

SUMMARY
Clostridioides difficile is the leading cause of nosocomial intestinal infections in the United States. Ingested C. difficile spores encounter host bile acids and other cues that are necessary for germinating into toxin-producing vegetative cells. While gut microbiota disruption (often by antibiotics) is a prerequisite for C. difficile infection (CDI), the mechanisms C. difficile employs for colonization remain unclear. Here, we pioneered the application of imaging mass spectrometry to study how enteric infection changes gut metabolites. We find that CDI induces an influx of bile acids into the gut within 24 h of the host ingesting spores. In response, the host reduces bile acid biosynthesis gene expression. These bile acids drive C. difficile outgrowth, as mice receiving the bile acid sequestrant cholestyramine display delayed colonization and reduced germination. Our findings indicate that C. difficile may facilitate germination upon infection and suggest that altering flux through bile acid pathways can modulate C. difficile outgrowth in CDI-prone patients.
Graphical abstract
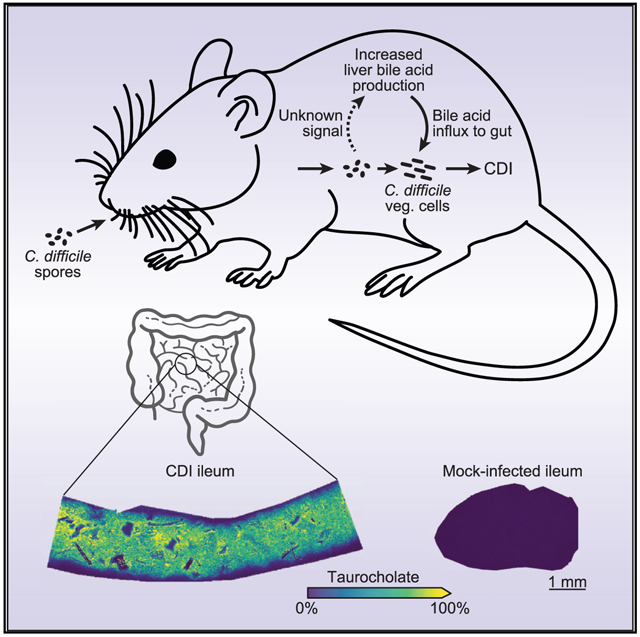
In brief
To illuminate how the gut environment changes during enteric infection, Wexler, Guiberson et al. measured the abundances and distributions of gut metabolites during C. difficile infection using imaging mass spectrometry. They find that C. difficile rapidly alters host bile acid physiology during colonization, which facilitates spore germination and outgrowth.
INTRODUCTION
Use of oral antibiotics disrupts the ecological networks that stabilize the gut microbiota (Hasegawa et al., 2012; Wexler and Goodman, 2017). This instability allows pathogens such as Clostridioides difficile to colonize and cause wide-ranging gastrointestinal disorders that vary in severity from diarrhea to fulminant colitis and death (Abt et al., 2016; Ducarmon et al., 2019; Leffler and Lamont, 2015). Our understanding of the factors that underlie C. difficile colonization is limited by a lack of in situ information about how pathogens restructure the intestinal environments they invade. The advent of powerful molecular imaging technologies allows for the visualization of host-microbe interactions within tissue microenvironments (Cassat et al., 2018; Knippel et al., 2018; Zackular et al., 2016). In particular, recent advances in imaging mass spectrometry (IMS) provide improved sensitivity, specificity, and spatial resolution, presenting a unique opportunity to illuminate these processes in situ and improve our understanding of enteric disease progression along the entire length of the intestinal tract (Buchberger et al., 2018; Niehaus et al., 2019; Prentice et al., 2020; Spraggins et al., 2019; Stoeckli et al., 1999).
The primary mediators of C. difficile infection (CDI) pathology are the toxins TcdA and TcdB, which disrupt host signaling and cause cell death (Shen, 2012). The epidemic C. difficile R20291 strain is considered hypervirulent, as it expresses toxins at higher levels and causes more severe disease than the laboratory-adapted strain 630 (Matamouros et al., 2007; Stabler et al., 2009). While CDI is the leading cause of nosocomial enteric infections, cases of community spread are on the rise, as is the emergence of additional hypervirulent strains (Furuya-Kanamori et al., 2015; Ghose, 2013; McDonald et al., 2018).
Patients that develop CDI do so after ingesting spores. While these spores are tolerant of harsh conditions found both outside and inside the host, they are metabolically dormant and cannot replicate or cause disease. However, once spores enter the small intestine, they sense chemical markers of this location, including the host bile acid taurocholate (TCA) (Francis et al., 2013; Paredes-Sabja et al., 2014). These factors are required to initiate spore germination, resulting in vegetative cells capable of replication and pathogenesis (Shen, 2015). Despite our growing understanding of the processes governing C. difficile germination and sporulation in vitro, less is known about the conditions that are necessary for these processes to occur during host colonization.
Bile acids are produced by the host in the liver before being secreted by the gallbladder into the proximal small intestine, where they aid the host in lipid and vitamin absorption (Ticho et al., 2019). Approximately 95% are reabsorbed by the host in the ileum, while the remainder pass into the large intestine, where reabsorption is minimal (Ticho et al., 2019). Although bile acids must undergo a conjugation step that appends either a glycine or taurine moiety (e.g., taurine appended to cholate to form TCA) to move into the gallbladder from the liver, members of the gut microbiota possess bile salt hydrolases that can deconju-gate these bile acids within the intestinal lumen (Ridlon et al., 2006). Approximately 95% of mouse bile acids are taurine conjugated (compared with ~35% in humans), and all cholate derivatives stimulate spore germination, including glycocholate (Garbutt et al., 1969; Giel et al., 2010; Sorg and Sonenshein, 2008).
Here, we pioneered the application of IMS to study how enteric infection changes gut metabolites. We find that CDI rapidly alters host physiology to increase the availability of germination factors by orders of magnitude within the gut, thereby supporting the colonization and outgrowth of C. difficile. Moreover, C. difficile toxins determine host bile acid levels in a manner that is independent of inflammation and pathology. In response to CDI-induced increases in bile acid levels, the host represses transcriptional flux through its de novo bile acid biosynthesis pathway in the liver. Blocking C. difficile from accessing this augmented pool of germination factors using the bile-acid-sequestering drug cholestyramine delays colonization and reduces spore germination. Together, these findings define major biochemical changes occurring within the host during enteric infection that underpin pathogen colonization and disease progression.
RESULTS
MALDI IMS reveals that CDI increases the abundance of ileal TCA
To determine how CDI alters the intestinal environment, we infected wild-type C57BL/6J mice by oral gavage with 105 C. difficile R20291 spores (or PBS for mock infections) following cefoperazone treatment. We monitored mice for 3 days post-infection (DPI) and subjected cryosections of intestinal tissue and luminal content to MALDI IMS, followed by hematoxylin and eosin (H&E) staining for histopathology. From MALDI IMS, we detect over 170 differentially abundant analytes between intestinal sections from CDI and mock-infected mice, representing a rich source of molecular information about how CDI changes the intestinal environment (Figures 1A, 1B, and S1A–S1C). The most prevalent analyte we detect in CDI mice has a mass-to-charge ratio (m/z) of 514.2946 in negative ionization mode (Figures 1A and S1B). This analyte is present at high intensity throughout the lumen of the distal small intestine of CDI mice but is largely absent from the same region in mock-infected controls (Figures 1B and S1B). Based on its exact mass from a high-resolution FT-ICR, we predicted this ion to be the bile acid TCA. To confirm its identity, we subjected a TCA standard and CDI intestinal tissue homogenate to liquid chromatography tandem mass spectrometry (LC-MS/MS). We observe a prominent ion at m/z 514.3, as well as a peak at m/z 124.0, in both the standard and the sample that correspond to a taurine-conjugated bile acid and the taurine moiety, respectively (Figure 1C). Together, these data suggest that CDI induces a substantial increase in intestinal TCA abundance.
Figure 1. MALDI IMS reveals elevated levels of TCA in the ileal lumen of CDI mice.
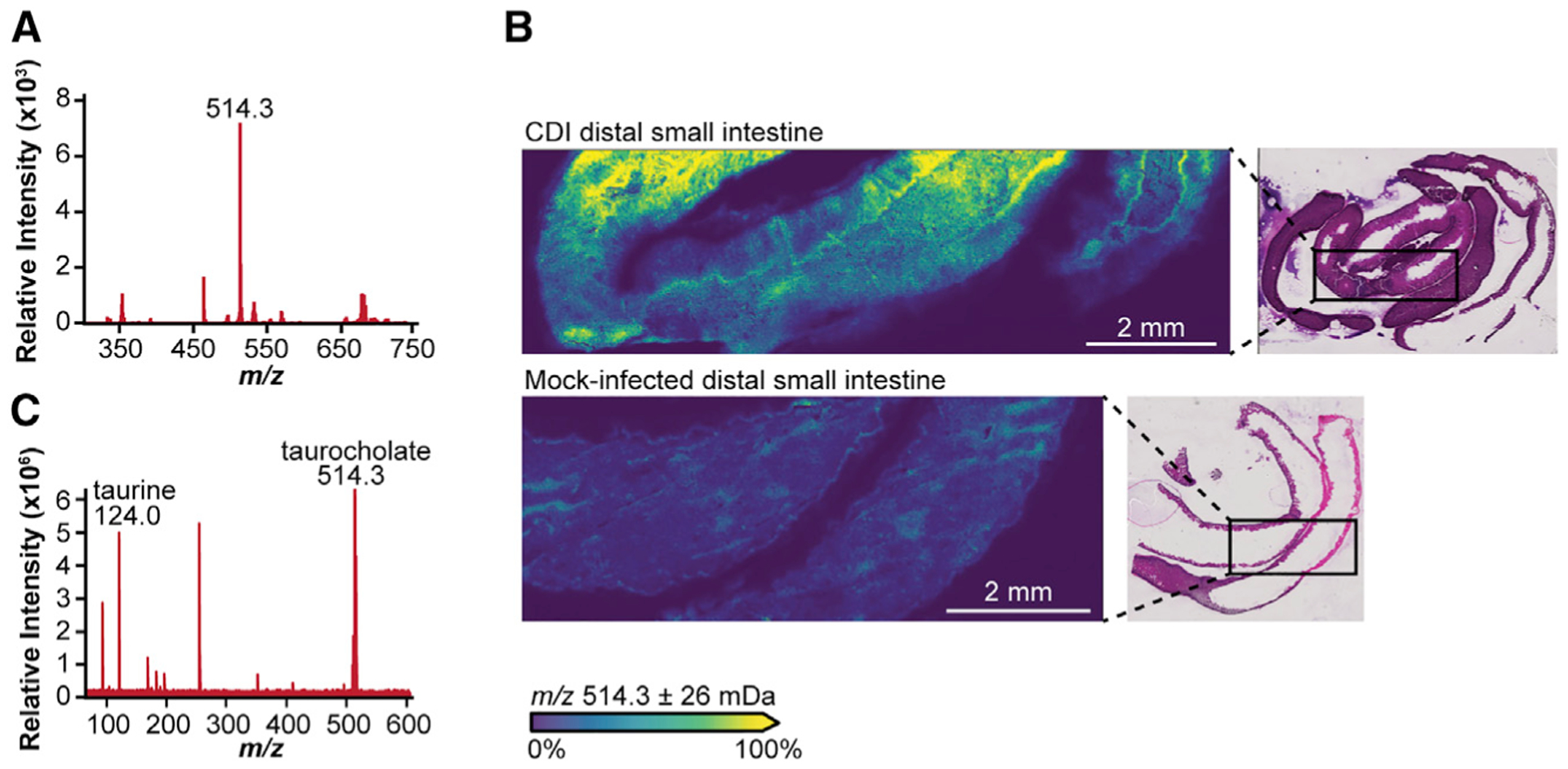
(A) Mass spectrum from MALDI IMS analysis of the ileal region and lumen of a CDI mouse at 3 DPI reveals over 170 analytes, with m/z 514.3 being the most abundant analyte detected.
(B) MALDI IMS analysis of the ileal region and lumen of CDI or mock-infected animals at 3 DPI reveals elevated levels of m/z 514.3 in CDI mice relative to mock-infected controls normalized by total ion current. Inset: H&E stain of MALDI-imaged cryosections.
(C) LC-MS/MS analysis of CDI distal small intestine tissue and lumen homogenate shows a peak at m/z 514.3, supporting the identification of m/z 514.3 in (A) and (B) as TCA.
Levels of primary bile acids rapidly rise in the gut following C. difficile colonization in a manner dependent on TcdAB but independent of inflammation
To determine the degree to which CDI increases intestinal TCA levels, we measured the abundance of other primary bile acids, including the unconjugated primary bile acids cholate (CA) and β-muricholate (bMCA), as well as their taurine-conjugates TCA and tauro-β-muricholate (TbMCA), in CDI and mock-infected mice; the primary bile acid chenodeoxycholate (CDCA) and its taurine-conjugated form were undetected in both groups of mice. We extracted bile acids from liver, ileal lumen, and cecal lumen samples to capture key points within the host bile acid cycle (de novo production, pre-reabsorption, and post-reabsorption, respectively), and quantified them using LC-MS/MS.
The abundance of CA (Figure 2A) and bMCA (Figure 2B) decrease markedly in the liver of CDI mice relative to mock-infected controls over time and are undetected in CDI mice by 3 DPI. This is despite the liver being the site of de novo bile acid biosynthesis, as well as the organ to which reabsorbed intestinal bile acids recirculate (Ticho et al., 2019). By contrast, the abundance of CA (Figure 2A) and bMCA (Figure 2B) increase more than 10-fold on average by 3 DPI in the ileum and by ~10-fold on average by 2 DPI in the cecum of CDI mice relative to mock-infected controls.
Figure 2. CDI induces a rapid influx of primary bile acids into the gut in a manner dependent on C. difficile toxins but independent of inflammation.
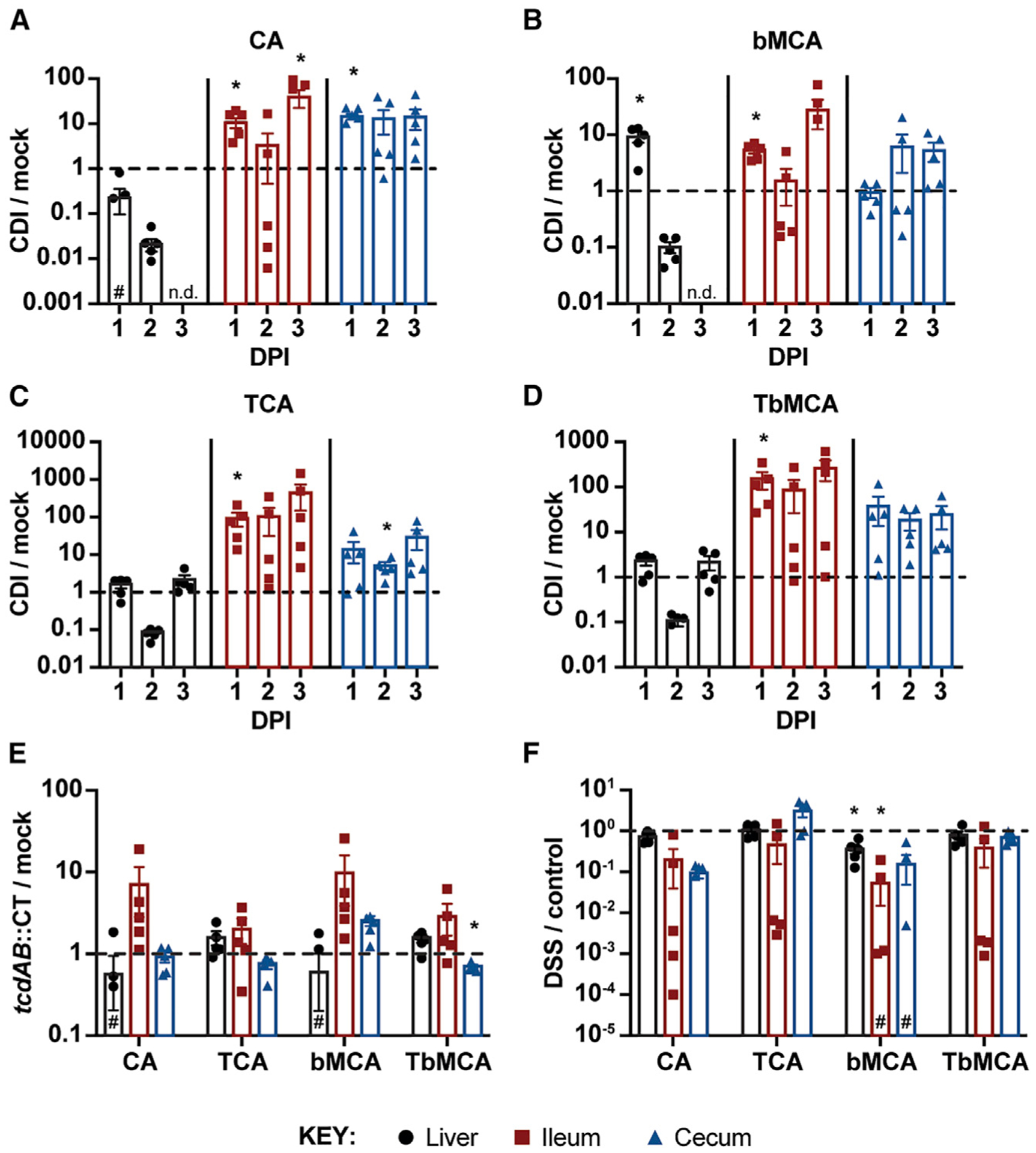
(A–D) Ratios of unconjugated (A and B) and taurine-conjugated (C and D) primary bile acids in the liver, ileum, and cecum of CDI versus mock-infected mice.
(E) Ratios of primary bile acids in the liver, ileum, and cecum of toxin-deficient (tcdAB::CT) versus mock-infected mice at 3 DPI.
(F) Ratios of primary bile acids in the liver, ileum, and cecum of DSS-treated versus untreated control mice after 2 days of treatment. Concentrations are adjusted based on sample weight (μM/g) prior to calculating LC-MS/MS-based ratios.
Data are represented as mean ± standard error of the mean (SEM); DPI, days post-infection; n.d., not detected in CDI mice; #, not detected in all mice; *p < 0.05, significance determined by unpaired two-sided t tests; n = 5 mice/group.
The abundance of TCA (Figure 2C) and TbMCA (Figure 2D) remain detectable in the livers of both groups of mice throughout the 3-day infection. Surprisingly, concentrations of TCA (Figure 2C) and TbMCA (Figure 2D) also remain higher in the ilea of CDI mice relative to mock-infected controls by ~100-fold or more on average from as early as 1 DPI. TCA (Figure 2C) and TbMCA (Figure 2D) concentrations are also elevated by 10-fold or more on average in the ceca of CDI mice relative to mock-infected controls.
Together, these data suggest that levels of primary bile acids rapidly rise in the gut following the onset of CDI. These data not only support our initial observations from MALDI IMS regarding elevated TCA abundance during CDI (Figures 1 and S1B) but also reveal that CDI increases TCA levels by orders of magnitude within 24 h of infection and renders CA and bMCA undetected in the liver by 3 DPI.
To determine the contribution of toxins TcdA and TcdB to elevated bile acid levels during CDI, we infected mice by oral gavage with 105 toxin-deficient C. difficile R20291 spores (tcdAB::CT) or PBS for mock infections following cefoperazone treatment, and measured bile acid abundance in tissues at 3 DPI. Contrary to wild-type CDI mice (Figures 2A–2D), the tcdAB::CT-infected mice display a negligible deviation from the mock-infected control group in terms of TCA and TbMCA abundance in all three tissue types, despite colonizing to a similar degree as wild-type C. difficile (Figures 2E, S2A, and S2B). Moreover, TcdA and TcdB individually are insufficient to significantly augment bile acid levels, as mice receiving a rectal infusion of 50 μg of either toxin largely did not deviate significantly from bile acid levels of PBS-infused mice after 8 h (Figures S2C–S2F). While the exposure time was limited to 8 h because of the toxicity of TcdA and TcdB, it may have been an insufficient duration, as mice gavaged with 105 C. difficile R20291 spores also did not exhibit a significant increase in bile acid levels after 8 h (Figures S2C–S2F).
Dextran sulfate sodium (DSS) is a chemical inducer of colitis that impacts the colonic epithelium of mice within 1 day of exposure, and causes severe inflammation within 3–4 days of treatment (Chassaing et al., 2014). To determine the contribution of pathogen-independent intestinal inflammation to host bile acid pool size, we treated mice with 3% DSS in their drinking water (or no DSS for control mice) for 2 days following cefoperazone treatment and measured bile acid levels in liver, ileal lumen, and cecal lumen samples by LC-MS/MS. Contrary to mice infected with wild-type C. difficile (Figures 2A–2D), the abundances of CA, TCA, bMCA, and TbMCA are similar or lower in DSS-treated mice than controls (Figure 2F). These data suggest that intestinal inflammation alone does not account for modulations to host bile acid physiology observed during CDI. Indeed, histological analysis of H&E-stained cryosections of the distal small intestine of CDI and mock-infected mice reveals no signs of inflammation or pathology at 3 DPI (Figures S3A–S3C), despite clear CDI symptomology (Figure S2A). Together, these data suggest that CDI induces changes to host bile acid physiology in a manner that is dependent on C. difficile toxins but independent of intestinal inflammation and pathology.
The host responds to CDI-induced elevations in bile acid levels by reducing flux through its de novo biosynthesis pathway
Primary bile acids are synthesized in the liver from cholesterol via the classical pathway, involving 18 steps to yield conjugated bile acid products such as TCA (Ticho et al., 2019). The first step converts cholesterol to 7α-hydroxycholesterol via the enzyme CYP7A1 and thus determines the overall bile acid pool size in the host (Figure 3A). The pathway later splits to yield either CDCA (and MCA in mice) or CA in a manner that depends on the presence and activity of CYP8B1, the activity of which leads to production of CA over CDCA/MCA (Figure 3A). Primary bile acids are later conjugated with either taurine or glycine by the enzyme bile acid-coenzyme A (CoA):amino acid N-acyltransfer-ase (BAAT) in the liver (Figure 3A) (Ticho et al., 2019).
Figure 3. The host responds to CDI-induced elevations in bile acid pools by reducing bile acid biosynthesis gene expression.
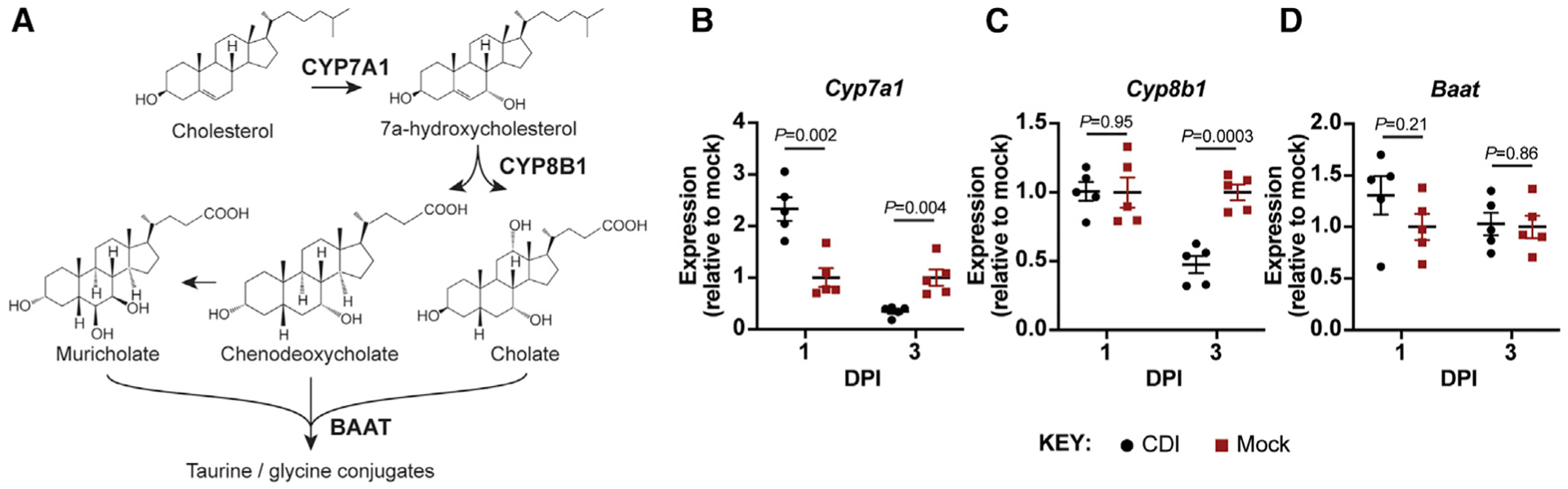
(A) Schematic of key steps in the bile acid biosynthesis pathway in the murine liver. CYP7A1 controls bile acid pool size, CYP8B1 determines the ratio of cholate to chenodeoxycholate and muricholate, and BAAT conjugates bile acids with taurine or glycine.
(B–D) Expression of Cyp7a1 (B), Cyp8b1 (C), and Baat (D) in the livers of CDI mice relative to mock-infected mice over time. All expression values were first normalized to β-actin expression. Data plots show mean ± SEM; significance determined by unpaired two-sided t tests; n = 5 mice/group.
Given that the levels of CA and bMCA in the liver of CDI mice decline over time and are undetected at 3 DPI (Figures 2A and 2B), and that the levels of conjugated bile acids TCA and TbMCA increase rapidly upon C. difficile colonization, we suspected CDI affects transcriptional flux through the bile acid biosynthesis pathway in the liver. To test this, we extracted RNA from the livers of CDI and mock-infected control mice at 1 and 3 DPI and measured expression of genes encoding for key enzymes within the de novo bile acid biosynthesis pathway: Cyp7a1, Cyp8b1, and Baat. Cyp7a1 expression was more than 2-fold higher on average in CDI mice relative to mock-infected controls at 1 DPI but fell to approximately half the expression level of mock-infected controls by 3 DPI (Figure 3B). Cyp8b1 expression in CDI mice remained similar to mock-infected controls at 1 DPI but also fell to approximately half the expression level of mock-infected controls by 3 DPI (Figure 3C). Baat expression was modestly but insignificantly elevated in CDI mice relative to mock-infected controls at 1 DPI but was indistinguishable between both groups at 3 DPI (Figure 3D).
These data suggest that CDI promptly boosts transcriptional flux through the host bile acid biosynthesis pathway in the liver but that this transcriptional flux is repressed by 3 DPI. Moreover, the unchanged expression of Baat in CDI mice suggests that reabsorbed bile acids from the intestinal tract are still conjugated in the liver during initial colonization and therefore can be recycled back into the intestine.
Reducing free intestinal bile acid levels with cholestyramine diminishes C. difficile colonization and spore germination
Cholestyramine is a bile-acid-sequestering drug often used for lowering a patient’s cholesterol—the molecule from which all host bile acids are derived (Figure 3A) (Scaldaferri et al., 2013). It functions by reducing bile acid reabsorption in the gut through sequestration, which causes the host to produce more bile acids and thus reduces the body’s cholesterol supply (Scaldaferri et al., 2013). To determine the contribution of elevated intestinal bile acid levels to C. difficile colonization and spore germination, we placed mice on a diet containing cholestyramine (20 g/kg) or a control diet without cholestyramine for 7 days prior to infection by oral gavage with 105 C. difficile R20291 spores and maintained them on their respective diets throughout the infection (Scaldaferri et al., 2013).
While both vegetative cells (average of ~109 colony-forming units [CFUs]/g; Figures 4A and 4B) and spores (average of ~107 CFUs/g; Figures 4B and 4C) are detected in fecal samples of mice on the control diet at 1 DPI, they are undetected in mice on the cholestyramine diet at 1 DPI. By 2 DPI, only 60% of cholestyramine-treated mice have detectable levels of vegetative cells (average of ~109 CFUs/g; Figures 4A and 4B), and 30% have detectable levels of spores (average of ~108 CFUs/g; Figures 4B and 4C). Interestingly, we find the vegetative cell/spore ratio (an indicator of germination frequency) in these three mice to be ~10-fold lower on average than the vegetative cell/spore ratio in the control diet mice at 2 DPI (Figure 4D). This ~10-fold ratio is maintained at 3 DPI and correlates with a lower average burden of vegetative cells in cholestyramine-treated mice (Figure 4A). Importantly, intestinal TCA abundance is similarly elevated in CDI mice on both the cholestyramine and control diets at 3 DPI relative to mock-infected mice on the control diet (Figures 4E and S4A–S4D); our extraction method is designed to capture both cholestyramine-bound and unbound bile acids.
Figure 4. Elevated bile acid pool size is necessary for rapid host colonization and spore germination.
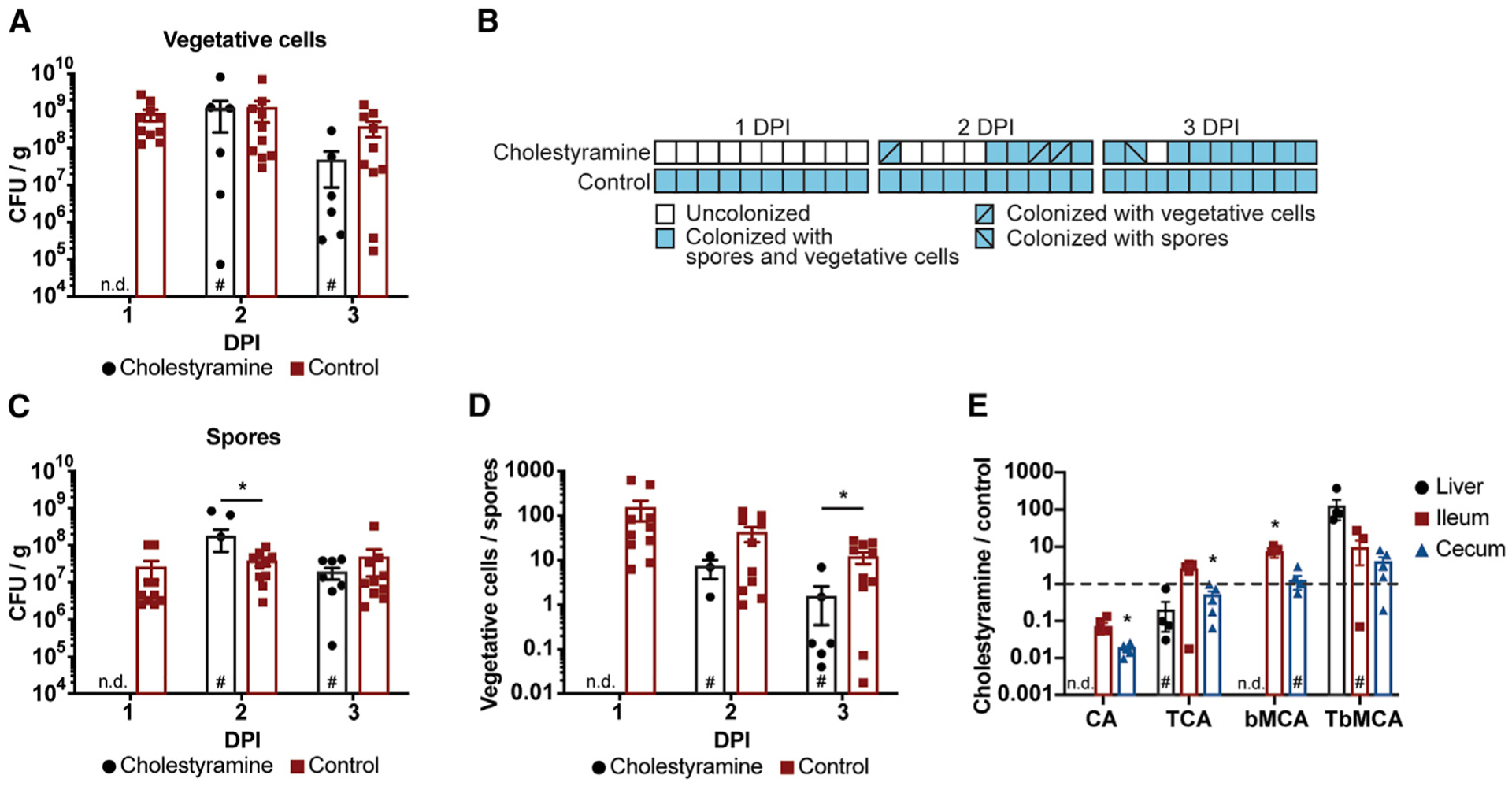
CDI mice provided cholestyramine in their diet display delayed colonization relative to CDI mice provided a control diet without cholestyramine.
(A) Vegetative cell counts from fecal samples collected over time.
(B) CDI mice on a cholestyramine diet display delayed colonization compared with CDI mice on a control diet.
(C and D) Spore counts and vegetative cell counts (A) from fecal samples collected over time (C) were used to determine the ratio (D) of vegetative cells to spores in cholestyramine and control mice.
(E) CDI mice on a cholestyramine diet carry similar levels of primary bile acids in their ilea and ceca compared with CDI mice on a control diet at 3 DPI.
Data are represented as mean ± SEM; n.d., not detected; #, not detected in all mice; *p < 0.05, significance determined by unpaired two-sided t tests; n = 10 mice/group.
These data show that blocking access to abundant intestinal bile acid levels using cholestyramine limits C. difficile colonization efficiency and spore germination during CDI. While the spore/vegetative cell ratio can be affected by vegetative cell growth rate, sporulation frequency, and germination rate, our findings suggest that the massive influx of primary bile acids into the gastrointestinal tract following host consumption of C. difficile spores drives spore germination, pathogen outgrowth, and CDI progression through either continued spore germination or another mechanism promoting colonization or potentiation of toxin activity.
DISCUSSION
Our study provides evidence that although C. difficile is physically restricted to the intestinal lumen, its influence on host physiology within the first 24 h of infection extends beyond this site. Our findings stem from the use of cutting-edge IMS technology that allows for untargeted discovery-based visualization of molecular species, revealing spatial localization and intensity patterns that would be impossible to detect with traditional LC-MS/MS alone (Figure S1). This approach has been useful in a variety of host-pathogen interaction studies, such as in Staphylococcus aureus infections (Guiberson et al., 2021). The incorporation of MALDI IMS to study S. aureus soft tissue abscesses allows for molecular profiling of the bacterial nidus and surrounding tissue, revealing molecular changes that occur in this region during infection (Cassat et al., 2018). IMS has also been applied to the intestines in a variety of studies outside the context of host-pathogen interactions (Nguyen et al., 2019). Most imaging experiments have probed discrete regions of the intestine for analysis due to the difficulty in preparing large sections of intestine for sectioning (Carter et al., 2019). Our study, however, underscores the importance of including luminal content in MALDI IMS analysis for a more representative analysis of the gut, as seen by the bile acid localization patterns previously not shown with traditional sample preparation approaches (Figure S1) (Genangeli et al., 2019).
Bile acid physiology is governed by complex networks of both redundant and counteracting regulatory pathways spanning multiple body sites, including the liver, gallbladder, intestine, and circulatory system (Dawson and Karpen, 2015; Ticho et al., 2019). The gut microbiota significantly affects bile acid composition, as germ-free mice exhibit impaired lipid absorption and many commensal species can modify primary (host-derived) into secondary (microbe-derived) bile acids (Martinez-Guryn et al., 2018; Ridlon et al., 2006). Secondary bile acids possess antimicrobial properties against C. difficile (Abt et al., 2016). Cefoperazone and other antibiotics disrupt the bile-acid-modifying activity of the gut microbiota and reduce the abundance of secondary bile acids (Theriot et al., 2016). Because the vast majority of gut microbes reside in the large intestine, downstream of ileal receptors for reabsorption, secondary bile acids predominantly exert their effects in the large intestine (Ridlon et al., 2006).
While the mechanism by which CDI initially disrupts bile acid regulatory networks is unclear, the consequences are significant. The bile acid sequestrant cholestyramine is not currently used to treat CDI, and in the few studies that have investigated its potential in limiting CDI incidences, the rationale for its use centered on its ability to inactivate TcdA and TcdB through sequestration. One study investigated whether cholestyramine could reduce CDI incidences in patients with Lyme disease receiving intravenous ceftriaxone (Puri et al., 2015). The authors reported a ~3.5-fold reduction in the incidence of CDI in their patient cohort relative to another published report monitoring CDI in a similar patient population that did not receive cholestyramine (Westh et al., 1991). The delayed colonization of C. difficile in mice on the cholestyramine diet observed in Figure 4 is more likely due to bile acid sequestration than toxin sequestration, since the toxins are necessary (but insufficient; Figures S2C–S2F) for increasing bile acid abundance during CDI (Figure 2E) but dispensable for colonization (Figure S2B). Our findings suggest that C. difficile’s access to bile acids can be modulated through the activity of therapeutic drugs. A deeper understanding of how pathogen colonization and outgrowth occurs in the human gut will be necessary for designing more targeted therapeutics to disrupt these processes in patients.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Eric Skaar (eric.skaar@vumc.org).
Materials availability
This study did not generate new unique reagents.
Data and software availability
All raw LC-MS/MS data are available on Mendeley Data: (https://doi.org/10.17632/rhbktnpyj2.1).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial strains
Bacterial strains used in this study are listed in the Key Resources Table. Wild-type C. difficile R20291 and C. difficile R20291 tcdAB::CT were grown at 37°C in an anaerobic chamber (90% nitrogen, 5% hydrogen, 5% carbon dioxide, Coy Lab Products) in brain-heart-infusion broth (BD Life Sciences) supplemented with 0.5% yeast extract (BD Life Sciences) and 0.1% cysteine (Sigma-Aldrich).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Clostridioides difficile R20291 | Stabler et. al., 2009 | N/A |
| Clostridioides difficile R20291 tcdAB::CT | This study | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Cefoperazone | Sigma-Aldrich | Cat#C4292 |
| Phosphate buffered saline | Sigma-Aldrich | Cat#79383 |
| 2.6% carboxymethylcellulose | Sigma-Aldrich | Cat#419273 |
| Dextran sulfate sodium | MP Biomedicals | Cat#02160110 |
| 9-aminoacridine | Sigma-Aldrich | Cat#A38401 |
| Ammonium formate | Sigma-Aldrich | Cat#516961 |
| 1,5- diaminonaphthalene | Sigma-Aldrich | Cat#D21200 |
| Hematoxylin | Sigma-Aldrich | Cat#H3136 |
| Eosin | Sigma-Aldrich | Cat#230251 |
| Beta-mercaptoethanol | Sigma-Aldrich | Cat#63689 |
| Phenol:chloroform:IAA, pH 6.7 | Sigma-Aldrich | Cat#P1944 |
| Cholestyramine | Sigma-Aldrich | Cat#11041 |
| iQ SYBR Green mix | Bio-Rad | Cat#25230 |
| Poly-l-lysine | Sigma-Aldrich | Cat#P8920 |
| Critical commercial assays | ||
| RNeasy Mini Kit | QIAGEN | Cat#74104 |
| iScript cDNA synthesis kit | Bio-Rad | Cat#1708890 |
| Wizard SV Gel and PCR clean-up system | Promega | Cat#A9281 |
| Deposited data | ||
| Raw LC-MS/MS data | This study | Mendeley Data: https://dx.doi.org/10.17632/rhbktnpyj2.1 |
| Experimental models: Organisms/strains | ||
| C57BL/6J mice | The Jackson Laboratory | Cat#664 |
| Oligonucleotides | ||
| Primers for qRT-PCR: Table S1 | N/A | N/A |
| Software and algorithms | ||
| Xcaliber Quan Browser | Thermo Fisher Scientific | Version 4.3 |
| Prism | Graph Pad | Version 8.2 |
| Canvas X16 | Canvas | Version 16 |
| Adobe Illustrator | Adobe | Version 16.0 |
| SCiLS | Bruker Daltonics | Version 2020 |
| Gen5 Data Analysis Software | BioTek | Gen5 3.0 |
| Other | ||
| Lysing Matrix D tubes | MP Biomedicals | Cat#116913050 |
| SecurityGuard C18 guard column (3.2 × 8 mm) | Phenomenex | Cat#AJ0-4286 |
| Ascentis Express HPLC C18 column (25 cm × 2.1 mm, 5 um particle size) | Supelco Analytical | Cat#5811305-U |
| AIN-93G ± 2% cholestyramine | Dyets, Inc. | Diet#103550 |
| Indium Tin Oxide Coated Glass Slides | Delta Technologies | Cat#NC1501896 |
Animal models
All animal experiments were performed using protocol M2000027–00 approved by the Vanderbilt University Medical Center Institutional Animal Care and Use Committee and in compliance with NIH guidelines, the Animal Welfare Act, and US Federal law. Animal studies were conducted using 8- to 12-week old male C57BL/6 (Jackson Laboratories) mice housed in groups of up to five at the Vanderbilt University Medical Center Animal Facilities. Briefly, C57BL/6 mice received cefoperazone in their drinking water (0.5 mg/ml) for 5 days. Following a 2-day recovery period, mice were gavaged orally with PBS or infected with 105 C. difficile spores to an endpoint of three days. CDI symptomology was monitored daily by weight loss. Mice were humanely euthanized using compressed CO2. Intestinal segments were dissected, embedded in 2.6% carboxymethylcellulose (CMC), and frozen in liquid nitrogen. Samples were stored at −80°C prior to sectioning. Mice provided a cholestyramine or control diet (Dyets, Inc., Bethlehem, PA) were placed on these diets at the same time they were given cefoperazone, and remained on their respective modified diets for the duration of the experiment. Mice provided 3% DSS in their drinking water for 2 days were first treated with cefoperazone for 5 days, followed by a 2-day recovery period. Mice receiving rectal infusion of purified, recombinant TcdA or TcdB (or PBS) were placed on 0.5 mg/ml cefoperazone in their drinking water for 5 days, followed by a 2-day recover period, before receiving 50 μg of either toxin (Markham et al., 2021). Mice were euthanized after 8 hours.
METHOD DETAILS
Determining bacterial burdens
C. difficile CFUs were quantified daily from fecal samples. Fresh samples were weighed, homogenized in PBS, serially diluted, and plated onto taurocholate cycloserine cefoxitin fructose agar (TCCFA) for enumeration as colony forming units per gram of feces (CFU/g). Vegetative cells were enumerated by plating fecal suspensions on cycloserine cefoxitin fructose agar without taurocholate, while spores were enumerated by heating fecal suspensions at 65°C for 20 minutes aerobically prior to plating onto TCCFA.
MALDI imaging mass spectrometry
Tissue and CMC were cryosectioned at 10 μm thickness using a Cryostar NX70 Cryostat (Thermo Scientific, Waltham, MA, USA) and thaw mounted onto 1% poly-l-lysine coated indium tin oxide coated glass slides (Delta Technologies, Loveland, CO, USA). All slides were kept at room temperature prior to mounting to ensure full thaw-mounting. Samples were brought to room temperature in a desiccator 30 minutes prior to matrix application. Samples were sprayed with 9-aminoacridine (9-AA) for bile acid analysis using an automated sprayer (TM Sprayer, HTX Technologies, Chapel Hill, NC, USA). 9-AA was prepared at a concentration of 5 mg/mL in 70% acetonitrile, and sprayed at 30°C, 400 mm/min, 0.04 mL/min for 8 passes. Following matrix application, high-mass resolution imaging mass spectrometry of intestinal samples were performed using either a Solarix 15T MALDI FT-ICR mass spectrometer (Bruker Daltonik, Billerica, MA, USA; Figures S1B and S4) or a rapifleX MALDI Tissuetyper (Bruker Daltonics, Billerica, MA, USA; Figure 1B). The rapifleX MALDI Tissuetyper was equipped with a Smartbeam 3D 10 kHz Nd:YAG (355 nm) laser. The FTICR was equipped with an Apollo II dual MALDI/ESI ion source and a Smartbeam II 2 kHz frequency tripled Nd:YAG laser (355 nm). Images collected on the rapifleX MALDI Tissuetyper were generated using the single spot laser setting with a pixel scan size of 20 μm in both x and y dimensions. Data were collected in negative ion mode from m/z 361–801 with 250 laser shots per pixel. Images collected on the 15T MALDI FT-ICR were generated using the small laser setting (~50 μm) with a pixel spacing of 50 μm. Data were collected with 650 laser shots per pixel at 60% power from m/z 250–800 with a transient length of 1.1534 s and a resolving power of 260,000 at m/z 512.2723. Data were visualized using SCiLS Lab (Bruker Daltonik, Dillerica, MA, USA). Samples for lipid analysis (Figure S1C) were washed with ammonium formate and distilled H2O prior to matrix applicated. DAN matrix for lipid analysis was sublimated onto the tissue surface (~1.0 mg/cm2). Briefly, slides were taped to the bottom of the condenser in a simple sublimation apparatus. Approximately 300 mg of DAN was sublimed at 130°C and 24 mTorr for 3 min. Imaging mass spectrometry was performed using a prototype timsTOF Pro equipped with a duel ESI/MALDI source with a SmartBeam 3D 10 kHz frequency tripled Nd:YAG laser (355 nm) (Spraggins et al., 2019). The system was operated in qTOF mode with TIMS deactivated. Images were collected at 25 μm spatial resolution using 400 laser shots per pixel and 40% laser power. Data were collected in negative ion mode from m/z 200–2000. All tentative lipid identifications were determined based on mass accuracy using the LIPIDMAPS lipidomics gateway (lipidmaps.org) with an accuracy threshold of 5 ppm. Data were visualized using SCiLS Lab 2020. (Spraggins et al., 2019). The system was operated in qTOF mode with TIMS deactivated. Images were collected at 25 μm spatial resolution using 400 laser shots per pixel and 40% laser power. Data were collected in negative ion mode from m/z 200–2000. All tentative lipid identifications were determined based on mass accuracy using the LIPIDMAPS lipidomics gateway (lipidmaps.org) with an accuracy threshold of 5 ppm. Data were visualized using SCiLS Lab 2020.
Histology analysis
Following MALDI IMS imaging, matrix was removed from samples using 100% ethanol, and the samples were rehydrated with graded ethanol washes and double distilled water. Tissue samples were then stained using hematoxylin and eosin (H&E) stain and coverslipped. After drying, brightfield microscopy images were collected using a Leica SCN400 Brightfield Slide Scanner at 20x magnification (Leica Microsystems, Buffalo Grove, IL, USA) and visually analyzed in a blinded fashion for signs of inflammation and tissue damage.
qRT-PCR
Liver samples from mice were flash frozen in liquid nitrogen and stored at −80°C prior to RNA extraction. Livers were homogenized in Lysing Matrix D tubes (MP Biomedicals) containing 700 μl RLT buffer with 1% (v/v) β-mercaptoethanol (QIAGEN) using a FastPrep-24 (MP Biomedicals) bead beater for 45 s at 6.5 m/s (repeated 3 times, incubating on ice for 1 minute in between rounds). Homogenates were centrifuged at 20,000 × g at 4°C for 5 minutes to pellet debris, RNA was extracted using phenol:chloroform:IAA, pH 6.7 (Sigma), and samples were mixed with 50% ethanol prior to being transferred to QIAGEN RNeasy columns, according to the manufacturer’s instructions. Cleaned RNA samples were subjected to DNase treatment (Invitrogen) prior to cDNA synthesis using an iScript cDNA synthesis kit (Bio-Rad). RNA was removed through the addition of 1 N NaOH at 65°C for 30 minutes, followed by the addition of an equal volume of 1 N HCl. cDNA was cleaned using a PCR clean-up kit (Promega) according to the manufacturer’s instructions. cDNA concentrations were adjusted to 1 ng/μl using the Synergy 2 with Gen 5 software (Bio-Tek) prior to qRT-PCR using iQ SYBR Green mix (Bio-Rad), gene-specific primers (Table S1), and a CFX96 qPCR cycler (Bio-Rad). Transcript abundance was calculated using the ΔΔCt method and data were normalized to β-actin gene expression.
Bile acid extractions
Liver, ileal lumen, and cecal lumen samples were harvested from mice, and flash frozen in liquid nitrogen. Samples were weighed prior to extraction, and homogenized in Lysing Matrix D tubes (MP Biomedicals) containing 2:1:1 mixture of ethanol:chloroform:methanol using a FastPrep-24 (MP Biomedicals) bead beater for 45 s at 6.5 m/s (repeated 3 times, incubating on ice for 1 minute in between rounds). Homogenates were centrifuged at 20,000 × g at 4°C for 15 minutes to pellet debris. Supernatant was transferred to a clean glass vial, and evaporated under nitrogen gas flow until dry. Samples were reconstituted in methanol prior to LC-MS/MS analysis.
Liquid chromatography tandem mass spectrometry
Bile acids from tissue and fecal extracts were injected in volumes of 5 μL and gradient eluted onto a SecurityGuard C18 guard column (3.2 × 8 mm, Phenomenex, Torrance, CA, USA) Ascentis Express HPLC C18 column (25 cm × 2.1 mm, 5 μm particle size, Supelco Analytical, Bellefonte, PA, USA). Mobile phase A consisted of H2O with 0.1% formic acid, and mobile phase B consisted of acetonitrile with 0.1% formic acid. Bile acids were eluted on a linear gradient of 20%–60% B for 15 minutes, followed by 60%–100% B for 15 minutes, and 100%–20% B for 30 minutes at a flow rate of 0.4 mL/min. All analytes were measured on a TSQ Quantum Triple Quadrupole mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA), with an identity transition. Optimal collision energies were determined empirically before each experiment. Quantification was determined using a calibration curve from 5 μL injections of 0, 0.1, 0.3, 1, 3, 10, and 30 μM bile acid standards using Xcaliber Quant Browser (Thermo Fisher Scientific, Waltham, MA, USA).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using GraphPad Prism 8 and Microsoft Excel. Statistical significance was assessed using an unpaired two-sided t tests. Significance was defined as p < 0.05, and the data were only excluded on the basis of technical errors associated with the experiment. Exact statistical tests used, significance values, group sizes, and dispersion and precision of measurements are defined in the figure legends.
Supplementary Material
Highlights.
Imaging mass spectrometry (IMS) is applied to study C. difficile infection (CDI)
IMS reveals a rapid influx of bile acids into the gut during CDI
The host then reduces transcriptional flux through bile acid biosynthesis pathways
Increased bile acids contribute to C. difficile spore germination and outgrowth
ACKNOWLEDGMENTS
We thank members of the Skaar, Spraggins, and Caprioli labs, as well as Drs. Borden Lacy and Maribeth Nicholeson, for their critical review of the manuscript. We thank Dr. Sarah Kuehne (University of Birmingham, UK) for creating and sharing the C. difficile R20291 tcdAB::CT mutant. This work was supported by National Institute of Allergy and Infectious Diseases grants R01AI073843 (to E.P.S.), R01AI45992 (to. E.P.S. and J.M.S.), and R01AI38581 (to E.P.S. and J.M.S.) and the National Institute of Diabetes and Digestive and Kidney Diseases grant P30DK058404 (to E.P.S.). A.G.W. was supported by a Helen Hay Whitney Foundation research fellowship and National Institute for Biomedical Imaging and Bioengineering training grant T32EB001628. W.N.B. is supported by American Heart Association postdoctoral fellowship 18POST34030426. The prototype MALDI timsTOF MS was developed as part of the National Science Foundation Major Research Instrument Program (CBET – 1828299 awarded to J.M.S. and R.M.C.), and the Bruker 15T solariX FT-ICR MS in the Mass Spectrometry Research Center at Vanderbilt University was acquired through the NIH Shared Instrumentation Grant Program (1S10OD012359 awarded to R.M.C.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109683.
REFERENCES
- Abt MC, McKenney PT, and Pamer EG (2016). Clostridium difficile colitis: pathogenesis and host defence. Nat. Rev. Microbiol 14, 609–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchberger AR, DeLaney K, Johnson J, and Li L (2018). Mass Spectrometry Imaging: A Review of Emerging Advancements and Future Insights. Anal. Chem 90, 240–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter CL, Hankey KG, Booth C, Tudor GL, Parker GA, Jones JW, Farese AM, MacVittie TJ, and Kane MA (2019). Characterizing the Natural History of Acute Radiation Syndrome of the Gastrointestinal Tract: Combining High Mass and Spatial Resolution Using MALDI-FTICR-MSI. Health Phys. 116, 454–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassat JE, Moore JL, Wilson KJ, Stark Z, Prentice BM, Van de Plas R, Perry WJ, Zhang Y, Virostko J, Colvin DC, et al. (2018). Integrated molecular imaging reveals tissue heterogeneity driving host-pathogen interactions. Sci. Transl. Med 10, eaan6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chassaing B, Aitken JD, Malleshappa M, and Vijay-Kumar M (2014). Dextran Sulfate Sodium (DSS)-Induced Colitis in Mice. Curr. Protoc. Immunol 104, 15.25.1–15.25.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson PA, and Karpen SJ (2015). Intestinal transport and metabolism of bile acids. J. Lipid Res 56, 1085–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducarmon QR, Zwittink RD, Hornung BVH, Schaik W, van, Young VB, and Kuijper EJ (2019). Gut Microbiota and Colonization Resistance against Bacterial Enteric Infection. Microbiol. Mol. Biol. R 83, e00007–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis MB, Allen CA, Shrestha R, and Sorg JA (2013). Bile acid recognition by the Clostridium difficile germinant receptor, CspC, is important for establishing infection. PLoS Pathog. 9, e1003356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuya-Kanamori L, Marquess J, Yakob L, Riley TV, Paterson DL, Foster NF, Huber CA, and Clements ACA (2015). Asymptomatic Clostridium difficile colonization: epidemiology and clinical implications. BMC Infect. Dis 15, 516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbutt JT, Heaton KW, Lack L, and Tyor MP (1969). Increased ratio of glycine- to taurine-conjugated bile salts in patients with ileal disorders. Gastroenterology 56, 711–720. [PubMed] [Google Scholar]
- Genangeli M, Heijens AMM, Rustichelli A, Schuit ND, Micioni Di Bonaventura MV, Cifani C, Vittori S, Siegel TP, and Heeren RMA (2019). MALDI-Mass Spectrometry Imaging to Investigate Lipid and Bile Acid Modifications Caused by Lentil Extract Used as a Potential Hypocholesterolemic Treatment. J. Am. Soc. Mass Spectrom 30, 2041–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghose C (2013). Clostridium difficile infection in the twenty-first century. Emerg. Microbes Infect 2, e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giel JL, Sorg JA, Sonenshein AL, and Zhu J (2010). Metabolism of bile salts in mice influences spore germination in Clostridium difficile. PLoS ONE 5, e8740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiberson ER, Weiss A, Ryan DJ, Monteith AJ, Sharman K, Gutierrez DB, Perry WJ, Caprioli RM, Skaar EP, and Spraggins JM (2021). Spatially Targeted Proteomics of the Host-Pathogen Interface during Staphylococcal Abscess Formation. ACS Infect. Dis 7, 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, Kamada N, Jiao Y, Liu MZ, Núñez G, and Inohara N (2012). Protective role of commensals against Clostridium difficile infection via an IL-1β-mediated positive-feedback loop. J. Immunol 189, 3085–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knippel RJ, Zackular JP, Moore JL, Celis AI, Weiss A, Washington MK, DuBois JL, Caprioli RM, and Skaar EP (2018). Heme sensing and detoxification by HatRT contributes to pathogenesis during Clostridium difficile infection. PLoS Pathog. 14, e1007486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leffler DA, and Lamont JT (2015). Clostridium difficile infection. N. Engl. J. Med 372, 1539–1548. [DOI] [PubMed] [Google Scholar]
- Markham NO, Bloch SC, Shupe JA, Laubacher EN, Thomas AK, Kroh HK, Childress KO, Peritore-Galve FC, Washington MK, Coffey RJ, and Lacy DB (2021). Murine Intrarectal Instillation of Purified Recombinant Clostridioides difficile Toxins Enables Mechanistic Studies of Pathogenesis. Infect. Immun 89, e00543–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Guryn K, Hubert N, Frazier K, Urlass S, Musch MW, Ojeda P, Pierre JF, Miyoshi J, Sontag TJ, Cham CM, et al. (2018). Small Intestine Microbiota Regulate Host Digestive and Absorptive Adaptive Responses to Dietary Lipids. Cell Host Microbe 23, 458–469.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matamouros S, England P, and Dupuy B (2007). Clostridium difficile toxin expression is inhibited by the novel regulator TcdC. Mol. Microbiol 64, 1274–1288. [DOI] [PubMed] [Google Scholar]
- McDonald LC, Gerding DN, Johnson S, Bakken JS, Carroll KC, Coffin SE, Dubberke ER, Garey KW, Gould CV, Kelly C, et al. (2018). Clinical Practice Guidelines for Clostridium difficile Infection in Adults and Children: 2017 Update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin. Infect. Dis 66, 987–994. [DOI] [PubMed] [Google Scholar]
- Nguyen H-N, Tanaka M, Li B, Ueno T, Matsuda H, and Matsui T (2019). Novel in situ visualisation of rat intestinal absorption of polyphenols via matrix-assisted laser desorption/ionisation mass spectrometry imaging. Sci. Rep 9, 3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehaus M, Soltwisch J, Belov ME, and Dreisewerd K (2019). Transmission-mode MALDI-2 mass spectrometry imaging of cells and tissues at sub-cellular resolution. Nat. Methods 16, 925–931. [DOI] [PubMed] [Google Scholar]
- Paredes-Sabja D, Shen A, and Sorg JA (2014). Clostridium difficile spore biology: sporulation, germination, and spore structural proteins. Trends Micro-biol. 22, 406–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentice BM, Ryan DJ, Grove KJ, Cornett DS, Caprioli RM, and Spraggins JM (2020). Dynamic Range Expansion by Gas-Phase Ion Fractionation and Enrichment for Imaging Mass Spectrometry. Anal. Chem 92, 13092–13100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri BK, Hakkarainen-Smith JS, and Monro JA (2015). The potential use of cholestyramine to reduce the risk of developing Clostridium difficile-associated diarrhoea in patients receiving long-term intravenous ceftriaxone. Med. Hypotheses 84, 78–80. [DOI] [PubMed] [Google Scholar]
- Ridlon JM, Kang D-J, and Hylemon PB (2006). Bile salt biotransformations by human intestinal bacteria. J. Lipid Res 47, 241–259. [DOI] [PubMed] [Google Scholar]
- Scaldaferri F, Pizzoferrato M, Ponziani FR, Gasbarrini G, and Gasbarrini A (2013). Use and indications of cholestyramine and bile acid sequestrants. Intern. Emerg. Med 8, 205–210. [DOI] [PubMed] [Google Scholar]
- Shen A (2012). Clostridium difficile toxins: mediators of inflammation. J. Innate Immun 4, 149–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen A (2015). A Gut Odyssey: The Impact of the Microbiota on Clostridium difficile Spore Formation and Germination. PLoS Pathog. 11, e1005157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorg JA, and Sonenshein AL (2008). Bile salts and glycine as cogerminants for Clostridium difficile spores. J. Bacteriol 190, 2505–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spraggins JM, Djambazova KV, Rivera ES, Migas LG, Neumann EK, Fuetterer A, Suetering J, Goedecke N, Ly A, Van de Plas R, and Caprioli RM (2019). High-Performance Molecular Imaging with MALDI Trapped Ion-Mobility Time-of-Flight (timsTOF) Mass Spectrometry. Anal. Chem 91, 14552–14560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stabler RA, He M, Dawson L, Martin M, Valiente E, Corton C, Lawley TD, Sebaihia M, Quail MA, Rose G, et al. (2009). Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol. 10, R102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckli M, Farmer TB, and Caprioli RM (1999). Automated mass spectrometry imaging with a matrix-assisted laser desorption ionization time-of-flight instrument. J. Am. Soc. Mass Spectrom 10, 67–71. [DOI] [PubMed] [Google Scholar]
- Theriot CM, Bowman AA, and Young VB (2016). Antibiotic-Induced Alterations of the Gut Microbiota Alter Secondary Bile Acid Production and Allow for Clostridium difficile Spore Germination and Outgrowth in the Large Intestine. MSphere 1, e00045, e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ticho AL, Malhotra P, Dudeja PK, Gill RK, and Alrefai WA (2019). Intestinal Absorption of Bile Acids in Health and Disease. Compr. Physiol 10, 21–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westh H, Iversen JT, and Gyrtrup HJ (1991). Clostridium difficile in faecal flora after perioperative prophylaxis with ampicillin or ceftriaxone. J. Infect 23, 347–350. [DOI] [PubMed] [Google Scholar]
- Wexler AG, and Goodman AL (2017). An insider’s perspective: Bacteroides as a window into the microbiome. Nat. Microbiol 2, 17026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zackular JP, Moore JL, Jordan AT, Juttukonda LJ, Noto MJ, Nicholson MR, Crews JD, Semler MW, Zhang Y, Ware LB, et al. (2016). Dietary zinc alters the microbiota and decreases resistance to Clostridium difficile infection. Nat. Med 22, 1330–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw LC-MS/MS data are available on Mendeley Data: (https://doi.org/10.17632/rhbktnpyj2.1).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.