

Abstract
Biallelic loss-of-function (LoF) of SLC13A5 (solute carrier family 13, member 5) induced deficiency in sodium/citrate transporter (NaCT) causes autosomal recessive developmental epileptic encephalopathy 25 with hypoplastic amelogenesis imperfecta (DEE25; MIM #615905). Many pathogenic SLC13A5 single nucleotide variants (SNVs) and small indels have been described; however, no cases with copy number variants (CNVs) have been sufficiently investigated. We describe a consanguineous Iraqi family harboring an 88.5 Kb homozygous deletion including SLC13A5 in Chr17p13.1. The three affected male siblings exhibit neonatal-onset epilepsy with fever-sensitivity, recurrent status epilepticus, global developmental delay/intellectual disability (GDD/ID) and other variable neurological features as shared phenotypical features of DEE25. Two of the three affected subjects exhibit hypoplastic amelogenesis imperfecta (AI), while the proband shows no evidence of dental abnormalities or AI at 2 years of age with apparently unaffected primary dentition. Characterization of the genomic architecture at this locus revealed a potential genomic instability mediated by an Alu-Alu mediated rearrangement (AAMR) which was confirmed through breakpoint junction Sanger sequencing. This multiplex family from a distinct population elucidates the phenotypic consequence of complete LoF of SLC13A5 and illustrates the importance of read-depth based CNV detection in comprehensive exome sequencing (ES) analysis to solve cases that otherwise remain molecularly unsolved.
Keywords: SLC13A5, homozygous deletion, developmental and epileptic encephalopathy, Alu-Alu mediated rearrangement, CNV alleles, genomic instability
Introduction
High-affinity sodium-dependent citrate transporter (NaCT), encoded by solute carrier family 13, member 5 (SLC13A5), plays a crucial role in citrate metabolism in the brain, teeth, and liver by acting as a primary regulator of the sodium/citrate concentrations in the cytosol (Gopal et al., 2007). Dysfunction of NaCT impairs citrate uptake and transport activity intracellularly, resulting in disturbance of neurotransmitter in the cerebrospinal fluid (Gopal et al., 2007; Hardies et al., 2015). Brain citrate level alteration and abnormality of hippocampus neuronal network excitability were also noted in Slc13a5-knockout mice (Henke et al., 2020). Biallelic loss-of-function (LoF) variants in SLC13A5 (MIM *608305) have been described as causing autosomal recessive (AR) developmental epileptic encephalopathy 25 with hypoplastic amelogenesis imperfecta (DEE25; MIM #615905), with a spectrum of developmental and neurological abnormalities (Hardies et al., 2015; Thevenon et al., 2014). Individuals diagnosed with DEE25 have been reported with neonatal seizures, variable neurological outcomes, and dental abnormalities. Notably, nine patients with the clinical diagnosis of Kohlschütter–Tönz syndrome (KTZS, #226750) had no pathogenic variants in the frequently causative gene ROGDI (MIM *614574), but rather were found to have biallelic variants in SLC13A5 (Schossig et al., 2017). ROGD1 encodes an atypical leucine zipper protein with high expression in the human brain and spinal cord, with a presynaptic site localization pattern (Riemann et al., 2017). KTZS is characterized by early-onset epilepsy, global developmental delay (GDD), and amelogenesis imperfecta. These clinical phenotypic features are shared between DEE25 and KTZS, and indicate that SLC13A5 could be associated with ROGDI-negative KTZS (Schossig et al., 2017) and perhaps their protein products act in the same network, interactome, or biochemical pathway.
More than 59 cases with biallelic pathogenic single nucleotide variants (SNV) in SLC13A5 have been described (Matricardi et al., 2020); 12 pathogenic and 17 likely pathogenic alleles have been documented in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar). Nevertheless, no cases with copy number variants (CNVs) have been clearly studied. Here, we describe a consanguineous family of Iraqi descent with a novel 88.5 Kb homozygous deletion in Chr17p13.11, resulting in a complete LoF of SLC13A5 in three affected male siblings, with the elucidation of detailed clinical and molecular genetic features. Breakpoint junction analysis demonstrates that the SLC13A5 deletion was generated by Alu-Alu mediated genomic rearrangement (AAMR)(Song et al., 2018).
Methods
Family-based exome sequencing (ES) and rare variant analyses for three affected siblings with DEE (Figure 1A–G) and their parents (pentad ES) were performed under an Institutional Review Board (IRB) approved research protocol (H-29697) at the Baylor-Hopkins Center for Mendelian Genomics and as part of analysis of a large Middle Eastern cohort with rare neurodevelopmental disorders. Pentad ES analysis for shared rare homozygous, compound heterozygous, and recurrent de novo (for parental mosaicism) variants did not identify any candidate SNV. CNV analysis using exome hidden Markov model (XHMM) and an in-house developed algorithm, HMZdelFinder (https://github.com/BCM-Lupskilab/HMZDelFinder), designed to identify rare intragenic homozygous and hemizygous deletions using ES data (Fromer & Purcell, 2014; Gambin et al., 2017), detected a shared novel homozygous deletion involving the whole-gene of SLC13A5 and first exon of the adjacent gene XAF1 in all three affected siblings (Z-score: −5.58) (Figure 1H).
Figure 1. Family pedigree, facial features, dentition, and bioinformatic characteristics of whole-gene deletion of SLC13A5 and exonic deletion of XAF1.
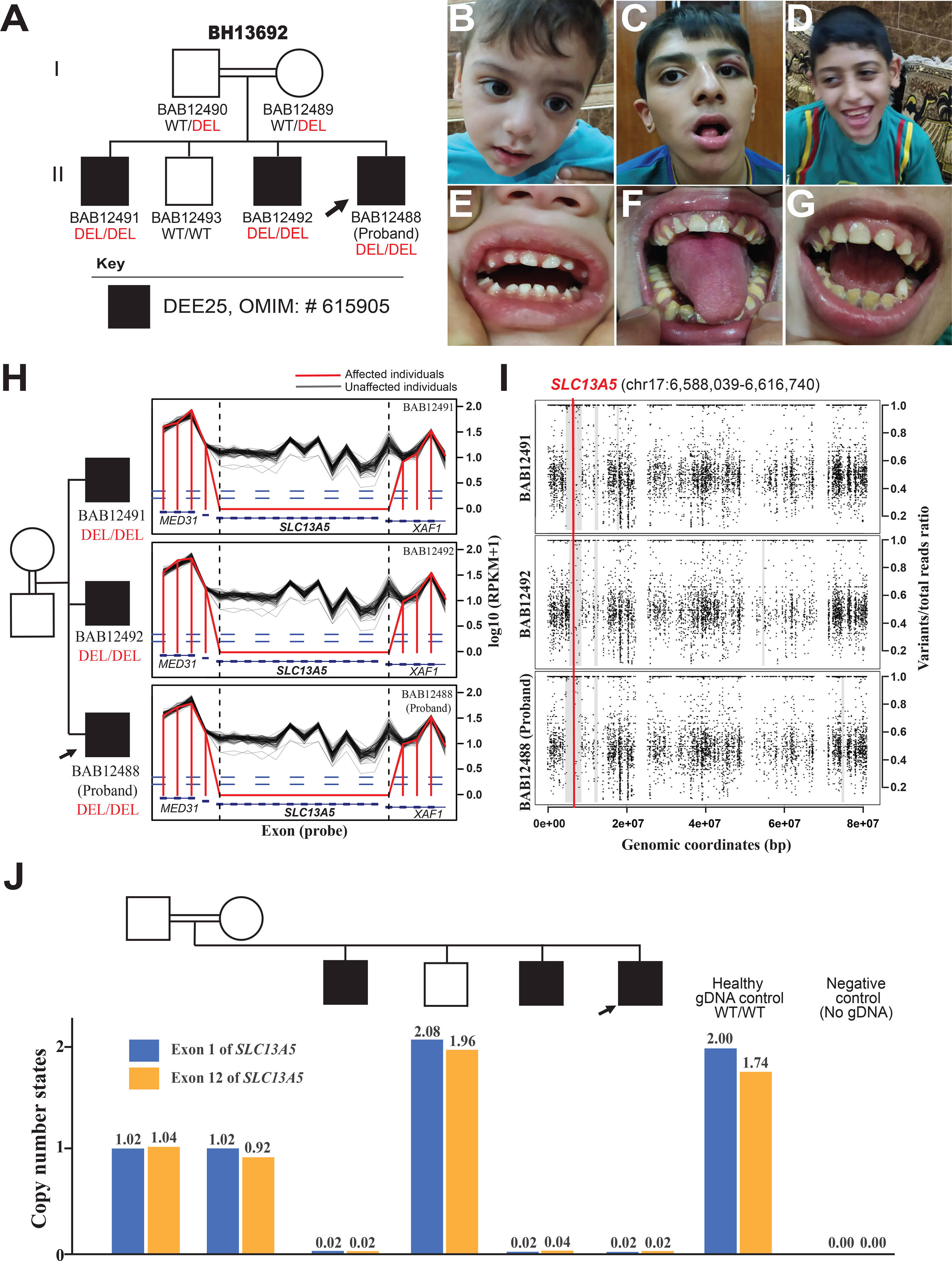
(A) Pedigree of the family showing the affected males as black shaded squares with the proband demarcated (arrow). The parents are second-degree cousins; extended multigenerational family pedigree analyses (Supplementary) reveals historical evidence for consanguineous relationships in previous generations. WT: Wild Type, DEL: Deletion.
(B-D) Facial photographs of the proband (B), the 15-year-old affected sibling (C), and the 11-year-old affected sibling (D) with no distinct dysmorphic features.
(E) A dental photograph of the proband (B) shows no evidence of dental abnormalities or AI at 2 years of age with appeared unaffected primary dentition.
(F) A dental photograph of the 15-year-old affected sibling (C) shows hypoplastic enamel, dark yellow color with severely worn enamel surfaces, generalized gingival inflammation, and marginal gingival overgrowth. AI type 1 (hypoplastic enamel) is highly suspected.
(G) A dental photograph of the 11-year-old affected sibling (D) shows defective and severely worn enamel surfaces, accumulation of higher calculus and plaque deposits, and dark color of the underlying dentin. Incipient caries at the lower left canine, and generalized gingival inflammation are also noted. These findings are consistent with AI type I (hypoplastic enamel).
(H) The log10 RPKM profile of targeted region from HMZDelFinder raw data; Designed exon capture region is marked as blue rectangles; RPKM values of the three affected siblings are marked as red lines, while black lines delineate RPKM value from individual ES samples from an internal database with similar experimental conditions.
(I) B-allele frequency of chromosome 17 for the proband and affected siblings showing a shared 2.5–2.7 Mb interval of Absence of heterozygosity (AOH) surrounding the homozygous SLC13A5 deletion and marked with a thick gray line. Few small unshared AOH blocks are also seen shaded with thin grey lines. The location of the CNV is marked with a red line across the shared AOH region.
(J) Copy number state of exon 1 and exon 12 of SLC13A5, determined by relative positive droplet ratios, are independently examined by digital droplet PCR (ddPCR) on all family members. According to relative positive droplet ratios, three affected individuals manifest zero-copy of both exons; both parents present as heterozygous carriers (single copy), while the unaffected brother shows two copies of both exons.
To investigate for genomic evidence of potential consanguinity, we used BafCalculator (https://github.com/BCM-Lupskilab/BafCalculator), an in-house developed bioinformatic tool (Gambin et al., 2017; Karaca et al., 2018). This tool uses ES variant call format (vcf) file to extract the B-allele frequency from unphased ES variant data to calculate the absence of heterozygosity (AOH), as a surrogate measure of runs of homozygosity (ROH) and reflecting identity-by-descent genomic segments. Analyses of the exomes from the proband (BAB12488) and two affected siblings (BAB12491 and BAB12492), revealed that the homozygous SLC13A5 and partial XAF1 deletion maps within 2.5 Mb, 2.7 Mb and 2.7 Mb genomic intervals of AOH/ROH with a total AOH size of 125.4 Mb, 108.5 Mb and 179 Mb, respectively (Figure 1I). The coefficient of consanguinity determined from total AOH and estimated as percent genome shared, and identical-by-descent is 0.033. By historical report there was known consanguinity of second-degree cousins with a predicted coefficient of consanguinity of 0.03125.
The deletion and heterozygous copy number state in the parents were subsequently validated by an orthogonal experimental method - droplet digital polymerase chain reaction (ddPCR) in all family members (Figure 1J). Further characterization of the structural variant by array-based comparative genomic hybridization (aCGH) targeting chromosome 17 allowed for a more precise genomic resolution of the size, extent, and genomic content of the deletion CNV (Figure 2A). Both parents were found to be heterozygous carriers of the deletion, while the unaffected sibling carried homozygous wild-type alleles in accordance with Mendelian expectations.
Figure 2. Molecular genomic architecture of SLC13A5 whole-gene deletion and XAF1 exonic deletion in the family mediated by AAMR.
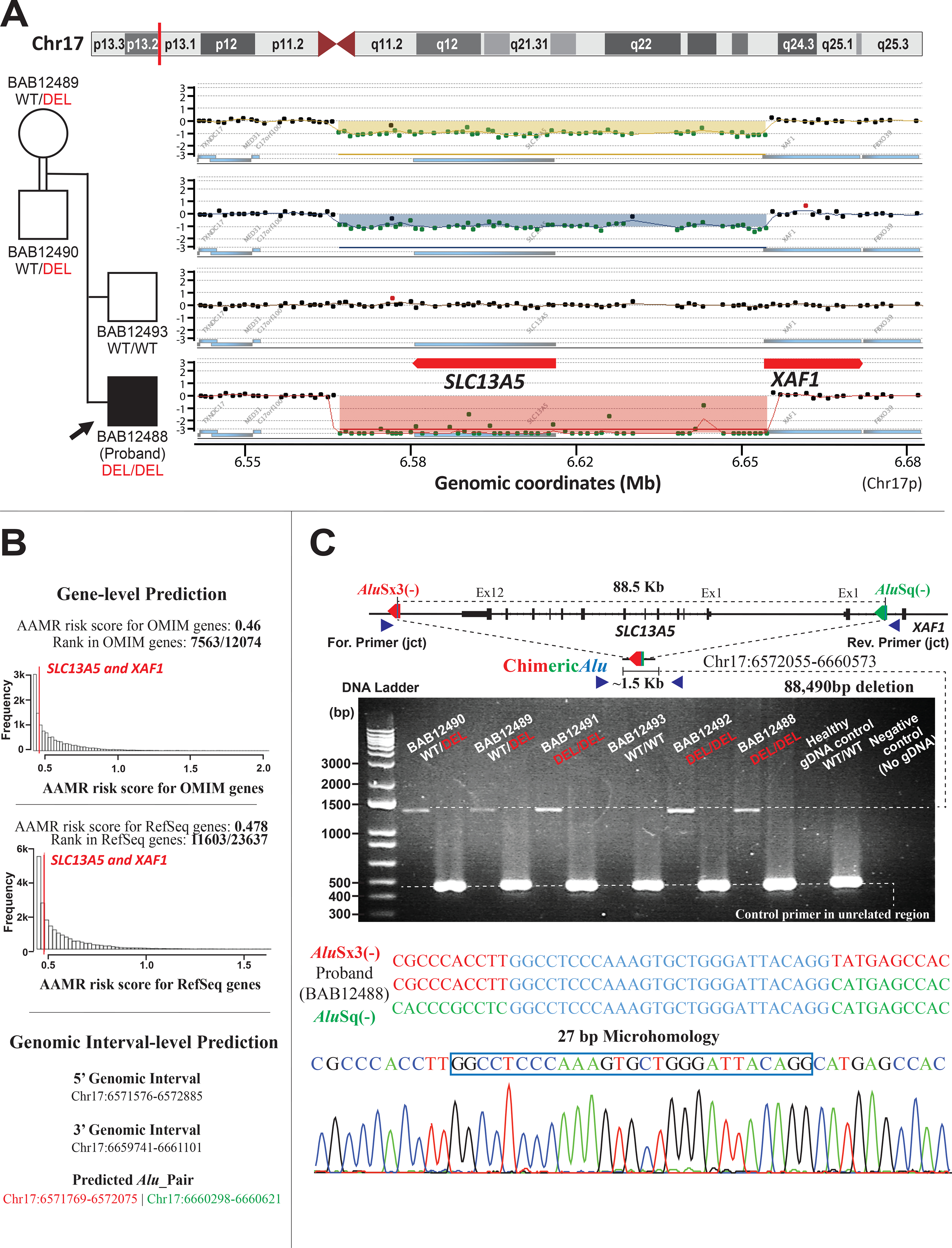
(A) A high-resolution array-based comparative genomic hybridization (aCGH) over Chr17p13.1 of the parents and unaffected brother, and proband, from top to bottom respectively, performed to evaluate for copy number status of SLC13A5 and XAF1. aCGH confirmed the homozygous deletion in the proband and the heterozygous deletion (carrier status) in both parents, while the unaffected brother was homozygous for the wild type (WT) allele (copy number neutral state).
(B) Gene-level prediction from AluAluCNVPredictor shows SLC13A5 and XAF1 have the same AAMR risk score in OMIM genes (0.46) and RefSeq genes (0.478), respectively. Genomic interval-level prediction successfully addresses this one CNV-Alu pair (AluSx3/AluSq), generating the deletion in this family with the information of genomic intervals acquired experimentally.
(C) A schematic of the genomic profile of AluSx3/AluSq pair mediated rearrangement and the primer design along with the gel electrophoresis of the PCR products. The junction primer pair was designed to produce an amplicon size of ~1.5 Kb for deleted alleles resulting from AAMR with the formation of a recombinant or chimeric Alu. Control primers were selected from a 450 bp unrelated genomic locus. Below the schematic is the 1% agarose gel electrophoresis of PCR products. From left to right of the panel: parents with lighter bands representing a heterozygous ~1.5 Kb deleted alleles; affected brother (BAB12491) with a thick band of ~1.5 Kb, consistent with the homozygous deleted allele; unaffected brother (BAB12493) showing no deleted band; affected brother (BAB12492) and proband with thick bands of ~1.5 Kb, consistent of homozygous deleted alleles; Healthy gDNA control with no deleted allele, and negative control (no template). Below the gel figure is an illustration of breakpoint junction analysis by Sanger sequencing revealed an 88,490 bp deletion resulting from recombination of AluSx3/AluSq pair in the same orientation (red and green arrow, respectively) encompassing a region of microhomology (blue line with each Alu). This results in a chimeric Alu with a 27 bp of microhomology (blue vertical line within chimeric Alu). The three sequences below the schematic from top to bottom represent the reference AluSx3 sequence (colored in red), the proband “chimeric” sequence, and the AluSq sequence (colored in green). The region of microhomology is colored in blue. The bottom image represents Sanger tracing of the breakpoint junction with the 27 bp of microhomology marked with a blue box.
We previously showed that Chr17p13 undergoes frequent genomic rearrangements that are often generated by Alu -Alu recombination (Carvalho et al., 2014; Gu et al., 2015). By examining the genomic intervals identified by aCGH, a directly oriented Alu pair intersecting the deleted genomic region was identified and AAMR was suspected as the potential mutational mechanism leading to the observed ~90 Kb deletion CNV. We also used AluAluCNVPredictor, an in-house developed bioinformatic tool, and the same Alu pair was predicted as the only CNV-Alu pair for the given genomic interval delineated from the aCGH data, with an AAMR risk score of 0.46 for MIM (Mendelian Inheritance in Man; omim.org) genes and 0.478 for RefSeq gene transcripts for both SLC13A5 and XAF1 (Figure 2B)(Song et al., 2018). This hypothesis of AAMR mechanism driven deletion CNV formation for the derivation of this disease-causing allele was investigated experimentally and confirmed by breakpoint junction analysis. Sanger sequencing revealed a chimeric Alu element derived from an AluSx3/AluSq recombinant, with 27 bp microhomology at the apparent recombinant joint, resulting in an 88,490 bp deletion (Figure 2C).
Following the molecular diagnosis, dental photos (Figure 1E–G) were obtained and reviewed by advanced practice and pediatric dentists (AF and GB coauthors) and the siblings were referred to a local pediatric dentist to be evaluated for the possibility of amelogenesis imperfecta (AI) and for dental management. Panoramic dental x-rays were not available for review. The z-score and standard deviation for all growth parameters were determined using Center for Disease Control and Prevention (CDC) growth charts.
All data included in this study can be de-identified and shared upon reasonable request to the corresponding author.
Clinical Family Case Reports
The proband (BAB12488, Figure 1A and B) is a 2-year-old male born to a healthy second degree-cousin consanguineous family at term following an uneventful pregnancy and elective Cesarean section delivery. Birth weight was 2.4 kg (−1.86 SD). He has two older affected brothers (BAB12491 and BAB12492) and one older unaffected brother (BAB12493); family history was positive for a stillborn infant born to the paternal grandmother, Sudden Infant Death Syndrome (SIDS) at 15 days of life in the non-identical twin of the stillborn infant, and epilepsy in one maternal cousin once removed and one paternal cousin once removed (Figure 1A–G, Figure S1).
The proband (Figure 1B) exhibited frequent episodes of generalized clonic seizures (GTCs) shortly after birth and was subsequently admitted to the neonatal intensive care unit (NICU) for 12 days, where he received phenobarbital and was seizure-free by the time of discharge. Seizures recurred in the subsequent weeks but were eventually controlled on a combination of antiepileptic drugs (AED) levetiracetam, phenytoin and clonazepam. Developmental delay was evident by 6 months of age. At the age of 8 months, phenobarbital was gradually weaned, with seizure recurrence after 2 months. This observed breakthrough seizure was described as generalized tonic stiffening of the limbs with deviation of the eyes and head to the left side, lasting 5 – 10 minutes; it occurred following a febrile illness. Carbamazepine was added to his AED regimen, and he became seizure free following dose escalation.
At his current age of 2 years, he demonstrates severe to profound GDD. He can roll, sit with support, and reach for objects. He does not have a pincer grasp and cannot feed himself. He can bear weight on his legs but cannot stand or walk independently. He can make monosyllables, knows two simple words only, smiles reciprocally and can recognize his parents. He is not toilet trained. Anthropometric measurements are as follows: OFC 46.3 cm (−1.65 SD), height 88 cm (+0.22 SD) and weight 11 kg (−1.37 SD). Physical examination showed an alert, playful, and socially interactive child. He displayed self-injurious behavior (finger biting). He also had esotropia, choreoathetotic movement of the limbs, hypertonia, brisk deep tendon reflexes throughout, and a flexor plantar response. His dental status (Figure 1E) revealed relatively normal dentition for age with no signs of delayed eruption, yellow teeth discoloration, worn molars or hypodontia. His lower canines were both present while upper canines have yet to erupt. There was no evidence of AI.
Clinical laboratory investigations at age 2 years included a normal complete blood count, electrolytes, liver and renal function test, and tandem mass spectroscopy on serum sample. Routine awake and asleep electroencephalogram was normal. Brain Magnetic Resonance Imaging (MRI) showed nonspecific enlargement of subarachnoid spaces with normal cerebrum, cerebellum, and myelination for age.
The oldest affected sibling (BAB12491, Figure 1C) is a 15-year-old male with GDD, profound intellectual disability (ID), and drug-resistant epilepsy (DRE). He was born at 37 weeks gestational age by uneventful vaginal delivery. The pregnancy was complicated by upper urinary tract infection at 20-week gestation which necessitated hospitalization and treatment with intravenous antibiotics. He exhibited GTC seizures at birth and was hospitalized and treated for 12 days in the NICU. Initially, seizures occurred twice a week, lasting between 5–15 minutes consistent with status epilepticus and mostly triggered by febrile illness. He was placed on carbamazepine and the seizures reduced in severity and frequency with age, with only occasional breakthrough seizures after 10 years of age triggered by fever. Delay in development was first evident at 6 months of life when he did not roll or demonstrate head support.
At his current age (15 years), he walks independently, can go up/downstairs, follows a few simple commands, speaks four simple monosyllable words only. He is not toilet trained, cannot use a spoon or cup to eat and drink independently, and is dependent on his family in all his activities of daily living (ADLs). His anthropometric measurements at current age (15 years) are as follows: OFC 52 cm (−1.95 SD), weight 38 kg (−2.29 SD) and height 147 cm (−2.76 SD). Examination showed an alert child with autistic behaviors including echolalia, impaired social interaction, and hand flapping. He has normal ocular movement. He had hypertonia, extensive burn-related scarring of the left side of the body causing skin contracture at the left anti-cubital area, and flexion posturing of the left elbow. He had normal deep tendon reflexes, ankle clonus bilaterally, flat feet, and genu varus deformity. His gait was ataxic. Examination of the dentition showed hypoplastic enamel supported by abnormally heavy plaque and calculus deposition (Figure 1F). The teeth had a dark yellow color due to the exposed dentin and the enamel surfaces were severely worn. There was no delayed eruption or hypodontia, while generalized gingival inflammation and marginal gingival overgrowth were present. AI type 1 (hypoplastic enamel) was highly suspected.
The second affected brother of the proband (BAB12492, Figure 1D) was an 11-year and 5-month-old male. He was born at 41 weeks gestational age by uneventful vaginal delivery. The pregnancy was complicated by maternal anemia at 16 weeks gestational age. Seizures first started on day of life three (DOL3) and he was admitted to the NICU where seizures were controlled with phenobarbital. The NICU admission was complicated by neonatal jaundice (maximum total serum bilirubin was 15 mg/dl) requiring phototherapy. He was eventually discharged on DOL13. He had occasional breakthrough seizures described as generalized tonic and usually triggered by fever. Seizure control improved after starting carbamazepine. Additionally, he exhibits sleep episodes of spontaneous arousal, screaming with loss of awareness concerning for seizures versus parasomnia. He started walking at the age of 3 years and can go up - and - down stairs. At 11 years of age, he is currently nonverbal but can follow a few simple commands. His comprehension is slightly better than his other affected siblings per parental report. He can use his hands, but not spoon, to eat and can drink from a cup. He is not toilet trained. On examination, growth parameters were as follows: OFC 50.5 cm (−2.08 SD), weight 31 kg (−1.16 SD), height 134 cm (−1.57 SD). He was alert and displayed mild autistic features and self-injurious behavior with evidence of thickening of the dorsum of the hands because of self-biting. Compared to his affected siblings, he was less hyperactive and was more socially engaged. He had mild hypertonia with normal deep tendon reflexes. His gait was ataxic. His dental examination (Figure 1G) showed defective and severely worn enamel surfaces, discolored teeth from exposed underlying dentin, and accumulation of higher calculus and plaque deposits. There were also incipient caries on the lower left canine, generalized gingival inflammation, malocclusion and flared anterior teeth (possible presence of anterior open bite). There is no delayed eruption or hypodontia on his dentition. Overall, these dental findings were consistent with AI type I (hypoplastic enamel).
Discussion
We report three affected siblings from a consanguineous Iraqi family with DEE25 due to an 88.5 Kb novel homozygous deletion involving SLC13A5 and XAF1, caused by AAMR.
Biallelic LoF variants in SLC13A5 cause DEE25 characterized by neonatal-onset epilepsy with fever-sensitivity, recurrent status epilepticus, global developmental delay/intellectual disability in addition to variable neurological features including ataxia, microcephaly, choreoathetosis and spasticity (Hardies et al., 2015; Klotz et al., 2016; Kopel et al., 2017; Matricardi et al., 2020; Schossig et al., 2017; Thevenon et al., 2014). The three affected siblings have the aforementioned features with profound GDD/ID although only one affected child, the eldest (BAB12492), had microcephaly (defined as OFC <−2 SD) while the other two had borderline microcephaly (− 1.65SD and z = − 1.95SD) thus fitting into the spectrum of SLC13A5-related disorders. The oldest affected sibling (BAB12491) also had short stature and failure to thrive. We have not observed any substantial phenotypic deviation from the SLC13A5 LoF spectrum on all three affected individuals in this family. Thus, the SLC13A5 deletion could explain all phenotypic features observed in these siblings. XAF1 is a XIAP-associated factor 1 (MIM *606717) with no associated human phenotype; its potential contribution to the phenotype in the siblings remains unclear.
Defects in tooth development such as hypoplasia, hypodontia and AI have been noted in many clinical reports as a pivotal sign associated with DEE25 (Hardies et al., 2015; Klotz et al., 2016; Kopel et al., 2017; Schossig et al., 2017). Yellow to orange discoloration of both primary and permanent teeth, as well as wide interdental spaces and abnormal crown formation, are major dental features of DEE25. Like that reported in Schossig et al, 2017, the two older siblings had evidence of AI type 1 (hypoplastic type). Interestingly, the proband had no evidence of dental abnormalities or AI at 2 years of age and his primary dentition appeared unaffected in contrast to previous reports (Matricardi et al., 2020; Schossig et al., 2017). However, dental X-rays were not available to evaluate for hypoplastic enamel.
CNVs account for a significant fraction of human genome variation and are increasingly recognized as causal for many Mendelian disorders (Dharmadhikari et al., 2019; Stankiewicz & Lupski, 2010; Yuan et al., 2020). Pathogenic or likely pathogenic CNVs and uniparental disomy (UPD) contribute to 10.6% of all molecularly diagnosed ES cases (Dharmadhikari et al., 2019). Biallelic CNVs, particularly small homozygous deletions (<1,000 Kb) embedded in a genomic block of AOH/ROH, are an under-recognized cause of autosomal recessive disease traits (AR-CNVs))(Yuan et al., 2020). These small intragenic exonic deletions often produce null alleles resulting in complete LoF of gene products (Alkuraya, 2015) and most affect only one gene. Such disease-causing alleles often arise de novo ancestrally as a “founder pathogenic variant” in a specific population and are ‘homozygosed’ through identity-by-descent, and thus are more frequently transmitted than dominant disease-causing alleles which have more substantial selection pressures (Lupski et al., 2011; Yuan et al., 2020). Given the high frequencies of structural variant (SV) mutagenesis at a locus, such CNV alleles can also appear as new mutations contributing to a compound heterozygous biallelic state to cause an AR disease trait rather than by a Clan Genomics derived allele subsequently inherited and brought to homozygosity by identity-by-descent (Lupski, 2007; Lupski et al., 2011).
CNV-calling methods using exome data, such as HMZDelFinder, demonstrate that sequence read-depth serves as a good indicator for such small CNVs; even those involving only a single exon. (Gambin et al., 2017). Such algorithms, when implemented can increase the molecular diagnostic ‘solved rate’ of ES by 3–8% and are best when integrated together with SNV analysis (Karaca et al., 2015; Pehlivan et al., 2019; Yuan et al., 2020). One such under-recognized mechanism for such exonic deletion alleles is AAMR (Boone et al., 2014), a form of microhomology mediated break induced replication (MMBIR) (Mayle et al., 2015); such AAMR derived deletion rearrangement alleles tend toward a genomic size range of 10s to 100s of Kb, and < 1 Mb; genomic sizes often overlooked in clinical CMA and ES studies (Song et al., 2018; Yuan et al., 2020). The AAMR risk score of SLC13A5 is 0.46 for MIM genes and 0.478 for RefSeq genes and is below the 0.6 threshold used to implicate genes that may be “at increased risk” for AAMR mediated genomic instability (Figure 2B), (Song et al., 2018). Yet, experimental evidence showed that this specific CNV-Alu rearrangement event was mediated by a pair of AluS elements, AluSx and AluSq (Figure 2C), located 88.5 Kb apart, with one AluS element residing in another adjacent gene, XAF1. Thus, this suggests that even genes with low-relative risk prediction scores could still be susceptible to formation of pathogenic CNVs through AAMR events; such genes can also be completely deleted using one Alu from the substrate pair mapping to the adjacent gene. Two pathogenic CNVs associated with phenotypes of DEE25 were previously submitted on ClinVar, encompassing the full-length of SLC13A5. Of note, one of the CNV, accession number VCV000442539.1, possesses a highly similar deletion boundaries to our case, including the full-length of SLC13A5 and first-exon of the XAF1. While further data regarding to the validation and break-point analysis of these two CNVs are not addressed on the ClinVar, these observations may provide additional evidence of genomic instability and potential recurrent copy number loss in this locus.
This case report further emphasizes the importance of utilizing read-depth based CNV detection tools such as HMZdelFinder as a necessary tool for comprehensive ES analysis (Lupski et al., 2020). Importantly, ES analysis following by a strategy of applying family-based experimental validations including aCGH, ddPCR and Sanger sequencing is not only necessary to elucidate molecular mechanisms underlying the genomic events via reaching to a single-base resolution, but also provide the footprints of given genomic events from familial segregation analysis. These benefits may not be achieved by performing whole-genome sequencing alone on the proband. The identification of such rare, pathogenic homozygous and compound heterozygous deletion alleles could facilitate better Mendelian disease gene discovery, molecular diagnosis, and more informed recurrence risk counseling.
Supplementary Material
Figure S1. Extended multigeneration pedigree of the family showing an affected paternal cousin once removed and an affected maternal cousin once removed with epilepsy. The molecular diagnosis could not be confirmed in these distant affected cousins due to inability to obtain their DNA samples.
Acknowledgement
We thank the family for taking part in this study.
Funding information
This study was supported in part by the U.S. National Human Genome Research Institute (NHGRI) and National Heart Lung and Blood Institute (NHLBI) to the Baylor-Hopkins Center for Mendelian Genomics (BHCMG, UM1 HG006542, J.R.L); NHGRI grant to Baylor College of Medicine Human Genome Sequencing Center (U54HG003273 to R.A.G.), U.S. National Institute of Neurological Disorders and Stroke (NINDS) (R35NS105078 to J.R.L.) and Muscular Dystrophy Association (MDA) (512848 to J.R.L.). D.M. is supported by a Medical Genetics Research Fellowship Program through the United States National Institute of Health (T32 GM007526-42). T.M. is supported by the Uehara Memorial Foundation. D.P. is supported by International Rett Syndrome Foundation (IRSF grant #3701-1). J.E.P. was supported by NHGRI K08 HG008986.
Footnotes
Conflict of interest
J.R.L. has stock ownership in 23andMe, is a paid consultant for Regeneron Pharmaceuticals, and is a co-inventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting. The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing conducted at Baylor Genetics (BG) Laboratories; JRL participates in the BG scientific advisory board. Other authors have no potential conflicts to report.
Reference
- Alkuraya FS (2015). Natural human knockouts and the era of genotype to phenotype. Genome Medicine, 7(1), 48. 10.1186/s13073-015-0173-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone PM, Yuan B, Campbell IM, Scull JC, Withers MA, Baggett BC, Beck CR, Shaw CJ, Stankiewicz P, Moretti P, Goodwin WE, Hein N, Fink JK, Seong MW, Seo SH, Park SS, Karbassi ID, Batish SD, Ordóñez-Ugalde A, … Lupski JR (2014). The Alu-rich genomic architecture of SPAST predisposes to diverse and functionally distinct disease-associated CNV alleles. American Journal of Human Genetics, 95(2), 143–161. 10.1016/j.ajhg.2014.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CMB, Vasanth S, Shinawi M, Russell C, Ramocki MB, Brown CW, Graakjaer J, Skytte AB, Vianna-Morgante AM, Krepischi ACV, Patel GS, Immken LD, Aleck K, Lim C, Cheung SW, Rosenberg C, Katsanis N & Lupski JR (2014). Dosage changes of a segment at 17p13.1 lead to intellectual disability and microcephaly as a result of complex genetic interaction of multiple genes. American Journal of Human Genetics, 95(5), 565–578. 10.1016/j.ajhg.2014.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharmadhikari AV, Ghosh R, Yuan B, Liu P, Dai H, Al Masri S, Scull J, Posey JE, Jiang AH, He W, Vetrini F, Braxton AA, Ward P, Chiang T, Qu C, Gu S, Shaw CA, Smith JL, Lalani S, … Bi W (2019). Copy number variant and runs of homozygosity detection by microarrays enabled more precise molecular diagnoses in 11,020 clinical exome cases. Genome Medicine, 11(1), 1–17. 10.1186/s13073-019-0639-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromer M & Purcell SM (2014). Using XHMM Software to Detect Copy Number Variation in Whole-Exome Sequencing Data. Current Protocols in Human Genetics, 81, 7.23.1–21. 10.1002/0471142905.hg0723s81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambin T, Akdemir ZC, Yuan B, Gu S, Chiang T, Carvalho CMB, Shaw C, Jhangiani S, Boone PM, Eldomery MK, Karaca E, Bayram Y, Stray-Pedersen A, Muzny D, Charng W-L, Bahrambeigi V, Belmont JW, Boerwinkle E, Beaudet AL, … Lupski JR (2017). Homozygous and hemizygous CNV detection from exome sequencing data in a Mendelian disease cohort. Nucleic Acids Research, 45(4), 1633–1648. 10.1093/nar/gkw1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal E, Miyauchi S, Martin PM, Ananth S, Srinivas SR, Smith SB, Prasad PD & Ganapathy V (2007). Expression and functional features of NaCT, a sodium-coupled citrate transporter, in human and rat livers and cell lines. American Journal of Physiology-Gastrointestinal and Liver Physiology, 292(1), G402–G408. 10.1152/ajpgi.00371.2006 [DOI] [PubMed] [Google Scholar]
- Gu S, Yuan B, Campbell IM, Beck CR, Carvalho CMB, Nagamani SCS, Erez A, Patel A, Bacino CA, Shaw CA, Stankiewicz P, Cheung SW, Bi W & Lupski JR (2015). Alu-mediated diverse and complex pathogenic copy-number variants within human chromosome 17 at p13.3. Human Molecular Genetics, 24(14), 4061–4077. 10.1093/hmg/ddv146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardies K, de Kovel CGF, Weckhuysen S, Asselbergh B, Geuens T, Deconinck T, Azmi A, May P, Brilstra E, Becker F, Barisic N, Craiu D, Braun KPJ, Lal D, Thiele H, Schubert J, Weber Y, van ‘t Slot R, Nürnberg P, … Consortium, on behalf of the autosomal recessive working group of the E. R. E. S. (2015). Recessive mutations in SLC13A5 result in a loss of citrate transport and cause neonatal epilepsy, developmental delay and teeth hypoplasia. Brain, 138(11), 3238–3250. 10.1093/brain/awv263 [DOI] [PubMed] [Google Scholar]
- Henke C, Töllner K, van Dijk RM, Miljanovic N, Cordes T, Twele F, Bröer S, Ziesak V, Rohde M, Hauck SM, Vogel C, Welzel L, Schumann T, Willmes DM, Kurzbach A, El-Agroudy NN, Bornstein SR, Schneider SA, Jordan J, … Löscher W (2020). Disruption of the sodium-dependent citrate transporter SLC13A5 in mice causes alterations in brain citrate levels and neuronal network excitability in the hippocampus. Neurobiology of Disease, 143(July), 105018. 10.1016/j.nbd.2020.105018 [DOI] [PubMed] [Google Scholar]
- Karaca E, Harel T, Pehlivan D, Jhangiani SN, Gambin T, Coban Akdemir Z, Gonzaga-Jauregui C, Erdin S, Bayram Y, Campbell IM, Hunter JV, Atik MM, Van Esch H, Yuan B, Wiszniewski W, Isikay S, Yesil G, Yuregir OO, Tug Bozdogan S, … Lupski JR (2015). Genes that Affect Brain Structure and Function Identified by Rare Variant Analyses of Mendelian Neurologic Disease. Neuron, 88(3), 499–513. 10.1016/j.neuron.2015.09.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaca E, Posey JE, Coban Akdemir Z, Pehlivan D, Harel T, Jhangiani SN, Bayram Y, Song X, Bahrambeigi V, Yuregir OO, Bozdogan S, Yesil G, Isikay S, Muzny D, Gibbs RA & Lupski JR (2018). Phenotypic expansion illuminates multilocus pathogenic variation. Genetics in Medicine, 20(12), 1528–1537. 10.1038/gim.2018.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klotz J, Porter BE, Colas C, Schlessinger A & Pajor AM (2016). Mutations in the Na+/citrate cotransporter NaCT (SLC13A5) in pediatric patients with epilepsy and developmental delay. Molecular Medicine, 22(5), 310–321. 10.2119/molmed.2016.00077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopel JJ, Bhutia YD, Ramachandran S, Lawrence JJ, Neugebauer V & Ganapathy V (2017). Opinion Tooth Hypoplasia for Differential Diagnosis of Childhood Epilepsy Associated with SLC13A5 Mutations-International Journal of Neurological Disorders SCIRES Literature-Volume 1 Issue 2- www.scireslit.com. Int J Neurol Dis, 1(2), 033–037. www.scireslit.com [Google Scholar]
- Lupski JR (2007). Genomic rearrangements and sporadic disease. Nature Genetics, 39(7S), S43–S46. 10.1038/ng2084 [DOI] [PubMed] [Google Scholar]
- Lupski JR, Belmont JW, Boerwinkle E & Gibbs RA (2011). Clan genomics and the complex architecture of human disease. Cell, 147(1), 32–43. 10.1016/j.cell.2011.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR, Liu P, Stankiewicz P, Carvalho CMB & Posey JE (2020). Clinical genomics and contextualizing genome variation in the diagnostic laboratory. Expert Review of Molecular Diagnostics, 20(10), 995–1002. 10.1080/14737159.2020.1826312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matricardi S, De Liso P, Freri E, Costa P, Castellotti B, Magri S, Gellera C, Granata T, Musante L, Lesca G, Oertel J, Craiu D, Hammer TB, Møller RS, Barisic N, Abou Jamra R, Polster T, Vigevano F & Marini C (2020). Neonatal developmental and epileptic encephalopathy due to autosomal recessive variants in SLC13A5 gene. Epilepsia, 61(11), 2474–2485. 10.1111/epi.16699 [DOI] [PubMed] [Google Scholar]
- Mayle R, Campbell IM, Beck CR, Yu Y, Wilson M, Shaw CA, Bjergbaek L, Lupski JR & Ira G (2015). Mus81 and converging forks limit the mutagenicity of replication fork breakage. Science, 349(6249), 742 LP–747. 10.1126/science.aaa8391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehlivan D, Bayram Y, Gunes N, Coban Akdemir Z, Shukla A, Bierhals T, Tabakci B, Sahin Y, Gezdirici A, Fatih JM, Gulec EY, Yesil G, Punetha J, Ocak Z, Grochowski CM, Karaca E, Albayrak HM, Radhakrishnan P, Erdem HB, … Lupski JR (2019). The Genomics of Arthrogryposis, a Complex Trait: Candidate Genes and Further Evidence for Oligogenic Inheritance. The American Journal of Human Genetics, 105(1), 132–150. 10.1016/j.ajhg.2019.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riemann D, Wallrafen R & Dresbach T (2017). The Kohlschütter-Tönz syndrome associated gene Rogdi encodes a novel presynaptic protein. Scientific Reports, 7(1), 1–14. 10.1038/s41598-017-16004-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schossig A, Bloch-Zupan A, Lussi A, Wolf NI, Raskin S, Cohen M, Giuliano F, Jurgens J, Krabichler B, Koolen DA, de Macena Sobreira NL, Maurer E, Muller-Bolla M, Penzien J, Zschocke J & Kapferer-Seebacher I (2017). SLC13A5 is the second gene associated with Kohlschütter–Tönz syndrome. Journal of Medical Genetics, 54(1), 54 LP–62. 10.1136/jmedgenet-2016-103988 [DOI] [PubMed] [Google Scholar]
- Song X, Beck CR, Du R, Campbell IM, Coban-Akdemir Z, Gu S, Breman AM, Stankiewicz P, Ira G, Shaw CA & Lupski JR (2018). Predicting human genes susceptible to genomic instability associated with Alu/Alu-mediated rearrangements. Genome Research, 28(8), 1228–1242. 10.1101/gr.229401.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P & Lupski JR (2010). Structural variation in the human genome and its role in disease. Annual Review of Medicine, 61, 437–455. 10.1146/annurev-med-100708-204735 [DOI] [PubMed] [Google Scholar]
- Thevenon J, Milh M, Feillet F, St-Onge J, Duffourd Y, Jugé C, Roubertie A, Héron D, Mignot C, Raffo E, Isidor B, Wahlen S, Sanlaville D, Villeneuve N, Darmency-Stamboul V, Toutain A, Lefebvre M, Chouchane M, Huet F, … Rivière JB (2014). Mutations in SLC13A5 cause autosomal-recessive epileptic encephalopathy with seizure onset in the first days of Life. American Journal of Human Genetics, 95(1), 113–120. 10.1016/j.ajhg.2014.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan B, Wang L, Liu P, Shaw C, Dai H, Cooper L, Zhu W, Anderson SA, Meng L, Wang X, Wang Y, Xia F, Xiao R, Braxton A, Peacock S, Schmitt E, Ward PA, Vetrini F, He W, … Bi W (2020). CNVs cause autosomal recessive genetic diseases with or without involvement of SNV/indels. Genetics in Medicine, 22(10), 1633–1641. 10.1038/s41436-020-0864-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Extended multigeneration pedigree of the family showing an affected paternal cousin once removed and an affected maternal cousin once removed with epilepsy. The molecular diagnosis could not be confirmed in these distant affected cousins due to inability to obtain their DNA samples.