

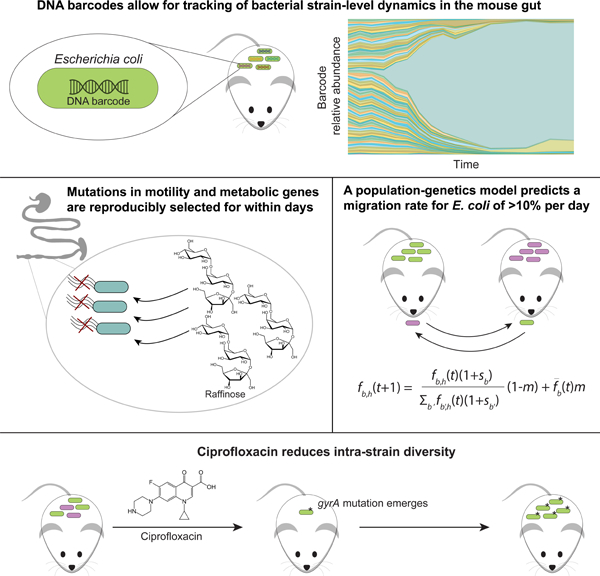
Summary
Due to limitations on high-resolution strain tracking, selection dynamics during gut-microbiota colonization and transmission between hosts remain mostly mysterious. Here, we introduced hundreds of barcoded Escherichia coli strains into germ-free mice and quantified strain-level dynamics and metagenomic changes. Mutations in genes involved in motility and metabolite utilization are reproducibly selected within days. Even with rapid selection, coprophagy enforced similar barcode distributions across co-housed mice. Whole-genome sequencing of hundreds of isolates revealed linked alleles that demonstrate between-host transmission. A population-genetics model predicts substantial fitness advantages for certain mutants and that migration accounted for ~10% of the resident microbiota each day. Treatment with ciprofloxacin suggests interplay between selection and transmission. While initial colonization was mostly uniform, in two mice a bottleneck reduced diversity and selected for ciprofloxacin resistance in the absence of drug. These findings highlight the interplay between environmental transmission and rapid, deterministic selection during evolution of the intestinal microbiota.
Keywords: Escherichia coli, cross-housing, transmission, motility, metabolism, parallel evolution, metagenomics, population-genetics modeling, ciprofloxacin, gyrase
Graphical Abstract
eTOC Blurb
Vasquez et al. demonstrate the effectiveness of DNA barcodes for quantifying bacterial intra-species colonization dynamics, selection, and transmission in the mammalian gut, and the effects of antibiotic treatment. In combination with a population-genetics model, the migration rate of gut bacteria is estimated to be ~10% per day.
Introduction
The gut microbiota is important for many aspects of host physiology, including resistance to pathogen invasion (Litvak and Baumler, 2019), immune-system modulation (Pickard et al., 2017), and metabolism (Visconti et al., 2019). The close relationship between mammalian hosts and microbes has emerged through extensive co-evolution (Moeller et al., 2016). While substantial progress has been made in correlating microbiota composition to host health and disease, the complexity and rapid turnover of this ecosystem has limited our understanding of colonization dynamics, as well as of the magnitude and importance of transmission between hosts and seeding from the environment.
Comprehensive studies of genetic variability within commensal species have revealed substantial strain-level variation (Poyet et al., 2019) that potentially reflects a range of adaptations. Diet has been associated with particular strains of the commensal Prevotella copri (De Filippis et al., 2019), due to mutations in polysaccharide-utilization loci (Fehlner-Peach et al., 2019). Genetic variation of the pathogen Listeria monocytogenes has been connected to differences in ecology and virulence potential, leading to diagnostic applications (Ragon et al., 2008). Genetic bottlenecking of Yersinia pestis arises because this species can infect with only a few cells (Perry and Fetherston, 1997). In humans, resident populations of gut bacteria exhibit a relatively clonal structure, with a few strains of each species at intermediate or high frequencies (Garud et al., 2019; Truong et al., 2017); it is currently unclear whether this phenomenon is typically caused by strong colonization bottlenecks, the transient invasion of external strains, or stable ecological sub-structure within species. Thus, a deeper understanding of selection and migration, and the time scales over which genetic variation is established within a host would have broad relevance regarding gut microbiota dynamics.
Laboratory long-term evolution experiments have provided quantitative insights into the interplay between genetic variation and selection. During repeated in vitro passaging for >60,000 generations, Escherichia coli experienced large increases in fitness (Wiser et al., 2013), cell size (Philippe et al., 2009), and metabolic potential (Grosskopf et al., 2016). Metagenomic sequencing revealed that microecologies (multiple co-existing strains) can persist within these evolved populations for >10,000 generations, with relative frequencies that are continually perturbed by the accumulation of additional mutations (Good et al., 2017). During in vitro passaging of budding yeast, the use of DNA barcodes (genomic integration of unique sequences for subsequent identification) and sequencing to monitor the relative frequencies of many competing lineages revealed rapid and reproducible evolutionary dynamics (Levy et al., 2015), and large-scale isolation of adapted strains permitted genotype-fitness mapping (Venkataram et al., 2016) and uncovered growth-phase tradeoffs (Li et al., 2019).
Several previous studies have capitalized on barcoding approaches to study selection in the mouse gut. Colonization of conventional mice with two E. coli strains harboring different fluorescent proteins found ubiquitous clonal interference (competition among lineages arising from mutations that appeared independently) (Barroso-Batista et al., 2014). Evolution of E. coli in ex-germ-free mice depended on selection based on amino-acid consumption and niche availability (Barroso-Batista et al., 2020). Clonal interference was also observed in conventional mice infected with eight Salmonella Typhimurium strains with distinct DNA barcodes; only one or a few strains persisted in the gut after 35 days (Lam and Monack, 2014). Notably, distinct sets of barcodes emerged in individual mice, indicating either stochastic colonization, selection, or post-colonization drift (Lam and Monack, 2014). It is difficult to determine whether lack of barcode diversity is inherent to Salmonella colonization or was due to the presence of other microbiota members. Moreover, the individual housing of mice and/or limited number of barcodes in these previous studies precluded quantification of transmission between hosts sharing the same environment. Perturbations such as antibiotic treatment (Ng et al., 2019) or osmotic diarrhea (Tropini et al., 2018) can apply selective pressures that lead to long-term changes in microbiota diversity and composition as well as the emergence of resistant strains (Ng et al., 2019). It has yet to be determined how such perturbations affect intraspecies genetic diversity and migration between hosts, which could affect recovery of the microbiota and the host. Barcodes provide a powerful tool to quantify these fundamental behaviors.
Here, we utilized heritable DNA barcodes to simultaneously track, at high temporal resolution, the evolutionary dynamics and genetic adaptations of nearly 200 isogenic E. coli strains in ex-germ-free mice. Colonization was initially even across most of these mono-colonized mice, followed by multiple waves of selection. In all cages, migration between hosts enforced similar abundances. Strain dominance was dependent on the fitness conferred by newly acquired mutations. Using metagenomic and whole-genome sequencing, we repeatedly identified selection for mutations that affect motility and metabolism, and isolates with multiple mutations in common demonstrated within-cage transfer. Nearly identical evolutionary trajectories were identified in a replicate experiment. Metabolomic analysis of mouse intestinal and fecal contents and phenotypic profiling of isolates suggested selection for mutations that enable raffinose utilization. Using a population-genetics model, we determined that migration accounts for a substantial fraction of the resident microbiota, which homogenizes barcode composition across mice in the same cage and quantitatively predicts transmission kinetics. Treatment with ciprofloxacin created a large bottleneck that allowed for transmission between cages and selected for a single resistant strain, demonstrating that strain-level variation can be altered dramatically by environmental perturbations. Unexpectedly, the resistance mutation was pre-existing due to a bottleneck in two mice during colonization. This study demonstrates the power of genetic barcoding for uncovering rates of selection and transmission across environments.
Results
Stable colonization of ex-germ-free mice by a barcoded E. coli library
To determine the kinetics of gut colonization and community assembly at the intra-species level, we sought to investigate how subpopulations of the enteric bacterium E. coli would colonize, compete, and evolve within gnotobiotic mice in the absence of other species. A previous study generated a library of high-copy plasmids that each carried a unique barcode (Figure 1A) (Cira et al., 2018). We transformed the laboratory wild-type strain MG1655 with 191 distinct barcoded plasmids. Our choice of MG1655 was motivated by the goal of investigating rapid selection, since we surmised that lab-adapted MG1655 would undergo accelerated evolution in vivo within the mammalian gut. All transformed strains had similar growth kinetics in vitro (Figure S1A). We split the 191 strains into sets of 95 (set 1) and 96 (set 2) to separately colonize mice, enabling study of both evolution within mice and transmission from one group of mice into another.
Figure 1: Colonization of germ-free mice by a barcoded E. coli library is initially even, but a single strain dominates within one week.
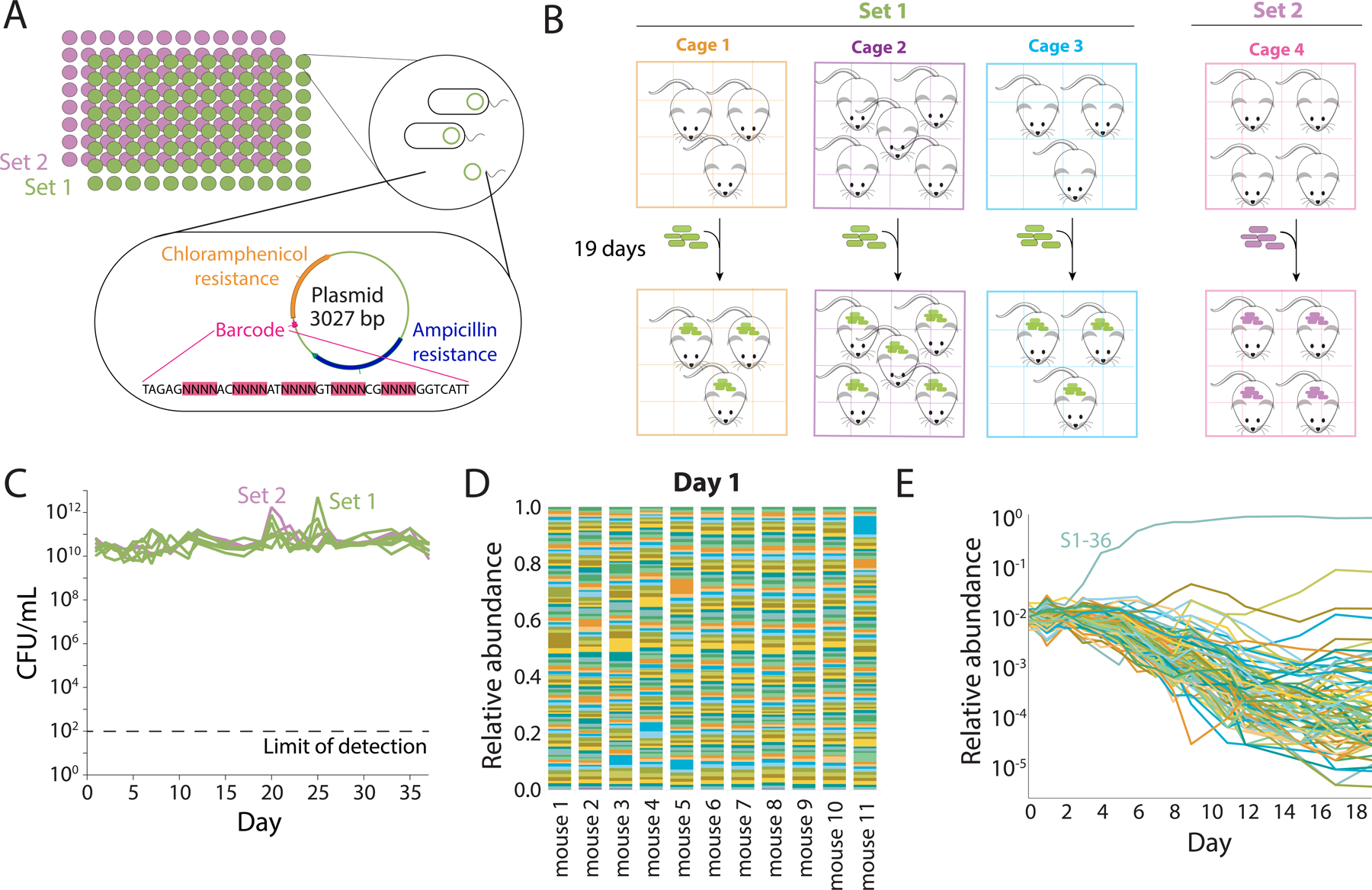
A) Plasmids containing a unique 28-bp DNA barcode and encoding resistance cassettes to chloramphenicol and ampicillin were used to transform E. coli MG1655, a common laboratory wild-type strain. We isolated 191 unique strains, sequenced them to verify the presence of a single, unique barcode surrounded by common primer sequences, and split them into two sets of 95 (S1) and 96 (S2) strains that were used to independently colonize germ-free mice. The abundance of each strain was tracked via sequencing after barcode amplification.
B) Schematic of housing layout of mice colonized with barcoded E. coli. Three cages with 3, 5, and 3 mice were colonized with S1 strains, and one cage with 4 mice was colonized with S2 strains.
C) After mice were colonized, fecal samples were collected daily. Mice gavaged with S1 strains were colonized to similar levels as mice gavaged with S2 strains (~1010−1011 CFUs/mL), and bacterial loads remained stable over the entire experiment.
D) One day after gavage with the barcoded strains, the 11 mice with S1 strains were approximately evenly colonized with the 95 strains.
E) A single barcode can become dominant within one week. After day 4, most strains in mouse 2 continuously decreased in relative abundance as barcode S1–36 took over, although a few maintained their level of relative abundance or experienced a resurgence after day 10.
We gavaged germ-free mice on day 0 with either set 1 (S1) or set 2 (S2) barcoded strains, (Figure 1B). Feces were collected daily beginning on day 1 to enable barcode tracking at high temporal resolution, and DNA was extracted for barcode and metagenomic sequencing. We measured CFUs/mL from each sample and found that mice were colonized with ~1010–1011 CFUs/mL by day 1, with this level maintained thereafter (Figure 1C).
Barcode amplification and sequencing enabled quantification of the relative abundances of each barcode throughout the experiment. The initial loss of some strains would represent a strong bottleneck during colonization. However, we found that every barcode was present in S1 mice on day 1, with an approximately even distribution of relative abundances (mean coefficient of variation of 0.44; Figure 1D). In a representative mouse, this even distribution was maintained for several days, before apparent selection of a more fit strain (S1–36) starting on day 4 (Figure 1E). Thus, we conclude that the entire set of barcodes transited through the mouse intestinal tract and colonized with approximately equal fitness.
Co-housed mice maintain highly similar barcoded strain abundances despite rapid adaptation within hosts
As expected from previous studies (Barroso-Batista et al., 2014), we observed rapid expansion of individual barcodes within a few days, suggesting that some lineages had started to acquire adaptive mutations. Given our co-housing design, we sought to determine whether the dynamics of these barcodes would be independent in different mice (e.g. due to independent mutation events), or whether between-host transmission would lead to similar barcode trajectories across all mice in the same cage. While isolated examples of strain transmission have previously been observed in E. coli (Barroso-Batista et al., 2015; Lescat et al., 2017), it remains unclear whether this process can effectively homogenize the gut microbiota on similar timescales to the emergence of mutations within individual hosts. Our hierarchical design allowed us to test this question directly, by comparing the trajectories of the S1 barcodes across mice from the same versus different cages.
We found that a single strain (S1–36) rapidly expanded in all S1 mice by day 3, and became the dominant strain in all but one mouse by day 10. The early timing of this expansion, combined with the consistency of the S1–36 trajectories within and between cages, suggests that this event was likely driven by growth of a pre-existing mutation that was present at low frequency in the initial S1–36 stock (Levy et al 2014). We investigate this hypothesis further below.
By contrast to the S1–36 lineage, many other strains that reached higher frequencies tended to expand in all mice in the same cage, but did not expand at all in other cages (Figure 2A, S2A). Notable examples include the S1–57 lineage in cage 1, the S1–46 lineage in cage 2, and the S1–86 lineage in cage 3. Such cage-level differences were likely driven by de novo mutations that arose in individual mice, and rapidly spread to the other mice that shared the same cage. These phenomena suggest that strain transmission between co-housed mice (rather than the highly unlikely independent emergence of the same barcodes in each mouse) was an important factor in establishing community composition in vivo.
Figure 2: Cage-specific subsets of barcoded strains quickly expand in abundance due to selection of mutations in motility- and metabolism-related genes.
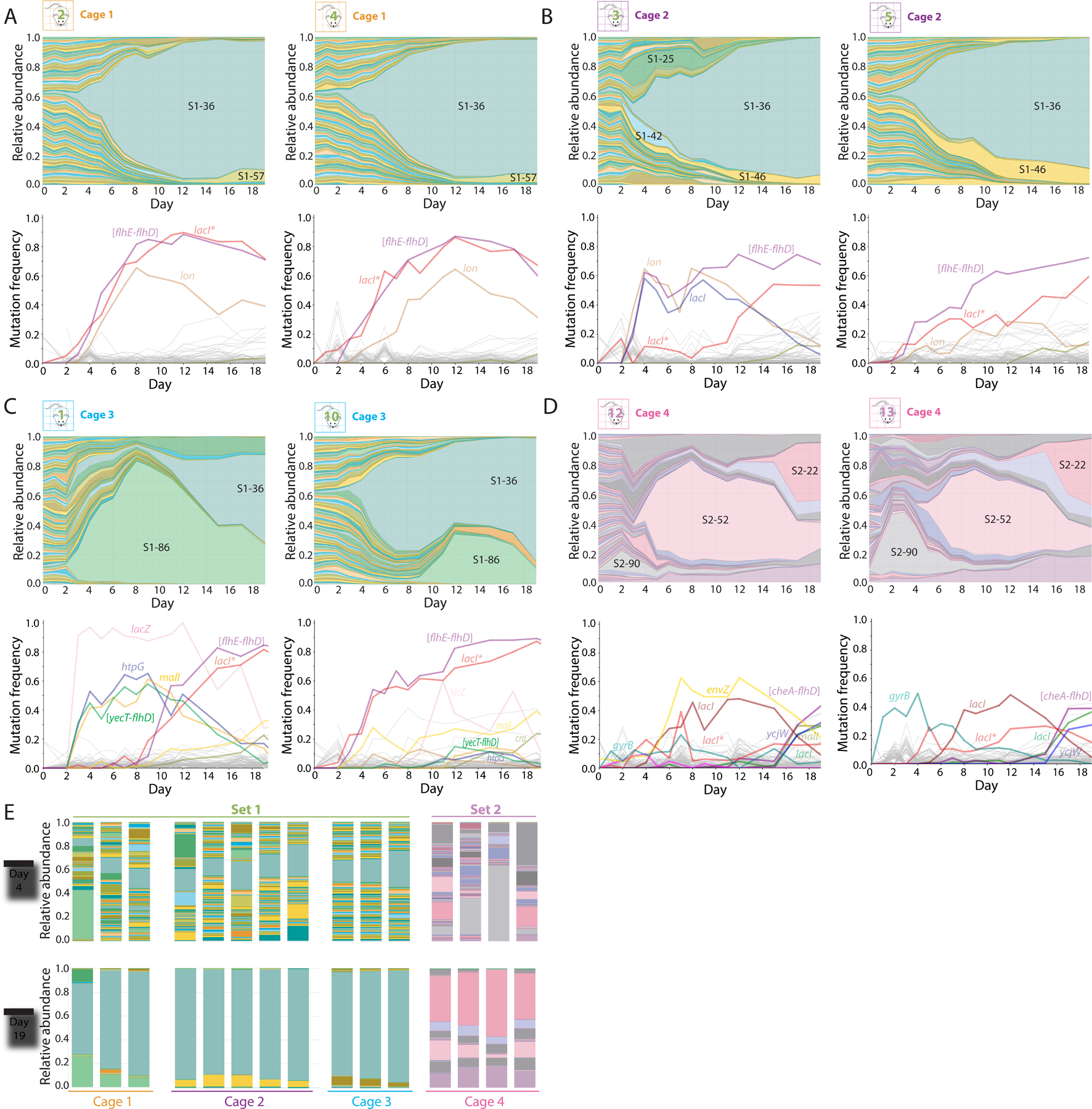
A-C) Relative abundances of barcodes (top) and mutations (bottom) in two representative mice within each cage of S1 mice. In all three cages, S1–36 took over; this strain harbors mutations in lacI and large motility deletions such as [flhE-flhD]. The other strains that bloomed were cage-specific.
D) Relative abundances of barcodes (top) and mutations (bottom) in two representative S2 mice. Multiple waves of partial barcode takeover occurred, with several barcodes persisting at >5% by day 19.
E) On day 4, relative abundances between mice in the same cage were much more variable compared to on day 19, despite differences among cages.
We also observed examples of imperfect homogenization within cages. For example, the S1–25 and S1–42 lineages expanded in mouse 3, but not in any of the other 4 co-housed mice in the same cage (Figure 2B). Similarly, the early expansion of S1–86 in Mouse 1 occurred almost ~6 days before it started to expand in mouse 10 (Figure 2C). These examples highlight the interplay of within-host selection and between-host transmission on these short evolutionary timescales.
Metagenomic sequencing allowed us to link barcode dynamics with specific mutations (Table S1). All mice in cage 1 exhibited high frequencies of three identical mutations: a 4-bp frameshift insertion in lacI, which encodes the Lac operon repressor (henceforth “lacI*”); a 15,897-bp IS-mediated deletion in the flagellar operon from flhE to flhD (henceforth “[flhE-flhD]”); and an IS insertion in the promoter region of lon, which encodes the Lon protease. The [flhE-flhD] mutation has been observed in previous mouse colonization experiments involving E. coli (Gauger et al., 2007) and the lacI* mutation has been observed in in vitro evolution experiments (Quan et al., 2012). The lacI* and [flhE-flhD] mutations reached >60% frequency by day 6, indicating that they were nearly fixed within the S1–36 lineage by this time and were potentially drivers of the early dominance of this strain (Figure 2A, S2A).
Identical lacI* and [flhE-flhD] mutations were also observed during the expansion of S1–36 lineages in the other two cages of S1 mice (Figure 2B,C, S2B,C), even though neither were detectable from metagenomic sequencing of the original S1–36 stock. Notably however, the [flhE-flhD] mutation in mouse 3 was initially accompanied by a separate point mutation in lacI (G272V), which was eventually outcompeted by the lacI* mutation around day 12 (Figure 2B). Thus, the lacI mutations likely occurred within the S1–36 lineage at some point after the initial [flhE-flhD] mutation. These discoveries highlight the synergy between metagenomic and barcode sequencing data.
In the third cage of S1 mice, the transient dominance of the S1–86 lineage allowed us to identify the mutations associated with this strain (Figure 2C, S2C): a related motility deletion ([yecT-flhD]), which includes the same genes as [flhE-flhD] in S1–36, as well as additional mutations in lacZ, htpG, and malI. Notably, all four mutations later appeared in the other two mice in cage 3 around the same time that the S1–86 lineage started to expand there. These data provide further evidence that expansion of S1–86 was driven by the transmission of adaptive mutations.
To determine whether a group of mice colonized with different barcodes would exhibit similar selection dynamics as the S1 mice, we used sequencing to characterize the S2 mice (Figure 2D). In two of the four S2 mice (#12 and #14), the initial abundances of the 96 barcoded strains were approximately uniformly distributed (Figure S2D), as in the S1 mice (Figure 1D). However, in the other two S2 mice (#13 and #15), the initial distribution of abundances was highly uneven, with a handful of barcodes at high frequencies (>5%) and a large fraction below the limit of detection (0.1%) (Figure S2D). On day 1, >95% of the bacteria in mouse 15 were accounted for by just three strains, all of which were also present at >5% in mouse 13. This high degree of overlap suggests that the colonization bottlenecks in these two mice were accompanied by strong selection pressures, which independently selected for pre-existing mutations in a subset of the lineages. Nonetheless, the undetectable lineages quickly reappeared in these mice by day 4, while the high-frequency barcodes quickly declined to lower frequencies (Figure 2E, S2E). These data suggest that exchange of bacteria between mice effectively eliminated the initial unevenness from a transient colonization bottleneck.
Subsequent waves of adaptation in S2 mice were again accompanied by the emergence of metabolic and motility-related mutations, including the lacI* frameshift and another IS-mediated deletion in the flagellar operon. We also observed two other lacI mutations distinct from lacI* and lacIG272V (Figure 2D, S2F). The genetic variation exhibited by S2 strains suggests a wider diversity of adaptive mutations that likely did not emerge in S1 mice due to early dominance of the lacI* and [flhE-flhD] combination in S1 mice. These data demonstrate the capacity of a host to support several strains despite multiple waves of expansion and competition, with transmission from environmental reservoirs establishing a common composition among co-housed mice.
The gut environment reproducibly selects for the same set of mutations
It was clear in S2 mice that a wider range of mutations could be maintained without the presence of a strong competitor like S1–36. Thus, we repeated the experiment by gavaging three new cages of germ-free mice (n=9 total) with the original S1 barcode pool except for S1–36, to ensure a different evolutionary trajectory; we refer to these as S136− mice. All S136− barcodes were detected in the mice on day 1 (Figure 3A, S3A–C), and the library colonized to similar CFUs/mL (Figure S3D) as in our first experiment (Figure 1C).
Figure 3: Similar mutations are repeatedly selected for in the mouse gut, likely driven at least in part by the presence of raffinose, an abundant carbon source in the gut.
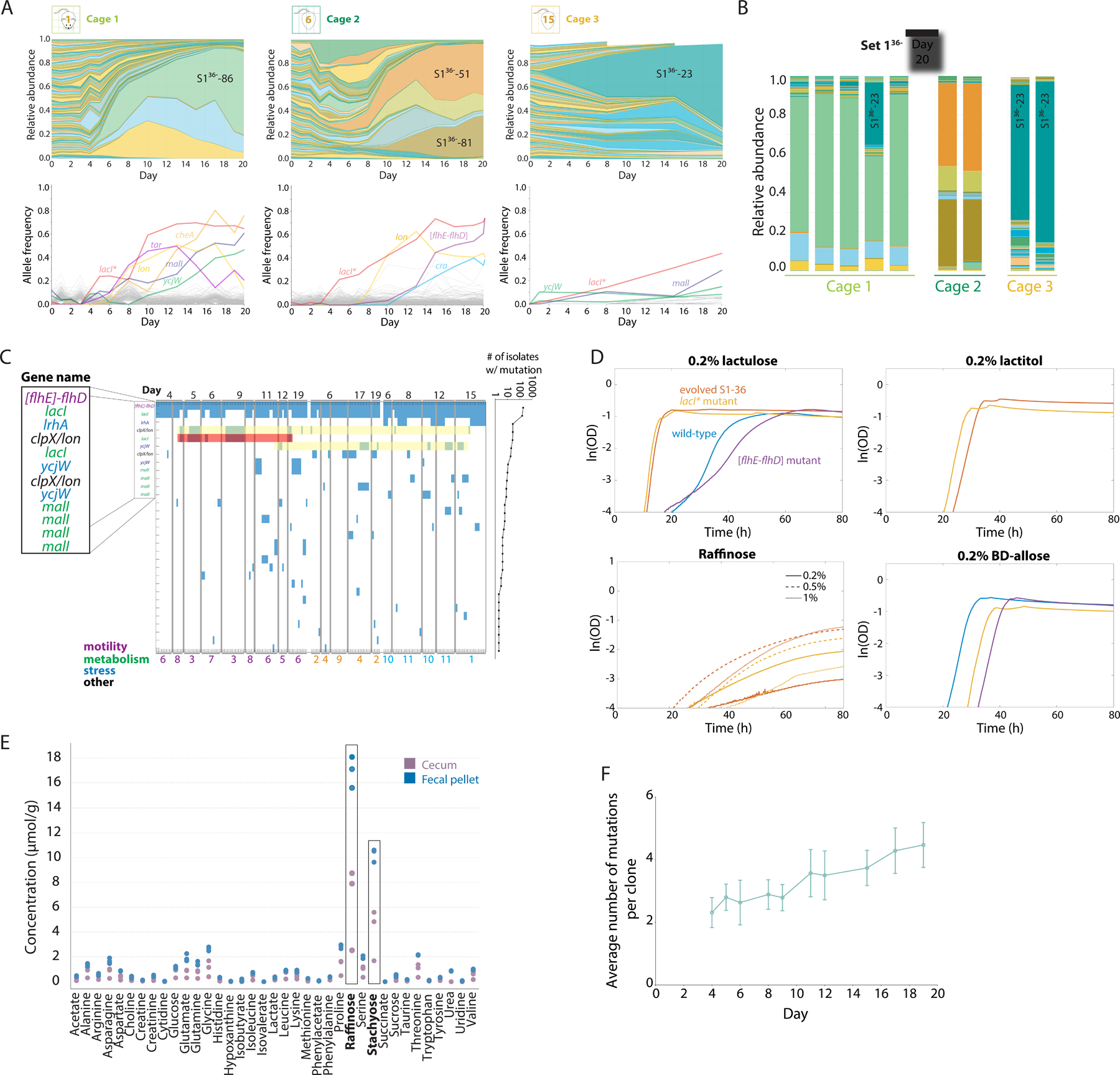
A) All S1 strains except S1–36 were used to gavage 9 germ-free mice in three cages on day 0 and tracked via daily fecal sampling. Shown are relative abundances (top) and mutations identified from metagenomic sequencing (bottom) of the barcoded E. coli populations from a representative mouse in each cage over the first 20 days of colonization. Different barcodes emerged in each cage, with similar mutations in cage 1 and 2 but not cage 3.
B) By day 20, relative abundances of the S136− barcodes were similar within each cage, despite differences across cages.
C) Map of mutations identified via whole-genome sequencing of 189 S1–36 clones isolated from various mice organized by cage throughout the experiment. The most prevalent mutations, which include the [flhE-flhD] deletion (purple) and mutations mostly in metabolism-related genes (green), are labeled with the associated gene. Yellow regions highlight a lon promoter and ycjW alleles that co-occurred in multiple cages, indicating parallel evolution. The red region highlights a lacIG272V mutation that co-occurred with the lon promoter mutation in cage 2 mice and hence is more likely due to within-cage transfer than parallelism.
D) Growth curves of the wild-type parent, evolved S1–36, a lacI* mutant, and an [flhD-flhE] mutant in M9 supplemented with four carbon sources. In lactulose, lactitol, and raffinose, evolved S1–36 and the lacI* mutant grew much faster and with a shorter lag than the parent or the [flhD-flhE] mutant; in lactitol and raffinose, the parent and [flhD-flhE] mutant could not grow at all, suggesting that the ability to utilize raffinose is conferred by the lacI* mutation. In β-D allose, the parent grew better than the lacI or [flhD-flhE] mutants; evolved S1–36 exhibited no growth and hence is not shown.
E) Concentrations of various amino acids and carbon sources in the feces and ceca of germ-free mice measured using NMR. Feces and ceca exhibited high concentrations of raffinose and stachyose (boxes, n=3 mice).
F) The number of mutations detected in S1–36 clones increased over the duration of the experiment, suggesting continued selection. Data are mean values and error bars represent 1 standard deviation, with n=10–38 isolates on each day.
After 20 days, barcode relative abundances were again similar across mice in each cage (Figure 3B). Notably, the subsets of barcodes that expanded were essentially nonoverlapping with the first experiment or across cages, illustrating the stochastic nature of evolution with regard to the barcoded strains that take over. Mirroring our first experiment, by day 20 the three cages contained alleles at >10% frequency in motility, metabolism, and stress-response genes (Figure 3A, S3A–C). The most abundant strain in cage 2 (S136−-51) contained the lacI* and [flhE-flhD] allele combination identified in our first experiment (Figure 3A), emphasizing the parallel nature of selection across experiments. As before, we observed substantial overlap in allele frequencies among mice in the same cage, likely reflecting within-cage transmission and takeover of higher-fitness mutants.
Dense isolate sequencing distinguishes parallel evolution, strain transmission, and selection on standing variation
Since the S1–36 strain began to take over early on (by day 5) in most S1 mice and metagenomic sequencing showed that the lacI* and motility mutations arose approximately concurrently (Figure 2A–C), we sought to disentangle the order in which these mutations appeared and potential co-occurrence between alleles. We isolated and sequenced the genomes of >750 colonies from a range of mice across experimental time points reflecting various blooms of specific barcodes, including many in which S1–36 dominated. In total, we ended up with 189 S1–36 isolates from before cross-housing across 11 mice and 10 time points.
All S1–36 isolates contained the [flhE-flhD] motility deletion (Figure 3C), consistent with the hypothesis that this mutation existed at low frequency in the original S1–36 stock. As predicted from our metagenomic data, many S1–36 isolates also carried the lacI* mutation, but 26 clones from three mice in cage 2 contained lacIG272V instead (Figure 3C). These two lacI mutations were mutually exclusive, and clones from the two classes cohabited some mice (Figure 3C). Moreover, six S1–36 clones from days 6–12 lacked any lacI mutations (Figure 3C), further supporting the hypothesis that the motility deletion was the first to emerge.
To identify phenotypes associated with these early mutations, we isolated or reconstructed strains with all combinations of the [flhE-flhD] and lacI* alleles. We grew these four strains on a broad range of carbon sources and identified differential growth curves in several conditions (Figure 3D, S3E). Although previous studies speculated that lacI mutations were linked to the presence of lactose in the environment, we found that the largest growth rate differences occurred in raffinose (a non-digestible dietary carbohydrate) in which only the lacI* mutants could grow (Figure 3D). NMR analysis of germ-free mouse intestines revealed high concentrations of raffinose (Figure 3E), suggesting that this diet-derived metabolite was responsible for the strong growth advantage and widespread genetic parallelism of the lacI* mutation in our experiments.
In addition to these early mutations, many other alleles were prevalent across the isolates. Of the 68 alleles we identified, 31 (45.6%) were observed in two or more isolates; 23 (33.8%) were observed in isolates from different mice, and 12 (17.6%) were observed in isolates from different cages. The most frequently observed mutations tended to occur in metabolic genes (Figure 3C), consistent with previous studies of E. coli evolution in the mouse gut (Barroso-Batista et al., 2020; Barroso-Batista et al., 2014; Ghalayini et al., 2019). The number of mutations in each isolate increased steadily with time, with an average of 5 mutations per clone by day 20 (Figure 3F).
The co-occurrence patterns of highly prevalent mutations provided a unique opportunity to distinguish instances of strain transmission from parallel evolution of the same mutations. For example, the IS-insertion in the lon promoter was observed in 27 isolates from 6 different mice in 3 different cages (Figure 3C, yellow), consistent with strong parallel evolution. However, all occurrences of this allele in cage 2 mice were accompanied by lacIG272V (Figure 3C, red), while its presence in other cages was accompanied by lacI* instead. These observations suggest that a transmission event allowed the lon alleles to spread within cage 2 mice, while the same alleles in other cages were likely generated by parallel evolution. Similarly, identical ycjW alleles from 3 different cages were associated with different malI alleles, while similar combinations of these mutations were observed in different mice from the same cage. These examples show that co-occurrence patterns can be used to identify strain transmission events in the midst of widespread parallel evolution.
Cross-housing rapidly remodels strain abundances and allows quantification of migration rates between hosts
The separation of barcodes into two sets afforded the opportunity to study migration of gut bacteria between hosts. In the first experiment, on day 19 we moved two S1 mice and two S2 mice into a new cage and cross-housed them for 18 days (Figure 4A) to quantify the ability of S1 strains to colonize S2 mice and vice versa. After the initiation of cross-housing, both sets of mice experienced invasion from the opposite set (Figure 4B, S4A). Among S1 mice, the S1–36 strain remained dominant, with the total relative abundance of S2 strains peaking at ~5–10% on day 21 and decreasing thereafter (Figure 4B, left). By contrast, S1–36 rapidly expanded in S2 mice after cross-housing, reaching >80% by day 28 (Figure 4B, right). These asymmetric outcomes suggest that migration from other hosts (likely via coprophagy) introduces a sizeable fraction of the community into each host, and that the evolved S1–36 strain had a substantial fitness advantage over the evolved S2 strains that enabled successful engraftment in new hosts and displacement of the previous residents.
Figure 4: Population-genetics model recapitulates experimental cross-housing data and predicts a high rate of migration.
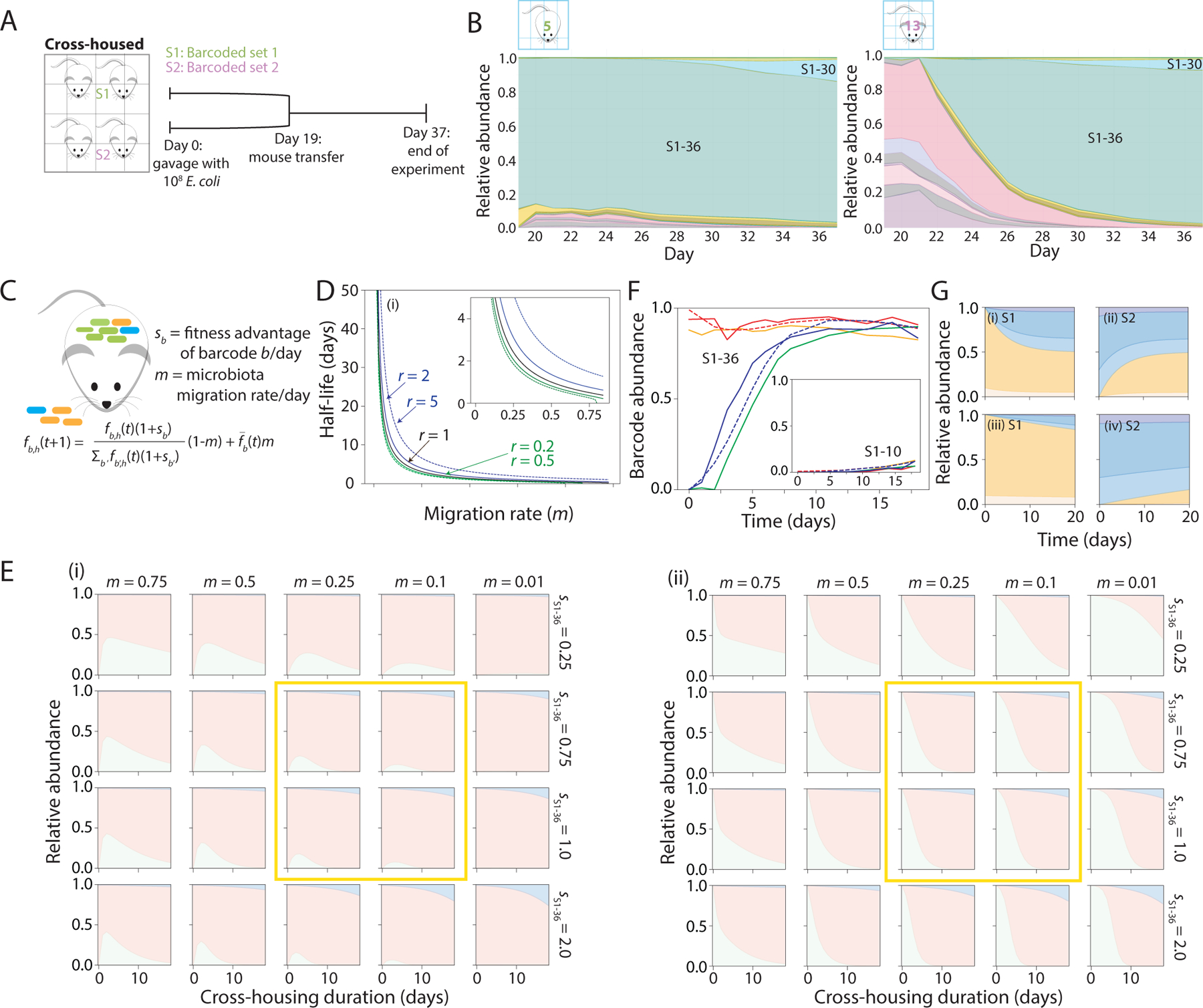
A) Schematic of S1 and S2 mice cross-housing. Mice colonized with S1 strains were separately housed from mice colonized with S2 strains for 19 days, then transferred into the same cage. Fecal pellets were collected daily until day 37.
B) Relative abundances of S1 (shades of green) and S2 (shades of pink) barcodes in a mouse initially colonized with S1 strains (left) or S2 strains (right). S1–36 remained dominant in the S1 mouse, and progressively expanded in the S2 mouse over ~2 weeks.
C) In the model, the frequency of barcode b in host h on day t+1 (fb,h(t+1)) is determined by the fitness (excess growth/day) of barcode b compared with other barcodes (sb) and the microbiota migration fraction each day due to coprophagy is the relative abundance of barcode b on day t averaged over all hosts, which is assumed to represent the relative load of barcode b in feces that is taken in via coprophagy.
D) When mice harboring distinct barcode abundances are cross-housed, the model predicts that coprophagy drives all mice to the same barcode composition on a timescale (half-life) that increases strongly with the migration rate m. In the absence of fitness advantages, the time scale is ~ − 1/ln(1 − m) (black line). If mice are predisposed to consume their own feces r times as often as the feces of other mice, then the half-life is not strongly affected if 0.2<r<5 (green and blue lines).
E) Simulations of the model with three barcodes, one each representing S1–36 (pink) and S1–10 (blue) and one representing all S2 barcodes (green), recapitulated experimental data (Figure 3E,F) when fS1−36 = 0.75–1.0, fS1−10 = 1.33fS1−36, and m = 0.1–0.25 (yellow boxes). Inset shows predicted and actual abundances of S1–10 from the time of cross-housing.
F) Parameter estimates fS1−36 = 0.75, fS1−10 = 1.0, and m = 0.12 were obtained from the simulations in (E) by minimizing the squared deviation of the model (dashed lines) from the dynamics of barcodes S1–36 and S1–10 averaged across the four mice in Figure 3E,F (S1: solid red and orange lines; S2: solid blue and green lines).
G) Simulations show that the equilibrium composition is reached within 20 days for two mice with highly distinct starting compositions (i,ii) when m = 0.2 and with no fitness advantages among barcodes, but not when m = 0.02 (iii,iv).
To estimate the underlying rates of migration and selection, we developed a population-genetics model to fit the observed barcode abundance dynamics within and across mice that share the same cage. Motivated by our observations, we assumed that metapopulation dynamics could be described by a generalized ‘island model’ (Latter, 1973; Wright, 1943)
| (1) |
that captures the interplay between local competition within hosts and migration from a common reservoir (Figure 4C). fb,h(t) represents the frequency of barcode b in host ℎ at day t, is the average frequency across all hosts, and m is the migration rate. The Sb,h(t) and terms represent fitness differences among barcodes during within-host competition and migration, respectively, which will generally vary over time (and across hosts) as mutations accumulate. If all barcodes have the same fitness, the model predicts that barcode frequencies will equilibrate across mice within a cage on a timescale of ~1/m days; this estimate is insensitive to the predisposition r for hosts to consume their own feces (Figure 4D). When barcodes differ in fitness, this equilibration must compete with local amplification via natural selection, which occurs on a timescale ~1/s. Thus, emergent evolutionary dynamics within the metapopulation will strongly depend on the relative magnitudes of m and Sb,h(t).
Estimating these parameters can be challenging due to the time dependence of within-host fitnesses, Sb,h(t), which can potentially vary across hosts in an idiosyncratic manner due to clonal interference and frequency-dependent selection. Fortunately, our cross-housing experiment provides an opportunity to directly measure the migration rate, independently of Sb,h(t), by focusing on the invasion of new barcodes immediately after cross-housing. For example, on the first day after the time of cross-housing (tc), the frequencies of S2 barcodes in S1 mice can be directly attributed to migration:
which allowed us to infer migration rates through a simple application of linear regression. We fit this model to our cross-housing data, in which essentially all S2 barcodes migrated into S1 mice (Figure S4B). Typical migration rates were on the order of 10% per day across a broad range of initial frequencies, suggesting that stochastic transmission bottlenecks do not play a major role in this frequency range (f>0.001). These data also revealed some global variation in m across mice, as well as some systematic variation in transmission efficiency between barcodes . Nonetheless, higher frequency barcodes did not exhibit systematically higher migration fitness, and differences across mice averaged out within 1–2 days (Figure S4C). Together, these data suggest that between-host migration is reasonably approximated by an island model with average migration rate m~10% per day.
Interestingly, these short-term estimates of the migration rate are qualitatively consistent with the equilibration timescales in our experiments (Figure 2E, 3B), typically ~5–10 days. To make this comparison more explicit, we asked whether we could quantitatively recapitulate our experimental data using a coarse-grained version of Eq. 1, in which we assign a constant fitness advantage to a few key lineages. For example, in the cross-housing experiment, we assume that all S2 barcodes and all S1 barcodes except S1–36 and S1–30 had similar (low) fitness and thus could be treated as a single barcode. Fits to barcode abundances in S1 and S2 cross-housed mice were excellent for SS1−36 = 0.75, SS1−30 = 1.0, suggesting that the S1–36 and S1–30 genotypes confer a ~75% and ~100% growth advantage per day, respectively, and m = 0.12 (Figure 4E,F), a similar estimate for migration rate in the first few days after cross-housing as obtained from simulating the equilibration of co-housed S2 mice (Figure 4G). Predictions based on these parameters (Figure 4E) remained consistent with barcode abundances at day 62 (Figure S4D), suggesting that any subsequent mutations did not dramatically affect the relative fitness advantage of S1–30 over S1–36. Taken together, our dynamic barcode measurements combined with mathematical modeling permitted robust, quantitative estimates of key parameters dictating assembly and evolution within mouse intestines.
Evolution and cross-invasion after recolonization with a strain of increased fitness
Since strain S1–36 became dominant so quickly in our first experiment, we were hampered in our ability to use barcode sequencing to track further evolution among the population. To overcome this obstacle, we isolated an S1–36 clone on day 37, cured it of its plasmid, and transformed the resulting strain with 86 S2 plasmids. This new set of strains, which we refer to as S2* strains, allowed us to restart the evolutionary process from a higher-fitness genotype. We gavaged three cages of germ-free mice with the S2* strains and tracked them for 43 days. Distinct barcodes dominated the three cages, yet mutations in the same genes emerged, all related to metabolism or motility (Figure 5A, S5A). After 20 days, within each cage the relative abundance of each barcode was roughly uniform across mice (Figure 5B). The dominant strain in cage 1 (S2*−62) had mutations in ycjW, malI, and an intergenic region near lrhA (Figure 5A), all of which also occurred in S136− mice (Figure 3A).
Figure 5: Cross-housing of mice repeatedly demonstrates the strength of the motility and lacI allele combination.
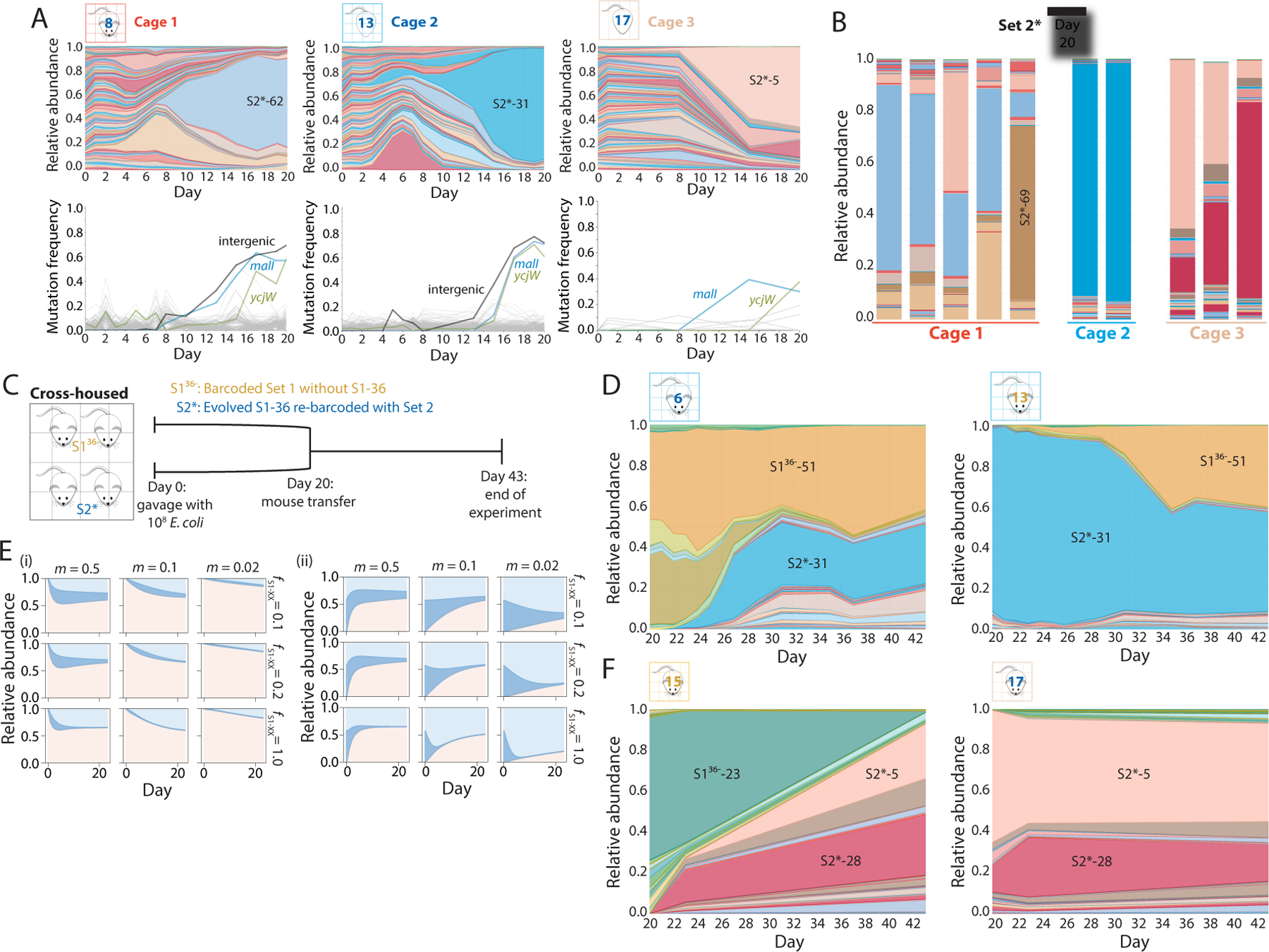
A) S2 barcodes were reintroduced into an evolved clone of S1–36 (S2* strains), used to gavage 10 germ-free mice in three cages on day 0, and tracked via daily fecal sampling for 37 days. Shown are relative abundances (top) and mutations identified from metagenomic sequencing (bottom) of the barcoded E. coli populations from a representative mouse in each cage over the first 20 days of colonization. Various barcodes emerged in each cage, but all three cages hosted bacteria with similar mutations.
B) By day 20, the relative abundances of the S2* barcodes were similar within each cage, despite differences across cages.
C) Schematic of cross-housing of S136− and S2* mice from day 20 to day 43.
D) Relative abundances of all barcodes in representative cross-housed S136− (left) and S2* (right) mice from cage 1. In the S136− mouse, S2* barcodes invaded and stabilized at ~50% relative abundance, and vice versa for the S2* mouse, suggesting that the two barcode sets have approximately equal fitness.
E) The population-genetics model applied to four cross-housed mice with three barcodes, one representing S136−-51 (light blue), one representing all S2* barcodes (pink) with the same fitness as S136−-51, and one representing all other S1 barcodes (blue), recapitulates the coexistence of S136− and S2* barcodes (D) when SS1−51 = SS2 = 0.1–1.0 and m ≈ 0.1–0.25, illustrating the general applicability of the model.
F) Relative abundances of all barcodes in representative cross-housed S136− (left) and S2* (right) mice from cage 2. In the S136− mouse, S2* barcodes invaded and took over by day 43, while S2* barcodes were able to maintain colonization in the S2* mice, indicating S2* barcodes were more fit than the S136− barcodes in this cage.
Our initial cross-housing experiment with S1 and S2 mice combined strains that had evolved for similar amounts of time, but had nevertheless acquired different mutations. By contrast, the high degree of overlap in mutations present in S136− and S2* mice led us to hypothesize that their cross-housing could lead to distinct outcomes compared to S1 and S2 cross-housing; note that S136− and S2* colonization experiments were carried out concurrently. To test this hypothesis, we performed independent cross-housing experiments with S136− and S2* mice from two different cages starting on day 20 and tracked barcode and metagenomic dynamics for an additional 17 days (Figure 5C). These data revealed dramatically different outcomes in the two experiments.
In the first case, strain S136−-51 was at ~50% relative abundance at the time of cross-housing in mice 6 and 7, and remained at ~50% or slightly higher throughout cross-housing (Figure 5D, S5B). Meanwhile, the S2* barcodes increased to ~50% in cross-housed S136− mice (Figure 5D, S5B); conversely, in S2* mice, S136−-51 increased over the first ~10 days and stabilized at ~50% (Figure 5D). These dynamics suggested coexistence of strains from the two sets with similar fitness, and indeed metagenomic sequencing indicated that S136−-51 had the lacI* mutation, along with [cheA-flhD] and mutations in malI and ycjW, similar to the S2* strains. Thus, we assumed that S136−-51 and all persistent S2* barcodes had the same fitness advantage over all other S136− barcodes, and used our model to simulate the dynamics of three strains representing S136−-51, all other S136− strains, and all high-fitness S2* strains. The data were consistent with SS136−−51 ≈ SS2∗ ≈ 0.2 (Figure 5E), indicating that the motility operon deletions in other S2* strains narrowed the fitness difference compared with the parental strain, against which S136−-51 had a fitness benefit >0.75. While frequency-dependent selection could also play a role in the expansion leading up to coexistence, notably these data were best fit with a similar value for migration rate (m~0.1) as our other co-housing and cross-housing experiments.
The second cross-housing experiment of S136− and S2* mice led to a qualitatively distinct outcome from coexistence. In this case, the S2* strains displayed a clear fitness advantage over S136− strains (Figure 5F, S5C), which contained the lacI* mutation but not a large motility deletion (Figure 3A, mouse 15). These data suggest that the lacI* mutation alone is unable to compete with strains that also have the [flhE-flhD] allele. Taken together, these data illustrate the potential for repeated selection of the same allele combinations and their importance in determining coexistence or out-competition.
Treatment with ciprofloxacin decreases intrastrain diversity and stimulates transmission
Antibiotic treatment causes massive disruptions to the mammalian microbiota that can vary across bacterial species (Ng et al., 2019). To understand how such a disruption would affect bacterial population dynamics at the intra-species level, we treated five S1 mice and two S2 mice with 5 mg of ciprofloxacin once daily on days 19, 20, and 21 (Figure 6A). This treatment effectively reduces the number of bacteria in humanized mice (Ng et al., 2019). To probe the effects of an antibiotic perturbation on transmission, on the same day as the first ciprofloxacin treatment, two S1 mice and two S2 mice were moved into a new cage and cross-housed for 18 days (Figure 6A). These two cages shared a different isolator from non-antibiotic-treated mice.
Figure 6: Ciprofloxacin treatment leads to complete loss of substrain-level genetic variation.
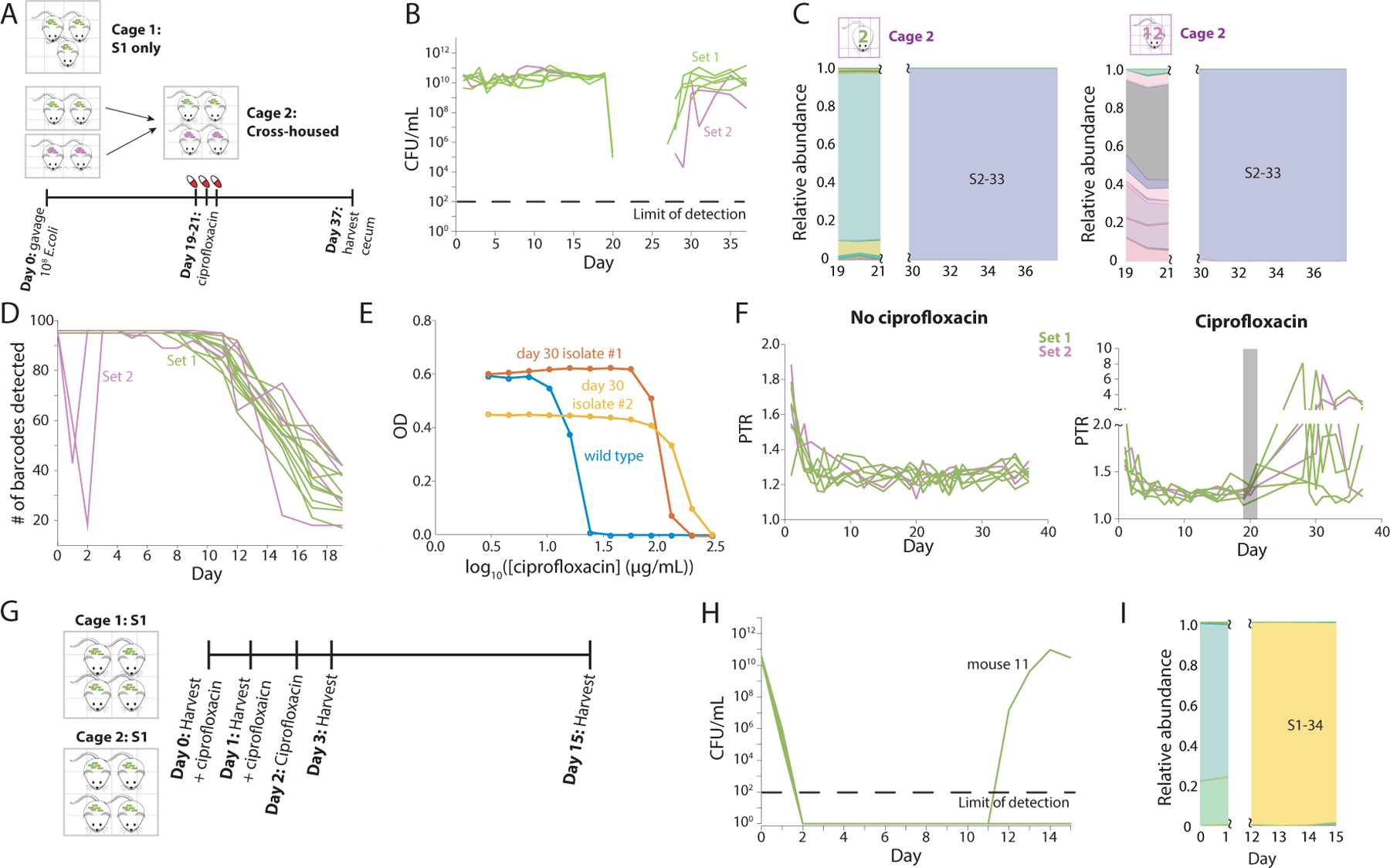
A) Schematic of ciprofloxacin treatment protocol involving five and two germ-free mice gavaged with S1 and S2 strains, respectively, on day 0. On day 19, two S1 mice were cross-housed with two S2 mice in cage 2. At the same time, the mice in cage 1 (the remaining three S1 mice) and cage 2 began three days of ciprofloxacin treatment.
B) CFUs/mL decreased below the limit of detection during ciprofloxacin treatment and did not recover until several days after treatment ended.
C) S2–33 was the only strain that recovered in both cages of mice. Omitted days represent days in which bacterial load was too low for accurate sequencing (and hence results resembled water controls).
D) S2 mice with uneven barcode abundance distributions on day 1 (Figure S2D) exhibited a large decrease in the total number of detectable barcodes on days 1 and 2, signifying a bottleneck. These two mice quickly recovered the other barcodes, presumably due to coprophagy. All other mice maintained the full set of barcodes until day ~5, when barcodes such as S1–36 started to become dominant.
E) Two S2–33 clones isolated on day 30 from mouse 14 displayed higher IC50 values than wild type, indicating decreased susceptibility. One clone also grew to lower maximum OD600, suggesting a growth tradeoff for the resistance mutation.
F) Metagenomic data were used to calculate PTR in ciprofloxacin-treated (left) and untreated (right) mice over the duration of the experiment. Gray rectangle represents the interval of ciprofloxacin treatment. Data for the 7 days after treatment are not shown due to low read counts (Figure S7A).
G) Schematic of second ciprofloxacin treatment and harvest protocol in which 8 mice were colonized with S1 barcodes for 2 months before ciprofloxacin treatment daily on days 0, 1, and 2. Mice were also harvested on days 0, 1, and 2, leaving two singly housed mice for tracking until day 15.
H) Only mouse 11 showed bacterial recovery, starting on day 12. The other mouse was likely returned to a germ-free state.
I) S1–34 was the only strain that recovered in mouse 11.
Culturable densities decreased sharply after one day of treatment (Figure 6B), and then dropped below the limit of detection (102 CFUs/mL) and remained undetectable for ~7 days in all mice. Around day 28 (7 days after the last dose), E. coli densities began to recover (Figure 6B). Recent studies utilized the ratio of the number of metagenomic reads at the origin versus the terminus as a proxy for the rate of replication, with fast-growing cells having a higher ‘peak-to-trough ratio’ (PTR) (Brown et al., 2016; Korem et al., 2015). Using our metagenomics data, we calculated the PTR for all mice over time. PTR decreased gradually from ~1.8 on day 1 (corresponding to a growth rate of ~1 h−1 for in vitro culturing (Korem et al., 2015), which is similar to previous estimates of E. coli growth in germ-free mice (Barroso-Batista et al., 2015; Rang et al., 1999)) to ~1.2 by day 5 (corresponding to early stationary phase in vitro (Korem et al., 2015)) (Figure 6F), consistent with the rapid initial increase in CFUs/mL. After treatment, concurrent with the recovery in CFUs, PTR jumped to >2 in most mice, in some cases remaining high for several days (Figure 6F). Thus, recovery after treatment likely signifies higher growth rates during re-expansion. These data demonstrate the wide range of E. coli growth rates in vivo depending on whether the microbiota is at steady state or recovering from a perturbation.
As soon as densities recovered, the population in all cross-housed mice was dominated by strain S2–33 (Figure 6C, left). Interestingly, strain S2–33 also took over after antibiotic treatment in the cage of only S1 mice (Figure 6C, right), indicating the potential for transmission between cages during antibiotic treatment, presumably due to the opening of niches when the resident bacterial population is diminished. Sequencing of S2–33 clones on day 37 revealed a resistance mutation (D87G) in gyrA, which encodes a subunit of type II topoisomerase DNA gyrase. To our surprise, S2–33 clones isolated from as early as day 1 contained this same mutation, suggesting that this resistant strain had persisted at low frequencies in S2 mice since the beginning of the experiment. We also found that another barcode (S2–90) contained a missense mutation in gyrB (E466D) on day 1. No other mutations were identified in these clones. Relative to the parent, the IC50 to ciprofloxacin increased by 5- and 3.5-fold in the S2–33 and S2–90 isolate, respectively (Figure S6B). Metagenomic sequencing of the initial S2–33 and S2–90 stocks did not identify any mutations in gyrA or gyrB (or the rest of the genome).
As noted above, the initial barcode unevenness in mice 13 and 15 (Figure S2D) suggested the occurrence of a bottleneck during colonization. The observation that distinct gyrase-related mutations arose specifically in the two strains that were enriched during colonization suggested that the conditions that generated the strong bottleneck also selected for gyrase mutants. We tested whether the gyrase mutations conferred an advantage in acidic or high osmolarity environments, to mimic conditions a bacterium might experience during passage through the stomach and gut, but observed no significant advantages in growth or survival (Figure S6C–E). Thus, the nature of the bottleneck in these two mice remains unknown. The advantage experienced by the gyrase mutants was short-lived; after one week their relative abundance dropped below 1% and the diversity of barcodes detected was similar in all S2 mice after day 3 (Figure 6D), although the enrichment of S2–33 after ciprofloxacin treatment demonstrates its continued presence at low levels (Figure 2D, S2F). Thus, bottlenecks (likely created by host physiological differences) can select for gyrase mutations in the absence of ciprofloxacin that confer increased resistance to the drug, but microbiota composition quickly returns to the expected trajectory after the bottleneck.
In the ciprofloxacin experiments above, all mice were in an isolator with S2 mice (even if they were in a different cage) and hence were exposed to S2–33 (which harbored pre-existing gyrase mutations). Thus, we repeated ciprofloxacin treatment in the absence of S2 strains using eight S1 mice. We began treatment with 5 mg ciprofloxacin daily for 3 days (Figure 6G), and given the dramatic reduction in CFUs in our first experiment, we sought to determine whether bacteria were still present in the gut and/or cecum but were not being excreted in the feces. We sacrificed six of the eight mice on days 0, 1, 3 during ciprofloxacin treatment and harvested the large intestine and cecum. Upon plating, no colony growth was observed (data not shown).
For the remaining two mice (which were singly housed after day 3), we tracked CFUs/mL in the feces daily. In one mouse, no colonies were detected for 13 days after ciprofloxacin treatment (Figure 6G). To test whether there were reservoirs of viable E. coli within the gut that did not get incorporated into feces, we sacrificed the mouse on day 15 after treatment and ground up the entire intestine before plating. We did not detect any colonies (data not shown), indicating that these mice were likely returned to a germ-free state by ciprofloxacin treatment.
In the other mouse, recovery took substantially longer than in our previous experiment (Figure 6B), with CFUs/mL only increasing above 102 ten days after treatment ended (day 12) (Figure 6H). The population consisted of a single barcode (S1–34) after day 12 (Figure 6I), and this strain showed no prior sign of fitness advantage: levels were <0.01% prior to antibiotic treatment (Figure 6I). Whole-genome sequencing revealed a gyrAA119K mutation that was distinct from those of the S2 barcodes in the first experiment and conferred an even larger increase in IC50 (Figure S6F). The mutation was likely already present at very low levels at the onset of treatment, as it would have been >10,000 times more likely for a de novo mutation to emerge in the more abundant barcodes instead. The extended interval before recovery may be due to the lower population sizes of S1–34, or to the absence of coprophagic exchange with other hosts due to single housing that would reinforce recovery. Regardless, these data highlight the possibility for recovery even when poised at the boundary of extinction.
Discussion
Here, we showcased the power of DNA barcodes for tracking the evolution of nearly 200 E. coli strains during the colonization of a live mammalian host. In the future, introducing a larger number of barcoded bacterial strains into mice may permit quantification of the fitness of more barcoded strains (Levy et al., 2015) prior to population takeover. Nonetheless, the rapid dynamics that we observed in this study suggest that a large fraction of the barcodes may be driven extinct within a few weeks regardless of the initial library size.
Barcodes can be employed to study processes that are otherwise difficult to analyze due to the inability to distinguish phenotypically and/or genotypically identical populations. Here, we used barcoding to quantify the extent and effects of microbial transmission between mammalian hosts. Our findings suggest that coprophagy is a strong homogenizing force on the microbiota, such that the microbiota of mice in the same cage are highly non-independent, as has been shown in previous studies (Barroso-Batista et al., 2015; Lescat et al., 2017). It is likely that migration rates are lower in humans, given the lack of coprophagy; this study suggests that singly housed mice may be better than co-housed mice for modeling the human microbiota. Future studies of barcoding in other commensals, particularly obligate anaerobes, will be useful to reveal whether the same degree of homogenization occurs when survival in the environment is more challenging (due to the presence of oxygen) than for the facultative anaerobe E. coli. In the future, barcoding could also be used to shed light on microbial exchange with environmental reservoirs through spike-ins, and to determine whether mice engage in selective coprophagy (e.g. as a function of mouse genotype).
Our metagenomic analyses of >1,300 samples yielded complementary insights into evolutionary dynamics, providing mechanistic insights into why certain barcoded strains took over the community. In some cases, the same mutation emerged in multiple barcoded strains that continued to coexist (Figure 5D), suggesting either that the two strains are spatially segregated but subject to the same selective pressures or existence of a microecology involving the two strains. In addition, we sometimes observed competing lineages within the same barcode (clonal interference, Figure 2B), presumably due to rapid and continued evolution of strains throughout our experiments. Disentangling these intra-barcode dynamics was greatly assisted by sequencing of large numbers of isolates, a strategy that has also been exploited during in vitro studies (Li et al., 2019).
By coupling daily barcode tracking data and metagenomic sequencing, we discovered rapid and repeatable waves of selection of mutations involved in motility and metabolism (Figure 2A–D, 3A). Our NMR analysis of gut and cecal contents of germ-free mice on a standard diet revealed an abundance of the trisaccharide raffinose (Figure 3E), which the evolved S1–36 strain but not the parental strain had the ability to metabolize (Figure 3D). These findings are similar to a recent study demonstrating adaptations that increase E. coli’s ability to compete for amino acids during monocolonization (Barroso-Batista et al., 2020). During colonization of pregnant mice and their offspring, a human gut commensal E. coli acquired mutations in the lac operon (particularly lacI) that deactivate repression (Ghalayini et al., 2019), and mutations in maltose metabolism were identified during colonization of germ-free mice by E. coli (De Paepe et al., 2011).Together, these studies show that nutritional competition is a major selective pressure in intra-species interactions driving E. coli evolution in the mouse gut, and suggest (perhaps unsurprisingly) that diet is a strong selective force. If so, identification of available metabolic niches may be a good predictor of evolutionary dynamics; this idea is consistent with previous findings that porphyran was sufficient to bias the microbiota toward a Bacteroides species with exclusive access (Shepherd et al., 2018). The lacI* mutation was selected for in a previous long-term evolution experiment involving in vitro passaging in minimal medium with glucose/lactose (Quan et al., 2012); curiously, our measurements indicate that lacI* confers higher fitness on lactitol and lactulose but not lactose (Figure 3D). Previous efforts successfully selected for raffinose utilization through UV mutagenesis, and determined that expression of the alpha-galactosidase necessary for raffinose utilization requires constitutive beta-galactosidase expression (Lester and Bonner, 1957). We were unable to spontaneously evolve raffinose utilization in vitro after a week of passaging, either on plates or in liquid culture, perhaps due to the rare occurrence of the 4-bp insertion. The repeated emergence of the lacI* mutation in vivo may be due to larger population sizes in vivo than in vitro, a larger selective advantage, fewer tradeoffs, and/or a higher mutation rate in vivo compared with in vitro. Regardless, our findings indicate that the presence of an unoccupied niche such as raffinose utilization can serve as a strong evolutionary driver, motivating future studies of barcoded library colonization of mice (or humans) fed various diets to identify nutrient-dependent changes to the trajectory and rate of evolution.
The high sensitivity of E. coli to ciprofloxacin meant that treatment eliminated almost all cells (Figure 6B,H), leading to selection of a single barcode after a surprisingly long recovery time (Figure 6C,I). These findings indicate that even if certain species appear to recover from antibiotics, intrastrain-level heterogeneity that is important for host and microbiota health may be eliminated during treatment, highlighting yet another negative consequence of antibiotics. Notably, when mice were singly housed, the recovery period was extended relative to co-housed mice (Figure 6H), with one mouse seemingly reverting to a germ-free state. This recovery delay may be due to the lack of microbiota reinforcement from other mice; these findings are reminiscent of recent work showing that single housing leads to heterogeneous antibiotic recovery trajectories (Ng et al., 2019; Oliveira et al., 2020). In the future, the magnitude of disruption necessary to cause a decrease in intrastrain diversity due to a perturbation can be quantified via the extinction of barcoded strains.
In this study, we chose a laboratory strain of E. coli to accelerate evolution, presuming that MG1655 has evolved away from the host environment due to its longstanding use as a laboratory model organism. Given the rapid pace of E. coli evolution in vivo, do other commensals exhibit evolutionary dynamics on a similar timescale, or do these “wild” species already carry high fitness adaptations? Notably, colonization of germ-free mice with a Bacteroides thetaiotaomicron transposon library revealed metabolic mutations that increase fitness in vivo (Goodman et al., 2009; Liu et al., 2021), suggesting the existence of evolutionary potential. However, despite the vast array of monocolonization experiments that have been carried out with various gut commensals, there is very little data on the frequency of adaptive mutations in these species. A second aspect that may affect the pace of evolution is the presence of other species; for example, a recent study found that bicolonization of mice with E. coli and Blautia coccoides altered the gut metabolome and the evolutionary trajectory (Barroso-Batista et al., 2020). Our results suggest that another scenario that would likely alter the evolutionary trajectory of E. coli is if another species such as Bacteroides ovatus (Gherardini et al., 1985) can fill the raffinose-utilization niche. Regardless, the prevalence of metabolic mutations suggests that identification of genetic variation within an individual’s microbiota could provide key information for personalized dietary recommendations.
Overall, our study demonstrates that barcoded libraries will be a powerful resource for future interrogation of microbiota function; importantly, barcodes provide a high-dimensional window into each species’s evolution, without any obvious phenotypic cost. The continued development of genetic tools for gut commensals (Dodd et al., 2017; Guo et al., 2019; Shiver et al., 2019) should drive the creation of phylogenetically diverse barcoded libraries. Important insight can arise from coupling barcode and metagenomic dynamics to metabolomics to better understand metabolic selective pressures; given the high degree of repeatability in our experiments, the ability to tune the metabolic environment may allow engineering of a particular evolutionary trajectory. More generally, using barcodes to determine and quantify selective forces will reveal important insights into adaptation to the gut environment.
STAR METHODS
Resource Availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Kerwyn Casey Huang (kchuang@stanford.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request. All original code is available at https://bitbucket.org/kchuanglab/bacterial_transmission_model/src/master/. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental Model and Subject Details
Mice
All mouse experiments were conducted in accordance with the Administrative Panel on Laboratory Animal Care, Stanford University’s IACUC. Experiments involved female Swiss-Webster mice 6–12 weeks of age. For all experiments, mice were co-housed unless stated otherwise. For monocolonization experiments, mice were gavaged with 108 cells of an equal mixture of E. coli cells and fed normal mouse chow (LabDiet 5010) ad libitum throughout the experiment. To evaluate the necessity of antibiotic selection for plasmid maintenance, after day 7, drinking water was supplemented with 0.2 mg/mL ampicillin; ampicillin was given only after day 7 to avoid possible disruption of the initial colonization dynamics. No substantial changes in CFUs/mL were observed once ampicillin was added. Mice were euthanized with CO2 and death was confirmed via cervical dislocation.
Escherichia coli strains and culture
Escherichia coli MG1655 wild-type, mutant, and evolved strains were grown in LB broth and incubated aerobically, shaking orbitally (225 rpm) at 37 °C.
Barcode library creation
Barcoded plasmids were generated in (Cira et al., 2018). To generate the barcoded library in E. coli MG1655, plasmid DNA was extracted from TOP10 E. coli cells (ThermoFisher) with a miniprep kit according to the manufacturer’s instructions (Macherey-Nagel). Plasmids were transformed into chemically competent E. coli MG1655 cells via heat shock and plated on LB agar plates containing chloramphenicol (35 μg/mL) and ampicillin (100 μg/mL). Colonies were picked, grown separately in LB broth with chloramphenicol (35 μg/mL) and ampicillin (100 μg/mL), and banked as glycerol freezer stocks at −80 °C.
lacI* mutant creation
E. coli MG1655 cultures were grown from a glycerol stock for 12 h in LB at 30 °C. The next day, cultures were diluted 1:100 and grown at 32 °C to a final OD600 of 0.4–0.6. Cultures were incubated at 42 °C for 15 min and then placed in an ice slurry for 5–10 min. Cultures were washed in ice-cold water three times in decreasing volumes of 50 mL, 800 µL, and 200 µL. Fifty microliters of competent cells were mixed with 2 µL of 100 µM oligo (C*C*A* T*TA AGT TCT GTC TCG GCG CGT CTG CGT CTG GCT GGC TGG CTG GCA TAA ATA TCT CAC TCG CAA TCA AAT TCA GCC GAT AGC GGA ACG, where * indicates a phosphorothioated DNA base) and electroporated at 1.8 kV. Finally, cells were rescued in fresh LB for 2 h at 37 °C and plated on LB agar. Mutants were identified via growth on M9+0.2% raffinose and confirmed through Sanger sequencing of the lacI gene using primers 5’-TGG CTG GCT GGC TGG CAT AAA T (forward) and 5’-CGC AGC CCG GAC TCG GTA AT (reverse).
Measurements of population growth
Cultures were grown from glycerol stock for 12 h in LB with chloramphenicol (35 μg/mL) and ampicillin (100 μg/mL) at 37 °C with constant shaking. Then, cultures were diluted to 5×105 cells/mL and 200 µL of the dilutions were transferred to clear-bottom transparent 96-well plates. For measurements of antibiotic IC50, cells were diluted into fresh LB medium with 3–300 ng/mL ciprofloxacin. Plates were sealed with transparent film pierced with a laser cutter to create ~0.5-mm holes that allowed aeration in each well. Absorbance was measured at 600 nm in an Epoch2 plate reader (BioTek Instruments). Plates were shaken between readings with linear and orbital modes for 145 s each. Growth rates and lag times were quantified using custom code in MATLAB (Mathworks) that can be found at https://github.com/sv1714/Plate_reader_analysis.
pH and osmolarity measurements
S2–33 and S2–90 strains isolated from days 1 and 4, respectively, along with the parental strain were grown in LB and then resuspended in minimal medium without a carbon source at a range of pH values <7. Colony counting indicated that viability after 9 h was similar to the parent across the entire pH range (Figure S7C). These strains were also grown in LB supplemented with a range of concentrations of the osmolyte sucrose. Again, we observed no advantage for the mutants (Figure S7D); in unsupplemented LB, the lag time of the mutants during aerobic growth was actually longer than that of wild type (Figure S7E).
Plasmid curing of evolved strain
A gentamicin plasmid with the same origin of replication as the barcoded plasmid was used to transform an E. coli S1–36 isolate from a mouse from day 37 of our first experiment. Cells were plated on LB agar containing 200 μg/mL gentamicin. Colonies were picked, grown separately in LB broth with 200 μg/mL gentamicin, and replated on LB agar plates containing 35 μg/mL chloramphenicol to identify colonies that had lost the barcoded plasmid. Chloramphenicol-sensitive clones were confirmed to lack the barcoded plasmid through Sanger sequencing. One of these clones was transformed with set 2 barcoded plasmids as described above, and recovered on LB agar plates containing 35 μg/mL chloramphenicol. 86 of the 96 barcoded plasmids were successfully used for transformation, creating the S2* strains. Loss of the gentamicin plasmid was achieved by growing the strains in LB with 200 µg/mL gentamicin, and Sanger sequencing confirmed gentamicin plasmid loss.
DNA extraction and sequencing
DNA was extracted from whole fecal pellets with PowerSoil and PowerSoil-htp kits (Qiagen). For barcode sequencing, extracted DNA was amplified using a two-stage platinum Pfx DNA polymerase PCR (Thermo). In a one-cycle PCR, adapter regions and unique molecular identifiers (barcodes) were added to each template. This step generates one uniquely labeled functional template per initial template plasmid molecule. The initial primers were then removed using Ampure XP Beads at a ratio of 1:1 (Beckman Coulter). A second PCR was run for 35 cycles to amplify these labeled templates. During this reaction, known DNA indices were attached to the product to allow informatic demultiplexing of pooled libraries.
First PCR: 94 °C for 2 min, 2X (94 °C for 15 s, 53 °C for 30 s, 68 °C for 30 s), 68 °C for 5 min, hold at 4 °C.
Primers #1:
5’- ACACTCTTTCCCTACACGACGCTCTTCCGATCTNNNNNNNNTCGCTAAGGATG ATTTCTGGA-3’
5’- GACTGGAGTTCAGACGTGTGCTCTTCCGATCTNNNNNNNNNNNNNNNNTCGC TTGGACTCCTGTTGAT-3’
NNN = unique molecular identifier (barcode)
Second PCR: 94 °C for 2 min, 35X (94 °C for 15 s, 72 °C for 30 s, 68 °C for 30 s), 68°C for 5 min, hold at 4 °C.
Primers #2:
5’- AATGATACGGCGACCACCGAGATCTACACnnnnnnAAACACTCTTTCCCTACACG ACGCTCTTCCGATCT-3’
5’- CAAGCAGAAGACGGCATACGAGATAAnnnnnnGTGACTGGAGTTCAGACGTGTG CTCTTCCGATCT-3’
nnnnnn = multiplexing indices
Successful reactions were confirmed via agarose gel electrophoresis and pooled in equal abundance. The sequencing library was then finalized via purification with Ampure XP Beads at a ratio of 1:1. Sequencing was performed on an Illumina MiSeq with read length 2×75 bp and an average of 100,000 reads per sample.
For metagenomic and whole-genome sequencing, extracted DNA was arrayed into 384-well plates and concentrations were quantified and normalized using the PicoGreen dsDNA quantitation kit (ThermoFisher). DNA was added to a tagmentation reaction, incubated for 10 min at 55 °C, and immediately neutralized. Mixtures were then added to 10 cycles of a PCR that appended Illumina primers and identification barcodes to allow for mixing of samples during sequencing. Wells were mixed, using 1 µL per well, and the pooled library was purified twice using Ampure XP beads to select the appropriately sized bands. Finally, library concentration was quantified using a Qubit (Thermo Fisher). Sequencing was performed on a NovaSeq S4 with read lengths of 2×146 bp.
Sequence analysis
Barcode sequences were analyzed with SeqPrep v.1.3.2 (https://github.com/jstjohn/SeqPrep) and custom MATLAB scripts, and plotted using Tableau Desktop v. 10.4. Genome sequences were analyzed using a previously described pipeline (Good et al., 2017).
Some barcode sequences that were not included in some samples nevertheless had very small numbers of reads assigned to them, and applying more stringent thresholds by counting only exact matches to species barcodes did not remedy this apparent noise, so it is unlikely that the noise was due to sequencing errors that resulted in misclassification of one species barcode as another. Instead, this noise likely arose from index hopping that occurs when multiplexing samples on Illumina sequencers. Thus, we acknowledged a noise floor below which reads could not be reliably detected. Based on previous work with these barcodes (Cira et al., 2018), we applied a 0.1% threshold for barcode abundances. While this threshold is conservative, it ensures that detected barcodes were actually present in the experiment.
Quantification of bacterial densities
Bacterial densities were quantified by spot plating in duplicate on plates with LB only and with LB supplemented with ampicillin (100 μg/mL) and chloramphenicol (35 μg/mL). Plates were incubated in a 37 °C warm room.
Preparation of mouse fecal pellets for metabolomics
Cecal and fecal contents of germ-free mice were collected and quickly stored at −80 °C. We performed 1H-NMR on aqueous extracts of cecal or fecal contents as specified. Cecal contents of germ-free mice were diluted 50% (w/v) in deuterated water (D2O, Sigma-Aldrich). Fecal samples of germ-free mice were diluted in 1 mL of D2O. For all samples, extraction was performed as follows: ~0.3 g of 0.1-mm glass beads (Scientific Industries, SI-BG01) were added to each tube and samples were bead-beaten using a QIAGEN Tissuelyser II (Retsch) for 2 min with 30 rev/s pulses. Large debris and the glass beads were pelleted by centrifugation at 14,000 rpm for 30 min at 4 °C. Supernatant was collected and filtered through a 0.22-μm filter (EMD Millipore), followed by another filtration with a 3 KDa filter (Vivaspin 500) using centrifugation at 15000g and 4 °C for 3 h or until 150 μL of filtrate was obtained. Samples were stored at −80 °C until spectra were acquired. For acquisition, samples (150 μL of filtrate) were thawed at room temperature for 10 to 15 min and then mixed with 60 μL of 350 mM phosphate buffer (pH 7.09) with 2% NaN3, 10 μL of a 0.05% (w/v) 3-(Trimethylsilyl)propionic-2,2,3,3-d4 (TSP-d4, Sigma-Aldrich) solution, and 380 μL of D2O (for a total volume of 600 μL). This mixture was transferred to a 5-mm glass NMR tube. All solutions were prepared in D2O. Samples were homogenized by inversion and spectra were acquired after pH measurement. Acquisitions were performed on a Bruker AVANCE II+ 500 MHz instrument equipped with Cryo TCI (F) (Prodigy) 5-mm probehead with z-gradients. 1H-NMR spectra were acquired using a 1D NOESY pulse sequence with pre-saturation (noesypr1d) under the following conditions: 90 degrees pulse for excitation with mixing time 100 ms, acquisition time 4 s, and relaxation delay 1 s. All spectra were acquired with 200 scans at 25 °C, with 48,000 data points and 6002 Hz (12 ppm) spectral width (Chenomx acquisition parameters). The recorded 1H-NMR spectra were phase-corrected using Bruker TopSpin v. 3.2 and spectra were then processed using Chenomx NMR Suite v. 8.1. Compounds were identified by manually fitting reference peaks to spectra in the database Chenomx 500 MHz v. 10. Quantification was based on internal standard peak integration (TSP-d4).
PTR (peak-to-trough ratio) analysis
Metagenomic sequencing data were used to compute PTR. In brief, DNA sequencing reads were mapped to complete genome sequences and the differences in sequencing coverage at the origin and terminus of replication were quantified. The sequencing data, along with the NCBI E. coli MG1655 sequence were used as input to the bPTR algorithm (https://github.com/christophertbrown/iRep) with default settings.
Quantification and statistical analyses
Data were plotted using Matlab and Python. In figures with error bars, the central point represents the mean and the top and bottom edges indicate one standard error of the mean or one standard deviation, as indicated.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Escherichia coli strain MG1655 | Huang lab strain collection | N/A |
| Evolved S1-36 | This paper | N/A |
| lacI* mut | This paper | N/A |
| [flhE-flhD] mutant | This paper | N/A |
| gyrBD87G mutant | This paper | N/A |
| gyrAE466D mutant | This paper | N/A |
| Critical commercial assays | ||
| Biolog PM1 and PM2A microplates | Biolog Inc. | Cat. #12111,12112 |
| PicoGreen dsDNA quantification kit | Thermo Fisher | Cat. #P7589 |
| MiniPrep kit | Macherey-Nagel | Cat. #740588.50 |
| Oligonucleotides | ||
| lacI mutant oligo | C*C*A* T*TA AGT TCT GTC TCG GCG CGT CTG CGT CTG GCT GGC TGG CTG GCA TAA ATA TCT CAC TCG CAA TCA AAT TCA GCC GAT AGC GGA ACG, (where * indicates a phosphorothioated base) | N/A |
| lacI Sanger sequencing forward primer | Fwd: 5’-TGGCTGGCTGGCTGGCATAAAT | N/A |
| lacI Sanger sequencing reverse primer | Rev: 5’-CGCAGCCCGGACTCGGTAAT | N/A |
| Barcode sequencing forward primer #1 | Fwd: 5’-ACACTCTTTCCCTACACGACGCTCTTCCGATCTNNNNNNNNTCGCTAAGGATGATTTCTGGA NNN = unique molecular identifier |
N/A |
| Barcode sequencing reverse primer #1 | Rev: 5’-GACTGGAGTTCAGACGTGTGCTCTTCCGATCTNNNNNNNNNNNNNNNNTCGCTTGGACTCCTGTTGAT NNN = unique molecular identifier |
N/A |
| Barcode sequencing forward primer #2 | Fwd: 5’-AATGATACGGCGACCACCGAGATCTACACnnnnnnAAACACTCTTTCCCTACACGACGCTCTTCCGATCT nnnnnn = multiplexing indices |
N/A |
| Barcode sequencing reverse primer #2 | Rev: 5’-CAAGCAGAAGACGGCATACGAGATAAnnnnnnGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT nnnnnn = multiplexing indices |
N/A |
| Experimental models: Organisms/strains | ||
| Swiss Webster Mice – Germ Free | Taconic | SW GF |
| Recombinant DNA | ||
| DNA barcode plasmids | (Cira et al., 2018) | N/A |
| Gentamicin cassette containing plasmid (pUC18-mini-Tn7T-Gm-lux) | (Choi et al., 2005) | Addgene Ref. 64963 |
| Software and algorithms | ||
| MATLAB | Mathworks | v. 2017a |
| SeqPrep | https://github.com/jstjohn/SeqPrep | v. 1.2 |
| Tableau Desktop | Tableau | v. 2018.3 |
| Breseq | https://github.com/barricklab/breseq | v. 0.33.1 |
| PTR | https://github.com/christophertbrown/iRep | v. 1.10 |
| Transmission model code | https://bitbucket.org/kchuanglab/bacterial_transmission_model/src/master/ | N/A |
| Growth rate and lag time analysis code | https://github.com/sv1714/Plate_reader_analysis | N/A |
| Other | ||
| PowerSoil-htp kit | Qiagen | Ref. 12955-4 |
| Platinum Pfx DNA polymerase | Thermo Fisher | Ref. 11708-021 |
| Ampure XP Beads | Beckman Coulter | Ref. A63881 |
| MiSeq | Illumina | N/A |
| NovaSeq S4 | Illumina | N/A |
Highlights.
DNA barcodes allow tracking of bacterial strain-level dynamics in the mouse gut
Mutations in motility and metabolic genes are reproducibly selected for within days
A population-genetics model predicts a bacterial migration rate of >10% per day
Ciprofloxacin treatment decreases strain diversity and stimulates transmission
Acknowledgments
The authors thank the Huang and Sonnenburg labs for helpful discussions and Jason Peters for providing the pUC18 plasmid. The authors acknowledge support from the Allen Discovery Center at Stanford on Systems Modeling of Infection (to K.C.H.), NIH RM1 Award GM135102 (to K.C.H.), and a Friedrich Wilhelm Bessel Award from the Humboldt Foundation (to K.C.H.). J.L.S. and K.C.H. are Chan Zuckerberg Biohub Investigators. This work was also supported in part by the National Science Foundation under Grant PHYS-1066293 and the hospitality of the Aspen Center for Physics. NMR data were acquired at CERMAX, ITQB-NOVA, Oeiras, Portugal with equipment funded by FCT, project AAC 01/SAICT/2016.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Barroso-Batista J, Demengeot J, and Gordo I (2015). Adaptive immunity increases the pace and predictability of evolutionary change in commensal gut bacteria. Nat Commun 6, 8945. 10.1038/ncomms9945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barroso-Batista J, Pedro MF, Sales-Dias J, Pinto CJG, Thompson JA, Pereira H, Demengeot J, Gordo I, and Xavier KB (2020). Specific Eco-evolutionary Contexts in the Mouse Gut Reveal Escherichia coli Metabolic Versatility. Curr Biol 30, 1049–1062 e1047. 10.1016/j.cub.2020.01.050. [DOI] [PubMed] [Google Scholar]
- Barroso-Batista J, Sousa A, Lourenco M, Bergman ML, Sobral D, Demengeot J, Xavier KB, and Gordo I (2014). The first steps of adaptation of Escherichia coli to the gut are dominated by soft sweeps. PLoS Genet 10, e1004182. 10.1371/journal.pgen.1004182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CT, Olm MR, Thomas BC, and Banfield JF (2016). Measurement of bacterial replication rates in microbial communities. Nat Biotechnol 34, 1256–1263. 10.1038/nbt.3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cira NJ, Pearce MT, and Quake SR (2018). Neutral and selective dynamics in a synthetic microbial community. Proc Natl Acad Sci U S A 115, E9842–E9848. 10.1073/pnas.1808118115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Filippis F, Pasolli E, Tett A, Tarallo S, Naccarati A, De Angelis M, Neviani E, Cocolin L, Gobbetti M, Segata N, and Ercolini D (2019). Distinct Genetic and Functional Traits of Human Intestinal Prevotella copri Strains Are Associated with Different Habitual Diets. Cell Host Microbe 25, 444–453 e443. 10.1016/j.chom.2019.01.004. [DOI] [PubMed] [Google Scholar]
- De Paepe M, Gaboriau-Routhiau V, Rainteau D, Rakotobe S, Taddei F, and Cerf-Bensussan N (2011). Trade-off between bile resistance and nutritional competence drives Escherichia coli diversification in the mouse gut. PLoS Genet 7, e1002107. 10.1371/journal.pgen.1002107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd D, Spitzer MH, Van Treuren W, Merrill BD, Hryckowian AJ, Higginbottom SK, Le A, Cowan TM, Nolan GP, Fischbach MA, and Sonnenburg JL (2017). A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature 551, 648–652. 10.1038/nature24661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehlner-Peach H, Magnabosco C, Raghavan V, Scher JU, Tett A, Cox LM, Gottsegen C, Watters A, Wiltshire-Gordon JD, Segata N, et al. (2019). Distinct Polysaccharide Utilization Profiles of Human Intestinal Prevotella copri Isolates. Cell Host Microbe 26, 680–690 e685. 10.1016/j.chom.2019.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garud NR, Good BH, Hallatschek O, and Pollard KS (2019). Evolutionary dynamics of bacteria in the gut microbiome within and across hosts. PLoS Biol 17, e3000102. 10.1371/journal.pbio.3000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghalayini M, Magnan M, Dion S, Zatout O, Bourguignon L, Tenaillon O, and Lescat M (2019). Long-term evolution of the natural isolate of Escherichia coli 536 in the mouse gut colonized after maternal transmission reveals convergence in the constitutive expression of the lactose operon. Mol Ecol 28, 4470–4485. 10.1111/mec.15232. [DOI] [PubMed] [Google Scholar]
- Gherardini F, Babcock M, and Salyers AA (1985). Purification and characterization of two alpha-galactosidases associated with catabolism of guar gum and other alpha-galactosides by Bacteroides ovatus. J Bacteriol 161, 500–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good BH, McDonald MJ, Barrick JE, Lenski RE, and Desai MM (2017). The dynamics of molecular evolution over 60,000 generations. Nature 551, 45–50. 10.1038/nature24287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman AL, McNulty NP, Zhao Y, Leip D, Mitra RD, Lozupone CA, Knight R, and Gordon JI (2009). Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe 6, 279–289. 10.1016/j.chom.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosskopf T, Consuegra J, Gaffe J, Willison JC, Lenski RE, Soyer OS, and Schneider D (2016). Metabolic modelling in a dynamic evolutionary framework predicts adaptive diversification of bacteria in a long-term evolution experiment. BMC Evol Biol 16, 163. 10.1186/s12862-016-0733-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo CJ, Allen BM, Hiam KJ, Dodd D, Van Treuren W, Higginbottom S, Nagashima K, Fischer CR, Sonnenburg JL, Spitzer MH, and Fischbach MA (2019). Depletion of microbiome-derived molecules in the host using Clostridium genetics. Science 366. 10.1126/science.aav1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korem T, Zeevi D, Suez J, Weinberger A, Avnit-Sagi T, Pompan-Lotan M, Matot E, Jona G, Harmelin A, Cohen N, et al. (2015). Growth dynamics of gut microbiota in health and disease inferred from single metagenomic samples. Science 349, 1101–1106. 10.1126/science.aac4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam LH, and Monack DM (2014). Intraspecies competition for niches in the distal gut dictate transmission during persistent Salmonella infection. PLoS Pathog 10, e1004527. 10.1371/journal.ppat.1004527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latter B (1973). The island model of population differentiation: a general solution. Genetics 73, 147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lescat M, Launay A, Ghalayini M, Magnan M, Glodt J, Pintard C, Dion S, Denamur E, and Tenaillon O (2017). Using long-term experimental evolution to uncover the patterns and determinants of molecular evolution of an Escherichia coli natural isolate in the streptomycin-treated mouse gut. Mol Ecol 26, 1802–1817. 10.1111/mec.13851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester G, and Bonner DM (1957). Genetic control of raffinose utilization in Escherichia coli. J Bacteriol 73, 544–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy SF, Blundell JR, Venkataram S, Petrov DA, Fisher DS, and Sherlock G (2015). Quantitative evolutionary dynamics using high-resolution lineage tracking. Nature 519, 181–186. 10.1038/nature14279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Petrov DA, and Sherlock G (2019). Single nucleotide mapping of trait space reveals Pareto fronts that constrain adaptation. Nat Ecol Evol 3, 1539–1551. 10.1038/s41559-019-0993-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvak Y, and Baumler AJ (2019). The founder hypothesis: A basis for microbiota resistance, diversity in taxa carriage, and colonization resistance against pathogens. PLoS Pathog 15, e1007563. 10.1371/journal.ppat.1007563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Shiver AL, Price MN, Carlson HK, Trotter VV, Chen Y, Escalante V, Ray J, Hern KE, Petzold CJ, et al. (2021). Functional genetics of human gut commensal Bacteroides thetaiotaomicron reveals metabolic requirements for growth across environments. Cell Rep 34, 108789. 10.1016/j.celrep.2021.108789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller AH, Caro-Quintero A, Mjungu D, Georgiev AV, Lonsdorf EV, Muller MN, Pusey AE, Peeters M, Hahn BH, and Ochman H (2016). Cospeciation of gut microbiota with hominids. Science 353, 380–382. 10.1126/science.aaf3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng KM, Aranda-Diaz A, Tropini C, Frankel MR, Van Treuren WW, O’Laughlin C, Merrill BD, Yu FB, Pruss KM, Oliveira RA, et al. (2019). Recovery of the gut microbiota after antibiotics depends on host diet and environmental reservoirs. Cell Host Microbe [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira RA, Ng KM, Correia MB, Cabral V, Shi H, Sonnenburg JL, Huang KC, and Xavier KB (2020). Klebsiella michiganensis transmission enhances resistance to Enterobacteriaceae gut invasion by nutrition competition. Nat Microbiol 5, 630–641. 10.1038/s41564-019-0658-4. [DOI] [PubMed] [Google Scholar]
- Perry RD, and Fetherston JD (1997). Yersinia pestis--etiologic agent of plague. Clin Microbiol Rev 10, 35–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe N, Pelosi L, Lenski RE, and Schneider D (2009). Evolution of penicillin-binding protein 2 concentration and cell shape during a long-term experiment with Escherichia coli. J Bacteriol 191, 909–921. 10.1128/JB.01419-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickard JM, Zeng MY, Caruso R, and Nunez G (2017). Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol Rev 279, 70–89. 10.1111/imr.12567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poyet M, Groussin M, Gibbons SM, Avila-Pacheco J, Jiang X, Kearney SM, Perrotta AR, Berdy B, Zhao S, Lieberman TD, et al. (2019). A library of human gut bacterial isolates paired with longitudinal multiomics data enables mechanistic microbiome research. Nat Med 25, 1442–1452. 10.1038/s41591-019-0559-3. [DOI] [PubMed] [Google Scholar]
- Quan S, Ray JC, Kwota Z, Duong T, Balazsi G, Cooper TF, and Monds RD (2012). Adaptive evolution of the lactose utilization network in experimentally evolved populations of Escherichia coli. PLoS Genet 8, e1002444. 10.1371/journal.pgen.1002444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragon M, Wirth T, Hollandt F, Lavenir R, Lecuit M, Le Monnier A, and Brisse S (2008). A new perspective on Listeria monocytogenes evolution. PLoS Pathog 4, e1000146. 10.1371/journal.ppat.1000146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rang CU, Licht TR, Midtvedt T, Conway PL, Chao L, Krogfelt KA, Cohen PS, and Molin S (1999). Estimation of growth rates of Escherichia coli BJ4 in streptomycin-treated and previously germfree mice by in situ rRNA hybridization. Clin Diagn Lab Immunol 6, 434–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd ES, DeLoache WC, Pruss KM, Whitaker WR, and Sonnenburg JL (2018). An exclusive metabolic niche enables strain engraftment in the gut microbiota. Nature 557, 434–438. 10.1038/s41586-018-0092-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiver AL, Culver R, Deutschbauer AM, and Huang KC (2019). Rapid ordering of barcoded transposon insertion libraries of anaerobic bacteria. BioRxiv, 780593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropini C, Moss EL, Merrill BD, Ng KM, Higginbottom SK, Casavant EP, Gonzalez CG, Fremin B, Bouley DM, Elias JE, et al. (2018). Transient Osmotic Perturbation Causes Long-Term Alteration to the Gut Microbiota. Cell 173, 1742–1754 e1717. 10.1016/j.cell.2018.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong DT, Tett A, Pasolli E, Huttenhower C, and Segata N (2017). Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res 27, 626–638. 10.1101/gr.216242.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataram S, Dunn B, Li Y, Agarwala A, Chang J, Ebel ER, Geiler-Samerotte K, Herissant L, Blundell JR, Levy SF, et al. (2016). Development of a Comprehensive Genotype-to-Fitness Map of Adaptation-Driving Mutations in Yeast. Cell 166, 1585–1596 e1522. 10.1016/j.cell.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visconti A, Le Roy CI, Rosa F, Rossi N, Martin TC, Mohney RP, Li W, de Rinaldis E, Bell JT, Venter JC, et al. (2019). Interplay between the human gut microbiome and host metabolism. Nat Commun 10, 4505. 10.1038/s41467-019-12476-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiser MJ, Ribeck N, and Lenski RE (2013). Long-term dynamics of adaptation in asexual populations. Science 342, 1364–1367. 10.1126/science.1243357. [DOI] [PubMed] [Google Scholar]
- Wright S (1943). Isolation by Distance. Genetics 28, 114–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request. All original code is available at https://bitbucket.org/kchuanglab/bacterial_transmission_model/src/master/. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.