (mice) package.32 Longitudinal annualized progression was estimated by construction of a generalized linear mixed-effects model in R studio using the ‘lme4’ package.33 Total PSPRS, mPSPRS, MMSE or ACE-R scores were included in separate models as dependent variables, with fixed effects of the first corresponding score and time interval in years for each follow-up visit from baseline (without interaction terms). Random effects were tested, under the assumption that intercepts and slope may differ between subjects. Neither normality nor homoscedasticity of residual plots was deviated from. For each clinical/cognitive outcome, the annual rate of change is represented by the estimated slope of the linear relationship between test-specific scores and time, accounting for individual differences.
The Shapiro–Wilk test was used to assess for normality of data distribution. Comparison of means was performed using the independent samples t-test when normally distributed and the Kruskal–Wallis test when not normally distributed. Mean values of continuous data are expressed with their associated standard deviations and mean values of progression data with associated 95% confidence intervals (CIs). Categorical data were compared using the Chi square test. Variables within the survival model are detailed along with their associated hazard ratios (HRs) and 95% CI. The log-rank test was used to compare survival curves. For all analyses, significance level was adjusted by Bonferroni’s correction and a P-value <0.05 was considered significant. The ‘survival’34 and ‘survminer’35 packages in R studio were used to construct Cox Survival regression models. The ‘BayesFactor’36 package was used for supplementary Bayesian analysis.
Data availability
Anonymized derived data will be available upon reasonable request to the senior author, for academic non-commercial purposes, subject to potential limitations to protect participant confidentiality and maintain General Data Protection Regulation compliance.
Results
Demographics and visit statistics
Demographic data are presented in Table 1 and Supplementary Table 2 with frequentist tests. The Bayesian analysis of demographic data is available in Supplementary Table 3. The median number of assessments was 3, maximum 12. Sixty patients had only one PSPRS recorded during the data capture period. Such single attenders were older [73.5 ± 7.7 years versus 71.2 ± 6.7 years, χ2 (1) = 4.4, P = 0.04], and more severe at presentation [PSPRS 41.9 ± 16.1 versus 36.2 ± 13.7, χ2 (1) = 5.5, P = 0.02] and were more likely to have a Richardson’s syndrome diagnosis [88% versus 73%, χ2 (1) = 5.9, P = 0.02] than multiple attenders. Single and multiple attenders were similar in time from symptom onset to first attendance [3.6 ± 2.1 versus 3.5 ± 2.3 years, t(114) = −0.3, P = 0.8] and sex [60% male versus 58% male, χ2 (1) = 0.1, P = 0.8]. Single attenders were included in the cohort analysis to represent our natural history population more accurately. ‘Variant PSP’ case diagnoses are shown in Fig. 1. Of these, the majority had converted clinically to Richardson’s syndrome by study end-point (41/53, 77%).
Table 1.
Clinical and demographic characteristics of the study population by disease phenotype
| Clinical diagnosis of PSP |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| All PSP | PSPRS | All Variant | Cortical | Subcortical | RS vs variant P-value |
RS vs cortical P-value |
RS vs subcortical P-value |
Cortical vs subcortical | |
| Mean ± SD | Mean ± SD | Mean ± SD | Mean ± SD | Mean ± SD | |||||
| (n = 227) | (n = 174) | (n = 53) | (n = 20) | (n = 28) | |||||
| Age at first visit (years) | 71.8 ± 7.0 | 71.8 ± 7.4 | 71.8 ± 5.9 | 72.3 ± 5.7 | 71.5 ± 6.2 | 1 | 0.7 | 0.8 | 0.7 |
| Age at onset (years) | 68.4 ± 7.0 | 68.8 ± 7.1 | 67.4 ± 6.7 | 68.9 ± 6.1 | 66.3 ± 7.1 | 0.3 | 0.9 | 0.1 | 0.2 |
| Age range at first visit (years) | 48–92 | 48–92 | 59–82 | 60–80 | 59–82 | ||||
| Duration from onset to first visit (years) | 3.4 ± 2.0 | 3.1 ± 1.7 | 4.4 ± 2.8 | 3.4 ± 1.5 | 5.2 ± 3.3 | 0.001 | 0.3 | <0.001 | 0.01 |
| Duration of clinic follow-up (months) | 21.6 ± 15.6 | 20.0 ± 14.3 | 26.4 ± 18.5 | 21.1 ± 11.9 | 30.0 ± 22.1 | 0.05 | 0.4 | 0.1 | 0.2 |
| Sex (male/female) (%) | 133/94 (59/41) | 99/75 (57/43) | 34/19 (64/36) | 13/7 (65/35) | 19/9 (68/32) | 0.4 | 0.6 | 0.4 | 1 |
|
| |||||||||
| Survival according to clinical diagnosis | |||||||||
|
|
|||||||||
| All PSP | PSPRS | Variant | Cortical | Subcortical |
RS vs variant
P-value |
RS vs cortical
P-value |
RS vs subcortical
P-value |
Cortical vs subcortical
P-value |
|
| Mean ± SD | Mean ± SD | Mean ± SD | Mean ± SD | Mean ± SD | |||||
| (n = 177) | (n = 141) | (n = 36) | (n = 15) | (n = 17) | |||||
|
| |||||||||
| Duration from onset to death (years) | 6.0 ± 2.6 | 5.6 ± 2.0 | 7.3 ± 3.9 | 6.1 ± 2.3 | 8.7 ± 5.0 | 0.02 | 0.7 | <0.001 | 0.07 |
| Duration from first visit to death (years) | 2.5 ± 1.5 | 2.5 ± 1.5 | 2.6 ± 1.7 | 2.5 ± 1.4 | 3.0 ± 2.1 | 0.6 | 1.0 | 0.4 | 0.4 |
| Age at death (years) | 74.3 ± 6.7 | 74.2 ± 6.8 | 74.6 ± 6.2 | 75.2 ± 6.3 | 74.0 ± 6.7 | 0.7 | 0.9 | 1 | 0.6 |
| Sex (male/female) (%) | 104/73 (59/41) | 82/59 (58/42) | 22/14 (61/39) | 8/7 (53/47) | 12/5 (71/29) | 0.9 | 0.9 | 0.5 | 0.5 |
PSP = progressive supranuclear palsy; RS = Richardson’s syndrome; SD = standard deviation.
Figure 1.

Variation in clinical diagnosis between presentation and end-point of study. Predominantly cortical and subcortical categories are indicated along with overall number of patients and percentage of whole in individual segments. bvFTD = behavioural variant frontotemporal dementia; CBS = corticobasal syndrome; F = frontal; P =Parkinsonism; PGF = predominant gait freezing; PI = postural instability; PSP = progressive supranuclear palsy; RS = Richardson’s syndrome; SL = speech and language.
One hundred and twenty-nine of the 227 participants (56%) were deemed likely to have met criteria for the Phase II clinical trial at first presentation. The principal reasons for not meeting criteria were a variant diagnosis at first presentation, lack of independent ambulation, presence of PSP symptoms for more than 5 years and MMSE <20. Forty-nine people had post-mortem confirmation of PSP pathology. One individual had post-mortem confirmation of an alternative primary pathology (alpha-synuclein) and was excluded from analysis.
PSP onset and survival
Symptom duration and survival data are displayed in Table 1 and Supplementary Table 2. Variant diagnoses presented to clinic over a year later after symptom onset than Richardson’s syndrome but the duration of follow-up to last clinical review was similar. This effect was driven by the subcortical group which presented on average 2 years later than the Richardson’s syndrome group whereas the cortical group presented to clinic after a similar interval. Similar statistically significant absolute differences were observed in the pathologically confirmed subgroup between subcortical and Richardson’s syndrome diagnoses.
One hundred and seventy-seven patients (78%) had died by the census date, with a global mean survival of 6 years. Variant and Richardson’s syndrome groups were similar in terms of age at death and duration from first visit to death, although survival from first symptom was longer in the variant group, again driven by the subcortical group. Longer survival was also observed in the subcortical group of pathologically confirmed subset versus Richardson’s syndrome.
Re-analysis applied to those meeting clinical trial entry criteria is presented in Table 2 and Supplementary Table 2. One hundred and twenty-nine patients (57%) met eligibility criteria at first visit. Trial-eligible patients (‘Trial’) were younger at first visit, presented to clinic sooner after first symptom onset and had a shorter survival after first symptom onset than not-eligible (‘Non-trial’) patients. These between-group differences persisted when comparing trial-eligible Richardson’s syndrome to trial-not-eligible Richardson’s syndrome cases (n = 45) who also had a shorter follow-up and shorter survival after first visit.
Table 2.
Clinical and demographic characteristics of the study population by clinical trial eligibility
| Clinical trial eligibility |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| All PSP Mean ± SD (n = 227) | Trial cases Mean ± SD (n = 129) | Non-trial Mean ± SD (n = 98) | Non-trial RS Mean ± SD (n = 45) | Non-trial variant Mean ± SD (n = 53) | Trial vs non-trial P-value | Trial vs non-trial RS P-value | Trial vs non-trial variant, P-value | Non-trial variant vs non-trial RS P-value | |
| Age at first visit (years) | 71.8 ± 7.0 | 70.9 ± 7.1 | 73.1 ± 6.7 | 74.6 ± 7.4 | 71.8 ± 5.9 | 0.03 | 0.006 | 0.7 | 0.1 |
| Age at onset (years) | 68.4 ± 7.0 | 68.4 ± 7.0 | 68.5 ± 7.1 | 69.9 ± 7.4 | 67.4 ± 6.7 | 0.8 | 0.4 | 0.7 | 0.2 |
| Age range at first visit (years) | 48–92 | 48–84 | 57–92 | 57–92 | 59–82 | ||||
| Duration from onset to first visit (years) | 3.4 ± 2.0 | 2.5 ± 1.0 | 4.5 ± 2.5 | 4.7 ± 2.1 | 4.4 ± 2.8 | <0.0001 | <0.001 | <0.001 | 0.2 |
| Duration of clinic follow-up (months) | 21.6 ± 15.6 | 19.6 ± 14.2 | 25.2 ± 17.2 | 10.4 ± 15.1 | 19.4 ± 19.7 | 0.4 | 0.02 | 0.3 | 0.02 |
| Sex (male/female) (%) | 133/94 (59/41) | 73/56 (57/43) | 60/38 (61/39) | 26/19 (58/42) | 34/19 (64/36) | 0.6 | 1 | 0.4 | 0.7 |
|
| |||||||||
| Survival according to clinical trial eligibility | |||||||||
|
|
|||||||||
| All PSP Mean ± SD (n = 177) | Trial Mean ± SD (n = 100) | Non-trial Mean ± SD (n = 77) | Non-trial RS Mean ± SD (n = 36) | Non-trial variant Mean ± SD (n = 41) | Trial vs non-trial P-value | Trial vs non-trial RS P-value | Trial vs non-trial variant P-value | Non-trial variant vs non-trial RS P-value | |
|
| |||||||||
| Duration from onset to death (years) | 6.0 ± 2.6 | 5.2 ± 1.6 | 6.9 ± 3.3 | 6.6 ± 2.6 | 7.3 ± 3.9 | <0.001 | 0.005 | <0.001 | 0.4 |
| Duration from first visit to death (years) | 2.5 ± 1.5 | 2.7 ± 1.4 | 2.3 ± 1.6 | 1.9 ± 1.3 | 2.6 ± 1.7 | 0.07 | 0.007 | 0.6 | 0.1 |
| Age at death (years) | 74.3 ± 6.7 | 73.4 ± 6.6 | 75.4 ± 6.8 | 76.0 ± 7.2 | 74.6 ± 6.2 | 0.06 | 0.1 | 0.6 | 0.6 |
| Sex (male/female) (%) | 104/73 (59/41) | 58/42 (58/42) | 46/31 (60/40) | 22/14 (61/39) | 24/17 (59/41) | 0.9 | 1 | 0.9 | 1 |
PSP = progressive supranuclear palsy; RS = Richardson’s syndrome; SD = standard deviation; Trial cases refers to the subset deemed likely eligible to recent Phase II clinical trial.
Kaplan–Meier curves and log-rank comparisons for all patients from symptom onset to death with breakdown according to phenotype are presented in Fig. 2. Variant subtype (HR 0.47, 95% CI 0.33–0.66, P < 0.001) and the subcortical phenotype (HR 0.37, 95% CI 0.20–0.68, P = 0.001) were strong predictors of improved survival versus Richardson’s syndrome. No additional effects of age, sex or first PSPRS score were observed. Comparing those who did, versus did not, meet criteria for clinical trial, there was a survival advantage for members of the non-trial group (HR 0.38, 95% CI 0.27–0.53, P < 0.001), indicating that those typical of clinical trials have a more aggressive disease course. Male sex (HR 1.93, 95% CI 1.02–3.69, P = 0.044) was associated with poorer survival in the group with neuropathology.
Figure 2.

Survival analysis from symptom onset to death. Analysis was performed using a Cox regression model split according to diagnostic groups with associated number at risk tables below each plot. Displayed P-values represent pairwise log-rank comparisons with correction for multiple comparisons. (A) PSP-Richardson’s syndrome (PSPRS) and variant groups. (B) PSPRS, PSP-cortical (PSP-C) and PSP-subcortical (PSP-SC). (C) Trial eligible and trial-not-eligible groups. (D) Trial eligible Richardson’s syndrome [Trial (RS)], trial-not-eligible Richardson’s syndrome [Non-trial (RS)] and trial-not-eligible variant syndrome [Non-trial (Variant)].
Rate of progression
Progression data are displayed in Tables 3–5 and Supplementary Table 4. In terms of the PSPRS, the Richardson’s syndrome patients progressed faster than variant patients by ∼2 points/year. Cortical patients progressed ∼2 points/year faster than subcortical patients. Similar rates were found in the pathologically confirmed cases of Richardson’s syndrome and variants. Faster progression in Richardson’s syndrome versus variant phenotypes was not significant in the analysis of all patients but it was significant for patients with more than 12 months follow-up. Progression in the cortical group was neither significantly different from Richardson’s syndrome nor the cortical group. Progression of subscales of the PSPRS did not differ between groups.
Table 3.
Rates of progression in disease severity by clinical phenotype
| Clinical diagnosis of PSP |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Baseline Mean ± SD (n = 227) | All patients SPE, 95% CI (n = 227) | RS SPE, 95% CI (n = 174) | Variant SPE, 95% CI (n = 53) | Cortical SPE, 95% CI (n = 20) | Subcortical SPE, 95% CI (n = 28) | RS vs variant P-value | RS vs cortical P-value | RS vs subcortical P-value | Cortical vs subcortical | |
| PSPRS | ||||||||||
| History | 7.9 ± 4.4 | 1.8 (1.5–2.2) | 2.0 (1.6–2.4) | 1.3 (0.7–1.9) | 2.0 (0.9–3.2) | 0.7 (−0.02 to 1.4) | 0.1 | 0.6 | 0.05 | 1 |
| Mentation | 3.4 ± 2.9 | 0.8 (0.5–1.1) | 0.7 (0.4–1.1) | 1.0 (0.4–1.5) | 0.5 (−0.2 to 1.1) | 0.9 (0.1–1.7) | 0.8 | 1 | 0.9 | 1 |
| Bulbar | 2.3 ± 1.7 | 0.9 (0.7–1.1) | 1.0 (0.8–1.2) | 0.7 (0.5–1.0) | 0.9 (0.3–1.4) | 0.6 (0.3–0.9) | 0.7 | 1 | 1 | 1 |
| Ocular | 7.8 ± 3.6 | 1.6 (1.2–1.9) | 1.5 (1.2–1.9) | 1.7 (1.0–2.4) | 2.7 (1.2–4.2) | 0.9 (0.2–1.7) | 0.9 | 1 | 0.4 | 0.9 |
| Limb | 4.3 ± 2.8 | 0.9 (0.7–1.2) | 1.1 (0.8–1.4) | 0.6 (0.2–0.9) | 0.4 (−0.1 to 0.9) | 0.6 (−0.01 to 1.1) | 0.1 | 0.3 | 0.4 | 1 |
| Gait | 11.1 ± 4.9 | 2.4 (2.0–2.7) | 2.6 (2.1–3.0) | 1.7 (1.0–2.5) | 2.1 (0.8–3.3) | 1.3 (0.4–2.3) | 0.1 | 1 | 0.2 | 1 |
| Total | 36.8 ± 14.8 | 8.4 (7.3–9.6) | 9.0 (7.6–10.3) | 7.1 (4.9–9.4) | 7.7 (4.3–11.2) | 5.2 (2.5–7.8) | 0.2 | 1 | 0.13 | 1 |
| Total (>6 months follow-up) | (n = 143) | (n = 143) | (n = 106) | (n = 37) | (n = 14) | (n = 20) | ||||
| 34.6 ± 13.6 | 8.3 (7.2–9.5) | 8.9 (7.6–10.2) | 6.8 (4.6–9.1) | 6.9 (3.5–10.4) | 4.9 (2.3–7.6) | 0.06 | 0.9 | 0.04 | 1 | |
| Total (>12 months follow-up) | (n = 109) | (n = 109) | (n = 80) | (n = 29) | (n = 10) | (n = 16) | ||||
| 34.7 ± 13.6 | 8.1 (6.9–9.2) | 8.7 (7.5–10.0) | 6.3 (4.1–8.4) | 5.7 (2.6–8.8) | 5.2 (2.4–7.9) | 0.04 | 0.2 | 0.1 | 1 | |
| mPSPRS | ||||||||||
| History | 3.7 ± 1.8 | 0.7 (0.5–0.9) | 0.8 (0.5–1.0) | 0.5 (0.3–0.8) | 0.6 (0.1–1.0) | 0.3 (0.01–0.6) | 0.2 | 1 | 0.2 | 1 |
| Mentation | 0.9 ± 0.5 | 0.2 (0.1–0.2) | 0.2 (0.1–0.3) | 0.1 (0.1–0.2) | 0.3 (0.1–0.5) | 0.1 (0.01–0.2) | 0.6 | 1 | 1 | 1 |
| Bulbar | 2.0 ± 1.0 | 0.5 (0.4–0.6) | 0.4 (0.3–0.5) | 0.6 (0.4–0.9) | 0.5 (0.2–0.9) | 0.5 (0.1–1.0) | 1 | 1 | 0.8 | 1 |
| Ocular | 0.9 ± 0.6 | 0.1 (0.1–0.2) | 0.1 (0.1–0.2) | 0.2 (0.1–0.2) | 0.2 (0.04–0.3) | 0.1 (0.02–0.2) | 0.6 | 1 | 1 | 0.7 |
| Limb | 1.1 ± 0.6 | 0.2 (0.1–0.2) | 0.2 (0.1–0.3) | 0.1 (−0.04 to 0.2) | 0.1 (−0.1 to 0.2) | 0.1 (−0.1 to 0.2) | 0.1 | 0.9 | 0.9 | 1 |
| Gait | 5.7 ± 2.0 | 0.9 (0.7–1.0) | 0.9 (0.7–1.2) | 0.7 (0.4–1.0) | 0.8 (0.3–1.3) | 0.6 (0.1–1.0) | 0.3 | 1 | 0.6 | 1 |
| Total | 14.4 ± 4.7 | 2.6 (2.2–3.0) | 2.7 (2.2–3.2) | 2.3 (1.5–3.1) | 2.7 (1.4–3.9) | 1.8 (0.7–2.9) | 0.4 | 1 | 0.3 | 1 |
mPSPRS = modified Progressive Supranuclear Palsy Rating Scale; PSP = progressive supranuclear palsy; PSPRS = Progressive Supranuclear Palsy Rating Scale; RS = Richardson’s Syndrome; SD = standard deviation; SPE = 1-year progression estimate.
Table 5.
Rates of progression in cognition by phenotype and clinical trial eligibility
| Clinical diagnosis of PSP |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | All patients | RS | Variant | Cortical | Subcortical | RS vs variant | RS vs cortical | RS vs subcortical | Cortical vs subcortical | |
| Mean ± SD | SPE, 95% CI | SPE, 95% CI | SPE, 95% CI | SPE, 95% CI | SPE, 95% CI | |||||
| (n = 227) | (n = 227) | (n = 174) | (n = 53) | (n = 20) | (n = 28) | P-value | P-value | P-value | P-value | |
| ACE-R | ||||||||||
| Attention | 16.3 ± 2.5 | −0.9 (−1.1 to −0.6) | −1.0 (−1.3 to −0.7) | −0.6 (−1.0 to −0.3) | −0.5 (−0.9 to −0.1) | −0.5 (−1.0 to −0.1) | 0.1 | 0.7 | 0.2 | 1 |
| Memory | 20.3 ± 4.7 | −0.6 (−1.0 to −0.3) | −0.7 (−1.2 to −0.3) | −0.5 (−1.0 to 0.1) | −0.5 (−1.3 to 0.3) | −0.4 (−1.2 to 0.4) | 0.3 | 1 | 0.9 | 1 |
| Fluency | 5.0 ± 3.4 | −0.5 (−0.6 to −0.3) | −0.5 (−0.8 to −0.3) | −0.5 (−0.8 to −0.2) | −0.4 (−0.8 to −0.03) | −0.5 (−0.8 to −0.1) | 0.1 | 1 | 0.07 | 0.9 |
| Language | 22.9 ± 3.7 | −0.9 (−1.2 to −0.6) | −1.2 (−1.6 to −0.8) | −0.3 (−0.6 to −0.1) | −0.4 (−0.7 to −0.1) | −0.3 (−0.6 to −0.03) | 0.0003 | 0.05 | 0.1 | 1 |
| Visuospatial | 12.5 ± 3.4 | −1.7 (−1.9 to −1.4) | −2.0 (−2.3 to −1.6) | −0.9 (−1.4 to −0.5) | −1.3 (−2.0 to −0.5) | −0.6 (−1.2 to −0.01) | 0.002 | 0.4 | 0.001 | 0.8 |
| MMSE Total | 26.1 ± 3.8 | −1.5 (−1.9 to −1.1) | −1.8 (−2.4 to −1.2) | −0.9 (−1.3 to −0.4) | −0.8 (−1.3 to −0.2) | −0.8 (−1.4 to −0.2) | 0.005 | 0.2 | 0.03 | 1 |
| ACE-R total | 77.0 ± 13.6 | −4.6 (−5.4 to −3.7) | −5.3 (−6.4 to −4.2) | −3.0 (−4.2 to −1.8) | −2.9 (−4.6 to −1.3) | −2.5 (−4.2 to −0.8) | 0.01 | 0.3 | 0.0007 | 0.6 |
|
| ||||||||||
| Clinical trial eligibility | ||||||||||
|
|
||||||||||
| Baseline | All patients | Trial | Non-trial | Non-trial RS | Non-trial Variant | Trial vs non-trial | Trial vs non-trial RS | Trial vs non-trial variant | Non-trial variant vs non-trial RS | |
| Mean ± SD | SPE, 95% CI | SPE, 95% CI | SPE, 95% CI | SPE, 95% CI | SPE, 95% CI | |||||
| (n = 227) | (n = 227) | (n = 129) | (n = 98) | (n = 45) | (n = 53) | P-value | P-value | P-value | P-value | |
|
| ||||||||||
| ACE-R | ||||||||||
| Attention | 16.3 ± 2.5 | −0.9 (−1.1 to −0.6) | −0.9 (−1.2 to −0.5) | −0.9 (−1.2 to −0.5) | −1.6 (−2.7 to −0.4) | −0.6 (−1.0 to −0.3) | 0.5 | 1 | 0.2 | 0.05 |
| Memory | 20.3 ± 4.7 | −0.6 (−1.0 to −0.3) | −0.7 (−1.2 to −0.2) | −0.6 (−1.0 to −0.2) | −0.9 (−1.8 to −0.03) | −0.5 (−1.0 to 0.1) | 0.9 | 1 | 0.5 | 0.3 |
| Fluency | 5.0 ± 3.4 | −0.5 (−0.6 to −0.3) | −0.6 (−0.9 to −0.3) | −0.5 (−0.7 to −0.3) | −0.5 (−0.8 to −0.1) | −0.5 (−0.8 to −0.2) | 0.1 | 1 | 0.3 | 1 |
| Language | 22.9 ± 3.7 | −0.9 (−1.2 to −0.6) | −1.1 (−1.6 to −0.7) | −0.7 (−1.1 to −0.3) | −1.6 (−2.7 to −0.6) | −0.3 (−0.6 to −0.1) | 0.4 | 0.2 | 0.1 | 0.002 |
| Visuospatial | 12.5 ± 3.4 | −1.7 (−1.9 to −1.4) | −1.8 (−2.2 to −1.5) | −1.4 (−1.9 to −1.0) | −2.5 (−3.3 to −1.7) | −0.9 (−1.4 to −0.5) | 0.8 | 0.04 | 0.04 | 0.001 |
| MMSE total | 26.1 ± 3.8 | −1.5 (−1.9 to −1.1) | −1.6 (−2.1 to −1.0) | −1.3 (−1.9 to −0.8) | −2.8 (−4.8 to −0.8) | −0.9 (−1.3 to −0.4) | 0.5 | 0.7 | 0.08 | 0.01 |
| ACE-R total | 77.0 ± 13.6 | −4.6 (−5.4 to −3.7) | −4.9 (−6.1 to −3.8) | −4.2 (−5.5 to −2.8) | −6.7 (−10.0 to −3.4) | −3.0 (−4.2 to −1.8) | 0.3 | 1 | 0.01 | 0.01 |
ACE-R = revised Addenbrooke’s Cognitive Examination; MMSE = Mini-Mental State Examination; PSP = progressive supranuclear palsy; RS = Richardson’s Syndrome; SD = standard deviation; Trial cases refers to the subset deemed likely eligible to recent Phase II clinical trial; SPE = 1-year progression estimate.
Table 4.
Rates of progression in disease severity by clinical trial eligibility
| Clinical trial eligibility |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Baseline Mean ± SD (n = 227) | All patients SPE, 95% CI (n = 227) | Trial SPE, 95% CI | Non-trial SPE, 95% CI (n = 98) | Non-trial RS SPE, 95% CI (n = 45) | Non-trial variant SPE, 95% CI (n = 53) | Trial vs non-trial P-value | Trial vs non-trial RS P-value | Trial vs non-trial variant P-value | Non-trial variant vs non-trial RS P-value | |
| PSPRS | ||||||||||
| History | 7.9 ± 4.4 | 1.8 (1.5–2.2) | 2.3 (1.9–2.8) | 1.0 (0.5–1.5) | 0.4 (−0.3 to 1.1) | 1.3 (0.7–1.9) | 0.0004 | 0.4 | 0.3 | 1 |
| Mentation | 3.4 ± 2.9 | 0.8 (0.5–1.1) | 0.9 (0.5–1.2) | 0.7 (0.3–1.1) | 0.1 (−0.6 to 0.9) | 1.0 (0.4–1.5) | 0.4 | 1 | 1 | 1 |
| Bulbar | 2.3 ± 1.7 | 0.9 (0.7–1.1) | 1.0 (0.8–1.3) | 0.7 (0.5–0.9) | 0.7 (0.3–1.1) | 0.7 (0.5–1.0) | 0.2 | 1 | 1 | 1 |
| Ocular | 7.8 ± 3.6 | 1.6 (1.2–1.9) | 1.7 (1.3–2.1) | 1.3 (0.8–1.8) | 0.7 (0.2–1.2) | 1.7 (1.0–2.4) | 0.1 | 0.1 | 1 | 1 |
| Limb | 4.3 ± 2.8 | 0.9 (0.7–1.2) | 1.2 (0.8–1.5) | 0.7 (0.3–1.0) | 1.0 (0.2–1.8) | 0.6 (0.2–0.9) | 0.1 | 1 | 1 | 1 |
| Gait | 11.1 ± 4.9 | 2.4 (2.0–2.7) | 3.0 (2.4–3.5) | 1.5 (1.0–2.0) | 1.0 (0.4–1.7) | 1.7 (1.0–2.5) | 0.0006 | 0.4 | 0.1 | 1 |
| Total | 36.8 ± 14.8 | 8.4 (7.3–9.6) | 10.1 (8.6–11.5) | 6.1 (4.3–7.8) | 3.9 (1.3–6.5) | 7.1 (4.9–9.4) | 0.001 | 0.1 | 0.2 | 1 |
| Total (>6 months follow-up) | (n = 143) | (n = 143) | (n = 85) | (n = 58) | (n = 21) | (n = 37) | ||||
| 34.6 ± 13.6 | 8.3 (7.2–9.5) | 9.9 (8.5–11.3) | 6.0 (4.3–7.7) | 4.7 (2.3–7.1) | 6.8 (4.6–9.1) | 0.0005 | 0.005 | 0.04 | 1 | |
| Total (>12 months follow-up) | (n = 109) | (n = 109) | (n = 64) | (n = 45) | (n = 16) | (n = 29) | ||||
| 34.7 ± 13.6 | 8.1 (6.9–9.2) | 9.6 (8.2–11.1) | 5.8 (4.2–7.4) | 5.0 (2.6–7.4) | 6.3 (4.1–8.4) | 0.0007 | 0.01 | 0.03 | 1 | |
| mPSPRS | ||||||||||
| History | 3.7 ± 1.8 | 0.7 (0.5–0.9) | 0.9 (0.7–1.2) | 0.4 (0.2–0.6) | 0.1 (−0.5 to 0.6) | 0.5 (0.3–0.8) | 0.006 | 0.7 | 0.3 | 1 |
| Mentation | 0.9 ± 0.5 | 0.2 (0.1–0.2) | 0.3 (0.2–0.3) | 0.1 (0.06–0.2) | 0.1 (−0.1 to 0.2) | 0.1 (0.1–0.2) | 0.4 | 1 | 1 | 1 |
| Bulbar | 2.0 ± 1.0 | 0.5 (0.4–0.6) | 0.5 (0.4–0.6) | 0.5 (0.3–0.7) | 0.3 (0.2–0.5) | 0.6 (0.4–0.9) | 0.3 | 1 | 1 | 1 |
| Ocular | 0.9 ± 0.6 | 0.1 (0.1–0.2) | 0.1 (0.1–0.2) | 0.1 (0.1–0.2) | 0.1 (−0.01 to 0.2) | 0.2 (0.1–0.2) | 0.6 | 1 | 1 | 1 |
| Limb | 1.1 ± 0.6 | 0.2 (0.1–0.2) | 0.3 (0.2–0.3) | 0.1 (−0.03 to 0.1) | 0.04 (−0.1 to 0.2) | 0.1 (−0.04 to 0.2) | 0.01 | 1 | 0.04 | 0.4 |
| Gait | 5.7 ± 2.0 | 0.9 (0.7–1.0) | 1.1 (0.9–1.3) | 0.5 (0.3–0.8) | 0.3 (0.1–0.5) | 0.7 (0.4–1.0) | 0.006 | 0.3 | 0.4 | 1 |
| Total | 14.4 ± 4.7 | 2.6 (2.2–3.0) | 3.1 (2.6–3.6) | 1.9 (1.2–2.5) | 0.7 (−0.4 to 1.9) | 2.3 (1.5–3.1) | 0.005 | 0.2 | 0.4 | 1 |
mPSPRS = modified Progressive Supranuclear Palsy Rating Scale; PSP = progressive supranuclear palsy; PSPRS = Progressive Supranuclear Palsy Rating Scale; RS = Richardson’s Syndrome; SD = standard deviation; Trial cases refers to the subset deemed likely eligible to recent Phase II clinical trial; SPE = 1-year progression estimate.
Using the mPSPRS, the Richardson’s syndrome phenotype again progressed faster than the variant group and cortical variants progressed faster than subcortical variants, although not reaching statistical significance. Absolute differences were <1 point/year between groups. Similar rates were found in the pathologically confirmed cases of Richardson’s syndrome versus variants (∼1 point/year) with significant differences in the Limb score between Richardson’s syndrome and variant groups (<1 point/year) and the total score between Richardson’s syndrome and subcortical groups (∼3 points/year).
On cognitive measures, progression was significantly faster in Richardson’s syndrome than the variant group for ACE-R total score (∼2 points/year) and MMSE (∼1 point/year). Visuospatial and Language domain progression differed between Richardson’s syndrome and variant groups, both by ∼1 point/year. Baseline Fluency was poor across all groups (global mean score 5/14). ACE-R progression in the subcortical group was slower than the Richardson’s syndrome group (∼3 points/year) with significant differences in Visuospatial subscore (∼1 point/year). MMSE progression was slower in the subcortical group than Richardson’s syndrome (∼1 point/year). No significant differences were observed between Richardson’s syndrome and cortical groups. Similar rates were observed in the pathologically confirmed cohort although apart from the Visuospatial subscore between Richardson’s syndrome and variant groups (∼1 point/year) and the Fluency score between Richardson’s syndrome and subcortical groups (∼1 point/year), these did not reach statistical significance.
In the comparison of those eligible versus not-eligible for the trial, we found PSPRS progression rates were faster in trial-eligible patients compared to trial-not-eligible patients by ∼4 points per year. This difference persisted if considering only those with >6 and >12 months follow up. Subgroup analysis demonstrated slower progression in PSPRS by ∼5 points/year between trial-eligible and trial-not-eligible Richardson’s syndrome in the groups with at least 6 months and at least 12 months follow-up. Analysis of the PSPRS subscores showed faster progression in History and Gait in eligible patients (both ∼1 point/year). Total mPSPRS progression was faster in trial-eligible patients compared to trial-not-eligible patients with differences in History, Limb and Gait subscores (all <1 point/year). No differences were found in ACE-R progression between eligible and not-eligible patients as a whole. Progression in MMSE (∼2 points/year) and ACE-R (∼4 points/year) total scores and Language and Visuospatial subscores (∼1 point/year) was faster in the trial-not-eligible Richardson’s syndrome patients compared to the trial-not-eligible variant patients. Similar trends were seen in the pathologically confirmed subgroup.
Discussion
The principal results of this longitudinal study are that (i) variant presentations of PSP progress more slowly than the Richardson’s syndrome phenotype. Subcortical presentations presented later to clinic, had slowest motor and cognitive progression and longest survival; and (ii) only half of patients were considered likely to have met clinical trial entry criteria, and this group showed faster motor and cognitive progression and shorter survival than those not meeting trial criteria. A fifth of the cohort had pathological confirmation of the PSP diagnosis, among whom the subgroup progression rates were similar to the main study group, suggesting that diagnostic error is not the cause of differential progression rates.
The effects of subgroup on progression were similar for the 14-item mPSPRS compared to the 28-item PSPRS. History and Gait subscores contributed most to the accelerated progression in trial-eligible patients versus trial-not-eligible patients. It is encouraging that the shorter mPSPRS score, focused on clinically meaningful disease milestones, performed similarly to the PSPRS in quantifying differential progression rates between our study groups although did not reach statistical significance.
The PSPRS progression rate is in line with previous observational studies and clinical trials of Richardson’s syndrome.5,15,17,37,38 However, clinical trial entry criteria from these studies would have excluded almost half of the current clinical PSP population at first presentation. Nonetheless, for those meeting criteria, we confirmed similar progression rates as in earlier reports. The challenge, however, is to understand the progression of the cases that would not have met entry criteria in past—or imminent—Phase II/III clinical trials. This trial-not-eligible group is heterogeneous, including variant PSP phenotypes and many with Richardson’s syndrome. The Richardson’s syndrome population within the trial-not-eligible group progressed slower than their trial-eligible counterparts, suggesting that the difference observed is not solely driven by variant diagnoses in the not-eligible group. The identification of slower progression in this group has implications for clinical trial representativeness and the application of findings from a successful clinical trial to the PSP population as a whole: if not-eligible cases have a slower rate of progression, caution would be needed when attempting to generalize risk–benefit and cost–benefit analyses from clinical trials to the whole PSP population. The use of clinical trial cohort data as a proxy for natural history progression, or for simulation and planning of clinical trials and drug development, may be inaccurate with respect to a significant proportion of patients with PSP.
Richardson’s syndrome constituted the majority of our dataset (174/227, 77%) at first presentation, as in previous studies.6,39 The majority of other, variant, presentations converted clinically to Richardson’s syndrome during the follow-up period (41/53, 77%) confirming Richardson’s syndrome as the predominant eventual phenotype during the full disease course (215/227, 95%). Those presenting with Richardson’s syndrome presented to clinic earlier after symptom onset but progressed faster than variant patients. This finding was particularly apparent when considering the Richardson’s syndrome population eligible for current clinical trials. The reason for this earlier presentation is not known. It may reflect unique features of the UK healthcare system’s referral pathways, but awareness of PSP is low globally, and awareness of variant phenotypes even more so. Historically, poor recognition of early features and delayed referral to specialist services have been blamed,40 although a marginal improvement in time to diagnosis has been observed in recent decades.9,40 Misdiagnosis of PSP (as Parkinson’s disease, depression or stroke among others) is common internationally.41,42
Variant presentations took longer to present to clinic, indicating a lost window of opportunity for disease-modifying therapies. This may in part be related to poorer recognition of such phenotypes prior to publication of the revised diagnostic criteria in 2017. However, they then went on to have slower rates of progression and longer survival during follow-up. In keeping with a multicentre longitudinal study,5 subcortical presentations of PSP had distinct disease characteristics demonstrable on multiple assessment methods of cognitive and motor function. We confirm the survival benefit of this subcortical group, as demonstrated in a recent multicentre brain-bank study.43
The PSPRS includes several questions related to cognitive function and behaviour. It is unfortunate that these were removed from the mPSPRS, even though cognitive and behavioural changes are major influences on quality of life,44,45 carer burden46,47 and survival.26,27 Indeed, seven of the nine cases originally described by Steele et al.48 had severe cognitive impairment. Cognitive progression has previously been described with many screening tools, including the MMSE,49,50 Montreal Cognitive Assessment50 and the ACE-R,17,20 as well as more in-depth neuropsychological assessments. Here, ACE-R and MMSE progression was similar to previous work in Richardson’s syndrome,17,20,49 but there was slower decline in variant presentations compared to RS. The subcortical group showed slower decline than Richardson’s syndrome, and the cortical group. Impairment in fluency, long established as a distinguishing feature between PSP and Parkinson’s disease20,51 is very poor at presentation across all phenotypes suggesting perhaps that this is a very early marker of deterioration and a focus for further work.
An advantage of this study is the embedding in a regional healthcare service with referral pathways drawing on movement disorders clinics, cognitive disorders clinics, general neurology and gerontology. The main cohort is therefore broadly representative of the regional PSP population as a whole rather than a subset meeting clinical trials criteria and was strengthened by neuropathological confirmation in 49 patients.
The study has several limitations, in phenotyping, subgrouping for analysis and assessment tools. Since in most cases, several years had passed from first symptom to first assessment, important phenotype evolution may already have occurred. Moreover, the date of symptom onset is challenging to estimate, especially for cognitive and behavioural change: a first fall may be clearly recalled, but subtler changes in personality or fluency may not be. The lack of clarity over a start date will affect the estimation of survival from onset to death. We also rely on clinical diagnosis for the majority of cases, with pathological confirmation obtained in only a fifth of participants. The PSPRS is the most widely used severity scale for PSP, but it may not be equally sensitive across the array of symptoms present in variant syndromes.52 The proposed focus on patient-centred meaningful outcomes in the mPSPRS does not address this particular issue. There are analysis issues to consider. Our statistical methods are similar to those used previously16,49 although we incorporated more predictor variables in the linear mixed model, with improved model fit. Our model included participants with single assessments, although confining to those with a follow-up period >6 or 12 months had a limited effect on absolute progression rates in Richardson’s syndrome but did improve the distinction between Richardson’s syndrome and variant presentations. Nonetheless, even our broadly inclusive longitudinal design does not account for phenotype soon after symptom onset. This study draws on data from a single site. Across different sites, differential referral pathways may affect a clinics’ balance of phenotypes, where, for example, the case mix in cognitive disorders clinics may vary from clinics specialized in movement disorders or Parkinson’s disease. Familiarity with PSP variants, and the tools used to assess them, may also affect diagnostic accuracy or delay. The differential timelines for diagnosis between Richardson’s and variant syndromes may itself be a function of the type of centre to which a patient is referred: our centre receives referrals from regional cognitive and movement disorders clinics, which may have influenced the range of variant cases. In view of limited sample sizes, we grouped variant cases into one of two broad categories: ‘cortical’ and ‘subcortical’. This division is not novel,5,7 but nonetheless represents an over-simplification of diverse clinical syndromes.
A greater difficulty lies in trying to determine retrospective potential eligibility to a clinical trial. Only half of our cohort nominally met trial criteria—and we acknowledge that we did not take participants through the invasive screening tests (e.g. electrocardiogram, bloods) or consent processes, that may have raised additional exclusions. Moreover, there are differences in inclusion/exclusion criteria between previous Phase II/III trials (NCT00211224, NCT01187888, NCT01110720), related in part to the trial compounds and tolerance of study procedures. However, more recent trials (NCT03068468, NCT04253132) have much in common in their criteria, selecting those with Richardson’s syndrome and an upper limit on time from symptom onset (e.g. <5 years). As such, our trial-eligibility criterion is only an approximation, but one that is sufficient to highlight the problem of prognostic bias. New trials may benefit from greater inclusivity of the full spectrum of PSP, with consideration of adjusted risk–benefit analyses and alternative outcome measures for variant phenotypes.
In conclusion, variant presentations of PSP progress more slowly than Richardson’s syndrome, and are not currently well represented in clinical trial cohorts. People with PSP but outside of clinical trial criteria constitute approximately half of the PSP population as a whole at first visit.4,5 Future clinical trialists need to consider phenotypic variance of PSP, and the impact of case selection on applicability of the outcome of future trials. We hope that the results of this study assist in modelling and planning of future trials, to ensure the maximal benefit to the full spectrum of people affected by this devastating disease.
Supplementary material
Supplementary material is available at Brain Communications online.
Supplementary Material
Acknowledgements
The authors thank all the participants with progressive supranuclear palsy, their families and carers for their contributions to this study.
Funding
This study was co-funded by the National Institute for Health Research (NIHR) Biomedical Research Centre at Cambridge University Hospitals NHS Foundation Trust and the University of Cambridge (BRC-1215-20014) and the Cambridge Brain Bank; the Holt Fellowship (RG86564); the Medical Research Council (MR/P01271X/1; SUAG051/G101400 and SUAG004/051/RG91365); Fitzwilliam College and the Cambridge University Centre for Parkinson-plus (RG95450). The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care or of other funders.
Competing interests
The authors report no competing interests with the current work. Dr Street, Dr Malpetti, Dr Rittman, Dr Murley, Dr Coyle-Gilchrist and Dr Passamonti report no disclosures. Dr Ghosh provides consultancy to NICE, UCB and Biogen and sits on the research committee of the PSP Association (UK). Professor Rowe serves as editor to Brain, and is a non-remunerated trustee of the Guarantors of Brain and the PSP Association; he provides consultancy to Asceneuron, Biogen, UCB, SVHealth and Wave, and has research grants from AZ-Medimmune, Janssen, Lilly as industry partners in the Dementias Platform UK.
Glossary
- ACE-R =
revised Addenbrooke’s Cognitive Examination
- CI =
confidence interval
- HR =
hazard ratio
- MMSE =
Mini-Mental State Examination
- mPSPRS =
modified Progressive Supranuclear Palsy Rating Scale
- NHS =
National Health Service
- PSP =
progressive supranuclear palsy
- PSPRS =
Progressive Supranuclear Palsy Rating Scale
Contributor Information
Duncan Street, Department of Clinical Neurosciences, Cambridge University Hospitals NHS Trust, University of Cambridge, Cambridge CB2 0SZ, UK.
Maura Malpetti, Department of Clinical Neurosciences, Cambridge University Hospitals NHS Trust, University of Cambridge, Cambridge CB2 0SZ, UK.
Timothy Rittman, Department of Clinical Neurosciences, Cambridge University Hospitals NHS Trust, University of Cambridge, Cambridge CB2 0SZ, UK.
Boyd C P Ghosh, Department of Clinical Neurosciences, Cambridge University Hospitals NHS Trust, University of Cambridge, Cambridge CB2 0SZ, UK; Wessex Neurological Centre, University Hospitals Southampton NHS Foundation Trust, Southampton SO16 6YD, UK.
Alexander G Murley, Department of Clinical Neurosciences, Cambridge University Hospitals NHS Trust, University of Cambridge, Cambridge CB2 0SZ, UK.
Ian Coyle-Gilchrist, Department of Clinical Neurosciences, Cambridge University Hospitals NHS Trust, University of Cambridge, Cambridge CB2 0SZ, UK; Norfolk and Norwich NHS Foundation Trust, Norwich NR4 7UY, UK.
Luca Passamonti, Department of Clinical Neurosciences, Cambridge University Hospitals NHS Trust, University of Cambridge, Cambridge CB2 0SZ, UK; Consiglio Nazionale delle Ricerche (CNR), Istituto di Bioimmagini e Fisiologia Molecolare (IBFM), Milano, 20090 Segrate (MI), Italy.
James B Rowe, Department of Clinical Neurosciences, Cambridge University Hospitals NHS Trust, University of Cambridge, Cambridge CB2 0SZ, UK; Medical Research Council Cognition and Brain Sciences Unit, University of Cambridge, Cambridge CB2 7EF, UK.
References
- 1. Höglinger GU, Respondek G, Stamelou M, et al. ; Movement Disorder Society-endorsed PSP Study Group. Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov Disord. 2017;32(6):853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gazzina S, Respondek G, Compta Y, et al. Neuropathological validation of the MDS-PSP criteria with PSP and other frontotemporal lobar degeneration. bioRxiv 520510. 2019;1–44. 10.1101/520510 (Accessed 07 September 2021). [DOI] [Google Scholar]
- 3. Grimm MJ, Respondek G, Stamelou M, et al. ; Movement Disorder Society-Endorsed PSP Study Group. Clinical conditions “suggestive of progressive supranuclear palsy”—diagnostic performance. Mov Disord. 2020;35(12):2301–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Picillo M, Cuoco S, Tepedino MF, et al. ; PSP Salerno Study Group. Motor, cognitive and behavioral differences in MDS PSP phenotypes. J Neurol. 2019;266(7):1727–1735. [DOI] [PubMed] [Google Scholar]
- 5. Jabbari E, Holland N, Chelban V, et al. Diagnosis across the spectrum of progressive supranuclear palsy and corticobasal syndrome. JAMA Neurol. 2020;77(3):377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Respondek G, Stamelou M, Kurz C, et al. ; Movement Disorder Society-endorsed PSP Study Group. The phenotypic spectrum of progressive supranuclear palsy: A retrospective multicenter study of 100 definite cases. Mov Disord. 2014;29(14):1758–1766. [DOI] [PubMed] [Google Scholar]
- 7. Kovacs GG, Lukic MJ, Irwin DJ, et al. Distribution patterns of tau pathology in progressive supranuclear palsy. Acta Neuropathol. 2020;140(2):99–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Glasmacher SA, Leigh PN, Saha RA. Predictors of survival in progressive supranuclear palsy and multiple system atrophy: A systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2017;88(5):402–411. [DOI] [PubMed] [Google Scholar]
- 9. dell'Aquila C, Zoccolella S, Cardinali V, et al. Predictors of survival in a series of clinically diagnosed progressive supranuclear palsy patients. Park Relat Disord. 2013;19(11):980–985. [DOI] [PubMed] [Google Scholar]
- 10. Coyle-Gilchrist ITS, Dick KM, Patterson K, et al. Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology. 2016;86(18):1736–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bensimon G, Ludolph A, Agid Y, Vidailhet M, Payan C, Leigh PN. Riluzole treatment, survival and diagnostic criteria in Parkinson plus disorders: The NNIPPS Study. Brain. 2009;132(1):156–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Murley AG, Rouse MA, Coyle-Gilchrist ITS, et al. Predicting loss of independence and mortality in frontotemporal lobar degeneration syndromes. J Neurol Neurosurg Psychiatry. 2021;92(7):737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Malpetti M, Passamonti L, Simon Jones P, et al. PET markers of neuroinflammation and tau pathology predict disease progression in progressive supranuclear palsy. J Neurol Neurosurg Psychiatry. 2021;92(7):769–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boxer AL, Yu JT, Golbe LI, Litvan I, Lang AE, Höglinger GU. Advances in progressive supranuclear palsy: New diagnostic criteria, biomarkers, and therapeutic approaches. Lancet Neurol. 2017;16(7):552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. VandeVrede L, Ljubenkov PA, Rojas JC, Welch AE, Boxer AL. Four-repeat tauopathies: Current management and future treatments. Neurotherapeutics. 2020;17(4):1563–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Golbe LI, Ohman-Strickland PA. A clinical rating scale for progressive supranuclear palsy. Brain. 2007;130(6):1552–1565. [DOI] [PubMed] [Google Scholar]
- 17. Ghosh BCP, Carpenter RHS, Rowe JB. A longitudinal study of motor, oculomotor and cognitive function in progressive supranuclear palsy. PLoS One. 2013;8(9):e74486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Grötsch M, Respondek G, Colosimo C, et al. A modified progressive supranuclear palsy rating scale. Mov Disord. 2021;36(5):1203–1215. [DOI] [PubMed] [Google Scholar]
- 19. Brown RG, Lacomblez L, Landwehrmeyer BG, et al. ; NNIPPS Study Group. Cognitive impairment in patients with multiple system atrophy and progressive supranuclear palsy. Brain. 2010;133(Pt 8):2382–2393. [DOI] [PubMed] [Google Scholar]
- 20. Rittman T, Ghosh BC, McColgan P, et al. The Addenbrooke’s Cognitive Examination for the differential diagnosis and longitudinal assessment of patients with parkinsonian disorders. J Neurol Neurosurg Psychiatry. 2013;84(5):544–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gerstenecker A, Duff K, Mast B, Litvan I; ENGENE-PSP Study Group. Behavioral abnormalities in progressive supranuclear palsy. Psychiatry Res. 2013;210(3):1205–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pellicano C, Assogna F, Cellupica N, et al. Neuropsychiatric and cognitive profile of early Richardson’s syndrome, progressive supranuclear palsy-parkinsonism and Parkinson’s disease. Park Relat Disord Internet. 2017;45:50–56. [DOI] [PubMed] [Google Scholar]
- 23. Peterson KA, Patterson K, Rowe JB. Language impairment in progressive supranuclear palsy and corticobasal syndrome. J Neurol. 2021;268(3):796–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chaithra SP, Prasad S, Holla VV, et al. The Non-motor symptom profile of progressive supranuclear palsy. J Mov Disord. 2020;13(2):118–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kaat LD, Boon AJW, Kamphorst W, Ravid R, Duivenvoorden HJ, Van Swieten JC. Frontal presentation in progressive supranuclear palsy. Neurology. 2007;69(8):723–729. [DOI] [PubMed] [Google Scholar]
- 26. Lansdall CJ, Coyle-Gilchrist ITS, Vázquez Rodríguez P, et al. Prognostic importance of apathy in syndromes associated with frontotemporal lobar degeneration. Neurology. 2019;92(14):E1547–E1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Murley AG, Coyle-Gilchrist I, Rouse MA, et al. Redefining the multidimensional clinical phenotypes of frontotemporal lobar degeneration syndromes. Brain. 2020;143(5):1555–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Respondek G, Kurz C, Arzberger T, et al. ; Movement Disorder Society-Endorsed PSP Study Group. Which ante mortem clinical features predict progressive supranuclear palsy pathology? Mov Disord. 2017;32(7):995–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Grimm MJ, Respondek G, Stamelou M, et al. ; Movement Disorder Society-endorsed PSP Study Group. How to apply the movement disorder society criteria for diagnosis of progressive supranuclear palsy. Mov Disord. 2019;34(8):1228–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)—A metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Harris PA, Taylor R, Minor BL, et al. ; REDCap Consortium. The REDCap consortium: Building an international community of software platform partners. J Biomed Inform. 2019;95:103208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van Buuren S, Groothuis-Oudshoorn K. mice: Multivariate imputation by chained equations in R. J Stat Softw. 2011;45(3):1–67. [Google Scholar]
- 33. Bates D, Mächler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. J Stat Softw. 2015;67(1):1–48. [Google Scholar]
- 34. Therneau T. A Package for Survival Analysis in R [Internet]. 2020. https://cran.r-project.org/package=survival (Accessed 07 September 2021).
- 35. Kassambara A, Kosinski M, Biecek P. survminer: Drawing Survival Curves using “ggplot2” [Internet]. 2020. https://cran.r-project.org/package=survminer (Accessed 07 September 2021).
- 36. Morey Richard D, Rouder JN. Computation of Bayes Factors for Common Designs [Internet]. 2018. https://cran.r-project.org/package=BayesFactor (Accessed 07 September 2021).
- 37. Boxer AL, Lang AE, Grossman M, et al. Davunetide in patients with progressive supranuclear palsy: A randomised, double-blind, placebo-controlled phase 2/3 trial. Lancet Neurol. 2014;13(7):676–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tolosa E, Litvan I, Höglinger GU, et al. ; TAUROS Investigators. A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Mov Disord. 2014;29(4):470–478. [DOI] [PubMed] [Google Scholar]
- 39. Williams DR, Lees AJ. Progressive supranuclear palsy: Clinicopathological concepts and diagnostic challenges. Lancet Neurol. 2009;8(3):270–279. [DOI] [PubMed] [Google Scholar]
- 40. Golbe LI, Davis PH, Schoenberg BS, Duvoisin RC. Prevalence and natural history of progressive supranuclear palsy. Neurology. 1988;38(7):1031–1031. [DOI] [PubMed] [Google Scholar]
- 41. Burn DJ, Lees AJ. Progressive supranuclear palsy: Where are we now? Lancet Neurol. 2002;1(6):359–369. [DOI] [PubMed] [Google Scholar]
- 42. Xie T, Kang UJ, Kuo SH, Poulopoulos M, Greene P, Fahn S. Comparison of clinical features in pathologically confirmed PSP and MSA patients followed at a tertiary center. Parkinsons Dis. 2015;1(February):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Guasp M, Molina-Porcel L, Painous C, et al. Association of PSP phenotypes with survival: A brain-bank study. Parkinsonism Relat Disord. 2021;84:77–81. [DOI] [PubMed] [Google Scholar]
- 44. Schrag A, Selai C, Davis J, Lees AJ, Jahanshahi M, Quinn N. Health-related quality of life in patients with progressive supranuclear palsy. Mov Disord. 2003;18(12):1464–1469. [DOI] [PubMed] [Google Scholar]
- 45. Pekmezović T, Ječmenica-Lukić M, Petrović I, Špica V, Tomić A, Kostić VS. Quality of life in patients with progressive supranuclear palsy: One-year follow-up. J Neurol. 2015;262(9):2042–2048. [DOI] [PubMed] [Google Scholar]
- 46. Burrell JR, Hodges JR, Rowe JB. Cognition in corticobasal syndrome and progressive supranuclear palsy: A review. Mov Disord. 2014;29(5):684–693. [DOI] [PubMed] [Google Scholar]
- 47. Wedderburn C, Wear H, Brown J, et al. The utility of the Cambridge Behavioural Inventory in neurodegenerative disease. J Neurol Neurosurg Psychiatry. 2008;79(5):500–503. [DOI] [PubMed] [Google Scholar]
- 48. Steele JC, Richardson JC, Olszewski J. Progressive supranuclear palsy. Arch Neurol. 1964;10:333–359. [DOI] [PubMed] [Google Scholar]
- 49. Litvan I, Kong M. Rate of decline in progressive supranuclear palsy. Mov Disord. 2014;29(4):463–468. [DOI] [PubMed] [Google Scholar]
- 50. Fiorenzato E, Weis L, Falup-Pecurariu C, et al. Montreal Cognitive Assessment (MoCA) and Mini-Mental State Examination (MMSE) performance in progressive supranuclear palsy and multiple system atrophy. J Neural Transm. 2016;123(12):1435–1442. [DOI] [PubMed] [Google Scholar]
- 51. Rittman T, Coyle-Gilchrist IT, Rowe JB. Managing cognition in progressive supranuclear palsy. Neurodegener Dis Manag. 2016;6(6):499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Piot I, Schweyer K, Respondek G, et al. ; DescribePSP Study Group. The progressive supranuclear palsy clinical deficits scale. Mov Disord. 2020;35(4):650–661. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Anonymized derived data will be available upon reasonable request to the senior author, for academic non-commercial purposes, subject to potential limitations to protect participant confidentiality and maintain General Data Protection Regulation compliance.