

Graphical abstract

Keywords: COVID-19, Oxazole, Macrocycles, SARS-CoV-2, Main protease
Abstract
We have discovered a family of synthetic oxazole-based macrocycles to be active against SARS-CoV-2. The synthesis, pharmacological properties, and docking studies of the compounds are reported in this study. The structure of the new macrocycles was confirmed by NMR spectroscopy and mass spectrometry. Compounds 13, 14, and 15a-c were evaluated for their anti-SARS-CoV-2 activity on SARS-COV-2 (NRC-03-nhCoV) virus in Vero-E6 cells. Isopropyl triester 13 and triacid 14 demonstrated superior inhibitory activities against SARS-CoV-2 compared to carboxamides 15a-c. MTT cytotoxicity assays showed that the CC50 (50% cytotoxicity concentration) of 13, 14, and 15a-c ranged from 159.1 to 741.8 μM and their safety indices ranged from 2.50 to 39.1. Study of the viral inhibition via different mechanisms of action (viral adsorption, replication, or virucidal property) showed that 14 had mild virucidal (60%) and inhibitory effects on virus adsorption (66%) at 20 μM concentrations. Compound 13 displayed several inhibitory effects at three levels, but the potency of its action is primarily virucidal. The inhibitory activity of compounds 13, 14, and 15a-c against the enzyme SARS-CoV-2 Mpro was evaluated. Isopropyl triester 13 had a significant inhibition activity against SARS-CoV-2 Mpro with an IC50 of 2.58 µM. Large substituents on the macrocyclic template significantly reduced the inhibitory effects of the compounds. Study of the docking of the compounds in the SARS CoV-2-Mpro active site showed that the most potent macrocycles 13 and 14 exhibited the best fit and highest affinity for the active site binding pocket. Taken together, the present study shows that the new macrocyclic compounds constitute a new family of SARS CoV-2-Mpro inhibitors that are worth being further optimized and developed.
1. Introduction
The severe acute respiratory syndrome coronavirus 2 (SARS-COV-2) virus was first detected in China's Wuhan province at the end of 2019 and has greatly affected human societies due to the global pandemic. The new virus has now infected over 120 million people around the world and resulted in over 2.6 million deaths [1], [2], [3], [4], [5]. SARS-CoV-2, like SARS-CoV and MERS-CoV, belongs to the genus Beta coronavirus and is responsible for the infection of the lower respiratory tract in particular, resulting in the disease COVID-19 [1], [6]. SARS-CoV-2 is a 30,000-base pair single stranded RNA virus, which possesses an envelope containing spike (S) proteins on its surface which binds with high affinity to the human angiotensin-converting enzyme 2 (ACE2) embedded in the membrane of cells found in the lungs, heart, kidneys, intestines, and arteries. This results in the infection of these cells by the virus [1], [7]. The incubation time can vary from 2 days up to 24 days and is asymptomatic in many cases, which facilitated the virus transmission [6]. Several efficient and safe vaccines have rapidly been developed by several pharmaceutical companies [7]. The production of sufficient doses for the vaccination of the world population has proved challenging with the concomitant threat of the emergence of vaccine-resistant variants of the virus.
Very few known drugs have proved effective in reducing the gravity of the disease in severely affect patients with COVID-19 [2], [6]. COVID-19-related research has been able to elucidate many druggable SARS-CoV-2 targets, including the spike (S) protein, the 3-chymotrypsin-like protease/main protease (3CLpro/Mpro), the papain-like protease (PLpro), and the RNA-dependent polymerase [3], [7]. Of these, viral proteases PLpro and Mpro constitute the most promising targets for the development of effective drugs against SARS-CoV-2. Viral proteases in coronaviruses are responsible for the development of non-structural proteins (nsps) by processing viral RNA translated polyproteins [3], [4], [7]. Thus, Mpro and PLpro have acknowledged a great deal of publicity for their significant role in the enzymatic operation leading to their post-translational treatment of replicate polyproteins that are essential to the life cycle of the corona virus [8], [9], [10], [11], [12], [13], [14], [15], [16]. In the last year, several peptide-like derivatives were introduced as COVID-19 Mpro inhibitors I-IX as shown in Fig. 1 [17], [18], [19], [20], [21]. Virtual screening and docking studies have played a pivotal role in the discovery of these kinds of inhibitors.
Fig. 1.

Peptide-like inhibitors of COVID-19 Mpro.
On the other hand, more than 40 cyclic peptides are used clinically in treatment of different diseases [22]. These peptides came from natural and synthetic origin. Takagi et al. have reported a series of cyclic peptide as NS2B-NS3 protease inhibitor [23]. The design of these peptides was done to mimic the substrate of NS2B-NS3 protease. Among this series, the cyclic peptide X (Fig. 2 ) exhibited inhibitory activity of NS2B-NS3 protease of dengue virus at IC50 of 0.95 μM. Aspergilli peptide D XI is a natural product isolated from Aspergillus sp and exhibited inhibitory activity against herpes simplex virus type 1 at IC50 value of 9.5 μM [24].
Fig. 2.

Cyclic peptides with antiviral activity.
The eight-residue cyclic D,L-α-peptide XII exhibited antiviral activity against adenovirus infection without an apparent adverse effect on cell viability [25]. The plausible mechanism of action of this compound could be mediated by targeting endosomal compartments in pH-dependent viral infections. Liang et al. have reported a series of cyclic peptide XIII with significant antifungal and obvious antiviral activity against herpes simplex virus [26]. Among these derivatives, Simplicilliumtide J XIV exhibited significant antiviral activities against herpes simplex virus type 1 (IC50 = 14.0 μM) [26].
The cyclic peptide ICU-1 (XI) was designed as potential anti-COVID-19 activity [27]. The design was performed using SARS coronavirus (SARS-CoV) main protease bound to a peptide substrate, which was modified to give a cyclic peptide inhibitor VI. A molecular docking study was used to illustrate the binding interactions of the designed peptide with main protease of both SARS-CoV and SARS-CoV-2.
Macrocyclic peptides are notoriously promising privilege structures in drug discovery due to their improved selectivity and stability against proteolytic enzymes in addition to their higher membrane permeability compared to their linear counterparts [28], [29].
Motivated by the aforementioned data and over the course of the screening of in-house collections of molecules we discovered that a family of oxazole-based macrocycles, previously developed for other applications [30], [31], [32] possessed significant activity against SARS-CoV Mpro. The large macrocyclic structures were made following the work of Rebek et al who developed this type of nature-inspired molecules for applications in supramolecular chemistry [30] and tackle the inhibition of protein–protein interactions [31], [32] (Fig. 3 ).
Fig. 3.

Structures of marine natural product dolastatin E, Rebek platform, galanin receptor agonist Galmic, and the bioactive oxazole-based macrocycles studied herein.
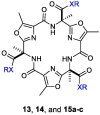
We report herein the inhibitory activity of the new macrocyclic compounds 13, 14, and 15a-c (Fig. 3) against the SARS-COV-2 main protease, their pharmacological properties in infected Very-E6 cells, and results of docking studies in the SARS-COV-2 main protease active site.
2. Results and discussion
2.1. Chemistry
The synthesis of the macrocyclic compounds is largely inspired from the synthesis of Rebek triacid platform and Galmic [30], [31], [32]. However, we prepared the required oxazole building blocks having the opposite absolute configuration to that required for the synthesis of Galmic, leading to a slightly simplified synthesis of the macrocycles.
Enantiomerically pure amino acid 6 was prepared in five steps from commercially available amino diester 1 (Scheme 1 ). Boc-protection of this compound followed by α-methylation, and kinetic resolution using pig liver esterase gave enantiomerically pure Boc-protected amino acid 4. This compound was then esterified as an isopropyl ester and the methyl ester was hydrolyzed to afford the required compound 6 in good overall yield.
Scheme 1.

Synthesis of carboxylic acid 6. Reagents and conditions: (i) Boc2O, NaHCO3, NaCl, DCM/H2O, reflux for 1.5 h, 92%; (ii) CH3I, CH3ONa, CH3OH, stir at 50 ◦C for 2 h, 79%; (iii) PLE, K2HPO4, KH2PO4, H2O/CH3CN, stir 5 days at rt, 91%; (iv) isopropyl alcohol, DCC, DMAP, stir at rt for 20 h, 77%; (v) LiOH, THF/H2O, stir at rt for 2 h, 94%.
Oxazole isopropyl amino ester building block 12 was then obtained from compound 6 by performing a sequence of reactions similar to those described for the synthesis of the Rebek platform (Scheme 2 ). Coupling of Boc-amino acid 6 with commercially available l-threonine benzyl ester 7 using EDCI and HOBt in the presence of DIPEA gave dipeptide 8. Oxidation of the secondary alcohols of 8 with the Dess-Martin reagent followed by cyclodehydration using iodine/triphenylphosphine in the presence of 4-dimethyl aminopyridine (DMAP) and TEA afforded oxazole 10. IR spectrum of oxazole 10 showed a stretching NH band at 3424 cm−1 and C O stretching bands at 1755, 1740, and 1710 cm−1, consistent with the proposed structure and literature data on similar compounds [26]. The 1HNMR spectrum of oxazole 10 showed signals for the benzyl group at 7.45–7.28 ppm (m, 5H, Ar-H) and 5.34 (s, 2H, PhCH2), isopropyl group at 5.02 ppm (h, J = 6.3 Hz, 1H, CH(CH3)2) and 1.16 (dd, J = 13.1, 6.3 Hz, 6H, CH(CH3)2), and finally a singlet signal at 2.56 ppm corresponding to the oxazole CH3 groups. The compound was further characterized by HRESI-MS with the presence of a [M + H]+ ion at m/z 447.2129 which correlates with the molecular weight and molecular formula C23H31N2O7 of 10. Hydrogenation of the benzyl ester group was then performed using Pd/C (10%) in EtOAc with vigorous stirring at room temperature under hydrogen atmosphere to yield free carboxylic acid 11. Deprotection of the Boc group was carried out under acidic conditions by treatment of 11 with 4 M HCl solution in dioxane to afford a quantitative yield of 12 as a hydrochloride salt.
Scheme 2.

Synthesis of oxazole building block 12. Reagents and conditions: (i) HOBt, EDC, DIPEA, DMF, rt, overnight, 74%; (ii) Dess-Martin, DCM, rt, 4 h; (iii) PPh3, I2, TEA, DMAP, DCM, rt, 5 h, 84%; (iv) Pd/C (10%), H2, EtOAc, rt, overnight, 87%; (v) 4 M HCl in dioxane, rt, 1.5 h, greater than 99%.
The syntheses of triacid macrocycle 14 and the triamide macrocycles 15a-c are depicted in Scheme 3 . Triester macrocycle 13 was directly obtained in good yield through treatment of the salt 12 with BOP in the presence of DIPEA. The isopropyl ester groups of 13 were subsequently saponified using 5% NaOH to furnish triacid 14 after acidic workup. Coupling of 14 with appropriate amines using BOP in the presence of DIPEA afforded the desired carboxamides 15a-c in good yields. The 1H NMR of 15a (as a representative example), shows singlet signals at 8.79 ppm (3H, NHCO), 3.30 ppm (9H, oxazole-CH3), and 1.97 ppm (9H, quaternary C-CH3). The phenethyl group signals appear at 3.51 ppm (q, J = 7.2 Hz, 6H, 3 × NHCH2CH2) and 2.75 ppm (t, J = 7.2 Hz, 6H, 3 × NHCH2CH2). HRESI-MS analysis confirmed the molecular formula of 15a HRESI-MS m/z calcd for [M + H]+ C48H52N9O9: 898.3883, found: 898.3884, see supplementary file.
Scheme 3.

Synthesis of macrocycles 13, 14 and 15a-c. Reagents and conditions:(i) BOP, DIPEA, DMF, 24 h, rt, 60%; (ii) 5% NaOH, EtOH, 0 °C, 1 h, quantitative; (iii) Appropriate amine, BOP, DIPEA, DCM, overnight, rt, 70–75%.
2.2. Biological activities
2.2.1. Detection of in vitro cytotoxicity
A colorimetric MTT assay [33] was used to assess the cytotoxicity of 13, 14, and 15a-c on Vero-E6 cells. The results showed that their CC50 ranged from 159.1 to 741.8 μM and their safety indices ranged from 2.50 to 39.1 (Table 1 , Fig. 4 a). Two of the best SARS-COV-2 inhibitors are 13 and 14 with IC50 of 18.3 and 18.95 μM and SI of 20.8 and 39.1.
Table 1.
Anti-SARS-COV-2 activity, cytotoxicity, and selectivity indices of compounds 13, 14, and 15a-c.
 | ||||||
|---|---|---|---|---|---|---|
| Compd. | X | R | CC50a(μM) | IC50b(μM) | SIc | IC50 SARS-CoV-2 Mpro(μM) |
| 13 | O | isopropyl | 380.7 | 18.3 | 20.8 | 2.6 ± 0.1 |
| 14 | O | H | 741.8 | 18.9 | 39.1 | 3.8 ± 0.1 |
| 15a | NH | phenethylamine | 558.5 | 24.7 | 22.6 | 7.3 ± 0.2 |
| 15b | NH | 4-(piperidin-1-yl)phenylethylamine | 672.6 | 61.7 | 10.9 | 15.4 ± 0.5 |
| 15c | NH | 4-(N,N-dimethylamine)phenylethylamine | 159.1 | 63.3 | 2.5 | 18.3 ± 0.5 |
CC50: half maximal cytotoxic concentration of test compounds within Vero-E6 cells
IC50: half maximal inhibitory concentration of test compounds on SARS-COV-2 (NRC-03-nhCoV) virus in Vero-E6 cells
SI: Safety index = CC50/IC50 for the compound.
Fig. 4.

CC50 (50% cytotoxic concentration) of the tested macrocycles in Vero-E6 cells (Fig. 4a) and IC50 (50% inhibitory concentration) against NRC-03-nhCoV virus in Vero-E6 cells (4b). The CC50 and IC50 were plotted for each tested compound using Graph Pad Prism and were calculated from the non-linear regression curve-fit analysis, relative to the virus and cell controls.
2.2.2. Anti-SARS-CoV-2 activity
The inhibitory effect of compounds 13, 14, and 15a-c on SARS-COV-2 (NRC-03-nhCoV) virus in Vero-E6 cells were evaluated as previously described [34], [35], [36], [37]. Results from Table 1 showing that most of the compounds tested significantly inhibited the replication of the NRC-03-nhCoV virus in a dose-dependent profile. Triester macrocycle 13 and triacid 14 demonstrated superior inhibitory activity compared to carboxamides 15a-c ( Table 2 , Fig. 4 b). The strongest anti-SARS-COV-2 compounds are 13, 14 and 15a, which showed IC50s of 18.3, 18.95 and 24.69 μM, respectively. The triester macrocycle compound 13 (R = isopropyl) was the most effective synthesized derivatives with an IC50 value of 18.3 µM against Vero-E6 cells. The unsubstituted triacid derivative 14 (R = H) was comparable to 13 showing IC50 18.95 µM. Replacement of the isopropyl group in 13 by phenethylamine, 4-(piperidin-1-yl) phenylethylamine and 4-(N,N-dimethylamine) phenylethylamine in compounds 15a, 15b and 15c respectively resulted in a decrease of at least 1.4-fold in the IC50 value, suggesting the significance of the isopropyl group (less hindered group) for SARS-COV-2 replication inhibitory activity in Vero-E6 cells.
Table 2.
Mechanisms of action of compounds 13, 14, and 15a.
| Compound | Conc. (µM) |
Mode of action* |
||
|---|---|---|---|---|
| Viral adsorption | Viral replication | virucidal | ||
| 13 | 20 | 52% | 53% | 57% |
| 10 | 12% | 23% | 18% | |
| 5 | 00 | 6% | 12% | |
| 2.5 | 00 | 00 | 13% | |
| 14 | 20 | 66% | 38% | 60% |
| 10 | 24% | 36% | 28% | |
| 5 | 16% | 30% | 12% | |
| 2.5 | 00 | 11% | 6% | |
| 15a | 20 | 11% | 51% | 89% |
| 10 | 9% | 19% | 58% | |
| 5 | 00 | 5% | 00 | |
| 2.5 | 00 | 00 | 00 | |
The mechanism of action of the three compounds were done at concentrations higher than the half-maximal inhibitory effect “IC50” to better resolve the mechanism of action.
2.2.3. Mechanism of Anti-SARS-CoV-2 activity
The percent inhibition of different mechanisms of action (viral adsorption, replication, and virucidal) for the most active compounds 13, 14 and 15a is shown in Table 2 [36]. Compound 15a has a virucidal effect of up to 90%, meaning that it acts directly on the virion and inactivates it. Furthermore, it inhibited viral replication by up to 50% at a concentration of 20 µM with a marginal effect on viral adsorption. Compound 14 had a mild viral replication effect with a 60% inhibitory effect and a 65% inhibitory effect on virus adsorption. Finally, Compound 13 displayed several inhibitory effects at three levels, but the potency of its action is primarily a virucidal influence.
2.2.4. Inhibitory activity against SARS-CoV-2 Mpro
Several potential target proteins in coronavirus (COVID-19) have been isolated and are now available through protein data bank. Protease is one of these important proteins that has been targeted by a panel of inhibitors with different chemical scaffolds and were suggested as potential drugs to be developed against this challenging virus [38], [39]. Interestingly, there is no similarity in cleavage specificity of this enzyme and the corresponding human proteases which in turn makes it an ideal antiviral target with high margins of safety and selectivity [4]. Inspired by these findings, it was of interest to investigate the potential protease inhibitory activities of compounds 13, 14, and 15a-c against COVID-19.
The inhibitory activity of compounds 13, 14, and 15a-c against SARS-CoV-2 Mpro were evaluated following the previously published procedure [40]. Results recorded in Table 1 demonstrated that the isopropyl triester 13 (R = isopropyl) had a promising inhibition activity against SARS-CoV-2 Mpro with an IC50 of 2.58 µM. Replacement of the isopropyl group in compound 13 by (H) in triacid 14 (R = H) decreases the activity of 14 (IC50 = 3.82 μM) which indicated the importance of the hydrophobic isopropyl group for activity. Large substituents at the R position such as phenethylamine (15a), 4-(piperidin-1-yl)phenethylamine (15b), and 4-(N,N-dimethylamine)phenethylamine (15c) groups decrease the activity of the compounds with IC50s of 7.35 µM,15.34 µM, and 18.32 µM, respectively.
2.3. Molecular docking study
Given the promising activity of new macrocyclic compounds 13, 14, and 15a-c against COVID-19, we conducted a molecular docking study to investigate possible binding patterns and interactions of the compounds in the active site of COVID-19 main protease (SARS-CoV-2 Mpro). The 3-dimentional crystal structure of SARS-CoV-2 Mpro (PDB ID: 6LU7) in complex with covalent inhibitor N3, was downloaded from the protein data bank (https://www.rcsb.org/structure/6LU7) and the docking simulation process was accomplished using AutoDock Vina [41] as docking module embedded in AutoDockTools (ADT) [42], [43]. Ligands and protein files were prepared using Discovery Studio Visualizer [44], AutoDockTools, and docked to SARS-CoV-2 Mpro (PDB ID: 6LU7) with AutoDock Vina.
Validation of the docking was performed using the recently reported X-ray structure of SARS-CoV-2 Mpro in complex with non-covalent inhibitor Mcule-5948770040 (PDB ID: 7LTJ) [45]. Examination of the crystal structure indicated that Mcule-5948770040 binding position is probably not significantly influenced by bridging water molecules, contrary to some other non-covalent inhibitors. The shapes of the binding sites in 6LU7 and 7LTJ are highly identical and structure 7LTJ was deemed appropriate for performing the docking validation as Mcule-5948770040 is non-covalent inhibitor. Remarkably in this case, the highest scoring pose (−9.4 kcal/mol) for the re-docking of Mcule-5948770040 was found to have an almost identical binding position and conformation to that of the ligand in the crystal structure (RMDS of 0.1325 Å) [Fig. 5 ]. This validated the use of Autodock Vina in the present docking study assuming that there are no major water-mediated interactions between the enzyme and the macrocyclic compounds.
Fig. 5.

Comparison of crystal structure of Mcule-5948770040 bound to SARS-CoV-2 Mpro [PDB ID: 7LTJ] (grey carbon, blue nitrogen, and green chlorine atoms) and highest scoring pose (yellow atoms) found with AutoDock Vina. The root-mean-square deviation of atomic positions (RMSD) is 0.1325 Å. The hydrogen atoms have been omitted for clarity. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Rebek triacid platform as well as ester and amide derivatives are known to adopt a quasiplanar structure stabilized by six bifurcated hydrogen bonds provided by the alternating oxazole (hydrogen-bond acceptor) and trans amide (hydrogen-bond donor) groups of the macrocycle [46], [47]. The structures of compounds 13, 14 and 15a-c were prepared in Discovery Studio Visualizer using the MMFF94 force field [48] and submitted to docking via ADT on SARS-CoV-2 Mpro (PDB ID: 6LU7) with the ligand N3 and water molecules being removed.
Docking was initially performed using triester 13, which was found to dock in cavities of SARS-CoV-2 Mpro, in particular the enzyme active site, site 2 and site 3 [Fig. 6 ]. However, the docking scores for the poses docked in the active site were significantly better. This also proved true for triacid and triamides 15a-c. We therefore focused on the binding mode of these compounds in the enzyme active site.
Fig. 6.

Electrostatic surface representation of COVID-19 main protease showing possible binding sites for: (A) Ligand N3 in active site as published by Jin et al (PDB code: 6LU7) [49]. (B) Compound 13 docked in active site (yellow), site 2 (green, brown, and purple poses) and site 3 (blue). The ligands were rendered using the stick representation. Non-polar hydrogen atoms were omitted for clarity. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 7A shows a close-up view of the bound conformation of ligand N3 in PDB 6LU7 in comparison to the docked structure of triester 13 in the same enzyme active site (Fig. 7 B). The large surface area of compound 13 complements well the relatively flat surface area of this part of the active site defined by Phe-140, His-163, Met-165, Glu-166, and Pro-168, with one of the hydrophobic isopropyl groups of the compound protruding into the recognition site of the benzyl group of N3. The methyl substituents of the oxazole rings and two methyl groups of the macrocyclic scaffold appear to complement small pockets (Fig. 7 C and D). Discovery studio visualizer suggests the presence of a significant anion-π interaction between glutamate-166 and one of the oxazole rings of the ligand (Fig. 7 E). The docking score for this pose was −7.5 kcal/mol. Glu-189 and Asp-144 side chains also appear to lock the molecule in place. In fact, re-docking of the molecule with the enzyme where these side chains were rotated to offer additional hydrogen-bonding interactions with the ligand yielded a higher docking score of −7.8 kcal/mol (Fig. 7 F). Additional hydrophobic interactions are also seen in this case with Pro-168.
Fig. 7.

(A) and (B): Electrostatic surface representation of COVID-19 main protease showing the position of ligand N3 and docked triester 13, respectively, within the enzyme active site. (C) and (D): Solvent access surface (yellow, probe radius of 1.4 Å) representation of COVID-19 main protease binding site, side view and top view (respectively) for the best binding pose of triester 13. (E) 2D interaction diagram for the best docking pose of triester 13. Docking score of −7.5 kcal/mol (F) 2D interaction diagram for the best docking pose of triester 13 after rotation of Gln189 and Asn142 side chains and re-docking showing additional interactions between the ligand and protein (docking score of −7.8 kcal/mol). The ligands were rendered using the stick representation. Non-polar hydrogen atoms were omitted for clarity. CH-π and hydrophobic interactions are shown in pink, conventional hydrogen bonds are shown in green, anion-π interactions are shown in orange. The strength of the interactions is approximately proportional to the size of the blue circles. Filled blurry purple circles indicate groups in contact with the solvent. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Following the docking of triester 13, which has the best anti-COVID activity among the macrocycles tested, we studied the possible interactions that the enzyme could make with the other bioactive compounds triacid 14 and amide 15a (Fig. 8 ). AutoDock Vina docking results suggest that the triacid 14 and triester 13 can bind to the active site in an almost identical way. We can visualize the same shape complementarity of the platform with the binding site including almost identical hydrophobic interactions, the presence of the uncommon anion-π interaction, and the apparition of a weak hydrogen bond between one of the carboxylic acid groups and Asn-142. However, there are no protruding groups in this case to fill the N3 benzyl group binding site. Nevertheless, the binding energy obtained by AutoDock Vina for the triacid 14 (-7.9 kcal/mol) was found to be comparable to that obtained for the triester 13 (-7.5 kcal/mol).
Fig. 8.

Electrostatic surface representation of COVID-19 main protease showing the active site binding modes of: (A) triacid 14 and (B) triamide 15a. The ligands were rendered using the stick representation. Non-polar hydrogen atoms were omitted for clarity. Ligand-protein interactions are shown as dashed lines. CH-π and hydrophobic interactions are shown in pink, conventional hydrogen bonds are shown in green, CH-O hydrogen bonds are shown in light green, anion-π interactions are shown in orange, and sigma-π interactions are shown in red violet. The strength of the interactions is approximately proportional to the size of the filled blue circles. Blurry light purple circles indicate chemical groups making contact with the bulk solvent. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Triamide 15a was also found to adopt a similar position as triester 13 and triacid 14 in the binding site, with essentially similar interactions with the platform. Additional interactions can also be seen, including an excellent size and shape complementarity of the V3 benzyl group binding site with one of the phenethyl groups of 15a, interacting with His-41, Leu-27 and Cys-145. The other phenethyl groups, however, cannot adopt low-energy extended conformations and this would cause a significant decrease in binding affinity. One of these groups would also make extensive contact with the bulk water affecting the overall binding. Despite this, AutoDock Vina estimated a binding energy of −8.3 kcal/mol.
Collectively, the docking study, along with in vitro SARS-CoV-2 Mpro results, seem to indicate that the active site of the protease can accommodate large molecules. The docking study suggests that derivatives of triester 13, which feature elements of triacid 14 and triamide 15a, as well as modifications of the tris(oxazole) platform using appropriate substituents could lead to highly active SARS-CoV-2 Mpro inhibitors.
3. Conclusion
A small set of new oxazole-based macrocycles 13, 14, and 15a-c synthesized, and tested for their anti-SARS-COV-2 activity. Compounds 13, 14 and 15a showed promising inhibitory properties for the SARS-COV-2 (NRC-03-nhCoV) virus in Vero-E6 cells, and the SARS-CoV-2 main protease assay revealed significant inhibitory effects of these compounds. This mechanism of action is further supported by docking experiments which have shown that most of the tested compounds fit well within the SARS CoV-2-Mpro active pocket. The results of the docking study are consistent with the in vitro SARS CoV-2-Mpro inhibitory activities of the compounds. Taken together, these oxazole-based macrocyclic compounds could constitute promising leads for further optimization and development against SARS-CoV main proteases, and open new avenues to help defeat future infections by coronavirus variants.
4. Experimental
4.1. Chemistry
General details: See Appendix A
4.1.1. Dimethyl 2-(tert-butoxycarbonylamino)malonate (2)
To a stirred suspension of dimethyl amino malonate hydrochloride (11.9 g, 64.26 mmol, 1 eq) in CHCl3 (50 mL), NaHCO3 (6.4 g, 1.2 eq) dissolved in H2O (10 mL), NaCl (4.54 g, 1.2 eq), and Boc2O (17 g, 1.2 eq) were added to reaction mixture with refluxing for 1.5 h. After cooling, the organic layer was separated, dried over anhydrous Na2SO4, and evaporated under reduced pressure to give 18.5 g (92%) of 2 as yellow oil after purification by flash chromatography using EtOAc/ hexane (1:2) as an eluent.
1H NMR (250 MHz, CDCl3) δ 5.54 (s br, 1H, NH), 4.95 (d, J = 8.0 Hz, 1H, NHCH), 3.75 (s, 6H, 2 × OCH3), 1.38 (s, 9H, C(CH3)3). 13C NMR (62.5 MHz, CDCl3) δ 167.0 (C O), 154.74 (NHC = O), 85.1, 57.2, 53.3, 28.2.
4.1.2. Dimethyl 2-(tert-butoxycarbonylamino)-2-methylmalonate (3)
A mixture of 2 (18 g, 72.8 mmol, 1 eq) and CH3ONa (15.64 mL) in CH3OH (20 mL) were charged to double necked flask equipped with condenser. Methyl iodide (5.45 mL, 1.2 eq) was added dropwise to the reaction mixture over period of 30 mins with stirring at 50 °C and the stirring was continued for another 1.5 h at the same temperature. After removal of solvent under reduced pressure, saturated solution NH4Cl (10 mL) were added and reaction mixture was extracted with EtOAc, washed with brine, dried over MgSO4, and evaporated under reduced pressure to give 14 g (79 %) of 3 as a colorless oil. ν max (neat, film)/cm−1 3434 ((NH), 2981, 2850, 1750 (C O), 1716 (C O), 1488, 1369, 1290, 1171, 1104, 1062. 1H NMR (250 MHz, CDCl3) δ 5.95 (s br, 1H, NH), 3.76 (s, 6H, 2 × OCH3), 1.71 (s, 3H, CH3), 1.40 (s, 9H, C(CH3)3). 13C NMR (62.5 MHz, CDCl3) δ 169.4, 153.6, 80.1, 62.9, 53.5, 28.2, 21.7. HRESI-MS m/z calcd for [M + H]+ C11H20NO6: 262.1285, found: 262.1287.
4.1.3. (R)-2-(tert-butoxycarbonylamino)-3-methoxy-2-methyl-3-oxopropanoic acid (4)
In three-liter beaker with magnetic stirrer, K2HPO4 (31.23 g, 3.5 eq) and KH2PO4 (17.42 g, 2.5 eq) were dissolved in water (1800 mL). Then PLE (0.495 g, 5000 units Sigma) was added with vigorous stirring for 15 min at rt. A solution of 3 (13.3 g, 51.67 mmol, 1 eq) dissolved in CH3CN (450 mL) was added to reaction mixture with the vigorous stirring for 5 days. pH of the reaction mixture was maintained during reaction time between 7.5 and 8.5. After adjusted of pH = 3 by 1 N HCl, EtOAc and NaCl were successively added with stirring at rt for 5 mins. The separated organic layer was dried over anhydrous MgSO4 and evaporated under reduced pressure to give 11.2 g (91%) of 4 as a white solid, mp 112–114 °C. ν max (KBr disc)/cm−1 3350 (OH and NH), 2950, 1732 (C O), 1694 (C O), 1651 (C O), 1118, 1059, 968, 902, 774, 640. 1H NMR (400 MHz, CDCl3) δ 8.09 (s, 1H, NH), 3.78 (s, 3H, OCH3), 1.78 (s, 3H, CH3), 1.41 (s, 9H, C(CH3)3). 13C NMR (101 MHz, CDCl3) δ 171.6 (COOH), 169.1, 156.2, 82.8, 63.3, 52.8, 28.1, 21.9.HRESI-MSm/z calcd for [M−H]- C10H16NO6: 246.0983, found: 246.0976.
4.1.4. (S)-1-isopropyl 3-methyl 2-(tert-butoxycarbonylamino)-2-methylmalonate (5)
To a stirred solution of 4 (6 g, 24.27 mmol, 1 eq) in DCM (100 mL), isopropyl alcohol (3 mL, 1.5 eq), DCC (5.55 g, 1.1 eq), and DMAP (0.3 g, 0.1 eq) were added at 0 ◦C then the temperature was warmed to rt with stirring for 20 h. The urea precipitate was filtered, and the DCM was removed under reduced pressure to give 5.4 g (77%) of 5 as a colourless oil after chromatographed on silica gel using EtOAc/hexane (1:8) as an eluent. ν max (neat, film)/cm−1 3434 (NH), 2981, 2850, 1760 (C O), 1752 (C O), 1720 (C O), 1488, 1444, 1287, 1170, 1063. 1H NMR (400 MHz, CDCl3) δ 5.89 (s, 1H, NH), 5.03 (h, J = 6.3 Hz, 1H, CH(CH3)2), 3.73 (s, 3H, OCH3), 1.68 (s, 3H, CCH3), 1.39 (s, 9H, C(CH3)3), 1.19 (d, J = 6.4 Hz, 6H, CH(CH3)2).13C NMR (101 MHz, CDCl3) δ 169.4, 168.2, 153.8, 80.1, 70.2, 62.9, 53.1, 28.2, 21.4, 21.3.HRESI-MS m/z calcd for [M + H]+ C13H24NO6: 290.1598, found: 290.1602.
4.1.5. (S)-2-(tert-butoxycarbonylamino)-3-isopropoxy-2-methyl-3-oxopropanoic acid (6)
To a solution of 5 (3.91 g, 13.52 mmol, 1 eq) in a mixture of THF and H2O (2:1, 12 mL), LiOH (0.422 g, 1.3 eq) was added with stirring for 2 h at rt. The residue after removal of solvent under reduced pressure was then taken into water, precipitated out at pH = 1 using 5% HCl, extracted twice with EtOAc, dried over MgSO4, and evaporated under reduced pressure to give 3.5 g (94%) of 6 as a colourless oil. ν max (neat, film)/cm−1 3427 (br, OH, NH), 2980, 1760 (C O), 1732 (C O), 1670 (C O), 1489, 1286, 1064. 1H NMR (250 MHz, CDCl3) δ 7.9 (s, 1H, NH), 5.06 (m, 1H, CH(CH3)2), 1.73 (s, 3H, CCH3), 1.39 (s, 9H, C(CH3)3), 1.23 (d, J = 5.25 Hz, 6H, CH(CH3)2). 13C NMR (62.5 MHz, CDCl3) δ 172.2, 168.0, 156.1, 82.7, 70.0, 63.3, 28.2, 21.9, 21.4. HRESI-MS m/z calcd for [M + H]+ C12H22NO6: 276.1442, found: 276.1442.
4.1.6. Benzyl 2-((S)-2-(tert-butoxycarbonylamino)-3-isopropoxy-2-methyl-3-oxo-propan amido)-3-hydroxybutanoate (8)
To a stirred solution of 6 (5 g, 18.16 mmol, 1 eq) in DMF (70 mL), HOBt (1.93 g, 1 eq), EDC (1.93 g, 1 eq), l-threonine benzyl ester 7 (6.44 g, 1.2 eq) were added and reaction mixture was cooled to 0 °C before dropwise addition of DIPEA (1.05 mL, 1.2 eq) then the reaction mixture was stirred overnight at rt. The reaction mixture was diluted with EtOAc and successively washed twice with H2O. The organic layer was washed with 5% aqueous HCl, saturated NaHCO3, and finally with brine. The organic phase was dried over MgSO4, and the solvent was removed under reduced pressure to yield 6.10 g (74%) of 8 as a colourless oil which solidify to a white solid after chromatographed on silica gel using EtOAc/ hexane (1:1) as an eluent. ν max (neat, film)/cm−1 3407 (br, OH, NH), 2980, 2936, 1760 (C O), 1750 (C O), 1720 (C O), 1685 (C O), 1491, 1450, 1376, 1282, 1102, 1061, 753, 699. 1H NMR (400 MHz, CDCl3) δ 7.48–7.00 (m, 6H, Ar-H, NHC = O), 6.02 (s, 1H, NHCOO), 5.16 (d, J = 5.0 Hz, 2H, PhCH2), 5.05 (h, J = 6.3 Hz, 1H, CH(CH3)2), 4.57 (dd, J = 8.9, 2.7 Hz, 1H, NHCH), 4.34–4.30 (m, 1H, CHOH), 2.69 (s, 1H, OH), 1.71 (s, 3H, CCH3), 1.41 (s, 9H, C(CH3)3), 1.21 (d, J = 6.3 Hz, 6H, CH(CH3)2), 1.14 (d, J = 6.4 Hz, 3H, CHCH3). 13C NMR (101 MHz, CDCl3) δ 170.5, 170.2, 168.6, 154.3, 135.2, 128.6, 128.4, 128.2, 70.5, 67.9, 67.3, 62.9, 57.9, 28.2, 21.5, 21.4, 19.9. HRESI-MS m/z calcd for [M + H]+ C23H35N2O8: 467.2388, found: 467.2394.
4.1.7. Benzyl 2-((S)-2-((tert-butoxycarbonyl)amino)-3-isopropoxy-2-methyl-3-oxo- propanamido)-3-oxobutanoate (9)
To a solution of 8 (3.6 g, 7.74 mmol, 1 eq) in DCM (70 mL), Dess-Martin periodinane (4.26 g, 1.3 eq) was added with stirring at 0 °C for 30 mins then the temperature was warmed to rt for 4 h. Saturated solution of Na2S2O3 (20 mL) and saturated solution of NaHCO3 (20 mL) were added to reaction mixture with stirring at rt for 15 mins. The organic layer was separated, dried over Na2SO4,and evaporated under reduced pressure to yield a crude product which was used for next step without further purification.
4.1.8. (S)-benzyl 2-(2-(tert-butoxycarbonylamino)-1-isopropoxy-1-oxopropan-2-yl)-5-methyloxazole-4-carboxylate (10)
To a solution of 9 (2.48 g, 5.34 mmol, 1 eq) in DCM (100 mL), triethylamine (TEA, 2.96 mL, 4 eq), and DMAP (0.064 g, 0.1 eq) were added with stirring for 1 h at rt. Triphenylphosphine (PPh3, 1.68 g, 1.2 eq) was added to reaction mixture followed by portion wise addition of iodine (1.63 g, 1.2 eq) at 0 °C. The cooling bath was removed after 30 mins and stirring was continued at rt for 5 h. The solvent was removed under reduced pressure. The residue was diluted with Et2O to enhance the precipitation of triphenylphosphine oxide (OPPh3) which was filtered out. Pure oxazole was obtained after flash chromatography of crude product using a mixture of ethyl acetate, hexanes (1:4) as an eluent to give 2 g (84%) of 10 as a colourless oil. ν max (neat, film)/cm−1 3424 (NH), 2980, 1755 (C O), 1740 (C O), 1719 (C O), 1618, 1491, 1458, 1368, 1280, 1170, 753, 699. 1H NMR (400 MHz, CDCl3) δ 7.45–7.28 (m, 5H, Ar-H), 6.14 (s, 1H, NH), 5.34 (s, 2H, PhCH2), 5.02 (h, J = 6.3 Hz, 1H, CH(CH3)2), 2.56 (s, 3H, oxazole-CH3), 1.94 (s, 3H, CCH3), 1.38 (s, 9H, C(CH3)3), 1.16 (dd, J = 13.1, 6.3 Hz, 6H, CH(CH3)2). 13C NMR (101 MHz, CDCl3) δ 168.7, 161.9, 160.1, 157.0, 153.8, 135.7, 128.5, 128.4, 128.3, 127.6, 80.2, 70.6, 66.5, 58.9, 28.2, 21.4, 21.4, 12.2. HRESI-MS m/z calcd for [M + H]+ C23H31N2O7: 447.2126, found: 447.2129.
4.1.9. (S)-2-(2-(tert-butoxycarbonylamino)-1-isopropoxy-1-oxopropan-2-yl)-5-methyl- oxazole-4-carboxylic acid (11)
To a vigorous stirred solution of oxazole 10 (1.2 g, 2.88 mmol) in EtOAc (15 mL) in two-necked flask purge with N2, Pd/C (10%, 0.26 g) was added. The reaction mixture was stirred overnight under balloon of hydrogen at rt. Pd/C was filtered through double filter paper and EtOAc was removed under reduced pressure to give 0.83 g (87%) of pure corresponding acid 11 as a colourless oil. ν max (neat, film)/cm−1 3419 (br, OH, NH), 2984, 1750 (C O), 1655 (C O), 1627 (C O), 1491, 1369, 1285, 1110, 752. 1H NMR (400 MHz, CDCl3) δ 6.52 (s, 1H, NH), 5.03 (h, J = 6.3 Hz, 1H, CH(CH3)2), 2.60 (s, 3H, oxazole-CH3), 1.94 (s, 3H, CCH3), 1.36 (s, 9H, C(CH3)3), 1.16 (dd, J = 17.7, 6.2 Hz, 6H, CH(CH3)2). 13C NMR (101 MHz, CDCl3) δ 168.7, 165.5, 160.2, 157.8, 154.0, 127.4, 70.8, 60.4, 58.8, 28.2, 21.4, 21.3 for (CH3)2, 21.0, 12.2. HRESI-MS m/z calcd for [M + H]+ C16H25N2O7: 357.1656, found: 357.1653.
4.1.10. (S)-2-(4-carboxy-5-methyloxazol-2-yl)-1-isopropoxy-1-oxopropan-2-aminium chloride (12)
4 M HCl in dioxane (50 mL) was added to carboxylic acid building bock 11 (0.7 g, 0.62 mmol) at 0 °C under N2 atmosphere with stirring for 10 mins then the temperature was warmed to rt for 1.5 h. The reaction mixture was concentrated by rotary evaporation under high vacuum then the residue was washed with Et2O three times to give 0.63 g (95%) of HCl salt of 12 as white solid. ν max (neat, film)/cm−1 3425 (br, OH, NH3), 2932, 1751 (C O), 1719 (C O), 1618, 1450, 1297, 1232, 1185, 1100. 1H NMR (400 MHz, DMSO‑d 6) δ 9.63 (s, 1H, COOH), 5.01 (h, J = 6.3 Hz, 1H, CH(CH3)2), 2.59 (s, 3H, oxazole-CH3), 1.89 (s, 3H, CCH3), 1.19 (dd, J = 21.4, 6.3 Hz, 6H, CH(CH3)2). 13C NMR (101 MHz, DMSO‑d 6) δ 166.5, 162.8, 157.9, 156.2, 128.6, 72.2, 57.5, 21.5, 21.5 for (CH3)2, 20.6, 12.3. HRESI-MS m/z calcd for [M−Cl]+ C11H17N2O5: 257.1137, found: 257.1132.
4.1.11. Synthesis of triester macrocycle (13)
To a stirred solution of 12 (0. 6 g, 2.054 mmol, 1 eq) in DMF (80 mL), BOP (2.73 g, 2 eq) and DIPEA (2.5 mL, 7 eq) was added at 0 °C with stirring for 1 h, then the temperature was warmed to rt for 24 h. The reaction mixture was diluted with EtOAc and successively washed with H2O to separate EtOAc layer. The EtOAc layer was washed with 5% HCl, saturated NaHCO3, and brine. The organic phase was dried over MgSO4, and the solvent was removed under reduced pressure to yield 0.29 g (60%) of pure triester macrocycle 13 as a white solid after chromatographed on silica gel using EtOAc/ hexane (2:3) as an eluent, mp 250–252 °C. ν max (KBr disc)/cm−13390 (NH), 2965, 2850, 1754 (C O), 1686 (C O), 1515, 1443, 1262, 1187, 1102, 917. 1H NMR (400 MHz, CDCl3) δ 8.98 (s, 3H, 3 × NH), 5.13 (h, J = 6.3 Hz, 3H, 3 × CH(CH3)2), 2.64 (s, 9H, 3 xoxazole-CH3), 2.00 (s, 9H, 3 × CCH3), 1.24 (dd, J = 6.3, 4.2 Hz, 18H, 3 × CH(CH3)2). 13C NMR (101 MHz, CDCl3) δ 167.0, 159.8, 159.1, 155.2, 128.4, 70.7, 59.1, 22.5, 21.5, 21.5 for (CH3)2, 11.7. HRESI-MS m/z calcd for [M + H]+ C33H43N6O12: 715.2933, found: 715.2930.
4.1.12. Synthesis of triacid platform (14)
A mixture of triester macrocycle 13 (0.2 g, 0.32 mmol) and 5% NaOH (8 mL) in ethanol (20 mL) was stirred at 0 °C for 1 h. The residue after removal of ethanol under reduced pressure was then taken into water, precipitated out at pH = 1 using 5% HCl, extracted twice with EtOAc, dried over MgSO4, and evaporated under vacuo to give 0.18 g (95%) of pure triacid platform 14 as a white solid, mp 268–270 °C. ν max (KBr disc)/cm−1 3378 (br, OH and NH), 3012, 2956, 1749 (C O), 1683 (C O), 1581, 1520, 1441, 1304, 1239, 1188, 1133, 915, 780, 738, 656. 1H NMR (250 MHz, CD3OD): δ 9.06 (br, 3H, 3 × NH), 2.66 (s, 9H, 3 × oxazole-CH3), 2.03 (s, 9H, 3 × CCH3). 13C NMR (62.5 MHz, CD3OD):δ 170.4, 161.9, 160.1, 157.3, 129.6, 60.4, 22.7, 11.7. HRESI-MS m/z calcd for [M + NH4]+ C24H28N7O12: 606.1790, found: 606.1786.
4.1.13. General procedure for synthesis of macrocyclic carboxamides (15a-c)
A mixture of the triacid platform 14 (0. 05 g, 0.085 mmol, 1 eq), BOP (0.14 g, 3.6 eq.), and DIPEA (0.1 mL, 6 eq.) in DCM (10 mL) was stirred for 10 mins at rt before addition of an appropriate amine (3.4 eq) e.g., phenethylamine, 4-(piperidin-1-yl)phenylethylamine, 4-(N,N-dimethylamine)phenylethylamine and the resulting reaction mixture was stirred overnight at rt. After removing of the solvent in vacuo, the residue was extracted with EtOAc, washed with 5% HCl, saturated NaHCO3 solution, brine, dried over MgSO4, and evaporated under reduced pressure to give a crude product which was purified by flash chromatography on silica gel using EtOAc/ hexane (1:2) as an eluent to afford the corresponding carboxamides.
4.1.13.1. Macrocycle carboxamide (15a).
mp 168–170 °C as a white solid. ν max (KBr disc)/cm−1 3340 (NH), 2960, 1683 (C O), 1634 (C O), 1506, 1453, 1187, 841, 749, 701. 1H NMR (400 MHz, DMSO‑d 6) δ 8.74 (s, 3H, 3 xNHC = O), 8.27 (t, J = 5.7 Hz, 3H, 3 × NHCH2), 7.23–7.06 (m, 15H, 3 xAr-H), 3.27 (q, J = 7.2 Hz, 6H, 3 xNHCH2CH2), 2.68 (t, J = 7.1 Hz, 6H, 3 xNHCH2CH2), 2.57 (s, 9H, 3 xoxazole-CH3), 1.95 (s, 9H, 3 × CCH3). 13C NMR (101 MHz, DMSO‑d 6) δ 166.6, 160.4, 159.5, 155.0, 139.8, 129.0, 128.6, 128.3, 126.4, 59.7, 41.7, 35.1, 21.8, 11.9. HRESI-MS m/z calcd for [M + H]+ C48H52N9O9: 898.3883, found: 898.3884.
4.1.13.2. Macrocycle carboxamide (15b)
mp 180–182 °C as a white solid. 1H NMR (400 MHz, DMSO‑d 6) δ11.70 (s, 3H, 3 × NHC = O), 8.84 (t, J = 5.5 Hz, 3H, 3 × NHCH2), 7.10 (d, J = 8.0 Hz, 6H, Ar-H), 6.84 (d, J = 8.0 Hz, 6H, Ar-H), 3.51 (q, J = 7.2 Hz, 6H, 3 × NHCH2CH2), 3.30 (s, 9H, 3 × oxazole-CH3), 3.05 (t, J = 5.2 Hz, 12H, piperidin-H), 2.75 (t, J = 7.2 Hz, 6H, 3 × NHCH2CH2), 1.97 (s, 9H, 3 × CCH3) 1.62–1.47 (m, 18H, piperidin-H).13C NMR (101 MHz, DMSO‑d 6) δ 167.5, 161.4, 159.0, 154.8, 148.2, 129.5, 129.2, 127.9, 111.9, 55.8, 40.1, 36.5, 25.6, 25.4, 24.2, 11.5. HRESI-MS m/z calcd for [M + H]+ C63H79N12O9: 1147.6087, found: 1147.6089.
4.1.13.3. Macrocycle carboxamide (15c)
mp 178–180 °C as a white solid. 1H NMR (400 MHz, DMSO‑d 6) δ 8.76 (s, 3H, 3 × NHC = O), 8.22 (t, J = 5.8 Hz, 3H, 3 × NHCH2), 6.91 (d, J = 8.5 Hz, 6H, Ar-H), 6.56 (d, J = 8.5 Hz, 6H, Ar-H), 3.20 (q, J = 6.6 Hz, 6H, 3 × NHCH2CH2), 2.78 (s, 18H, 3 × N(CH3)2), 2.66 – 2.50 (m, 15H, 3 × NHCH2CH2, 3 × oxazole-CH3), 1.94 (s, 9H, 3 × CCH3). 13C NMR (101 MHz, DMSO‑d 6) δ 166.5, 160.4, 159.6, 155.0, 149.3, 129.5, 129.4, 128.3, 113.0, 59.7, 42.1, 40.8, 34.1, 21.8, 11.8. HRESI-MS m/z calcd for [M + H]+C54H67N12O9: 1027.5148, found: 1027.5143.
4.2. Biology
In vitro bioassay of cytotoxicity [33], antiviral activity [34], [35], [36], [37], mode of action(s) [36], and inhibitory bioassay against SARS-CoV-2 Mpro [40] were performed according to previously reported procedures. See Appendix A
4.3. Molecular docking
4.3.1. Preparation of receptor molecular coordinate files
The crystallography coordinates of the complexes were downloaded as pdb files. The files were loaded in AutoDockTools, the ligand(s) and water molecules were removed, hydrogens and charges were added, and the structures were saved as pdbqt files.
4.3.2. Preparation of the ligands’ molecular coordinate files
The ligands were drawn in Discovery Studio Visualizer, energy minimized by molecular mechanics (MMFF force field) and saved as mol2 files. These ligand files were loaded in AutoDockTools, hydrogen atoms and charges were added, all the relevant bonds were made rotatable, and the files were saved as pdbqt files.
4.3.3. Docking
A Grid Box was manually defined in AutoDockTools to perform docking in the enzyme active site. A config.txt files was prepared indicating the receptor and ligand file names, the Grid Box coordinates and size, the energy range (4 kcal/mol) and exhaustiveness of the docking (set to 8). AutoDock Vina docking was started from the AutoDockTools built-in GUI interface. Each docking was run five times. The docking poses (in pdbqt format) and scores were visualized in AutoDockTools, and were exported for further analyses in Discovery Studio Visualizer.
Funding
This research was funded by the Deanship of Scientific Research at Princess Nourah bint Abdulrahman University through the Fast-track Research Funding Program.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bioorg.2021.105363.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- 1.Tay M.Z., Poh C.M., Rénia L., MacAry P.A., Ng L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020;20:363–374. doi: 10.1038/s41577-020-0311-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Estrada E. Topological analysis of SARS CoV-2 main protease. Chaos. 2020;30 doi: 10.1063/5.0013029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dai W., Zhang B., Jiang X.-M., Su H., Li J., Zhao Y., Xie X., Jin Z., Peng J., Liu F., Li C., Li Y., Bai F., Wang H., Cheng X.i., Cen X., Hu S., Yang X., Wang J., Liu X., Xiao G., Jiang H., Rao Z., Zhang L.-K., Xu Y., Yang H., Liu H. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368(6497):1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.WHO Coronavirus Disease (COVID-19) Dashboard, https://covid19.who.int/ [accessed on2nd Feb. 2021].
- 6.Florindo H.F., Kleiner R., Vaskovich-Koubi D., Acúrcio R.C., Carreira B., Yeini E., Tiram G., Liubomirski Y., Satchi-Fainaro R. Immune-mediated approaches against COVID-19. Nat. Nanotechnol. 2020;15(8):630–645. doi: 10.1038/s41565-020-0732-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amin S.A., Jha T. Fight against novel coronavirus: A perspective of medicinal chemists. Eur. J. Med. Chem. 2020;201:112559. doi: 10.1016/j.ejmech.2020.112559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choudhury C. Fragment tailoring strategy to design novel chemical entities as potential binders of novel corona virus main protease. J. Biomol. Struct. Dyn. 2020;1–14 doi: 10.1080/07391102.2020.1771424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Douangamath A., Fearon D., Gehrtz P., Krojer T., Lukacik P., Owen C.D., Resnick E., Strain-Damerell C., Aimon A., Ábrányi-Balogh P., Brandão-Neto J., Carbery A., Davison G., Dias A., Downes T.D., Dunnett L., Fairhead M., Firth J.D., Jones S.P., Keeley A., Keserü G.M., Klein H.F., Martin M.P., Noble M.E.M., O'Brien P., Powell A., Reddi R.N., Skyner R., Snee M., Waring M.J., Wild C., London N., von Delft F., Walsh M.A. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat. Commun. 2020;11:5047. doi: 10.1038/s41467-020-18709-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rut W., Lv Z., Zmudzinski M., Patchett S., Nayak D., Snipas S.J., El Oualid F., Huang T.T., Bekes M., Drag M., Olsen S.K. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: A framework for anti-COVID-19 drug design. Sci. Adv. 2020;6(42):eabd4596. doi: 10.1126/sciadv.abd4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McClain C.B., Vabret N. SARS-CoV-2: The many pros of targeting PLpro. Signal Transduct. Target. Ther. 2020;5:223. doi: 10.1038/s41392-020-00335-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.G. Derosa, P. Maffioli, A. D'Angelo, F. Di Pierro, A role for quercetin in coronavirus disease 2019 (COVID-19), Phytother. Res. 35 (2020) 1-7. [DOI] [PMC free article] [PubMed]
- 13.Gao Xiaopan, Qin Bo, Chen Pu, Zhu Kaixiang, Hou Pengjiao, Wojdyla Justyna Aleksandra, Wang Meitian, Cui Sheng. Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharm. Sin B. 2021;11(1):237–245. doi: 10.1016/j.apsb.2020.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vuong W., Khan M.B., Fischer C., Arutyunova E., Lamer T., Shields J., Saffran H.A., McKay R.T., van Belkum M.J., Joyce M.A., Young H.S., Tyrrell D.L., Vederas J.C., Lemieux M.J. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2020;11:4282. doi: 10.1038/s41467-020-18096-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu Wei, Xu Miao, Chen Catherine Z., Guo Hui, Shen Min, Hu Xin, Shinn Paul, Klumpp-Thomas Carleen, Michael Samuel G., Zheng Wei. Identification of SARS-CoV-2 3CL protease inhibitors by a quantitative high-throughput screening. ACS Pharmacol. Transl. Sci. 2020;3(5):1008–1016. doi: 10.1021/acsptsci.0c00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loffredo M., Lucero H., Chen D.-Y., O’Connell A., Bergqvist S., Munawar A., Bandara A., De Graef S., Weeks S.D., Douam F., Saeed M., Munawar A.H. The effect of famotidine on SARS-CoV-2 proteases and virus replication. Sci. Rep. 2021;11:5433–5441. doi: 10.1038/s41598-021-84782-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sacco Michael Dominic, Ma Chunlong, Lagarias Panagiotis, Gao Ang, Townsend Julia Alma, Meng Xiangzhi, Dube Peter, Zhang Xiujun, Hu Yanmei, Kitamura Naoya, Hurst Brett, Tarbet Bart, Marty Michael Thomas, Kolocouris Antonios, Xiang Yan, Chen Yu, Wang Jun. Structure, and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against M(pro) and cathepsin L. Sci. Adv. 2020;6(50):eabe0751. doi: 10.1126/sciadv.abe0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang K.S., Ma X.R., Ma Y., Alugubelli Y.R., Scott D.A., Vatansever E.C., Drelich A.K., Sankaran B., Geng Z.Z., Blankenship L.R., Ward H.E., Sheng Y.J., Hsu J.C., Kratch K.C., Zhao B., Hayatshahi H.S., Liu J., Li P., Fierke C.A., Tseng C.-T.K., Xu S., Liu W.R. A Quick Route to Multiple Highly Potent SARS-CoV-2 Main Protease Inhibitors. ChemMedChem. 2020;15:1–8. doi: 10.1002/cmdc.202000924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin Zhenming, Du Xiaoyu, Xu Yechun, Deng Yongqiang, Liu Meiqin, Zhao Yao, Zhang Bing, Li Xiaofeng, Zhang Leike, Peng Chao, Duan Yinkai, Yu Jing, Wang Lin, Yang Kailin, Liu Fengjiang, Jiang Rendi, Yang Xinglou, You Tian, Liu Xiaoce, Yang Xiuna, Bai Fang, Liu Hong, Liu Xiang, Guddat Luke W., Xu Wenqing, Xiao Gengfu, Qin Chengfeng, Shi Zhengli, Jiang Hualiang, Rao Zihe, Yang Haitao. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582(7811):289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 20.Qiao Jingxin, Li Yue-Shan, Zeng Rui, Liu Feng-Liang, Luo Rong-Hua, Huang Chong, Wang Yi-Fei, Zhang Jie, Quan Baoxue, Shen Chenjian, Mao Xin, Liu Xinlei, Sun Weining, Yang Wei, Ni Xincheng, Wang Kai, Xu Ling, Duan Zi-Lei, Zou Qing-Cui, Zhang Hai-Lin, Qu Wang, Long Yang-Hao-Peng, Li Ming-Hua, Yang Rui-Cheng, Liu Xiaolong, You Jing, Zhou Yangli, Yao Rui, Li Wen-Pei, Liu Jing-Ming, Chen Pei, Liu Yang, Lin Gui-Feng, Yang Xin, Zou Jun, Li Linli, Hu Yiguo, Lu Guang-Wen, Li Wei-Min, Wei Yu-Quan, Zheng Yong-Tang, Lei Jian, Yang Shengyong. SARS-CoV-2 M(pro) inhibitors with antiviral activity in a transgenic mouse model. Science. 2021;371(6536):1374–1378. doi: 10.1126/science.abf1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fu L., Ye F., Feng Y., Yu F., Wang Q., Wu Y., Zhao C., Sun H., Huang B., Niu P., Song H., Shi Y., Li X., Tan W., Qi J., Gao G.F. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 2020;11:4417. doi: 10.1038/s41467-020-18233-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abdalla M.A., McGaw L.J. Natural cyclic peptides as an attractive modality for therapeutics: a mini review. Molecules. 2018;23:2080–2099. doi: 10.3390/molecules23082080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takagi Youhei, Matsui Kouhei, Nobori Haruaki, Maeda Haruka, Sato Akihiko, Kurosu Takeshi, Orba Yasuko, Sawa Hirofumi, Hattori Kazunari, Higashino Kenichi, Numata Yoshito, Yoshida Yutaka. Discovery of novel cyclic peptide inhibitors of dengue virus NS2B-NS3 protease with antiviral activity. Bioorg. Med. Chem. Lett. 2017;27(15):3586–3590. doi: 10.1016/j.bmcl.2017.05.027. [DOI] [PubMed] [Google Scholar]
- 24.Ma Xuan, Nong Xu-Hua, Ren Zhe, Wang Jie, Liang Xiao, Wang Lu, Qi Shu-Hua. Antiviral peptides from marine gorgonian-derived fungus Aspergillus sp. SCSIO 41501. Tetrahedron Lett. 2017;58(12):1151–1155. [Google Scholar]
- 25.Horne W.S., Wiethoff C.M., Cui C., Wilcoxen K.M., Amorin M., Ghadiri M.R., Nemerow G.R. Antiviral cyclic D, L-alpha-peptides: targeting a general biochemical pathway in virus infections. Bioorg. Med. Chem. 2005;13:5145–5153. doi: 10.1016/j.bmc.2005.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liang Xiao, Nong Xu-Hua, Huang Zhong-Hui, Qi Shu-Hua. Antifungal and antiviral cyclic peptides from the deep-sea-derived fungus Simplicillium obclavatum EIODSF 020. J. Agric. Food Chem. 2017;65(25):5114–5121. doi: 10.1021/acs.jafc.7b01238. [DOI] [PubMed] [Google Scholar]
- 27.S. Zhang, M. Krumberger, M.A. Morris, C.M.T. Parrocha, J.H. Griffin, A. Kreutzer, J.S. Nowick, Structure-based drug design of an inhibitor of the SARS-CoV-2 (COVID-19) main protease using free software: a tutorial for students and scientists., ChemRxiv Prepr. Serv. Chem. (2020). Zhang S, Krumberger M, Morris MA, et al. Structure-Based Drug Design of an Inhibitor of the SARS-CoV-2 (COVID-19) Main Protease Using Free Software: A Tutorial for Students and Scientists. Preprint. ChemRxiv. 2020;10.26434/chemrxiv.12791954.v1.
- 28.Giordanetto F., Kihlberg J. Macrocyclic drugs and clinical candidates: what can medicinal chemists learn from their properties? J. Med. Chem. 2014;57:278–295. doi: 10.1021/jm400887j. [DOI] [PubMed] [Google Scholar]
- 29.Driggers Edward M., Hale Stephen P., Lee Jinbo, Terrett Nicholas K. The exploration of macrocycles for drug discovery–an underexploited structural class. Nat. Rev. Drug Discov. 2008;7(7):608–624. doi: 10.1038/nrd2590. [DOI] [PubMed] [Google Scholar]
- 30.Haberhauer Gebhard, Somogyi László, Rebek Julius. Synthesis of a second-generation pseudopeptide platform. Tetrahedron Lett. 2000;41(26):5013–5016. [Google Scholar]
- 31.Bartfai T., Lu X., Badie-Mahdavi H., Barr A.M., Mazarati A., Hua X.-Y., Yaksh T., Haberhauer G., Ceide S.C., Trembleau L., Somogyi L., Krock L., Rebek J. Galmic, a nonpeptide galanin receptor agonist, affects behaviors in seizure, pain, and forced-swim tests. PNAS. 2004;101(28):10470–10475. doi: 10.1073/pnas.0403802101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ceide S.C., Trembleau L., Haberhauer G., Somogyi L., Lu X., Bartfai T., Rebek J. Synthesis of galmic: A nonpeptide galanin receptor agonist. PNAS. 2004;101:16727–16732. doi: 10.1073/pnas.0407543101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 34.Mostafa A., Abdelwhab E.M., Mettenleiter T.C., Pleschka S. Zoonotic potential of influenza a viruses: a comprehensive overview. Viruses. 2018;10:497–534. doi: 10.3390/v10090497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alnajjar Radwan, Mostafa Ahmed, Kandeil Ahmed, Al-Karmalawy Ahmed A. Molecular docking, molecular dynamics, and in vitro studies reveal the potential of angiotensin II receptor blockers to inhibit the COVID-19 main protease. Heliyon. 2020;6(12):e05641. doi: 10.1016/j.heliyon.2020.e05641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.A. Mostafa, A. Kandeil, Y. AMME, O. Kutkat, Y. Moatasim, A. A. Rashad, FDA-Approved Drugs with Potent In Vitro Antiviral Activity against Severe Acute Respiratory Syndrome Coronavirus 2, Pharma.13 (2020) 443-466. [DOI] [PMC free article] [PubMed]
- 37.Al-Rabia Mohammed, Alhakamy Nabil, Ahmed Osama, Eljaaly Khalid, Aloafi Ahmed, Mostafa Ahmed, Asfour Hani, Aldarmahi Ahmed, Darwish Khaled, Ibrahim Tarek, Fahmy Usama. Repurposing of sitagliptin- melittin optimized nanoformula against SARS-CoV-2; antiviral screening and molecular docking studies. Pharmaceutics. 2021;13(3):307. doi: 10.3390/pharmaceutics13030307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.L. Dong, S. Hu, J. Gao, discovering drugs to treat coronavirus disease 2019 (COVID-19), Drug Discov. Ther. 14 (2020) 58–60. [DOI] [PubMed]
- 39.Singh A.K., Singh A., Shaikh A., Singh R., Misra A. Chloroquine, and hydroxychloroquine in the treatment of COVID-19 with or without diabetes: A systematic search and a narrative review with a special reference to India and other developing countries. Diabetes Metab. Syndr. 2020;14:241–246. doi: 10.1016/j.dsx.2020.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma Chunlong, Sacco Michael Dominic, Hurst Brett, Townsend Julia Alma, Hu Yanmei, Szeto Tommy, Zhang Xiujun, Tarbet Bart, Marty Michael Thomas, Chen Yu, Wang Jun. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020;30(8):678–692. doi: 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanner M.F. Python: A programming language for software integration and development. J. Mol. Graphics Mod. 1999;17:57–61. [PubMed] [Google Scholar]
- 43.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. Autodock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;16:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.BIOVIA, Dassault Systèmes, Discovery Studio Visualizer v16.1.0.15350, San Diego: Dassault Systèmes, 2015.
- 45.A. Clyde, S. Galanie, D. W. Kneller, H. Ma, Y. Babuji, B. Blaiszik, A. Brace, T. Brettin, K. Chard, R. Chard, L. Coates, I. Foster, D. Hauner, V. Kertesz, N. Kumar, H. Lee, Z. Li, A. Merzky, J. G. Schmidt, L. Tan, M. Titov, A. Trifan, M. Turilli, H. Van Dam, S. C. Chennubhotla, S. Jha, A. Kovalevsky, A. Ramanathan, M. S. Head, R. Stevens; High Throughput Virtual Screening and Validation of a SARS-CoV-2 Main Protease Non-Covalent Inhibitor. bioRxiv 2021.03.27.437323; doi: https://doi.org/10.1101/2021.03.27.437323.
- 46.Mink D., Mecozzi S., Rebek J., Jr. Natural products analogues as scaffolds for supramolecular and combinatorial chemistry. Tetrahedron Lett. 1998;39:5709–5712. [Google Scholar]
- 47.Haberhauer Gebhard, Drosdow Eugen, Oeser Thomas, Rominger Frank. Structural investigation of westiellamide analogues. Tetrahedron. 2008;64(8):1853–1859. [Google Scholar]
- 48.Halgren Thomas A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996;17(5-6):490–519. [Google Scholar]
- 49.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L.W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.