

Abstract
Guillain–Barré syndrome (GBS) is an acute inflammatory and immune‐mediated demyelinating disease of the peripheral nervous system (PNS). Macrophages play a central role in its animal model, experimental autoimmune neuritis (EAN), which has been well accepted. Additionally, nuclear factor (NF)‐κB inhibitors have been used to treat cancers and have shown beneficial effects. Here, we investigated the therapeutic effect of M2 macrophage and the NF‐κB pathway’s correlation with macrophage activation in EAN in C57BL/6 mice. We demonstrate that M2 macrophage transfusion could alleviate the clinical symptoms of EAN by reducing the proportion of M1 macrophage in the peak period, inhibiting the phosphorylation of NF‐κB p65. The NF‐κB inhibitor (BAY‐11‐7082) could alleviate the clinical symptoms of EAN and shorten the duration of symptoms by reducing the proportion of M1 macrophages and the expression of proinflammatory cytokines. Consequently, BAY‐11‐7082 exhibits strong potential as a therapeutic strategy for ameliorating EAN by influencing the balance of M1/M2 macrophages and inflammatory cytokines.
Keywords: BAY11‐7082, EAN, GBS, macrophages, NF‐κB
M2 macrophages were induced in vitro successfully. M2 macrophages were transferred to EAN and ameliorated the severity of EAN. Percentage of M1 macrophages was reduced and the M2 macrophages were increased in spleen MNCs of M2 macrophages‐treated group compared to the control group. M2 macrophages treatment had reduced the level of p‐p65 and decreased pro‐inflammatory cytokines greatly compared to the control group.

INTRODUCTION
Guillain–Barré syndrome (GBS) is a common autoimmune disease characterized pathologically by inflammation and demyelination in the peripheral nervous system (PNS) [1, 2]. Experimental autoimmune neuritis (EAN) is a classic animal model of GBS, which has been widely used in basic research of GBS [3, 4]. After activation, macrophages can be divided into proinflammatory M1 macrophages and anti‐inflammatory M2 macrophages, and new research has recently found that the M3 switch phenotype also exists [5, 6]. In GBS and EAN, M1 macrophages are involved in the inflammatory impairment of the myelin sheath through promoting cellular cytotoxicity and producing T helper type 1 (Th1) cytokines. M2 macrophages contribute to repairing myelin and axon by promoting the Th2 immune response and secretion of anti‐inflammatory cytokines [7, 8, 9]. Several studies have demonstrated that the switch from M1 to M2 could effectively ameliorate the severity of EAN [7, 10, 11].
Nuclear factor kappa B (NF‐κB) is an inducible transcription factor expressed in a large number of cells and is involved in immune and inflammatory responses [12]. Moreover, the activated form p65 of NF‐κB has been observed in the sural nerve macrophages of acute and chronic inflammatory demyelinating polyneuropathy (AIDP, CIDP) and the sciatic nerves of rats with EAN, suggesting that NF‐κB is attributed to inflammatory demyelination. Currently, there have been very few studies demonstrating the role of NF‐κB in the course of polarized macrophages [13, 14]. However, research examining the relationship between macrophages and NF‐κB in this process has been controversial.
The objective of this study was to assess the effect of M2 macrophages in treating EAN and to explore the role of the NF‐κB regulating macrophage subtype. Our results show that M2 macrophages were effective in treating EAN by inhibiting the activation of NF‐κB p65 and the production of proinflammatory cytokines. The NF‐κB inhibitor, BAY11‐7082, attenuated the severity of EAN by inhibiting the NF‐κB pathway and the polarization of M1 macrophages.
MATERIALS AND METHODS
Animals
Male C57BL/6J mice, 5–6 weeks old, were purchased from the Beijing Vital River Laboratory Animal Technology Co., Ltd (Beijing, China). Mice were housed in pathogen‐free conditions and fed with food and water ad libitum. The study was approved by the local ethics committee of The First Hospital of Jilin University, Changchun, China, and all experimental procedures complied with the regulations for the management of laboratory animals in the Jilin Province (Ethical approval number: 2017‐216).
Induction and clinical evaluation of EAN
EAN was induced by immunizing mice twice (days 0 and 8) by subcutaneous injection inoculum containing 150 μg P0 peptides 180–199 (GenScript, Piscataway, New Jersey, USA) and 0.5 mg Mycobacterium tuberculosis (strain H37 RA; Difco, Franklin Lakes, New Jersey, USA) in 25 ml saline and 25 ml Freund’s incomplete adjuvant (FIA; ICN Biomedicals, Costa Mesa, California, USA) into the backs of the mice. All mice were injected with 400, 300 and 300 ng pertussis toxin (PTX; Merck, Whitehouse Station, New Jersey, USA) via tail veins on days −1, +1 and +3, respectively. Using a blinded protocol, the clinical signs of EAN were scored immediately before immunization (day 0) by two different examiners as follows until day 30: 0 = normal; 1 = less lively, reduced tonus of the tail; 2 = flaccid tail; 3 = abnormal gait; 4 = gait ataxia; 5 = mild paraparesis; 6 = moderate paraparesis; 7 = severe paraparesis; and 0.5 = intermediate clinical signs. The control group is treated in the same way as mentioned above, but without adding P0 peptides 180–199. Experiments were replicated three times (n = 10).
BAY11‐7082 prevented and treated EAN
BAY11‐7082 (Beyotime, Nantong, China) was dissolved in 1% dimethyl sulfoxide (DMSO) in phosphate‐buffered saline (PBS). EAN mice were divided into three groups (control, preventative and therapeutic). For preventative treatment, BAY11‐7082 solution was administered by intraperitoneal injection (20 mg/kg) from days 2 to 8 post‐immunization (p.i.) every 2 days. For therapeutic treatment, BAY11‐7082 solution was administered in the same way as the preventative treatment at the same dose from the day on which the onset of symptoms (day 10) was observed to day 16 every 2 days. Control animals with EAN received the same volume of the vehicle solution (1% DMSO in PBS). Clinical signs in the mice were assessed using the same protocol described as above. Experiments were replicated three times (n = 10).
Flow cytometry
The spleens were removed under aseptic conditions at different phases of disease (onset, peak and recovery), and splenocytes were harvested after lysing red blood cells. Spleen mononuclear cells (MNCs) with 1 × 106 resuspended in 100 μl PBS 1% bovine serum albumin (BSA) were first stained with F4/80 (Biolegend, San Diego, California, USA), CD11b (BD Pharmingen, San Diego, California, USA), CD206 (Biolegend), Arg‐1 (R&D Systems, Minneapolis, Minnesota, USA) and CD40 (BD Pharmingen), fixed and permeabilized with the fixation/permeabilization solution kit for 20 min (BD Pharmingen), and then stained with antibody for intracellular inducible nitric oxide synthase (iNOS) (eBioscience, San Diego, California, USA).
In order to observe the M2 macrophage proportion before the treated M2, the cultured macrophages were harvested and resuspended at 1 × 106 cells in PBS, then stained by F4/80‐phycoerythrin‐cyanin 7 (PE‐Cy7) (Biolegend), CD11b‐fluorescein isothiocyanate (FITC) (BD Pharmingen), CD206‐BV650 (Biolegend) and arginase 1‐allophycocyanin (Arg‐1‐APC) (R&D Systems) for 45 min at room temperature (RT). Flow cytometric data were acquired using a fluorescence activated cell sorter (FACS) Aria™ flow cytometry (BD Biosciences, San Jose, California, USA) and analyzed with FlowJo software version 7.6.1 (flowjo.com).
In FACS analysis, both F4/80 and CD11b‐positive were calculated as macrophages. In macrophages, both iNOS‐ and CD40‐positive were identified as M1 macrophages, and either CD206‐ or Arg‐1‐positive was identified as M2 macrophages.
Macrophages cultured in vitro and transferred to EAN mice
After lysis of the red blood cells, the collected bone marrow cells from femur and tibia were grown in complete RPMI‐1640 medium (gibco, Waltham, Massachusetts, USA) containing 10% fetal bovine serum (FBS, Sigma Aldrich, St Louis, Missouri, USA), penicillin (100 U/ml; Hyclone, Logan, Utah, USA) and streptomycin (100 U/ml; Hyclone). A total of 1 × 106 cells were seeded into 96‐well plates, then stimulated with macrophage colony‐stimulating factor (M‐CSF) (10 ng/ml; Pepro‐Tech, Rocky Hill, New Jersey, USA) for 48 h, followed by lipopolysaccharide (LPS) (10 ng/ml; Solarbio, Beijing, China) for M1 macrophage or IL‐4 (10 ng/ml; PeproTech) for M2 macrophage was added and incubated for another 48 h. Thereafter, cells were washed three times by PBS to prepare for flow cytometry. The staining procedure was similar to that of the spleen cells, as mentioned above. Cultured cells (M2 macrophages) stimulated with M‐CSF and IL‐4, respectively, were harvested and incubated with P0 (20 µg/ml) peptide for 4 h at 37°C. After washing with PBS twice, the cells were transferred into EAN mice (0.1 ml, 1 × 106 cells for each mouse) via the caudal vein.
The other cultured cells (M1 and M2 macrophages) were added by Bay11‐7082 (1 µM) and incubated for 48 h, then 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) solution and dimethyl sulfoxide (150 µl) were added to each hole.
Cytokine cytometric bead array (CBA)
Serum was obtained by centrifugation of blood samples at 1509g for 15 min at 4°C. The levels of IL‐1β, IL‐4, IL‐6, IL‐10, IL‐12p70, IL‐17A and TNF‐α were measured using CBA kits (BD Biosciences), as per the manufacturer’s instructions.
Western blot analysis
The samples of protein from sciatic nerves were loaded into 12% sodium dodecyl sulfide–polyacrylamide gel electrophoresis (SDS‐PAGE) gel, then transferred to polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, Massachusetts, USA). The following antibodies were used: rabbit anti‐NF‐κB p65 and anti‐NF‐κB p‐p65 monoclonal antibody (Cell Signaling Technology, Danvers, Massachusetts, USA, 1:1000); β‐actin (Bioss, Beijing, China, 1:2000) and the corresponding horseradish peroxidase (HRP)‐conjugated secondary antibodies (Bioss; 1:1000). The blots were developed with the enhanced chemiluminescence kit (Amersham Imager 600; GE Healthcare, Chicago, Illinois, USA). Densitometric analysis of Western blots were performed using ImageJ software.
Statistical analysis
All experiments were repeated three times with 10 mice in each group and data were expressed as mean ± standard error of the mean (SEM). Student’s t‐test was used for comparisons between two groups. Two‐way analysis of variance (ANOVA) was used to compare the score and weight of the two groups. For all statistical analyses, the level of significance was set at p < 0.05.
RESULTS
Clinical course of EAN
All mice immunized with P0 peptides 180–199 in combination with complete Freund’s adjuvant (FCA)‐acquired EAN. In our experiment, the onset of clinical symptoms of EAN in mice occurred on day 10 p.i. After the peak phase, the severity scores of EAN decreased. On day 14 EAN mice began to recover. The other mice immunized with PBS and FCA as controls did not show symptoms of EAN (Figure 1).
FIGURE 1.
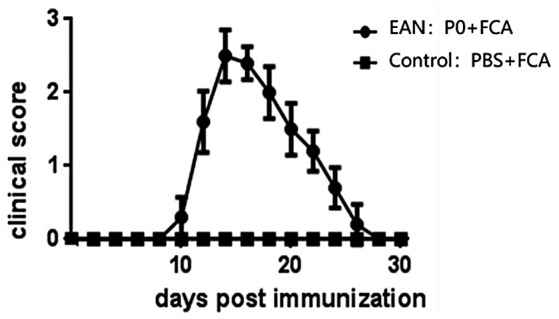
The clinical scores of experimental autoimmune neuritis (EAN) after immunization. EAN was induced in C57BL/6 mice by immunization with P0 peptides 180–199 in combination with complete Freund’s adjuvant (FCA). The mice in the control group received FCA only. The statistically significant differences occurred from days 10 to 15 post‐immunization (p.i.) (p < 0.05 at each time‐point). The mean peak clinical scores were 2.6 ± 0.47 in the EAN group
M2 macrophage treatment attenuated clinical severity in EAN
On the 8th day after culture of M2 macrophages induced by M‐CSF and IL‐4 in vitro, we evaluated the proportions of macrophages and M2 macrophages by flow cytometric analysis. The proportions of macrophages and M2 macrophages were identified as 98.48 ± 0.189% and 94.60 ± 1.407%, respectively (Figure 2a).
FIGURE 2.
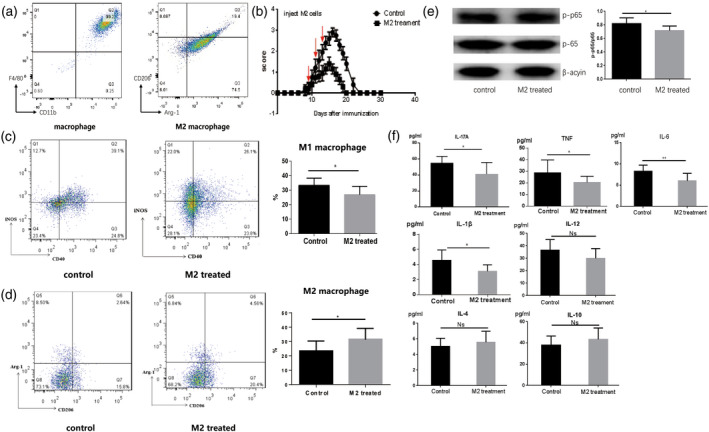
M2 macrophage‐treated experimental autoimmune neuritis (EAN). (a) Macrophage colony‐stimulating factor (M‐CSF) and interleukin (IL‐4)‐induced M2 macrophages. On the 8th day after in‐vitro culture, we evaluated the proportions of macrophages and M2 macrophages. According to flow cytometric analysis, the proportions of macrophages and M2 macrophages were 98.48 ± 0.189% and 94.60 ± 1.407%, respectively. (b) M2 macrophages treatment ameliorated the severity of EAN. We administered M2 macrophages (0.1 ml, 1 × 106 cells) to EAN rats via the caudal vein from days 8 to 14. The scores for the severity of EAN decreased in the M2‐treated group compared to rats in the control group (p < 0.05). Notably, in the M2‐treated group, the duration of clinical symptoms was shorter than the control group. (c–d) The percentage of M1 macrophages was reduced and the percentage of M2 macrophages was increased in spleen mononuclear cells (MNCs) of the M2 macrophage‐treated group compared to the control group. (e) M2 macrophage treatment reduced the level of p‐p65 in sciatic nerves of EAN mice during the peak phase when compared to the control group. (f) The levels of interleukin (IL)‐17A, IL‐1β, IL‐6 and tumor necrosis factor (TNF)‐α decreased greatly compared to the control group. No marked differences appeared for the expression of IL‐12, IL‐4 and IL‐10 among the two groups, but the levels of IL‐4 and IL‐10 increased, although there was no significance
M2 macrophages were administered via the caudal vein from days 8 to 14 p.i. (0.1 ml, 1 × 106 cells, once every 2 days) to treated EAN mice. Our results showed that that the clinical scores of EAN were significantly lighter in the M2 macrophage‐treated group from days 8 to 20 p.i. compared to the control EAN group (p < 0.05) (Figure 2b). Additionally, the duration of symptoms was markedly shorter in the M2 macrophage‐treated group than the control group receiving PBS.
M2 macrophage treatment reduced the expression of M1 macrophages and reduced the activation of NF‐κB in EAN
The flow cytometric data displayed a reduced expression of M1 macrophages (both CD40‐ and iNOS‐positive) compared to the control group (p < 0.05) and elevated expression of M2 macrophages (either CD206‐ or Arg‐1‐positive) from spleen MNCs (p < 0.05) (Figure 2c,d).
Western blotting results indicated that M2 macrophage treatment reduced the expression of NF‐κB p‐p65 in sciatic nerves of EAN mice during the peak phase compared to the control group (p < 0.05) (Figure 2e).
M2 macrophage treatment inhibited the levels of inflammatory cytokines in serum
When compared to the control group, a significant reduction in the expression of inflammatory cytokines, including IL‐1β, IL‐17A, TNF‐α and IL‐6, were observed in the M2 macrophage treatment group (p < 0.05). In contrast, the levels of the anti‐inflammatory cytokines IL‐4 and IL‐10 in M2 macrophage treatment group tended to rise. However, there was no significant difference regarding the expression of IL‐4, IL‐10 and IL‐12 between the control and M2 treatment groups (Figure 2f).
BAY11‐7082 treatment suppressed the severity of EAN in mice
BAY11‐7082 was applied using two different patterns to treat EAN mice (Figure 3a). For the preventative group, BAY11‐7082 was administered from day 2 to the onset of clinical signs of EAN (day 8 p.i). For the therapeutic group, the same dose of BAY11‐7082 was administered from days 10 to 16. Our results showed that the EAN severity scores were decreased in both the preventative and treated groups with BAY11‐7082 compared to control EAN groups, and the preventative group clinical score was more significant. However, the onset of EAN was delayed in the preventative group, and the duration of symptoms was shorter than both therapeutic and control groups (Figure 3a).
FIGURE 3.
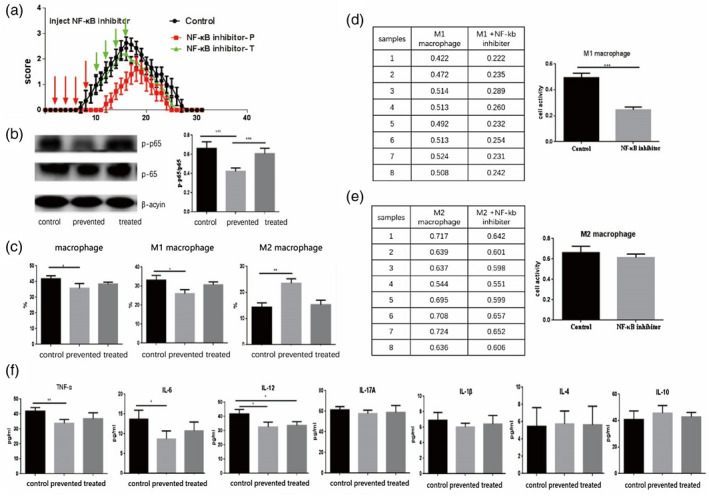
BAY11‐7082 treatment ameliorated the severity of experimental autoimmune neuritis (EAN). In a preventative treatment paradigm, BAY11‐7082 was administered from days 2 to 8 (20 mg/kg, once every 2 days) to EAN mice. The same dosage was applied in a therapeutic treatment paradigm from days 10 to 16. (a) EAN clinical scores were markedly better in BAY11‐7082‐preventative groups. The mice in the preventative group displayed notably better clinical scores from days 8 to 16 with the control group, but there no marked differences appeared for the clinical scores between the therapeutic and control groups. (b) BAY11‐7082 treatment inhibited production of p‐p65 in sciatic nerves of EAN mice. The BAY11‐7082‐preventative group had a significantly decreased expression of p‐p65 compared to BAY11‐7082 therapeutic and control groups. The expression of p‐p65 in the BAY11‐7082 therapeutic group was reduced when compared to the control group, but the differences were not significant. (c) The BAY11‐7082‐preventative group reduced the percentage of M1 macrophages and increased the number of M2 macrophages compared to the therapeutic and control groups. (d,e) BAY11‐7082 inhibited the proliferation of M1 macrophages, whereas the proliferation of M2, despite the differences, was not significant compared to control group in‐vitro studies. (f) The levels of tumor necrosis factor (TNF)‐α, interleukin (IL)‐12 and IL‐6 decreased greatly in the preventative treatment group compared to the control and therapeutic treatment groups. No marked differences appeared for the expression of IL‐17, IL‐1β, IL‐4 and IL‐10 among the three groups, but the levels of IL‐4 and IL‐10 increased although there was no significance
According to the Western blotting results, the expression of p‐NF‐κB p65 was inhibited in the preventative group treated by BAY11‐7082 compared to the control group (Figure 3b).
BAY11‐7082 promoted M2 macrophage polarization in EAN mice
The proportion of M1 and M2 cells in spleens of EAN mice received BAY11‐7082 in treatment/preventative groups was investigated via flow cytometry analysis. The results indicated that the proportion of M2 was elevated significantly in the preventative group compared to the control group (p < 0.01). Interestingly, the proportion of M1 was reduced significantly in the preventative group compared to the control group (p < 0.05), suggesting that BAY11‐7082 was able to promote macrophage polarization to M2 type (Figure 3c).
To explore the mechanisms behind the effects of BAY11‐7082 suppressing EAN, M1 and M2 macrophages were cultured in vitro, respectively, and half of them were added by BAY11‐7082 as an experimental group. The activity and proliferation of M1 and M2 macrophages were evaluated by the MTT method. The results showed that BAY11‐7082 inhibited the activity and proliferation of M1 macrophages significantly compared to the control cultures without BAY11‐7082 (p < 0.001), whereas BAY11‐7082 up‐regulated the activity and proliferation of M2 in spite of the differences and was not significant compared to the control culture group (Figure 3d,e).
BAY11‐7082 altered the expression of cytokines
The expression of inflammatory cytokines shapes the outcome of EAN [15]. We used CBA to confirm the relevant changes in cytokines of EAN after treatment with BAY11‐7082; that is, the expressions of TNF‐α, IL‐6 and IL‐12 were greatly reduced in the BAY11‐7082 preventative group compared to control and therapeutic groups (p < 0.01) (Figure 3f). However, there were no marked differences for the expression of IL‐4, IL‐10, IL‐1β and IL‐17A among the three groups (Figure 3f). Also, differences regarding the expression of cytokines mentioned above in the control and therapeutic groups were meaningless.
DISCUSSION
In this study, we used EAN mice to explore the effects of M2 macrophages and BAY11‐7082 as an NF‐κB inhibitor on this disease. Our results showed that M2 macrophages ameliorated the clinical signs and reduced the duration of symptoms of EAN by inhibiting M1 macrophage and cytokine accumulation and increasing M2 macrophage polarization via inhibiting the NF‐κB signaling pathway. BAY11‐7082 could also diminish EAN symptoms and delay the onset of EAN by reducing M1 macrophage and proinflammatory cytokine accumulation and increasing M2 macrophage proportion in preventive group. In vitro, BAY11‐7082 inhibited the activity and proliferation of M1 macrophages, which was similar to other results [16]. Overall, these results demonstrated that the NF‐κB signaling pathway may be involved in the pathogenesis of EAN by regulating the polarization of macrophages and inhibiting the expression of inflammatory cytokines. The results of adoptive transfer of M2 macrophages into EAN mice displayed the marked beneficial effects on EAN by inhibiting the activation of NF‐κB.
Macrophages play very important roles in GBS, which are broadly divided into two phenotypes: proinflammatory macrophages (M1) and anti‐inflammatory macrophages (M2) [14, 17]. M1 macrophages secrete proinflammatory cytokines that cause tissue damage and disease development, whereas M2 macrophages express high levels of anti‐inflammatory molecules to reduce inflammation and promote disease recovery [14, 18]. Macrophages play either a pro‐ or anti‐inflammatory role in the different stages of GBS [19]. M1 cells can promote the expression of major histocompatibility complex II (MHC‐II), adhesion molecules, reactive oxygen intermediates (ROI), amplify Th1 response via exosomes and inflammatory cytokines, resulting in inflammation, broken brood–nerve barrier (BNB) and demyelination [3, 20]. In contrast, M2 macrophages exert a neuroprotective role in the pathogenicity of EAN [21]. M2 macrophages may contribute to the spontaneous remyelination and regeneration of the axon [22, 23] by promoting T cell apoptosis, suppressing inflammatory responses [9], clearing myelin and axonal debris [18] and inducing the secretion of anti‐inflammatory cytokines such as IL‐10 and transforming growth factor (TGF)‐β [24].
In agreement with those findings, the present study showed that the improved outcome of EAN was associated with a higher proportion of M2 macrophages and the M2 macrophage polarization was increased in the M2 treatment group. In addition, the role of BAY11‐7082 inhibiting EAN is clearly related to shifting macrophages from M1 to M2 type, as shown by the evidence, with a higher percentage of M2 macrophages and a lower percentage of M1 macrophages in EAN treated by BAY11‐7082.
NF‐κB is an inducible transcription factor expressed in a large number of cells and involved in immune and inflammatory responses. It plays a critical role in cell differentiation and apoptosis, as well as oncogenesis. NF‐κB, as a proinflammatory signaling pathway, facilitates the inflammatory reaction by up‐regulation of NF‐κB target genes encoding proinflammatory cytokines, chemokines and adhesion molecules. These signals lead to the recruitment and activation of neutrophils, macrophages and leukocytes to sites of inflammation [25].
NF‐κB can modulate the inflammatory response in EAN, as several studies have demonstrated that the activated p65 of NF‐κB was observed in peripheral nerve macrophages in patients with AIDP or CIDP and EAN [26, 27]. The activated p65 of NF‐κB in T cells and macrophages has a higher intensity at the peak of EAN than the control. The activation of NF‐κB is induced by a large number of potent stimuli, such as lipopolysaccharide (LPS), TNF‐α and IL‐1. Activated NF‐κB is responsible for the expression of many proinflammatory cytokines (TNF, IL‐1, IL‐6 and IL‐8), chemokines [28], adhesion molecules, prostaglandins, reactive oxygen species [29] and matrix metalloproteinases [30]. P65 is required for leukocyte recruitment and macrophage activation during the onset of inflammation [31]. However, recent studies have found that NF‐κB activation promoted neuronal survival by inducing the transcription of anti‐apoptotic genes and a number of growth factors [32, 33, 34]. P65/RelA over‐expression induces the expression of anti‐apoptotic gene and protects neurons from death. NF‐κB protected neurons against amyloid β‐peptide toxicity, glutamate‐induced toxicity and excitotoxic or oxidative stress [35].
In this work, we found that the application of BAY11‐7082 could reduce the phosphorylation of NF‐κB p65, and at the same time reduce M1 macrophages, increase M2 macrophages and reduce proinflammatory factors in vivo. In‐vitro culture of macrophages found that BAY11‐7082 could reduce the production of macrophages, especially M1 macrophages, while it had little effect on M2 macrophages. Therefore, we considered that BAY11‐7082 could inhibit the phosphorylation process of NF‐κB p65, reduce the production of proinflammatory cytokines and, at the same time, inhibit the formation of macrophages, especially M1 macrophages, although its effect on M2 macrophages was not clear. In addition, previous studies have reported that inhibition of NF‐κB can prolong the inflammatory process and maintain leukocyte activation [36]. Therefore, the possible effects of NF‐κB mediating macrophage polarization in EAN warrant further studies.
CONCLUSIONS
Elevated M2 macrophages ameliorated the clinical severity of EAN by down‐regulating the activation of NF‐κB p65 and the accumulation of proinflammatory M1 macrophages and cytokines. Furthermore, BAY11‐7082 as an inhibitor of NF‐κB attenuated EAN through mediating the phenotypical shift in macrophages from M1 to M2 cells. The anti‐inflammatory effects of BAY11‐7082 on EAN probably restore the balance of M1–M2 macrophages and their cytokines. The NF‐κB inhibitor may be a potent candidate for the treatment of polyneurotic diseases in future.
AUTHOR CONTRIBUTIONS
Donghui Shen and Chunrong Li finished the experiment; Fengna Chu and Yue Lang prepared the manuscript; Chao Zheng and Kangding Liu provided views and revised the manuscript; Jie Zhu designed the framework of manuscript, prepared and finalized the manuscript. All authors agreed to approve the final manuscript.
ACKNOWLEDGEMENTS
This study was supported by the National Natural Science Foundation (no. 81471216, no. 81671186, no. 81671177) as well as from the Swedish Research Council (2015‐03005).
Shen D, Chu F, Lang Y, Zheng C, Li C, Liu K, et al. Nuclear factor kappa B inhibitor suppresses experimental autoimmune neuritis in mice via declining macrophages polarization to M1 type. Clin Exp Immunol. 2021;206:110–117. 10.1111/cei.13637
Donghui Shen and Fengna Chu contributed equally to this work.
DATA AVAILABILITY STATEMENT
The data used to support the findings of this study are available from the corresponding author upon request.
REFERENCES
- 1.Nyati KK, Prasad KN. Role of cytokines and Toll‐like receptors in the immunopathogenesis of Guillain–Barre syndrome. Mediat Inflamm. 2014;2014:758639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jasti AK, Selmi C, Sarmiento‐Monroy JC, Vega DA, Anaya JM, Gershwin ME. Guillain–Barre syndrome: causes, immunopathogenic mechanisms and treatment. Exp Rev Clin Immunol. 2016;12:1175–89. [DOI] [PubMed] [Google Scholar]
- 3.Zhang HL, Zheng XY, Zhu J. Th1/Th2/Th17/Treg cytokines in Guillain–Barre syndrome and experimental autoimmune neuritis. Cytokine Growth Factor Rev. 2013;24:443–53. [DOI] [PubMed] [Google Scholar]
- 4.Duan RS, Zhang XM, Mix E, Quezada HC, Adem A, Zhu J. IL‐18 deficiency inhibits both Th1 and Th2 cytokine production but not the clinical symptoms in experimental autoimmune neuritis. J Neuroimmunol. 2007;183:162–7. [DOI] [PubMed] [Google Scholar]
- 5.Banerjee S, Cui H, Xie N, Tan Z, Yang S, Icyuz M, et al. miR‐125a‐5p regulates differential activation of macrophages and inflammation. J Biol Chem. 2013;288:35428–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malyshev I, Malyshev Y. Current concept and update of the macrophage plasticity concept: intracellular mechanisms of reprogramming and M3 macrophage ‘Switch’ phenotype. Biomed Res Int. 2015;2015: 341308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Han R, Xiao J, Zhai H, Hao J. Dimethyl fumarate attenuates experimental autoimmune neuritis through the nuclear factor erythroid‐derived 2‐related factor 2/hemoxygenase‐1 pathway by altering the balance of M1/M2 macrophages. J Neuroinflammation. 2016;13:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nyati KK, Prasad KN, Rizwan A, Verma A, Paliwal VK. TH1 and TH2 response to Campylobacter jejuni antigen in Guillain–Barre syndrome. Arch Neurol. 2011;68:445–52. [DOI] [PubMed] [Google Scholar]
- 9.Kiefer R, Kieseier BC, Stoll G, Hartung HP. The role of macrophages in immune‐mediated damage to the peripheral nervous system. Prog Neurobiol. 2001;64:109–27. [DOI] [PubMed] [Google Scholar]
- 10.Jin T, Yu H, Wang D, Zhang H, Zhang B, Quezada HC, et al. Bowman–Birk inhibitor concentrate suppresses experimental autoimmune neuritis via shifting macrophages from M1 to M2 subtype. Immunol Lett. 2016;171:15–25. [DOI] [PubMed] [Google Scholar]
- 11.Han R, Gao J, Zhai H, Xiao J, Ding Y, Hao J. RAD001 (everolimus) attenuates experimental autoimmune neuritis by inhibiting the mTOR pathway, elevating Akt activity and polarizing M2 macrophages. Exp Neurol. 2016;280:106–14. [DOI] [PubMed] [Google Scholar]
- 12.Hayden MS, West AP, Ghosh S. NF‐kappaB and the immune response. Oncogene 2006;25:6758–80. [DOI] [PubMed] [Google Scholar]
- 13.Pires B, Silva R, Ferreira G, Abdelhay E. NF‐kappaB: two sides of the same coin. Genes 2018;9:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu J, Mix E, Link H. Cytokine production and the pathogenesis of experimental autoimmune neuritis and Guillain–Barre syndrome. J Neuroimmunol. 1998;84:40–52. [DOI] [PubMed] [Google Scholar]
- 16.Chu F, Shi M, Lang Y, Chao Z, Jin T, Cui L, et al. Adoptive transfer of immunomodulatory M2 macrophages suppresses experimental autoimmune encephalomyelitis in C57BL/6 mice via blockading NF‐κB pathway. Clin Exp Immunol. 2021;204:199–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koike H, Katsuno M. Macrophages and autoantibodies in demyelinating diseases. Cells. 2021;10:844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol. 2013;229:176–85. [DOI] [PubMed] [Google Scholar]
- 19.Shen D, Chu F, Lang Y, Geng Y, Zheng X, Zhu J, et al. Beneficial or harmful role of macrophages in Guillain–Barre syndrome and experimental autoimmune neuritis. Mediators Inflamm. 2018;2018:4286364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Du T, Yang CL, Ge MR, Liu Y, Zhang P, Li H, et al. M1 macrophage derived exosomes aggravate experimental autoimmune neuritis via modulating Th1 response. Front Immunol. 2020;11:1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laskin DL. Macrophages and inflammatory mediators in chemical toxicity: a battle of forces. Chem Res Toxicol. 2009;22:1376–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang HL, Hassan MY, Zheng XY, Azimullah S, Quezada HC, Amir N, et al. Attenuated EAN in TNF‐alpha deficient mice is associated with an altered balance of M1/M2 macrophages. PLOS ONE. 2012;7:e38157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu G, Ma H, Qiu L, Li L, Cao Y, Ma J, et al. Phenotypic and functional switch of macrophages induced by regulatory CD4+CD25+ T cells in mice. Immunol Cell Biol. 2011;89:130–42. [DOI] [PubMed] [Google Scholar]
- 24.McWhorter FY, Davis CT, Liu WF. Physical and mechanical regulation of macrophage phenotype and function. Cell Mol Life Sci. 2015;72:1303–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghosh S, May MJ, Kopp EB. NF‐kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–60. [DOI] [PubMed] [Google Scholar]
- 26.Andorfer B, Kieseier BC, Mathey E, Armati P, Pollard J, Oka N, et al. Expression and distribution of transcription factor NF‐kappaB and inhibitor IkappaB in the inflamed peripheral nervous system. J Neuroimmunol. 2001;116:226–32. [DOI] [PubMed] [Google Scholar]
- 27.Laura M, Mazzeo A, Aguennouz M, Santoro M, Catania MA, Migliorato A, et al. Immunolocalization and activation of nuclear factor‐kappaB in the sciatic nerves of rats with experimental autoimmune neuritis. J Neuroimmunol. 2006;174:32–8. [DOI] [PubMed] [Google Scholar]
- 28.Barnes PJ, Karin M. Nuclear factor‐kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–71. [DOI] [PubMed] [Google Scholar]
- 29.Hayden MS, Ghosh S. NF‐kappaB in immunobiology. Cell Res. 2011;21:223–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Al‐Sayeqh AF, Loughlin MF, Dillon E, Mellits KH, Connerton IF. Campylobacter jejuni activates NF‐κB independently of TLR2, TLR4, Nod1 and Nod2 receptors. Microb Pathog. 2010;49:294–304. [DOI] [PubMed] [Google Scholar]
- 31.Lawrence T. The nuclear factor NF‐kappaB pathway in inflammation. Cold Spring Harbor Perspect. Biol. 2009;1:a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiarugi A. Characterization of the molecular events following impairment of NF‐κB‐driven transcription in neurons. Mol Brain Res. 2002;109:179–88. [DOI] [PubMed] [Google Scholar]
- 33.Nickols JC, Valentine W, Kanwal S, Carter BD. Activation of the transcription factor NF‐κB in Schwann cells is required for peripheral myelin formation. Nat Neurosci. 2003;6:161. [DOI] [PubMed] [Google Scholar]
- 34.Kassed CA, Butler TL, Patton GW, De Mesquita DD, Navidomskis MT, Mémet S, et al. Injury‐induced NF‐κB activation in the hippocampus: implications for neuronal survival. FASEB J. 2004;18:723–4. [DOI] [PubMed] [Google Scholar]
- 35.Bhakar AL, Tannis L‐L, Zeindler C, Russo MP, Jobin C, Park DS, et al. Constitutive nuclear factor‐κB activity is required for central neuron survival. J Neurosci. 2002;22:8466–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alcamo E, Mizgerd JP, Horwitz BH, Bronson R, Beg AA, Scott M, et al. Targeted mutation of TNF receptor I rescues the RelA‐deficient mouse and reveals a critical role for NF‐kappa B in leukocyte recruitment. J Immunol. 2001;167:1592–600. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.