

Abstract
Prostate cancer is one of the most prominent malignancies of elderly men in many Western countries including Europe and the United States with increasing trend worldwide. The growth of normal prostate as well as of prostate carcinoma cells depends on functional androgen receptor (AR) signaling. AR manifests the biological actions of androgens and its transcriptional activity is known to be influenced by signal transduction pathways. Here we show that Src, a nonreceptor tyrosine kinase, is overexpressed in androgen-independent prostate carcinoma C4-2 cells. Interestingly, the expression of Src was found to progressively increase (up to threefold) in transgenic adenocarcinoma of mouse prostate mice as a function of age and cancer progression. Blocking Src kinase function by a specific inhibitor, PP2, resulted in decreased AR transactivation function on two different reporters, mouse mammary tumor virus (MMTV) and prostate-specific antigen (PSA). Consistent with this, overexpression of a functional Src mutant also led to a dramatic decrease in AR transactivation potential in a hormone-dependent manner. Interference with Src function in C4-2 cells led to decreased recruitment of AR on the target gene PSA enhancer and also resulted in the abrogation of hormone-dependent PSA transcript induction. Src inhibition also led to a dramatic decrease in the cell invasion in addition to decreasing the cellular growth. We suggest that targeting Src kinase could be an effective strategy to inhibit prostate cancer growth and metastasis.
Keywords: androgen receptor, casodex, invasion, prostate cancer, PP2, Src kinase
Introduction
Prostate cancer (PCa) is an endocrine malignancy, which initially depends on male sex hormones, testosterone and dihydrotestosterone for growth. Androgen receptor (AR), which is a member of nuclear hormone receptor superfamily, manifests the biological action of male hormones. AR has a modular structure containing the N terminus harboring transcriptional activation domain(s) (or transactivation domain), a central DNA-binding domain (DBD) and a C-terminal ligand-binding domain (LBD). Binding of androgens to the LBD induces conformational changes in the AR and subsequently results in the shuttling of the receptor to the target cell nucleus where it forms homodimer that is recruited on the androgen response element (ARE) present in the regulatory element on the target genes such as prostate-specific antigen (PSA) and regulates growth of prostate gland by modulating the expression of target genes implicated in growth and proliferation (Young et al., 1992; Perry et al., 1996).
To block AR function, androgen ablation using antiandrogens, in particular by Casodex (bicalutamide), remains the hormone therapy for PCa. This treatment unfortunately proves ineffective as the androgen-independent tumor eventually emerges in a short span of 3–4 semesters for which there is no curative therapy (Isaacs, 2000). It is noteworthy that hormone-refractory PCa cells nevertheless depend on functional AR signaling for growth as their growth is severely compromised if AR is depleted in cells using RNAi or ribozyme (Chen et al., 1998, 2004; Haag et al., 2005; Liao et al., 2005). This makes AR a prime drug target and AR-regulated genes as potential biomarkers in the treatment and diagnosis of advanced PCa, respectively.
It has been proposed that in the absence of a ligand, the AR activation could take place by cross talk with various growth factors known to be components of protein kinase pathways. For example, it has been demonstrated that epidermal growth factor and its receptor, keratinocyte growth factor, insulin-like growth factor-1, protein kinase A (PKA), mitogen-activated protein kinase (MAPK), as well as interleukin 6 (IL-6) could activate the AR signaling (Culig et al., 1994; Abreu-Martin et al., 1999; Craft et al., 1999; Sadar, 1999; Jenster, 2000). An additional mechanism underlying ligand-dependent activation of the AR by these alternative pathways may involve phosphorylation of either the AR or its associated cofactor proteins (Sadar, 1999; Dotzlaw et al, 2002; Ueda et al, 2002).
In this study we demonstrate that Src, a nonreceptor tyrosine kinase, is overexpressed in PCa and interference with Src function results in inhibition of PCa growth via regulation of AR function and decreased cell invasion. We also demonstrate that Src inhibition results in the interference of AR functioning. We suggest that Src inhibition, either by relatively safer chemical inhibitors or by natural diet-based compounds may be an effective strategy for inhibition of PCa growth and invasion.
Results
Src kinase is overexpressed in androgen-independent prostate carcinoma cells
Western blot analysis was performed to screen for endogenous Src expression in a panel of normal as well as PCa cells. Here, in addition to normal prostate epithelial cells (PrEC), androgen-dependent LNCaP prostate carcinoma, androgen independently growing PC3, 22Rν1 and C4-2 cells were included. Interestingly, C4-2 cells are derivatives of LNCaP cells and express functional endogenous AR; they can grow in an androgen-independent manner making them an excellent model representing transition of the initial androgen-dependent disease to an androgen-independent state (Thalmann et al, 1994). The data indicate that the expression of Src kinase was modest in PrEC normal human PCa cells (Figure 1a). The expression of Src was significantly higher in androgen-independent PCa cell lines PC3 (two fold), C4-2 (three fold) and slightly higher in 22Rν1 cells when compared with normal PrEC (Figure 1a).
Figure 1.

Src kinase is overexpressed in androgen independently growing C4-2 prostate carcinoma cells and its blockade induces apoptosis in C4-2 cells. (a) Western blot analysis of endogenous Src expression was carried out as described in ‘Materials and methods’. Equal loading of protein was confirmed by stripping the membrane and reprobing it with β-actin. Values on top of bands represent relative densities normalized to β-actin. (b) Immunofluorescence analysis to detect apoptotic cells based on the detection of annexin V was carried out as described in ‘Materials and methods’. (c) Quantification of apoptosis is presented. (d) A total of 200 000 C4-2 cells were cultured in 10 cm tissue culture dishes in T-media containing 10% normal serum for a period of 6 weeks. Cells were then provided media twice a week along with 1 μM PP2. After 6 weeks cells were counted and photographed. The bar graph shows relative cell number. Experiments were repeated three times with similar observations and representative figures are shown.
Interestingly, we also found that Src expression is progressively increased during the progression of PCa in transgenic adenocarcinoma of mouse prostate (TRAMP) model that spontaneously develops prostate tumor with increasing age (Greenberg et al., 1995). A progressive increase in the Src protein expression and its phosphorylation at Tyr418 in the activation loop of kinase domain that upregulates its activity was observed in TRAMP mice as cancer progressed from not detectable cancer at 8 weeks to well-differentiated carcinoma at 16 weeks and finally to moderately differentiated carcinoma at 24 weeks (Figure 2a). The expression of Src protein in nontransgenic littermates was found to be constant (data not shown). Similar results were observed by immunohistochemical analysis of the prostate tissue of TRAMP mice (Figure 2b). Mice with moderately differentiated (24 weeks) exhibited strong staining for Src, with negligible staining in their nontransgenic counterparts. This staining was especially seen in the epithelia of prostatic acini and also in the stroma. These results clearly indicate the progressive increase in Src expression during the progression of PCa.
Figure 2.

Expression of Src in the dorsolateral prostate during progressive stages of prostate cancer development in transgenic adenocarcinoma of mouse prostate (TRAMP) mice. (a) Protein levels of Src and phospho-Src (Tyr 416) by immunoblot analysis. Equal loading of protein was confirmed by stripping the blot and reprobing with β-actin antibody. Western blot analysis was conducted in five animals in each group, and only representative blots are shown. Values on top of bands represent relative densities normalized to β-actin. (b) Immunohistochemical analysis as detailed in ‘Materials and methods’, the protein levels were determined in the dorsolateral prostate of 8-, 16-, and 24-week-old TRAMP mice. Immunostaining data were confirmed in two slides from five animals. Photomicrographs (magnification, × 20 and × 40) represent immunohistochemical staining for Src in TRAMP mice with moderately differentiated carcinoma (24 weeks).
Inhibition of Src kinase induces apoptosis and in combination with Casodex further enhances apoptotic response in androgen-independent prostate cancer C4-2 cells
To address the role of Src overexpression in PCa cells, C4-2 cells, which showed a robust expression of Src, were chosen for further studies. Cells were treated with PP2, a specific chemical inhibitor of Src kinase. Treatment of C4-2 cells with potent antiandrogens, in particular by Casodex, had marginal effect on cellular growth similar to untreated control cells indicating their androgen-refractory/independent behavior (Figures 1b and c). However, PP2 treatment led to a dramatic increase (seven fold) in the apoptosis induction identified based on the increased expression of annexin V, an early marker of apoptosis. C4-2 cells co-treated with PP2 and Casodex exhibited a further heightened apoptosis induction (ten fold; Figures 1b and c). To address the role of Src kinase in growth promotion, a cell growth assay was performed. Incubation of C4-2 cells with Src inhibitor led to a 50% decrease in cell growth (Figure 1d). These findings indicate that Src overexpression in androgen-independent PCa can serve to defy response to antiandrogen therapy.
Src specifically potentiates AR transactivation function in C4-2 cells
AR transactivation is critical for PCa progression and the antiandrogen therapy is known to inactivate AR function by decreasing its transactivation. We therefore determined the possible role of Src in modulating AR transactivation function using luciferase reporter assays. C4-2 cells were transfected with MMTV-Luc and PSA-Luc androgen-responsive reporters. We found that AR-mediated transactivation repressed to almost 50% on both mouse mammary tumor virus (MMTV) and PSA reporter elements by treatment of cells with Src inhibitor PP2 (Figure 3a). This suggests that in androgen independently growing PCa cells, AR is still functional on its target genes and that its transactivation function is potentiated by endogenously overexpressed Src kinase. To test, whether PP2-mediated decrease in target gene expression was specific to androgen-responsive promoters and was not due to decrease in global transcription, another control reporter plasmid pCMV-LacZ, which is not responsive to androgens was used. As expected, the reporter activity remained unaffected by Src inhibition (data not shown) suggesting that PP2 treatment specifically decreases AR transactivation on its target genes.
Figure 3.

Src kinase inhibition attenuates androgen receptor (AR) transactivation on reporter genes in C4-2 cells. (a) C4-2 cells were seeded out in 10% serum containing T-media and transfected with mouse mammary tumor virus (MMTV)-Luc or prostate-specific antigen (PSA)-Luc reporter plasmids as detailed in ‘Materials and methods’ section. Luciferase values obtained without the inhibitor in each case were set as 1. The graph represents the activation of reporter expression as LacZ normalized relative luciferase units. (b) C4-2 grown in T media supplemented with 10% charcoal-stripped fetal bovine serum (FBS) and transfected with MMTV-Luc reporter along with expression plasmid Src-wt or dominantnegative Src. Cells were treated as described in the ‘Materials and methods’ section. Luciferase values were normalized with renilla luciferase internal control. Values obtained without the inhibitor and ligand were set as 1. The graph represents the activation of reporter as fold hormone induction. Experiment was repeated three times with similar observations, a representative figure is shown.
To further confirm these findings reporter assays were performed with ectopically expressed Src-wt (wild-type) or Src dominant-negative mutant in C4-2 cells. As compared to control, where a robust hormone induction was observed (Figure 3b), PP2 co-treatment led to a decrease in AR transactivation. However, ectopic expression of Src-wt did not further increase hormone-dependent AR transactivation suggesting that both ectopic wt-Src and endogenous kinases act through the similar pathway to enhance AR transactivation. Consistent with the inhibitor data, interference with wt-Src function by ectopic overexpression of dominant-negative Src mutant led to a potent decrease in AR transactivation in a hormone-dependent manner, in line with the inhibitor data (Figure 3a). This indicates that enhanced transactivation by AR observed in C4-2 cells is in part boosted by overexpression of Src kinase as evident from PP2 chemical as well as Src mutant-mediated inhibition of wt-Src function, in both cases it led to a decrease in AR transactivation function.
Src kinase inhibition decreases AR recruitment on PSA enhancer and modulates PSA expression
To determine the effect of Src inhibition on AR recruitment to its target genes, chromatin immunoprecipitation (ChIP) assay was performed with C4-2 cells. A substantial recruitment of AR was observed on androgen-responsive PSA enhancer in the absence of androgen agonist R1881 (Figure 4a). Treatment of C4-2 cells with R1881 led to a further increase in AR recruitment indicating hormone responsiveness of these cells. Intriguingly, C4-2 cells pretreated with Src inhibitor exhibited drastic loss of AR recruitment on PSA enhancer in the absence of R1881. However, AR was strongly re-recruited upon treatment of PP2-pretreated cells with R1881.
Figure 4.
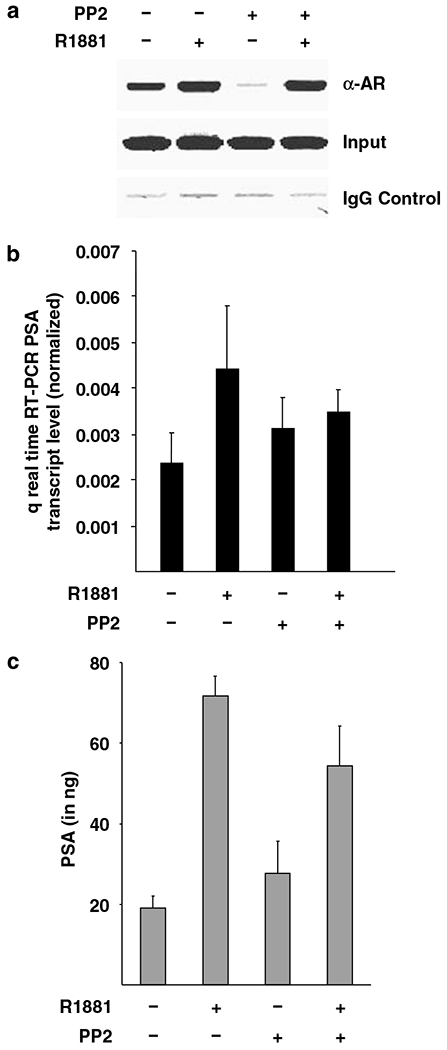
Src inhibition modulates the recruitment of androgen receptor (AR) on prostate-specific antigen (PSA) enhancer and attenuates PSA transcript induction in C4-2 cells. (a) A total of 750 000 C4-2 cells were grown for 4 days in 10% fetal bovine serum (FBS) containing media and were further grown for 48 h in hormone-depleted FBS containing T-media and treated with 1 μM PP2. Before lysis, cells were treated with ligands R1881 (10−8M) for 1 h after which chromatin immunoprecipitation (ChIP) procedure was followed as described in the ‘Materials and methods’. Purified DNA was PCR amplified by specific primers spanning the enhancer region of PSA containing ARE III and pictured. (b) In total 200000 C4-2 cells per well were seeded out in hormone-depleted FBS containing T-media in six-well tissue culture dishes. After 24 h cells were treated with R1881 (10−10M) for 48 h. Afterwards total cellular RNA was isolated, reverse transcribed to cDNA and amplified by light cycler using specific primers and control primers for actin. The graph represents the actin-normalized values of the PSA transcript. (c) Media supernatant from the above experiment was used to test for PSA secretion using kit as described in ‘Materials and methods’. Experiment was repeated three times with similar observations, a representative figure is shown.
To test the implication of the modulation of AR recruitment by Src blockade on target gene expression, real-time RT-PCR and enzyme-linked immunosorbent assay (ELISA) for PSA were performed. Treatment with R1881 led to further three fold induction of PSA gene transcripts, in agreement of enhanced AR recruitment under that condition (Figure 4b). R1881-mediated induction of PSA mRNA expression was lost in C4-2 cells pretreated with PP2. Consistent with this, the induction of PSA secretion in response to hormone was also abolished in C4-2 cells pretreated with PP2 (Figure 4c). Taken together, results suggest that a basal level of PSA transcripts are expressed in C4-2 cells in the absence of agonist. Agonist R1881 leads to enhanced AR transactivation function on target genes reflected in the induction of PSA gene transcription. PP2 treatment (the extent blockade was not tested, therefore a milder expression) suggests that induction of PSA transcription by AR in response to agonist is significantly contributed by Src signal transduction pathway, inhibiting Src activity therefore decreased AR target gene expression and hormone-responsive induction. Treatment of C4-2 cells growing in androgen-depleted T-media with Src kinase inhibitor PP2 alone however, leads to loss of AR recruitment, surprisingly no further decrease in PSA gene transcript was observed. This suggests that the C4-2 cells express basal level of PSA mRNA independent of AR transactivation function. However, induction of PSA expression by AR requires not only enhanced AR occupancy but also Src-mediated signaling plays an important role in AR transactivation leading to enhanced PSA expression.
Src kinase inhibition suppresses colony formation in C4-2 cells
As shown above (Figure 1d) incubation of C4-2 cells with Src inhibitor led to a 50% decrease in cell growth. The long-term functional consequence of PP2-mediated decrease in AR transactivation and reduced target gene expression in response to agonist was tested in relation to cellular growth. C4-2 cells stably expressing either wt-Src or Src dominant-negative mutants were therefore generated. C4-2 cells stably overexpressing Src-wt formed large-sized colonies indicating their accelerated growth (Figure 5), which was comparable to colonies derived from C4-2 stably transfected with empty vector. However, stable clones of C4-2 overexpressing the dominant-negative mutant form of Src showed a marked reduction in colony size indicating the growth-promoting effect of Src kinase. Taken together these results indicate that androgen independently growing cells overexpress Src kinase that is associated with enhanced growth.
Figure 5.

Src inhibition decreases growth of C4-2 cells. The stable clones of C4-2 overexpressing Src-wt or Src-mut were generated as described in ‘Materials and methods’. Experiment was repeated three times with similar observations, a representative photomicrograph is shown.
Src inhibition decreases C4-2 cell invasion
As the tumor metastasis by invading cells remains a major cause of therapy failure in PCa patients, an invasion assay was performed to test whether inhibition of Src may modulate invasive potential of C4-2 cells, which are known to have high metastatic potential (Thalmann et al., 2000). To rule out the fact that a decrease in cell number may result in decreased invasion, a time frame of 20 h at which no apoptosis was detectable was selected. While untreated and Casodex-treated C4-2 cells showed a vigorous migration through extracellular matrix, cells treated with PP2 showed a potent inhibition of their migratory capability (Figure 6a). The combination of PP2 and antiandrogen Casodex did not further decrease the invasion by these cells suggesting that Src-assisted invasion of C4-2 cells may occur independent of genomic action of AR function. Nevertheless, it may involve nongenomic AR actions whereby MAPK–Ras–Raf axis stimulation by AR signaling (Peterziel et al., 1999) may play a role in invasion and metastasis. To further strengthen these findings, stable clones overexpressing wt or mut Src were generated and the impact of Src kinase was tested on cell migration through extra cellular matrix. Here, no change in cell migration was observed between cells overexpressing either empty vector or Src-wt (Figure 6b), interestingly however, C4-2 cells overexpressing mutant form of Src showed drastic reduction in their migratory capacity (Figure 6b, last panel).
Figure 6.

Src inhibition decreases invasive potential of C4-2 cells. (a, b) A total of 70 000 wt-C4-2 cells or Src stable clones were suspended in culture media and the experiment was performed as described in ‘Materials and methods’. Wt C4-2 cells were treated with PP2 and allowed to invade for 20 h followed by staining as per manufacturer’s protocol. Experiment was repeated three times with similar observations and representative photomicrographs of invading cells are shown.
Discussion
The transcriptional activation function of AR is not only essential for the normal sexual development in men but is also implicated in the progression of PCa (Chen et al., 2004; Notini et al., 2005). Src kinase is activated in breast carcinoma where it has been shown to promote agonistic action of tamoxifen (Shah and Rowan, 2005). However, its role remains poorly understood in PCa. Our observation that Src expression proportionates with enhanced androgenic independence suggests that Src kinase may play a nonredundant function in potentiating AR function in PCa cells and thereby may serve as an important therapeutic target. Also, in TRAMP model expression and activating phosphorylation of Src is enhanced indicating its association with PCa growth that is regulated by AR. It is evident from the experiment that C4-2 cells show a basal leaky expression of PSA gene transcripts in the absence of agonist R1881 (Figure 3b), which may be explained largely by the agonist-independent recruitment of AR on PSA enhancer or may due to the androgen-independent leaky expression of PSA.
The AR is synthesized as a single 110 kDa protein, which becomes rapidly phosphorylated to a 112 kDa protein (Brinkmann et al., 1992). A recent report has suggested that AR tyrosine phosphorylation is in fact induced by growth factors and elevated in hormone-refractory prostate tumors and that mutation in these tyrosine residues significantly inhibits the growth of PCa cells under androgen-depleted conditions (Guo et al., 2006). The study further showed a positive correlation of AR tyrosine phosphorylation with Src activity in human PCa. Src kinase is known to be overexpressed in PCa and enhances the AR function at very low concentration of agonist R1881 (Castoria et al., 2003). In the androgen-depleted environment, the recruitment of AR on PSA enhancer was completely abolished without altering basal expression. It is possible that phosphorylation of various domains may have different functional consequences on receptor activation, suggesting that Src enhances the transactivation function of AR and may also cooperate in its ability to bind to target genes.
Another set of co-regulatory molecules such as AR co-repressors repress AR transactivation function and therefore inhibit PCa cell growth and their expression is decreased in some PCa cells (Baniahmad, 2005; Wang et al., 2005; Dehm and Tindall, 2007). We have recently shown that stable overexpression of co-repressors alien in LNCaP cells leads to an antihormone-dependent decrease in cell growth (Moehren et al., 2007). Also, PKA pathway leads to reduced co-repressor SMRT binding to AR and thus could be a pathway by which PKA activates AR (Dotzlaw et al., 2002) and a similar possibility might apply also for Src pathway. The decrease in the AR transactivation observed may therefore be due to the enhanced interaction of corepressors to AR and thereby a decrease in AR transactivation and local chromatin condensation associated with lower accessibility of transcription factors.
In addition a decrease in the phosphorylation of critical AR residues in the amino terminus by Src inhibition may also explain the observed decrease in AR transactivation. C4-2 cells also express PSA at a basal level, which could be attributed to the androgen-independent marginal recruitment of AR allowing the expression of PSA, in line with a report suggesting PSA basal expression in androgen independently growing cells do not require AR binding (Jia and Coetzee, 2005). However, induction of PSA mRNA by R1881 is associated with enhanced AR binding (Figure 4a). This induction of AR-mediated PSA expression may require Src kinase signaling blocking that leads to decreased target gene expression analysis (Figures 4b and c). Long-term PP2 treatment leads to growth inhibition of C4-2 cells in androgen-containing growth factor rich T-media, suggesting that PP2-mediated Src blockade not only affects induction of target gene expression, but also leads to decreased growth of C4-2 cells presumably by interfering with the expression of target genes that play important function in cell growth and proliferation. Another important finding of this work is the promotion of cell invasion by Src kinase. As a result, interfering with Src function either chemically or by expressing dominant-negative Src leads to robust decrease in cell invasion. Blocking Src function could therefore serve as an important strategy against metastasis of PCa and thereby may result in more effective treatment of a prostate-confined disease either by radiotherapy or surgery. In addition, Src function may be explored to understand and better treat other endocrine malignancies. We suggest that employment of Src inhibitors may therefore offer a great therapeutic advance by prostate-specific confinement of the tumor, thereby making the disease manageable by currently available therapies, both androgen ablation and surgical management.
Materials and methods
Materials
AR and Src antibodies were obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA) and Cell Signaling (Danvers, MA, USA), respectively. Anti-mouse and anti-rabbit secondary horseradish peroxidase (HRP) conjugates were from Amersham Life Science Inc. (Piscataway, NJ, USA), R1881 from PerkinElmer (Waltham, MA, USA). Annexin V-Fluos-based immunofluorescent apoptosis staining kit was from Roche (Berkeley, CA, USA). Guanidium HCl and hematoxylin were from Sigma Chemicals (St Louis, MO, USA). RNA purification kit was from Qiagen (Valencia, CA, USA). BCA protein assay kit was obtained from Pierce (Rockford, IL, USA). Luciferase assay reagent was purchased from Promega (Madison, WI, USA). The human PSA ELISA kit was purchased from Anogen (Ontario, Canada). Diaminobenzidine was obtained from DakoCytomation (Carpinteria, CA, USA) and Novex precast Tris-glycine gels from Invitrogen. PP2 was procured from Calbiochem (San Diego, CA, USA). Src-wt and mutant plasmids were kind gift from Dr Sarah Courtneidge.
Protein extraction and western blotting
Protein lysates from cultured cells were prepared, 40 μg protein was subjected to SDS–polyacrylamide gel electrophoresis (PAGE) and western blot was performed as previously described (Siddiqui et al., 2006). The extraction of tumor proteins from TRAMP mice has been described previously (Adhami et al., 2004). Densitometric measurements of the bands in western blot analysis were done using digitalized scientific software program, UN-SCAN-IT, purchased from Silk Scientific Corporation (Orem, UT, USA).
Immunohistochemical analysis
Sections (4 μm) were cut from paraffin-embedded prostate tissues. Immunostaining was performed using specific antibodies with appropriate dilutions and was replaced with either normal host serum or block for negative controls, followed by staining with appropriate HRP-conjugated secondary antibodies. The slides were developed in diaminobenzidine and counter stained with a weak solution of hematoxylin stain as described previously (Adhami et al., 2004). The stained slides were dehydrated and mounted in Permount and visualized on a Zeiss-Axiophot DM HT microscope (Zeiss-Axiophot, Jena, Germany). Images were captured with an attached camera linked to a computer.
Apoptosis detection by fluorescence microscopy
The cells were grown on cell culture slides (BD Biosciences, Rockville, MD, USA) and then treated with 1 μM of PP2 for 48 h period of time. The cells were then incubated for 10 min with Annexin V-Fluos labeling reagent to detect annexin, an early marker of apoptosis and analysed using fluorescence microscopy (excitation at 450–500 nm and detection at 515–565 nm). Cells were visualized by Zeiss confocal microscope and green fluorescent cells were scored as apoptotic.
Cell culture and transient transfection
LNCaP cells were grown as previously described (Moehren et al., 2007). PC3 and 22Rν1 were grown in RPMI-1640 medium containing 10% fetal bovine serum (FBS). Human PrEC were obtained from Cambrex Bioscience (Walkersville, MD, USA) and grown in prostate epithelial basal cell medium (Cambrex Bioscience) according to the manufacturer’s instructions and the C4-2 cells were in T-media (Invitrogen) containing 10% FBS (American type culture collection) and 1% penicillin–streptomycin solution (Invitrogen). Transfections were performed using a modified calcium-phosphate method (Dotzlaw et al., 2002). In total, 300 000 C4-2 cells were seeded out in six-well dishes and transfected with 1 μg of reporter plasmid MMTV-Luc or PSA-Luc; 0.2 μg either of pCMV-LacZ or renilla luciferase plasmid served as internal control; 1 μg either of wt-Src vector or the dominant-negative Src was used for transfection. Cells were treated with the agonist R1881 (10−9M) and harvested 72 h posttransfection using passive lysis buffer provided with luciferase assay reagent to measure both luciferase activity and β-galactosidase or renilla luciferase activity. Independent triplicate experiments were performed each time and were repeated at least three times. Error bars represent the deviation of the mean value.
Chromatin immunoprecipitation assay
C4-2 ChIP experiment was performed essentially as described previously (Moehren et al., 2007). ChIP experiments were repeated at least three times with similar results.
Real-time RT-PCR
Isolation of mRNA and the real-time PCR was performed as described earlier (Moehren et al., 2007). A total of 200 000 C4-2 cells per well were seeded out in charcoal-stripped serum containing T-media in six-well tissue culture dishes. After 24 h cells were treated with R1881 (10−10M) for 48 h, total cellular RNA was isolated, 1 μg RNA was reverse-transcribed to cDNA and was subjected to amplification by light cycler using specific primers and control primers against actin.
Enzyme-linked immunosorbent assay
The human PSA ELISA kit was used for the quantitative analysis of PSA levels in culture medium as per manufacturer’s protocol. C4-2 cells were treated with Src inhibitor PP2 and/or Casodex, and 200 μl media was assayed.
Generation of stable clones
A total of 200 000 C4-2 cells were transfected with wt-Src or mutant Src along with pETE-Hyg plasmid in 5:1 molar ratio with (total amount being 10 μg) using electroporation kit from AMAXA (Gaithersburg, MD, USA). The medium was changed and replaced with fresh T-media 18 h posttransfection. Stable clones were selected as described previously (Moehren et al., 2007).
Invasion assay
The assay was performed following the manufacturer’s protocol. Briefly, 500 μl of complete T-media was added in lower chamber as chemoattractant and 70 000 cells suspended in serum free medium were applied to the inner chamber. Treatment with Src inhibitor and Casodex was given in the inner chamber either alone or in combination and the cells were incubated for 20 h in humidified incubator. Invading cells were stained and photomicrographed as per manufacturer’s instruction.
Statistical analysis
All assays were repeated in three independent experiments, and only representative blots are presented. Immunoblots were scanned by HP PrecisionScan Pro 3.13 (Hewlett-Packard, Palo Alto, CA, USA). Densitometry measurements of the scanned bands were done using digitalized scientific software program UN-SCAN-IT. Data were normalized to β-actin and expressed as mean ± s.e. followed by appropriate statistical analysis.
Acknowledgements
We thank Dr GP Reddy for gift of Casodex, Dr GN Thalmann for providing C4-2 cells and Dr Sarah Courtneidge for mammalian expression vectors of wt and mutant Src. This work was supported by US PHS Grants RO1CA78809; RO1CA101039, RO1CA120451 and O’Brian center Grant P50DK065303-01 to HM and Association of International Cancer research (AICR, UK) and DFG-BA1457/3 grant to AB.
References
- Abreu-Martin MT, Chari A, Palladino AA, Craft NA, Sawyers CL. (1999). Mitogen activated protein kinase kinase kinase 1 activates androgen receptor-dependent transcription and apoptosis in prostate cancer. Mol Cell Biol 19: 5143–5154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhami VM, Siddiqui IA, Ahmad N, Gupta S, Mukhtar H. (2004). Oral consumption of green tea polyphenols inhibits insulin-like growth factor-I-induced signaling in an autochthonous mouse model of prostate cancer. Cancer Res 64: 8715–8722. [DOI] [PubMed] [Google Scholar]
- Baniahmad A. (2005). Nuclear hormone receptor co-repressors. J Steroid Biochem Mol Biol 93: 89–97. [DOI] [PubMed] [Google Scholar]
- Brinkmann AO, Jenster G, Kuiper GG, Ris C, van Laar JH, van der Korput JA et al. (1992). The human androgen receptor: structure/function relationship in normal and pathological situations. J Steroid Biochem Mol Biol 41: 361–368. [DOI] [PubMed] [Google Scholar]
- Castoria G, Lombardi M, Barone MV. (2003). Androgen-stimulated DNA synthesis and cytoskeletal changes in fibroblasts by a nontranscriptional receptor action. J Cell Biol 161: 547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R et al. (2004). Molecular determinants of resistance to antiandrogen therapy. Nat Med 10: 33–39. [DOI] [PubMed] [Google Scholar]
- Chen S, Song CS, Lavrovsky Y, Bi B, Vellanoweth R, Chatterjee B et al. (1998). Catalytic cleavage of the androgen receptor messenger RNA and functional inhibition of androgen receptor activity by a hammerhead ribozyme. Mol Endocrinol 12: 1558–1566. [DOI] [PubMed] [Google Scholar]
- Craft N, Shostak Y, Carey M, Sawyers CL. (1999). A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER2/neu tyrosine kinase. Nat Med 5: 280–285. [DOI] [PubMed] [Google Scholar]
- Culig Z, Hobisch A, Cronauer MV, Radmayr C, Trapman J, Hittmair A et al. (1994). Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res 54: 5474–5478. [PubMed] [Google Scholar]
- Dehm SM, Tindall DJ. (2007). Androgen receptor structural and functional elements: role and regulation in prostate cancer. Mol Endocrinol 21: 2855–2863. [DOI] [PubMed] [Google Scholar]
- Dotzlaw H, Moehren U, Mink S, Cato AC, Iniguez Lluhi JA, Baniahmad A. (2002). The amino terminus of the human AR is target for corepressor action and antihormone agonism. Mol Endocrinol 16: 661–673. [DOI] [PubMed] [Google Scholar]
- Greenberg NM, DeMayo F, Finegold MJ, Medina D, Tilley WD, Aspinall JO et al. (1995). Prostate cancer in a transgenic mouse. Proc Natl Acad Sci USA 92: 3439–3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Dai B, Jiang T, Xu K, Xie Y, Kim O et al. (2006). Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell 10: 309–319. [DOI] [PubMed] [Google Scholar]
- Haag P, Bektic J, Bartsch G, Klocker H, Eder IE. (2005). Androgen receptor down regulation by small interference RNA induces cell growth inhibition in androgen sensitive as well as in androgen-independent prostate cancer cells. J Steroid Biochem Mol Biol 96: 251–258. [DOI] [PubMed] [Google Scholar]
- Isaacs JT. (2000). Apoptosis: translating theory to therapy for prostate cancer. J Natl Cancer Inst 92: 1367–1369. [DOI] [PubMed] [Google Scholar]
- Jenster G. (2000). Ligand-independent activation of the androgen receptor in prostate cancer by growth factors and cytokines. J Pathol 191: 227–228. [DOI] [PubMed] [Google Scholar]
- Jia L, Coetzee GA. (2005). Androgen receptor-dependent PSA expression in androgen-independent prostate cancer cells does not involve androgen receptor occupancy of the PSA locus. Cancer Res 65: 8003–8008. [DOI] [PubMed] [Google Scholar]
- Liao X, Tang S, Thrasher JB, Griebling TL, Li B. (2005). Small-interfering RNA-induced androgen receptor silencing leads to apoptotic cell death in prostate cancer. Mol Cancer Ther 4: 505–515. [DOI] [PubMed] [Google Scholar]
- Moehren U, Papaioannou M, Reeb CA, Hong W, Baniahmad A. (2007). Alien interacts with the human androgen receptor and inhibits prostate cancer cell growth molecular endocrinology. Mol Endocrinol 21: 1039–1048. [DOI] [PubMed] [Google Scholar]
- Notini AJ, Davey RA, McManus JF, Bate KL, Zajac JD. (2005). Genomic actions of the androgen receptor are required for normal male sexual differentiation in a mouse model. J Mol Endocrinol 35: 547–555. [DOI] [PubMed] [Google Scholar]
- Perry JE, Grossmann ME, Tindall DJ. (1996). Androgen regulation of gene expression. Prostate Suppl 6: 79–81. [PubMed] [Google Scholar]
- Peterziel H, Mink S, Schonert A, Becker M, Klocker H, Cato AC. (1999). Rapid signalling by androgen receptor in prostate cancer cells. Oncogene 18: 6322–6329. [DOI] [PubMed] [Google Scholar]
- Sadar MD. (1999). Androgen-independent induction of prostate-specific antigen gene expression via cross-talk between the androgen receptor and protein kinase A signal transduction pathways. J Biol Chem 274: 7777–7783. [DOI] [PubMed] [Google Scholar]
- Shah YM, Rowan BG. (2005). The Src kinase pathway promotes tamoxifen agonist action in Ishikawa endometrial cells through phosphorylation-dependent stabilization of estrogen receptor (alpha) promoter interaction and elevated steroid receptor coactivator 1 activity. Mol Endocrinol 19: 732–748. [DOI] [PubMed] [Google Scholar]
- Siddiqui IA, Zaman N, Aziz MH, Reagan-Shaw SR, Sarfaraz S, Adhami VM et al. (2006). Inhibition of CWR22Rnu1 tumor growth and PSA secretion in athymic nude mice by green and black teas. Carcinogenesis 27: 833–839. [DOI] [PubMed] [Google Scholar]
- Thalmann GN, Anezinis PE, Chang SM, Zhau HE, Kim EE, Hopwood VL et al. (1994). Androgen-independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer. Cancer Res 54: 2577–25781. [PubMed] [Google Scholar]
- Thalmann GN, Sikes RA, Wu TT, Degeorges A, Chang SM, Ozen M et al. (2000). LNCaP progression model of human prostate cancer: androgen-independence and osseous metastasis. Prostate 44: 91–103. [DOI] [PubMed] [Google Scholar]
- Ueda T, Mawji NR, Bruchovsky N, Sadar MD. (2002). Ligand-independent activation of the androgen receptor by interleukin-6 and the role of steroid receptor coactivator-1 in prostate cancer cells. J Biol Chem 277: 38087–38094. [DOI] [PubMed] [Google Scholar]
- Wang L, Hsu CL, Ni J, Wang PH, Yeh S, Keng P, Chang C. (2004). Human checkpoint protein hRad9 functions as a negative coregulator to repress androgen receptor transactivation in prostate cancer cells. Mol Cell Biol 24: 2202–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young CY, Andrews PF, Montgomery BT, Tindall DJ. (1992). Tissue-specific and hormonal regulation of human prostate-specific glandular kallikrein. Biochemistry 31: 818–824. [DOI] [PubMed] [Google Scholar]