

Summary:
Cardiac macrophages represent a heterogeneous cell population with distinct origins, dynamics, and functions. Recent studies have revealed that C-C Chemokine Receptor 2 positive (CCR2+) macrophages derived from infiltrating monocytes regulate myocardial inflammation and heart failure pathogenesis. Comparatively little is known about the functions of tissue resident (CCR2−) macrophages. Herein, we identified an essential role for CCR2− macrophages in the chronically failing heart. Depletion of CCR2− macrophages in mice with dilated cardiomyopathy accelerated mortality and impaired ventricular remodeling and coronary angiogenesis, adaptive changes necessary to maintain cardiac output in the setting of reduced cardiac contractility. Mechanistically, CCR2− macrophages interacted with neighboring cardiomyocytes via focal adhesion complexes and were activated in response to mechanical stretch through a transient receptor potential vanilloid 4 (TRPV4) dependent pathway that controlled growth factor expression. These findings establish a role for tissue resident macrophages in adaptive cardiac remodeling and implicate mechanical sensing in cardiac macrophage activation.
Keywords: Macrophages, C-C chemokine receptor 2 (CCR2), heart failure, dilated cardiomyopathy, cardiac remodeling, angiogenesis, transient receptor potential vanilloid 4 (TRPV4)
Graphical Abstract
eTOC blurb
Resident cardiac macrophages are key regulators of heart development and homeostasis, however, the role of these cells during disease is presently unclear. Wong, Mohan, Kopecky et al. reveal that resident cardiac macrophages orchestrate adaptive remodeling and survival of the failing heart by sensing mechanical stimuli through a TRPV4 dependent mechanism.
Introduction:
Macrophage populations arise from distinct developmental origins including extraembryonic and definitive hematopoietic progenitors and are maintained through differing mechanisms (Davies et al., 2013; Epelman et al., 2014b; Hashimoto et al., 2013; Hettinger et al., 2013; Hoeffel et al., 2015; Wynn et al., 2013; Yona et al., 2013). Microglia found within the brain are originate from extraembryonic hematopoiesis and maintained through local proliferation independent of monocyte input (Ginhoux et al., 2010), while intestinal macrophages are derived from definitive hematopoietic progenitors and are continually replenished by recruited monocytes (Bain et al., 2013). Most organs including the heart, lung, liver, and skin contain admixtures of macrophages with differing development origins, morphologies, and population dynamics (Epelman et al., 2014a; Guilliams et al., 2013; Hoeffel et al., 2012; Theret et al., 2019). These findings have raised the possibility that individual macrophage subsets may execute unique and context specific functions.
Resident macrophages have important functions in shaping and remodeling tissues throughout development and adulthood. These cells are essential for maturation of the nervous and vascular systems, contribute to bone and tooth morphogenesis, clear apoptotic cells within the embryo, and orchestrate regeneration of cardiac and appendage tissue (Aurora et al., 2014; Fantin et al., 2010; Godwin et al., 2017; Godwin et al., 2013; Lavine et al., 2014; Munoz-Espin et al., 2013; Parkhurst et al., 2013; Petrie et al., 2014; Storer et al., 2013; Theret et al., 2019). In the adult, resident macrophages maintain organ homeostasis and physiology including iron metabolism and transport, regulation of hematopoiesis, clearance of airway debris and surfactant, and facilitation of electrical impulses through the heart (Chow et al., 2011; Hashimoto et al., 2013; Hulsmans et al., 2017; Soares and Hamza, 2016). Little is understood regarding the functions of these cells in disease.
Heart failure is a common disease that occurs as a result of cardiomyocyte loss or intrinsic deficits in cardiac contractility. The failing heart undergoes robust geometric changes characterized by thickening and dilation of the left ventricle (LV). This process is initially beneficial as it maintains cardiac output by increasing stroke volume (volume of blood ejected) and reducing LV wall stress and is distinct from maladaptive remodeling, which contributes to heart failure progression through cardiomyocyte hypertrophy, cell death, and fibrosis (Burchfield et al., 2013; Xie et al., 2013). Beneficial components of remodeling mimic changes seen in the athlete’s heart, where LV chamber dilation and eccentric hypertrophy represent physiological adaptations to exercise conditioning. This physiological form of hypertrophy is associated with coronary angiogenesis and cardiomyocyte lengthening. Fibrosis and contractile dysfunction are typically not present (Nakamura and Sadoshima, 2018). The cell types and molecular mechanisms that regulate adaptive and maladaptive remodeling in chronic heart failure have remained elusive (Cohn et al., 2000; Patel et al., 2017).
Cardiac resident macrophages are poised to govern remodeling. Under homeostatic conditions, the heart contains a heterogeneous population of resident macrophages that can be divided into two functionally distinct subsets based on the cell surface expression of C-C chemokine receptor 2 (CCR2) (Epelman et al., 2014a). CCR2+ macrophages are derived from definitive hematopoietic progenitors, replenished by monocyte recruitment and subsequent proliferation, and initiate inflammatory cascades. In response to cardiomyocyte death, CCR2+ macrophages produce inflammatory cytokines, orchestrate neutrophil and monocyte recruitment, generate damaging oxidative products, and consequently, contribute to heart failure progression through collateral myocardial injury and adverse remodeling (Bajpai et al., 2019; Lavine et al., 2014; Li et al., 2016; Patel et al., 2018). CCR2+ macrophage abundance is predictive of and associated with adverse remodeling in advanced heart failure patients (Bajpai et al., 2018). CCR2− macrophages are largely derived from embryonic (yolk sac and fetal liver) hematopoietic progenitors, maintained independent of monocyte recruitment through local proliferation, and orchestrate coronary vascular maturation, growth, and neonatal heart regeneration (Lavine et al., 2014; Leid et al., 2016). CCR2− macrophages suppress inflammatory responses following acute myocardial injury (Bajpai et al., 2019).
Herein, we employ a mouse model of dilated cardiomyopathy harboring a causative human mutation to define the composition and dynamics of macrophages residing within the chronically failing heart. We have revealed an essential role for CCR2− macrophages in adaptive remodeling, coronary angiogenesis, maintenance of cardiac output, and survival of mice with dilated cardiomyopathy. Furthermore, we have provided evidence that mechanical sensing through a transient receptor potential vanilloid 4 (TRPV4) dependent pathway regulates growth factor expression in CCR2− cardiac macrophages.
Results:
Distinct Macrophage Populations Reside within the Failing Heart.
To investigate cardiac macrophage composition and function in chronic heart failure, we chose to focus on a mouse model of dilated cardiomyopathy. Previously, mice were generated that harbor a causative mutation (ΔK210) in the endogenous Troponin T2 (Tnnt2) locus (Du et al., 2007). The Tnnt2ΔK210 mutation has been identified in familial and sporadic dilated cardiomyopathy patients and is considered clinically as a pathogenic variant (McNally and Mestroni, 2017). Incorporation of the Tnnt2ΔK210 mutant protein into sarcomeres leads to reduced thin filament calcium sensitivity and cardiomyocyte contractility (Clippinger et al., 2019; Morimoto et al., 2002).
Homozygous mice (Tnnt2ΔK210/ΔK210) develop dilated cardiomyopathy with profound LV remodeling (dilation and eccentric hypertrophy), reduced LV systolic function (ejection fraction), and early mortality (Fig. 1A–B and Fig. S1). Serial echocardiography revealed that Tnnt2ΔK210/ΔK210 mice were born with reduced LV ejection fraction reflective of intrinsic impairment in cardiomyocyte contractility. LV dilation and eccentric hypertrophy were first evident at 2 weeks of age, increased progressively over time (Table S1), and correlated with initial preservation of stroke volume. Cardiac catheterization revealed reduced contractility in Tnnt2ΔK210/ΔK210 mice compared to controls as early as 4 weeks of age (dP/dt max: 6258 ± 674 vs. 2496 ± 1059 mmHg/sec, p<0.001) These findings suggest that early remodeling represents a compensatory response to reduced LV contractility that serves to maintain cardiac output and systemic perfusion. At later time points (16 weeks of age), we observed blunted LV dilation and growth, further reduction in LV ejection fraction, and resultant decreased in stroke volume, indicating that adaptive LV remodeling can be overcome by severe and progressive decline in contractility.
Figure 1. Cardiac macrophages in a mouse model of dilated cardiomyopathy.
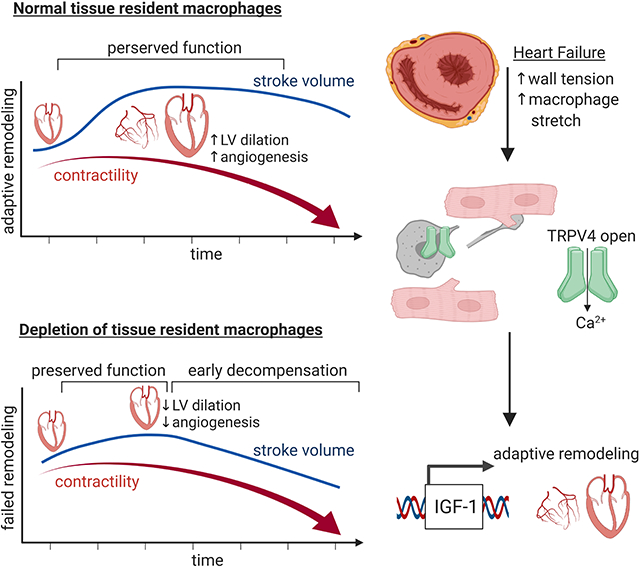
A, End-diastolic echocardiographic images of 8-week-old control and Tnnt2ΔK210/ΔK210 mice. B, Quantification of ejection fraction, LV diastolic dimension, LV mass index, and stroke volume. * denotes p<0.05 (ANOVA, Post-hoc Tukey) compared to controls, ** p<0.05 compared to Tnnt2ΔK210/ΔK210 at 8 weeks. n=10–11 per group. C, H&E (top) and immunostaining images (bottom, CD68-white, cardiac actin-green) of 8-week-old control and Tnnt2ΔK210/ΔK210 mice. n=7 per group. Scale bar: 20μm. D, Flow cytometry plots of CD45+Ly6G−CD64+ macrophages in control and Tnnt2ΔK210/ΔK210 mice at 8 weeks of age. n=4 per group. E-F, Quantification of CD68 immunostaining and flow cytometry. * denotes p<0.05 (Mann-Whitney test) compared to controls. G, CCR2 and CCR5 PET/CT of control and Tnnt2ΔK210/ΔK210 mice (6–10 weeks of age). n=4–17 per group. PET: positron emission tomography, CT: computed tomography. H, Quantification of CCR2 and CCR5 tracer uptake within the heart. * denotes p<0.05 (Mann-Whitney test) compared to controls. I, MA plot and pathway analysis of RNA sequencing data comparing control to Tnnt2ΔK210/ΔK210 hearts. n=5–6 per group. See also Figure S1–3, Table S1, Data S1.
To investigate the influence of chronic heart failure on cardiac macrophage abundance and composition, we examined histological sections obtained from control and Tnnt2ΔK210/ΔK210 hearts. CD68 immunostaining revealed increased macrophage abundance at both 1 week and 8 weeks of age (Fig. 1C, E and Fig. S1). Flow cytometry demonstrated significant shifts in cardiac macrophage composition over time. Tnnt2ΔK210/ΔK210 hearts contained increased abundance of CCR2+ macrophages, CCR2− MHC-IIlo macrophages, CCR2− MHC-IIhi macrophages, and monocytes at 8 weeks of age compared to controls. At 12 weeks of age, we observed a progressive increase in CCR2+ macrophages and marked increase in monocyte abundance (Fig. 1D, F, Fig. S1).
To non-invasively assess cardiac macrophage composition in intact mice, we took advantage of a positron emission tomography (PET) based molecular imaging strategy that detects CCR2+ macrophages (CCR2 PET) and total macrophages (CCR5 PET) (Heo et al., 2019; Luehmann et al., 2014). We observed robust CCR2 PET signal in the hearts of Tnnt2ΔK210/ΔK210 mice compared to controls. Inclusion of Tnnt2ΔK210/ΔK210 Ccr2−/− ruled out the possibility that increased CCR2 PET signal was a result of expanded blood pool size. Consistent with greater numbers of total cardiac macrophages in Tnnt2ΔK210/ΔK210 hearts, we observed increased CCR5 PET signal in the hearts of Tnnt2ΔK210/ΔK210 mice compared to controls (Fig. 1G–H).
Analysis of RNA sequencing data comparing control and Tnnt2ΔK210/ΔK210 hearts showed marked differences in gene expression that included pathways associated with collagen deposition, extracellular matrix organization, cell migration, and immune functions (Fig. 1I). Numerous differentially expressed genes have been implicated in macrophage activation and function (Cd44, Mrc2, Nr4a1, Tlr4, Lbp, Csf2ra, Jun, Fos, Irf6, Socs2, Chil1, Ctgf, Gdf15, Ifngr1, Maff) (Fig. S1).
Cardiac Macrophages in Heart Failure have Distinct Origins and Functions.
To delineate the contribution of monocytes to CCR2− and CCR2+ macrophage subsets in the chronically failing heart, we bred Tnnt2ΔK210/ΔK210 mice to Ccr2GFP genetically targeted mice to generate the following experimental groups: Ccr2GFP/+ (control), Ccr2GFP/GFP (Ccr2 deletion), Tnnt2ΔK210/ΔK210 Ccr2GFP/+ (heart failure), Tnnt2ΔK210/ΔK210 Ccr2GFP/GFP (heart failure, Ccr2 deletion). Ccr2GFP mice allow visualization of CCR2+ cells in the absence of CCR2 protein expression (Satpathy et al., 2013). Immunostaining demonstrated increased abundance of both CCR2− and CCR2+ macrophages in Tnnt2ΔK210/ΔK210 hearts compared to controls at 8 weeks of age. Ccr2 deletion led to significant reductions in the abundance of CCR2+ macrophages in control and Tnnt2ΔK210/ΔK210 hearts. No effect on CCR2− macrophages was observed. Flow cytometric analysis at 8 weeks of age confirmed selective reduction in CCR2+ macrophages in Tnnt2ΔK210/ΔK210 Ccr2GFP/GFP hearts compared to Tnnt2ΔK210/ΔK210 Ccr2GFP/+ hearts (Fig. S2). These data indicate that during this stage of heart failure, CCR2− macrophages are maintained in the absence of monocyte input, whereas, CCR2+ macrophages require ongoing monocyte recruitment. Cell proliferation as assessed by Ki67 staining was exclusively increased in CCR2− macrophages in Tnnt2ΔK210/ΔK210 hearts compared to controls (Fig. S2).
To delineate the developmental origin of cardiac macrophages in the chronically failing heart, we crossed Tnnt2ΔK210/ΔK210 mice to Flt3-cre Rosa26-tdTomato mice. Flt3-cre is selectively active in definitive hematopoietic stem cells, and thus labels monocytes and macrophages derived from definitive hematopoiesis (Boyer et al., 2011). This strategy has extensively been used to distinguish macrophages derived from extraembryonic hematopoiesis from macrophages derived from definitive hematopoiesis (Epelman et al., 2014a; Lavine et al., 2014; Leid et al., 2016). >90% of CCR2+ macrophages in control hearts were tdTomato+ at both 1 and 12 weeks of age. Conversely, <40% of CCR2− macrophages in control hearts were tdTomato+. The frequency of tdTomato+ positivity did not differ between control and Tnnt2ΔK210/ΔK210 hearts (Fig. S2). These findings indicate that in chronic heart failure, CCR2− macrophages are mixed population with contributions from extraembryonic and definitive hematopoiesis and are maintained by local proliferation rather than monocyte input. CCR2+ macrophages are exclusively derived from definitive hematopoiesis and maintained through monocyte input.
To gain insights into functional differences between CCR2− and CCR2+ macrophage in the chronically failing heart, we performed gene expression profiling on macrophage subsets isolated from Tnnt2ΔK210/ΔK210 Flt3-Cre Rosa26-tdTomato hearts: Ly6Chi monocytes, CCR2−MHC-IIlotdTomato−, CCR2−MHC-IIhitdTomato−, CCR2−MHC-IIlotdTomato+, CCR2−MHC-IIhitdTomato+, and CCR2+ (CCR2+MHC-IIhitdTomato+) macrophages. Hierarchical clustering and principal component analysis revealed that the largest differences existed between CCR2− macrophages, CCR2+ macrophages, and Ly6Chi monocytes. CCR2+ macrophages clustered close to monocytes, consistent with ontological relationship between cell types. CCR2− macrophage subsets clustered together suggesting a high degree of similarity in gene expression amongst those populations (Fig. S3).
We identified 893 genes differentially expressed between CCR2− and CCR2+ macrophages (>1.5-fold change, FDR p-value<0.05) highlighting the marked divergence between these populations. Few differences were observed between individual CCR2− macrophage subsets. Comparison of CCR2− macrophages derived from definitive and extra-embryonic hematopoiesis revealed a single differentially expressed gene. 13 genes were differentially expressed between CCR2−MHC-IIhi and CCR2−MHC-IIlo populations, many of which were MHC-II alleles. Pathway analysis demonstrated that CCR2+ macrophages expressed genes associated with antigen presentation, immune, inflammatory response, T-cell co-stimulation, integrin signaling, and angiogenesis. Examples of genes upregulated in CCR2+ macrophages included Il1b, Gdf3, Lgals3, Ccl17, Cxcl19, Itgax, Itgb7, Itgax, Traf1, Tnip3, Tnfsf14, Timp1, Mmp12, Mmp19, Vegfa, Pgf, Col4a1, Col3a1, and Fn1. In contrast, CCR2− macrophages showed enrichment of pathways associated with endocytosis, transport, nervous system development, cell adhesion, and migration. CCR2− macrophages expressed a paucity of inflammatory mediators, and instead, expressed growth factors and genes associated with sensing mechanical stimuli including Igf1, Hbegf, Bmp2, Cyr61, Pdgfc, Fgf9, Trpv4, CD33, and Rhob (Fig. S3).
CCR2− Macrophages are Required for Survival, Adaptive Tissue Remodeling, and Maintenance of Cardiac Output in Chronic Heart Failure.
We chose to focus on CCR2− macrophages, given their absolute abundance and gene expression signatures. We utilized CD169-DTR mice to selectively deplete CCR2− macrophages from the heart during the adaptive remodeling phase. We generated the following experimental groups: control, CD169-DTR, Tnnt2ΔK210/ΔK210, and Tnnt2ΔK210/ΔK210 CD169-DTR mice. Consistent with our previous findings (Bajpai et al., 2019), daily diphtheria toxin (DT) injections led to marked reduction in cardiac macrophage density and selective elimination of CCR2− macrophages (Fig. 2A–D). Neutrophil, monocyte, and CCR2+ macrophage abundance was not impacted. CCR2− macrophage depletion did not increase CCR2+ macrophage chemokine or cytokine expression or result in alteration in peripheral blood cell counts, serum chemistries, or cytokines (Fig. S4, Table S2).
Figure 2. CCR2− macrophages influence survival and LV remodeling in dilated cardiomyopathy.
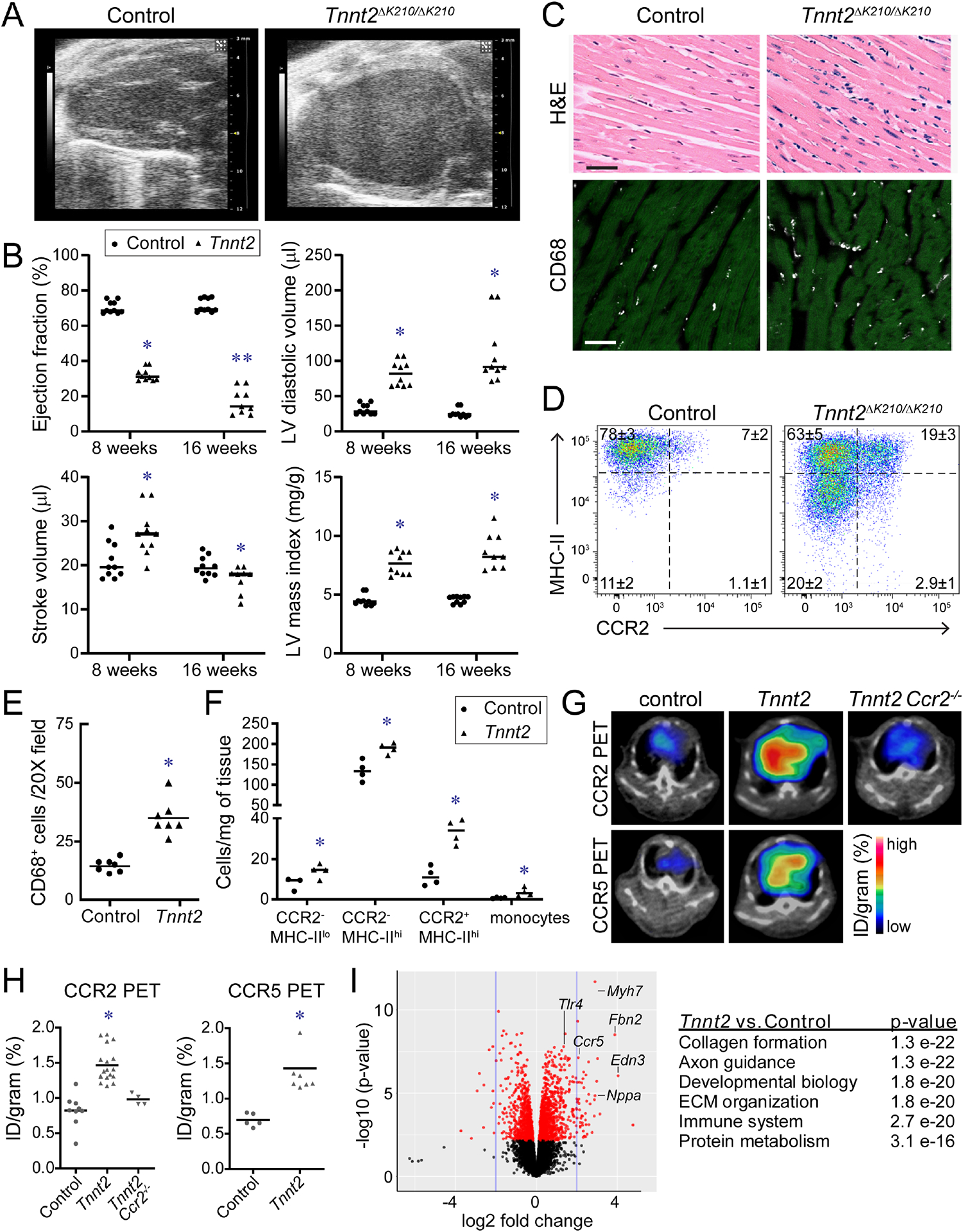
A, Schematic of experimental groups, CCR2− macrophage depletion strategy, and endpoints. B, CD68 immunostaining of control, CD169-DTR, Tnnt2ΔK210/ΔK210, and Tnnt2ΔK210/ΔK210 CD169-DTR hearts after 3 weeks of DT treatment. n=4 per group. Scale bar: 20μm. C, Quantification of CD68 immunostaining. ** denotes p<0.05 (ANOVA, Post-hoc Tukey) compared to all other groups. D, Flow cytometry of CD45+Ly6G−CD64+ macrophages showing specific depletion of CCR2− macrophages in Tnnt2ΔK210/ΔK210 CD169-DTR hearts. n=4 per group. E, Kaplan-Meier analysis of survival in control, CD169-DTR, Tnnt2ΔK210/ΔK210, and Tnnt2ΔK210/ΔK210 CD169-DTR mice after DT treatment. n=12–15 per group. F-I, LV ejection fraction, relative wall thickness, LV volumes (μl), and stroke volume in control, CD169-DTR, Tnnt2ΔK210/ΔK210, and Tnnt2ΔK210/ΔK210 CD169-DTR mice 3 weeks after DT treatment. n=4–8 per group. * denotes p<0.05 (ANOVA, Post-hoc Tukey) compared to controls. # denotes p<0.05 compared to Tnnt2ΔK210/ΔK210 mice. J, Pressure volume loops showing reduced stroke volume in Tnnt2ΔK210/ΔK210 CD169-DTR compared to Tnnt2ΔK210/ΔK210 mice. n=4 per group. K, Invasive hemodynamics: LV dP/dt max, heart rate (HR), and LV end systolic pressure (LVESP) at baseline and during peak infusion of dobutamine (64 ng/min) in control, CD169-DTR, Tnnt2ΔK210/ΔK210, and Tnnt20394K210/ΔK210 CD169-DTR mice 3 weeks after DT treatment. n=5 per group. * denotes p<0.05 (ANOVA, Post-hoc Tukey) compared to controls. # denotes p<0.05 compared to Tnnt2ΔK210/ΔK210 mice. See also Figure S4–6, Tables S2–S3.
We treated control, CD169-DTR, Tnnt2ΔK210/ΔK210, and Tnnt2ΔK210/ΔK210 CD169-DTR mice with DT beginning at 6 weeks of age. The primary endpoints included a Kaplan-Meier survival analysis and echocardiographic assessment of LV function and remodeling performed at 9 weeks of age (3 weeks of DT treatment). Kaplan-Meier analysis revealed reduced survival of Tnnt2ΔK210/ΔK210 CD169-DTR mice compared to Tnnt2ΔK210/ΔK210 mice. No mortality was observed in control or CD169-DTR mice (Fig. 2E). Echocardiography demonstrated similar reductions in LV ejection fraction between Tnnt2ΔK210/ΔK210 and Tnnt2ΔK210/ΔK210 CD169-DTR mice compared to controls (Fig. 2F). Tnnt2ΔK210/ΔK210 mice displayed robust measures of remodeling including LV dilation, reduced relative wall thickness, and increased stroke volume compared to controls. Tnnt2ΔK210/ΔK210 CD169-DTR mice displayed attenuated LV dilation, increased relative wall thickness, and reduced stroke volume compared to Tnnt2ΔK210/ΔK210 mice. (Fig. 2G–I). No differences were observed between control and CD169-DTR mice for all echocardiographic variables examined.
Simultaneous measurement of LV pressure and volume confirmed reduced stroke volume in Tnnt2ΔK210/ΔK210 CD169-DTR compared to Tnnt2ΔK210/ΔK210 mice (Fig. 2J). LV catheterization and dobutamine infusion revealed reduced LV end systolic pressure, myocardial contractility, and contractile reserve in Tnnt2ΔK210/ΔK210 mice compared to controls. CCR2− macrophage depletion did not impact measurements of myocardial contractility in either control or Tnnt2ΔK210/ΔK210 backgrounds (Fig. 2K, Table S3). Collectively, these findings indicate that CCR2− macrophage depletion blunts LV remodeling in Tnnt2ΔK210/ΔK210 mice without impacting myocardial contractility.
CCR2− Macrophages are Required for Myocardial Reorganization and Coronary Angiogenesis.
To examine whether structural differences contribute to alterations in LV remodeling observed in Tnnt2ΔK210/ΔK210 CD169-DTR mice, we performed histological analysis following 3 weeks of DT treatment. Compared to controls, Tnnt2ΔK210/ΔK210 hearts displayed evidence of myocardial reorganization including circumferential enlargement of the LV, loss of trabecular myocardium, and expansion of compact myocardium. Tnnt2ΔK210/ΔK210 CD169-DTR hearts displayed reduced circumferential LV enlargement, persistent trabecular myocardial tissue, and failed to expand the compact myocardium. CD169-DTR mice displayed no significant changes compared to controls (Fig. 3A–C). To verify these structural alterations by a second method, we performed X-Ray microscopy (XRM), a variant of micro-computed tomography that provides full volume datasets. Surface projection and virtual histology images of the LV chamber revealed the presence of smooth appearing walls and precise alignment of muscle fibers in Tnnt2ΔK210/ΔK210 hearts. In contrast, the LV walls of Tnnt2ΔK210/ΔK210 CD169-DTR hearts displayed a ribbon-like appearance and impaired muscle fiber alignment (Fig. 3D). Collectively, these findings indicate that CCR2− macrophages influence LV remodeling through alterations in myocardial tissue organization.
Figure 3. CCR2− macrophages orchestrate myocardial tissue adaptations in dilated cardiomyopathy.
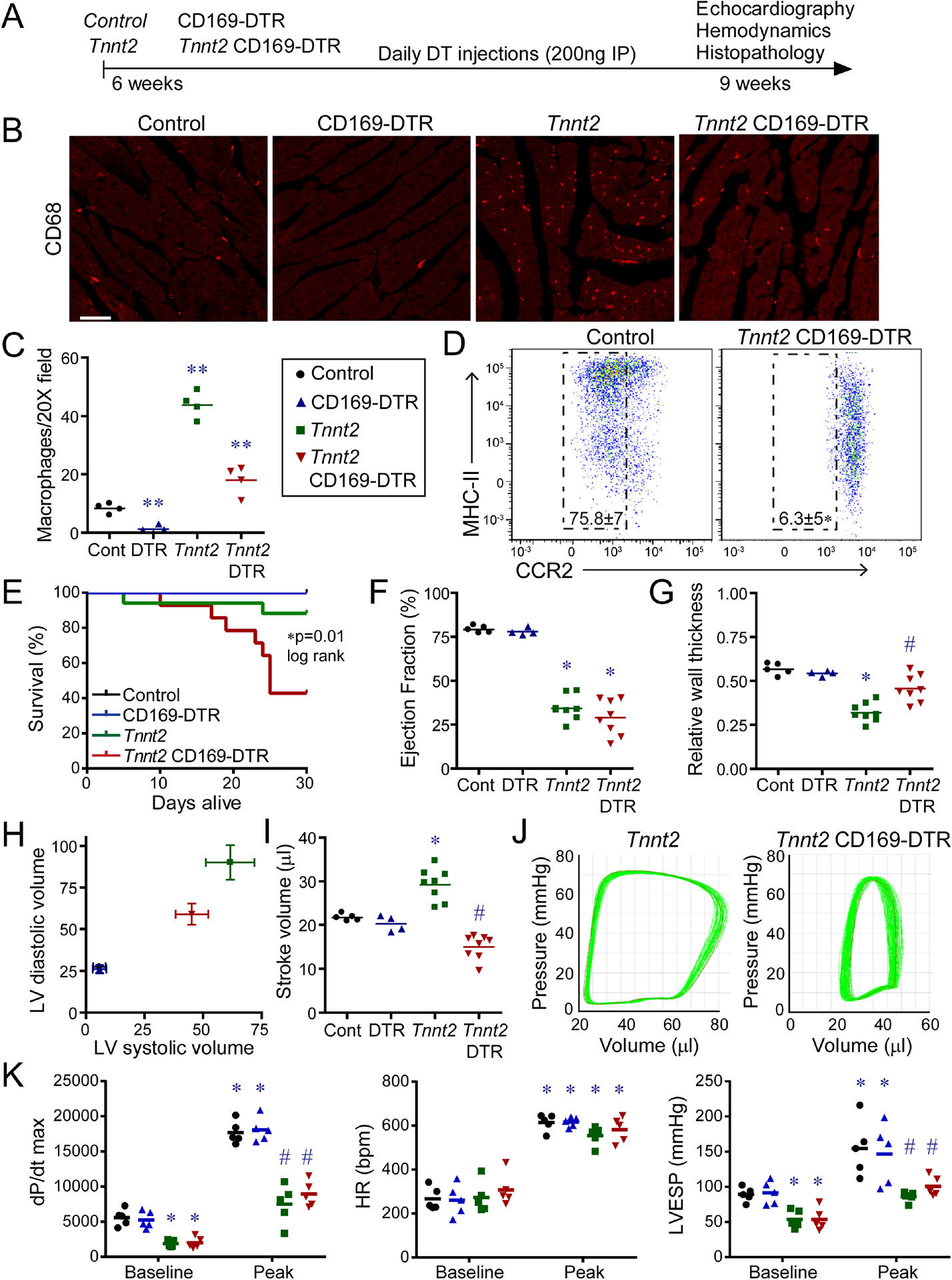
A, Low magnification H&E images of control, CD169-DTR, Tnnt2ΔK210/ΔK210, and Tnnt2ΔK210/ΔK210 CD169-DTR hearts after 3 weeks of DT treatment. n=4–9 per group. Scale bar: 200μm. B, Quantification of the ratio of compact to trabecular myocardium in control, CD169-DTR, Tnnt2ΔK210/ΔK210, and Tnnt2ΔK210/ΔK210 CD169-DTR hearts. C, Wheat germ agglutinin (WGA, red) staining showing alignment of trabecular cardiomyocytes in control, Tnnt2ΔK210/ΔK210, and Tnnt2ΔK210/ΔK210 CD169-DTR hearts. n=4–9 per group. Scale bar: 40μm. D, X-ray microscopy and virtual histology images comparing myocardial architecture of Tnnt2ΔK210/ΔK210 and Tnnt2ΔK210/ΔK210 CD169-DTR hearts following 3 weeks of DT injection. n=4 per group. Scale bar: 200μm. E, Images of cardiomyocytes digested from control, Tnnt2ΔK210/ΔK210, and Tnnt2ΔK210/ΔK210 CD169-DTR hearts. Hearts were relaxed in potassium prior to fixation. Scale bar: 20μm. F, Quantification of cardiomyocyte area, length, and width. * denotes p<0.05 (ANOVA, Post-hoc Tukey) compared to controls. ** denotes p<0.05 compared to control and Tnnt2ΔK210/ΔK210 mice. See also Figure S4.
To examine whether CCR2− macrophages affect cardiomyocyte size, we performed a morphometric analysis of cardiomyocytes isolated from control, Tnnt2ΔK210/ΔK210, and Tnnt2ΔK210/ΔK210 CD169-DTR hearts. Tnnt2ΔK210/ΔK210 and Tnnt2ΔK210/ΔK210 CD169-DTR cardiomyocytes had increased 2-dimensional area compared to controls. The extent of cell enlargement was greater in Tnnt2ΔK210/ΔK210 compared to Tnnt2ΔK210/ΔK210 CD169-DTR cardiomyocytes. Tnnt2ΔK210/ΔK210 cardiomyocytes displayed increases in both cell width and length compared to controls. Tnnt2ΔK210/ΔK210 CD169-DTR cardiomyocytes displayed similar increases in cell width, but less cell lengthening (Fig. 3E–F). These data suggest that modulation of cardiomyocyte length might also be involved in CCR2− macrophage dependent remodeling.
To evaluate whether CCR2− macrophages influence adverse remodeling, we examined cardiomyocyte cross-sectional area, interstitial fibrosis, and mRNA expression. Wheat germ agglutinin staining revealed similarly increased cardiomyocyte area in both Tnnt2ΔK210/ΔK210 and Tnnt2ΔK210/ΔK210 CD169-DTR hearts compared to controls. Minimal interstitial fibrosis was evident at the stages examined. Nppa, Nppb, and Myh7 mRNA expression was increased to a similar degree in Tnnt2ΔK210/ΔK210 and Tnnt2ΔK210/ΔK210 CD169-DTR hearts compared to controls. Previous studies have implicated matrix metalloproteinase (MMP) activity in LV dilation and remodeling after myocardial infarction (Ducharme et al., 2000; Heymans et al., 1999). We observed negligible MMP9 activity in the hearts of control and Tnnt2ΔK210/ΔK210 mice. Robust MMP9 activity was present following DT-mediated cardiomyocyte ablation (Fig. S4).
CCR2− macrophages regulate coronary angiogenesis in the embryonic and neonatal heart (Lavine et al., 2014; Lavine et al., 2013; Leid et al., 2016). We evaluated whether CCR2− macrophages also modulate coronary angiogenesis in chronic heart failure. Visualization of the coronary arterial vasculature using Microfil casting demonstrated marked expansion in Tnnt2ΔK210/ΔK210 hearts compared to controls. Minimal coronary artery expansion was observed in Tnnt2ΔK210/ΔK210 CD169-DTR hearts (Fig. 4A). Measurement of coronary microvascular density revealed robust increases in Tnnt2ΔK210/ΔK210 hearts compared to controls and Tnnt2ΔK210/ΔK210 CD169-DTR hearts (Fig. 4B–C).
Figure 4. CCR2− macrophages are essential for coronary angiogenesis in dilated cardiomyopathy.
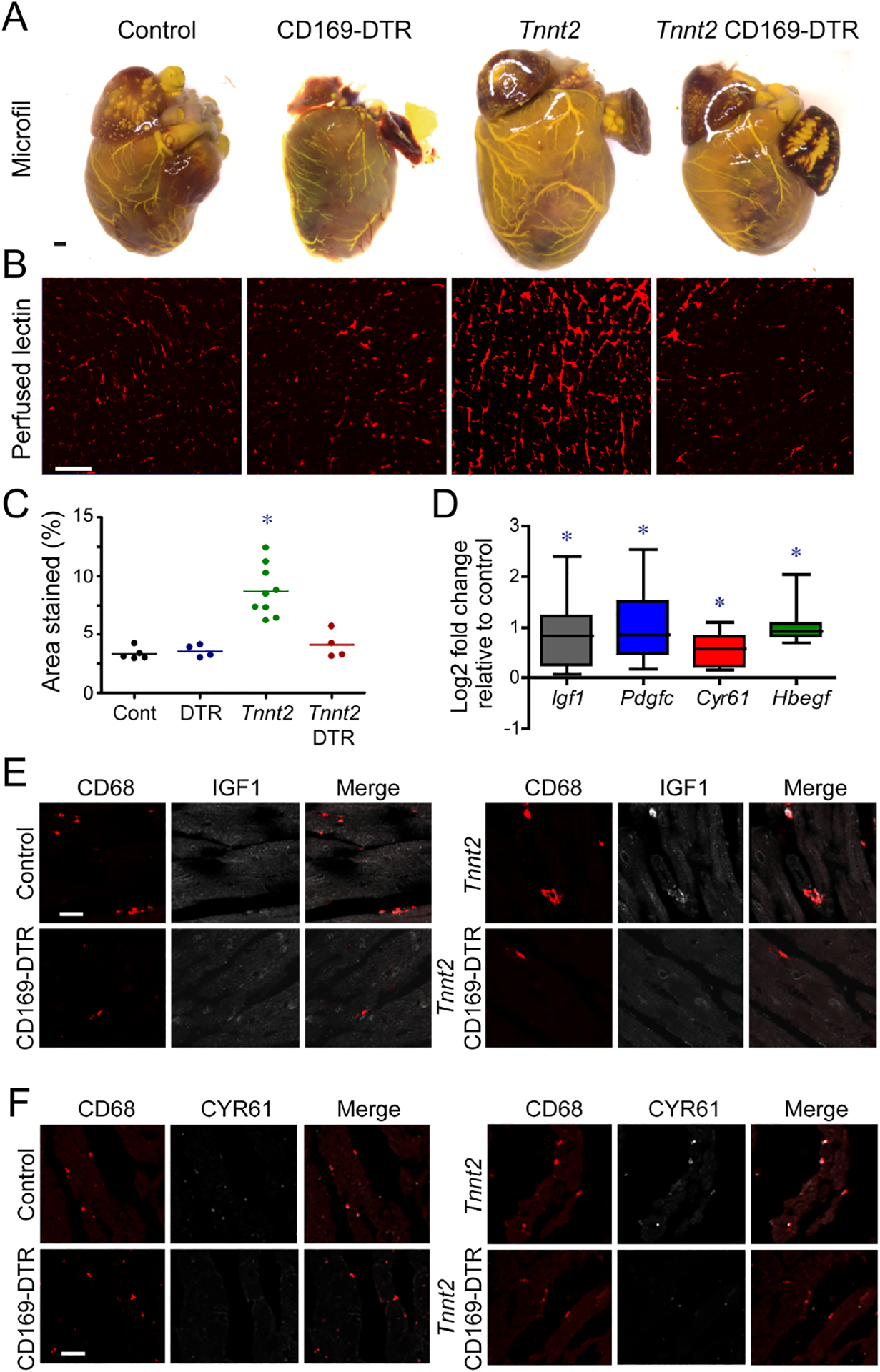
A, Microfil vascular casting of control, CD169-DTR, Tnnt2ΔK210/ΔK210, and Tnnt2ΔK210/ΔK210 CD169-DTR hearts after 3 weeks of DT treatment. n=4 per group. Scale bar: 200μm. B, Perfused lectin staining of control, CD169-DTR, Tnnt2ΔK210/ΔK210, and Tnnt2ΔK210/ΔK210 CD169-DTR hearts after 3 weeks of DT treatment.. n=4–9 per group. Scale bar: 20μm. C, Quantification of the coronary microvascular in control, CD169-DTR, Tnnt2ΔK210/ΔK210, and Tnnt2ΔK210/ΔK210 CD169-DTR hearts. * denotes p<0.05 (ANOVA, Post-hoc Tukey) compared to controls. D, CCR2− macrophages from Tnnt2ΔK210/ΔK210 hearts display increased Igf1, Pdgfc, Cyr61, and Hbegf mRNA expression compared to controls. n=5 per group. * denotes p<0.05 (Mann-Whitney test) compared to controls. E-F, IGF1 (E, white) and CYR61 (F, white) immunostaining of control, CD169-DTR, Tnnt2ΔK210/ΔK210, and Tnnt2ΔK210/ΔK210 CD169-DTR hearts after 3 weeks of DT treatment. Red: CD68. n=5 per group. Scale bars: 10μm (E) and 20μm (F).
To explore potential mechanisms by which CCR2− macrophages promote coronary angiogenesis, we measured pro-angiogenic growth factor expression in CCR2− macrophages. Igf1, Pdgfc, Cyr61, and Hbegf mRNA expression was increased in CCR2− macrophages from Tnnt2ΔK210/ΔK210 hearts compared to controls (Fig. 4D).
Immunostaining revealed that macrophage IGF1 and CYR61 expression was increased in Tnnt2ΔK210/ΔK210 hearts compared to controls. Increased macrophage IGF1 and CYR61 expression was not evident in Tnnt2ΔK210/ΔK210 CD169-DTR hearts, presumably due to the absence of CCR2− macrophages (Fig. 4E–F).
Previous work has suggested that cardiac macrophages have the potential to influence propagation of electrical signals through the atrioventricular node (Hulsmans et al., 2017). To assess whether alterations in electoral conduction occurred following depletion of CCR2− macrophages from control and Tnnt2ΔK210/ΔK210 mice, we analyzed surface electrocardiograms obtained from anesthetized control, CD169-DTR, Tnnt2ΔK210/ΔK210, and Tnnt2ΔK210/ΔK210 CD169-DTR mice treated with DT for 3 weeks. We did not observe significant differences in RR (heart rate), PR (atrioventricular node conduction), or QRS (intraventricular conduction) intervals between experimental groups. Tnnt2ΔK210/ΔK210 and Tnnt2ΔK210/ΔK210 CD169-DTR mice displayed similar prolongation of the QT (ventricular repolarization) interval compared to control and CD169-DTR mice. While these results indicate that defects in electrical propagation are unlikely to account for increased mortality observed in Tnnt2ΔK210/ΔK210 CD169-DTR mice, they do not rule out the possibility that CCR2− macrophages contribute to optimal cardiac conduction. We found that both CCR2− and CCR2+ macrophages were located within the AV node potentially accounting for the lack of an overt electrical phenotype (Fig. S5).
A recent study suggests that cardiac macrophages regulate myocardial metabolism through effects on mitochondrial homeostasis (Nicolas-Avila et al., 2020). To examine whether this might contribute to the phenotype of Tnnt2ΔK210/ΔK210 CD169-DTR mice, we isolated mitochondria from control, CD169-DTR, Tnnt2ΔK210/ΔK210, and Tnnt2ΔK210/ΔK210 CD169-DTR hearts following 3 weeks of DT treatment. We did not detect differences in mitochondrial respiration across experimental groups (Fig. S5), indicating that alterations in mitochondrial function are unlikely responsible for the cardiac phenotype of Tnnt2ΔK210/ΔK210 CD169-DTR mice.
CCR2− Macrophages Promote Adaptive Remodeling following Pressure Overload.
To decipher whether CCR2− macrophages influence remodeling in a separate model of heart failure, we subjected control and CD169-DTR mice to either sham surgery or transaortic constriction (pressure overload induced heart failure). In control mice, transaortic constriction resulted in reduced LV systolic function, eccentric remodeling, myocardial fibrosis, and expansion of the coronary vasculature. Transaortic constriction of CD169-DTR mice led to greater reductions in LV systolic function, attenuated LV dilation, exaggerated myocardial fibrosis, and diminished coronary microvascular growth. Measurement of LV wall thickness revealed that control and CD169-DTR mice underwent distinct forms of LV remodeling. Cordinated increases in LV dimension and mass observed in control mice were consistent with eccentric hypertrophy, a process that maintains stroke volume. Conversely, CD169-DTR mice displayed attenuated LV dilation despite an equal increase in LV mass, consistent with concentric hypertrophy, a process associated with diminished LV stroke volume and elevated Anp, Bnp, and Myh6 mRNA expression. We did not observe any differences between sham treated control and CD169-DTR mice (Fig. S6). These findings indicate that CCR2− macrophages preserve LV systolic function, contribute to adaptive remodeling (eccentric hypertrophy, angiogenesis), and suppress maladaptive remodeling (concentric hypertrophy, fibrosis) in the setting of pressure overload.
CCR2− Macrophages Physically Interact with Neighboring Cardiomyocytes.
To gain insights into how CCR2− macrophages influence myocardial remodeling and angiogenesis, we examined macrophage localization and structure in control and Tnnt2ΔK210/ΔK210 hearts using confocal microscopy. Under baseline and heart failure conditions, CCR2− macrophages were observed within close proximity to cardiomyocytes and extended processes that contacted adjacent cardiomyocytes. CCR2+ macrophages extended processes within the interstitial space (Fig. 5A–B). Electron microscopy of Tnnt2ΔK210/ΔK210 Ccr2GFP/+ hearts stained with anti-CD68 and anti-GFP antibodies confirmed that CCR2− macrophages physically contacted neighboring cardiomyocytes (Fig. 5C). CCR2+ macrophages extended processes into the interstitial space, but did not directly contact cardiomyocytes (Fig. 5D). The projection length of CCR2− macrophages was greater in Tnnt2ΔK210/ΔK210 hearts compared to controls. The projection length of CCR2+ macrophages was indistinguishable between control and Tnnt2ΔK210/ΔK210 Ccr2GFP/+ hearts (Fig. 5E).
Figure 5. CCR2− macrophages interact with neighboring cardiomyocytes through focal adhesion complexes.
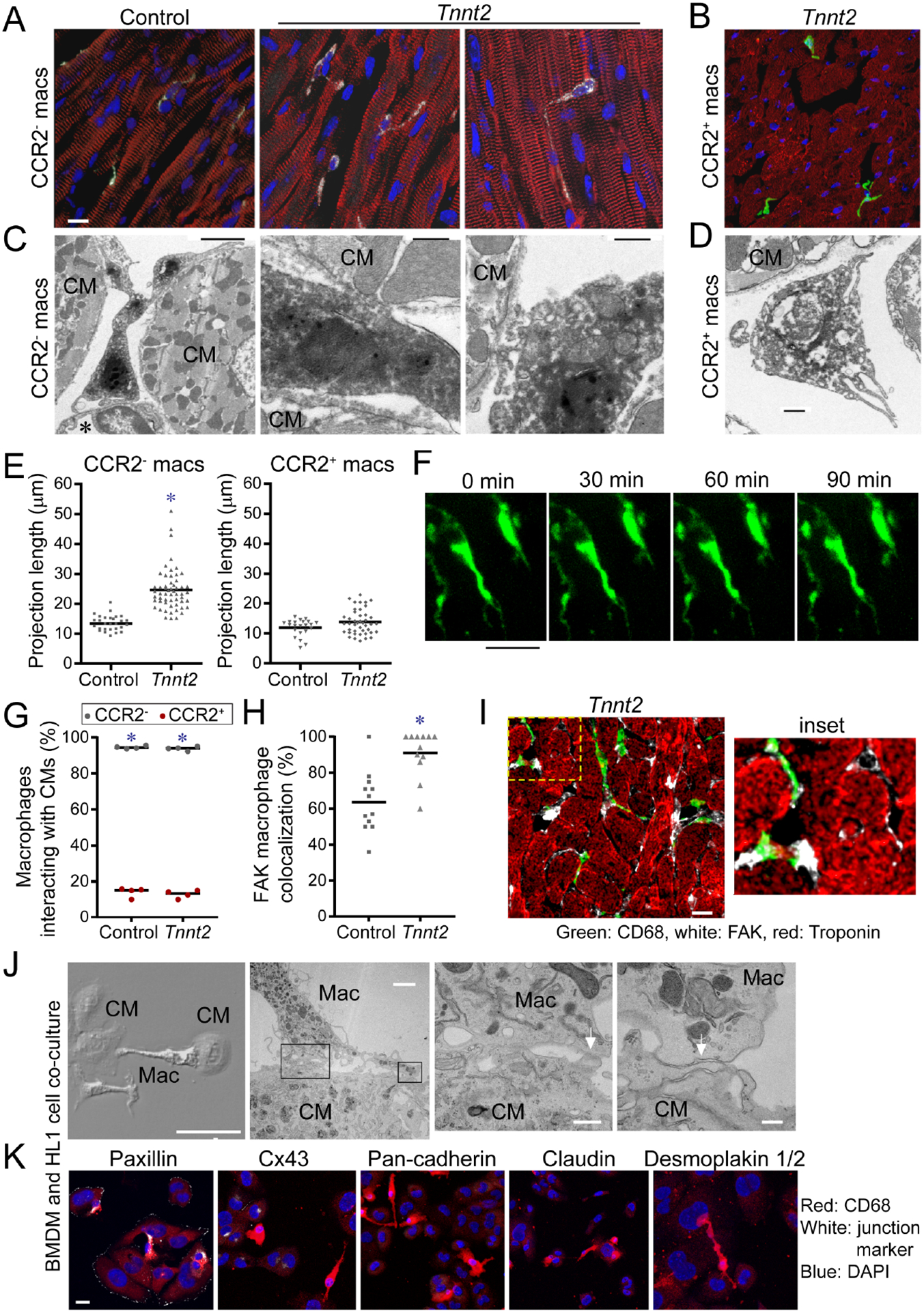
A-B, Compressed Z-stack images of CCR2− (A) and CCR2+ (B) cardiac macrophages of a 8-week-old control Ccr2GFP/+ and Tnnt2ΔK210/ΔK210 Ccr2GFP/+ hearts. n=4–6 per group. CD68: white, GFP: green, α-actinin, red, DAPI: blue. Scale bar: 10μm. C-D, Electron microscopy of CCR2− (C) and CCR2+ (D) cardiac macrophages in Tnnt2ΔK210/ΔK210 Ccr2GFP/+ hearts. CCR2− macrophage are adjacent to endothelial cells (*) and contact cardiomyocytes (CM). n=4 per group. Scale bars: 2μm (C, left), 500nm (C, center; C, right, D). E, Measurement of projection length in CCR2− and CCR2+ macrophages of 8-week-old Ccr2GFP/+ and Tnnt2ΔK210/ΔK210 Ccr2GFP/+ hearts. * denotes p<0.05 (Mann-Whitney test) compared to controls. F, 2-photon microscopy of live Cx3cr1GFP/+ Ccr2RFP/+ papillary muscle cell preparations (n=4) over 90 minutes. CCR2− macrophages (green). Scale bar: 25μm. G, Quantification of the percent of CCR2− (black data points) and CCR2+ (red data points) macrophages interacting with cardiomyocytes in control and Tnnt2ΔK210/ΔK210 hearts. n=4 per group. * denotes p<0.05 (Mann-Whitney test) comparing CCR2− to CCR2+ macrophages. H-I, Immunostaining for CD68 (green), FAK (white), and troponin (red). Scale bar: 10μm. n=12 per group. * denotes p<0.05 (Mann-Whitney test) compared to controls. J, Electron microscopy of bone marrow-derived macrophages (MΦ) and HL1 cardiomyocytes (CM) after 4 hours of co-culture. Physical interactions (white arrows) between macrophages and cardiomyocytes. Right center right panels: high magnification of boxed areas shown in left center panel. Scale bars: 50μm, 2μm, 500nm, 200nm. K, Immunostaining of cell junction markers. Blue-DAPI, red-CD68, white-cell junction markers: Paxillin (focal adhesion complex) Cx43 (gap junction), pan-Cadherin (adherins junction), Claudin (tight junction), Desmoplakin I/II (desmosome). Representative images from 4 independent experiments. Scale bar: 10μm. See also Figure S6, Supplemental movie 1–2.
To characterize the temporal dynamics of interactions between CCR2− macrophages and cardiomyocytes, we performed two-photon microscopy on isolated mouse papillary muscle preparations. Papillary muscles were isolated from Cx3cr1GFP/+ Ccr2RFP/+ mice and imaged for 1–2 hours in a temperature-controlled imaging chamber containing oxygenated media. Cellular processes were observed that extended from CCR2− macrophages and formed stable contacts with neighboring cardiomyocytes. These processes did not further extend or retract over the 90-minute time course of imaging (Fig. 5F, supplemental movie). Predominately CCR2− macrophages interacted with cardiomyocytes (Fig. 5G).
We did not observe electron densities indicative of desmosomes, adherens junctions, or tight junctions between CCR2− macrophages and cardiomyocytes. We found that FAK (focal adhesion complex marker) was frequently present between CCR2− macrophages and cardiomyocytes (Fig. 5H–I). We then utilized an in vitro system to delineate whether focal adhesion complexes were responsible for macrophage-cardiomyocyte interactions. We found that HL-1 cardiomyocytes and bone marrow-derived macrophages formed spontaneous interactions when co-cultured. Electron microscopy revealed evidence of physical interaction between HL1-cells and cardiomyocytes. Electron densities consistent with desmosomes, adherens, tight, or gap junctions were not evident. Immunostaining showed presence of Paxillin staining at sites of interactions between HL-1 cells and bone marrow-derived macrophages (Fig. 5J–K).
Previous studies have established that β-integrins are essential for focal adhesion complexes (Parsons et al., 2010). Addition of antibodies that blocked either β1-integrin or β2-integrin binding was sufficient to disrupt interactions between HL-1 cells and bone marrow-derived macrophages. Application of β1-integrin neutralizing antibodies also resulted in loss of Paxillin staining within macrophages at sites of cardiomyocyte-macrophage interaction (Fig. S7). Collectively, these findings indicate that CCR2− macrophages physically interact with cardiomyocytes through focal adhesion complexes.
TRPV4 Regulates Growth Factor Expression in Macrophages.
We considered the possibility that elevated LV pressure and resultant increased myocardial wall stress may be sensed by CCR2− macrophages through interactions with neighboring cardiomyocytes. To explore whether CCR2− macrophages may be activated by mechanical cues, we assayed the expression of known mechanoresponsive factors and found that Trpv4 mRNA was abundantly expressed in CCR2− macrophages (Fig. 6A, Fig. S7). Ratiometric calcium assays demonstrated that the TRPV4 channel was active in cardiac macrophages. Treatment of CCR2− and CCR2+ macrophages isolated from the heart by flow cytometry with a specific TRPV channel activator (GSK101) or TRPV4 channel inhibitor (GSK219) confirmed functional TRPV4 protein in both CCR2− and CCR2+ macrophages within the LV myocardium (Fig. 6B, Fig. S7). Immunostaining and flow cytometry of Trpv4-GFP BAC transgenic mice provided evidence the TRPV4 was predominately expressed in cardiac macrophages and neutrophils (Fig. 6C–D, Fig. S7). We detected TRPV4 activity in cardiac macrophages in situ using Cx3cr1-ertCre Rosa26-GCaMP6s Rosa26-tdtomato reporter mice (Madisen et al., 2015), which allows selective visualization of macrophage cytoplasmic calcium. 2-photon imaging of Cx3cr1-ertCre Rosa26-GCaMP6s Rosa26-tdtomato papillary muscles placed under axial tension revealed GCaMP signal in cardiac macrophages. Application of a TRPV4 inhibitor (GSK219) suppressed GCaMP signal, indicating that TRPV4 channel activity is responsible for the observed rise in macrophage cytoplasmic calcium (Fig. 6E–F).
Figure 6. TRPV4 regulates growth factor expression in macrophages.
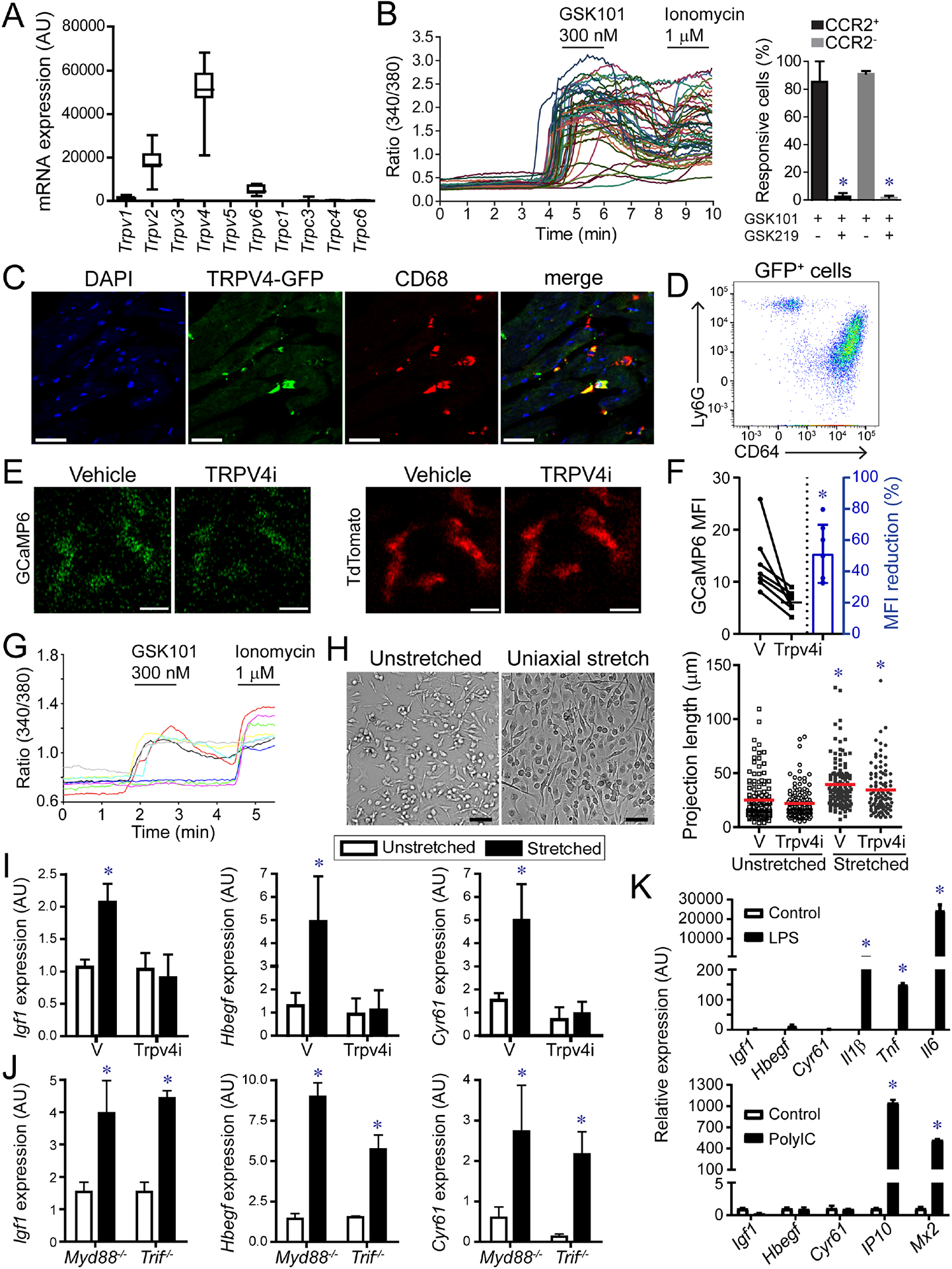
A, mRNA expression of TRP channels in CCR2− macrophages. n=20 samples. B, Ratiometric calcium assay demonstrating that cardiac macrophages have active TRVP4 channels. GSK101: TRPV4 agonist, GSK219: TRPV4 antagonist. * denotes p<0.05 (ANOVA, Post-hoc Tukey) comparing GSK101 treated cells with GSK101 and GSK219 treated cells. C, Immunostaining of Trpv4-GFP BAC transgenic mice showing GFP (green) expression in CD68+ macrophages (red). Scale bar: 20μm. D, Flow cytometry of cardiac CD45+GFP+ leukocytes isolated from Trpv4-GFP hearts. n=4 per group. E, 2-photon imaging of GFP (green) and tdTomato (red) in papillary muscle preparations harvested from Cx3cr1-ertCre; Rosa26-GCaMP6/tdTomato mice treated with either vehicle or the TRPV4 inhibitor GSK219 (TRPV4i). Scale bar: 10μm. F, Quantification of GCaMP6 signal. n=6 per group. * denotes p<0.05 (paired t-test) compared to vehicle. G, Ratiometric calcium assay showing that bone marrow derived macrophages express active TRPV4. GSK101: TRPV4 agonist, Ionomycin: calcium ionophore. H, Cyclic uniaxial stretch (1 Hz, 10% deformation, 24 hours) promotes elongation of bone marrow derived macrophages. n=4 independent experiments. Scale bar: 50μm. * denotes p<0.05 (ANOVA, Post-hoc Tukey) compared to vehicle unstretched. I, Quantitative RT-PCR measuring Igf1, Hbegf, and Cyr61 mRNA expression in stretched macrophages treated with vehicle or TRPV4 inhibitor. n=4 independent experiments. * denotes p<0.05 compared to vehicle treated unstretched cells (ANOVA post-hoc Tukey). J, Quantitative RT-PCR measuring Igf1, Hbegf, and Cyr61 mRNA expression in stretched control, Myd88−/−, and Trif−/− macrophages. n=4 independent experiments. * denotes p<0.05 compared to vehicle treated unstretched cells (ANOVA post-hoc Tukey) (F-H). K, Quantitative RT-PCR of macrophages stimulated with vehicle control, LPS, or polyIC. * denote p<0.05 (Mann-Whitney). n=4 independent experiments. See also Figure S6, Data S2.
We utilized bone marrow-derived macrophages to examine whether TRPV4 mediates macrophage activation by mechanical cues. Ratiometric calcium assays confirmed that bone marrow-derived macrophages express functional TRPV4 channels (Fig. 6G). We then subjected bone marrow-derived macrophages to cyclic mechanical stretch. Cells were cultured on silicone membranes and subjected to uniaxial stretch stretched (10% deformation, 1 Hz) for 24 hours in the presence of vehicle control or GSK219. Following 24 hours of stretch, both vehicle and TRPV4 inhibitor treated macrophages elongated along the axis of membrane deformation (Fig. 6H). Quantitative RT-PCR revealed increased Igf1, Hbegf, and Cyr61 mRNA expression in response to mechanical stretch. Application of the TRPV4 channel inhibitor blocked this response (Fig. 6I). To assess the contribution of canonical macrophage signaling, we subjected control, Myd88−/− and Trif−/− bone marrow derived-macrophages to uniaxial cyclic stretch. Quantitative RT-PCR demonstrated that deletion of Myd88 or Trif had no impact on the expression of Igf1, Hbegf, or Cyr61 (Fig. 6J). Activators of MYD88 and TRIF signaling (LPS, PolyIC) were unable to increase the expression of Igf1, Hbegf, or Cyr61 and mechanical stretch did not induce the expression of inflammatory cytokines (Fig. 6K). These observations indicate that mechanical stretch promotes growth factor expression from macrophages through a TRPV4 dependent mechanism that is independent of MYD88 and TRIF.
TRPV4 regulates IGF1 expression in CCR2− macrophages and is required for coronary angiogenesis.
To assess whether TRPV4 regulates cardiac macrophage IGF1 expression in vivo, we treated control and Tnnt2ΔK210/ΔK210 mice with either vehicle, TRPV4 inhibitor, or TRPV4 agonist beginning at 6 weeks of age. Immunostaining for CD68 and IGF1 after 2 days of treatment revealed that TRPV4 increases cardiac macrophage IGF1 expression. Compared to controls, cardiac macrophages in Tnnt2ΔK210/ΔK210 hearts expressed IGF1 at higher frequency and increased mean value. Treatment of Tnnt2ΔK210/ΔK210 mice with the TRPV4 inhibitor was sufficient to reduce cardiac macrophage IGF1 expression (frequency, mean value). Mice treated with the TRPV4 agonist displayed increased IGF1 expression in cardiac macrophages (Fig. 7A–B).
Figure 7. TRPV4 regulates IGF1 expression in CCR2− macrophages and is required for coronary angiogenesis.
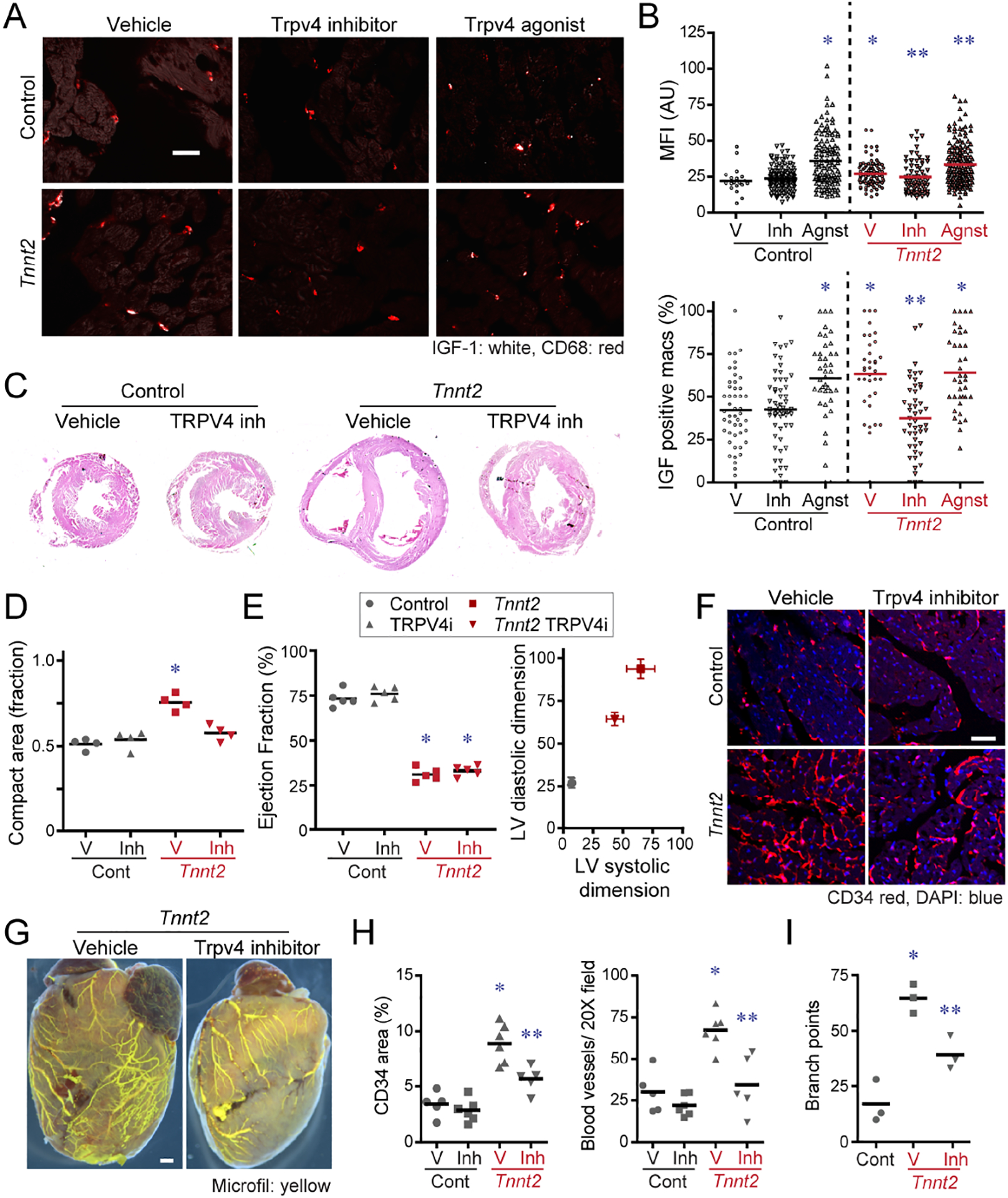
A, Immunostaining for IGF1 (white) and CD68 (red) inf control and Tnnt2ΔK210/ΔK210 hearts treated with vehicle, TRPV4 inhibitor, or TRPV4 agonist. Scale bar: 20μm. B, Quantification of IGF1 protein expression (% IGF1+ macrophages, IGF1 MFI) in control and Tnnt2ΔK210/ΔK210 hearts treated with vehicle, TRPV4 inhibitor, or TRPV4 agonist. MFI: mean florescent intensity. n=5 per group. * denotes p<0.05 compared to vehicle treated control hearts. ** denotes p<0.05 compared to vehicle treated Tnnt2ΔK210/ΔK210 hearts. (ANOVA, post-hoc Tukey). C, Low magnification H&E images of control and Tnnt2ΔK210/ΔK210 hearts treated with vehicle or TRPV4 inhibitor for 2 weeks beginning at 6 weeks of age. n=4 per group. Scale bar: 200μm. D, Quantification of the ratio of compact to trabecular myocardium in control and Tnnt2ΔK210/ΔK210 hearts treated with vehicle or TRPV4 inhibitor. * denotes p<0.05 compared to vehicle treated control hearts (ANOVA, post-hoc Tukey). E, LV ejection fraction and LV chamber dimensions in control and Tnnt2ΔK210/ΔK210 mice treated with vehicle or TRPV4 inhibitor for 2 weeks beginning at 6 weeks of age. n=5 per group. * denotes p<0.05 compared to vehicle treated control hearts (ANOVA, post-hoc Tukey). F, CD34 (red) immunostaining of control and Tnnt2ΔK210/ΔK210 hearts treated with vehicle or TRPV4 inhibitor. Scale bar: 40μm. G, Microfil vascular casting showing that TRPV4 is necessary for expansion of coronary microvasculature in Tnnt2ΔK210/ΔK210 hearts. Scale bar: 200μm. H-I, Quantification of coronary microvasculature (G) and coronary microvasculature (H). n=5 per egroup. * denotes p<0.05 compared to vehicle treated control. ** denotes p<0.05 compared to all other groups (ANOVA, Post-hoc Tukey).
To determine whether TRPV4 inhibition impairs adaptive remodeling, we treated control and Tnnt2ΔK210/ΔK210 mice with a TRPV4 inhibitor daily for 2 weeks beginning at 6 weeks of age. Histology demonstrated persistence of trabecular myocardium in Tnnt2ΔK210/ΔK210 hearts treated with the TRPV4 inhibitor, a phenotype that resembles depleting CCR2− macrophages (Fig. 7C–D). While TRPV4 inhibition had no impact on ejection fraction, we observed attenuated LV dilation in Tnnt2ΔK210/ΔK210 mice treated with the TRPV4 inhibitor (Fig. 7E). Vehicle treated Tnnt2ΔK210/ΔK210 mice displayed evidence of coronary angiogenesis at the microvascular and macrovascular levels compared to control mice. Tnnt2ΔK210/ΔK210 mice treated with the TRPV4 inhibitor displayed marked reductions in microvascular density within the myocardium and reduced large coronary artery complexity and branching compared to vehicle treated Tnnt2ΔK210/ΔK210 hearts. The TRPV4 inhibitor had no impact on coronary microvascular density in control mice (Fig. 7F–I). These data demonstrate that TRPV4 channel activity is necessary for adaptive remodeling and coronary angiogenesis in dilated cardiomyopathy and suggest mechanical sensing as a mechanism by which tissue resident cardiac macrophages are activated in the failing heart.
Discussion:
Previous studies have supported a division of labor between cardiac macrophage subsets (Bajpai et al., 2018; Epelman et al., 2014a; Pinto et al., 2016; Pinto et al., 2012). Here, we provide evidence that resident CCR2− cardiac macrophages represent a protective population that mediates adaptive remodeling and survival of the chronically failing heart. By employing a mouse model of dilated cardiomyopathy harboring a causative human mutation, we demonstrate that CCR2− macrophages maintain adequate cardiac output in the setting of reduced cardiac contractility by promoting LV enlargement and expansion of the coronary system at the macrovascular and microvascular levels. We observed a similar requirement for CCR2− macrophages in pressure overload induced heart failure. These observations are consistent with findings from an accompanying manuscript demonstrating that tissue resident cardiac macrophage maintain cardiac function following angiotensin II infusion (Zyman et al. Immunity-D-20-00713).
CCR2− macrophages stably interact with neighboring cardiomyocytes through focal adhesion complexes and sense mechanical stretch. The contribution of focal adhesion complexes to mechanosensing is well established (Geiger et al., 2009). It is not yet clear whether macrophages interact with the cardiomyocyte basement membrane or directly with cardiomyocytes themselves. Regardless, these structures are stable over time and have the potential to serve as sensors of mechanical deformations such as increased wall tension that may occur in the context of elevated preload or afterload.
We found that mechanical stretch serves as a stimulus for pro-angiogenic growth factor expression, a process that was dependent on TRPV4. Inhibition of TRPV4 reduced CCR2− macrophage pro-angiogenic growth factor expression and prevented coronary angiogenesis and myocardial tissue remodeling. These findings establish an unanticipated role for cardiac macrophages in adaptive remodeling of the chronically failing heart and introduce a mechanism of cardiac macrophage activation through sensing of myocardial stretch. Future work will clarify the breadth of hemodynamic stimuli that might activate CCR2− macrophages, and identify the exact signaling pathways triggered by TRPV4 channel activity. Mechanical stretch also regulates the proliferation of cardiac macrophages recruited to the injured heart, however the role of TRPV4 in this process remains to be explored (Sager et al., 2016).
As TRPV4 has been implicated in alveolar and intestinal macrophages (Hamanaka et al., 2010; Luo et al., 2018; Pairet et al., 2018), sensing of mechanical tissue deformation may constitute a conserved role of tissue resident macrophages. TRPV4 is also essential for phagocytosis-induced inflammation, a mechanism involved in clearance of cellular debris following tissue injury (Dutta et al., 2020; Goswami et al., 2019; Mannaa et al., 2018; Scheraga et al., 2016). We found that activation of TRPV4 by cyclic mechanical stretch was not dependent on MYD88 or TRIF signaling. A recent manuscript has suggested that Piezo1 activation may trigger TRPV4 channel opening providing a more direct link to mechanical stimulation (Swain et al., 2020).
These findings have several implications for the cardiovascular field. First, it is widely recognized that LV dilation is a strong predictor of heart failure outcomes including mortality (Merlo et al., 2011). Whether this represents an associative or causative relationship is not clear and ultimately may depend on the underlying pathology. Our observations indicate that LV dilation may be adaptive in some scenarios as it preserved cardiac output through augmentation of stroke volume. Second, previous studies have demonstrated that CCR2− macrophages remain abundant within the myocardium of patients with chronic heart failure (Bajpai et al., 2018). However, their function within this context was unknown. Using a mouse model of genetic dilated cardiomyopathy, we reveal an indispensable role for CCR2− macrophages in orchestrating myocardial tissue reorganization, cardiomyocyte lengthening, LV chamber enlargement, and coronary angiogenesis. These data exemplify the division of labor between CCR2− and CCR2+ cardiac macrophage populations and highlight dichotomous contributions to disease pathogenesis and protective adaptations, respectively (Bajpai et al., 2019; Dick et al., 2019; Epelman et al., 2014a; Hulsmans et al., 2018; Lavine et al., 2014; Leid et al., 2016). Therapeutically, these findings indicate strategies that preserve or enhance the functions of CCR2− macrophages may provide additive benefit to established medications for heart failure (ACE inhibitors, ARNIs, beta blockers, aldosterone antagonists), which target a separate mechanism (adverse remodeling).
Recent work has implicated cardiac macrophages in facilitating electrical conduction through the atrioventricular node (Hulsmans et al., 2017; Sugita et al., 2021). Surface electrocardiography did not identify arrhythmias or alterations in conduction following CCR2− macrophage depletion. Dedicated electrophysiology studies are required to definitely address the electrophysiological functions of CCR2− macrophages. We did observe CCR2+ macrophages in the AV node. CCR2− and CCR2+ macrophages each express numerous ion channels, suggesting the possibility that CCR2+ macrophages may participate in aspects of cardiac pacemaker function and electrical propagation.
Limitations of Study
We focused on a genetic and pressure overload mouse models. It remains to be shown whether CCR2− macrophages function in a similar manner in other heart failure models. Based on available literature, we chose to employ CD169-DTR mice to deplete CCR2− macrophages. This line depletes other macrophage populations outside of the heart and we cannot rule out the possibility that extra-cardiac macrophages contribute to some of the observed phenotypes. Finally, while CCR2− macrophages represent the most abundant cardiac cell type that express TRPV4, we cannot exclude the possibility that TRPV4 may influence the function of other cell types.
In conclusion, we establish a role for resident CCR2− macrophages in adaptive cardiac remodeling and survival of the failing heart. Furthermore, we provide initial evidence that mechanical sensing through a TRPV4 dependent pathway contributes to the activation of CCR2− macrophages within the heart.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kory Lavine (klavine@wustl.edu)
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data will be made immediately available upon request. Gene expression data will be available within the data supplement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals used in this study
Mice were bred and maintained at the Washington University School of Medicine and all experimental procedures were done in accordance with the animal use oversight committee. Mouse strains utilized included Tnnt2ΔK210 (Du et al., 2007), Rosa26-tdTomato (Madisen et al., 2010), Rosa26-GCaMP6 (Madisen et al., 2015), Flt3-cre (Boyer et al., 2011), Ccr2GFP (Satpathy et al., 2013), CD169-DTR (Miyake et al., 2007), Tnnt2-DTR (Bajpai et al., 2019), Cx3cr1GFPCcr2RFP (Jung et al., 2000; Saederup et al., 2010), Myd88−/− (Hou et al., 2008), Trif/Ticam1−/− (Hoebe et al., 2003), and Trpv4-GFP (Mutant Mouse Regional Resource Centers, MMRRC). All mice were on the C57/B6 background and genotyped according to established protocols. Equal numbers of male and female mice were included in all experiments. CCR2- macrophage depletion was induced by administering 200ng diphtheria toxin (Sigma) via intraperitoneal injection daily to CD169-DTR mice beginning at 6 weeks of age. GSK2193874 (10mg/kg) and GSK1016790A (0.1mg/kg) were administered to mice via IP injection daily for 2 weeks starting at 6 weeks of age.
METHOD DETAILS
Echocardiography, Invasive Hemodynamics, Electrocardiography, and Transaortic Constriction
Mouse echocardiography was performed in the Washington University Mouse Cardiovascular Phenotyping Core facility using the VisualSonics 770 Echocardiography System. Avertin (0.005 ml/g) was used for sedation based on previously established methods of infarct quantification (Kanno et al., 2002). 2D and M-mode images were obtained in the long and short axis views. Ejection fraction (EF) and LV dimensions were calculated using edge detection software and standard techniques. Measurements were performed on 3 independently acquired images per animal, by investigators who were blinded to experimental group. Each experimental group included at least 5 animals.
For invasive hemodynamic measurements, mice were anesthetized with 1% isoflurane and a 1.2 French pressure-volume catheter was position into the LV (ADVantage PV system, Scisense). Pressure and volume data were recorded using a Scisense 404-16 Bit Four Channel Recorder (Scisense) and analyzed with LabScribe2 Software (Scisense). Each experimental group included at least 5 animals.
For dobutamine cardiac catheterization studies, mice were anesthetized with a mixture of xylazine (10 mg/kg) and ketamine (100 mg/kg) administered i.p. Mice were then ventilated using a Harvard MicroVent. Open chest cardiac catheterization was performed by opening the chest wall just above the diaphragm and visualizing the apex of the heart. A small hole was formed by puncture with a 28 gauge needle and a 1.4 Fr Scisense catheter was inserted and advanced into the left ventricle of the mouse. The catheter was secured in the ventricle by a loop of suture on the chest wall. Hemodynamic measurements were then recorded. After acquisition of stable baseline data, dobutamine was serially infused at rates of 2, 4, 8, 16, 32, and 64 ng . g BW−1 . min −1 Data was analyzed using the Lab Scribe software for comparison of contractile performance between groups at each infusion rate.
Surface electrocardiography was performed using a MouseMonitor system (INDUS, Instruments). Recordings were obtained for 1 hour under inhaled anesthesia (1% isoflurane). Temperature was monitored and maintained using a heating pad. For data analysis, .csv files were imported into MATLAB with channels for time and voltage outputs for leads I, II, and III. The remainder of the analysis was performed using data from lead II. The signal was first filtered using Savitzky-Golay filtering based on previously established methods for ECG signal de-noising (Samann and Schanze, 2019). This filtering was performed twice so that both high frequency and low frequency noise could be filtered. The wavelet transform was then computed using the maximal overlap discrete wavelet transform from the MATLAB wavelet toolbox. The peaks of the QRS complexes were identified from the square of this transformed signal. For each QRS complex, the locations of the peak and minima on either side were identified. The largest peak above a specified threshold and within a specified range before each QRS complex was identified as the p-wave. A similar procedure was applied to capture the peak of the t-wave in the range following a QRS complex. Local minima were captured for signals determined by inspection to have inverted t-waves. After the P, Q, R, S, and T waves were identified, the RR, PR, and QT intervals as well as the QRS width were computed. Each experimental group included at least 4 animals.
Transaortic constriction was performed on mice 8–12 weeks of age. Mice were anesthetized with xylazine (10 mg/kg) and ketamine (100 mg/kg), and either the sham or TAC procedure was performed by a surgeon blinded to genotype. Briefly, the chest was opened and, following blunt dissection, the thoracic aorta was identified. A 7-0 silk suture was placed around the transverse aorta and tied around a 26 gauge blunt needle that was subsequently removed. The chest was closed with a purse-string suture. At the end of the procedure, the incision was closed in two layers with an interrupted suture pattern. The mouse was kept on a heating pad until responsive to stimuli. Echocardiography was performed 4 weeks after transaortic constriction and mice were subsequently euthanized for pathologic and gene expression analysis.
Micro-PET/CT
The CCR2 (ECL1i: LGTFLKC) and CCR5 (DAPTA: D-A1STTTNYT) targeting peptides were synthesized from D-form amino acids by CPC Scientific (Sunnyvale, CA). Maleimido-mono-amide-DOTA was purchased from Macrocyclics, Inc (Dallas, TX). DOTA-ECL1i and DOTA-DAPTA was synthesized as reported (Heo et al., 2019; Liu et al., 2017; Luehmann et al., 2014). Probes were radiolabeling with 64Copper (64Cu). The radiolabeled compound was analyzed using radio-HPLC to ensure more than 95% radiochemical purity prior to animal studies. Mice were anesthetized with isoflurane and injected with 3.7 MBq of 64Cu-DOTA-ECL1i or 3.7 MBq 64Cu-DOTA-DAPTA in 100 μL of saline via the tail vein. Small animal PET scans (0 to 60 min dynamic scan) were performed on either microPET Focus 220 (Siemens, Malvern, PA) or Inveon PET/CT system (Siemens, Malvern, PA). The microPET images were corrected for attenuation, scatter, normalization, and camera dead time and co-registered with microCT images. All of the PET scanners were cross-calibrated periodically. The microPET images were reconstructed with the maximum a posteriori (MAP) algorithm and analyzed by Inveon Research Workplace. The uptake was calculated as the percent injected dose per gram (%ID/gram) of tissue in three-dimensional regions of interest (ROIs) without the correction for partial volume effect.
Histology, Immunostaining, Picrosirius Red, and Wheat Germ Agglutinin Staining.
For histological analyses, tissues were fixed in 2% PFA overnight at 4°C, dehydrated in 70% ethyl alcohol, and embedded in paraffin. 4-μm sections were cut and stained with Picrosirius red using standard techniques. Picrosirius red staining was quantified using Image J software. To quantify cardiomyocyte cross-sectional area paraffin sections were stained with rhodamine conjugated WGA (Vector labs), visualized on a Zeiss confocal microscope, and measurements preformed using Zeiss Axiovision software. For all immunostaining assays, tissues were fixed in 2% PFA overnight at 4°C, embedded in OCT, infiltrated with 30% sucrose, frozen, and 12-μm cryosections cut. Primary antibodies used were: CD68 clone FA-11 1:400 (Biolegend Cat# 137001), GFP (Abcam Cat# ab13970), RFP (Abcam Cat# ab62341), cardiac actin clone AC1-20.4.2 (Sigma Cat# A9357), α-actinin clone BM-75.2 (Sigma Cat# A5044), Ki67 (Abcam Cat# ab15580), CD34 clone MEC14.7 (Abcam Cat# ab8158), Cx43 (Cell Signaling Cat# 3512), Paxillin clone Y113 (Abcam Cat# ab32084), Pan-Cadherin (Cell Signaling Cat# 4068), Claudin I (Cell Signaling cat# 13255), Desmoplakin I/II clone DP2.15 (Abcam Cat# ab16434), IGF1 (R&D systems Cat# AF791), CYR61 (R&D systems Cat# AF4055), Ly6G clone 1A8 (BD Cat# 551459), HCN4 (Fisher Cat# PA5-111878), FAK (Abcam, Cat# ab76496), and TRPV4 clone 1B2.6 (Millipore Cat# MABS466).
Immunofluorescence was visualized using appropriate secondary antibodies on a Zeiss confocal microscopy system. For all experiments, at least 4 sections from 4 independent samples were analyzed in blinded fashion. Antibody specificity was validated using appropriate no primary and isotype controls. Co-localization was assessed by scoring images for the percent of macrophages expressing each junction marker at sites of cardiomyocyte interaction. Scoring was performed by 2 independent evaluators blinded to sample designation.
Cardiomyocyte Morphometry
Mouse hearts were harvested, cut into 1–2 mm tissue blocks, and fixed with 2% PFA at overnight at 4 degrees C. Tissue blocks were washed in PBS and then digested in Collagenase B (1.8 mg/mL) and Collagenase D (2.4 mg/mL) at 37 degrees C overnight (Han et al., 2020). Isolated cells were washed in PBS, fixed in 2% PFA, and imaged using an invested bright field microscope. Cardiomyocyte dimensions were measured using Image J.
Coronary Vascular Perfusion and Casting
To visualize the coronary arterial tress, Microfil perfusion reagent (yellow, Flow tech) was perfused retrograde into the ascending aorta per the manufacturer’s specifications. The hearts were then fixed and imaged on a Zeiss Discovery V.12 stereomicroscope. Branching points were quantified as a surrogate measure of vascular complexity. To visualize the coronary microvasulature, anesthetized mice were injected IV with biotinylated tomato lectin (Vector labs) 5 minutes prior to tissue harvest. The hearts were then fixed, embedded in OCT, and cryosections stained with streptavidin-FITC (Vector labs).
Electron Microscopy
200 micron thick vibratome sections were taken from the heart tissue obtained from Cx3cr1GFP/+Ccr2RFP/+ mice (VT1200S, Leica Biosystems, Vienna, Austria). Samples were immersion fixed in 2% paraformaldehyde overnight and immunolabeled with GFP (Abcam cat# ab13970) or RFP (Abcam cat# ab62341) antibodies. Antibody staining was visualized with DAB and tissues were then submerged in a mixture of 2.5% glutaraldehyde and 2% paraformaldehyde in 0.15 M cacodylate buffer (pH 7.4) containing 2 mM calcium chloride and incubated overnight at 4°C. Sample were rinsed in 0.15 M cacodylate buffer 3 times for 10 minutes each, and subjected to a secondary fixation step for one hour in 1% osmium tetroxide containing 1.5% potassium ferrocyanide in cacodylate buffer on ice. Following fixation, samples were then washed in ultrapure water 3 times for 10 minutes each and en bloc stained for 1 hour with 2% aqueous uranyl acetate. After staining was complete, samples were briefly washed in ultrapure water, dehydrated in a graded acetone series (50%, 70%, 90%, 100% x2) for 10 minutes in each step, infiltrated with microwave assistance (Pelco BioWave Pro, Redding, CA) into LX112 resin, embedded in silicone molds, and cured in an oven at 60°C for 48 hours. Once the resin was cured, each block was trimmed and faced using a diamond trim tool. 70nm longitudinal sections were obtained and imaged on a FE-SEM (Zeiss Crossbeam 540, Oberkochen, Germany) using the aSTEM detector. The SEM was operated at 28 KeV with a probe current of 0.9 nA, and the STEM detector was operated with the annular rings inverted for additional sample contrast. Large sample areas were imaged at a resolution of 4096 × 3072 pixels with a pixel size of 5.582 nm. At least 40 macrophages from 4 independent samples were included in the analysis.
X-Ray Microscopy
Whole hearts were extracted from mice, immediately bisected and immersion fixed in 2.5% glutaraldehyde and 2% paraformaldehyde in 0.15 M cacodylate buffer (pH 7.4) containing 2 mM calcium chloride overnight at 4°C. Post-fixation, each heart was stained by immersion into Lugol’s Iodine solution for a period of 5 days. Post-staining, hearts were mounted in a support matrix of 2% low-melt agarose within an Eppendorf tube to mitigate any movement during scanning. The Eppendorf tube was subsequently fixed onto a stub which was then mounted in the rotation holder within the X-Ray Microscope (Versa 520 XRM, Zeiss Microscopy, Pleasanton, CA). The samples were then imaged in the XRM at 80kV with an exposure time of 4 seconds, a pixel pitch of 5.41 μm with 2301 individual images acquired through a 360 degree rotation. Tomographic reconstruction was performed using the Zeiss 3DXMViewer software and rendered using ORS Dragonfly (ORS, Montreal, Quebec. Canada).
Papillary Muscle Preparations and Live Two-Photon Microscopy
Papillary muscles were dissected from the hearts of Cx3cr1GFP/+Ccr2RFP/+ or Cx3cr1-ertCre; Rosa26-GCaMP6 Rosa26-tdTomato mice as previously described (Uhl et al., 2015). Briefly, mouse hearts were harvested and perfused in oxygenated Krebs-Henseleit (KH) buffer preparation solution on ice. Left ventricles were exposed and papillary muscles were dissected from the left ventricular free walls. Dissected papillary muscles were transported in KH buffer on ice, covered from light to the Washington University Center for Cellular Imaging.
For static imaging, papillary muscles were placed into a 60mm glass bottom dish (Cellvis D60-30-1.5-N) and held in place with a slice hold-down (Warner Instruments 64-1419). Mounted papillary muscles were continuously bathed in pre-warmed (32°C) KH buffer with 95% oxygen/5% carbon dioxide. Pre-warmed KH buffer was gravity fed into one edge of the imaging dish while being continuously vacuum suctioned from the other end of the dish. Time-lapse imaging was performed on a Zeiss 880 inverted two-photon microscope. GFP+ macrophages were imaged with a 920nm laser (12% laser power) and captured with an LD Plan-neofluar 20x/0.4 objective (2–3x zoom, 4-line average) and Airyscan detectors. Z-stacks of ~80–100μM thickness were collected every 30 seconds for the duration of the time-lapse (90 minutes). Post-image processing was performed with ZEN3.0 (Blue version).
To apply axial tension, we generated a custom apparatus using both commercially available supplies (Mitutoyo 0.01mm 0–25mm micrometer, Thor Lab [XRN25/M, VC1/M, XRN-XZ/M, DT12A] supplies) and custom-made inserts. After transport to the imaging core in ice cold KH buffer, papillary muscles were carefully snagged without tension on microtweezers. Tension was then applied and papillary muscles imaged. Time-lapse imaging was performed on a Zeiss 880 inverted two-photon microscope. GFP+ macrophages (GCaMP6) were imaged with a 488nm laser (2.7% laser power) and TdT+ macrophages were imaged with a 561nm laser (3% power) and captured with an LD Plan-neofluar 20x/0.4 objective (2–3x zoom, 4-line average) and Airyscan detectors. Z-stacks of ~80–100μM thickness were collected every 30 seconds for the duration of the time-lapse (30 minutes). Baseline images were obtained over 5 minutes, and then vehicle (DMSO) was placed directly in the imaging dish and in the KH buffer and imaged for 10 minutes. After baseline and vehicle imaging, the feedline was flushed and TrpV4 inhibitor (GSK219 300nM) was directly added to the dish and to a second aliquot of pre-warmed KH buffer and papillary muscles imaged for an additional 10 minutes. Post-acquisition processing was performed in ZEN 3.0 SR (black edition). At least 30 macrophages from 4 independent samples were included in the analysis. Regions of interest were identified based on tdTomato staining and relative intensities of GFP (GCaMP6) were normalized against background and recorded over the duration of the experiment. The GFP intensity of each macrophage was averaged over the duration of baseline/vehicle imaging and compared to the averaged GFP intensity of axially stretched macrophages after administration of TrpV4 inhibitor.
Flow cytometry
Single cell suspensions were generated from saline perfused hearts by finely mincing and digesting them in DMEM with Collagenase 1 (450 U/ml), Hyaluronidase (60 U/ml) and DNase I (60 U/ml) for 1 hour at 37°C. All enzymes were purchased from Sigma. To deactivate the enzymes samples were washed with HBSS that was supplemented with 2% FBS and 0.2% BSA and filtered through 40 μM cell strainers. Red blood cell lysis was performed with ACK lysis buffer (Thermo Fisher Scientific). Samples were washed with HBSS and resuspended in 100 μL of FACS buffer (DPBS with 2% FBS and 2 mM EDTA). Cells were stained with monoclonal antibodies at 4°C for 30 minutes in the dark. All the antibodies were obtained from Biolegend. A complete list of antibodies is provided below. Samples were washed twice, and final resuspension was made in 300 μL FACS buffer. DAPI or LIVE/DEADTM Aqua dyes were used for exclusion of dead cells. Immune cells were first gated as CD45+. Neutrophils were gated as Ly6GhiCD64−. Monocytes were gated as CD64intLy6ChiCCR2+MHCIIlo. Macrophages were gated as Ly6G−CD64hiLy6Clo cells. Flow cytometric analysis and sorting were performed on BD LSRII, BD FACS ARIAIII, BD FACS Melody platforms.
CD45-PerCP/Cy5.5, clone 30-F11 (Biolegend cat# 103131)
CD64-APC, PE, PE/Cy7 clone X54-5/7.1 (Biolegend cat# 139305, 139303, 139313)
CCR2-BV421, clone: SA203G11 (Biolegend cat# 150605)
MHCII-APC/Cy7, clone M5/114.15.2 (Biolegend cat# 107627)
Ly6G-PE/Cy7, clone 1A8 (Biolegend cat# 127617)
Ly6C-APC and FITC, clone HK1.4 (Biolegend cat# 128015, 128005)
CD31-APC, clone 390 (Biolgend cat# 102409)
Anti-Feeder-PE, clone mEFSK4 (Miltenyi cat# 130-120-166)
RT-PCR
RNA was extracted from sorted cells or myocardial tissue using the RNeasy RNA mini kit and Tissue Lyser II (Qiagen). RNA concentration was measured using a nanodrop spectrophotometer (ThermoFisher Scientific). cDNA synthesis was performed using the High Capacity RNA to cDNA synthesis kit (Applied Biosystems). For sorted macrophages, cDNA was synthesized using the iScript™ Reverse Transcription Supermix (Bio-Rad) and pre-amplified using the Sso Advanced PreAmp Supermix kit (Bio-Rad). Quantitative real time PCR reactions were prepared with sequence-specific primers (IDT) with PowerUP™ Syber Green Master mix (ThermoFisher Scientific) in a 20 μL volume. Real time PCR was performed using QuantStudio 3 (ThermoFisher Scientific). mRNA expression was normalized to 36B4. All RT-PCR assays were performed using validated primer sets (IDT) with appropriate quality controls including melt curves and negative controls.
RNA sequencing
Total RNA was harvested from by disrupting samples in Trizol (Thermo) using the Qiagen TissueLyser homogenizer with stainless steel beads. Following phenol chloroform extraction, RNA was purified using the Ambion Purelink miniprep kit. RNA integrity was quantified on an Agilent Bioanalyzer and samples with RIN>8 utilized for RNA sequencing. Ribosomal RNAs were depleted with Ribo-Zero, cDNA libraries generated, and samples sequenced (1X50bp reads) on an Illumina HiSeq 3000 instrument. Sequence alignment, normalization, and differential expression analysis was carried out in the McDonnel Genome Institute at Washington University using Limma-Voom software. PCA and Hierarchical cluster analysis was performed in Partek Genomics. GO Pathway analysis was performed using DAVID. Transcripts with >10 reads in 50% of samples demonstrating a fold change >1.5 fold at an FDR<0.05 were included in the pathway analysis. Differential gene expression was performed using START (Nelson et al., 2017) and Partek Genomics analysis packages.
Microarray
To isolated RNA, macrophages were directly sorted into QLT buffer containing 2-mercaptoethanol and RNA isolated using the RNeay micro kit (Qiagen) per manufacturer’s instructions. Gene expression profiling was performed using microarray analysis in collaboration with the McDonnell Genome Institute at Washington University. RNA was amplified using the WTA (Sigma) system and hybridized to Agilent 8X60 gene chips. Data analysis was performed using Partek genome suite software.
MMP assay
Protein was extracted from mouse hearts using EDTA-free RIPA buffer and the Tissue Lyser II (Qiagen). Equal concentrations of protein were loaded onto zymogram gels (Novex) and run at 125 V constant for 90 minutes on the XCell SureLock Mini-Cell. The gels were then developed and incubated in 1X Zymogram Renaturing Buffer and 1X Zymogram Developing Buffer (Novex) per the manufacturer’s instructions. Gels were then stained with SimplyBlue Safestain (Invitrogen), imaged, and quantified using Image J software.
Cell Culture
To generate bone-marrow derived macrophages (BMDMs), isolated bone marrow cells from mouse femurs and tibiae were cultured for 7 days in DMEM supplemented with 10% FBS, 5% M-CSF, 5% horse serum, 1% streptomycin and 1% sodium pyruvate. On day 8 the media was replaced with M-CSF free media. For cell stretch experiments, BMDMs were plated on fibronectin and collagen coated silicone membranes and cultured overnight to facilitate adherence. Seeded membranes were then subjected to cyclic unaxial stretch (1 Hz, 10% deformation) for 24–48 hours. GSK2193874 was used at 1 μM. LPS and polyIC were used at 100 ng/ml and 20 μg/ml, respectively.
For cardiomyocyte-macrophage co-culture experiments, 6-well plates were coated with fibronectin and incubated at 37°C overnight. HL-1 cells were then cultured in Claycomb media (Sigma) supplemented with 2% L-glutamine for 24 hours. BMDMs were then co-cultured at a 1:6 ratio of BMDMs:HL-1 cells, at which point the co-culture was either treated with Itgb1/CD29 clone HM β1-1 (10 μM) or CD18 clone M18/2 (20 μM). Four hours after co-culture the cells were imaged at 20x and the number of BMDM-HL-1 interactions per 20x field were quantified. Cells were defined as interacting if a measurable BMDM projection made contact with an HL-1 cardiomyocyte.
Ratiometric Calcium assays
CCR2− and CCR2+ macrophages isolated from the heart by flow cytometry were resuspended in culture medium containing 10 % FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, plated on 5 mm coverslips coated with poly-L-lysine (10 μg/mL) and cultured under a humidified atmosphere of 5 % CO2 / 95% air at 37°C for 2 h. Then, cultured cardiac macrophages were loaded with 4 μM Fura-2 AM (Invitrogen, Carlsbad, CA) in culture medium at 37°C for 60 min as previous described (Luo et al., 2018). Cells were washed three times and incubated in HBSS at room temperature for 30 min before use. Fluorescence at 340 and 380 nm excitation wavelengths was recorded on an inverted Nikon Ti-E microscope equipped with 340 and 380 nm excitation filter wheels using NIS Elements imaging software (Nikon). Fura-2 ratios (F340/F380) were used to reflect changes in intracellular Ca2+ upon stimulation. The threshold of cellular activation was defined as 20% above the baseline. GSK1016790A and GSK2193874 were used at 300 nM. Ionomycin (1 μM) was used as a positive control.
Mitochondrial Isolation and Respiration Assays
One day prior to mitochondrial isolation, XFe96 Sensor cartridges were hydrated by adding 180uL ultrapure H2O to each well and incubating the plate in a non CO2 incubator overnight. Mitochondria were isolated from mouse heart tissue as previously described (Frezza et al., 2007). Following hemodynamic study, mice were sacrificed through cervical dislocation. Using a surgical scalpel and scissors, hearts were removed rapidly and immersed in a small beaker containing 5 mL of ice-cold PBS supplemented with 10mM EDTA. Heart tissue was minced into small pieces using scissors, washed thrice with ice-cold PBS supplemented with 10mM EDTA over a 40um filter, spun down at 200g for 5 min, and the supernatant discarded. Heart tissue was then resuspended in 1mL IBm1 (Prepare 100 ml of IBm1 by mixing 6.7 ml of 1M sucrose, 5ml of 1M Tris/HCl, 5 ml of 1M KCl, 1 ml of 1M EDTA, and 2 ml of 10% BSA. Adjust pH to 7.4. Bring the volume to 100 ml with distilled water) and hand homogenized using a pre-cooled Teflon pestle ten times. The homogenate was then transferred to a 1.7mL microcentrifuge tube and centrifuged at 700g for 10 min at 4oC. Supernatant was then transferred to a new 1.7mL microcentrifuge tube and centrifuged at 8,000g for 10 min at 4oC. Remaining supernatant was removed, and the pellet was resuspended in 1mL IBm2 (Prepare 100 ml of IBm2 by mixing 25 ml of 1 M sucrose, 3 ml of 0.1 M EGTA/Tris, and 1 ml of 1 MTris/HCl. Adjust pH to 7.4. Bring the volume to 100 ml with distilled water). The suspension was centrifuged at 8,000g for 10 min at 4oC and the supernatant discarded. The residual supernatant was used to resuspend the pellet, and mitochondrial concentration was measured using BCA Protein Assay.
Mitochondrial Respiration was measured as follows. Mitochondria were resuspended in MAS1 buffer (Prepare MAS1 by dissolving 220 mM of d-Mannitol, 70 mM of sucrose, 10 mM of KH2PO4, 5 mM of MgCl2, 2 mM of HEPES, 1 mM of EGTA, and 0.2% (w/v) of fatty acid-free BSA in ultrapure H2O and adjust the pH to 7.2 with KOH at 37 °C) containing either 10mM glutamate & 5mM malate (Complex I) or 5mM succinate & 2uM rotenone (Complex II). A volume of 50uL Mitochondria were plated on Seahorse XFe96 microculture plates at a concentration of 4ug mitochondria per well. Plates were centrifuged using a swinging bucket-microplate adaptor at 2000g for 20 min at 4oC. Seahorse sensor cartridges were loaded with 10X aliquots (ADP, oligomycin, FCCP, antimycin & rotenone) and calibrated during this step. Mitochondria were inspected by microscopy. Following centrifugation, 130uL MAS1 with substrates (glutamate/malate or succinate/rotenone) were added to each well for a total volume of 180uL. Plate was warmed at 37oC in a non-CO2 incubator for 5–10 min, after which it was placed into the Seahorse Bioanalyzer.
Blood cell counts and Serum Chemistry.
Blood and serum was collected by LV puncture prior to mouse harvest. Blood counts, chemistries and electrolytes were measured in the Washington University School of Medicine Division of Comparative Medicine Research Animal Diagnostic Laboratory using a clinical hematology analyzer and AMS Liasys 330 Clinical Chemistry System.
STATISTICAL ANALYSIS
Data were analyzed by using software (Prism, version 6.0–7.0; GraphPad, La Jolla, Calif). Differences between groups were compared by using Mann-Whitney U test. Multiple means were compared by using 1-way ANOVA (analysis of variance) with the post-hoc Tukey test. p<0.05 (two-sided) was indicative of a statistically significant difference. Bonferroni correction was performed when multiple hypotheses were tested. Data are presented as dot plots or box whisker plots generated in PRISM. The sample size used to calculate statistical significance is stated in the appropriate figure legend. Each data point denotes individual animals. Error bars represent standard deviation.
Supplementary Material
Data S1: Normalized RNA sequencing data (counts per million). Related to Figure 1
Data S2: Microarray expression data. Related to Figure 6
Supplemental Video 1: Zoom in time lapse 2-photon imaging of a CCR2- macrophage in papillary muscle preparations. Related to Figure 5
Supplemental Video 2: Zoom out time lapse 2-photon imaging of a CCR2- macrophage in papillary muscle preparations. Related to Figure 5
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD68 | Biolegend | Cat# 137001 |
| GFP | Abcam | Cat# ab13970 |
| RFP | Abcam | Cat# ab62341 |
| Cardiac actin | Sigma | Cat# A9357 |
| α-actinin | Sigma | Cat# A5044 |
| Ki67 | Abcam | Cat# ab15580 |
| Paxillin | Abcam | Cat# ab32084 |
| Cx43 | Cell Signaling | Cat# 3512 |
| Pan-Cadherin | Cell Signaling | Cat# 4068 |
| Claudin I | Cell Signaling | Cat# 13255 |
| Desmoplakin I/II | Abcam | Cat# ab16434 |
| Itgb1/CD29 | BD | Cat# 562219 |
| Itgb2/CD18 | BD | Cat# 557437 |
| TRPV4 | Millipore | Cat# MABS466 |
| IGF1 | R&D systems | Cat# AF791 |
| CYR61 | R&D systems | Cat# AF4055 |
| CD34 | Abcam | Cat# ab8158 |
| MHCII-APC/Cy7 | Biolegend | Cat# 107627 |
| CCR2-BV421 | Biolegend | Cat# 150605 |
| CD45-PerCP/Cy5.5 | Biolegend | Cat# 103131 |
| CD64-PE/Cy7, -PE, -APC | Biolegend | Cat# 139305, 139303, 139313 |
| Ly6G-PE/Cy7 | Biolegend | Cat# 127617 |
| Ly6C-APC, -FITC | Biolegend | Cat# 128015, 128005 |
| CD31-APC | Biolegend | Cat# 102409 |
| MEFSK4-PE | Miltenyi | Cat# 130-120-166 |
| HCN4 | Fisher | Cat# PA5-111878 |
| Ly6G | BD | Cat# 551459 |
| FAK | Abcam | Cat# ab76496 |
| Bacterial and Virus Strains | ||
| none | ||
| Biological Samples | ||
| none | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| RGD peptides | Selleckchem | Cat# S8008 |
| GSK1016790A | Selleckchem | Cat# S8107 |
| GSK2193874 | Tocris | Cat# 5106 |
| Ionomycin | Sigma | Cat# I9657 |
| Microfil | Flow Tech Inc. | Cat# MV-122 |
| Biotinylated Tomato Lectin | Vector labs | Cat# B-1175 |
| Wheat Germ Agglutinin | Vector labs | Cat# RL-1022 |
| Critical Commercial Assays | ||
| none | ||
| Deposited Data | ||
| Raw RNA sequencing data | deposited in GEO at time of publication | |
| Raw microarray data | deposited in GEO at time of publication | |
| Experimental Models: Cell Lines | ||
| HL1 cells | ||
| Experimental Models: Organisms/Strains | ||
| Mouse: B6;129-Tnnt2tm2Mmto | CARD | CARD: 1585 |
| Mouse: B6;129-Siglec1<tm1(HBEGF)Mtka> | RIKEN BRC | RBRC04395 |
| Mouse: B6.129-Tg(Flt3-cre)#Ccb/Ieg | EMMA | EMMA: 17790 |
| Mouse: B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J | Jackson Laboratory | JAX: 007909 |
| Mouse: B6(C)-Ccr2tm1.1Cln/J | Jackson Laboratory | JAX: 027619 |
| Mouse: Tg(Trpv4-EGFP)MT43Gsat/Mmucd | Mutant Mouse Resource and Research Centers |
MMRRC: 032771-UCD |
| Mouse: B6.129(Cg)-Cx3cr1tm1Litt Ccr2tm2.1Ifc/JernJ | Jackson Laboratory | JAX: 032127 |
| Mouse: B6.129P2(SJL)-Myd88tm1.1Defr/J | Jackson Laboratory | JAX: 009088 |
| Mouse: C57BL/6J-Ticam1Lps2/J | Jackson Laboratory | JAX: 005037 |
| Mouse: B6J.Cg-Gt(ROSA)26Sortm95.1(CAG-GCaMP6f)Hze/MwarJ | Jackson Laboratory | JAX: 028865 |
| Oligonucleotides | ||
| IGF1 | IDT | Mm.PT.58.5811533 |
| HB-EGF | IDT | Mm.PT.58.10448014 |
| CYR61 | IDT | Mm.PT.58.10039559 |
| IL-1β | IDT | Mm.PT.58.41616450 |
| TNF | IDT | Mm.PT.58.12575861 |
| IL6 | IDT | Mm.PT.58.10005566 |
| IP10 | IDT | Mm.PT.58.43575827 |
| MX2 | IDT | Mm.PT.58.29837402 |
| NPPA | IDT | Mm.PT.58.12973594 |
| NPPB | IDT | Mm.PT.58.8584045 |
| MYH7 | IDT | Mm.PT.58.17465550 |
| Recombinant DNA | ||
| none | ||
| Software and Algorithms | ||
| Partek Genomics | ||
| START | Nelson et al., 2017 |
https://doi.org/10.1093/bioinformatics/btw624
|
| Graph Pad PRISM | ||
| ImageJ | Schneider et al., 2012 |
https://imagej.nih.gov/ij/
|
| ZEN 3.0 SR (black edition) | ||
| Zeiss 3DXMViewer | ||
| ORS Dragonfly | ||
Highlights.
The failing heart contains heterogeneous subsets of resident and recruited macrophages
Resident cardiac macrophages promote adaptation and survival of the failing heart
Resident cardiac macrophages interact with cardiomyocytes via focal adhesion complexes
Resident cardiac macrophages sense mechanical stretch through TRPV4 channels
Acknowledgments
KL is supported by funding provided from the NHLBI (R01 HL138466, R01 HL139714, R01 HL151078), Leducq Foundation Network (#20CVD02), Burroughs Welcome Fund (1014782), Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital (CH-II-2015-462, CH-II-2017-628, PM-LI-2019-829), Foundation of Barnes-Jewish Hospital (8038-88). D.K. is supported by NIH P01AI116501 and R01 HL094601, Veterans Administration Merit Review grant 1I01BX002730 and the Foundation for Barnes-Jewish Hospital. H.H. was supported by grants from the NIH, R01DK103901, R01AR077183, and R01AA027065, the Department of Anesthesiology at Washington University School of Medicine. Y.L. is supported by NIH R35HL145212 and R01HL131908. We acknowledge the McDonnel Genome Institute, DDRCC histology core (P30 DK52574), and WUCCI (NIH OD021694,CDI-CORE-2015-505, CDI-CORE-2019-813, Foundation for Barnes-Jewish Hospital) for their valuable assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interest Statement
The authors have no financial or competing interests to disclose.
REFERENCES:
- Aurora AB, Porrello ER, Tan W, Mahmoud AI, Hill JA, Bassel-Duby R, Sadek HA, and Olson EN (2014). Macrophages are required for neonatal heart regeneration. J Clin Invest 124, 1382–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain CC, Scott CL, Uronen-Hansson H, Gudjonsson S, Jansson O, Grip O, Guilliams M, Malissen B, Agace WW, and Mowat AM (2013). Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol 6, 498–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajpai G, Bredemeyer A, Li W, Zaitsev K, Koenig AL, Lokshina I, Mohan J, Ivey B, Hsiao HM, Weinheimer C, et al. (2019). Tissue Resident CCR2− and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury. Circ Res 124, 263–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajpai G, Schneider C, Wong N, Bredemeyer A, Hulsmans M, Nahrendorf M, Epelman S, Kreisel D, Liu Y, Itoh A, et al. (2018). The human heart contains distinct macrophage subsets with divergent origins and functions. Nat Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer SW, Schroeder AV, Smith-Berdan S, and Forsberg EC (2011). All hematopoietic cells develop from hematopoietic stem cells through Flk2/Flt3-positive progenitor cells. Cell Stem Cell 9, 64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burchfield JS, Xie M, and Hill JA (2013). Pathological ventricular remodeling: mechanisms: part 1 of 2. Circulation 128, 388–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow A, Lucas D, Hidalgo A, Mendez-Ferrer S, Hashimoto D, Scheiermann C, Battista M, Leboeuf M, Prophete C, van Rooijen N, et al. (2011). Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J Exp Med 208, 261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clippinger SR, Cloonan PE, Greenberg L, Ernst M, Stump WT, and Greenberg MJ (2019). Disrupted mechanobiology links the molecular and cellular phenotypes in familial dilated cardiomyopathy. Proc Natl Acad Sci U S A 116, 17831–17840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn JN, Ferrari R, and Sharpe N (2000). Cardiac remodeling--concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol 35, 569–582. [DOI] [PubMed] [Google Scholar]
- Davies LC, Jenkins SJ, Allen JE, and Taylor PR (2013). Tissue-resident macrophages. Nat Immunol 14, 986–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick SA, Macklin JA, Nejat S, Momen A, Clemente-Casares X, Althagafi MG, Chen J, Kantores C, Hosseinzadeh S, Aronoff L, et al. (2019). Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat Immunol 20, 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du CK, Morimoto S, Nishii K, Minakami R, Ohta M, Tadano N, Lu QW, Wang YY, Zhan DY, Mochizuki M, et al. (2007). Knock-in mouse model of dilated cardiomyopathy caused by troponin mutation. Circ Res 101, 185–194. [DOI] [PubMed] [Google Scholar]
- Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde LE, Schoen FJ, Kelly RA, Werb Z, Libby P, et al. (2000). Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest 106, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta B, Arya RK, Goswami R, Alharbi MO, Sharma S, and Rahaman SO (2020). Role of macrophage TRPV4 in inflammation. Lab Invest 100, 178–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, Brija T, Gautier EL, Ivanov S, Satpathy AT, et al. (2014a). Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 40, 91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epelman S, Lavine KJ, and Randolph GJ (2014b). Origin and functions of tissue macrophages. Immunity 41, 21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantin A, Vieira JM, Gestri G, Denti L, Schwarz Q, Prykhozhij S, Peri F, Wilson SW, and Ruhrberg C (2010). Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood 116, 829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, and Scorrano L (2007). Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc 2, 287–295. [DOI] [PubMed] [Google Scholar]
- Geiger B, Spatz JP, and Bershadsky AD (2009). Environmental sensing through focal adhesions. Nat Rev Mol Cell Biol 10, 21–33. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godwin JW, Debuque R, Salimova E, and Rosenthal NA (2017). Heart regeneration in the salamander relies on macrophage-mediated control of fibroblast activation and the extracellular landscape. NPJ Regen Med 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godwin JW, Pinto AR, and Rosenthal NA (2013). Macrophages are required for adult salamander limb regeneration. Proc Natl Acad Sci U S A 110, 9415–9420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami R, Arya RK, Biswas D, Zhu X, and Rahaman SO (2019). Transient Receptor Potential Vanilloid 4 Is Required for Foreign Body Response and Giant Cell Formation. Am J Pathol 189, 1505–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, Deswarte K, Malissen B, Hammad H, and Lambrecht BN (2013). Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med 210, 1977–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamanaka K, Jian MY, Townsley MI, King JA, Liedtke W, Weber DS, Eyal FG, Clapp MM, and Parker JC (2010). TRPV4 channels augment macrophage activation and ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol 299, L353–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Choudhury S, Mich-Basso JD, Ammanamanchi N, Ganapathy B, Suresh S, Khaladkar M, Singh J, Maehr R, Zuppo DA, et al. (2020). Lamin B2 Levels Regulate Polyploidization of Cardiomyocyte Nuclei and Myocardial Regeneration. Dev Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, Becker CD, See P, Price J, Lucas D, et al. (2013). Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 38, 792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo GS, Kopecky B, Sultan D, Ou M, Feng G, Bajpai G, Zhang X, Luehmann H, Detering L, Su Y, et al. (2019). Molecular Imaging Visualizes Recruitment of Inflammatory Monocytes and Macrophages to the Injured Heart. Circ Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hettinger J, Richards DM, Hansson J, Barra MM, Joschko AC, Krijgsveld J, and Feuerer M (2013). Origin of monocytes and macrophages in a committed progenitor. Nat Immunol 14, 821–830. [DOI] [PubMed] [Google Scholar]
- Heymans S, Luttun A, Nuyens D, Theilmeier G, Creemers E, Moons L, Dyspersin GD, Cleutjens JP, Shipley M, Angellilo A, et al. (1999). Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med 5, 1135–1142. [DOI] [PubMed] [Google Scholar]
- Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, et al. (2003). Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature 424, 743–748. [DOI] [PubMed] [Google Scholar]
- Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, Beaudin AE, Lum J, Low I, Forsberg EC, et al. (2015). C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 42, 665–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffel G, Wang Y, Greter M, See P, Teo P, Malleret B, Leboeuf M, Low D, Oller G, Almeida F, et al. (2012). Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J Exp Med 209, 1167–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou B, Reizis B, and DeFranco AL (2008). Toll-like receptors activate innate and adaptive immunity by using dendritic cell-intrinsic and -extrinsic mechanisms. Immunity 29, 272–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, Hucker WJ, Wulfers EM, Seemann G, Courties G, et al. (2017). Macrophages Facilitate Electrical Conduction in the Heart. Cell 169, 510–522 e520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulsmans M, Sager HB, Roh JD, Valero-Munoz M, Houstis NE, Iwamoto Y, Sun Y, Wilson RM, Wojtkiewicz G, Tricot B, et al. (2018). Cardiac macrophages promote diastolic dysfunction. J Exp Med 215, 423–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, and Littman DR (2000). Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol 20, 4106–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno S, Lerner DL, Schuessler RB, Betsuyaku T, Yamada KA, Saffitz JE, and Kovacs A (2002). Echocardiographic evaluation of ventricular remodeling in a mouse model of myocardial infarction. J Am Soc Echocardiogr 15, 601–609. [DOI] [PubMed] [Google Scholar]
- Lavine KJ, Epelman S, Uchida K, Weber KJ, Nichols CG, Schilling JD, Ornitz DM, Randolph GJ, and Mann DL (2014). Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc Natl Acad Sci U S A 111, 16029–16034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavine KJ, Kovacs A, Weinheimer C, and Mann DL (2013). Repetitive myocardial ischemia promotes coronary growth in the adult mammalian heart. J Am Heart Assoc 2, e000343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leid J, Carrelha J, Boukarabila H, Epelman S, Jacobsen SE, and Lavine KJ (2016). Primitive Embryonic Macrophages are Required for Coronary Development and Maturation. Circ Res 118, 1498–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Hsiao HM, Higashikubo R, Saunders BT, Bharat A, Goldstein DR, Krupnick AS, Gelman AE, Lavine KJ, and Kreisel D (2016). Heart-resident CCR2(+) macrophages promote neutrophil extravasation through TLR9/MyD88/CXCL5 signaling. JCI Insight 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Gunsten SP, Sultan DH, Luehmann HP, Zhao Y, Blackwell TS, Bollermann-Nowlis Z, Pan JH, Byers DE, Atkinson JJ, et al. (2017). PET-based Imaging of Chemokine Receptor 2 in Experimental and Disease-related Lung Inflammation. Radiology 283, 758–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luehmann HP, Pressly ED, Detering L, Wang C, Pierce R, Woodard PK, Gropler RJ, Hawker CJ, and Liu Y (2014). PET/CT imaging of chemokine receptor CCR5 in vascular injury model using targeted nanoparticle. J Nucl Med 55, 629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Qian A, Oetjen LK, Yu W, Yang P, Feng J, Xie Z, Liu S, Yin S, Dryn D, et al. (2018). TRPV4 Channel Signaling in Macrophages Promotes Gastrointestinal Motility via Direct Effects on Smooth Muscle Cells. Immunity 49, 107–119 e104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Garner AR, Shimaoka D, Chuong AS, Klapoetke NC, Li L, van der Bourg A, Niino Y, Egolf L, Monetti C, et al. (2015). Transgenic mice for intersectional targeting of neural sensors and effectors with high specificity and performance. Neuron 85, 942–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannaa M, Marko L, Balogh A, Vigolo E, N’Diaye G, Kassmann M, Michalick L, Weichelt U, Schmidt-Ott KM, Liedtke WB, et al. (2018). Transient Receptor Potential Vanilloid 4 Channel Deficiency Aggravates Tubular Damage after Acute Renal Ischaemia Reperfusion. Sci Rep 8, 4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally EM, and Mestroni L (2017). Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ Res 121, 731–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlo M, Pyxaras SA, Pinamonti B, Barbati G, Di Lenarda A, and Sinagra G (2011). Prevalence and prognostic significance of left ventricular reverse remodeling in dilated cardiomyopathy receiving tailored medical treatment. J Am Coll Cardiol 57, 1468–1476. [DOI] [PubMed] [Google Scholar]
- Miyake Y, Asano K, Kaise H, Uemura M, Nakayama M, and Tanaka M (2007). Critical role of macrophages in the marginal zone in the suppression of immune responses to apoptotic cell-associated antigens. J Clin Invest 117, 2268–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto S, Lu QW, Harada K, Takahashi-Yanaga F, Minakami R, Ohta M, Sasaguri T, and Ohtsuki I (2002). Ca(2+)-desensitizing effect of a deletion mutation Delta K210 in cardiac troponin T that causes familial dilated cardiomyopathy. Proc Natl Acad Sci U S A 99, 913–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Espin D, Canamero M, Maraver A, Gomez-Lopez G, Contreras J, Murillo-Cuesta S, Rodriguez-Baeza A, Varela-Nieto I, Ruberte J, Collado M, et al. (2013). Programmed cell senescence during mammalian embryonic development. Cell 155, 1104–1118. [DOI] [PubMed] [Google Scholar]
- Nakamura M, and Sadoshima J (2018). Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol 15, 387–407. [DOI] [PubMed] [Google Scholar]
- Nelson JW, Sklenar J, Barnes AP, and Minnier J (2017). The START App: a web-based RNAseq analysis and visualization resource. Bioinformatics 33, 447–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas-Avila JA, Lechuga-Vieco AV, Esteban-Martinez L, Sanchez-Diaz M, Diaz-Garcia E, Santiago DJ, Rubio-Ponce A, Li JL, Balachander A, Quintana JA, et al. (2020). A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell 183, 94–109 e123. [DOI] [PubMed] [Google Scholar]
- Pairet N, Mang S, Fois G, Keck M, Kuhnbach M, Gindele J, Frick M, Dietl P, and Lamb DJ (2018). TRPV4 inhibition attenuates stretch-induced inflammatory cellular responses and lung barrier dysfunction during mechanical ventilation. PLoS One 13, e0196055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, Lafaille JJ, Hempstead BL, Littman DR, and Gan WB (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons JT, Horwitz AR, and Schwartz MA (2010). Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol 11, 633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel B, Bansal SS, Ismahil MA, Hamid T, Rokosh G, Mack M, and Prabhu SD (2018). CCR2(+) Monocyte-Derived Infiltrating Macrophages Are Required for Adverse Cardiac Remodeling During Pressure Overload. JACC Basic Transl Sci 3, 230–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel MD, Mohan J, Schneider C, Bajpai G, Purevjav E, Canter CE, Towbin J, Bredemeyer A, and Lavine KJ (2017). Pediatric and adult dilated cardiomyopathy represent distinct pathological entities. JCI Insight 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie TA, Strand NS, Yang CT, Rabinowitz JS, and Moon RT (2014). Macrophages modulate adult zebrafish tail fin regeneration. Development 141, 2581–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R, Chandran A, Wang L, Arora K, Rosenthal NA, et al. (2016). Revisiting Cardiac Cellular Composition. Circ Res 118, 400–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto AR, Paolicelli R, Salimova E, Gospocic J, Slonimsky E, Bilbao-Cortes D, Godwin JW, and Rosenthal NA (2012). An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PLoS One 7, e36814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saederup N, Cardona AE, Croft K, Mizutani M, Cotleur AC, Tsou CL, Ransohoff RM, and Charo IF (2010). Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS One 5, e13693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sager HB, Hulsmans M, Lavine KJ, Moreira MB, Heidt T, Courties G, Sun Y, Iwamoto Y, Tricot B, Khan OF, et al. (2016). Proliferation and Recruitment Contribute to Myocardial Macrophage Expansion in Chronic Heart Failure. Circ Res 119, 853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpathy AT, Briseno CG, Lee JS, Ng D, Manieri NA, Kc W, Wu X, Thomas SR, Lee WL, Turkoz M, et al. (2013). Notch2-dependent classical dendritic cells orchestrate intestinal immunity to attaching-and-effacing bacterial pathogens. Nat Immunol 14, 937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheraga RG, Abraham S, Niese KA, Southern BD, Grove LM, Hite RD, McDonald C, Hamilton TA, and Olman MA (2016). TRPV4 Mechanosensitive Ion Channel Regulates Lipopolysaccharide-Stimulated Macrophage Phagocytosis. J Immunol 196, 428–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares MP, and Hamza I (2016). Macrophages and Iron Metabolism. Immunity 44, 492–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe J, et al. (2013). Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155, 1119–1130. [DOI] [PubMed] [Google Scholar]
- Sugita J, Fujiu K, Nakayama Y, Matsubara T, Matsuda J, Oshima T, Liu Y, Maru Y, Hasumi E, Kojima T, et al. (2021). Cardiac macrophages prevent sudden death during heart stress. Nat Commun 12, 1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain SM, Romac JM, Shahid RA, Pandol SJ, Liedtke W, Vigna SR, and Liddle RA (2020). TRPV4 channel opening mediates pressure-induced pancreatitis initiated by Piezo1 activation. J Clin Invest 130, 2527–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theret M, Mounier R, and Rossi F (2019). The origins and non-canonical functions of macrophages in development and regeneration. Development 146. [DOI] [PubMed] [Google Scholar]
- Uhl S, Freichel M, and Mathar I (2015). Contractility Measurements on Isolated Papillary Muscles for the Investigation of Cardiac Inotropy in Mice. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA, Chawla A, and Pollard JW (2013). Macrophage biology in development, homeostasis and disease. Nature 496, 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Burchfield JS, and Hill JA (2013). Pathological ventricular remodeling: therapies: part 2 of 2. Circulation 128, 1021–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, et al. (2013). Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1: Normalized RNA sequencing data (counts per million). Related to Figure 1
Data S2: Microarray expression data. Related to Figure 6
Supplemental Video 1: Zoom in time lapse 2-photon imaging of a CCR2- macrophage in papillary muscle preparations. Related to Figure 5
Supplemental Video 2: Zoom out time lapse 2-photon imaging of a CCR2- macrophage in papillary muscle preparations. Related to Figure 5
Data Availability Statement
All data will be made immediately available upon request. Gene expression data will be available within the data supplement.