

Abstract
The use of allenes and 1,3-dienes as chiral allylmetal pronucleophiles in intermolecular catalytic enantioselective reductive additions to aldehydes, ketones, imines, carbon dioxide and other C=X electrophiles is exhaustively catalogued together with redox-neutral hydrogen auto-transfer processes. Coverage is limited to processes that result in both C-H and C-C bond formation. The use of alkynes as latent allylmetal pronucleophiles and multicomponent C=X allylations involving allene and dienes are not covered. As illustrated in this review, the ability of allenes and 1,3-dienes to serve as tractable non-metallic pronucleophiles has evoked many useful transformations that have no counterpart in traditional allylmetal chemistry.
Keywords: Enantioselective, Iridium, Ruthenium, Copper, Carbonyl Addition
Graphical Abstract
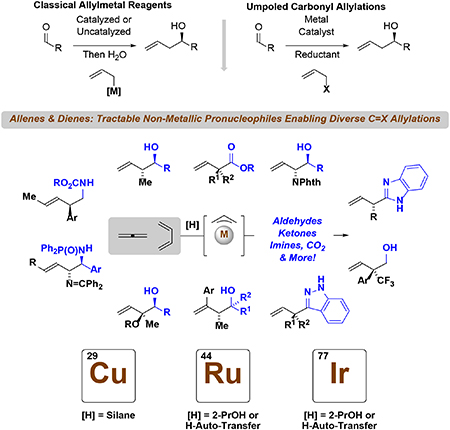
Beyond Preformed Allylmetal Reagents. In this review, the emerging use of allenes and dienes as chiral allylmetal pronucleophiles in catalytic enantioselective reductive additions to aldehydes, ketones, imines, carbon dioxide and other C=X (X = C, O, N) electrophiles is exhaustively catalogued.
I. Introduction and Historical Perspective:
The asymmetric allylation of C=X bonds has found broad use in chemical synthesis and innovation in this area to continues to unlock unique volumes of chemical space.[1] The arc of science traced from the parent allylation processes to the state-of-the-art in asymmetric catalysis reveals a progression from reactions that employ stoichiometric quantities of highly reactive allylmetal reagents to the use of tractable non-metallic pronucleophiles that deliver allylmetal species as transient catalytic intermediates (Figure 1). The very first examples of carbonyl allylation involved allylzinc reagents generated from allyl iodide and elemental zinc, as demonstrated by Zaitsev (1876)[2a–c] in allylations of ethyl formate and acetone, respectively. In 1904, carbonyl allylations using the eponymous allylmagnesium reagents developed by Grignard were reported by Béhal and Sommelet.[2d] The use of isolable allylmetal reagents followed. Mikhailov and Bubnov (1964) reported the first carbonyl allylations using isolable allylboron reagents.[3a] The first carbonyl allylations using isolable allyltin reagents were reported by Konig and Neumann (1967)[3b] and carbonyl allylations using isolable allylsilanes were reported by Hosomi and Sakurai (1976).[3c]
Figure 1.
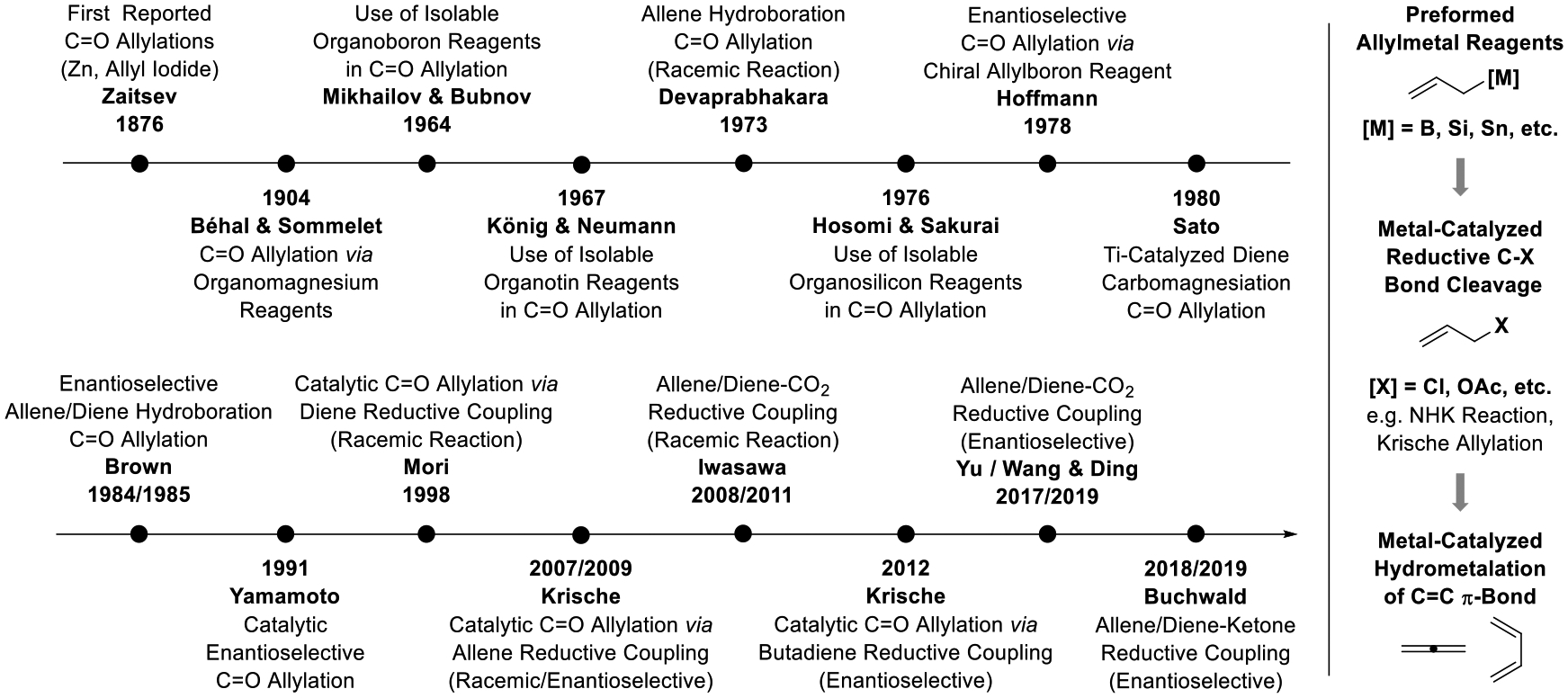
Selected milestones in carbonyl allylation as related to the use of allenes and dienes as allylmetal pronucleophiles in enantioselective metal-catalyzed carbonyl reductive coupling.
In a landmark study by Hoffmann (1978), the first enantioselective carbonyl allylations were developed using a chiral allylborate derived from camphor.[4a,b] This work was followed by numerous asymmetric carbonyl allylation protocols mediated by allylmetal reagents bearing chiral auxiliaries, including the work of Kumada (1982, Si),[4c] Brown (1983, B),[4d] Roush (1985, B),[4e] Reetz (1988, B),[4f] Masamune (1989, B),[4g] Corey (1989, B),[4h] Seebach (1987, Ti),[4i] Duthaler (1989, Ti),[4j] Panek (1991, Si),[4k] Leighton (2002, Si)[4l,m] Chong (2004, B)[4n] and Soderquist (2005, B).[4o] The development of catalytic enantioselective carbonyl allylations by Yamamoto (1991, Si)[5a] represents another major milestone. An abundance of distinctly different catalytic enantioselective methods for carbonyl allylation followed, as illustrated in work by Umani-Ronchi (1993, Sn)[5b] Keck (1993, Sn)[5c] Denmark (1994, Si),[5d] Shibasaki (2004, B),[5e] Schaus (2006, B)[5f] and Antilla (2010, B),[5g] as well as enantioselective Nozaki-Hiyama-Kishi (NHK) allylations (1999).[6,7]
These advances in asymmetric carbonyl allylation informed parallel explorations into the chemistry of allenes (Figure 1). Devaprabhakara (1973) reported the reaction of disiamylborane with 3-phenyl-1,2-butadiene to generate allylboron species, which upon treatment with aldehydes or ketones generated products carbonyl allylation.[8a] Brown (1984) reported asymmetric variants of this process using diisopinocampheylborane (Ipc2BH) using dimethylallene as the pronucleophile.[8b] Related allene hydroalumination- and hydrozirconation-carbonyl allylation sequences were subsequently developed,[9] along with metal-catalyzed allene hydrofunctionalizations of to form discrete allyltin[10a] and allylaluminum[10b] reagents that engage in carbonyl allylation. Finally, catalytic allene-carbonyl reductive couplings to form racemic[11a,b,f,12a–g] and enantiomerically enriched[11c–e,g–j,12h] products of carbonyl allylation were developed by the present author (2007, 2009).[11,12] Notably, these processes employ elemental hydrogen[11a] or 2-propanol[11b–e,h,j,12a,b,e,g,h] as reductants, or can be conducted via hydrogen auto-transfer using alcohol proelectrophiles as reductants.[11b,c,e–g,i,12c,d,f,h] This work and related alcohol-mediated carbonyl reductive couplings of allyl acetate (2008),[13,14] represent the first allylations of unactivated carbonyl compounds that do not require stoichiometric (organo)metallic reagents/reductants.
A similar chain of events is observed in the development of diene-mediated carbonyl allylations. Following observations by Takase (1970) that “diene-magnesium compounds” react with aldehydes and ketones to form products of carbonyl allylation in low yield,[15] Sato (1980) described highly efficient titanocene-catalyzed carbomagnesiations of butadiene and isoprene to form allylic Grignard reagents along with examples of acetone allylation.[16] Other metal-catalyzed diene hydrofunctionalizations to form discrete allylsilicon (1987),[17a] allylboron (1989),[17b] allyltin (1994)[17c] and allylchromium (1998)[17d] reagents that were shown to engage in carbonyl allylation were subsequently reported. The first enantioselective diene-mediated carbonyl allylations were reported by Brown (1985) and involved the reaction of cyclohexadiene with Ipc2BH-mediated hydroboration-carbonyl allylation reactions to form enantiomerically enriched products.[18] The first catalytic diene-carbonyl reductive couplings to form racemic products of allylation were reported by Mori (1998), who employed a nickel-silane catalyst system in combination with 1-phenylbutadiene.[19a] Achiral catalysts for reductive carbonyl allylation using the abundant chemical feedstock butadiene (12 × 106 tons/year) based on titanium (2005),[20] ruthenium (2008),[21] rhodium (2009)[22a] and iridium (2010)[22c] were subsequently reported, along with iridium-catalyzed reductive allylations of cyclohexadiene (2008).[22b] An enantioselective coupling of 1,4-diphenylbutadiene was subsequently reported by Sato (2007),[19b] but it was not until 2012 that butadiene itself was employed as a pronucleophile in catalytic enantioselective carbonyl allylation, as described by the present author.[23b,c]
In recent years, several significant advances in enantioselective metal-catalyzed allene/diene-C=X reductive coupling were made. Following the work of Iwasawa (2008, 2011) on allene/diene-CO2 reductive coupling,[24] Yu (2017)[25a] and Wang and Ding (2019)[25b] reported enantioselective variants of these processes that deliver products of hydroxymethylation. Guided by the pioneering work by Shibasaki and Kanai (2006),[26] Riant (2006)[27] and Lam (2012)[28] on enantioselective copper-catalyzed reductive aldol-type couplings to ketones, Buchwald (2018, 2019)[29] reported enantioselective copper-catalyzed allene/diene-ketone reductive couplings. The use of allenes and dienes as chiral allylmetal pronucleophiles continues to expand and many useful transformations that have no counterpart in traditional allylmetal chemistry have emerged. In this Mini-Review, the use of allenes and conjugated dienes as chiral allylmetal pronucleophiles in intermolecular catalytic enantioselective reductive additions to aldehydes, ketones, imines, carbon dioxide and other C=X electrophiles is exhaustively catalogued together with redox-neutral hydrogen auto-transfer processes.[30] Coverage is limited to processes that result in both C-H and C-C bond formation. For use of alkynes as latent allylmetal pronucleophiles in catalytic enantioselective C=X allylation[31] and multicomponent C=X allylations involving allene and diene pronucleophiles[32] the reader is referred to the review literature.[31,32] This brief survey omits detailed descriptions of reaction mechanism.
II. Allenes as Allylmetal Pronucleophiles in Catalytic Enantioselective C=X Addition
Pursuant to the development of the first allene-mediated carbonyl allylations to form racemic adducts, which were catalyzed by iridium[11a,b,e] and ruthenium[12a–g] complexes, the seminal report on corresponding catalytic enantioselective processes was disclosed (Scheme 1, eq. A).[11c] Specifically, using 1,1-dimethylallene as pronucleophile and the cyclometallated π-allyliridium-C,O-benzoate complex modified by SEGPHOS as catalyst, highly enantioselective carbonyl tert-prenylation could be achieved from alcohol reactants (via hydrogen auto-transfer) or aldehyde reactants (via 2-propanol mediated reductive coupling). Corresponding catalytic enantioselective tert-prenylation of isatins[11d] and furan methanols[11e] were subsequently developed (not shown). Using related π-allyliridium-C,O-benzoate complexes modified by H8-BINAP or tol-BINAP, analogous couplings of phthalimido-allene (Scheme 1, eq. B)[11i] and the parent allene (Scheme 1, eq. C),[11j] which is an abundant chemical feedstock, were described. These processes occur through a common mechanism involving hydrogen transfer from the primary alcohol proelectrophile to the allene pronucleophile to generate aldehyde-allyliridium pairs. For reactions conducted from the aldehyde oxidation level, hydrogen transfer from 2-propanol enables generation of the allyliridium nucleophile. Related cationic π-allyliridium species are potent electrophiles.[33] The π-allyliridium C,O-benzoate species are neutral, which confers amphiphilic character; that is, they are capable of catalyzing nucleophilic[13,14] or electrophilic allylation.[34] Indeed, neutral cyclometallated iridium complexes of PhanePhos also are capable of catalyzing enantioselective allene-mediated carbonyl allylations, as illustrated in couplings of CF3-allenes with methanol, an abundant feedstock alcohol (30 × 106 tons/yr) (Scheme 1, Eq. D),[11g] as well as iridium-(PhanePhos)-catalyzed couplings of 1,1-disubstituted allene with aqueous fluoral hydrate (Scheme 1, Eq. E).[11h] These processes enable enantioselective formation of acyclic quaternary carbon stereocenters.[35]
Scheme 1.
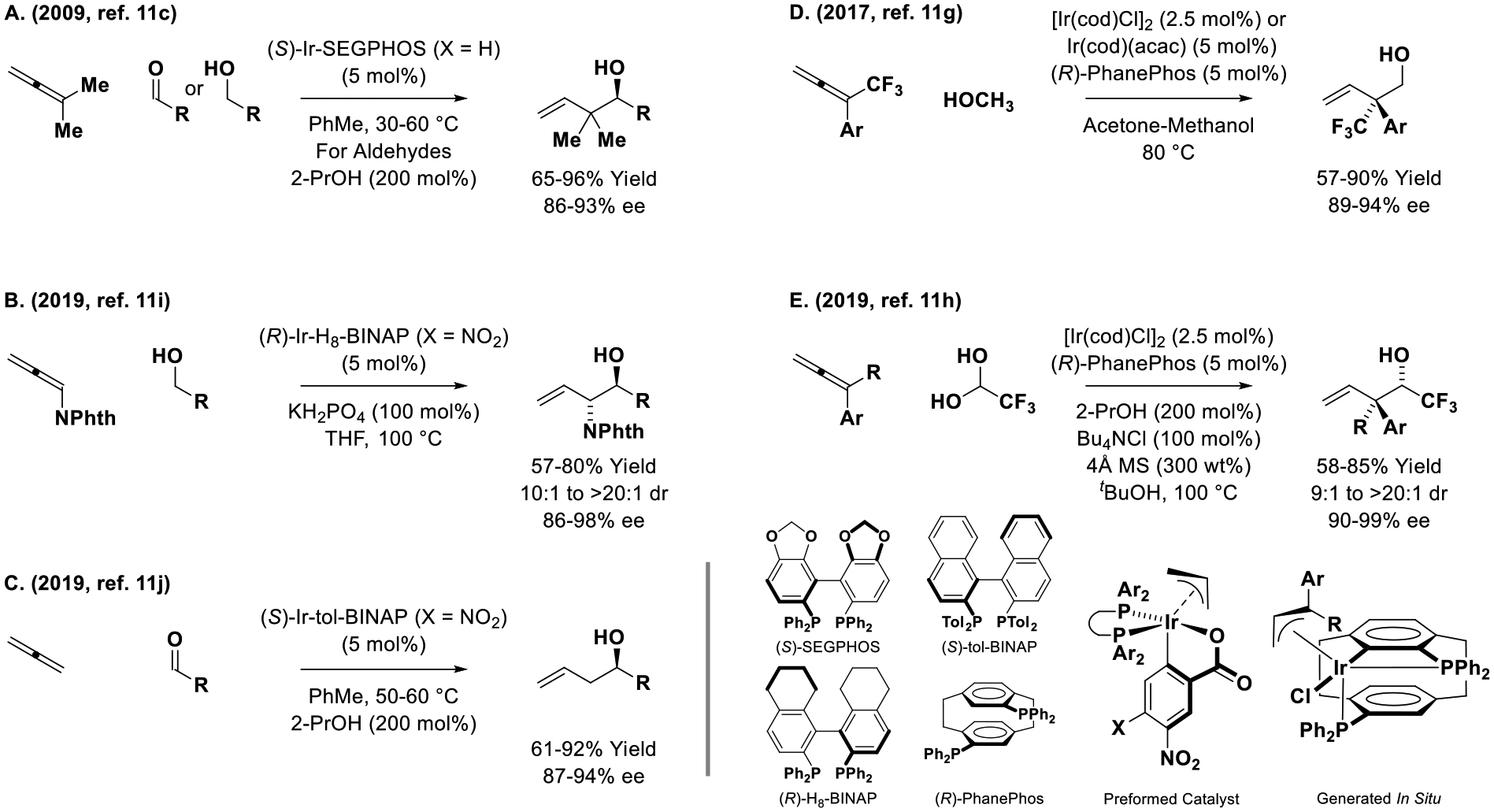
Allenes as chiral allylmetal pronucleophiles in iridium-catalyzed enantioselective aldehyde addition.
Copper is more electropositive than iridium (Pauling electronegativities of 1.9 vs 2.2), which enhances the nucleophilicity of hydrido- and organocopper species. Due to this effect, attempted copper-catalyzed aldehyde reductive couplings suffer from competing aldehyde reduction.[36] Conversely, the pronounced nucleophilicity of allylcopper species unlocks diverse enantioselective allene-C=X reductive couplings beyond aldehyde addition (Scheme 2).[25b,29a,37a–d] For example, following work on enantioselective copper-catalyzed acrylate-ketone[26,27] and vinyl azine-ketone[28] reductive couplings mediated by silane, Buchwald and coworkers reported highly diastereo- and enantioselective allene-mediated ketone allylations using (MeO)2MeSiH as reductant (Scheme 2, Eq. A).[29a] The next year (2019), this catalyst system was adapted to the use of gaseous allene (Scheme 2, Eq. B).[37a] The use of gaseous allene as an achiral allylmetal pronucleophile in metal-catalyzed carbonyl reductive coupling was first reported in 2007.[11b] Following Yu’s report of enantioselective copper-catalyzed diene-CO2 reductive coupling (Scheme 8, eq. A),[25a] Ding reported enantioselective copper-catalyzed allene-CO2 reductive couplings (Scheme 2, Eq. C).[25b] Under these conditions, excess (MeO)2MeSiH reduces the initially formed β,γ-unsaturated carboxylic acid to the primary alcohol. Identical compounds are obtained via redox-neutral iridium-(PhanePhos)-catalyzed C-C couplings of dienes with methanol,[46] which do not require an exogenous reductant (Scheme 7). Using a copper-(Ph-BPE) catalyst, allenes participate in (MeO)2MeSiH-mediated SN2′-type reductive couplings with N-(benzoyloxy)indazoles to furnish enantiomerically enriched C3-allyl indazoles (Scheme 2, Eq. D).[37b] Finally, allene-derived allylcopper species are capable of engaging in enantioselective acyl substitutions, as demonstrated in reductive couplings with anhydrides (Scheme 2, Eq. E)[37c] and fluoroformates (Scheme 2, Eq. F).[37d], as described by Ma and Buchwald, respectively. Catalytic enantioselective allene-mediated reductive imine allylation remains an unresolved challenge.[38]
Scheme 2.
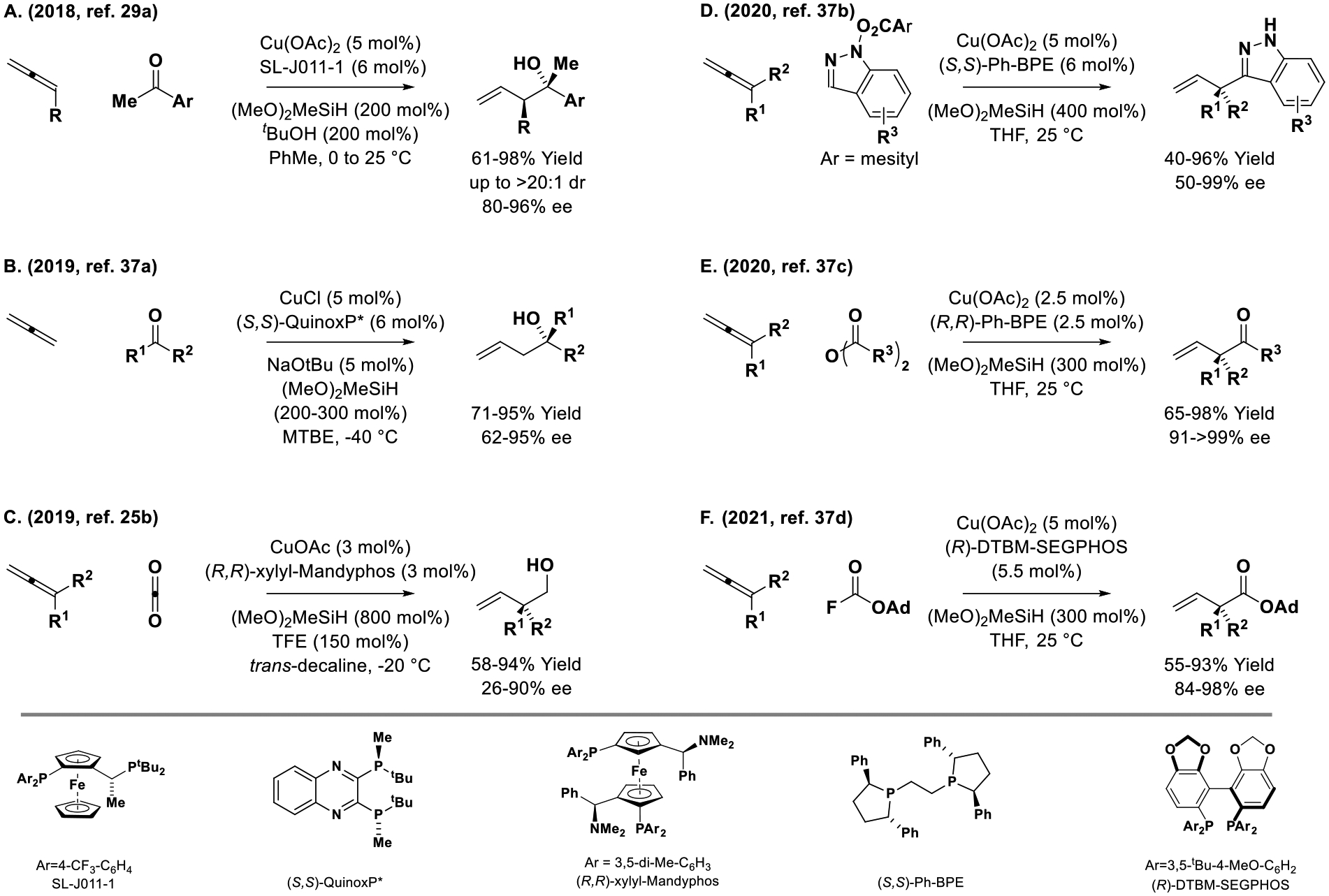
Allenes as chiral allylmetal pronucleophiles in copper-catalyzed enantioselective reductive couplings to C=X (X = O, N) π-electrophiles.
Scheme 8.
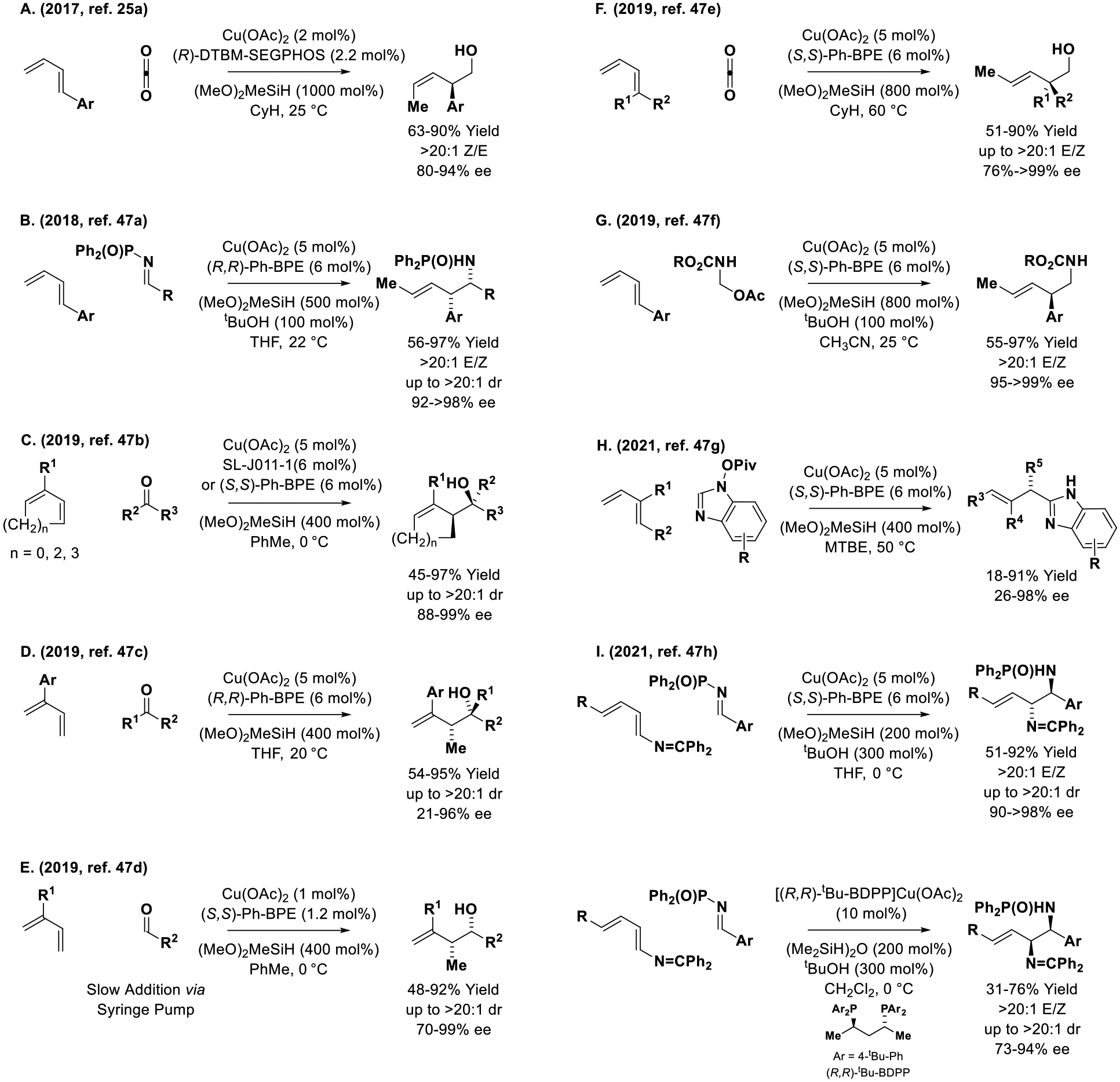
Dienes as chiral allylmetal pronucleophiles in copper-catalyzed enantioselective reductive couplings to C=X (X = O, N) π-electrophiles.
Scheme 7.
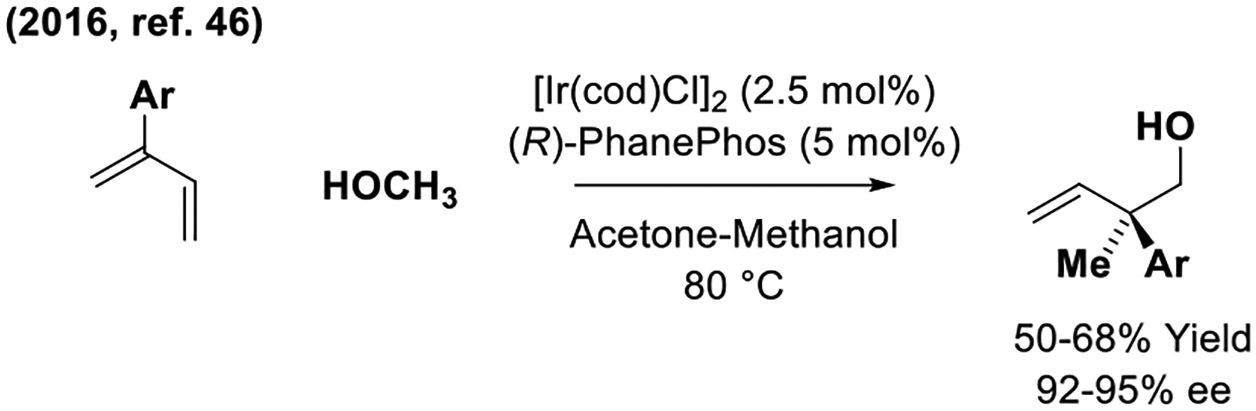
Dienes as chiral allylmetal pronucleophiles in iridium-catalyzed enantioselective methanol-mediated formaldehyde addition.
Two reports of copper-catalyzed reductive SN2′ reactions of allene pronucleophiles appeared contemporaneously in 2019 (Scheme 3).[39] In work by Xiong, allenes and allylic phosphates were exposed to a chiral copper catalyst modified by the indicated NHC ligand in the presence of TMDS, (Me2HSi)2O, to furnish 1,5-dienes bearing allylic stereocenters (Scheme 3, Eq. A).[39a] In work by Hoveyda, the same copper-NHC catalyst was used to reductively coupling allenylboron pronucleophiles (Scheme 3, Eq. B).[39b]
Scheme 3.
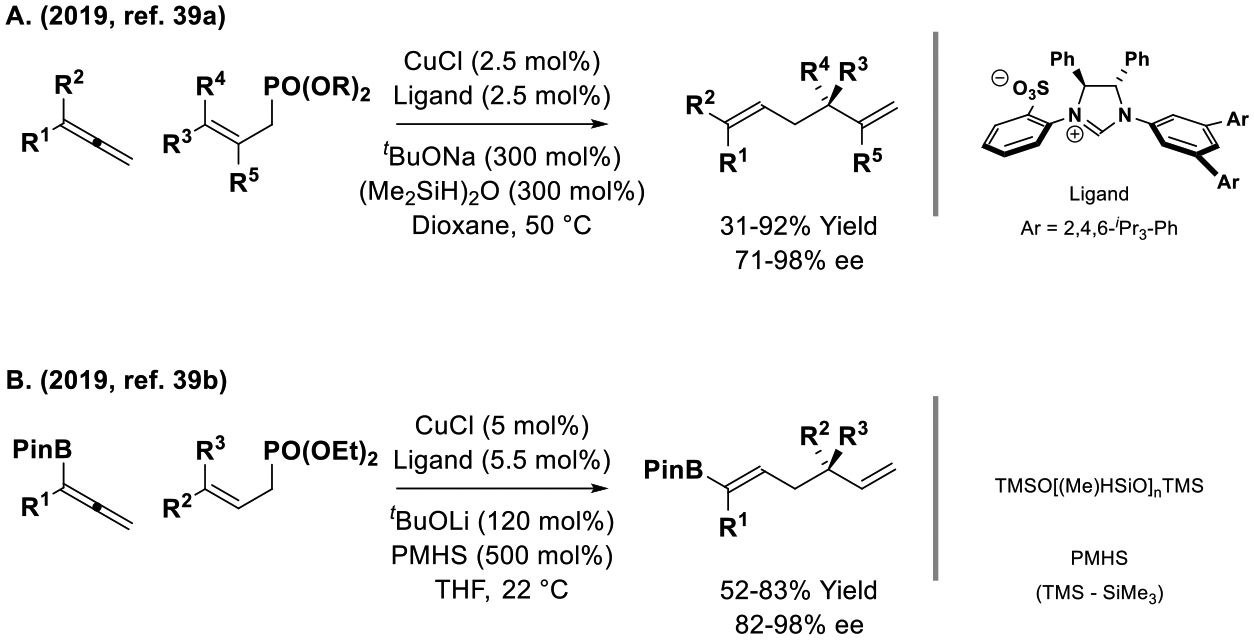
Enantioselective copper-catalyzed reductive SN2′ reactions of allene pronucleophiles.
Beyond iridium and copper, only two other metals have been employed as catalysts for enantioselective allene-mediated C=X allylation (Scheme 4). As described by the present author, ruthenium-(BINAP) catalysts promote reductive or redox-neutral C-C couplings of the indicated 1,1-disubstituted alkoxyallene with aldehydes or primary alcohols, respectively, to form enantiomerically enriched monoprotected syn-sec,tert-diols (Scheme 4, Eq. A).[12h] The observed syn-diastereoselectivity is unusual and was believed to arise via internal chelation of ruthenium to the benzhydryl oxygen, which directs intervention of (Z)-σ-alkoxyallylruthenium isomers that engage in stereospecific aldehyde addition by way Zimmerman-Traxler transition structures. Using a chiral palladium catalyst, Breit and coworkers developed a photo-mediated decarboxylative coupling of N-phenyl α-amino acids with alkoxyallenes to form vicinal amino alcohols with moderate levels of regio-, diastereo- and enantioselectivity (Scheme 4, Eq. B).[40] Although this reaction was not postulated to occur by way of nucleophilic allylpalladium species, formal products of imine allylation were obtained.
Scheme 4.
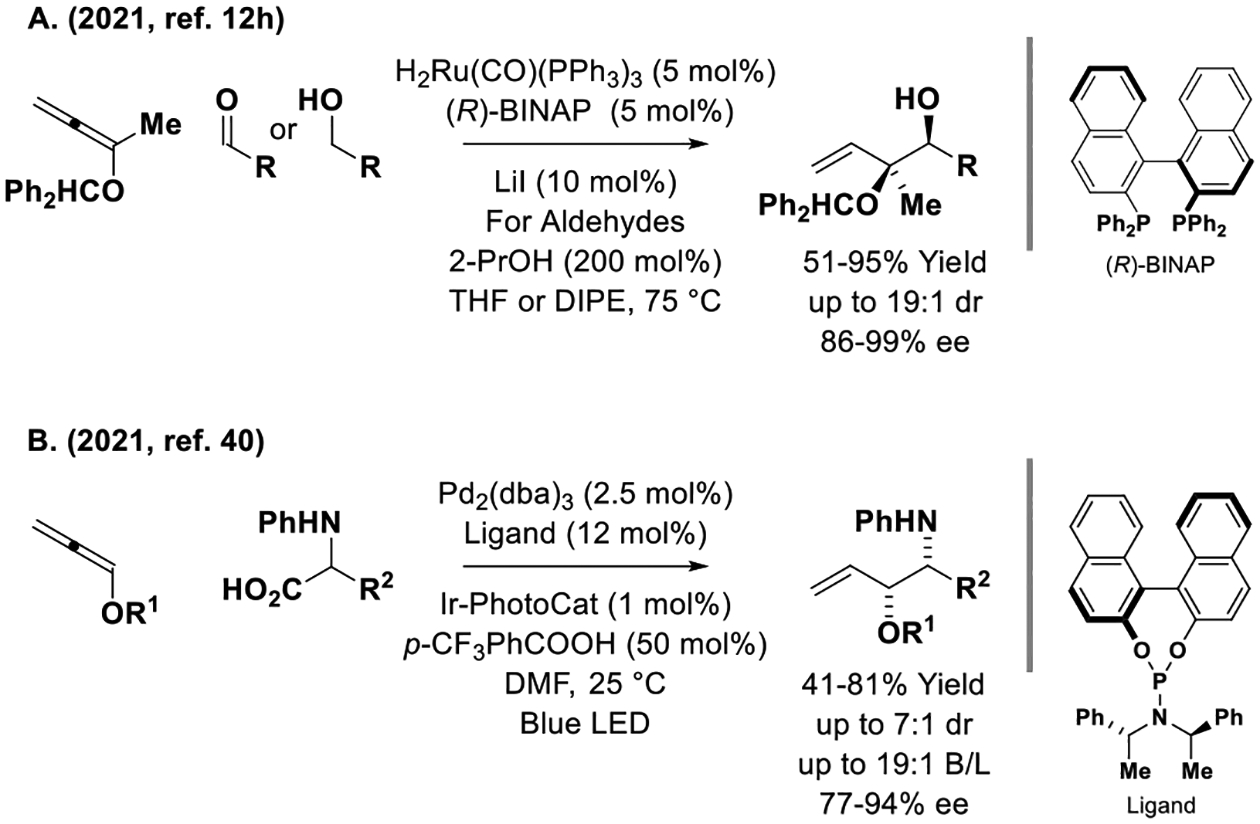
Enantioselective ruthenium- and palladium-catalyzed C-C coupling of allenes to C=X (X = O, NR) π-electrophiles.
III. Dienes as Allylmetal Pronucleophiles in Catalytic Enantioselective C=X Addition
The very first enantioselective diene-mediated reductive carbonyl allylation, which was reported by Sato in 2007, was conducted using a nickel catalyst modified by a C2-symmetric NHC-ligand (Scheme 5).[41] This silane-mediated diene-aldehyde reductive coupling delivers (Z)-homoallylic silyl ethers with complete control of alkene stereochemistry, however, high levels of enantiomeric enrichment were only observed using the esoteric diene, 1,4-diphenylbutadiene. The reader should note there exists a large body of nickel-catalyzed reductive diene-C=X (X = O, NR) homo-allylations, which fall outside the purview of this monograph.[42,43] The use of feedstock dienes (butadiene, isoprene, myrcene) in enantioselective nickel(0)-catalyzed diene-aldehyde reductive coupling remains an unmet challenge.
Scheme 5.
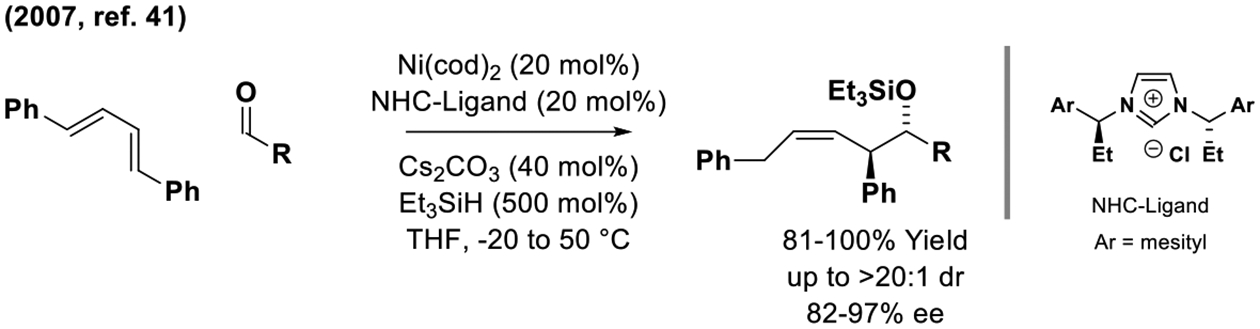
Dienes as chiral allylmetal pronucleophiles in nickel-catalyzed enantioselective aldehyde addition.
Informed by the development of achiral ruthenium catalysts for diene-mediated carbonyl allylation,[21] related enantioselective processes were devised (Scheme 6).[23] 2-Trialkylsilyl-butadienes undergo hydroruthenation to form geometrically defined allylruthenium nucleophiles that engage in stereospecific carbonyl addition to form products of crotylation with high levels of syn-diastereo- and enantioselectivity (Scheme 6, Eq. A).[23a] Brimble applied this method to the asymmetric syn-crotylation of chiral alcohols to form polyketide stereotriads, bypassing discrete formation of stereochemically labile chiral α-stereogenic aldehydes.[44] In 2012, the first catalytic enantioselective allylations employing butadiene were reported.[23b] Specifically, using a BINOL-derived phosphate counterion, catalytic carbonyl crotylation could be achieved with good levels of anti-diastereo- and enantioselectivity (Scheme 6, Eq. B).[23b] In contrast, the corresponding syn-diastereomers are formed using a chiral ruthenium catalyst modified by a TADDOL-phosphate counterion (Scheme 6, Eq. C).[23c] Computational studies on the origins of stereoselectivity in the latter process reveal intervention of a hydrogen bond between the aldehyde formyl C-H and phosphoryl oxygen as a key stereo-determining interaction.[45]
Scheme 6.
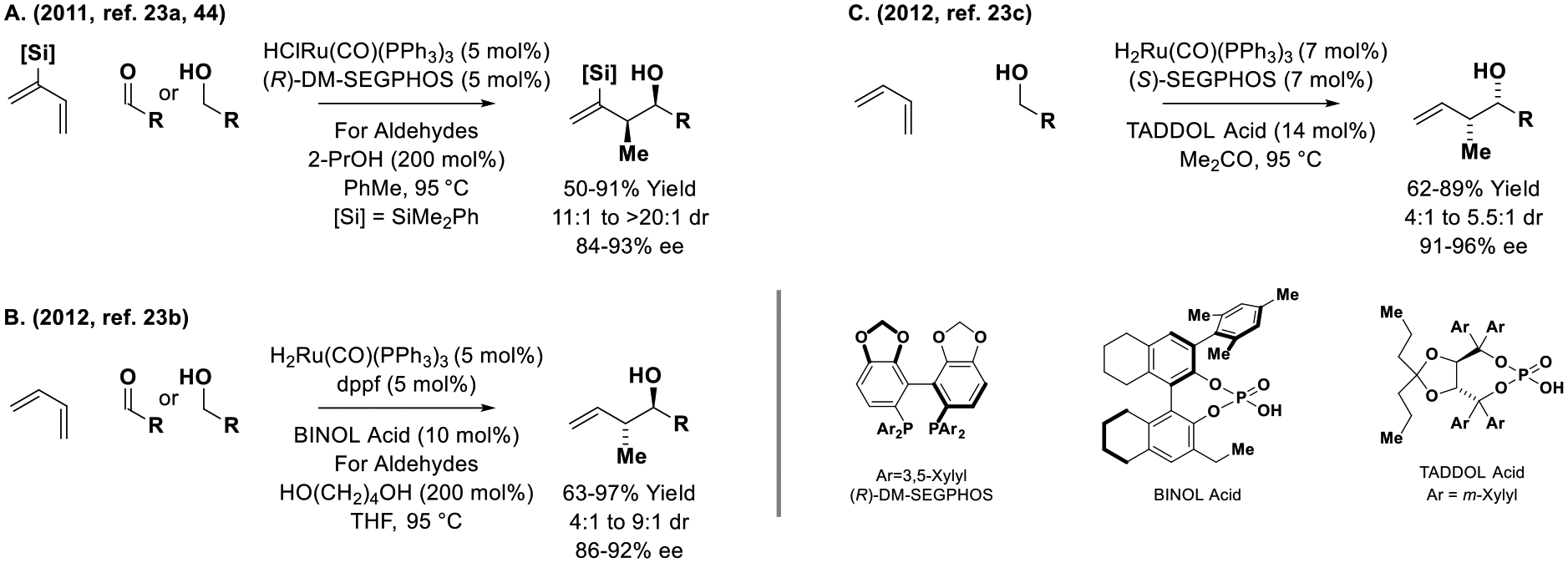
Dienes as chiral allylmetal pronucleophiles in ruthenium-catalyzed enantioselective aldehyde addition.
A single study of enantioselective iridium-catalyzed diene-mediated carbonyl allylation was reported by the present author in 2016 (Scheme 7).[46] In these processes, an iridium complex modified by PhanePhos promotes hydrogen transfer from 2-aryl butadienes to methanol to generate allyliridium-aldehyde pairs, which combine in a regioselective manner to form enantiomerically enriched products of hydrohydroxymethylation bearing quaternary carbon stereocenters. As determined in 2019,[11h] the active catalyst in this process is the cyclometallated iridium-(PhanePhos) complex depicted in Scheme 1.
Beginning in 2017 with the work of Yu,[25a] an impressive series of copper-catalyzed diene-mediated reductive C=X allylation reactions were reported that employ (MeO)2MeSiH as reductant (Scheme 8). In Yu’s study, 1-aryl butadienes were exposed to a copper-(DTBM-SEGPHOS) catalyst and (MeO)2MeSiH under an atmosphere of CO2 to furnish (Z)-β,γ-unsaturated carboxylic acids that are reduced further to the (Z)-2-arylpentenols (Scheme 8, Eq. A).[25a] In 2019, this method was extended to the use of 1,1-disubstituted butadienes, enabling formation of enantiomerically enriched acyclic quaternary carbon stereocenters (Scheme 8, Eq. F).[47e] Remarkably, for these two processes a ligand-dependent inversion of olefin geometry was observed. In 2018, Meng devised a related syn-diastereo- and enantioselective copper-(Ph-BPE)-catalyzed reductive coupling of 1-aryl butadienes with N-diphenylphosphoryl aldimines to form phenethylamines (Scheme 8, Eq. B).[47a] The following year, Buchwald and coworkers reported diastereo- and enantioselective copper-(Ph-BPE)-catalyzed reductive couplings of butadiene and higher cyclic dienes with ketones (Scheme 8, Eq. C).[47b] A report by Xiong on copper-(Ph-BPE)-catalyzed diene-ketone reductive coupling under essentially identical conditions appeared contemporaneously (Scheme 8, Eq. D).[47c] The copper-(Ph-BPE) catalyst system is also effective for reductive coupling of dienes with aldehydes, provided the aldehyde is introduced slowly via syringe pump to suppress competing reduction (Scheme 8, Eq. E).[47d] Pursuant to reports of achiral ruthenium-catalysts for diene-formaldimine reductive coupling to form products of hydroaminomethylation,[21d] Ge disclosed related enantioselective copper-(Ph-BPE)-catalyzed processes (Scheme 8, Eq. G).[47f] Buchwald applied the copper-(Ph-BPE) catalyst system to reductive SN2′ reactions of N-OPiv benzimidazoles to provide C2-allylated adducts with modest to excllent control of enantioselectivity (Scheme 8, Eq. H).[47g] In very recent work, Shao and Malcolmson reported reductive couplings of terminally disubstituted dienes bearing a N=CPh2 group that display a remarkable ligand-dependent divergence in diastereoselectivity (Scheme 8, Eq. I).[47h] Whereas copper-(Ph-BPE) promotes formation differentially protected anti-diamines, copper-(tBu-BDPP) catalyst promotes formation of the syn-diamines. In both cases, coupling occurs with high levels of regio-, diastereo- and enantioselectivity.
IV. Summary and Outlook
Although preformed carbanions have proven useful in numerous traditional methods for C-C bond formation, organometallic reagents often pose liabilities vis-à-vis safety, waste generation, functional group compatibility and cost. With ever-increasing frequency, it has been shown that the products of many classical C-C bond forming reactions are accessible via enantioselective metal-catalyzed reductive coupling, thus enabling replacement of stoichiometric organometallic reagents with safer and more tractable π-unsaturated feedstocks.[30,48] In the specific case of C-C bond forming allyl transfer reactions, a progression from stoichiometric allylmetal reagents to catalytic processes involving reductive generation of transient allylmetal species has begun to emerge. The majority of work in this area has involved allylmetal pronucleophiles that are activated via reductive C-X bond cleavage, for example, allyl halides or allyl acetates used in NHK-allylations[7] or Krische allylations.[14] As summarized in this Mini-Review, allenes and dienes represent a new and rapidly growing class of chiral allylmetal pronucleophiles that are unlocking transformations that have no counterpart in the classical lexicon of allyl metal chemistry. It is the authors’ hope that this survey of catalytic enantioselective allene- and diene-mediated allyl transfer processes will facilitate further progress in this burgeoning field of research.[49,50]
Acknowledgments
The Welch Foundation (F-0038) and the NIH (RO1-GM069445) are acknowledged for partial support of this research.
Biographies
Biographies and Photos

Professor Michael J. Krische obtained a B.S. degree in Chemistry from the University of California at Berkeley (1989), where he performed research under Professor Henry Rapoport. After a year abroad as a Fulbright Fellow, he initiated doctoral studies at Stanford University with Professor Barry Trost as a Veatch Graduate Fellow. Following receipt of his Ph.D. degree (1996), he joined the laboratory of Professor Jean-Marie Lehn at the Université Louis Pasteur as an NIH Post-Doctoral Fellow. In 1999, he joined the faculty at the University of Texas at Austin. He was promoted directly to the rank of full professor (2004) and shortly thereafter was appointed the Robert A. Welch Chair in Science (2007). Professor Krische has pioneered a broad, new class of C-C bond formations that merge the characteristics of carbonyl addition and catalytic hydrogenation, transfer hydrogenation and hydrogen auto-transfer.

Ming Xiang obtained his M.Sc. in Organic Chemistry from the Sichuan University, China (2017), where he conducted research under the supervision of Professor Jason J. Chruma. In Fall 2017, Ming entered the doctoral degree program at the University of Texas at Austin in the laboratory of Professor Michael J. Krische. Ming’s research is focused on iridium and ruthenium-catalyzed enantioselective carbonyl reductive couplings.

Dana E. Pfaffinger obtained a B.Sc. in Chemistry from Loyola Marymount University in 2017. After two years working at MD Anderson Cancer Center, Dana entered the doctoral degree program at the University of Texas at Austin as a Provost’s Graduate Excellence Fellow in Fall 2019. Dana is presently investigating the application of ruthenium-catalyzed reductive couplings to the total synthesis of type I polyketide natural products under the mentorship of Professor Michael J. Krische.
References
- [1].For selected reviews on asymmetric carbonyl allylation, see:; (a) Denmark SE, Fu J, Chem. Rev 2003, 103, 2763–2794; [DOI] [PubMed] [Google Scholar]; (b) Hall DG, Synlett. 2007, 11, 1644–1655; [Google Scholar]; (c) Hargaden GC, Guiry PJ, Adv. Synth. Catal 2007, 349, 2407–2424; [Google Scholar]; (d) Yus M, González-Gómez JC, Foubelo F, Chem. Rev 2011, 111, 7774–7854; [DOI] [PubMed] [Google Scholar]; (e) Huo H-X, Duvall JR, Huang M-Y, Hong R, Org. Chem. Front 2014, 1, 303–320; [Google Scholar]; (f) Spielmann K, Niel G, de Figueiredo RM, Campagne J-M, Chem. Soc. Rev 2018, 47, 1159–1173. [DOI] [PubMed] [Google Scholar]
- [2].(a) Saytzeff M, Saytzeff A, Sorokin Chem B. Ber. 1876, 9, 33–34; [Google Scholar]; (b) Kanonnikoff J, Saytzeff A, Ann. 1877, 185, 148–150; [Google Scholar]; (c) Saytzeff M, Saytzeff A, Ann. 1877, 185, 151–169; [Google Scholar]; (d) Béhal A, Sommelet M, Academie des Sciences, 1904, 138, 92. [Google Scholar]
- [3].(a) Mikhailov BM, Bubnov YN, Izv. Akad. Nauk SSSR, Ser. Khim 1964, 1874; [Google Scholar]; (b) König K, Neumann WP, Tetrahedron Lett. 1967, 8, 495–498; [Google Scholar]; (c) Hosomi A, Sakurai H, Tetrahedron Lett. 1976, 16, 1295–1298. [Google Scholar]
- [4].For selected examples of enantioselective carbonyl allylation mediated by chiral allylmetal reagents, see:; (a) Herold T, Hoffmann RW, Angew. Chem. Int. Ed. Engl 1978, 17, 768–769; [Google Scholar]; (b) Hoffmann RW, Herold T, Chem. Ber 1981, 114, 375–383; [Google Scholar]; (c) Hayashi T, Konishi M, Kumada M, J. Am. Chem. Soc 1982, 104, 4963–4965; [Google Scholar]; (d) Brown HC, Jadhav PK, J. Am. Chem. Soc 1983, 105, 2092–2093; [Google Scholar]; (e) Roush WR, Walts AE, Hoong LK, J. Am. Chem. Soc 1985, 107, 8186–8190; [Google Scholar]; (f) Reetz M, Pure Appl. Chem 1988, 60, 1607–1614; [Google Scholar]; (g) Short RP, Masamune S, J. Am. Chem. Soc 1989, 111, 1892–1894; [Google Scholar]; (h) Corey EJ, Yu C-M, Kim SS, J. Am. Chem. Soc 1989, 111, 5495–5496; [Google Scholar]; (i) Seebach D, Beck AK, Imwinkelzied R, Roggo S, Wonnacott A, Helv. Chim. Acta 1987, 70, 954–974; [Google Scholar]; (j) Riediker M, Duthaler RO, Angew. Chem., Int. Ed. Engl 1989, 28, 494–495; [Google Scholar]; (k) Panek JS, Yang M, J. Am. Chem. Soc 1991, 113, 6594–6600; [Google Scholar]; (l) Kinnaird JWA, Ng PY, Kubota K, Wang X, Leighton JL, J. Am. Chem. Soc 2002, 124, 7920–7921; [DOI] [PubMed] [Google Scholar]; (m) Hackman BM, Lombardi PJ, Leighton JL, Org. Lett 2004, 6, 4375–4377; [DOI] [PubMed] [Google Scholar]; (n) Wu TR, Shen L, Chong JM, Org. Lett 2004, 6, 2701–2704; [DOI] [PubMed] [Google Scholar]; (o) Burgos CH, Canales E, Matos K, Soderquist JA, J. Am. Chem. Soc 2005, 127, 8044–8049. [DOI] [PubMed] [Google Scholar]
- [5].For selected examples of catalytic enantioselective carbonyl allylation mediated by achiral allylmetal reagents, see:; (a) Furuta K, Mouri M, Yamamoto H, Synlett 1991, 561–562; [Google Scholar]; (b) Costa AL, Piazza MG, Tagliavini E, Trombini C, Umani-Ronchi A, J. Am. Chem. Soc 1993, 115, 7001–7002; [Google Scholar]; (c) Keck GE, Tarbet KH, Geraci LS, J. Am. Chem. Soc 1993, 115, 8467–8468; [Google Scholar]; (d) Denmark SE, Coe DM, Pratt NE, Griedel BD, J. Org. Chem 1994, 59, 6161–6163; [DOI] [PubMed] [Google Scholar]; (e) Wada R, Oisaki K, Kanai M, Shibasaki M, J. Am. Chem. Soc 2004, 126, 8910–8911; [DOI] [PubMed] [Google Scholar]; (f) Lou S, Moquist PN, Schaus SE, J. Am. Chem. Soc 2006, 128, 12660–12661; [DOI] [PubMed] [Google Scholar]; (f) Jain P, Antilla JC, J. Am. Chem. Soc 2010, 132, 11884–11886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].For selected examples of enantioselective Nozaki-Hiyama-Kishi reactions, see:; (a) Bandini M, Cozzi PG, Melchiorre P, Umani-Ronchi A, Angew. Chem., Int. Ed 1999, 38, 3357–3359; [DOI] [PubMed] [Google Scholar]; (b) Inoue M, Suzuki T, Nakada M, J. Am. Chem. Soc 2003, 125, 1140–1141; [DOI] [PubMed] [Google Scholar]; (c) Berkessel A, Mench D, Sklorz CA, Schröder M, Paterson I, Angew. Chem. Int. Ed 2003, 42, 1062–1065; [DOI] [PubMed] [Google Scholar]; (d) Y Lee J, Miller JJ, Hamilton SS, Sigman MS, Org. Lett 2005, 7, 1837–1839; [DOI] [PubMed] [Google Scholar]; (e) McManus HA, Cozzi PG, Guiry PJ, Adv. Synth. Catal 2006, 348, 551–558; [Google Scholar]; (f) Xia G, Yamamoto H, J. Am. Chem. Soc 2006, 128, 2554–2555; [DOI] [PubMed] [Google Scholar]; (g) Hargaden GC, Müller-Bunz H, Guiry PJ, Eur. J. Org. Chem 2007, 4235–4243; [Google Scholar]; (h) Hargaden GC, O’Sullivan TP, Guiry PJ, Org. Biomol. Chem 2008, 6, 562–566; [DOI] [PubMed] [Google Scholar]; (i) Zhang Z, Huang J, Ma B, Kishi Y, Org. Lett 2008, 10, 3073–3076; [DOI] [PubMed] [Google Scholar]; (j) White JD, Shaw S, Org. Lett 2011, 13, 2488–2491. [DOI] [PubMed] [Google Scholar]
- [7].For a review of recent advances in the enantioselective Nozaki-Hiyama-Kishi reactions, see:; Tian Q, Zhang G, Synthesis 2016, 48, 4038–4049and reference 1c. [Google Scholar]
- [8].For allene hydroboration-carbonyl allylation to give racemic and enantiomerically enriched products, respectively, see:; (a) Mehrotra I, Devaprabhakara D, J. Organomet. Chem 1973, 51, 93–98; [Google Scholar]; (b) Brown HC, Jadhav PK, Tetrahedron Lett. 1984, 25, 1215–1218. [Google Scholar]
- [9].For allene hydroalumination- and hydrozirconation-carbonyl allylation to give racemic products, see:; (a) Nagahara S, Maruoka K, Doi Y, Yamamoto H, Chem. Lett 1990, 1595–1598; [Google Scholar]; (b) Chino M, Matsumoto T, Suzuki K, Synlett 1994, 5, 359–363. [Google Scholar]
- [10].(a) Chang H-M, Cheng C-H, Org. Lett 2000, 2, 3439–3442; [DOI] [PubMed] [Google Scholar]; (b) Lee S, Lee S, Lee Y, Org. Lett 2020, 22, 5806–5810. [DOI] [PubMed] [Google Scholar]
- [11].For iridium-catalyzed carbonyl allylations employing allene pronucleophiles, see:; (a) Skucas E, Bower JF, Krische MJ, J. Am. Chem. Soc 2007, 129, 12678–12679; [DOI] [PubMed] [Google Scholar]; (b) Bower JF, Skucas E, Patman RL, Krische MJ, J. Am. Chem. Soc 2007, 129, 15134–15135; [DOI] [PubMed] [Google Scholar]; (c) Han SB, Kim I-S, Han H, Krische MJ, J. Am. Chem. Soc 2009, 131, 6916–6917; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Itoh J, Han SB, Krische MJ, Angew. Chem. Int. Ed 2009, 48, 6313–6434; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Bechem B, Patman RL, Hashmi ASK, Krische MJ, J. Org. Chem 2010, 75, 1795–1798; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Moran J, Preetz A, Mesch RA, Krische MJ, Nature Chem. 2011, 3, 287–290; [DOI] [PubMed] [Google Scholar]; (g) Holmes M, Nguyen KD, Schwartz LA, Luong T, Krische MJ, J. Am. Chem. Soc 2017, 139, 8114–8117; [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Schwartz LA, Holmes M, Brito GA, Gonçalves TP, Richardson J, Ruble JC, Huang K-W, Krische MJ, J. Am. Chem. Soc 2019, 141, 2087–2096; [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Spielmann K, Xiang M, Schwartz LA, Krische MJ, J. Am. Chem. Soc 2019, 141, 14136–14141; [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Kim SW, Meyer CC, Mai BK, Liu P, Krische MJ, ACS Catal. 2019, 9, 9158–9163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].For ruthenium-catalyzed carbonyl and imine allylations employing allene pronucleophiles, see:; (a) Ngai M-Y, Skucas E, Krische MJ, Org. Lett 2008, 10, 2705–2708; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Skucas E, Zbieg JR, Krische MJ, J. Am. Chem. Soc 2009, 131, 5054–5055; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zbieg JR, McInturff EL, Krische MJ, Org. Lett 2010, 12, 2514–2516; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zbieg JR, McInturff EL, Leung JC, Krische MJ, J. Am. Chem. Soc 2011, 133, 1141–1144; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Sam B, Montgomery TP, Krische MJ, Org. Lett 2013, 15, 3790–3793; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Sam B, Luong T, Krische MJ, Angew. Chem. Int. Ed 2015, 54, 5465–5469; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Oda S, Sam B, Krische MJ, Angew. Chem. Int. Ed 2015, 54, 8525–8528; [DOI] [PubMed] [Google Scholar]; (h) Xiang M, Pfaffinger D, Ortiz E, Krische MJ, J. Am. Chem. Soc 2021, 143, 8849–8854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].(a) Kim IS, Ngai M-Y, Krische MJ, J. Am. Chem. Soc 2008, 130, 6340–6341; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kim IS, Ngai M-Y, Krische MJ, J. Am. Chem. Soc 2008, 130, 14891–14899; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kim IS, Han SB, Krische MJ, J. Am. Chem. Soc 2009, 131, 2514–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].For recent reviews of π-allyliridium-C,O-benzoate-catalyzed carbonyl reductive couplings of allyl acetate, see:; (a) Kim SW, Zhang W, Krische MJ, Acc. Chem. Res 2017, 50, 2371–2380; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Santana CG, Krische MJ, ACS Catal. 2021, 11, 5572–5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].(a) Yang M, Yamamoto K, Otake K, Ando M, Takahase K, Tetrahedron Lett. 1970, 11, 3843–3846; [Google Scholar]; (b) Yang M, Ando M, Takase K, Tetrahedron Lett. 1971, 12, 3529–3532. [Google Scholar]
- [16].Sato F, Ishikawa H, Sato M, Tetrahedron Lett. 1980, 21, 365–368. [Google Scholar]
- [17].(a) Kira M, Kobayashi M, Sakurai H, Tetrahedron Lett. 1987, 28, 4081–4084; [Google Scholar]; (b) Satoh M, Nomoto Y, Miyaura N, Suzuki A, Tetrahedron Lett. 1989, 30, 3789–3792; [Google Scholar]; (c) Masuyama Y, Tsunoda M, Kurusu Y, J. Chem. Soc. Chem. Commun 1994, 1451–1452; [Google Scholar]; (d) Takai K, Toratsu C,. J. Org. Chem 1998, 63, 6450–6451. [Google Scholar]
- [18].Brown HC, Jadhav PK, Bhat KS. J. Am. Chem. Soc 1985, 107, 2564–2565. [Google Scholar]
- [19].(a) Takimoto M, Hiraga Y, Sato Y, Mori M, Tetrahedron Lett. 1998, 39, 4543–4546; [Google Scholar]; (b) Sato Y, Hinata Y, Seki R, Oonishi Y, Saito N, Org. Lett 2007, 9, 5597–5599. [DOI] [PubMed] [Google Scholar]
- [20].Bareille L, Le Gendre P, Moïse C, Chem. Commun 2005, 775–777. [DOI] [PubMed] [Google Scholar]
- [21].(a) Shibahara F, Bower JF, Krische MJ, J. Am. Chem. Soc 2008, 130, 6338–6339; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Smejkal T, Han H, Breit B, Krische MJ, J. Am. Chem. Soc 2009, 131, 10366–10367; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Köpfer A, Sam B, Breit B, Krische MJ, Chem. Sci 2013, 4, 1876–1880; [Google Scholar]; (d) Oda S, Franke J, Krische MJ, Chem. Sci 2016, 7, 136–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].(a) Kimura M, Nojiri D, Fukushima M, Oi S, Sonoda Y, Inoue Y, Org. Lett 2009, 11, 3794–3797; [DOI] [PubMed] [Google Scholar]; (b) Bower JF, Patman RL, Krische MJ, Org. Lett 2008, 10, 1033–1035; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zbieg JR, Fukuzumi T, Krische MJ, Adv. Synth. Catal 2010, 352, 2416–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].(a) Zbieg JR, Moran J, Krische MJ, J. Am. Chem. Soc 2011, 133, 10582–10586; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zbieg JR, Yamaguchi E, McInturff EL, Krische MJ, Science 2012, 336, 324–327; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) McInturff EL, Yamaguchi E, Krische MJ, J. Am. Chem. Soc 2012, 134, 20628–20631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].(a) Takaya J, Iwasawa N, J. Am. Chem. Soc 2008, 130, 15254–15255; [DOI] [PubMed] [Google Scholar]; (b) Takaya J, Sasano K, Iwasawa N, Org. Lett 2011, 13, 1698–1701. [DOI] [PubMed] [Google Scholar]
- [25].(a) Gui Y-Y, Hu N, Chen X-W, Liao L-L, Ju T, Ye J-H, Zhang Z, Li J, Yu D-G, J. Am. Chem. Soc 2017, 139, 17011–17014; [DOI] [PubMed] [Google Scholar]; (b) Qui J, Gao S, Li C, Zhang L, Wang Z, Wang X, Ding K, Chem. Eur. J 2019, 25, 13874–13878. [DOI] [PubMed] [Google Scholar]
- [26].(a) Zhao D, Oisaki K, Kanai M, Shibasaki M, Tetrahedron Lett. 2006, 47, 1403–1407; [Google Scholar]; (b) Zhao D, Oisaki K, Kanai M, Shibasaki M, J. Am. Chem. Soc 2006, 128, 14440–14441. [DOI] [PubMed] [Google Scholar]
- [27].Deschamp J, Chuzel O, Hannedouche J, Riant O, Angew. Chem. Int. Ed 2006, 45, 1292–1297. [DOI] [PubMed] [Google Scholar]
- [28].Saxena A, Choi B, Lam HW, J. Am. Chem. Soc 2012, 134, 8428–8431. [DOI] [PubMed] [Google Scholar]
- [29].(a) Tsai EY, Liu RY, Yang Y, Buchwald SL, J. Am. Chem. Soc 2018, 140, 2007–2011; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li C, Liu RY, Jesikiewicz LT, Yang Y, Liu P, Buchwald SL, J. Am. Chem. Soc 2019, 141, 5062–5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].For selected reviews on allene/diene C=X reductive coupling, see:; (a) Metal Catalyzed Reductive C-C Bond Formation. Topics in Current Chemistry; Krische MJ, Ed.; Springer-Verlag: Berlin, Heidelberg, 2007; Vol. 279; [Google Scholar]; (b) Ketcham JM, Shin I, Montgomery TP, Krische MJ, Angew. Chem. Int. Ed 2014, 53, 9142–9150; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Nguyen KD, Park BY, Luong T, Sato H, Garza VJ, Krische MJ, Science 2016, 354, aah5133; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Holmes M, Schwartz LA, Krische MJ, Chem. Rev 2018, 118, 6026–6052; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Blieck R, Taillefer M, Monnier F, Chem. Rev 2020, 120, 13545–13598. [DOI] [PubMed] [Google Scholar]
- [31].For a review on the use of alkynes as electrophilic or nucleophilic allylmetal precursors in transition metal catalysis, see:; Haydl AM, Breit B, Liang T, Krische MJ, Angew. Chem. Int. Ed 2017, 56, 11312–11325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].For selected reviews on multi-component allene/diene C=X couplings, see:; (a) Montgomery J, Angew. Chem. Int. Ed 2004, 43, 3890–3908; [DOI] [PubMed] [Google Scholar]; (b) Semba K, Fujihara T, Terao J, Tsuji Y, Tetrahedron 2015, 71, 2183–2197; [Google Scholar]; (c) Pulis AP, Yeung K, Procter DJ, Chem. Sci 2017, 8, 5240–5247; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Xiong Y, Sun Y, Zhang G, Tetrahedron Lett. 2018, 59, 347–355; [Google Scholar]; (e) Whyte A, Torelli A, Mirabi B, Zhang A, Lautens M, ACS Catal. 2020, 10, 11578–11622; [Google Scholar]; (f) Talbot FJT, Dherbassy Q, Manna S, Shi C, Zhang S, Howell GP, Perry GJP, Procter DJ, Angew. Chem. Int. Ed 2020, 59, 20278–20289; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Perry GJP, Jia T, Procter DJ, ACS Catal. 2020, 10, 1485–1499; [Google Scholar]; (h) Das KK, Manna S, Panda S, Chem. Commun 2021, 57, 441–459. [DOI] [PubMed] [Google Scholar]
- [33].For a recent review on iridium-catalyzed allylic substitutions, see:; Cheng Q, Tu H-F, Zheng C, Qu J-P, Helmchen G, You S-L, Chem. Rev 2019, 119, 1855–1969. [DOI] [PubMed] [Google Scholar]
- [34].For π-allyliridium-C,O-benzoate-catalyzed allylic substitutions, see:; (a) Meza AT, Wurm T, Smith L, Kim SW, Zbieg JR, Stivala CE, Krische MJ, J. Am. Chem. Soc 2018, 140, 1275–1279; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kim SW, Schwartz LA, Zbieg JR, Stivala CE, Krische MJ, J. Am. Chem. Soc 2019, 141, 671–676; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kim SW, Schempp TT, Zbieg JR, Stivala CE, Krische MJ, Angew. Chem. Int. Ed 2019, 58, 7762–7766; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zhang T-Y, Deng Y, Wei K, Yang Y-R, Org. Lett 2021, 23, 1086–1089. [DOI] [PubMed] [Google Scholar]
- [35].For a review on catalytic enantioselective formation of acyclic quaternary carbon stereocenters, see:; Feng J, Holmes M, Krische MJ, Chem. Rev 2017, 117, 12564–12580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36]. As described in reference [47d], slow addition of aldehyde via syringe pump is required to suppress competing aldehyde reduction in copper-catalyzed reductive couplings.
- [37].(a) Liu RY, Zhou Y, Yang Y, Buchwald SL, J. Am. Chem. Soc 2019, 141, 2251–2256; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ye Y, Kevlishvili I, Feng S, Liu P, Buchwald SL, J. Am. Chem. Soc 2020, 142, 10550–10556; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yuan Y, Zhang X, Qian H, Ma S, Chem. Sci 2020, 11, 9115–9121; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Feng S, Buchwald SL. J. Am. Chem. Soc 2021, 143, 4935–4941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Liu RY, Yang Y, Buchwald SL, Angew. Chem. Int. Ed 2016, 128, 14077–14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].(a) Xu G, Fu B, Zhao H, Li Y, Zhang G, Wang Y, Xiong T, Zhang Q, Chem. Sci 2019, 10, 1802–1806; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sun Y, Zhou Y, Shi Y, del Pozo J, Torker S, Hoveyda AJ, J. Am. Chem. Soc 2019, 141, 12087–12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zheng J, Nikbakht A, Breit B, ACS Catal. 2021, 11, 3343–3350. [Google Scholar]
- [41].Sato Y, Hinata Y, Seki R, Oonishi Y, Saito N, Org. Lett 2007, 9, 5597–5599. [DOI] [PubMed] [Google Scholar]
- [42].For a review on nickel-catalyzed reductive diene-C=X (X = O, NR) homoallylation, see:; Kimura M, Tamaru Y, Top. Curr. Chem 2007, 279, 173–207. [Google Scholar]
- [43].For enantioselective nickel-catalyzed homoallylation using 1,4-diphenylbutadiene, see:; Yang Y, Zhu S-F, Duan H-F, Zhou C-Y, Wang L-X, Zhou Q-L, J. Am. Chem. Soc 2007, 129, 2248–2249. [DOI] [PubMed] [Google Scholar]
- [44].Pantin M, Hubert JG, Söhnel T, Brimble MA, Furkert DP, J. Org. Chem 2017, 82, 11225–11229. [DOI] [PubMed] [Google Scholar]
- [45].Grayson MN, Krische MJ, Houk KN, J. Am. Chem. Soc 2015, 137, 8838–8850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Nguyen KD, Herkommer D, Krische MJ, J. Am. Chem. Soc 2016, 138, 14210–14213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].(a) Li M, Wang J, Meng F, Org. Lett 2018, 20, 7288–7292; [DOI] [PubMed] [Google Scholar]; (b) Li C, Liu RY, Jesikiewicz LT, Yang Y, Liu P, Buchwald SL, J. Am. Chem. Soc 2019, 141, 5062–5070; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Fu B, Yuan X, Li Y, Wang Y, Zhang Q, Xiong T, Zhang Q, Org. Lett 2019, 21, 3576–3580; [DOI] [PubMed] [Google Scholar]; (d) Li C, Shin K, Liu RY, Buchwald SL, Angew. Chem., Int. Ed 2019, 58, 17074–17080; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Chen X-W, Zhu L, Gui Y-Y, Jing K, Jiang Y-X, Bo Z-Y, Lan Y, Li J, Yu D-G, J. Am. Chem. Soc 2019, 141, 18825–18835; [DOI] [PubMed] [Google Scholar]; (f) You Y, Pham QV, Ge S, CCS Chem. 2019, 1, 455–463; [Google Scholar]; (g) Knippel JL, Ye Y, Buchwald SL, Org. Lett 2021, 23, 2153–2157; [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Zhou P, Shao X, Malcolmson SJ, ChemRxiv, DOI: 10.26434/chemrxiv.14347661.v1. [DOI] [Google Scholar]
- [48].For selected reviews on the use of π-unsaturated feedstocks as surrogates to preformed organometallic reagents in catalytic C-C coupling, see:; (a) Ngai M-Y, Kong J-R, Krische MJ, J. Org. Chem 2007, 72, 1063–1072; [DOI] [PubMed] [Google Scholar]; (b) Iida H, Krische MJ, Top. Curr. Chem 2007, 279, 77–104; [Google Scholar]; (c) Patman RL, Bower JF, Kim IS, Krische MJ, Aldrichimica Acta 2008, 41, 95–104; [PMC free article] [PubMed] [Google Scholar]; (d) Bower JF, Kim IS, Patman RL, Krische MJ, Angew. Chem. Int. Ed 2009, 48, 34–46; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Bower JF, Krische MJ, Top. Organomet. Chem 2011, 34, 107–138; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Sam B, Breit B, Krische MJ, Angew. Chem. Int. Ed 2015, 54, 3267–3274; [DOI] [PubMed] [Google Scholar]; (g) Ambler BR, Woo SK, Krische MJ, ChemCatChem. 2019, 11, 324–332; [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Doerksen RS, Meyer CC, Krische MJ, Angew. Chem. Int. Ed 2019, 58, 14055–14064; [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Meyer CC, Ortiz E, Krische MJ, Chem. Rev 2020, 120, 3721–3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].While outside the scope of this review, catalytic asymmetric C-C couplings of allenamides bearing chiral auxiliaries have been described:; (a) Klake RK, Gargaro SL, Gentry SL, Elele SO, Sieber JD, Org. Lett 2019, 21, 7992–7998; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gargaro SL, Klake RK, Burns KL, Elele SO, Gentry SL, Sieber JD, Org. Lett 2019, 21, 9753–9758; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Agrawal T, Martin RT, Collins S, Wilhelm Z, Edwards MD, Gutierrez O, Sieber JD, J. Org. Chem 2021, 86, 5026–5046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Although enantioselective borylative carbonyl allylations of allene and diene pronucleophiles are outside the scope of this review, the author would like to bring two early examples of such transformations to the attention of the reader:; (a) Morgan JB, Morken JP, Org. Lett 2003, 5, 2573–2575; [DOI] [PubMed] [Google Scholar]; (b) Woodward AR, Burks HE, Chan LM, Morken JP, Org. Lett 2005, 7, 5505–5507. [DOI] [PubMed] [Google Scholar]