

Abstract
Background:
Cebranopadol, a mixed nociceptin/opioid receptor full agonist, can effectively relieve pain in rodents and humans. However, it is unclear to what degree different opioid receptor subtypes contribute to its antinociception and whether cebranopadol lacks acute opioid-associated side effects in primates. We hypothesized that coactivation of nociceptin receptors and mu receptors produces analgesia with reduced side effects in non-human primates.
Methods:
The antinociceptive, reinforcing, respiratory depressant, and pruritic effects of cebranopadol in adult rhesus monkeys (n = 22) were compared with mu receptor agonists fentanyl and morphine using assays, including acute thermal nociception, intravenous drug self-administration, telemetric measurement of respiratory function, and itch scratching responses.
Results:
Subcutaneous cebranopadol (ED50 [95% CI]: 2.9 [1.8–4.6] μg/kg) potently produced antinociception compared to fentanyl (15.8 [14.6–17.1] μg/kg). Pretreatment with antagonists selective for nociceptin and mu receptors, but not delta and kappa receptor antagonists, caused rightward shifts of the antinociceptive dose-response curve of cebranopadol with dose ratios of 2 and 9, respectively. Cebranopadol produced fentanyl-comparable reinforcing effects, but with decreased reinforcing strength, i.e., cebranopadol (mean ± SD: 7 ± 3 injections) versus fentanyl (12 ± 3 injections) determined by a progressive ratio schedule of reinforcement. Unlike fentanyl (8 ± 2 breaths/minute), systemic cebranopadol at higher doses did not decrease the respiratory rate (17 ± 2 breaths/minute). Intrathecal cebranopadol (1 μg) exerted full antinociception with minimal scratching responses (231 ± 137 scratches) in contrast to intrathecal morphine (30 μg; 3,009 ± 1,474 scratches).
Conclusions:
In non-human primates, the mu receptor mainly contributed to cebranopadol-induced antinociception. Similar to nociceptin/mu receptor partial agonists, cebranopadol displayed reduced side effects, such as a lack of respiratory depression and pruritus. Although cebranopadol showed reduced reinforcing strength, its detectable reinforcing effects and strength warrant caution, which is critical for the development and clinical use of cebranopadol.
Introduction
Mu receptor agonists are the most widely used analgesics in clinics.1 However, the side effects associated with these drugs, including abuse liability, respiratory depression, constipation, and itch (pruritus), have resulted in a clear need for safe yet efficacious analgesics with better side effect profiles.2,3 Several scientific approaches have been proposed to ameliorate mu receptor-mediated side effects while preserving analgesic efficacy.4 Based on the pharmacological studies of the functional interactions between nociceptin receptors and mu receptors, the development of mixed nociceptin/mu receptor agonists is of particular interest.5 Mounting evidence strongly suggests that the coactivation of the nociceptin and mu receptors might provide synergistic analgesic effects and simultaneously counteract mu receptor-mediated side effects.6–10 Mixed nociceptin/mu receptor agonists are currently being pursued as promising novel analgesics.
Several mixed nociceptin/mu receptor agonists have been reported. BU08028 and BU10038 bind with reasonable affinity to all opioid receptor subtypes; however, both show only partial efficacy at nociceptin and mu receptors.8,11 Similarly, AT-121 displays a high affinity to nociceptin and mu receptors but only partial agonistic efficacy at both receptors.7 In preclinical pain models, these compounds showed potent antinociceptive effects with favorable side effect profiles, including reduced or lack of respiratory depression, reinforcing effects, physical dependence, and tolerance development.6–8 In comparison with these nociceptin/mu receptor partial agonists, cebranopadol stands out as a unique mixed nociceptin/opioid receptor agonist which displays full efficacy at mu, nociceptin and delta receptors, and partial efficacy at kappa receptors.12,13 The antinociceptive effects of cebranopadol have been demonstrated in various rodent pain models.14,15 This phenomenon has been translated to human clinical trials, showing promising efficacy in patient suffering from acute or chronic pain.14,15 However, the receptor components contributing to cebranopadol-induced antinociception in primates remain unknown. Given that nociceptin receptor activation counters mu receptor-mediated antinociception in rodents,5,10 it is worth investigating the antinociceptive effects of cebranopadol with receptor-selective antagonists in non-human primates.
In addition to evaluating the effectiveness of cebranopadol in relieving pain, the side effects typically associated with opioids have also been examined in rodents and humans.14,15 The absence of respiratory depression12,16 and low potential to produce physical dependence17,18 have been reported in rodent models and human clinical trials. However, there are equivocal reports of rewarding effects in the conditioned place preference paradigm in rodents.19,20 Given that abuse liability is one of the foremost drawbacks of opioid analgesics in clinical use and cebranopadol displays full efficacy at mu receptors, it is critical to evaluate the abuse potential of cebranopadol in non-human primate models with high translational relevance. IV drug self-administration in non-human primates is the gold standard for assessing the abuse potential of drugs.21,22 Data obtained from this experimental paradigm would be valuable for evaluating the abuse liability of cebranopadol. In addition, pruritus is a common side effect of spinal opioid analgesics that significantly compromises their pain relief value.23 Considering the full efficacy of cebranopadol at mu receptors, it is important to determine whether cebranopadol could elicit itch sensation.
Given the species differences in the functional and pharmacological profiles of nociceptin and mu receptor activation between rodents and non-human primates and the practicality of simulating the side effect profiles of mu receptor agonists in non-human primates,5,24,25 in the present study, we used non-human primate models to compare the functional profiles of cebranopadol with mu receptor agonists fentanyl and morphine in four aspects: 1) antinociceptive potency, 2) reinforcing effects and strength, 3) respiratory depressant effects, and 4) pruritic effects.
Materials and Methods
Animals
Adult rhesus monkeys (Macaca mulatta), 14 males and 8 females (n = 22, n refers to the number of animals), with a body weight of 6.4–12.1 kg and age of 10–18 years were used in the present study. The monkeys were housed at an indoor facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (Frederick, MD, USA), individually in cages with 6–12 square feet of floor space, with ceilings 2.7–5.4 feet high, in an environmentally controlled room (21–25°C, 40%–60% relative humidity) with a 12-h light/dark cycle (lights on: 6:30–18:30). The monkeys were provided with water and monkey chow (LabDiet St. Louis, MO, USA) and fresh fruit ad libitum. Primate enrichment devices and treats were provided daily. The animals were not subjected to any experiments or given opioid compounds for 1 month before the start of the study. The animals were assigned to each experiment based on the tasks they were trained to perform. All experiments followed a within-subject design (i.e., each group of animals served as its own control and all dosing conditions were randomized by a counterbalanced design). All experiments were conducted during the late mornings of weekdays until the time courses or testing sessions were completed. All animal care and experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals by the US National Institutes of Health (Bethesda, MD, USA) and were approved by the Institutional Animal Care and Use Committee of Wake Forest University (Winston-Salem, NC, USA). The present study was reported in accordance with the Animal Research: Reporting of In Vivo Experiments26 and designed in settings similar to those reported previously.27
Acute thermal nociception
Mu receptor agonists change nociceptive thresholds and produce antinociception in both non-human primates and humans. Warm water tail-withdrawal assays27,28 were conducted to examine the thermal antinociceptive effects of cebranopadol and fentanyl. Monkeys were seated in primate restraint chairs, and the lower parts of their shaved tails (~15 cm) were immersed in water maintained at 42°C, 46°C, or 50°C. Water at 42°C or 46°C was used as a non-noxious stimulus (i.e., no tail-withdrawal expected), and water at 50°C was used as an acute noxious stimulus (i.e., 2–3 s tail-withdrawal latency), but did not cause thermal injury. The primary outcome was tail-withdrawal latency. Monkeys were randomly assigned to the dosing condition. Experimenters unaware of the dosing conditions measured the tail-withdrawal latencies at each temperature randomly using a computerized timer. A maximum time of 20 s (the cutoff) was recorded if the monkey did not withdraw its tail within 20 s. The latencies were measured before and at multiple time points after subcutaneous or intrathecal administration of a single dose of the test compound. Tail-withdrawal latencies at 42°C and 46°C after exposure to 50°C water remained at 20 s. For dose-response curves, cebranopadol was subcutaneously administered by a cumulative dosing procedure with a 30 min inter-injection interval. Tail-withdrawal latencies were measured 20 min after each injection. To determine the involvement of the four opioid receptors in cebranopadol-induced antinociception, monkeys were given subcutaneously selective mu receptor antagonist naltrexone (0.03 mg/kg), selective nociceptin receptor antagonist J-113397 (0.1 mg/kg), or delta receptor antagonist naltrindole (1 mg/kg) 15 min before cebranopadol administration, whereas the kappa receptor antagonist 5′-guanidinonaltrindole (1 mg/kg) was given 24 h before cebranopadol administration. The doses and pretreatment time for these antagonists were chosen based on previous studies to show their selective receptor antagonism in rhesus macaques.29–32
Itch scratching responses
The scratching behavior31 of monkeys in their home cages was recorded to assess the itching sensation caused by the test compounds. Each 15-min recording session was conducted following subcutaneous or intrathecal administration of cebranopadol, fentanyl, or morphine. The primary outcome measure was the number of scratches. A scratch was defined as one brief (< 1 s) scraping on the skin surface of other body parts using the forepaw or hind paw. The total number of scratches was counted and summed for each 15-min period by experimenters blinded to the dosing conditions.
Drug self-administration
Six monkeys implanted with IV catheters were used in the drug self-administration procedure in the operant chamber under two different schedules of reinforcement. The primary outcome was the number of drug injections that animals received during the test session. The fixed ratio 30 schedule of reinforcement33,34 was used to determine whether cebranopadol had a reinforcing effect. The progressive ratio schedule of reinforcement was used to compare the reinforcing strengths of cebranopadol and fentanyl, which can differentiate reinforcing strengths of abused drugs that function as positive reinforcers.6,35,36 The ratio progression of the progressive ratio schedule was from 20 (1st injection), 25, 32, 40, 50, 62, 77, 95, 117, 144, 177, 218, 267, 328 to 402 (15th injection). The operant response was maintained at 3 μg/kg per injection of oxycodone until the response was stable (mean ± three injections for three consecutive sessions). Dose-effect curves were determined by substituting saline or various doses of cebranopadol (0.01‒0.06 μg/kg per injection) or fentanyl (0.03‒0.3 μg/kg per injection) for the maintenance dose in a random order under the fixed ratio 30 schedule. The dose range for the progressive ratio schedule was cebranopadol (0.03‒0.3 μg/kg per injection) and fentanyl (0.1‒0.6 μg/kg per injection). Doses were available for at least five consecutive sessions until the response was considered stable. On average, the animals were tested for four to five sessions. The blinding method was not used when collecting drug self-administration data, as these data were generated directly from the animals.
Respiratory responses
The acute effects of cebranopadol and fentanyl on respiratory function were evaluated in four freely moving monkeys implanted with the D70-PCTR telemetry transmitter6 (Data Sciences International, St. Paul, MN). Respiration data from 30 min before and 60 min after intramuscular administration of cebranopadol (0, 5.6, 10, and 18 μg/kg) or fentanyl (0, 30, and 56 μg/kg) were continuously collected and analyzed using Ponemah software version 5.2. The primary outcomes were respiration rate and minute volume. The mean value of each 5-min time block was generated from each animal to represent the measured outcome for each data point. The blinding method was not used when collecting telemetry data, as these data were generated directly from the telemetry device.
Surgical implantation
The surgical details regarding the implantation of telemetry devices and intrathecal catheterization have been reported previously.6,37 For preoperative care, before surgery, animals were administered atropine (0.04 mg/kg, intramuscular), buprenorphine (0.01–0.03 mg/kg, intramuscular), dexamethasone (2 mg/kg, IV), and cefotaxime (500 mg, IV) for pain relief and infection prevention. Animals were then anesthetized with ketamine (10 mg/kg, intramuscular) and intubated, and anesthesia was maintained via isoflurane inhalation (1%–2% in 1 L/min O2). Intraoperative monitoring was conducted to determine the depth of anesthesia and physiological status. Monkeys were administered postoperative buprenorphine (0.003–0.02 mg/kg, intramuscular) and meloxicam (0.15 mg/kg, subcutaneous) to alleviate pain and inflammation, and ceftiofur (2.2 mg/kg, intramuscular) to prevent infection. Postoperative care was performed daily until the veterinarians confirmed that the healing was complete. All animals were monitored daily by veterinarians and laboratory staff to ensure that they remained healthy throughout the study.
For intrathecal catheterization, hemilaminectomy was performed in the lateral aspect of the L4 or 5 vertebral body to expose the dura mater. The intrathecal catheter (3.0 Fr) was then inserted into the intrathecal space and advanced rostrally to place the catheter tip in the lumbar region L1–2. Confirmation of catheter placement within the intrathecal space was determined by observing the cerebrospinal fluid flow from the tip of the catheter. The catheter was routed subcutaneously from the hemilaminectomy site to the vascular access port site and attached to the port. The patency of the intrathecal catheter was confirmed using fluoroscopy after surgery. During the study period, the functionality of the catheter was evaluated based on the fluency of the injection and the response of the implanted monkey to intrathecal morphine. The longevity of the catheter varied from 2 to 4 years. The suspected malfunction of the catheter was investigated using fluoroscopy.
Drugs
Cebranopadol (CAS number: 863513-91-1, molecular weight: 378.5, logP: 4.7) was purchased from MedChemExpress (Monmouth Junction, NJ, USA). A concentrated stock solution of cebranopadol was formulated in dimethyl sulfoxide/Tween 80/5% glucose at a ratio of 1:1:18. The stock was diluted with sterile water to obtain the target working solution. The vehicle diluted by the same fold as the test compound was used as a control for both systemic and intrathecal administration. Fentanyl hydrochloride, morphine sulfate, oxycodone hydrochloride, naltrexone hydrochloride, and naltridole (National Institute on Drug Abuse, Bethesda, MD, USA) were dissolved in sterile water. 5′-Guanidinonaltrindole (National Institute on Drug Abuse) was dissolved in sterile saline. J-113397 was dissolved in dimethyl sulfoxide/Tween 80/sterile water at a ratio of 1:1:8. An injection volume of 0.1 mL/kg was used for systemic drug administration. For intrathecal administration,37 a total volume of 1 mL test compound or the control vehicle was administered through the subcutaneous access port, followed by 0.35 mL of saline to flush the dead volume of the port and catheter. For all systemic and intrathecal single-dosing procedures, drugs were administered at 1–2-week intervals.
Statistical analysis
The dose-response curves were analyzed using a previously reported method.38 Individual tail-withdrawal latencies were converted to the percent of maximum possible effect using the following formula: % maximum possible effect = [(test latency − control latency)/(cutoff latency − control latency)] × 100. The mean effective dose producing 50% of maximal effect (ED50) values were obtained after the log transformation of individual ED50 values, which were calculated by linear regression using the portion of the dose-effect curves spanning the 50% maximum possible effect, and 95% confidence interval (95% CI) were also determined. In addition, dose ratios were calculated by dividing the mean ED50 values in the presence of the antagonist by the baseline ED50 values. Significant shifts in dose-effect curves were defined when their 95% CI of ED50 values did not overlap.
GraphPad Prism version 9 software was used for statistical analysis. Blinding was not used to analyze the data. No statistical power calculations were performed prior to the study. The sample size was determined based on our previous experience with this design.7,37 Data are presented as mean values ± SD calculated by treatment and time using individual data from all studies. Comparisons were made for the same monkeys across all test sessions for the same experiment. For Figures 1, 3, and 4, the repeated measures analysis of variance (ANOVA) was used to compare the outcome measure (i.e., tail-withdrawal latency and the number of scratches) between two factors, dose and time (a two-tailed test). The interactions between the dose and time were also evaluated. For Figure 2, the mixed-effects model with random intercept was used to examine the association between the outcome measure (i.e., number of drug injections) and dose. Each monkey was subjected to different treatments; a mixed-effects model was used to handle the correlated structure. Dunnett’s multiple comparison test was used to correct for multiple tests. The significance level was set at P < 0.05. The assumptions of the ANOVA were verified by D’Agostino-Pearson omnibus (K2) tests for normality and Brown-Forsythe tests for homogeneity of variance. There were no missing data except for the drug self-administration experiment, in which one monkey missed some dosing conditions (Figure 2A: saline, cebranopadol 0.06 μg/kg per injection, fentanyl 0.03, and 0.3 μg/kg per injection; Figure 2B: saline, oxycodone, cebranopadol 0.03 and 0.3 μg/kg per injection) due to malfunction of its IV catheter that was not related to the testing drugs. A few potential outliers were identified based on scatter plots. Since these values represented real data, potential outliers were included in the evaluation, with unremarkable findings. Scatter plots showing the raw data from individual monkeys with the mean and SD imposed are presented in the supplemental figures.
Figure 1.
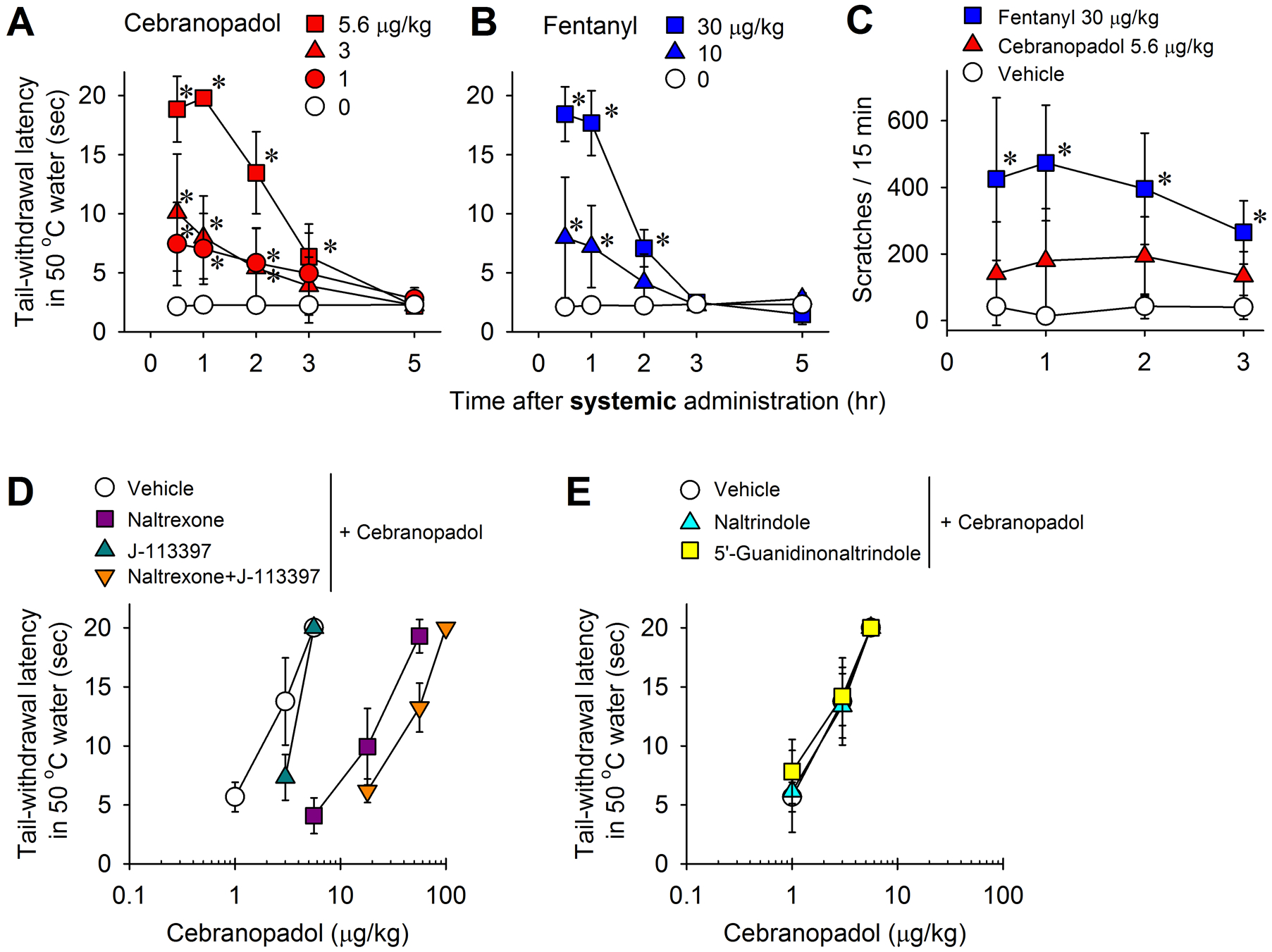
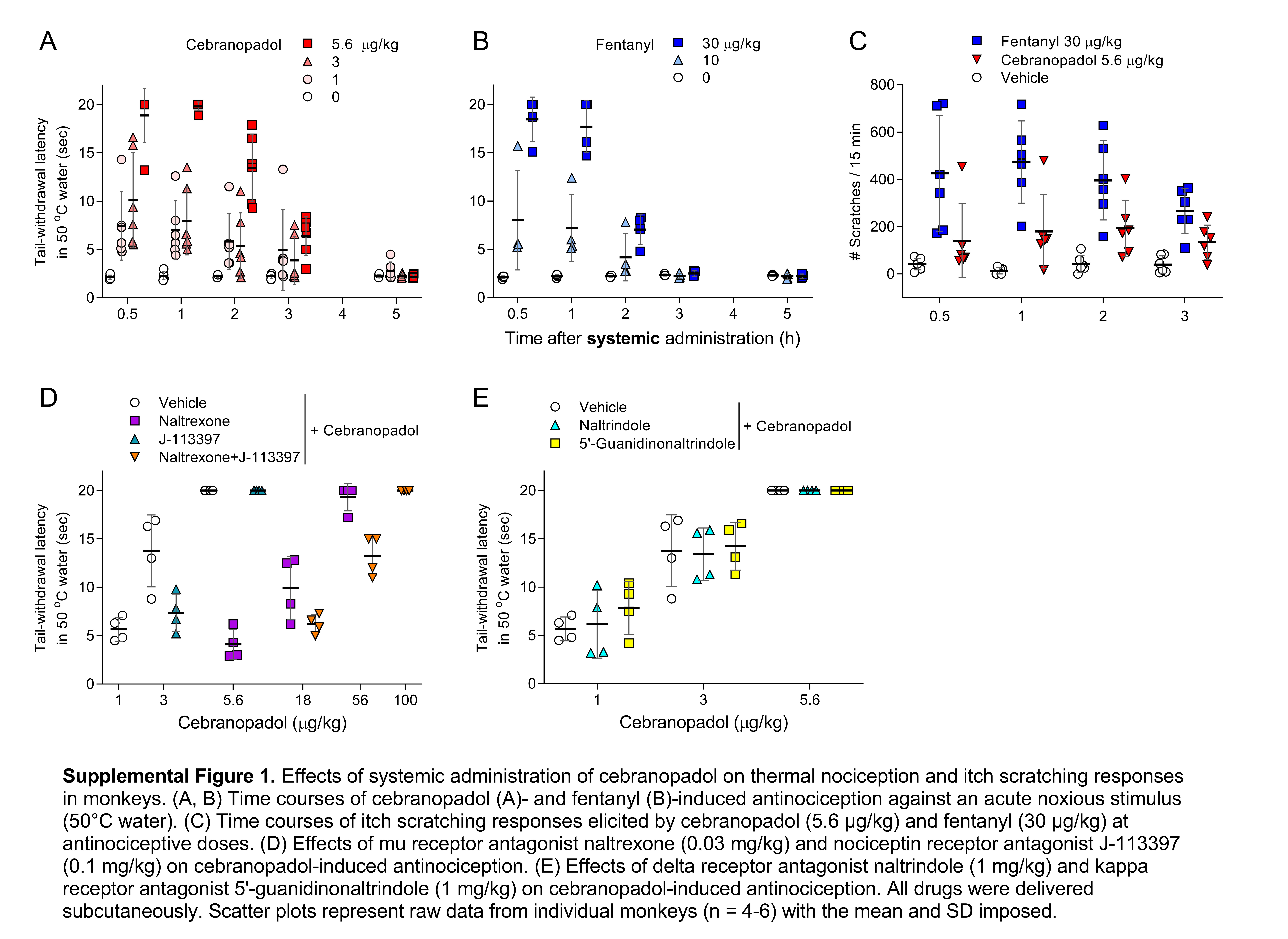
Effects of systemic administration of cebranopadol on thermal nociception and itch scratching responses in monkeys. (A, B) Time courses of cebranopadol (A)- and fentanyl (B)-induced antinociception against an acute noxious stimulus (50°C water). (C) Time courses of itch scratching responses elicited by cebranopadol (5.6 μg/kg) and fentanyl (30 μg/kg) at antinociceptive doses. (D) Effects of mu receptor antagonist naltrexone (0.03 mg/kg) and nociceptin receptor antagonist J-113397 (0.1 mg/kg) on cebranopadol-induced antinociception. (E) Effects of delta receptor antagonist naltrindole (1 mg/kg) and kappa receptor antagonist 5’-guanidinonaltrindole (1 mg/kg) on cebranopadol-induced antinociception. All drugs were delivered subcutaneously. Data represent the mean ± SD (n = 6 for A and C, and n = 4 for B, D, and E) and were analyzed by two-way repeated measures ANOVA followed by Dunnett’s multiple comparison test. *p < 0.05, significantly different from the vehicle condition.
Figure 3.
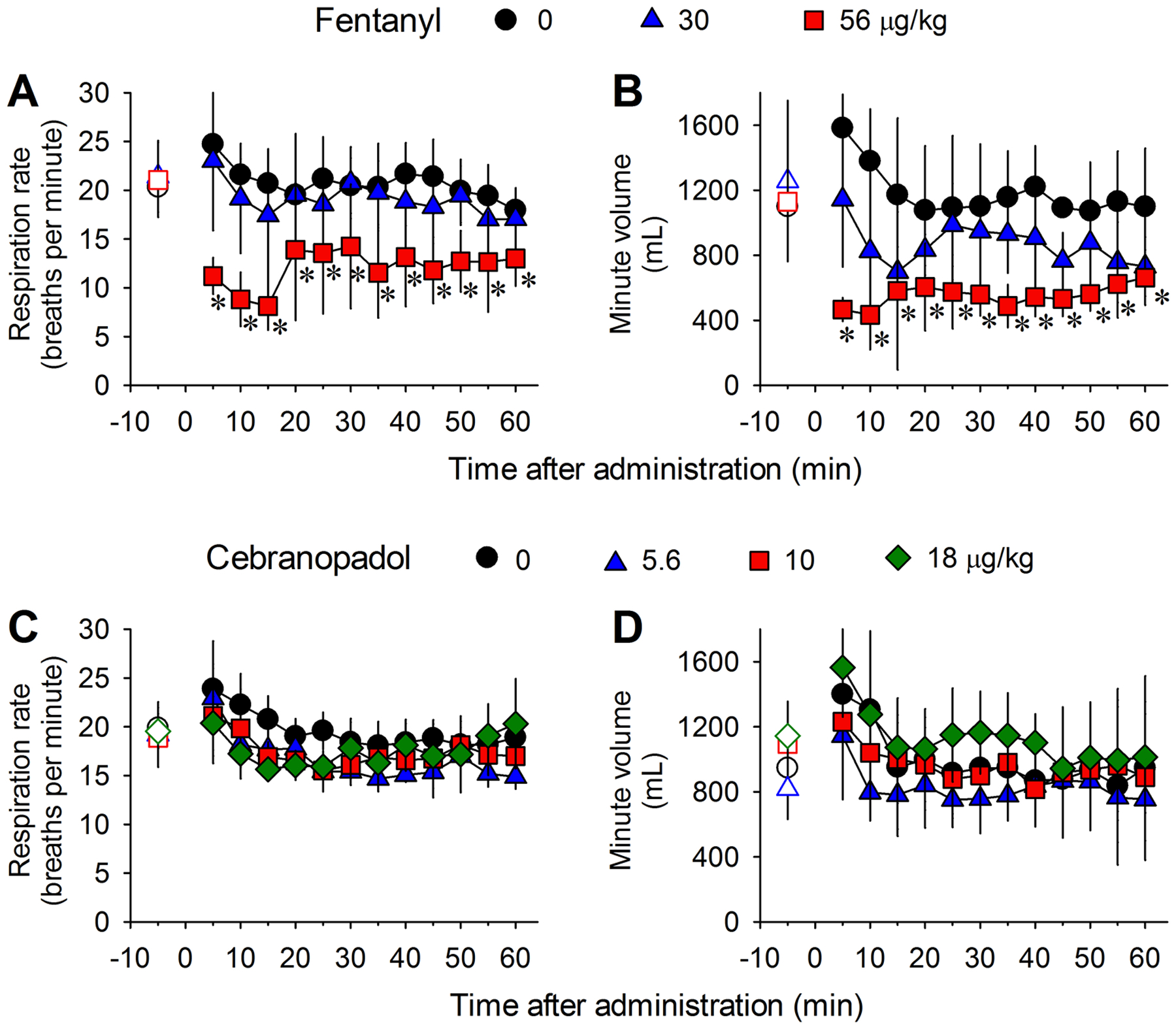
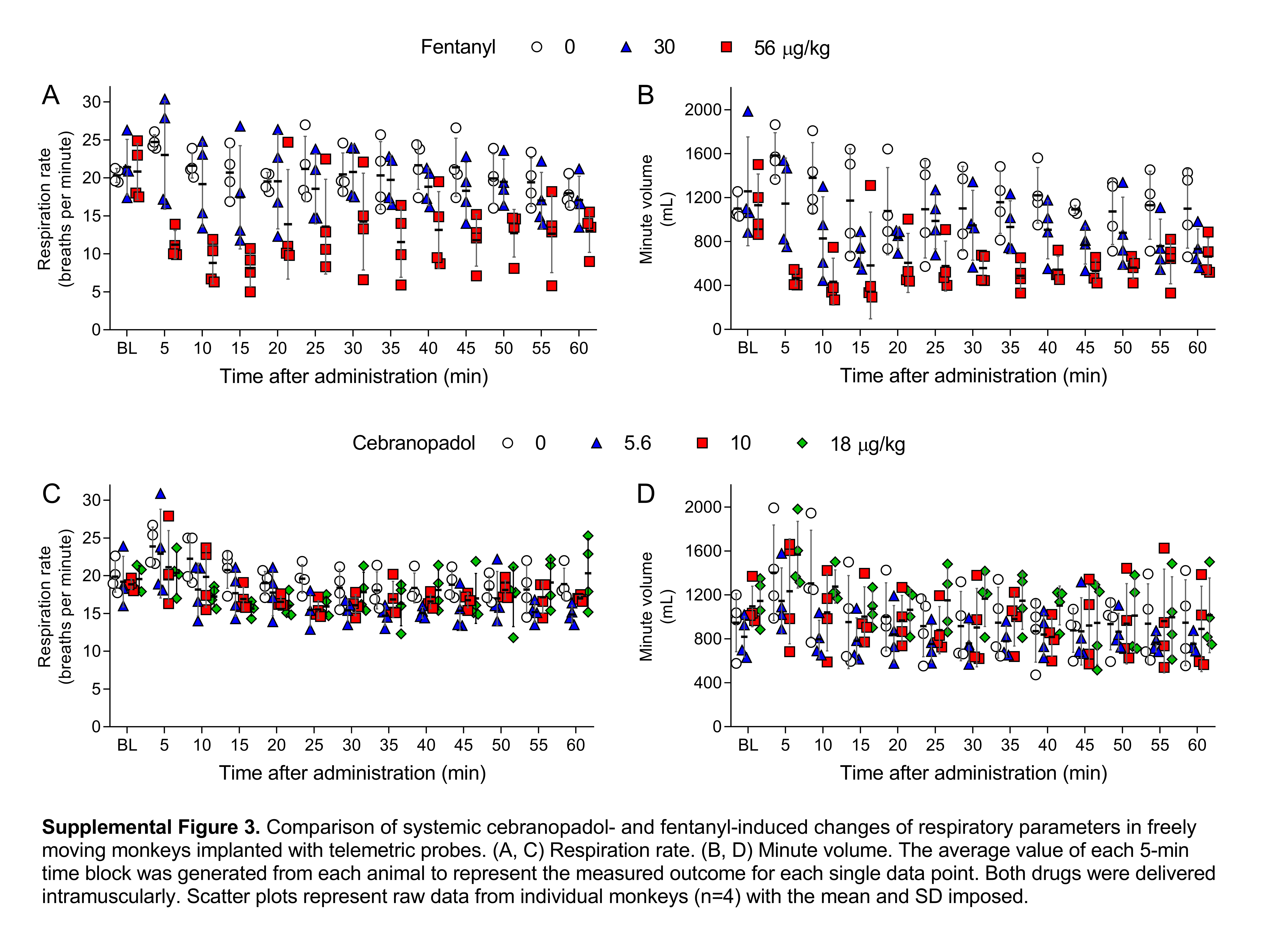
Comparison of systemic cebranopadol- and fentanyl-induced changes of respiratory parameters in freely moving monkeys implanted with telemetric probes. (A, C) Respiration rate. (B, D) Minute volume. Data represent the mean ± SD (n = 4) from each individual data averaged from a 5-min time block. Both drugs were delivered intramuscularly. Open symbols represent the baseline data of the different dosing conditions from the same monkeys before drug administration. Data were analyzed by two-way repeated measures ANOVA followed by Dunnett’s multiple comparison test. *p < 0.05, significantly different from the vehicle condition.
Figure 4.
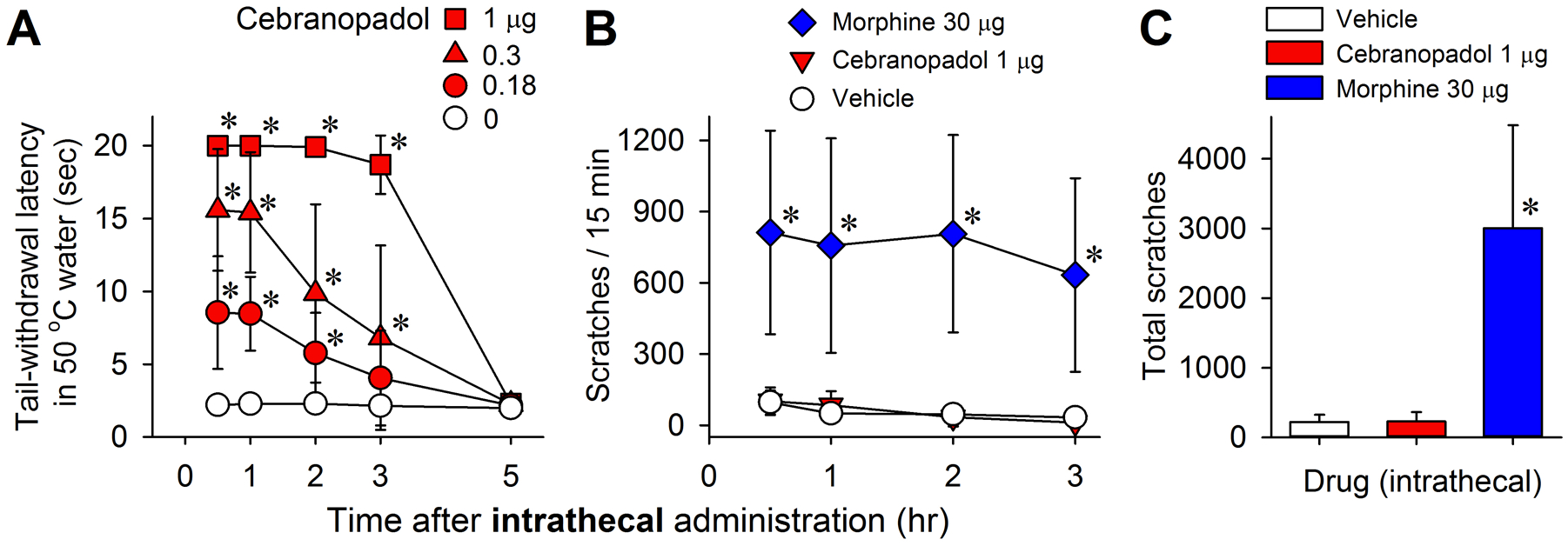
Effects of intrathecal administration of cebranopadol on thermal nociception and itch scratching responses in monkeys. (A) Time courses of cebranopadol-induced antinociception against an acute noxious stimulus (50°C water). (B) Time courses of itch scratching responses elicited by cebranopadol (1 μg) and morphine (30 μg) at antinociceptive doses. (C) Total number of scratches summed from the four time points shown in (B). Data represent the mean ± SD (n = 6) and were analyzed by two-way (A and B) or one-way (C) repeated measures ANOVA followed by Dunnett’s multiple comparison test. *p < 0.05, significantly different from the vehicle condition.
Figure 2.
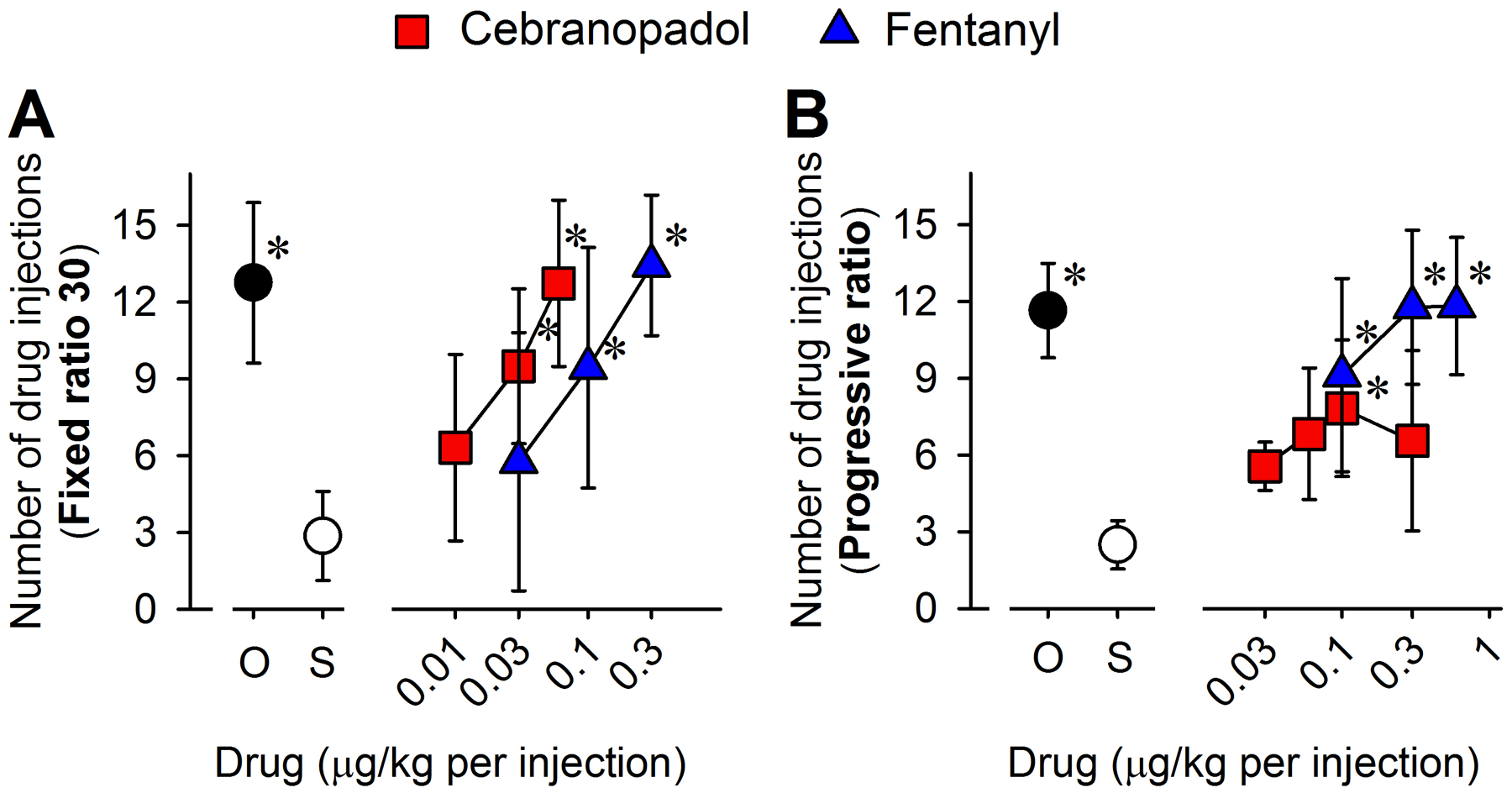
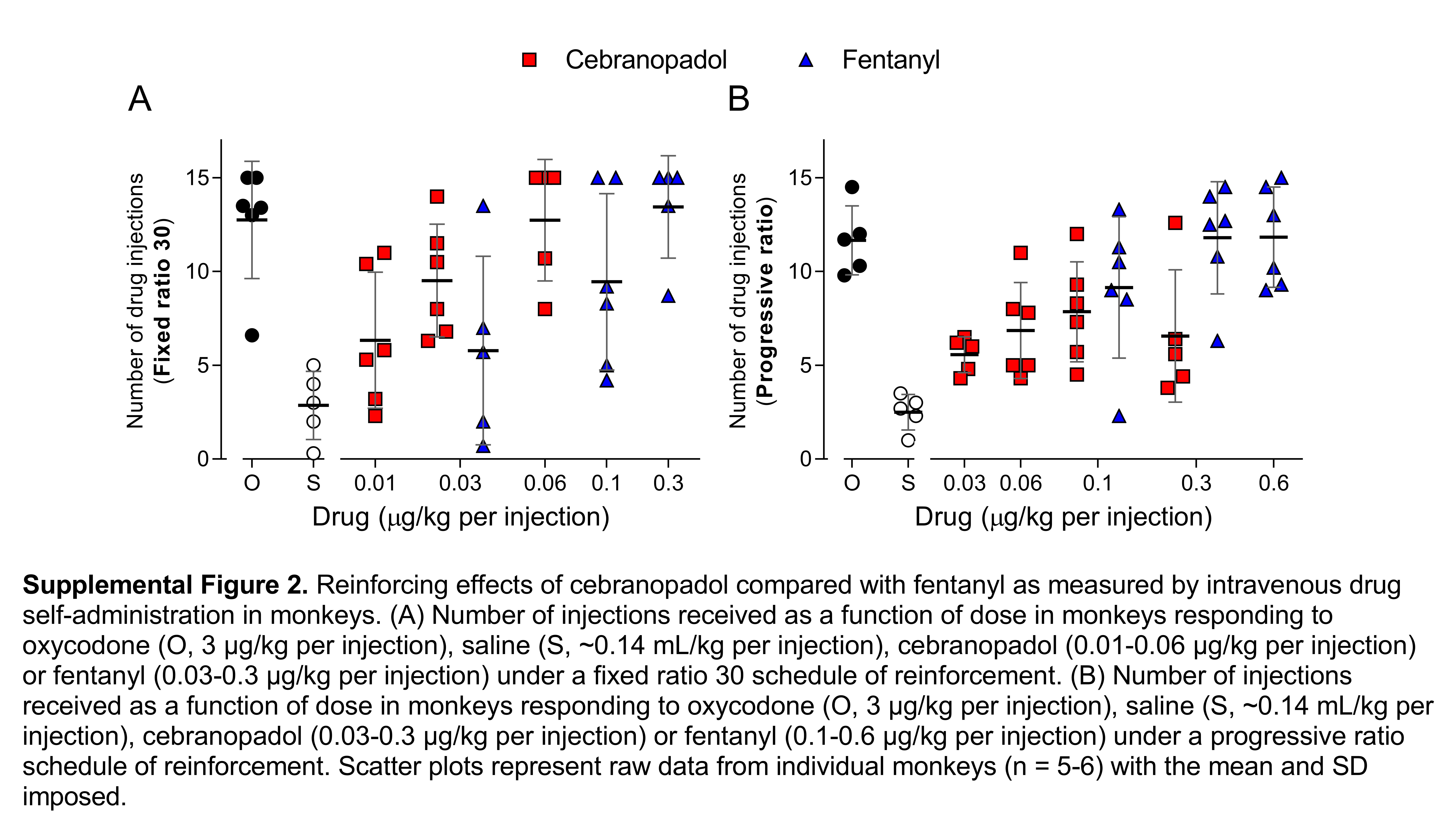
Reinforcing effects and strength of cebranopadol compared with fentanyl measured by intravenous drug self-administration in monkeys. (A) Number of injections received as a function of dose in monkeys responding to oxycodone (O, 3 μg/kg per injection, n = 6), saline (S, ~0.14 mL/kg per injection, n = 5), cebranopadol (0.01 [n = 6], 0.03 [n = 6], and 0.06 [n = 5] μg/kg per injection) or fentanyl (0.03 [n = 5], 0.1 [n = 6], and 0.3 [n = 5] μg/kg per injection) under a fixed ratio 30 schedule of reinforcement. (B) Number of injections received as a function of dose in monkeys responding to oxycodone (O, 3 μg/kg per injection, n = 5), saline (S, ~0.14 mL/kg per injection, n = 5), cebranopadol (0.03 [n = 5], 0.06 [n = 6], 0.1 [n = 6], and 0.3 [n = 5] μg/kg per injection) or fentanyl (0.1, 0.3, and 0.6 μg/kg per injection, n = 6) under a progressive ratio schedule of reinforcement. Data represent the mean ± SD and were analyzed by the mixed-effects model. *p < 0.05, significantly different from saline.
Results
Systemic cebranopadol produces potent antinociceptive effects but not itch scratching response
Subcutaneous cebranopadol (1‒5.6 μg/kg) produced antinociceptive effects in the acute thermal nociception assay in a dose-dependent (F3,15 = 22; P < 0.001) and time-dependent (F4,20 = 101.9; P < 0.001) manner with significant interaction between dose and time (F12,60 = 23.1; P < 0.001) (Fig. 1A). In comparison, fentanyl (10‒30 μg/kg) displayed antinociception in the same group of animals [dose (F2,6 = 18.6; P = 0.003), time (F4,12 = 176.7; P < 0.001 ), and dose × time interaction (F8,24 = 23.6; P < 0.001)] (Fig. 1B). The minimum effective dose of cebranopadol to produce full antinociception was 5.6 μg/kg (ED50 [95% CI] = 2.9 [1.8–4.6] μg/kg) (Fig. 1A), which was approximately 5-fold more potent than fentanyl, producing near full antinociception at 30 μg/kg (ED50 [95% CI] = 15.8 [14.6–17.1] μg/kg) (Fig. 1B). The duration of the antinociceptive action of cebranopadol (3 h) was slightly longer than that of fentanyl (2 h) (Fig. 1A and 1B). For the antinociceptive doses, cebranopadol 5.6 μg/kg did not significantly increase scratching responses, whereas fentanyl 30 μg/kg markedly increased the number of scratches in the same group of monkeys (F2,10 = 45.1; P < 0.001) (Fig. 1C).
Dose-response curves were generated for cebranopadol-induced thermal antinociception with vehicle pretreatment (ED50 [95% CI] = 2.1 [1.2–3.8] μg/kg). In the antagonist studies, pretreatment with mu receptor antagonist naltrexone 0.03 mg/kg and nociceptin receptor antagonist J-113397 0.1 mg/kg resulted in ED50 (95% CI) of 19 (11.1–32.5) and 3.6 (3.2–4) μg/kg, respectively, corresponding to approximately 9- and 2-fold rightward shifts of the dose-response curves. Additionally, combined pretreatment with naltrexone and J-113397 revealed a larger rightward shift (30-fold) with an ED50 (95% CI) of 41.8 (27.4–63.7) μg/kg for the cebranopadol dose-response curve (Fig. 1D). In contrast, pretreatment with delta receptor antagonist naltrindole 1 mg/kg (ED50 [95% CI] = 2 [0.9–4.3] μg/kg] or kappa receptor antagonist 5’-guanidinonaltrindole 1 mg/kg (ED50 [95% CI] = 1.7 [0.9–3.3] μg/kg) did not affect the dose-response curve. Therefore, mu and nociceptin receptors, but not delta and kappa receptors, contributed to the antinociceptive effects of cebranopadol. These findings suggest that systemic cebranopadol has a promising analgesic profile in primates that is pharmacologically distinct from classical mu opioid analgesics such as fentanyl.
Cebranopadol produces reinforcing effects with reduced reinforcing strength
In the IV drug self-administration paradigm, substitution of saline for the maintenance dose of oxycodone (3 μg/kg per injection) resulted in a much lower number of injections under both fixed ratio 30 (P < 0.001) and progressive ratio (P < 0.001) schedules. Under the fixed ratio 30 schedule, both cebranopadol (0.03 [P = 0.021] and 0.06 [P < 0.001] μg/kg per injection) and fentanyl (0.1 [P = 0.023] and 0.3 [P < 0.001] μg/kg per injection) functioned as reinforcers, showing a main effect of dose for both cebranopadol and fentanyl (Fig. 2A). Similar to the more potent antinociceptive effect described above, cebranopadol showed higher potency in producing reinforcing effects when compared to fentanyl (Fig. 2A). Under the progressive ratio schedule, both cebranopadol (0.1 μg/kg per injection [P = 0.011]) and fentanyl (0.1 [P = 0.001], 0.3 [P < 0.001], and 0.6 [P < 0.001] μg/kg per injection) showed a significantly higher reinforcing strength than saline; however, the reinforcing strength of cebranopadol was relatively lower than that of fentanyl (Fig. 2B). At the highest dose tested, monkeys earned 12 ± 3 injections (mean ± SD) of fentanyl (0.6 μg/kg per injection) but only 7 ± 3 injections of cebranopadol (0.3 μg/kg per injection) (Fig. 2B). These data demonstrated that cebranopadol produced a reinforcing effect and a relatively lower reinforcing strength than the selective mu receptor agonist fentanyl.
Higher doses of cebranopadol do not compromise respiratory function
Fentanyl at a dose of 30 μg/kg produced full antinociception but did not significantly change the respiratory parameters (Fig. 3A and 3B). However, as an opioid known to cause respiratory depression in humans, fentanyl caused drastic reductions in the respiration rate (F2,6 = 9.7; P = 0.013) and minute volume (F2,6 = 6.6; P = 0.031) in monkeys at a dose of 56 μg/kg, approximately 2-fold of its antinociceptive dose, showing a respiration rate of 8 ± 2 breaths per minute (mean ± SD) at 15 min after fentanyl administration (Fig. 3A and 3B). In contrast, cebranopadol, when given to the same group of monkeys at the antinociceptive dose 5.6 μg/kg or doses approximately 2–3-fold of its antinociceptive dose (10 and 18 μg/kg), did not significantly change the respiratory rate (F3,9 = 3.8; P = 0.053) or minute volume (F3,9 = 2.6; P = 0.115), showing a respiration rate of 17 ± 2 breaths per minute (mean ± SD) at 15 min after the administration of cebranopadol at a dose of 10 μg/kg (Fig. 3C and 3D). Therefore, cebranopadol may function as a safer analgesic than fentanyl.
Intrathecal cebranopadol produces potent antinociception but not itch sensation
Intrathecal cebranopadol (0.18‒1 μg) produced antinociceptive effects in the acute thermal nociception assay in a dose-dependent (F3,15 = 33.7; P < 0.001) and time-dependent (F4,20 = 160.4; P < 0.001) manner, with significant interaction between dose and time (F12,60 = 26.7; P < 0.001) (Fig. 4A). The minimum effective dose of cebranopadol to produce full antinociception was 1 μg. The antinociceptive action lasted approximately 3 h and subsided after 5 h. (Fig. 4A). Additionally, this dose of intrathecal cebranopadol (1 μg) did not significantly increase scratching responses. In contrast, intrathecal morphine (30 μg), an antinociceptive dose shown in a previous study,8 elicited robust scratching responses in the same group of monkeys (F2,10 = 19.3; P < 0.001) (Fig. 4B and 4C). The total number of scratches summed from the four 15-min recording sessions was 231 ± 137 (mean ± SD) for 1 μg of cebranopadol in contrast to 3,009 ± 1,474 for 30 μg of morphine (Fig. 4C). These data suggest that cebranopadol could serve as a promising spinal analgesic.
Discussion
Here, we documented the acute effects of cebranopadol following systemic and intrathecal administration. Although cebranopadol has been demonstrated to have analgesic efficacy in human studies,14,15 this non-human primate study provides additional information. Systemic cebranopadol produced antinociception, mainly mediated by mu receptors. It was safe and did not compromise respiratory functions at a dose 10-fold of its analgesic ED50 value. No pruritic effect was observed after either systemic or intrathecal administration of cebranopadol. Cebranopadol produced reduced reinforcing strength relative to fentanyl; however, it retained a certain degree of reinforcing effects and strength, implying its potential abuse liability. Overall, cebranopadol displayed analgesic efficacy similar to that of clinically used mu receptor agonists such as fentanyl and morphine, but with an improved side effect profile.
Subcutaneous cebranopadol more potently produced acute antinociception with a similar duration of action compared to fentanyl, yet without the accompanying itch scratching responses. The full efficacy and high potency of cebranopadol in the non-human primate model of acute pain were consistent with its analgesic efficacy in rodents and humans, whereas the duration of antinociceptive action (3 h) in non-human primates was shorter than that in the rat/mouse tail-flick assay.12,15 The antagonist studies revealed a larger mu receptor contribution than the nociceptin receptor and no involvement of delta and kappa receptors in cebranopadol-induced antinociception in the non-human primate model of acute pain. Delta and kappa receptor agonists are known to cause convulsions or sedation in non-human primates;24 therefore, the absence of convulsive and sedative behaviors after cebranopadol administration in our study is consistent with the lack of involvement of delta and kappa receptors. The nociceptin receptor antagonist had a weak influence on the antinociceptive effect of cebranopadol. This is different from the stronger nociceptin receptor antagonist effect toward AT-121-induced antinociception.7 Such difference may imply that mu receptor full agonists are more efficacious than agonists selective for other opioid receptor subtypes to suppress this nociceptive response in primates. A dual mu and nociceptin receptor agonism has been reported in a rat model of arthritis pain.39 In contrast, in a rat model of spinal nerve ligation, pretreatment with antagonists for mu, nociceptin, delta, and kappa receptors all attenuated the effect of cebranopadol to a similar degree and revealed a synergistic interaction of nociceptin receptor with mu/delta/kappa receptors.40,41 This difference might be attributed to differences in species (non-human primate vs. rodent) and pain modalities (acute pain, inflammatory pain, and neuropathic pain). The plasticity of the nociceptin ligand-receptor system in different pain states largely influences the functional expression and regulation of nociceptin receptors and their interaction with mu receptors.42 Nonetheless, the present study indicated that the mu receptor was the main driving force for the antinociceptive effect of cebranopadol in non-human primates. Thus, it should be cautious to use cebranopadol for acute pain management. It would be interesting to examine whether the effect of a nociceptin receptor antagonist on cebranopadol-induced analgesia changes in non-human primates under different pain states.
In addition to demonstrating efficacious analgesic effects, another critical aspect in developing novel analgesics is to evaluate whether they display favorable side effect profiles (e.g., devoid of abuse potential). Knowing that both fentanyl and cebranopadol are lipophilic13 and highly potent relative to other opioids, we conducted a side-by-side comparison between fentanyl and cebranopadol using an IV drug self-administration assay under two different schedules of reinforcement. Our results showed that cebranopadol produced fentanyl-comparable reinforcing effects under the fixed ratio 30 schedule; nevertheless, its reinforcing strength was lower than that of fentanyl under the progressive ratio schedule. This was different from rodent studies that showed ambiguous rewarding effects in the conditioned place preference paradigm.19,20 Although a human study showed that oral cebranopadol produced lower drug-liking effects than a mu receptor agonist hydromorphone,43 the reinforcing effects of cebranopadol were not observed with other reported nociceptin/mu receptor partial agonists, such as AT-121, BU08028, and BU10038 in the same experimental paradigm.6–8 It is difficult to conceive the potential use of “intravenous” cebranopadol for the management of pain compared to other nociceptin/mu receptor partial agonists that have no reinforcing strength. Considering that cebranopadol shows full efficacy, whereas the other three mixed agonists show only partial efficacy at both nociceptin and mu receptors, it is reasonable to conclude that although the reinforcing strength of cebranopadol was attenuated, nociceptin receptor activation might not be sufficient to completely block full mu receptor agonist-associated abuse potential. The balance between nociceptin and mu receptor efficacy could determine different pharmacological profiles of mixed nociceptin/mu receptor agonists, particularly their abuse potential. Given that a highly potent opioid fentanyl is widely abused in the US, the detectable reinforcing effect and strength of intravenous cebranopadol indicate its potential abuse liability and warrant caution for its clinical use.
Another side effect of classical opioid drugs is respiratory depression, which limits the therapeutic window of opioids and raises safety concerns. This is a major problem leading to increasing opioid overdose deaths during the opioid epidemic. We found that cebranopadol did not affect respiratory function at a dose 3-fold of the full analgesic dose, which was in clear contrast to fentanyl that caused significant decreases in respiration rate and minute volume by doubling the full analgesic dose, thus demonstrating a wider safety window for cebranopadol. The lack of a respiratory depressant effect of cebranopadol in non-human primates is consistent with observations in rodent and human studies,12,16 which could be attributed to the counterbalancing effect of nociceptin receptor agonist activity against mu receptor-dependent respiratory depression. Similar widened therapeutic windows have also been demonstrated with AT-121, BU08028, and BU10038.6–8 These findings support the research strategy to develop nociceptin/mu receptor agonists as innovative analgesics with improved safety profiles.
The spinal delivery of opioids, such as morphine, is a standard procedure for perioperative analgesia and is effectively used in different clinical contexts.44,45 However, its effectiveness in pain management is compromised by intense itch sensation.23,46 The non-human primate model of spinal morphine-induced itch has proven useful for evaluating the pruritic effects of drug candidates.31,37,47 In this model, intrathecal cebranopadol was more potent than morphine but did not elicit itch scratching responses. Spinal cebranopadol also showed high potency in producing antinociception and antihyperalgesia in rodents.48 These observations further strengthen the notion that simultaneous activation of nociceptin and mu receptors enhances the potency of analgesia without eliciting any common side effects. Delayed respiratory depression is associated with hydrophilic morphine rather than lipophilic neuraxial opioids.49 Although cebranopadol is lipophilic and systemic cebranopadol does not cause respiratory depression, it is important to further investigate whether this potential side effect is associated with spinal delivery of morphine versus cebranopadol. Systemic cebranopadol has been associated with several side effects, such as dizziness, vomiting, nausea, and constipation in clinical studies,17,43 making its use by a systemic route questionable. Given that intrathecal cebranopadol potently produced antinociception with good tolerability in non-human primates, intrathecal delivery of cebranopadol for pain management may limit the classical side effects observed with mu receptor agonists. These findings provide a pharmacological basis for the development of cebranopadol as a promising spinal analgesic.
Taken together, our study demonstrated that cebranopadol displayed analgesic efficacy with an improved side effect profile compared with the clinically used mu receptor agonists, fentanyl and morphine. It further supports nociceptin and mu receptor coactivation as a viable strategy to develop mixed nociceptin/mu receptor agonists as innovative analgesics with fewer side effects. However, cebranopadol (nociceptin/mu receptor full agonist) has higher abuse liability than AT-121 or other nociceptin/mu receptor partial agonists in non-human primate models,6–8 that is, nociceptin receptor activation suppresses the reinforcing strength mediated by partial, not full, mu receptor agonists. These findings indicate that nociceptin/mu receptor partial agonists might have a favorable side effect profile. In clinical studies, cebranopadol has shown encouraging efficacy in treating patients with chronic pain.17,50 Several rodent studies have suggested a slower tolerance development and lower potential to produce physical dependence after cebranopadol treatment.12,15,17 It is essential to further evaluate these outcome measures in non-human primates with chronic administration of cebranopadol. Nonetheless, these pharmacological studies in non-human primates document a major difference, i.e., abuse potential, between cebranopadol and nociceptin/mu receptor partial agonists and warrant caution on the clinical use of cebranopadol.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Ms. Kelsey Mabry and Brittany Kelly for their technical assistance with animal training and data collection, Dr. Heather DeLoid for performing intrathecal catheterization and telemetry device implantation, Drs. Nicole Bacarella and Erin Jackson and Animal Resources Program of Wake Forest University for veterinary care, and Editage for editing this manuscript.
Research support
The work was supported by grants from the National Institutes of Health and the National Institute on Drug Abuse (DA-053343, DA-049580, and DA-044450). The content is solely the responsibility of the authors and does not necessarily represent the official views of the U.S. federal agencies.
Footnotes
Competing interests
The authors declare no competing interests.
Prior presentation: Experimental Biology 2019 (American Society for Pharmacology and Experimental Therapeutics), April 7th, 2019, Orlando, Florida, USA.
References
- 1.Hewson DW, Struys M, Hardman JG: Opioids: refining the perioperative role of God’s own medicine. Br J Anaesth. 2019; 122:e93–e5 [DOI] [PubMed] [Google Scholar]
- 2.Brady KT, McCauley JL, Back SE: Prescription Opioid Misuse, Abuse, and Treatment in the United States: An Update. The American journal of psychiatry. 2016; 173:18–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Volkow ND, McLellan AT: Opioid Abuse in Chronic Pain--Misconceptions and Mitigation Strategies. The New England journal of medicine. 2016; 374:1253–63 [DOI] [PubMed] [Google Scholar]
- 4.Azzam AAH, McDonald J, Lambert DG: Hot topics in opioid pharmacology: mixed and biased opioids. Br J Anaesth. 2019; 122:e136–e45 [DOI] [PubMed] [Google Scholar]
- 5.Lin AP, Ko MC: The therapeutic potential of nociceptin/orphanin FQ receptor agonists as analgesics without abuse liability. ACS Chem Neurosci. 2013; 4:214–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ding H, Czoty PW, Kiguchi N, Cami-Kobeci G, Sukhtankar DD, Nader MA, Husbands SM, Ko MC: A novel orvinol analog, BU08028, as a safe opioid analgesic without abuse liability in primates. Proceedings of the National Academy of Sciences of the United States of America. 2016; 113:E5511–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ding H, Kiguchi N, Yasuda D, Daga PR, Polgar WE, Lu JJ, Czoty PW, Kishioka S, Zaveri NT, Ko MC: A bifunctional nociceptin and mu opioid receptor agonist is analgesic without opioid side effects in nonhuman primates. Science translational medicine. 2018; 10:eaar3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kiguchi N, Ding H, Cami-Kobeci G, Sukhtankar DD, Czoty PW, DeLoid HB, Hsu FC, Toll L, Husbands SM, Ko MC: BU10038 as a safe opioid analgesic with fewer side-effects after systemic and intrathecal administration in primates. Br J Anaesth. 2019; 122:e146–e56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lambert DG: Mixed mu-nociceptin/orphanin FQ opioid receptor agonists and the search for the analgesic holy grail. Br J Anaesth. 2019; 122:e95–e7 [DOI] [PubMed] [Google Scholar]
- 10.Kiguchi N, Ding H, Ko MC: Therapeutic potentials of nociceptin and mu receptor coactivation for the treatment of pain and opioid abuse. J Neurosci Res. 2020; doi: 10.1002/jnr.24624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khroyan TV, Polgar WE, Cami-Kobeci G, Husbands SM, Zaveri NT, Toll L: The first universal opioid ligand, (2S)-2-[(5R,6R,7R,14S)-N-cyclopropylmethyl-4,5-epoxy-6,14-ethano-3-hydroxy-6-meth oxymorphinan-7-yl]-3,3-dimethylpentan-2-ol (BU08028): characterization of the in vitro profile and in vivo behavioral effects in mouse models of acute pain and cocaine-induced reward. The Journal of pharmacology and experimental therapeutics. 2011; 336:952–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Linz K, Christoph T, Tzschentke TM, Koch T, Schiene K, Gautrois M, Schroder W, Kogel BY, Beier H, Englberger W, Schunk S, De Vry J, Jahnel U, Frosch S: Cebranopadol: a novel potent analgesic nociceptin/orphanin FQ peptide and opioid receptor agonist. The Journal of pharmacology and experimental therapeutics. 2014; 349:535–48 [DOI] [PubMed] [Google Scholar]
- 13.Schunk S, Linz K, Hinze C, Frormann S, Oberborsch S, Sundermann B, Zemolka S, Englberger W, Germann T, Christoph T, Kogel BY, Schroder W, Harlfinger S, Saunders D, Kless A, Schick H, Sonnenschein H: Discovery of a Potent Analgesic nociceptin and Opioid Receptor Agonist: Cebranopadol. ACS medicinal chemistry letters. 2014; 5:857–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Calo G, Lambert DG: Nociceptin/orphanin FQ receptor ligands and translational challenges: focus on cebranopadol as an innovative analgesic. Br J Anaesth. 2018; 121:1105–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tzschentke TM, Linz K, Koch T, Christoph T: Cebranopadol: A Novel First-in-Class Potent Analgesic Acting via nociceptin and Opioid Receptors. Handb Exp Pharmacol. 2019; 254:367–98 [DOI] [PubMed] [Google Scholar]
- 16.Dahan A, Boom M, Sarton E, Hay J, Groeneveld GJ, Neukirchen M, Bothmer J, Aarts L, Olofsen E: Respiratory Effects of the Nociceptin/Orphanin FQ Peptide and Opioid Receptor Agonist, Cebranopadol, in Healthy Human Volunteers. Anesthesiology. 2017; 126:697–707 [DOI] [PubMed] [Google Scholar]
- 17.Christoph A, Eerdekens MH, Kok M, Volkers G, Freynhagen R: Cebranopadol, a novel first-in-class analgesic drug candidate: first experience in patients with chronic low back pain in a randomized clinical trial. Pain. 2017; 158:1813–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruzza C, Holanda VA, Gavioli EC, Trapella C, Calo G: nociceptin agonist action of cebranopadol counteracts its liability to promote physical dependence. Peptides. 2019; 112:101–5 [DOI] [PubMed] [Google Scholar]
- 19.de Guglielmo G, Matzeu A, Kononoff J, Mattioni J, Martin-Fardon R, George O: Cebranopadol Blocks the Escalation of Cocaine Intake and Conditioned Reinstatement of Cocaine Seeking in Rats. The Journal of pharmacology and experimental therapeutics. 2017; 362:378–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen Q, Deng Y, Ciccocioppo R, Cannella N: Cebranopadol, a Mixed Opioid Agonist, Reduces Cocaine Self-administration through Nociceptin Opioid and Mu Opioid Receptors. Front Psychiatry. 2017; 8:234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ator NA, Griffiths RR: Principles of drug abuse liability assessment in laboratory animals. Drug Alcohol Depend. 2003; 70:S55–72 [DOI] [PubMed] [Google Scholar]
- 22.Mello NK, Negus SS: Preclinical evaluation of pharmacotherapies for treatment of cocaine and opioid abuse using drug self-administration procedures. Neuropsychopharmacology. 1996; 14:375–424 [DOI] [PubMed] [Google Scholar]
- 23.Ganesh A, Maxwell LG: Pathophysiology and management of opioid-induced pruritus. Drugs. 2007; 67:2323–33 [DOI] [PubMed] [Google Scholar]
- 24.Ding H, Ko MC: Translational value of non-human primates in opioid research. Exp Neurol. 2021; 338:113602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Phillips KA, Bales KL, Capitanio JP, Conley A, Czoty PW, t Hart BA, Hopkins WD, Hu SL, Miller LA, Nader MA, Nathanielsz PW, Rogers J, Shively CA, Voytko ML: Why primate models matter. Am J Primatol. 2014; 76:801–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG: Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010; 160:1577–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ding H, Kiguchi N, Perrey DA, Nguyen T, Czoty PW, Hsu FC, Zhang Y, Ko MC: Antinociceptive, reinforcing, and pruritic effects of the G-protein signalling-biased mu opioid receptor agonist PZM21 in non-human primates. Br J Anaesth. 2020; 125:596–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ko MC, Johnson MD, Butelman ER, Willmont KJ, Mosberg HI, Woods JH: Intracisternal nor-binaltorphimine distinguishes central and peripheral kappa-opioid antinociception in rhesus monkeys. The Journal of pharmacology and experimental therapeutics. 1999; 291:1113–20 [PMC free article] [PubMed] [Google Scholar]
- 29.Butelman ER, Ko MC, Traynor JR, Vivian JA, Kreek MJ, Woods JH: GR89,696: a potent kappa-opioid agonist with subtype selectivity in rhesus monkeys. The Journal of pharmacology and experimental therapeutics. 2001; 298:1049–59 [PubMed] [Google Scholar]
- 30.Cremeans CM, Gruley E, Kyle DJ, Ko MC: Roles of mu-opioid receptors and nociceptin/orphanin FQ peptide receptors in buprenorphine-induced physiological responses in primates. The Journal of pharmacology and experimental therapeutics. 2012; 343:72–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ko MC, Song MS, Edwards T, Lee H, Naughton NN: The role of central mu opioid receptors in opioid-induced itch in primates. The Journal of pharmacology and experimental therapeutics. 2004; 310:169–76 [DOI] [PubMed] [Google Scholar]
- 32.Sukhtankar DD, Lee H, Rice KC, Ko MC: Differential effects of opioid-related ligands and NSAIDs in nonhuman primate models of acute and inflammatory pain. Psychopharmacology (Berl). 2014; 231:1377–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ko MC, Terner J, Hursh S, Woods JH, Winger G: Relative reinforcing effects of three opioids with different durations of action. The Journal of pharmacology and experimental therapeutics. 2002; 301:698–704 [DOI] [PubMed] [Google Scholar]
- 34.Ko MC, Woods JH, Fantegrossi WE, Galuska CM, Wichmann J, Prinssen EP: Behavioral effects of a synthetic agonist selective for nociceptin/orphanin FQ peptide receptors in monkeys. Neuropsychopharmacology. 2009; 34:2088–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rowlett JK: A labor-supply analysis of cocaine self-administration under progressive-ratio schedules: antecedents, methodologies, and perspectives. Psychopharmacology (Berl). 2000; 153:1–16 [DOI] [PubMed] [Google Scholar]
- 36.Wang Z, Woolverton WL: Estimating the relative reinforcing strength of (+/−)-3,4-methylenedioxymethamphetamine (MDMA) and its isomers in rhesus monkeys: comparison to (+)-methamphetamine. Psychopharmacology (Berl). 2007; 189:483–8 [DOI] [PubMed] [Google Scholar]
- 37.Ding H, Hayashida K, Suto T, Sukhtankar DD, Kimura M, Mendenhall V, Ko MC: Supraspinal actions of nociceptin/orphanin FQ, morphine and substance P in regulating pain and itch in non-human primates. Br J Pharmacol. 2015; 172:3302–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ko MC, Divin MF, Lee H, Woods JH, Traynor JR: Differential in vivo potencies of naltrexone and 6beta-naltrexol in the monkey. The Journal of pharmacology and experimental therapeutics. 2006; 316:772–9 [DOI] [PubMed] [Google Scholar]
- 39.Schiene K, Schroder W, Linz K, Frosch S, Tzschentke TM, Jansen U, Christoph T: Nociceptin/orphanin FQ opioid peptide (nociceptin) receptor and micro-opioid peptide (mu) receptors both contribute to the anti-hypersensitive effect of cebranopadol in a rat model of arthritic pain. European journal of pharmacology. 2018; 832:90–5 [DOI] [PubMed] [Google Scholar]
- 40.Christoph T, Raffa R, De Vry J, Schroder W: Synergistic interaction between the agonism of cebranopadol at nociceptin/orphanin FQ and classical opioid receptors in the rat spinal nerve ligation model. Pharmacol Res Perspect. 2018; 6:e00444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rutten K, Schroder W, Christoph T, Koch T, Tzschentke TM: Selectivity profiling of nociceptin, mu, DOP and KOP receptor antagonists in the rat spinal nerve ligation model of mononeuropathic pain. European journal of pharmacology. 2018; 827:41–8 [DOI] [PubMed] [Google Scholar]
- 42.Schroder W, Lambert DG, Ko MC, Koch T: Functional plasticity of the N/OFQ-nociceptin receptor system determines analgesic properties of nociceptin receptor agonists. Br J Pharmacol. 2014; 171:3777–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gohler K, Sokolowska M, Schoedel KA, Nemeth R, Kleideiter E, Szeto I, Eerdekens MH: Assessment of the Abuse Potential of Cebranopadol in Nondependent Recreational Opioid Users: A Phase 1 Randomized Controlled Study. J Clin Psychopharmacol. 2019; 39:46–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bujedo BM, Santos SG, Azpiazu AU: A review of epidural and intrathecal opioids used in the management of postoperative pain. J Opioid Manag. 2012; 8:177–92 [DOI] [PubMed] [Google Scholar]
- 45.Caraway D, Walker V, Becker L, Hinnenthal J: Successful Discontinuation of Systemic Opioids After Implantation of an Intrathecal Drug Delivery System. Neuromodulation. 2015; 18:508–15; discussion 15–6 [DOI] [PubMed] [Google Scholar]
- 46.Waxler B, Dadabhoy ZP, Stojiljkovic L, Rabito SF: Primer of postoperative pruritus for anesthesiologists. Anesthesiology. 2005; 103:168–78 [DOI] [PubMed] [Google Scholar]
- 47.Lee H, Ko MC: Distinct functions of opioid-related peptides and gastrin-releasing peptide in regulating itch and pain in the spinal cord of primates. Sci Rep. 2015; 5:11676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tzschentke TM, Linz K, Frosch S, Christoph T: Antihyperalgesic, Antiallodynic, and Antinociceptive Effects of Cebranopadol, a Novel Potent Nociceptin/Orphanin FQ and Opioid Receptor Agonist, after Peripheral and Central Administration in Rodent Models of Neuropathic Pain. Pain Pract. 2017; 17:1032–41 [DOI] [PubMed] [Google Scholar]
- 49.Sultan P, Gutierrez MC, Carvalho B: Neuraxial morphine and respiratory depression: finding the right balance. Drugs. 2011; 71:1807–19 [DOI] [PubMed] [Google Scholar]
- 50.Eerdekens MH, Kapanadze S, Koch ED, Kralidis G, Volkers G, Ahmedzai SH, Meissner W: Cancer-related chronic pain: Investigation of the novel analgesic drug candidate cebranopadol in a randomized, double-blind, noninferiority trial. Eur J Pain. 2019; 23:577–88 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.