

Abstract
Ferroptosis plays a role in several diseases such as iron overload-induced liver diseases. Manipulation of ferroptosis has been explored as a potential therapeutic strategy to treat related diseases. Numerous antioxidants have been identified to control ferroptosis but the cell-autonomous mechanisms responsible for regulating ferroptosis remain elusive. In the present study, we found that iron overload promoted ferroptosis in hepatocytes by excessively inducing HO-1 expression, which contributed to the progression of liver injury and fibrosis, accompanied by the upregulation of the FGF21 protein level in vitro and in vivo. Interestingly, both recombinant FGF21 and Fgf21 overexpression significantly protected against iron overload-induced hepatocytes mitochondria damage, liver injury and fibrosis by inhibiting ferroptosis. In contrast, the loss of FGF21 aggravated iron overload-induced ferroptosis. Notably, FGF21-induced HO-1 inhibition (via the promotion of HO-1 ubiquitination and degradation) and NRF2 activation provide a mechanistic explanation for this phenomenon. Taken together, we identified FGF21 as a novel ferroptosis suppressor. Thus, FGF21 activation may provide an effective strategy for the potential treatment of iron overload-induced ferroptosis-related diseases, such as hereditary haemochromatosis (HH).
Keywords: Fibroblast growth factor 21 (FGF21), Ferroptosis, Oxidative stress, Lipid peroxidation, Ubiquitination
Highlights
-
•
Iron overload robustly induces hepatic FGF21 expression both in vitro and in vivo.
-
•
FGF21 suppresses iron overload-induced hepatocytes ferroptosis.
-
•
Constitutive HO-1 activation contributes to iron overload-induced ferroptosis in hepatocytes.
-
•
FGF21 protects hepatocytes from iron overload-induced ferroptosis by stimulating HO-1 ubiquitination and degradation.
Abbreviation
- ALT
alanine transaminase
- AST
aspartate transaminase
- FAC
ferric citrate
- FGF21
Fibroblast growth factor 21
- FTH/L
ferritin heavy/light chain
- GPX4
glutathione peroxidase 4
- GSH
glutathione
- HH
hereditary hemochromatosis
- HO-1
heme oxygenase 1
- LIP
labile iron pool
- MDA
malondialdehyde
- Keap1
kelch like ECH associated protein 1
- P62; ROS
reactive oxygen species
- NRF2
nuclear factor erythroid 2-related factor 2
- SLC3A2 (CD98)
solute carrier family 3 member 2
- SLC7A11
Solute carrier family 7 member 11
- TFR1
transferrin receptor 1
- TIBC
total iron binding capacity
- Ub
ubiquitin
- UIBC
unsaturated iron binding capacity
1. Introduction
Iron is an essential mineral element for almost all living cells and organisms. It is involved in numerous cellular processes, including DNA synthesis, mitochondrial respiration and oxygen transport during evolution [1]. In healthy individuals, the iron content in the body is delicately controlled by sophisticated systems that balance its absorption, regulation, utilisation and excretion [2]. Iron deficiency causes anaemia and growth arrest, whereas iron overload is toxic because of its redox reactivity, which promotes oxidative stress [3]. Hereditary haemochromatosis (HH) is the most common genetic iron overload disorder among Caucasians; it occurs in approximately one in 200–250 individuals [4,5]. The pathogenesis of HH involves the redox-active and toxic process of uncontrolled iron absorption and non-transferrin-bound iron (NTBI) accumulation, particularly in the liver [6]. Iron overload induces reactive oxygen species (ROS) through the Fenton reaction and results in oxidative damage and cell death, ultimately causing liver fibrosis, liver cirrhosis and even hepatocellular cancer [7,8]. If left untreated, HH can also result in multi-organ damage and failure [9]. The standard treatment for HH is life-long therapeutic phlebotomy [10]; however, it cannot reverse liver cirrhosis and hepatocellular cancer. Another drawback is that some patients are intolerant to this treatment [9]. Therefore, finding new therapeutic targets involved in iron-related pathological processes is essential for treating HH.
Ferroptosis is a form of regulated cell death marked by the iron-dependent accumulation of lipid peroxidase to lethal levels [11]. Emerging evidence suggests that ferroptosis is closely associated with numerous biological processes, such as the metabolism of iron, amino acids and polyunsaturated fatty acids and the biosynthesis of glutathione (GSH), NADPH and coenzyme Q10 [12]. Three key processes need to occur for iron-dependent death [13]: (1) the accumulation of free iron that causes oxidative stress through Fenton catalysis; (2) the consumption of the antioxidant GSH, leading to oxidative stress and (3) the accumulation of lipid oxidative damage, resulting in cell membrane degeneration. Each process must occur simultaneously in order for iron-dependent death to occur. Experimental evidence indicates that disruption of any of the above mentioned process would prevent ferroptosis. Numerous genes have been found to regulate ferroptosis or serve as markers for ferroptosis [14]. Glutathione peroxidase 4 (Gpx4) [15], ferroptosis suppressor protein 1 (Fsp1) [11,12], solute carrier family 7 member 11 (Slc7a11) [16] and nuclear factor erythroid 2-related factor 2 (Nrf2) [17] function as negative regulators of ferroptosis by limiting ROS production and reducing cellular iron uptake. In contrast, NADPH oxidase, transferrin receptor 1 (Tfr1), prostaglandin-endoperoxide synthase 2 (Ptgs2) and p53 act as positive regulators of ferroptosis by promoting ROS production, stimulating iron uptake and inhibiting Slc7a11 expression [18,19]. To date, ferroptosis has been implicated in multiple pathological cell death process, including tissue injury, cancer cell death and neurodegenerative diseases [14,20]. Therefore, regulation of ferroptosis has become a powerful therapeutic strategy. In addition, the identification of novel ferroptosis regulators is crucial to gain a better understanding of ferroptosis and to develop therapies for ferroptosis-related diseases.
Fibroblast growth factor 21 (FGF21), an endocrine member of the FGF family, plays an important role in glucose and lipid metabolism in order to maintain energy balance [21,22]. FGF21 is abundantly expressed in the liver, which is the source of more than 80% of circulating FGF21 [23]. Recent findings have suggested that intracellular stressors, such as autophagy defects, endoplasmic reticulum stress, calcium imbalance and mitochondrial dysfunction, can induce FGF21 expression [[24], [25], [26]]. Many chronic diseases are associated with increased intracellular oxidative stress, and FGF21 is considered a novel oxidative stress regulator in humans [24,[27], [28], [29]]. Despite the mechanism by which FGF21 responds to oxidative stress is still not well understood, FGF21 is considered to be an important stress response hormone [30]. However, the role of FGF21 in regulating ROS production and ferroptosis remains unclear.
In the present study, we investigated the role of FGF21 in iron overload-induced liver injury and fibrosis and found that iron overload robustly induced ferroptosis and FGF21 production in vitro and in vivo. Interestingly, FGF21 attenuated iron overload-induced ferroptosis by promoting HO-1 ubiquitination and inducing NRF2 expression, which protected the liver from iron overload-induced liver injury and fibrosis. These results indicate that FGF21 is a novel ferroptosis regulator and that the activation of the FGF21 pathway may provide an effective therapeutic strategy for iron overload-induced ferroptosis-related diseases, such as HH.
2. Materials and methods
2.1. Animal model
All experimental protocols were approved by the Institutional Animal Care and Use Committee of the Laboratory Animal Center at Sichuan Agricultural University (SICAU-2015-033). In the present study, 12-week-old C57BL/6J male mice (GemParmatech) were randomly divided into two groups with equal body weight (control and iron dextran injection groups). The mice in the iron dextran injection group were intraperitoneally injected with iron dextran (0.1 mg/g body weight per day) for 2 weeks, while those in the control group were injected with an equal volume of PBS.
In the adenovirus-mediated FGF21 over-expression mouse model, 12-week-old C57BL/6J male mice were divided into four groups: (1) EGFP vector overexpression + PBS injection group (n = 10), (2) EGFP vector overexpression + iron dextran injection group (n = 10), (3) FGF21–EGFP overexpression + PBS injection group (n = 10) and (4) FGF21–EGFP overexpression + iron dextran injection group (n = 10). The mice were administered PBS and iron dextran by intraperitoneal injection for 7 days.
2.2. Isolation and culture of mouse primary hepatocytes
Mouse primary hepatocytes were isolated and cultured as described [31,32]. Briefly, mouse primary hepatocytes were isolated by type Ⅱ collagenase (Gibco™, 17101015) digestion. Live hepatocytes were separated by percoll (Sigma-Aldrich, P1644) centrifugation and then were seed on collagen-coated 6-well or 12-well plates. Primary hepatocytes were cultured in DMEM culture medium for 24 h and washed once with PBS, fresh DMEM cell culture medium was added, and then cells were treated with different regents and harvested at the indicated time points for analysis.
2.3. Plasmid construction and cell expression
Mouse FGF21 cDNA was subcloned by PCR into the BamHI and XhoI restriction sites of the vector pcDNA3.1 (+)-myc-His A, and mouse HO-1 cDNA was subcloned by PCR into the EcoRI and BamHI restriction sites of the vector pLVX-Dsred-Monomer-N1. The plasmids were transfected into primary hepatocytes using Lipofectamine 3000 reagent (Invitrogen) according to the manufacturer's instructions.
2.4. Preparation of recombinant adenovirus
To construct the adenoviral vector for FGF21, the coding sequence of mouse FGF21 was amplified by PCR and cloned into the entry vector with an EGFP sequence at the 3′ end of FGF21. After sequence confirmation, the coding sequence was then recombined into the Gateway-based pAd-CMV DEST™ vector (Invitrogen) according to the manufacturer's instructions. Amplification of recombinant adenovirus was performed using HEK 293A cells according to the manufacturer's instructions (Invitrogen). Adenoviruses were purified using the ViraBind™ Adenovirus Purification Kit (Cell Biolabs) and quantified using the BioRad Protein Assay Kit (BioRad). The titer of the purified virus was examined to calculate concentrations of active viral particles.
2.5. FGF21 and HO-1 knockdown
siRNAs targeting mouse Fgf21 (sense: UGCUGGAGGACGGUUACAAUGTT and antisense: CAUUGUAACCGUCCUCCAGCATT) and Ho-1 (siRNA-1, sense: AAGACGAGAUAGAGCGCAATT and antisense: UUGCGCUCUAUCUCCUCUUTT; siRNA-2, sense: CAACAGUGGCAGU GGGAAUTT and antisense: AUUCCCACUGCCACUGUUGTT) and non-targeting control siRNA were purchased from Sangon Biotech. The siRNAs were transfected into primary hepatocytes using Lipofectamine 3000 reagent according to the manufacturer's instructions.
2.6. Immunoprecipitation (IP)
For IP experiments, HEK293T cells transfected with plasmids expressing Ub-Flag, HO-1–HA, FGF21 or empty vector for 36 h were harvested and lysed in lysis buffer. A total of 200 μg of cell lysate was incubated with 20 μl of protein A magnetic beads (CST, 73778) and 2.5 μg of anti-HA antibody (CST, 3724) overnight at 4 °C. The samples were washed three times with lysis buffer, and bound proteins were eluted by heating at 95 °C for 5 min in 2 × sample loading buffer and subjected to western blot analysis using appropriate antibodies.
2.7. Measurements of serum iron and tissue non-heme iron contents
Measurements of serum iron and tissue non-heme iron contents were performed as described previously [16].
2.8. Liver damage and fibrosis
Serum glutamic oxaloacetic transaminase (also known as aspartate transaminase, AST) and alanine aminotransferase (ALT) levels were measured using an automatic biochemistry analyser. Liver sections were stained with Sirius red and imaged using a polarised light microscope.
2.9. qPCR and western blotting
RNA samples were extracted from tissues or cells using TRIzol reagent (Invitrogen) according to the manufacturer's instructions and normalized to 1 μg/ul. The reaction solution was configured according to the instructions mentioned in the Reverse Transcription Kit (Takara). The relative gene expression was calculated using the ΔΔCT method; the results were normalized to housekeeping gene Hprt.
Western blotting were performed as described previously [33] using primary antibodies against CD98 (CST, 13180), SLC7A11 (CST, 98051), pNRF2 (Abcam, ab76026), NRF2 (Abcam, ab137550), HO-1 (Abcam; ab13248), P62 (Abcam, ab56416), GPX4 (Abcam, ab125066), TFR1 (Abcam, ab84036), Fpn (Abcam, ab78066), FTH/L (Abcam, ab75973) and FGF21 (Abcam, ab17194). Primary antibodies were diluted at 1:1000. HRP conjugated secondary goat anti-rabbit and goat anti-mouse antibodies (Santa Cruz, sc-2030 and sc-2031) were diluted at 1:3000.
2.10. Quantification of ROS and lipid ROS
For detection of ROS, 50 μM H2DCFDA (Sigma-Aldrich, D6883) was incubated with primary hepatocytes for 30 min at 37 °C. Lipid ROS was monitored by assessing the fluorescence levels upon staining with 5 μM CD11-BODIPY (Thermofisher, D3861) for 30 min at 37 °C. Subsequently, the cells were washed three times with PBS and analysed by flow cytometry.
2.11. Measurement of the labile iron pool (LIP)
LIP was measured as described previously. In brief, primary hepatocytes were incubated with 0.05 μM calcein acetoxymethyl ester (Sigma-Aldrich, 17783) for 30 min at 37 °C. The cells were then washed with PBS and incubated with deferiprone (DFO, 100 μM) for 1 h at 37 °C or left untreated. The cells were analysed using a flow cytometer. The difference in the mean cellular fluorescence with and without DFO incubation reflected the level of LIP.
2.12. Measurement of malondialdehyde (MDA) and GSH levels
The cells were treated as indicated in LIP protocol, and cellular MDA and GSH levels were assessed using MDA and GSH assay kits, respectively (Beyotime Biotechnology), according to the manufacturer's instructions.
2.13. Measurement of hydroxyl radical (OH.−) level
The tissue hydroxyl radical (OH.−) level was assessed using OH.− assay kits (Nanjing Jiancheng Bioengineering Institute, A018-1-1), according to the manufacturer's instructions.
2.14. Measurement of FGF21 level in supernatant
A total of 50 μl mouse serum or cell culture supernatant was used to measure the FGF21 level using the ELISA Kit (Biovendor R & D, E20-051) according to the manufacturer's instructions.
2.15. Transmission electron microscopy (TEM)
Transmission electron microscopy was performed using a Tecnai 10 microscope (FEI, Hillsboro, OR).
2.16. Statistical analysis
All data are expressed as the means ± standard errors (S.E.s). Groups were compared using unpaired two-tailed Student's t-test and/or one-way analysis of variance (ANOVA). P ≤ 0.05 was considered statistically significant (*P < 0.05, **P < 0.01 and ***P < 0.001). All results were plotted using GraphPad Prism 7 software.
3. Results
3.1. Iron overload-induced ferroptosis contributes to liver injury and fibrosis
Although iron is an essential micronutrient for almost all organisms, its high redox reactivity may be a source of ROS, making iron potentially cytotoxic. Iron dextran injection caused body weight loss (Fig. 1A and Fig. S1A), and iron accumulation in the liver (Fig. 1B) and spleen (Fig. S1B). Compared with the control group, iron metabolism-related genes and proteins and haematological parameters were remarkably altered in the iron dextran injection group, as evidenced by upregulated expression of Hamp1, Fth, Ftl and Fpn and downregulated expression of Tfr1 (Fig. 1C and Fig. S1C), increased serum iron concentration and total iron binding capacity (TIBC) and decreased unsaturated iron binding capacity (UIBC) (Fig. S1D). Moreover, iron dextran injection resulted in liver injury (Fig. 1D and E) and fibrosis (Fig. 1F and Fig. S1E).
Fig. 1.

Iron overload induces liver damage and fibrosis in mice.
12-week-old C57BL/6J male mice were intraperitoneally injected with Iron dextran (0.1 mg/g body weight per day) or equal volume of PBS for two weeks, serum and liver were collected for analyzing the following indexes. (A) The average body weight change of the control and iron dextran injected mice (n = 8 per group). (B) Liver nonheme iron concentrations. (C) Western blot analysis of iron metabolism relation proteins (TFR1, Fpn and FTH/L) in control and iron dextran injected mice. (D and E) Serum ALT and AST levels in control and iron dextran injected mice (n = 8). (F) The mRNA of hepatic pro-fibrogenic genes (Co11a1, Co13a1, Timp1 and ɑ-SMA) in control and iron-dextran injected mice (n = 8). (G to I) Liver OH.-, MDA and GSH content in control and iron dextran injected mice (n = 8). (J) Relative Ptgs2 (ferroptosis marker gene) mRNA expression in control and iron dextran injected mice liver (n = 8). (K) Expression of ferroptosis related proteins in control and iron dextran injected mice liver (n = 4).
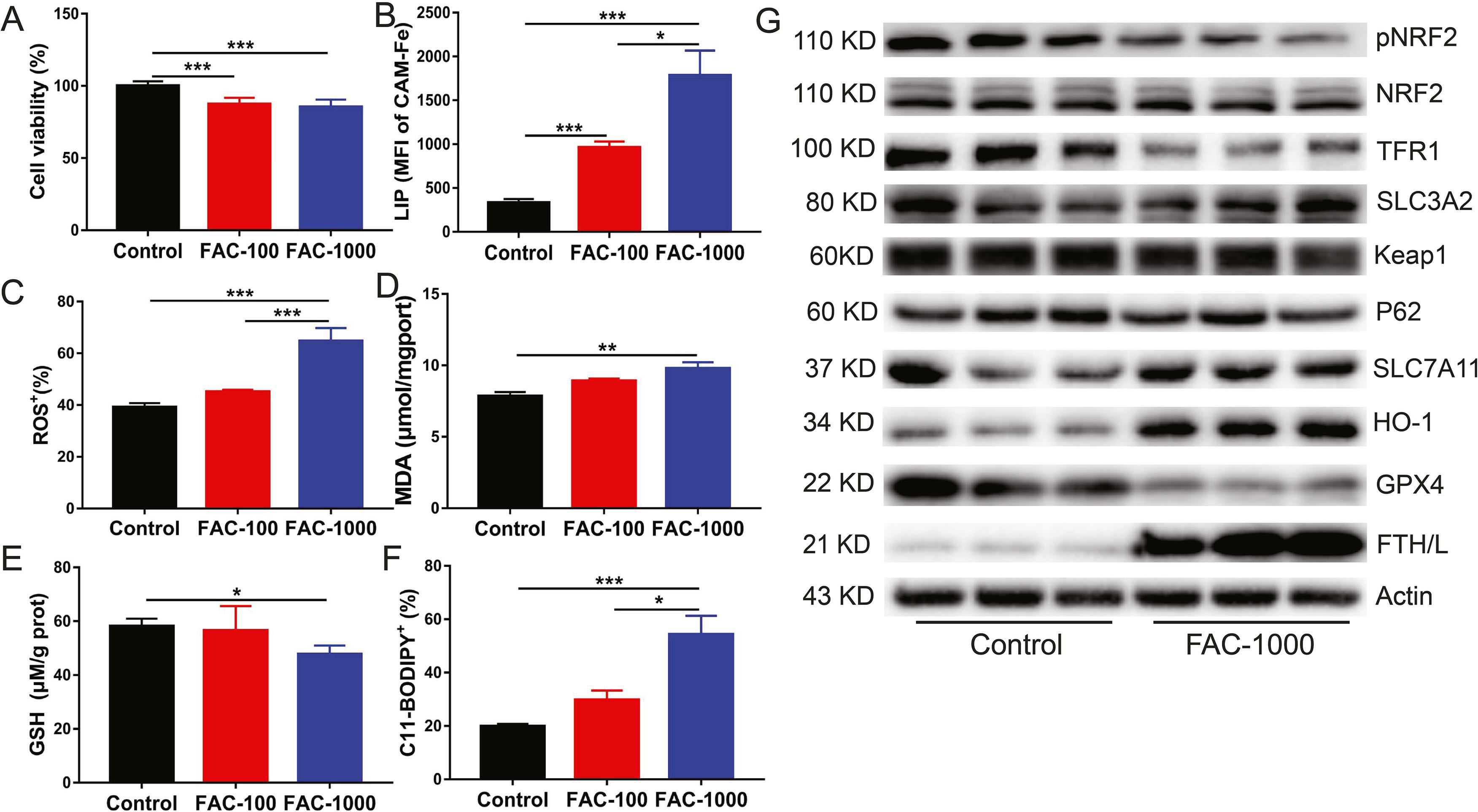
Ferroptosis is a form of regulated cell death that is caused by the iron-dependent peroxidation of lipids and implicated in the organ pathology. In the present study, to gain insight into the potential mechanisms for iron overload-induced liver injury and fibrosis, the key genes and pathways involved in ferroptosis were examined in the livers of mice with iron overload and in primary hepatocytes. The results showed that iron overload induced hydroxyl radical (OH.−), MDA (the product of lipid peroxidation) and GSH accumulation in the liver (Fig. 1G–I). In addition, iron overload significantly increased the mRNA expression of the ferroptosis-related gene Ptgs2 (Fig. 1J). In primary hepatocytes, iron overload resulted in cell death (Fig. S2A). This is potentially attributable to increased LIP levels (Fig. S2B) and subsequent elevated ROS production (Fig. S2C) and lipid peroxidation (Figs. S2D and F), which ultimately causing ferroptosis. Contrary to the in vivo results, iron overload slightly decreased the GSH level in primary hepatocytes treated with high-dose ferric citrate (FAC) (Fig. S2E). Subsequently, we found that iron overload robustly inhibited the phosphorylation of NRF2 (a master antioxidant response regulator that drives resistance to ferroptosis) and decreased the expression level of GPX4 (a ferroptosis repressor that provides an antioxidative defence mechanism to prevent lipid peroxidation) protein, which may lead to impaired ROS clearance ability in primary hepatocytes and in the liver of mice (Fig. 1K and Fig. S2G). In contrast, iron overload remarkably upregulated SLC7A11 and HO-1 protein levels in primary hepatocytes and in the liver of mice (Fig. 1K and Fig. S2G). The changes of other ferroptosis associated proteins (such as SLC3A2, p62 and Keap1) were not obvious (Fig. 1K and Fig. S2G). These results indicate that iron overload induces ferroptosis, which may contribute to subsequent liver injury and fibrosis.
3.2. Iron overload induces FGF21 expression in primary hepatocytes and in the liver of mice
FGF21 is considered to be a factor alleviating oxidative stress. Hence, mRNA and protein levels of FGF21 were examined in primary hepatocytes and in the liver of mice under iron overload conditions. When primary hepatocytes were treated with 100 μM or 1000 μM FAC for 24 h, mRNA and protein expression levels of FGF21 were dramatically increased compared to those in the control group (Fig. 2A–C). Consistent with the results of in vitro experiments, iron overload robustly enhanced FGF21 protein expression in the livers of iron dextran-injected mice (Fig. 2D).
Fig. 2.

Iron overload robustly enhances FGF21 expression in primary hepatocytes and in the liver of mice.
Primary hepatocytes were treated with 100 or 1000 μM FAC for 24 h (A to C) Fgf21 mRNA, supernatant FGF21 concentration and FGF21 protein levels were detected in primary hepatocytes following treatment (n = 4 per group). (D) FGF21 protein levels were detected in the liver of FAC and iron dextran injected mice (n = 8). (E) qRT-PCR analysis of iron metabolism and ferroptosis related gene expression in FAC (1000 μM) and/or FGF21 (200 ng/ml) recombinant protein treated primary hepatocytes (n = 4). (F to J) The LIP, ROS, MDA, GSH and CD11-BODIPY levels were detected in FAC (1000 μM) and/or FGF21 (200 ng/ml) recombinant protein treated primary hepatocytes (n = 4). (K) Transmission electron microscopy of primary hepatocytes treated with FAC (1000 μM) and/or FGF21 (200 ng/ml) recombinant protein for 24 h. Single white arrowheads: shrunken mitochondria.
3.3. FGF21 is a novel ferroptosis regulator and its activation inhibits ferroptosis in primary hepatocytes
To determine the role of FGF21 activation in iron overload-induced ferroptosis, FGF21 recombinant protein was added to primary hepatocytes culture medium supplemented with 1000 μM FAC. FGF21 treatment was found to further upregulate mRNA expression of Fpn (iron exporter), Fth and Ftl (iron storage proteins) compared to FAC treatment alone. Moreover, FGF21 addition rescued the iron overload-induced decrease in Nrf2 and Gpx4 mRNA expression (Fig. 2E). Interestingly, FGF21 downregulated mRNA expression of ferroptosis-related gene Ptgs2, which was induced by iron overload (Fig. 2E). These results indicate that FGF21 may suppress ferroptosis by altering cellular iron metabolism and redox balance. The results of flow cytometry (FACS) confirmed this finding. In FGF21-treated primary hepatocytes, FGF21 reduced cellular levels of LIP, ROS, MDA and lipid peroxidation but increased GSH, which potentially contribute to attenuation of iron overload-induced ferroptosis (Fig. 2F–J). Similar results were found in FGF21-over-expressing primary hepatocytes (Fig. S3). FGF21 over-expression attenuated high-dose FAC-induced cell death (Fig. S3A). In addition, FGF21 over-expression similarly deceased cellular levels of LIP, ROS, MDA and CD11-BODIPY but increased the GSH level in FAC-treated primary hepatocytes when compared to treatment with recombinant FGF21 protein (Figs. S3B–F). Furthermore, FGF21 significantly inhibited iron overload-induced HO-1 protein expression and increased FTH/L protein expression under iron overload conditions. FGF21 over-expression also upregulated NRF2 and SLC7A11 protein levels in primary hepatocytes with or without FAC treatment (Fig. S3G). These results suggest that FGF21 can regulate iron-dependent cell death (ferroptosis). The results of transmission electron microscopy also conformed this conclusion. Compared to control hepatocytes, FAC-treated hepatocytes had smaller, ruptured mitochondria. FAC-induced mitochondria damage and morphological changes can be completely rescued by FGF21 treatment (Fig. 2K).
To further clarify the role of FGF21 in iron overload-induced ferroptosis, FGF21 was knocked down in primary hepatocytes using FGF21 siRNA (Fig. 3A). In contrast to recombinant FGF21 protein treatment or adenovirus-mediated FGF21 over-expression, FGF21 knockdown significantly impaired cell viability (Fig. 3B) and dramatically increased cellular levels of LIP, ROS and C11-BODIPY (Fig. 3C–E). FGF21 knockdown also inhibited NRF2 phosphorylation and protein expression of NRF2 and GPX4. In contrast, FGF21 knockdown robustly increased HO-1 protein expression (Fig. 3F). FGF21-knockdown primary hepatocytes were subsequently treated with 1000 μΜ FAC for 24 h and ferroptosis was found to increase in FGF21 knock down cells compared to control cells (Fig. 3G and H). The HO-1 protein level was further increased by FAC treatment. However, FAC addition decreased FTH/L protein expression in FGF21-knockdown primary hepatocytes (Fig. 3I). Taken together, these results indicate that FGF21 is a novel ferroptosis suppressor.
Fig. 3.

FGF21 knockdown induces oxidative stress and worsen iron overload induced ferroptosis.
Fgf21 was knocked down in primary hepatocytes using Fgf21 siRNA, followed by treatment with or without FAC (1000 μM) for 24 h. (A) qRT-PCR analysis of Fgf21 mRNA expression in primary hepatocytes treated with control siRNA or Fgf21 siRNA. (B to E) Cell viability, LIP, ROS and CD11-BODIPY levels in the cells (n = 4). (F) Iron metabolism and ferroptosis related proteins levels in the cells (n = 3). (G–I) Cell viability, C11-BODIPY levels, and iron metabolism and ferroptosis related proteins expression in above primary hepatocytes treated with vehicle (mock) or 1000uM FAC.
3.4. Recombinant FGF21 protein rescues FGF21 knockdown-induced ferroptosis
Next, exogenous FGF21 treatment was applied to investigate whether it could attenuate FGF21 knockdown-induced ferroptosis. FGF21-knockdown primary hepatocytes were treated with 200 ng/ml FGF21 recombinant protein for 24 h, followed by ferroptosis assessment through measurement of LIP, ROS and lipid peroxidation (C11-BODIPY) levels. As expected, exogenous FGF21 treatment completely rescued FGF21 knockdown-induced ferroptosis and decreased cellular levels of LIP, ROS, lipid peroxidation, and MDA but increased GSH (Fig. 4A–E). Consistent with our previous findings, FGF21 treatment increased NRF2 phosphorylation and the total NRF2 protein level in both normal and FGF21-knockdown primary hepatocytes. Interestingly, exogenous FGF21 treatment could reverse the increase of the HO-1 protein level observed in FGF21-knockdown primary hepatocytes (Fig. 4F). However, the changes of other ferroptosis associated proteins (such as SLC3A2, p62 and Keap1) were not obvious (Fig. 4F).
Fig. 4.

Exogenous FGF21 rescues FGF21 knockdown-mediated ferroptosis.
Fgf21 was knocked down in the primary hepatocytes, followed by mock treatment or treatment with 200 ng/ml recombinant FGF21 for 24 h (A to E) Levels of LIP, ROS, GSH, MDA and C11-BODIPY in the cells (n = 4). (F) Iron metabolism and ferroptosis related proteins expression in the cells (n = 3).
3.5. FGF21 over-expression protects mice from iron overload-induced liver injury and fibrosis by inhibiting ferroptosis
To further clarify the role of FGF21 in iron overload-induced ferroptosis, a mouse model over-expressing FGF21 in the liver was constructed by using a recombinant adenovirus expression system. Compared to the control group, the Fgf21 mRNA level showed an approximately 80-fold increase in the livers of FGF21-overexpressing mice (Fig. 5A). FGF21 over-expression attenuated iron overload-induced body weight loss (Fig. 5B). Unexpectedly, compared to the EGFP control group mice which were administered with PBS, the serum and liver iron contents were decreased in the mice with hepatic FGF21 overexpression (Fig. S4A and Fig. 5C). Assessment of the Hamp1 mRNA level (Fig. S4B) and TFR1 protein level (Fig. 5I) also revealed that FGF21 over-expression improved iron metabolism in WT mice. However, the difference in iron metabolism between WT and FGF21-over-expressing mice disappeared after iron dextran administration (Fig. 5C and Fig. S4). Subsequently, FGF21 over-expression was found to decrease serum ALT and AST levels (Fig. 5D and E) and attenuate liver injury and fibrosis in mice overloaded with iron (Fig. 5F and Fig. S4D). In addition, FGF21 over-expression inhibited the gene expression of Ptsg2 (a marker of ferroptosis) (Fig. S4C). FGF21 over-expression also reduced hepatic OH.− and MDA levels under iron overload conditions (Fig. 5 G and H). Furthermore, FGF21 over-expression in the liver decreased HO-1 expression and increased FTH/L expression in PBS-administered mice (Fig. 5I). These results suggest that FGF21 over-expression can protect mice from developing iron overload-induced liver injury and fibrosis by inhibiting ferroptosis in WT mice.
Fig. 5.

FGF21 overexpression protects against iron overload-induced liver damage and fibrosis in mice.
C57/BL6 male mice were intravenously injected with AdEGFP or AdFGF21-EGFP. Two days later, mice were IP injected with PBS or iron dextran (0.1 mg/g body weight per day) daily for 7 days. Mice were sacrificed on day 7, and serum and liver were harvested. (A) Fgf21 mRNA levels in the liver (n = 10). (B) The body weight graph (n = 10). (C) Hepatic iron concentrations (n = 10). (D and E) Serum ALT and AST levels (n = 10). (F) The mRNA of hepatic pro-fibrogenic genes ɑ-SMA (n = 10). (G and H) Hepatic OH.- and MDA levels. (I) Iron metabolism and ferroptosis related protein expression in the liver.
3.6. FGF21 attenuates ferroptosis by promoting HO-1 ubiquitination and degradation
In general, HO-1 exhibits cytoprotective effects against various stress-related conditions. However, increasing evidence has shown that HO-1 acts as a critical mediator in the induction of ferroptosis and is responsible for the progression of several diseases [34,35]. To elucidate the role of FGF21-mediated HO-1 inhibition in the protection of ferroptosis in hepatocytes, primary hepatocytes were transfected with a plasmid expressing HO-1 (Fig. 6A). HO-1 over-expression was found to increase cell death and cellular levels of LIP, ROS and lipid peroxidation (Fig. 6B–E). Consistent with this observation, HO-1 knockdown decreased cell death (ferroptosis) and cellular levels of LIP, ROS, and lipid peroxidation in primary hepatocytes (Fig. 6F–J) even under iron overload. These results indicate that HO-1 is an activator of ferroptosis in hepatocytes.
Fig. 6.

HO-1 activation contributes to iron overload-induced ferroptosis.
Primary hepatocytes were transfected with HO-1 plasmid or Ho-1 siRNA for 36 h. (A) Western blot analysis of HO-1 expression in HO-1 over-expression primary hepatocytes. (B to E) Cell viability, LIP, ROS, and CD11-BODIPY in HO-1 over expression primary hepatocytes vs control cells (n = 4). (F) Western blot analysis of HO-1 expression in HO-1 knock down primary hepatocytes. (G to J) Cell viability, LIP, ROS, and C11-BODIPY in Ho-1 knock down primary hepatocytes (n = 4).
To reveal the potential mechanism by which FGF21 regulates HO-1 protein levels, Ho-1 mRNA expression levels were analysed in the livers of FGF21-overexpressing mice (Fig. 7A) and FGF21-treated primary hepatocytes (Fig. 7B). FGF21 did not change Ho-1 mRNA expression levels, indicating that FGF21 inhibits HO-1 expression in a transcription-independent manner. Next, cloheximide (CHX, a chemical protein synthesis inhibitor) was used to assess the transcription-independent mechanism underlying FGF21-mediated reduction of HO-1 protein expression. FGF21 was found to affect HO-1 protein stability and FGF21 treatment shortened the half-life of HO-1, thereby accelerating its degradation (Fig. 7C). Subsequently, FGF21 was also found to promote HO-1 ubiquitination (a post-translational modification, regulates a diverse range of proteins degradation). Compared with the vector control, FGF21 over-expression increased the ubiquitination of HA-tagged HO-1 protein in HEK293T cells (Fig. 7D). This provides a good explanation for FGF2-mediated HO-1 degradation. In addition, we used MG132 (a proteasome inhibitors) to treat hepatocytes and found that lower ubiquitination was responsible for HO-1 accumulation under iron overload (Fig. S5). Collectively, these findings indicate that FGF21 attenuates iron overload-induced ferroptosis by promoting HO-1 ubiquitination and degradation.
Fig. 7.

FGF21 attenuates iron overload-induced ferroptosis through promoting HO-1 ubiquitination.
(A) Ho-1 mRNA expression levels in the liver of FGF21 over-expression mice (n = 10). (B) Ho-1 mRNA expression levels in primary hepatocytes treated with recombinant FGF21 protein (n = 4). (C) HO-1 protein expression in primary hepatocytes treated with PBS (left) or 200 ng/ml recombinant FGF21 protein (right) for 4 h, followed by 20 μg/ml CHX for 3 h. (D) HEK293T cells were transfected with Flag-tagged ubiquitin cDNA (Ub-Flag) alone or together with HA-tagged Ho-1 cDNA (HO-1-HA) alone or together with FGF21 cDNA or vector plasmid for 36 h. Cell lysates were subjected to immunoprecipitation with anti-HA antibody, and the tag of flag was detected by western blot. (E) Model of proposed mechanism of FGF21 attenuated liver injury and fibrosis induced by iron overload. Iron overload induces hepatocyte ferroptosis, which contributes to liver injury and fibrosis in WT mice (Left panel). Mechanistically, iron overload robustly increases HO-1 expression, which catalyzes the conversion of heme into Fe2+, carbon monoxide and biliverdin. Notably, excess Fe2+ accumulating in the cells induces ROS generation which could be the direct reason for ferroptosis. In addition, iron overload inhibits the expression of NRF2, which is a master regulator of the antioxidant response. Of particular interest, FGF21 over expression (OE) attenuates liver injury and fibrosis induced by iron overload via suppressing hepatocytes ferroptosis (right panel). FGF21 not only enhances HO-1 ubiquitination and degradation to reduce Fe2+ generation in hepatocytes but also activates NRF2 expression to suppress iron overload-induced oxidative damage. Therefore, FGF21 is a novel regulator of ferroptosis and activation of FGF21 may provide an effective strategy for potential treatment in iron-mediated ferroptosis-related disease, such as HH.
4. Discussion
Diseases associated with iron deficiency or iron overload affect a large fraction of the world's population [36]. HH is a typical iron overload disease that involves uncontrolled iron absorption and iron overload in tissues, particularly in the liver [37]. As iron homeostasis needs to be strictly regulated in order to maintain health, it is not surprising that the disruption of iron balance results in various diseases, including HH [38]. We constructed an HH iron overload model by injecting iron dextran into WT mice and found that redox imbalance and lipid peroxidation induced by iron overload contribute to liver damage and fibrosis [16]. Increased ROS production due to iron overload causes apoptosis and autophagy; this has been suggested to be a major pathogenic mechanism underlying HH-associated complications, including liver fibrosis and cirrhosis [16,39]. However, this mechanism only partly explains the clinical observation in individuals with more serious complications.
Recently, ferroptosis has been implicated in multiple pathological cell death processes, including tissue injury, cancer cell death and neurodegenerative diseases [20,40]. Iron overload induced ferroptosis is associated with type 2 HH. Notably, high levels of iron overload serve as a driving factor in the induction of ferroptosis, as Hfe−/− mice with moderate iron overload have not been found to develop hepatic ferroptosis. However, serious hepatic ferroptosis has been observed in Hfe−/− mice fed on a high-iron diet [16]. In our HH iron overload mouse model, continuous iron dextran injection induced a 10-fold iron overload in the liver, which triggered severe hepatic ferroptosis (the level of the ferroptosis-related gene Ptsg2 was significantly up-regulated in the liver of mice with iron overload). In addition, the protein level of GPX4, a ferroptosis regulator that uses GSH to eliminate membrane phospholipid hydroperoxides in order to suppress ferroptosis [15], was dramatically decreased in the livers of mice with iron overload and in primary hepatocytes treated with FAC. Although GSH level was elevated in the livers of mice with iron overload, the loss of GPX4 could not effectively attenuate ROS-induced lipid peroxidation in these mice. The increase of the GSH level may be the result of the compensatory up-regulation of the expression of Slc7a11. Slc7a11 is a major component of the glutamate/cystine antiporter system xc−, which regulates the downstream synthesis of GSH, a key antioxidant that scavenges lipid peroxides and suppresses ferroptosis [19]. In order to eliminate the detrimental effects of iron overload-induced ROS, more cysteine is transported into cells by Slc7a11 to synthesize GSH. Consistent with previous findings, we found that iron overload could continuously increase the expression of Slc7a11 through the ROS–NRF2–ARE axis. Moreover, iron overload significantly inhibited the phosphorylation of NRF2, which is a master antioxidant response regulator, as many of its downstream target genes are involved in preventing or correcting redox imbalances in cells [41]. This could be another reason for iron overload-induced ferroptosis. As GPX4 is an established NRF2 transcriptional target [17], the NRF2–GPX4 pathway could provide potential therapeutic targets for iron overload-induced ferroptosis-related diseases, such as HH.
Notably, iron overload increases the expression of another NRF2 downstream target gene, Ho-1, in primary hepatocytes and in the liver of mice. Under normal conditions, high HO-1 expression helps to attenuate redox stress [35,42]; however, continuous HO-1 up-regulation can also trigger ferroptosis [34,43]. HO-1 catalyzes the conversion of heme into Fe2+, carbon monoxide and biliverdin. Excessive Fe2+ accumulation in cells is the direct cause for ferroptosis [44]. These results indicate that the dual role of HO-1 in ferroptosis regulation may depend on different pathological conditions. In the present study, we found that iron overload continued to increase HO-1 expression (approximately 500-fold up-regulation was noted in the livers of mice with iron overload), promoting ferroptosis in hepatocytes. Consistently, the inhibition of HO-1 expression decreased ferroptosis in the liver and mitigated liver injury and fibrosis. Constitutive HO-1 activation could further aggravate iron overload-induced ferroptosis in hepatocytes, particularly when NRF2 and GPX4 are inactive. Therefore, constant HO-1 elevation could be an explanation for iron overload-induced ferroptosis.
FGF21 is considered as a novel oxidative stress regulator in humans and rodents [29]. It can attenuate pulmonary and cardiac fibrogenesis by ameliorating oxidative stress in vivo and in vitro [28,45]. In our HH iron overload mouse model, we found that iron treatment robustly increases mRNA and protein expression of FGF21 in primary hepatocytes and in the liver of mice. Iron overload-induced ROS may account for the increase of FGF21 in the livers of mice and in primary hepatocytes, as both pathological stress and intracellular disturbances are associated with increased FGF21 expression [46,47]. In fact, FGF21 has been considered as an early predictor of numerous liver diseases, such as acute-on-chronic liver failure [48]. In general, serum FGF21 levels are up-regulated in these patients, probably to attenuate the inflicted damage [49,50]. Thus, FGF21 can be considered as a biomarker of iron overload-induced liver diseases.
We found that increased FGF21 levels can attenuate iron overload-induced liver injury and fibrosis. Iron overload induces FGF21 expression, which in turn ameliorates iron overload-induced oxidative damage. This finding supports the concept that FGF21 is an oxidative stress regulator [29]. Interestingly, ferroptosis was suppressed in hepatocytes and the liver of mice following FGF21 treatment or over-expression. This suggests that FGF21 is a novel ferroptosis suppressor. Considering our previous finding that ferroptosis contributes to iron overload-induced liver injury and fibrosis, it is reasonable to conclude that FGF21 can attenuate iron overload-induced liver injury and fibrosis. FGF21 is an endocrine hormone that plays an important role in glucose and lipid metabolism and in energy balance [51]. Although FGF21 is known to regulate cell development, proliferation, pyroptosis and autophagy [[52], [53], [54], [55], [56]], it has rarely been reported to regulate ferroptosis. Thus, the present finding further confirms that FGF21 is a cell death regulator.
Next, we found that FGF21 treatment up-regulates total and phosphorylated levels of NRF2 and the expression of its downstream target genes, Gpx4, Slac7a11, Ftl and Fth. FGF21 improves the body's antioxidant capacity and GSH synthesis ability by increasing GPX4 and Slc7a11 expression levels, which reduce lipid peroxidation. Similar results have been reported in atherosclerotic rats. FGF21 attenuates inflammation and oxidative stress in atherosclerotic rats by enhancing the NRF2–ARE signalling pathway [57]. In addition, FGF21 treatment promotes the expression of Ftl, Fth and Fpn genes, which can significantly reduce the intracellular content of free iron. It is very important for decreasing ROS production. These results are in agreement with previous findings that FGF21 can attenuate inflammation and oxidative stress. Thus, reducing either the free iron content or lipid peroxidation can help to suppress ferroptosis. However, the molecular mechanism by which FGF21 induces NRF2 and FH/L expression requires further investigation.
Finally, FGF21 treatment or over-expression inhibits HO-1 expression, which increases ferroptosis in primary hepatocytes and in the liver of mice. The molecular mechanism underlying FGF21- repressed HO-1 expression is through promoting HO-1 ubiquitination, which accelerates HO-1 degradation and subsequently decreases the conversion of heme to Fe2+, and ultimately suppresses ferroptosis. Therefore, potential therapeutic targets could arise from the FGF21–HO-1 pathway for treating iron overload-induced ferroptosis-related diseases, such as HH.
5. Conclusion
Collectively, the present study suggests that constitutive HO-1 activation is a novel mediator for iron overload-induced ferroptosis in hepatocytes, and FGF21 could protect hepatocytes from developing iron overload-induced ferroptosis by stimulating HO-1 ubiquitination and subsequent degradation (Fig. 7E). These findings indicate that the FGF21–HO-1 pathway could be targeted for treating iron overload-induced ferroptosis-related diseases, particularly HH.
Declaration of competing interest
The authors declare that they have no conflict of interest.
Acknowledgements
We thank Dr. Haiyan Xu at Merck & Co. Inc. for editing the manuscript. This work was supported by China National Key Research Project of 13th Five Year Plan (2016YFD0501204), China Agricultural Research System (CARS-35), Project funded by China Postdoctoral Science Foundation (2020T130455 and 2019M653470) and the 111 project (D17015).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2021.102131.
Contributor Information
Bing Yu, Email: ybingtian@163.com.
De Wu, Email: wude@sicau.edu.cn.
Daiwen Chen, Email: dwchen@sicau.edu.cn.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- 1.Ganz T. Systemic iron homeostasis. Physiol. Rev. 2013;93:1721–1741. doi: 10.1152/physrev.00008.2013. [DOI] [PubMed] [Google Scholar]
- 2.Finberg K.E. Regulation of systemic iron homeostasis. Curr. Opin. Hematol. 2013;20:208–214. doi: 10.1097/MOH.0b013e32835f5a47. [DOI] [PubMed] [Google Scholar]
- 3.Sonnweber T., Pizzini A., Tancevski I., Loffler-Ragg J., Weiss G. Anaemia, iron homeostasis and pulmonary hypertension: a review. Intern. Emerg. Med. 2020;15(4):573–585. doi: 10.1007/s11739-020-02288-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kawabata H. The mechanisms of systemic iron homeostasis and etiology, diagnosis, and treatment of hereditary hemochromatosis. Int. J. Hematol. 2018;107:31–43. doi: 10.1007/s12185-017-2365-3. [DOI] [PubMed] [Google Scholar]
- 5.Golfeyz S., Lewis S., Weisberg I.S. Hemochromatosis: pathophysiology, evaluation, and management of hepatic iron overload with a focus on MRI. Expet Rev. Gastroenterol. Hepatol. 2018;12:767–778. doi: 10.1080/17474124.2018.1496016. [DOI] [PubMed] [Google Scholar]
- 6.Brissot P., Ropert M., Le Lan C., Loreal O. Non-transferrin bound iron: a key role in iron overload and iron toxicity. Biochim. Biophys. Acta. 2012;1820:403–410. doi: 10.1016/j.bbagen.2011.07.014. [DOI] [PubMed] [Google Scholar]
- 7.Deugnier Y., Turlin B. Pathology of hepatic iron overload. Semin. Liver Dis. 2011;31:260–271. doi: 10.1055/s-0031-1286057. [DOI] [PubMed] [Google Scholar]
- 8.Kowdley K.V. Iron, hemochromatosis, and hepatocellular carcinoma. Gastroenterology. 2004;127:S79–S86. doi: 10.1016/j.gastro.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 9.Pantopoulos K. Inherited disorders of iron overload. Front. Nutr. 2018;5:103. doi: 10.3389/fnut.2018.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mariani R., Pelucchi S., Perseghin P., Corengia C., Piperno A. Erythrocytapheresis plus erythropoietin: an alternative therapy for selected patients with hemochromatosis and severe organ damage. Haematologica. 2005;90:717–718. [PubMed] [Google Scholar]
- 11.Bersuker K., Hendricks J.M., Li Z., Magtanong L., Ford B., Tang P.H. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688–692. doi: 10.1038/s41586-019-1705-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doll S., Freitas F.P., Shah R., Aldrovandi M., da Silva M.C., Ingold I. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693–698. doi: 10.1038/s41586-019-1707-0. [DOI] [PubMed] [Google Scholar]
- 13.Dixon S.J., Lemberg K.M., Lamprecht M.R., Skouta R., Zaitsev E.M., Gleason C.E. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li J., Cao F., Yin H.L., Huang Z.J., Lin Z.T., Mao N. Ferroptosis: past, present and future. Cell Death Dis. 2020;11:88. doi: 10.1038/s41419-020-2298-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang W.S., SriRamaratnam R., Welsch M.E., Shimada K., Skouta R., Viswanathan V.S. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang H., An P., Xie E., Wu Q., Fang X., Gao H. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology. 2017;66:449–465. doi: 10.1002/hep.29117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dodson M., Castro-Portuguez R., Zhang D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019;23:101107. doi: 10.1016/j.redox.2019.101107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stockwell B.R., Friedmann Angeli J.P., Bayir H., Bush A.I., Conrad M., Dixon S.J. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–285. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang L., Kon N., Li T., Wang S.J., Su T., Hibshoosh H. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62. doi: 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsurusaki S., Tsuchiya Y., Koumura T., Nakasone M., Sakamoto T., Matsuoka M. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 2019;10:449. doi: 10.1038/s41419-019-1678-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kharitonenkov A., Shiyanova T.L., Koester A., Ford A.M., Micanovic R., Galbreath E.J. FGF-21 as a novel metabolic regulator. J. Clin. Invest. 2005;115:1627–1635. doi: 10.1172/JCI23606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leng Y., Wang Z., Tsai L.K., Leeds P., Fessler E.B., Wang J. FGF-21, a novel metabolic regulator, has a robust neuroprotective role and is markedly elevated in neurons by mood stabilizers. Mol. Psychiatr. 2015;20:215–223. doi: 10.1038/mp.2013.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hondares E., Iglesias R., Giralt A., Gonzalez F.J., Giralt M., Mampel T. Thermogenic activation induces FGF21 expression and release in brown adipose tissue. J. Biol. Chem. 2011;286:12983–12990. doi: 10.1074/jbc.M110.215889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Henkel J., Buchheim-Dieckow K., Castro J.P., Laeger T., Wardelmann K., Kleinridders A. Reduced oxidative stress and enhanced FGF21 formation in livers of endurance-exercised rats with diet-induced NASH. Nutrients. 2019;11 doi: 10.3390/nu11112709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maruyama R., Shimizu M., Hashidume T., Inoue J., Itoh N., Sato R. FGF21 alleviates hepatic endoplasmic reticulum stress under physiological conditions. J. Nutr. Sci. Vitaminol. 2018;64:200–208. doi: 10.3177/jnsv.64.200. [DOI] [PubMed] [Google Scholar]
- 26.Manoli I., Sysol J.R., Epping M.W., Li L., Wang C., Sloan J.L. FGF21 underlies a hormetic response to metabolic stress in methylmalonic acidemia. JCI Insight. 2018;3 doi: 10.1172/jci.insight.124351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu Y., Bai F., Liu Y., Yang Y., Yuan Q., Zou D. Fibroblast growth factor (FGF21) protects mouse liver against D-galactose-induced oxidative stress and apoptosis via activating Nrf2 and PI3K/Akt pathways. Mol. Cell. Biochem. 2015;403:287–299. doi: 10.1007/s11010-015-2358-6. [DOI] [PubMed] [Google Scholar]
- 28.Zhang S., Yu D., Wang M., Huang T., Wu H., Zhang Y. FGF21 attenuates pulmonary fibrogenesis through ameliorating oxidative stress in vivo and in vitro. Biomed. Pharmacother. = Biomedecine & pharmacotherapie. 2018;103:1516–1525. doi: 10.1016/j.biopha.2018.03.100. [DOI] [PubMed] [Google Scholar]
- 29.Gomez-Samano M.A., Grajales-Gomez M., Zuarth-Vazquez J.M., Navarro-Flores M.F., Martinez-Saavedra M., Juarez-Leon O.A. Fibroblast growth factor 21 and its novel association with oxidative stress. Redox Biol. 2017;11:335–341. doi: 10.1016/j.redox.2016.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim K.H., Lee M.S. FGF21 as a stress hormone: the roles of FGF21 in stress adaptation and the treatment of metabolic diseases. Diabetes Metab. J. 2014;38:245–251. doi: 10.4093/dmj.2014.38.4.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Z., Zhang F., Guo X., An P., Tao Y., Wang F. Ferroportin1 in hepatocytes and macrophages is required for the efficient mobilization of body iron stores in mice. Hepatology. 2012;56:961–971. doi: 10.1002/hep.25746. [DOI] [PubMed] [Google Scholar]
- 32.Feng B., Jiao P., Helou Y., Li Y., He Q., Walters M.S. Mitogen-activated protein kinase phosphatase 3 (MKP-3)-deficient mice are resistant to diet-induced obesity. Diabetes. 2014;63:2924–2934. doi: 10.2337/db14-0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu A., Yu B., Zhang K., Xu Z., Wu, He J. Transmissible gastroenteritis virus targets Paneth cells to inhibit the self-renewal and differentiation of Lgr5 intestinal stem cells via Notch signaling. Cell Death Dis. 2020;11:40. doi: 10.1038/s41419-020-2233-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang L.C., Chiang S.K., Chen S.E., Yu Y.L., Chou R.H., Chang W.C. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Canc. Lett. 2018;416:124–137. doi: 10.1016/j.canlet.2017.12.025. [DOI] [PubMed] [Google Scholar]
- 35.Adedoyin O., Boddu R., Traylor A., Lever J.M., Bolisetty S., George J.F. Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am. J. Physiol. 2018:F702–F714. doi: 10.1152/ajprenal.00044.2017. Renal physiology 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gordan R., Wongjaikam S., Gwathmey J.K., Chattipakorn N., Chattipakorn S.C., Xie L.H. Involvement of cytosolic and mitochondrial iron in iron overload cardiomyopathy: an update. Heart Fail. Rev. 2018;23:801–816. doi: 10.1007/s10741-018-9700-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pietrangelo A. Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology. 2010;139:393–408. doi: 10.1053/j.gastro.2010.06.013. 408 e391-392. [DOI] [PubMed] [Google Scholar]
- 38.Powell L.W., Seckington R.C., Deugnier Y. Haemochromatosis. Lancet. 2016;388:706–716. doi: 10.1016/S0140-6736(15)01315-X. [DOI] [PubMed] [Google Scholar]
- 39.Prabhu A., Cargill T., Roberts N., Ryan J.D. Systematic review of the clinical outcomes of iron reduction in hereditary hemochromatosis. Hepatology. 2020;72:1469–1482. doi: 10.1002/hep.31405. [DOI] [PubMed] [Google Scholar]
- 40.Chen X., Li J., Kang R., Klionsky D.J., Tang D. Ferroptosis: machinery and regulation. Autophagy. 2020:1–28. doi: 10.1080/15548627.2020.1810918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kerins M.J., Ooi A. The roles of NRF2 in modulating cellular iron homeostasis. Antioxidants Redox Signal. 2018;29:1756–1773. doi: 10.1089/ars.2017.7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park S.Y., Park D.J., Kim Y.H., Kim Y., Kim S.G., Shon K.J. Upregulation of heme oxygenase-1 via PI3K/Akt and Nrf-2 signaling pathways mediates the anti-inflammatory activity of Schisandrin in Porphyromonas gingivalis LPS-stimulated macrophages. Immunol. Lett. 2011;139:93–101. doi: 10.1016/j.imlet.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 43.Kwon M.Y., Park E., Lee S.J., Chung S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget. 2015;6:24393–24403. doi: 10.18632/oncotarget.5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Valko M., Jomova K., Rhodes C.J., Kuca K., Musilek K. Redox- and non-redox-metal-induced formation of free radicals and their role in human disease. Arch. Toxicol. 2016;90:1–37. doi: 10.1007/s00204-015-1579-5. [DOI] [PubMed] [Google Scholar]
- 45.Ferrer-Curriu G., Redondo-Angulo I., Guitart-Mampel M., Ruperez C., Mas-Stachurska A., Sitges M. Fibroblast growth factor-21 protects against fibrosis in hypertensive heart disease. J. Pathol. 2019;248:30–40. doi: 10.1002/path.5226. [DOI] [PubMed] [Google Scholar]
- 46.Suomalainen A. Fibroblast growth factor 21: a novel biomarker for human muscle-manifesting mitochondrial disorders. Expert Opin. Med. Diagn. 2013;7:313–317. doi: 10.1517/17530059.2013.812070. [DOI] [PubMed] [Google Scholar]
- 47.Joe Y., Kim S., Kim H.J., Park J., Chen Y., Park H.J. FGF21 induced by carbon monoxide mediates metabolic homeostasis via the PERK/ATF4 pathway. Faseb. J. : Off. Publ. Feder. Am. Soc. Exper. Biol. 2018;32:2630–2643. doi: 10.1096/fj.201700709RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ruiz-Margain A., Pohlmann A., Ryan P., Schierwagen R., Chi-Cervera L.A., Jansen C. Fibroblast growth factor 21 is an early predictor of acute-on-chronic liver failure in critically ill patients with cirrhosis. Liver Transplant. 2018;24:595–605. doi: 10.1002/lt.25041. [DOI] [PubMed] [Google Scholar]
- 49.Zhang J., Li J., Ma J., Wang H., Yi Y. Human fibroblast growth factor-21 serves as a predictor and prognostic factor in patients with hepatitis B cirrhosis combined with adrenal insufficiency. Exper. Therap. Med. 2018;15:3189–3196. doi: 10.3892/etm.2018.5840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee K.J., Jang Y.O., Cha S.K., Kim M.Y., Park K.S., Eom Y.W. Expression of fibroblast growth factor 21 and beta-klotho regulates hepatic fibrosis through the nuclear factor-kappaB and c-jun N-terminal kinase pathways. Gut Liver. 2018;12:449–456. doi: 10.5009/gnl17443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lakhani I., Gong M., Wong W.T., Bazoukis G., Lampropoulos K., Wong S.H. Fibroblast growth factor 21 in cardio-metabolic disorders: a systematic review and meta-analysis. Metabolism. 2018;83:11–17. doi: 10.1016/j.metabol.2018.01.017. [DOI] [PubMed] [Google Scholar]
- 52.Wu S., Levenson A., Kharitonenkov A., De Luca F. Fibroblast growth factor 21 (FGF21) inhibits chondrocyte function and growth hormone action directly at the growth plate. J. Biol. Chem. 2012;287:26060–26067. doi: 10.1074/jbc.M112.343707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Salminen A., Kauppinen A., Kaarniranta K. FGF21 activates AMPK signaling: impact on metabolic regulation and the aging process. J. Mol. Med. 2017;95:123–131. doi: 10.1007/s00109-016-1477-1. [DOI] [PubMed] [Google Scholar]
- 54.Chen J.J., Tao J., Zhang X.L., Xia L.Z., Zeng J.F., Zhang H. Inhibition of the ox-LDL-induced pyroptosis by FGF21 of human umbilical vein endothelial cells through the TET2-UQCRC1-ROS pathway. DNA Cell Biol. 2020;39:661–670. doi: 10.1089/dna.2019.5151. [DOI] [PubMed] [Google Scholar]
- 55.Zeng Z., Zheng Q., Chen J., Tan X., Li Q., Ding L. FGF21 mitigates atherosclerosis via inhibition of NLRP3 inflammasome-mediated vascular endothelial cells pyroptosis. Exp. Cell Res. 2020;393:112108. doi: 10.1016/j.yexcr.2020.112108. [DOI] [PubMed] [Google Scholar]
- 56.Dai H., Hu W., Zhang L., Jiang F., Mao X., Yang G. FGF21 facilitates autophagy in prostate cancer cells by inhibiting the PI3K-Akt-mTOR signaling pathway. Cell Death Dis. 2021;12:303. doi: 10.1038/s41419-021-03588-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jia H., Cheng J., Zhou Q., Peng J., Pan Y., Han H. Fibroblast growth factor 21 attenuates inflammation and oxidative stress in atherosclerotic rat via enhancing the Nrf1-ARE signaling pathway. Int. J. Clin. Exp. Pathol. 2018;11:1308–1317. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.