

Abstract
We created optogenetic tools to control RhoA GTPase, a central regulator of actin organization and actomyosin contractility. RhoA GTPase, or its upstream activator ARHGEF11, were fused to BcLOV4, a photoreceptor that can be dynamically recruited to the plasma membrane by a light-regulated protein-lipid electrostatic interaction with the inner leaflet. Direct membrane recruitment of these proteins induced potent contractile signaling sufficient to separate adherens junctions with as little as one pulse of blue light. Induced cytoskeletal morphology changes were dependent on the alignment of the spatially patterned stimulation with the underlying cell polarization. RhoA-mediated cytoskeletal activation drove YAP nuclear localization within minutes and consequent mechanotransduction verified by YAP-TEAD transcriptional activity. These single-transgene tools do not require protein binding partners for dynamic membrane localization, and permit spatiotemporally precise control over RhoA signaling to advance the study of its diverse regulatory roles in cell migration, morphogenesis, and cell cycle maintenance.
Keywords: optogenetics, light-oxygen-voltage, RhoA
Graphical Abstract
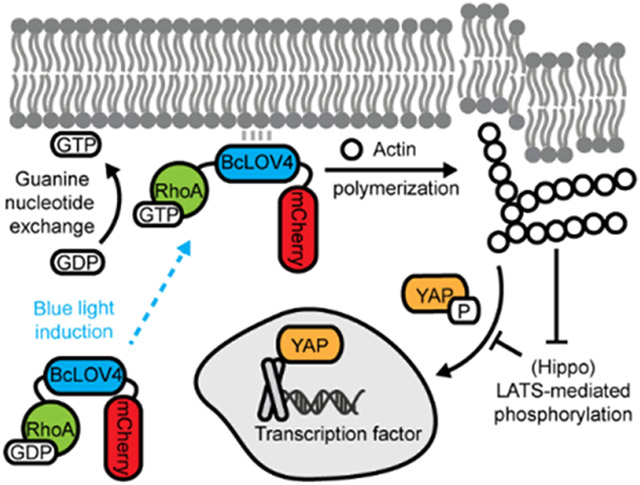
This article describes the creation of optogenetic tools for inducing RhoA-mediated signaling by dynamic membrane recruitment of either RhoA GTPase or its GEF activator upon blue light stimulation. The tools are simple to use because they only require the expression and visualization of one protein and can potently drive cytoskeletal contractility and consequent cytomechanical activation of YAP/TEAD-dependent transcriptional mechanotransduction.
1. Introduction
RhoA, a member of the Rho-family of small GTPases, centrally regulates actin organization and actomyosin contractility in cell migration, cell cycle maintenance, and developmental morphogenesis.[1-2] Key amongst its diverse roles, RhoA signaling dynamics coordinate actin stress fiber formation that determines how cells generate cytoskeletal tension to transmit mechanical forces across the cell, across neighboring cell-cell junctions, and to the extracellular matrix (ECM) via focal adhesions.[3] Thus, new tools for inducible control over RhoA activity may greatly enhance understanding of cytoskeletal dynamics and mechanotransduction.[4-5]
Optogenetics is highly attractive for this purpose owing to its high spatiotemporal precision vs. pharmacological and genetic techniques that can be encumbered by slow uptake/washout kinetics and pleotropic effects. Because small GTPases and their activating GEFs (guanine nucleotide exchange factors) signal at the plasma membrane, optogenetic membrane localization techniques are effective for inducible control over their function, where cytosol-sequestered proteins are dynamically recruited to the cytosol-facing inner leaflet of the plasma membrane to upregulate their signaling.[6] Based on earlier reported chemically inducible dimerization (CID)-based approaches for RhoA membrane recruitment,[7-8] optogenetic heterodimerization and photoactivatable chemically induced dimerization between a photosensory protein and a protein binding partner (one of which is membrane localized) have been widely used to control upstream RhoA-activating GEFs[9-14] and phosphatases.[15] The heterodimerization strategy is sensitive to the stoichiometry of the two components, and thus may require expression level-tuning by clonal cell line selection and/or multiple fluorescent tags (i.e. separate for each component) at the expense of optical bandwidth otherwise useful for visualizing other fluorescent probes.[16-18] Previously, we reported the direct optogenetic control over RhoA GTPase itself by another mechanism of inducible clustering of RhoA fused to an oligomerizing form of plant cryptochrome, which presumably increases the binding avidity of the GTPase to membrane GEFs. However, this system has limited spatial resolution due to extensive cytosolic diffusion beyond the optical stimulation field prior to stable membrane localization post-oligomerization.[19]
Recently, we reported that BcLOV4, a light-oxygen-voltage (LOV) flavoprotein from Botrytis cinerea, rapidly translocates to the plasma membrane in mammalian cells via a blue light-regulated electrostatic protein-lipid interaction (PLI) with the inner leaflet.[20-21] This direct interaction with the membrane itself is powerful for creating “single-component” tools for dynamic membrane recruitment of fused peripheral membrane proteins, without the obligate heterodimerization- or self-oligomerization protein partners of the aforementioned (PPI) protein-protein interaction-based systems.[22] We previously leveraged the intrinsic membrane-binding capability of BcLOV4 fused to Rho-family Cdc42-GEF and Rac1 GTPase proteins to induce filopodial and lamellipodial protrusions.[6,23] Here, we report the engineering of single-component optogenetic RhoA GTPase and ARHGEF11 to potently drive actomyosin contractility, stress fiber formation, and rapid activation of transcriptional mechanotransduction.
2. Results
2.1. Engineering of single-component tools
The genetic constructs were designed and engineered as previously described.[23] We screened domain arrangement combinations of BcLOV4, mCherry fluorescent reporter, and either RhoA or the catalytic DH (Dbl-homology) domain of RhoA-activating ARHGEF11. The domains were each separated by flexible glycine-serine-rich (GGGS)2 linkers, and the six respective domain arrangements were tested for their respective expression levels, subcellular distribution characteristics, and inducible translocation efficiency in HEK293T cells. For opto-RhoA, the most favorable domain arrangement was an N-terminal fusion to BcLOV4-mCherry (RhoA-BcLOV4-mCherry) (Figure 1 and S1), and in opto-RhoGEF, it was BcLOV4-ARHGEF11-mCherry (Figure S2 and S3).
Figure 1.

Single-component optogenetic control over RhoA signaling. a. Schematized induction of cytoskeletal changes and contractile signaling in response to opto-RhoA activation by dynamic membrane recruitment. b. Epifluorescence micrographs of HEK cells expressing opto-RhoA, visualized by mCherry. i. Trailing edge contraction in two adjacent cells. ii. Four-cell adherens junction separation. White box = spatially patterned blue light illumination field, stimulated at 1.6% duty ratio. Dotted yellow line = cell boundary mask in the dark-adapted state. Scale = 10 μm.
The cytoskeletal response to digital micromirror device (DMD)-patterned illumination was pronounced (Figure 1 and S3); sparse pulsatile stimulation (duty ratio φ = 1.6% or 1 s per minute; λ = 450 nm at 15 mW/cm2) potently induced contractility sufficient to separate intercellular adherens junctions (Figure 1b, and Video S1 and S2) and create membrane blebs, presumably from transient delamination of the membrane from the actomyosin network (Figure S3b). The duty ratio was initially chosen to ensure that the optogenetic activity was not photochemically limited, by providing one flavin photochemistry-saturating pulse per membrane association-dissociation cycle.
To our initial surprise, a single 5 s-pulse of whole-field or unpatterned stimulation was also sufficient to induce extensive morphological changes (Figure S4). This feature of opto-RhoA and -RhoGEF differs from our previous work with single-component Rac1 GTPase or Intersectin Cdc42-GEF, which do not produce as dramatic protrusions without DMD patterning or (ROI) region-of-interest selection and repeated stimulation. The ability to use unpatterned stimulation to drive morphological changes over a whole field-of-view (FOV) facilitated experimental throughput, statistical powering, and blinding for automated data analysis for systematic characterization of optogenetic efficacy (Figure 2a).
Figure 2.

Optogenetic induction of contractility in response to unpatterned wholefield stimulation. a. Image analysis workflow. (i) Whole FOV videos cropping (yellow) to contain one cell. (ii) Cell contour (black) and bounding rectangle (purple) definition from the initial frame. (iii) Iterative cell rotation (5° increments). The angle that maximizes rectangle height and positions the nucleus closer to the top (as a leading edge marker, see Supplementary Figure 5) is applied to all frames to align the y-axis with the cell polarity. (iv) Binary mask creation for initial and final timepoints for calculating cell areas, centroids, and lengths. B. Phalloidin stain intensity in dark-adapted vs. stimulated cells. Mean +/− standard error. N = 40 cells per condition. C. Box-and-whisker plot of cell area change upon wholefield stimulation. D. Box-and-whisker plot of cell length change upon wholefield stimulation. N = 82-93 cells per condition. E. Schematized calculation of angle of cell movement. Centroids of the initial (red) and final (blue) cell boundaries are calculated in OpenCV (moments function). The angle of movement between the cell polarization vector (green, dashed) and the centroid movement vector (red) is designated as β. F. Circumplex charts of the angle of movement relative to the polarization vector in cells expressing (i) BcLOV4, (ii) opto-RhoA, and (iii) opto-RhoGEF. N = 82-93 cells per condition. b-d. Two-sided Mann-Whitney U test: (**) p < 0.01; (***) p < 0.001; (****) p < 0.0001; (n.s.) not significant. c-d. Center line, median; “X”, mean; box limits, upper and lower quartiles; whiskers, 1.5x interquartile range; points, outliers.
2.2. Systematic characterization of optogenetically controlled activity
As primary measures of morphological dynamics (Figure 2b-f) upon blue light activation of opto-RhoA and opto-RhoGEF, we quantified the changes in cell area, cell length along the polarization axis (Figure S5), and cell centroid displacement vector, and the cytoskeletal tension levels by phalloidin staining of filamentous F-actin (Figure 2b). Light-dependent increases in filamentous actin levels were significant and of large effect size vs. BcLOV4-mCherry control, whereas the basal levels were similar for all constructs. The latter data suggests there is little leakiness in RhoA and ARHGEF11 signaling from diffusive membrane contact in the dark-adapted state, even at the over-expression levels supported by HEK cells.
Optogenetic activation drove cytoskeletal changes that were highly significant and of large effect size vs. the BcLOV4-mCherry control (Figure 2c-d). The cell centroid displacement vector pointed toward the leading edge with high preference (Figure 2e-f), meaning that the cytoskeletal retraction was predominately at the trailing edge and along the polarization axis upon stimulating the entire cell. This trend suggests that the tensile asymmetry introduced by the underlying cell polarization drives the cytoskeletal morphology changes upon optogenetic activation of stress fiber formation, and it is consistent with the fact that RhoA signaling complexes are most abundantly active in the cell rear during retraction.[24-26]
Cytoskeletal changes were largely abrogated in cells pre-treated with pharmacological inhibitors of RhoA-GEFs (Rhosin),[27-28] indicating that opto-RhoA-mediated cell contraction is GEF-dependent; therefore in dark-adapted opto-RhoA, the wildtype RhoA domain is in its inactive GDP-bound form before activation by endogenous RhoGEFs, as expected by its design. Inhibition of ROCK (Rho-associated protein kinase) signaling (with Y-27632)[29] also diminished cell contraction, confirming that observed cytoskeletal changes are RhoA-ROCK signaling-dependent (Figure S6). The kinetics of dynamic membrane association (τon-RhoA = 1.27 s, τon-GEF = 1.13 s) and undocking (τoff-RhoA = 114.2 s, τoff-GEF = 108.4 s), which were measured in the presence of Rhosin and Y-27632 to suppress RhoA signaling, was similar to those of BcLOV4-mCherry (Figure S7).
By all observed measures, GTPase-level stimulation by opto-RhoA was consistently more effective than GEF-level stimulation by opto-RhoGEF. However, the differences were modest (small effect size and marginal significance). No differences were observed in basal filamentous actin level or subcellular distribution patterns, although opto-RhoA expressed at slightly higher levels (Figure S8). While nuanced differences in signal integration may exist since GTPase-level control integrates the inputs of multiple GEFs, whereas GEF-level control can be subtype specific (as shown by others for Rac1[30]), for simplicity and experimental throughput, our further characterization focuses on opto-RhoA since it was generally more efficacious (including its ability to separate adherens junctions) and fewer tools exist for directly controlling RhoA GTPase than for its GEFs.
Note that we fused wildtype RhoA over constitutively active mutants to limit the probability of RhoA signal transduction in the dark by diffusive membrane contact alone, which was evident in cells expressing a BcLOV4-fused constitutively active GTP-bound RhoA(G14V) mutant, which had elevated basal stress fiber levels in the dark (Figure S9). Such dynamic range improvement by using wildtype GTPase was also observed with our previously reported opto-Rac1.[23]
2.3. Spatial determinants of RhoA induction and signaling
In cells responding to whole-field stimulation, the magnitude of the response was largest along the polarization axis with the trailing edge contracting toward the leading edge, which suggests that the cell polarization drove the vectorial change in cell shape. We next explored the effects of subcellularly patterned stimulation on RhoA-driven cell contraction, specifically whether the orientation of blue light stimulation with respect to the cell polarization axis affects the magnitude of change in cell morphology (Figure 3). We defined the “stimulation angle” as the angle between the polarization axis and a line segment between the cell centroid and the stimulated ROI (30° bins, ~25 μm box and ~25% fraction of membranes) (Figure 3a). The magnitude of induced change trended with the stimulation angle alignment with the polarization axis (Figure 3b-d). Stimulation at the leading and trailing edges led to largest constrictions, which was again consistent with the organization of the underlying stress fiber network and endogenous subcellular distribution of RhoA signaling complexes.[24-26] However, any stimulation angle, even completely orthogonal to the polarization axis, caused cytoskeletal constriction in cell area and length, and thus, the spatial patterning of the optical excitation is not required to align with the axis for a dramatic morphological change. These experimentally determined spatial relationships, between the input of optogenetic RhoA signaling induction and the downstream output of cytoskeletal contractility, will be useful for guiding experimental design and data interpretation across optical stimulation paradigms.
Figure 3.

Stimulation angle dependence of opto-RhoA-driven contraction. a. Schematic of stimulation angle (θ) calculation from the polarization axis, cell centroid, and centroid of the overlap region of the cell with the patterned stimulation field. b. Exemplar images of focal contraction of the trailing edge of a HEK cell after 10 minutes of pulsatile patterned stimulation (1.6% duty ratio). White box = illumination field. Scale = 10 μm. c. Box-and-whisker plot of change in cell area (relative to initial area) for binned stimulation angles. d. Box-and-whisker plot of change in cell length (relative to initial length) for binned stimulation angles. c-d. N = 10-35 independent videos per bin. Two-sided Mann-Whitney U test: (*) p < 0.05; (**) p < 0.01; (***) p < 0.001; (n.s.) not significant. Top row = vs. 0-30° leading edge bin; bottom row = vs. 150-180° trailing edge bin. Center line, median; “X”, mean; box limits, upper and lower quartiles; whiskers, 1.5x interquartile range.
2.4. RhoA-driven mechanotransduction
One way that RhoA regulates cellular dynamics is transcriptional mechanotransduction, the cytomechanical activation of gene expression (Figure 4a). RhoA-controlled stress fibers relay mechanical cues from the extracellular maxtrix (ECM) to the transcriptional co-activator Yes-associated protein, YAP (and its paralog, TAZ) that regulates the activity of other transcription factors, like TEAD-family transcription factors.[31-32] Previous work by others showed that optogenetic activation of ARHGEF11 could drive rapid nuclear import of YAP within ~5 minutes of the increased cytoskeletal tension.[9] We report that opto-RhoA activation drove nuclear import of GFP-tagged YAP (Figure 4b-d) on similarly fast timescales of ~3 minutes.
Figure 4.

Opto-RhoA induction of YAP-dependent mechanotransduction. a. Photoactivated opto-RhoA increases cytoskeletal tension to drive YAP nuclear import and transcriptional co-activation. b. Exemplar images of YAP-GFP nuclear import following blue-light photoactivation of opto-RhoA and BcLOV4 control. Scale bar = 10 μm. Arrows indicate nucleus position. c. Nuclear:cytosolic YAP in response to 1.6% duty ratio stimulation of opto-RhoA and BcLOV4 control. N = 30 cells each. d. Change in (i) nuclear YAP, (ii) cytosolic YAP, and (iii) nuclear:cytosolic YAP, normalized to region area. N = 30 cells each. e. Dual luciferase reporter assay showing increased YAP-coactivated TEAD-dependent transcription driven by opto-RhoA but not BcLOV4-only control. Luminescence was measured from firefly luciferase under a YAP-sensitive synthetic promoter (“8xGTIIC”), normalized to co-expressed Renilla luciferase. N = 8 wells per condition. c-e. mean +/− std err. Two-sided Mann-Whitney U test: (*) p < 0.05; (***) p < 0.001; (****) p < 0.0001; n.s. not significant.
Beyond nuclear localization, we confirmed YAP/TAZ-TEAD-dependent transcriptional activation using a previously described[5] luciferase reporter (Figure 4e) in serum-starved cells of low initial cytoskeletal tension. Thus, single-component optogenetic RhoA tools can potently drive downstream gene expression to study mechanotransduction as valuable complementary technologies to recently reported optogenetic YAP/TAZ domains[33-34] that shuttle into the nucleus when released from mitochondrial membranes into the cytosol by heterodimer dissociation. The RhoA-YAP relay is of particular importance in programming cell migration and tissue development.[35-37]
3. Discussion
We have expanded the repertoire of single-component optogenetic membrane recruitment-based tools to RhoA signaling, by fusing BcLOV4 to wildtype RhoA GTPase or the catalytic domain of ARHGEF11. While their optical induction mechanism of single-component dynamic membrane recruitment is the same, the tools signal in different ways where, opto-RhoA increases the local concentration of RhoA protein to be subsequently activated by the endogenous pool of all RhoGEF proteins, and opto-RhoGEF increases the local concentration of the catalytic DH domain of ARHGEF11 to stimulate the endogenous pool of RhoA protein. One can potentially consider the signaling by opto-RhoA as GEF-integrating and that of opto-RhoGEF as GEF-specific; the constitutively active RhoA(G14V) mutant, in which RhoA is locked in its active GTP-bound state by blunting its native GTPase ability, could be considered GEF-independent. These differences in signal integration may result in subtle contraction pattern variations based on the preferential subcellular distribution and balance of endogenous RhoA-activating GEFs and RhoA-inactivating GAPs (GTPase accelerating proteins).[38-39] For example, the G14V mutant should enhance contractile signaling uniformly in the entire membrane, whereas the wildtype domain of opto-RhoA will signal more strongly in regions of higher relative concentration of GEFs vs. GAPs.
Because the GTPase in opto-RhoA lacks a (“CAAX”) prenylation site,[40-41] it is less likely to be activated by the external stimuli that activate the endogenous cellular RhoA residing at the membrane, and thus, the occlusion of the C-terminus of RhoA GTPase in opto-RhoA also enhances its orthogonality to the endogenous GTPase. In addition, opto-RhoA lacks a GDI (guanosine nucleotide dissociation inhibitor) binding site because of the disrupted prenlyation, thus preventing the overexpressed opto-RhoA from destabilizing the endogenous pool of Rho-family GTPases by otherwise outcompeting it for regulatory interactions with the chaperone GDI. These features are useful for leaving the basal cytoskeletal physiology intact when opto-RhoA is inactive in its dark-adapted state.
Opto-RhoA and opto-RhoGEF exhibit rapid membrane association kinetics (τon ~ 1 sec) that ensures their spatiotemporal precision through fast induction of RhoA signaling and limited cytosolic diffusion during membrane recruitment. The signaling inactivation is more likely governed by RhoA signaling dynamics than the much faster membrane dissociation kinetics of BcLOV4, given that opto-RhoA accumulation is observable in DMD-patterned illumination fields, even well after the BcLOV4 membrane interaction period is complete. Such gradual accumulation of activated Rho-family GTPase is also observed with a heterodimerizing optogenetic Cdc42 created by others.[42-43] However, this accumulation is not as readily observed in control cells treated with GEF- and ROCK inhibitors, or in BcLOV4-mCherry controls, which suggests that the persistence originates from interactions amongst activated RhoA/actomyosin signaling complexes,[44-45] as opposed to homo-oligomerization into large photobodies, unlike the clustering mechanism of Cry2-RhoA we previously created.[18]
In summary, we have created high-performance single-component tools for optogenetic activation of RhoA GTPase and ARHGEF11 to control cell contractility and RhoA-driven transcriptional mechanotransduction. The accompanying characterization of how cytoskeletal changes depend on spatial patterning of the optical stimulation informs how tool performance should vary across different experimental setups and designs, and will also advance the study of diverse cell behaviors by connecting spatiotemporal patterns of focal RhoA signaling induction to the consequent cell-wide cytoskeletal responses. These tools further demonstrate the versatility of BcLOV4 technology for single-component optogenetic control over peripheral membrane proteins.
4. Experimental Section
Genetic constructs:
Domain arrangement combinations of BcLOV4, mCherry, and RhoA protein or catalytic DH domain of ARHGEF11 (with a flexible (GGGS)2 linker between each pair) were assembled by Gibson cloning using NEB HiFi DNA Assembly Master Mix (E2621) into the pcDNA3.1 mammalian expression vector under the CMV promoter. BcLOV4 and mCherry were amplified from their mammalian codon-optimized reported fusion (Addgene plasmid 114595).[20] Wildtype RhoA GTPase was amplified from CLPIT Cry2PHR-mCherry-RhoA (Addgene plasmid 42959) without the C-terminal “CAAX” motif to prevent prenylation.[19] The DNA sequence of ARHGEF11 (Genbank ID XP_011508491.1) was human codon-optimized using the Integrated DNA Technologies (IDT) Codon Optimization Tool and ordered as gBlocks®. The DH domain of the GEF, identified using the PROSITE ExPASy database, was amplified from these gBlocks. The RhoA constitutively active G14V mutant[46] was generated by QuikChange site-directed mutagenesis. All genetic constructs were transformed into competent E. coli (New England Biolabs, C2984H). All sequences were verified by Sanger sequencing. For the cell polarity assay, BFP nuclear marker (plasmid 55265) and miRFP703-tagged LifeAct (plasmid 79993) plasmids were acquired from Addgene. For mechanotransduction assays, EGFP-tagged YAP was acquired from Addgene (plasmid 17843), and YAP-sensitive promoter plasmid was acquired from Addgene (plasmid 34615). Plasmids for opto-RhoA and opto-RhoGEF will be distributed through Addgene (plasmids 164472 and 164473).
Mammalian culture and transduction:
HEK293T (ATCC, CRL-3216) cells were cultured in D10 media composed of Dulbecco’s Modified Eagle Medium with Glutamax (Invitrogen, 10566016), supplemented with 10% heat-inactivated fetal bovine serum (FBS) and penicillin-streptomycin at 100 U mL−1. Cells were maintained in a 5% CO2 water-jacketed incubator (Thermo/Forma 3110) at 37 °C. Cells were seeded onto poly-d-lysine-treated glass bottom dishes (MatTek, P35GC-1.5-14-C) or into 24-well glass bottom plates (Cellvis, P24-1.5H-N) at 15–20% confluency. Cells were transfected at ~30–40% confluency 24 hours later using the TransIT-293 transfection reagent (Mirus Bio, MIR2700) according to manufacturer instructions. Cells were imaged 24-48 h post-transfection.
Optical hardware:
Fluorescence microscopy was performed on an automated Leica DMI6000B fluorescence microscope under Leica MetaMorph control, with a sCMOS camera (pco.edge), an LED illuminator (Lumencor Spectra-X), and a 63× oil immersion objective. Excitation illumination was filtered at the LED source (mCherry imaging λ = 575/25 nm; GFP/AlexaFluor488 imaging or wide-field BcLOV4 stimulation λ = 470/24 nm; miRFP703 imaging λ = 632/22 nm). Fluorescent proteins were imaged with Chroma filters: mCherry (T585lpxr dichroic, ET630/75 nm emission filter, 0.2–0.5 s exposure), GFP (T495lpxr dichroic, ET 525/50 nm emission filter, 0.2 s exposure), miRFP703 (AT655dc dichroic, ET655 nm emission, 0.5 s exposure). Cells were imaged at room temperature in CO2-independent media (phenol-free HBSS supplemented with 1% l-glutamine, 1% penicillin–streptomycin, 2% essential amino acids, 1% nonessential amino acids, 2.5% HEPES pH 7.0, and 10% serum). The spatially patterned illuminator was custom-constructed from a digital light processor (DLP, Digital Light Innovations CEL5500), as previously described.[22]
Expression characteristics and membrane translocation:
For membrane recruitment quantification, prenylated GFP was co-transfected as a membrane marker with the BcLOV4 fusions as previously described.[20,22] Briefly, an mCherry fluorescence image (500 ms exposure) was captured to assess protein expression level and subcellular distribution. Cells were then illuminated with a 5 s-long blue light pulse to stimulate BcLOV4, during which time mCherry fluorescence images were also captured every 200 ms to monitor subcellular localization changes. The GFP membrane marker was imaged immediately after blue light stimulation for correlation analysis.[20] For membrane dissociation via thermal reversion of the photoactivated protein in the dark, cells were incubated with RhoGEF inhibitor Rhosin (Millipore-Sigma 555460, 25 μM) and ROCK inhibitor Y-27632 (Millipore-Sigma Y0503, 10 μM) for 24 hours prior to imaging to prevent cell contraction. mCherry was visualized every 5 s for 10-15 minutes in the absence of blue light stimulation. Membrane localization and dissociation were measured by line section analysis and correlation with prenylated GFP in ImageJ and MATLAB as previously described.[20]
DMD stimulation:
mCherry fluorescence was imaged every 15 s for 10 min. During this time, cells were stimulated for one second per minute (1.6% duty cycle) patterned illumination (25 μm-wide square encompassing ~25% of cell area). The stimulation angle was defined as the angle defined by the polarization axis and the line segment between the cell centroid and the centroid of the stimulated cell area.
Cell polarization marker analysis:
HEK293T cells were co-transfected with a nuclear marker (mTagBFP-Nucleus-7) and miRFP703-tagged LifeAct. The data analysis pipeline was applied to align the long axis of the cell with the y-axis, and to position the nucleus closer to the top of the cell as a putative marker of the leading edge. The actin arc was then located by identifying the brightest LifeAct-miRFP703 region of at least 5 μm2 in area. The angle between the long axis and the vector connecting the cell centroid to the actin arc centroid was then calculated to verify that the actin arc occurs at the computationally identified leading edge of the cell.
Phalloidin staining:
24 hours after transfection, cells were washed with PBS and the media was replaced with DMEM supplemented with penicillin-streptomycin and without FBS. Light-exposed 24-well tissue culture-treated plates (CytoOne, CC7682-75240) were incubated under Arduino-controlled blue strip LEDs (light intensity 15 mW/cm2) strobing at a 1.6% stimulation duty cycle in a 5% CO2 water-jacketed incubator for two hours. Cells were fixed with 4% paraformaldehyde in PBS at room temperature for 10 minutes, washed twice with PBS, and then permeabilized with 0.1% Triton-x-100 in PBS for 15 minutes. Cells were blocked with 1% BSA in PBS for 30 minutes, then stained with Alexa Fluor 488 Phalloidin (Invitrogen, A12379) diluted 1:400 in PBS. Plates were washed twice prior to imaging. Filamentous actin level was quantified by normalizing total cell Alexa Fluor 488 fluorescence to cell area, with N = 40 cells per condition.
Pharmacological inhibitors:
The GEF inhibitor Rhosin (Millipore-Sigma 555460) was added to cells at 25 uM final concentration upon transfection, 24 hours before imaging. ROCK inhibitor Y-27632 (Millipore-Sigma Y0503) was similarly added to cells at 10 uM final concentration.
YAP nuclear translocation:
HEK293T cells were initially plated at 75% confluency in 10 cm dishes to drive YAP to the cytosol. Cells were then passaged one day later and seeded at 25% confluency in 35 mm poly-d-lysine-treated glass bottom dishes. The next day, dishes were washed with PBS and media was replaced with DMEM supplemented with 2% heat-inactivated FBS and penicillin-streptomycin. Cells were then transfected with a 4:1 ratio of opto-RhoA-mCherry or BcLOV4-mCherry to EGFP-YAP. EGFP-YAP was imaged every 15 seconds using a 250 msec excitation pulse that also stimulated BcLOV4, and mCherry fluorescence was imaged every minute. Nuclear and cytosolic fluorescence normalized to the area of each region was calculated every minute by manual segmentation in ImageJ, with N = 20 cells per condition.
Transcriptional mechanotransduction:
YAP-TEAD-dependent transcriptional activity was quantified using a dual luciferase reporter system (Promega, E1910). HEK293T cells were co-transfected with a YAP-sensitive promoter driving firefly luciferase expression (8XGTIIC-luciferase, Addgene #34615), Renilla luciferase, and BcLOV4-mCherry or opto-RhoA-mCherry. At transfection, full media was replaced with DMEM supplemented with 2% heat-inactivated FBS and penicillin-streptomycin. Half the cells were incubated under pulsing blue light with a 1.6% stimulation duty cycle for 12 hours. Cells were lysed according to manufacturer instructions. Luminescence was measured in white 96-well plates (Corning, 3917) on a Tecan M200 spectrophotometer with a 10 second integration time. The firefly luminescence value for each sample was normalized to its Renilla luciferase readout. N = 8 lysate samples per condition.
Data analysis pipeline:
Change in cell area, cell length, and centroid movement were computed via a custom analysis Python script, as schematized in Figure 3a. For whole-field stimulation assays, videos of an entire field-of-view were cropped so that each contained only one cell. Contours of the cell membrane and nucleus were identified using the threshold function in OpenCV. An angle of rotation was identified by iterative (5° increments) rotation such that the long polarization axis of the cell aligned with the y-axis, and then a cell-bounding rectangle was calculated using the Imutils package with the short edge aligned with the x-axis; the angle of rotation was chosen such that the cell nucleus was closer to the top of the rectangle as a morphological marker for the cell leading edge, as confirmed by LifeAct imaging (Figure S5). Binary masks of the cell at initial (t = 0 min) and final (t = 10 min) timepoints were created for each cell. The change in cell area was calculated by finding the percent change in the area bounded by the cell contours at the final timepoint relative to its area at the initial timepoint. Change in cell length was calculated using the height of the bounding rectangle at the final and initial timepoints.
For experiments with spatially patterned illumination, a similar imaging processing workflow was followed. The angle of rotation was calculated as above, and binary masks of the cell at the initial and final timepoints and the stimulation zone were created and rotated such that the leading edge of the cell was positioned at the top of the image. Masks of the cell regions inside (the overlap) and outside (the non-overlap) the stimulation zone were created using the OpenCV bitwise operation functions “and” and “xor”, respectively. The centroids of the whole cell and the overlap region were computed at the initial and final timepoints. The angle of stimulation was defined as the angle between (i) the vector connecting the initial cell centroid and overlap region centroid and (ii) the vertical vector between the initial cell centroid and the leading edge of the cell.
Statistical analysis:
For whole-field stimulation assays, each cell was treated as a separate data point, with N = 82-93 cells from 10-12 field-of-view videos per condition, 6-8 cells per video. For spatially patterned stimulation assays, each data point was derived from one cell in an independent video, and cells were binned by angle of stimulation, with bin widths of 30 degrees spanning 0-180 degrees, N = 10-35 videos per bin. Statistical significance was assessed by the two-sided non-parametric Mann-Whitney U test (α = 0.05), uncorrected for multiple comparisons, using the Python SciPy Stats package. For column and line plots, data are presented as mean +/− standard error. Time constants and confidence intervals were calculated in MATLAB.
Supplementary Material
Acknowledgements
EEB designed genetic constructs, designed all experiments, and conducted all experiments. IAK created the patterned illumination system and assisted with the automated data analysis pipeline development. KY assisted with genetic construct design, engineering, and assays. BYC, JDB, and LJB coordinated all research. All authors contributed to experiment design, data analysis, and manuscript preparation. BYC acknowledges the support of National Science Foundation (NSF) Systems and Synthetic Biology (MCB 1652003), NIH/National Institute on Drug Abuse (R21 DA040434), and NIH/National Institute of Neurological Disorders and Stroke (NINDS) (R01 NS101106). EEB acknowledges the fellowship support of the National Institute of Neurological Disorders and Stroke of the National Institutes of Health (T32 NS091006). IAK acknowledges the fellowship support of the Paul and Daisy Soros Fellowship for New Americans and the NIH/National Institute of Mental Health F30 Award. LJB acknowledges the support of the NIH/National Institute of General Medical Sciences (R35 GM138211, R21 GM132831). JDB acknowledges support from the National Science Foundation, Division of Civil, Mechanical, and Manufacturing Innovation (CMMI 1548571) and the NIH/National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01 AR073809).
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Erin E. Berlew, Department of Bioengineering, University of Pennsylvania, 210 South 33rd Street, Philadelphia, PA 19104, USA
Ivan A. Kuznetsov, Department of Bioengineering, University of Pennsylvania, 210 South 33rd Street, Philadelphia, PA 19104, USA
Keisuke Yamada, Department of Electrical Engineering and Bioscience, Faculty of Science and Engineering, Waseda University, Tokyo, Japan.
Lukasz J. Bugaj, Department of Bioengineering, University of Pennsylvania, 210 South 33rd Street, Philadelphia, PA 19104, USA
Joel D. Boerckel, Department of Bioengineering, University of Pennsylvania, 210 South 33rd Street, Philadelphia, PA 19104, USA; Department of Orthopedic Surgery, University of Pennsylvania, Philadelphia, PA 19104, USA
Brian Y. Chow, Department of Bioengineering, University of Pennsylvania, 210 South 33rd Street, Philadelphia, PA 19104, USA
References
- [1].Etienne-Manneville S, Hall A, Nature 2002, 420, 629. [DOI] [PubMed] [Google Scholar]
- [2].Denk-Lobnig M, Martin AC, Small GTPases 2019, 10, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chrzanowska-Wodnicka M, Burridge K, J. Cell Biol 1996, 133. 1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Marjoram RJ, Lessey EC, Burridge K, Curr. Mol. Med 2014, 14, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S, Nature 2011, 474, 179. [DOI] [PubMed] [Google Scholar]
- [6].Hannanta-Anan P, Glantz ST, Chow BY, Curr. Opin. Struct. Biol 2019, 57, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Inoue T, Heo WD, Grimley JS, Wandless TJ, Meyer T, Nat. Methods 2005, 2, 415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].DeRose R, Miyamoto T, Inoue T, Pflugers Arch. 2013, 465, 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Valon L, Marin-Llaurado A, Wyatt T, Charras G, Trepat X, Nat. Commun 2017, 8, 14396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Oakes PW, Wagner E, Brand CA, Probst D, Linke M, Schwarz US, Glotzer M, Gardel ML, Nat. Commun 2017, 8, 15817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wagner E, Glotzer M, J. Cell Biol 2016, 213, 641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kotynkova K, Su KC, West SC, Petronczki M, Cell Rep. 2016, 17, 2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Izquierdo E, Quinkler T, De Renzis S, Nat. Commun 2018, 9, 2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chen X, Venkatachalapathy M, Kamps D, Weigel S, Kumar R, Orlich M, Garrecht R, Hirtz M, Niemeyer CM, Wu Y, Dehmelt L, Angew. Chem. Int. Ed 2017, 56, 5916. [DOI] [PubMed] [Google Scholar]
- [15].Guglielmi G, Barry JD, Huber W, De Renzis S, Dev. Cell 2015, 35, 646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Repina NA, Rosenbloom A, Mukherjee A, Schaffer DV, Kane RS, Annu. Rev. Chem. Biomol. Eng 2017, 8, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hannanta-Anan P, Chow BY, ACS Synth. Biol 2018, 7, 1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bugaj LJ, Sabnis AJ, Mitchell A, Garbarino JE, Toettcher JE, Bivona TG, A Lim W, Science 2018, 361, eaao3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bugaj LJ, Choksi AT, Mesuda CK, Kane RS, Schaffer DV, Nat. Methods 2013, 10, 249. [DOI] [PubMed] [Google Scholar]
- [20].Glantz ST, Berlew EE, Jaber Z, Schuster BS, Gardner KH, Chow BY, Proc. Natl. Acad. Sci. U. S. A 2018, 115, E7720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Glantz ST, Berlew EE, Chow BY, Methods Enzymol. 2019, 622, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].He L, Jing J, Zhu L, Tan P, Ma G, Zhang Q, Nguyen NT, Wang J, Zhou Y, Huang Y, Chem. Sci 2017, 8, 5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Berlew EE, Kuznetsov IA, Yamada K, Bugaj LJ, Chow BY, Photochem. Photobiol. Sci 2020, 19, 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Pertz O, Hodgson L, Klemke RL, Hahn KM, Nature 2006, 440, 1069. [DOI] [PubMed] [Google Scholar]
- [25].Bolado-Carrancio A, Rukhlenko OS, Nikonova E, Tsyganov MA, Wheeler A, Garcia-Munoz A, Kolch W, von Kriegsheim A, Kholodenko BN, eLife 2020, 9, e58165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wong K, Pertz O, Hahn K, Bourne H, Proc. Natl. Acad. Sci. U. S. A 2006, 103, 3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shang X, Marchioni F, Sipes N, Evelyn CR, Jerabek-Willemsen M, Duhr S, Seibel W, Wortman M, Zheng Y, Chem. Biol 2012, 19, 699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Shang X, Marchioni F, Evelyn CR, Sipes N, Zhou X, Seibel W, Wortman M, Zheng Y, Proc. Natl. Acad. Sci. U. S. A 2013, 110, 3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M, Narumiya S, Nature 1997, 389 990. [DOI] [PubMed] [Google Scholar]
- [30].Graziano BR, Gong D, Anderson KE, Pipathsouk A, Goldberg AR, Weiner OD, J. Cell Biol 2017, 216, 2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Li Z, Zhao B, Wang P, Chen F, Dong Z, Yang H, Guan K-L, Xu Y, Genes Dev. 2010, 24, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lin KC, Park HW, Guan KL, Trends Biochem. Sci 2017, 42, 862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Dowbaj AM, Jenkins RP, Hahn K, Montagner M, Sahai E, (Preprint)bioRxiv, 10.1101/2020.06.08.140228, submitted: June2020. [DOI] [Google Scholar]
- [34].Wang H, Vilela M, Winkler A, Tarnawski M, Schlichting I, Yumerefendi H, Kuhlman B, Liu R, Danuser G & Hahn KM, Nat. Methods 2016, 13, 755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mason DE, Collins JM, Dawahare JH, Nguyen TD, Lin Y, Voytik-Harbin SL, Zorlutuna P, Yoder MC, Boerckel JD, J. Cell Biol 2019, 218, 1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, Porrello ER, Mahmoud AI, Tan W, Shelton JM, Richardson JA, Sadek HA, Bassel-Duby R, Olson EN, Proc. Natl. Acad. Sci. U. S. A 2013, 110, 13839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kegelman CD, Mason DE, Dawahare JH, Horan DJ, Vigil GD, Howard SS, Robling AG, Bellido TM, Boerckel JD, FASEB J. 2018, 32, 2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Müller PM, Rademacher J, Bagshaw RD, Wortmann C, Barth C, van Unen J, Alp KM, Giudice G, Eccles RL, Heinrich LE, Pascual-Vargas P, Sanchez-Castro M, Brandenburg L, Mbamalu G, Tucholska M, Spatt L, Czajkowski MT, Welke R, Zhang S, Nguyen V, Rrustemi T, Trnka P, Freitag K, Larsen B, Popp O, Mertins P, Gingras A, Roth FP, Colwill K, Bakal C, Pertz O, Pawson T, Petsalaki E, Rocks O, Nat. Cell. Biol 202022, 498. [DOI] [PubMed] [Google Scholar]
- [39].Iwanicki MP, Vomastek T, Tilghman RW, Martin KH, Banerjee J, Wedegaertner PB, Parsons JT, J. Cell Sci 2008, 121, 895. [DOI] [PubMed] [Google Scholar]
- [40].Boulter E, Garcia-Mata R, Guilluy C, Dubash A, Rossi G, Brennwald PJ & Burridge K, Nat. Cell Biol 2010, 12, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Garcia-Mata R, Boulter E, Burridge K, Nat. Rev. Mol. Cell Biol 2011, 12, 493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bendezú FO, Vincenzetti V, Vavylonis D, Wyss R, Vogel H, Martin SG, PLoS Biol. 2015, 13, e1002097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lamas I, Merlini L, Vjestica A, Vincenzetti V, Martin SG, PLoS Biol. 2020, 18, 3000600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bagci H, Sriskandarajah N, Robert A, Boulais J, Elkholi IE, Tran V, Lin Z-Y, Thibault M-P, Dubé N, Faubert D, Hipfner DR, Gingras A-C & Côté J-F, Nat. Cell Biol 2020, 22, 120. [DOI] [PubMed] [Google Scholar]
- [45].Bement WM, Miller AL, von Dassow G, Bioessays 2006, 28, 983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Morin P, Flors C, Olson MF, Eur. J. Cell Biol 2009, 88, 495. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.