

Abstract
Atherosclerosis is an inflammatory disease with break‐down of homeostatic immune regulation of vascular tissues. As a critical initiator of host immunity, dendritic cells (DCs) have also been identified in the aorta of healthy individuals and atherosclerotic patients, whose roles in regulating arterial inflammation aroused great interest. Accumulating evidence has now pointed to the fundamental roles for DCs in every developmental stage of atherosclerosis due to their myriad of functions in immunity and tolerance induction, ranging from lipid uptake, efferocytosis and antigen presentation to pro‐ and anti‐inflammatory cytokine or chemokine secretion. In this study we provide a timely summary of the published works in this field, and comprehensively discuss both the direct and indirect roles of DCs in atherogenesis. Understanding the pathogenic roles of DCs during the development of atherosclerosis in vascular tissues would certainly help to open therapeutic avenue to the treatment of cardiovascular diseases.
Keywords: atherosclerosis, cytokines, dendritic cells, T cells
DCs play important roles in the development of atherosclerosis via both direct and indirect approaches, which could be either pro‐atherogenic or atheroprotective.
INTRODUCTION
Developed during the lifespan of an individual, atherosclerosis is inflammatory disease with immune destruction of the vascular tissues, involving both innate and adaptive immune responses in a sequential step. In the initial stage, activation of the endothelium by lipid deposition causes leucocyte recruitment, monocyte differentiation and foam cell formation, the death of which may further develop fibrotic plaques [1]. Later, in addition to the innate components of the immune cells such as monocytes/macrophages and antigen‐presenting dendritic cells (DCs), adaptive immunological components such as T and B cells were also found within atherosclerotic lesions, probably attracted by the chemokines secreted by these innate cells [2]. With the passage of time, both adventitia and intima significantly increase their thickness during the development of atherosclerosis because of various immune cell accumulations in both compartments [3, 4] and eventually form vascular inflammation. Like any non‐resolving peripheral inflammation, artery tertiary lymphoid organs (ATLO) emerge in the adventitia with the help of vascular smooth muscle cell lymphotoxin β receptors that do not affect secondary lymphoid organs. Interestingly, in these ATLOs regulatory T cells (Treg) were induced from naive T cells and antigen was presented by a unique set of DCs and B cells, orchestrating the immune cells in the vascular tissues for a regional immunity against atherogenesis [5, 6]. However, depending upon different pathological conditions, the production of soluble erosive factors, such as matrix proteases and cytokines by local immune cells, can sometimes eat away the surface of cellular aggregates, leading to the thinning of the fibrous cap on the top of the core and ruptures of the plaque, which can occlude the artery if formed thrombus in the blood flow [2, 3].
The existence of DCs in healthy human aortic tissues was first demonstrated by the morphological analysis of S‐100‐positive intimal cells, DC‐like cells found mainly accumulated in atherosclerosis‐prone areas of aortic walls, rather than evenly distributed throughout the vascular tissues [7]. During the development of atherosclerosis, large numbers of vascular DCs were identified inside the atherosclerotic plaques and in the adventitia [3, 4, 8], suggesting that vascular DCs may participate in the disease progression [8]. Because DCs are generally present in vasculatures that are easy to acquire an atherosclerotic plaque, such as bifurcations and curvatures [9, 10, 11], and regions that are exposed to high shear‐stress blood flow, the physical force of blood flow might contribute to the recruitment of DCs into those regions and probably also activation in the later stages. Collectively, anatomical presence in the atherosclerosis‐prone areas of vascular tissues and their increase in number with the development of disease make DCs a probable candidate to be involved in the regulation of inflammation in the aortic walls [9].
DCs work in subsets, and various DC subsets with differential expression of surface markers were found to be associated with the onset of atherosclerosis. DCs in normal intima were divided into at least two phenotypically distinct subsets, i.e. CD11c+CD11b−CD103+ and CD11c+CD11b+CD103− DCs [11], with the latter constituting 50–60% of DCs in healthy mouse aorta as the most abundant DC subset [9, 12]. This CD11b+ conventional DC (cDC) subset was also found to be predominant in the diseased plaque [13]. Dynamically, the CD11b+ DCs can exit atherosclerotic plaques under homeostatic conditions and migrate to draining lymph nodes via the afferent lymphatics [9, 14]. In human lower limb atherosclerosis occlusion syndrome (ASO), there were CD11clow CD11b+ tolerogenic DCs (TDC) whose number negatively correlated to the progress of the disease [15]. In addition, a strong inverse correlation was observed between CD11b+ cDCs and plaques [13], indicating a possible anti‐atherogenic role for this DC subset. Represented as an indigenous DC subset, vascular CD103+ cDCs rely upon fms‐like tyrosine kinase 3 (Flt3L) signalling for survival and development [9]. In Flt3L/low‐density lipoprotein receptor‐deficient (Flt3−/−Ldlr−/−) mice fed a high‐fat diet for 12 or 16 weeks, loss of this cDC subset caused reduced aortic Tregs, decreased anti‐inflammatory interleukin (IL)‐10) but enhanced proinflammatory cytokine [interferon (IFN)‐γ and tumour necrosis factor (TNF)‐α] production, which propagated atherosclerotic progression [9, 16], suggesting an anti‐atherogenic role in its presence. Interestingly, different from their common developmental precursor, a monocyte‐derived CD103+ DC subset was identified [17] which is also able to induce Tregs in the lesion to be atheroprotective [18]. Resided primarily in the arterial adventitia [19], the third DC subset that was reported to be associated with the development of atherosclerosis is CD45RA+ plasmacytoid DCs (pDCs) [20]. This pDC subset secretes large amounts of the type 1 interferons IFN‐α and IFN‐β, both of which are strong proinflammatory cytokines to facilitate the development of atherosclerosis [21]. The last, but not least, DC subset associated with the inflammatory vascular disease is C‐C motif chemokine ligand 17 (CCL17+) DCs that are also CD11b+, but they do not express colony‐stimulating factor 1 receptor (CSF1R) (CD115) or F4/80, distinguishing themselves from the conventional CD11b+ DC subset [22]. Interestingly, studies in CCL17− green fluorescent protein (GFP) reporter mice demonstrated that these CCL17+ DCs are only found in the intima and adventitia of atherosclerotic arteries, not in the arterial walls of healthy arteries [23]. Furthermore, the absence of this DC‐specific molecule in atherosclerotic apolipoprotein E (Apoe−/−) mice caused an expansion of Tregs but a decrease in macrophages in the plaques [23], assigning atherogenic roles to this DC subset.
With the development of modern technologies, better resolution of surface molecules has revealed much more complex heterogeneity in vascular myeloid cells, including DCs, than was understood previously with traditional methods, such as immunohistochemistry and conventional cytometry. Such modern approaches as high‐dimensional cytometry by time of flight (CyTOF) and single‐cell RNA sequencing (scRNAseq) now enable comprehensive mapping of various myeloid cells, the key players of the immune system, present in the plaques with better accuracy at single‐cell levels via both proteomic and genomic analysis. While CyTOF is good at discerning leucocyte subsets in the atherosclerotic aorta by assessing up to 42 cell surface and intracellular markers, scRNAseq provides more insight into their probable functions from expression matrices of thousands of genes per cell. The successful use of these advanced techniques were recently summarized in an excellent review which analyzed reported data from nine scRNAseq and two CyTOF studies [24, 25] in murine samples, although few of these techniques were applied to human atherosclerosis [26, 27].
The leucocytes infiltrate into atherosclerotic mouse aortas, as analyzed in the comprehensive meta‐analysis of nine scRNAseq studies, identifying four DC subsets with characteristic gene expression of Ccr1, Irf8, Cst3, Naaa, Naga and Plbd1 in cDC1; Cd209a, Lfitm1 and Kird1 in moDC/cDC2; Pla2g2d, Dnase1l3, C3 and Vcam1 in pDCs; and Ccr7 and Fascin1 in mature DCs [25]. Of the two studies employing CyTOF to characterize mouse aortic leucocytes in Apoe−/− mice [28, 29], both found cDC1 cells that expressed CD68, CD11c, major histocompatibility complex (MHC)‐II, CD103 and X‐C motif chemokine receptor 1 (XCR1) but little CD11b, and cDC2 that also expressed D68, CD11c and MHC‐II but not CD103. Instead, these cDC2 expressed CD11b, CD41 and CD172a. pDCs were identified only in one study defined by their expression of B220 and Siglec H [28]. Interestingly, in the other CyTOF study, a third DC subset that differed from cDC2 by lower expression of CD11b and absence of CD41 was identified [29]. Of note, cDC1 remains unchanged and cDC2 reduced; only pDC increased during the progression of atherosclerosis [28]. Collectively, these novel high‐dimensional techniques have provided a more detailed insight into DC phenotypical and transcriptional heterogeneity in atherosclerosis.
Given the various associations of different subset of DCs with the development of atherosclerosis, the roles of DCs in the disease progression could mechanistically be divided into either direct roles (phagocytose lipid/antigen, efferocytosis, present antigen to activate nearby T cells) or indirect roles (secrete cytokines/chemokines or other soluble factors for long distant effects) to mobilize other downstream immune cells, including T cells, macrophage, B cells and natural killer (NK) cells, etc. for vascular inflammation (Figure 1), of which T cells are focused upon as the major downstream effectors.
FIGURE 1.
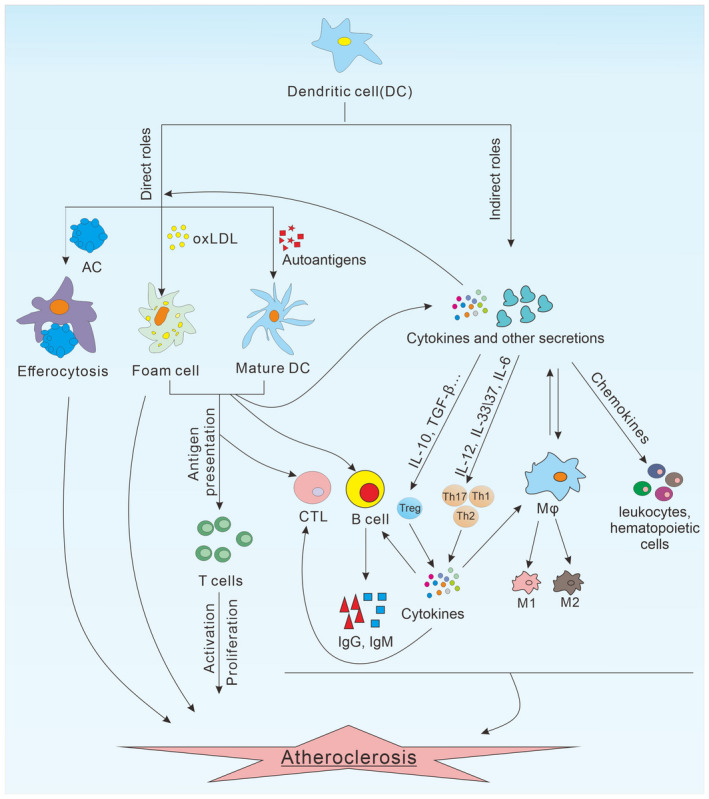
Multiple roles DCs during atherogenesis. DCs play an important role in the development of atherosclerosis through direct or indirect effects. DCs can uptake lipids directly to form foam cells, which are an important part of plaque at the early stage of the disease. Furthermore, the DCs that mature after exposure to AS‐associated autoantigens can present antigens to T cells to promote their activation and proliferation. In addition, DCs can also clear apoptotic cells in plaques by efferocytosis. Indirectly, DCs can regulate the function of other immune cells by cytokines and other soluble factors. DCs can secrete chemokines to recruit circulating leucocytes or haematopoietic cells to the vascular region. The production of conditioning cytokines from DCs control the differentiation of T cells into various T effectors [T helper type 1 (Th1), Th2, Th17 and regulatory T cells (Treg)], regulate the activation of B cells, polarize macrophages and activate CTLs, which ultimately result in immune destruction of vascular walls and onset of atherosclerosis. DCs = dendritic cells; AC = apoptotic cell; CTL = cytotoxic T lymphocyte; oxLDL = oxidized low‐density lipoprotein; Mφ = macrophage; M1 = M1 macrophages; M2 = M2 macrophages
DIRECT ROLE OF DCS IN ATHEROSCLEROSIS
Lipid uptake
Lipid deposition and accumulation in the vascular intima is a key event in atherosclerosis [30]. DCs home to the vessel walls, recognize and uptake antigens, of which oxidized LDL (oxLDL) and heat shock protein (HSP) 60/65 are major atherosclerosis‐related autoantigens [7, 31]. On one hand, oxLDL uptake might promote the presentation by the DCs of lipid and peptide antigens to NK T and T cells, respectively, further accelerating vascular inflammation [7]. On the other hand, the binding of DCs by oxLDL through scavenger receptors also stimulate DC themselves to secrete inflammatory cytokines for vascular destruction 7 [32]. In addition, lipid uptake at the early stage can transform DCs into foam cells, although the latter was often regarded as the role of macrophages in the vascular tissues. In line with this notion, DCs in the subendothelial space of the vascular walls were found to have substantial lipid contents and bear the markers of foam cells, resulting in further progression of atherosclerosis [11].
Scavenger receptor A (SR‐A), CD36 and lectin‐like ox‐LDL receptor‐1 (LOX‐1) are important scavenger receptors for phagocytes to uptake oxLDL before transformation into foam cells [33]. DCs in early atherosclerotic lesions uptake lipoproteins and lipid‐laden cells via these scavenger receptors to contribute to plaque foam cell pools in atherosclerosis. Sequestered in the artery walls, resident DCs were reported to respond to hyperlipidaemia‐inducing cholesterol‐rich diet and proliferate in situ in a granulocyte–macrophage colony‐stimulating factor (GM‐CSF)‐dependent manner. Subsequently, these DCs uptake lipid and develop morphology characteristic of foam cells [34]. Although the lipid uptake process of DCs may be related to their fluid‐phase endocytosis of native or modified LDL, the exact mechanism for the DCs to become foam cells is far from clear. However, the fact that DCs absorb lipid is solid. Elimination of DCs in CD11c‐diphtheria toxin receptor (DTR) mice via injection of diphtheria toxin reduced the lipid contents in nascent lesions, indicating that DCs are involved in lesional lipid accumulation [11]. Intriguingly, in addition to the role of DCs in lipid uptake in early lesions, several studies suggest that DCs from non‐vascular tissues might also participate in regulating systemic cholesterol levels. For example, treatment of the DC‐stimulating factor GM‐CSF reduced circulating cholesterol in hyperlipidaemic rabbits, as expected, [35] and this cholesterol‐reducing role of DCs may further influence the establishment of vascular inflammation during atherogenesis, because hypercholesterolaemia is a relevant factor for atherosclerosis. Similarly, when the DC lifespan was expanded by CD11c‐specific transgenic expression of the anti‐apoptotic protein Bcl‐2 instead of increasing their numbers by GM‐CSF treatment, both atherosclerotic Ldlr−/− andApoe−/− mice have demonstrated a decrease in the levels of VLDL and LDL cholesterol [36]. Conversely, elimination of DCs by injection of diphtheria toxin in CD11c‐DTR mice resulted in an increase in plasma cholesterol levels in the Ldlr−/− or Apoe−/− mice [36]. Although the working procedure that underlies these systemic effects on lipid levels is not clear, it was suggested that that DCs could have exerted their influence on cholesterol absorption from the intestine and faecal excretion of sterols [37]. Nevertheless, these results collectively emphasize that the importance of immune‐metabolism cross‐talk and underline the crucial role of immune cells in regulating metabolic disorders.
Present AS‐related autoantigens
Antigen presentation to activate adaptive immunity is the key function of DCs. Before contact with antigens, peripheral vascular DCs are immature, capable of capturing antigen in close proximity, and either interact directly with T cells in the adventitia or migrate from the vessel walls to distant lymphatic tissue for T cell activation. In addition, circulating antigens can also be directly ingested by DCs in the spleen or lymph node for T cell priming but not activation, as these DCs do not, as yet, up‐regulate MHC or the T cell co‐stimulatory molecules CD80, CD86 and CD40 on their surface. Following Toll‐like receptor (TLR)‐cross‐linking by pathogens, however, DCs experience a developmental process of maturation, accompanied by the down‐regulation of antigen uptakes in the forms of phagocytosis and efferocytosis but up‐regulation of MHC and the above‐mentioned co‐stimulatory molecules, so that they can either present antigens in situ to local T cells or migrate to lymphoid organs to stimulate systemic T cells.
The key issues regarding DC stimulation of T cell by antigen presentation for the development of atherosclerosis involve the nature of the atherosclerosis‐related autoantigen(s) and the location of DC–T cell interaction in vascular tissues or at lymphoid organs as described above. Currently, the lesional antigens identified as being associated with atherogenesis are oxidized LDL, Hsp60, Hsp65 and β2‐glycoprotein I [38]. It is possible that lesional DCs directly capture these and other antigens through macropinocytosis and scavenger receptor‐mediated uptake for MHC‐peptide presentation. In addition circulating DCs, even lymphoid‐tissue DCs outside the vascular walls, may also capture and then present circulating atherosclerosis‐relevant antigens for T cell activation and survival, like oxidized LDL. Notably, when DC maturation is disturbed in a mouse model, there was global Treg reduction in both atherosclerotic lesions and lymph nodes regardless of their draining or non‐draining natures, [39] suggesting that DCs may acquire certain periphery antigens by themselves.
Following uptake of atherosclerosis‐related antigens, vascular or circulating DCs become mature and are found to accumulate in the diseased plaques with active lesion in both human and animal studies [40]. DC maturation comes with up‐regulation of proinflammatory cytokine production and an enhanced capacity to activate naive T cells by presenting captured autoantigens, both of which turn out to be atherogenic [41]. DC maturation is also characterized by the up‐regulation of co‐stimulatory molecules, which was indicated to facilitate antigen presentation in inflammatory vascular disease, because atherosclerosis was found to be reduced in mice without CD80 and CD86 as well as in mice deficient for MHC‐II‐associated invariant chain CD74 that regulates antigen loading on MHC‐II [41, 42]. Therefore, DC maturation and subsequent antigen presentation to T cells are critical steps to the development of atherosclerosis. Similarly, stopping DC maturation by knocking down TLR signalling molecule myeloid differentiation factor 88 (MyD88) in DC lineage (Cd11cCre+Myd88fl/fl) was effectively utilized to reduce the potential of DCs to activate T cells [43]. Interestingly, crossed into the atherosclerotic model, these mice demonstrated compromised activation of both proinflammatory T cells and Tregs in lesions, although the ultimate outcome was the increased development of atherosclerotic lesions [39]. These data suggest that the role of DCs in antigen presentation is more important in the development of Tregs than that of pro‐atherogenic inflammatory T cells.
In addition to protein antigen presentation to T cells through MHC class molecules, DCs can also present AS‐related self or foreign lipid antigens through CD1d molecules to NK T cells [44]. This immunological pathway might also contribute to the development of atherosclerosis, which is supported by several experimental studies. For example, the MHC‐like lipid‐presenting molecule CD1d is expressed by antigen‐presenting cells (APCs) in mouse and human atherosclerotic lesions, [45, 46] and proatherogenic factors such as serum lipid up‐regulate the expression of CD1d on DCs [47]. Furthermore, NK T cells stimulated by lipid antigen α‐galactosylceramide (α‐GalCer) in Apoe−/− mice are associated with increased atherosclerosis [46], and administration of the α‐GalCer‐loaded mature DCs can lead to sustained expansion of NK T cells [48]. Finally, the relevance of the proatherogenic effect of α‐GalCer‐mediated NK T cell induction was confirmed in Apoe−/−CD1d−/− double knock‐out animals in which α‐GalCer injection failed to increase atherosclerosis [49]. As CD1d is predominantly expressed in APCs, including DCs, macrophages and B cells, conditional deletion of CD1d on CD11c+ cells in CD1dfl/flCD11cCre mice have revealed the importance of CD11c+ cells in controlling lipid‐dependent immunity in the intestinal compartment and demonstrated NK T cell–DC cross‐talk as a key mechanism for the regulation of gut homeostasis [50]. The significant effect of CD1d‐presented lipid by DCs on atherogenesis in this mouse model, however, awaits further investigation.
Although most of the data indicate that NK T cells are proatherogenic in animal models, the pathophysiological role of these cells in human is still uncertain. It was assumed that, like that of T cells to protein antigen, immature DCs maintain tolerance to the lipid antigen LDL by supporting the iNK T‐mediated development of immature DCs into tolerogenic DCs under physiological conditions. Under inflammatory conditions, however, NK T cell ligands may stimulate responses that result in NK T cell‐induced inflammation that disrupts the tolerance to LDL for the development of atherosclerosis [51].
Efferocytosis
The term ‘efferocytosis’ comes from the Grecian word effere (carrying the dead bodies to the grave), which refers to the engulfment of dead cells by phagocytic cells to inhibit secondary necrotic lysis of dying cells accumulated in an atherosclerotic plaque [52, 53, 54]. This term is specifically used to describe the phagocytosis of apoptotic cells (ACs), which is different from phagocytosis of other antigens in the body, such as autoantigens and pathogens [55] or lipid, as mentioned above. Since its first observation by a Russian scientist in the 1880s, efferocytosis has been extensively studied because it extends the action of ingesting and eliminating microbes to the clearance of apoptotic cells by phagocytes as part of the fundamental mechanism to maintain tissue homeostasis. Generally speaking, the process of efferocytosis can be briefly divided into four basic steps: (1) target recognition, (2) signalling activation for particle internalization, (3) phagosome formation and (4) phagosome maturation towards the phagolysosome [56].
Efferocytosis is separate from other forms of phagocytosis that remove foreign or aberrant substances rather than dying cells. Nearly all types of cells can engulf the apoptotic cells. However, some cells are extremely efficient at this task, and are thus called ‘professional efferocytic cells’ [55] such as DCs and macrophages. Non‐professional efferocytic cells include adjacent endothelial cells, single smooth muscle cells (SMCs), fibroblasts and phagocytes. Although both professional and non‐professional efferocytic cells are associated with the progression of atherosclerosis, DCs, the most potent antigen‐presenting cells, are specialized in receiving their environment signals and providing appropriate immune responses following the uptake of apoptotic cells, and thereby especially good at participating in this atherosclerosis‐related process. Moreover, when DCs efferocytose the apoptotic cells in the lesion they effectively prevent cellular necrosis, a major proinflammatory signal that accelerates atherosclerotic lesion development and necrotic core formation [54]. Most importantly, after clearing the apoptotic cells in the lesions, DCs do not remain in the diseased locus to become new necrotic cells. Instead, the DC‐mediated efferocytosis triggers their unique function of migration out of plaque to effectively eliminate the inflammatory debris. Interestingly, lipid‐containing form cells that often become apoptotic at later stages, therefore the role of DCs in engulfing lipid‐laden apoptotic cells as efferocytosis would play a critical role in eliminating oxidized lipids, cholesterol and other proinflammatory damage‐associated molecular patterns, demonstrating a distinguished atheroprotective attribute.
Mechanistically, efferocytosis of phagocytes in atherosclerosis requires not only an interplay between adenylyl cyclase (AC) ligands and phagocyte receptors, but also extracellular bridging molecules that link phagocytes to ACs. Immature DCs or CD103 subset of DCs express high levels of phagocyte receptors, and are therefore better at efferocytosis than their mature or other subset counterparts. Although the molecules required for efferocytosis by immature DCs in vivo are not well defined, there is a correlation of efferocytosis in vitro with efferocytosis receptor CD36 and αv integrins, [57] engulfment mer receptor tyrosine kinase (MERTK) [58] and the bridging molecule milk fat globule EGF‐like factor 8 (MFGE‐8), [59] indicating that these molecules could be mediators underlying DC properties of efferocytosis. Consistently, modulation of these efferocytosis‐related molecules has a significant impact upon the development of atherosclerosis. For example, the CD103+ DC subset preferentially expresses C‐type lectin receptor (Clec9A; also known as DNGR‐1) that recognizes a preformed signal exposed on dead cells as a phagocyte receptor and thus is required for cross‐presentation of dead cell‐associated antigen in vitro and immunogenicity in vivo [60]. Interestingly, in the context of atherogenesis, studies have demonstrated that the specific deletion of Clec9A significantly increases IL‐10 expression, reduces macrophage and T cell contents within the lesions and limits the development of atherosclerosis [61].
Activate T cells
Shared many characteristics with macrophages, including tissue distribution, environmental monitoring and adaptive immunity induction [62]. DCs, however, are unique in their special stellate or dendritic outlook and migration behaviour to lymphoid organs for T cell activation [62, 63]. Bridging between innate and adaptive immune systems, DCs are the most powerful cells to activate both naive and antigen‐experienced memory T cells. With the help of critical co‐stimulatory cell surface molecules such as CD86/80 and CD40, that are constitutively expressed and strongly up‐regulated upon antigen encounter, DCs potently activate both CD8+ and CD4+ T cells to drive them to proliferate and differentiate into effector cells [64, 65].
The activation of T cells and their subsequent polarization into effector cells by DCs are the hallmarks of vascular inflammation due to the immunological nature of the disease that requires molecular interaction at cell surface levels. For example, Ldlr−/− mice deficient for the invariant chain of MHC‐II molecules demonstrate compromised T cell activation in the lesion, and thus do not develop atherosclerosis [42]. T cell immune responses are initiated by antigen‐loaded DCs with complicated but balanced mechanisms for different outcomes, in which co‐stimulatory signals from the cell surface of the DCs can drive the T cell immunity into either an immunogenic or tolerogenic response during disease development [17]. For instance, the interaction of CD40 on DCs with the CD40 ligand on T cells leads to T helper type 1 (Th1) cell polarization, [66] and blocking this signal reduces atherosclerosis in mice and cynomolgus monkeys [67, 68]. CD80 and CD86 on DCs bind to the co‐stimulatory receptor CD28 on T cells for their activation, and the absence of CD80 and CD86 in atherosclerotic mice suppresses Th1 cell responses [69]. CD80 and CD86, at the later stage, also recognize cytotoxic T lymphocyte antigen 4 (CTLA‐4) expressed on Treg cells for T cell suppression, and over‐expression of CTLA‐4 in Apoe−/− mice mitigated the development of atherosclerosis compared with control mice [70]. Programmed cell death 1 ligand 1 (PD‐L1) on DCs and programmed cell death 1 (PD‐1) on T cells are paired ligand and receptor, whose specific interaction could have a major role in limiting early T cell activation and exhaustion in atherosclerosis most frequently demonstrated in chronic inflammation and cancer [71]. Notably, a higher expression of PD‐1 was found in T cells from human atherosclerotic plaques, and activation of the PD‐1–PL1 pathway turned out to limit pro‐atherogenic T cell responses in PD‐L1/2−/−LDLR−/− mice after 10 weeks of high‐cholesterol diet [66]. In addition, DCs also express TNF receptor superfamily member 4, or OX40 ligand, as co‐stimulatory molecule on the cell surface, whose cognate receptor is OX40 on T cells. Their interaction was found to increase Th2 cell responses, and disruption of this pathway in Ldlr−/− mice with atherosclerosis results in plaque regression [72]. Finally, co‐stimulatory signal derived from contact of CD137 ligand on DCs with CD137 on T cells enhanced T cell proliferation and survival to promote the development of atherosclerosis in mice with hyperlipidaemia [73].
At cellular levels, accumulating evidence suggest that DCs with different subsets can adjust T cell activation to control atherosclerosis. Primed by various DC subsets in both human and mouse samples, activated T cells accumulate in atherosclerotic lesions in the fibrous caps and the adventitia of older lesions [29, 43]. Different DC subsets in vascular tissues have been found to impact upon T cell activation in various settings. Isolated aortic CD11b+ DCs have the same capacity to efficiently activate allogeneic CD4+ T cells as that of splenic cDCs in vitro [9]. Intimal CD103+ cDCs demonstrate typical DC functions by showing a classical ‘dendritic’ morphology in the vascular tissues and the potent capacity to activate CD8+ T cells [16]. Specific deletion of MHC‐II on pDCs developed reduced atherosclerosis due to the impotent stimulation of ApoB100‐specific CD4+ T cells in response to native LDL, together with significant reductions of T cell‐derived IFN‐γ and lesion T cell infiltration [74]. Following stimulation by type A oligo dinucleotides (CpGs), pDCs in the plaque promoted the expression of TLR‐4, TNF‐γ and IL‐12 from myeloid DCs and elevated the functions of CD8+ T cells within human plaques [75]. Direct assessment of the T cell‐stimulatory ability of CCL17+ DCs demonstrated that these cells could prime OT‐II T cells in the presence of cognate antigen as effectively as did splenic cDCs, [23] indicating the pro‐atherogenic potential of this DC subset through MHC‐antigen presentation to some extent.
Given a plethora of evidence outlined above to indicate the strong activation of T cells by DCs in the atherosclerotic vascular tissues, where local chronic inflammation develops ATLO, it is possible that the vascular DCs could prime and activate T cells directly in the vascular tissues rather than in their draining lymph nodes. This possibility was recently verified by an excellent study from Maffia's group, in which they have shown the existence of naive CD4+ T cells in wild‐type (WT) and OTII murine aorta, and the local vascular T cell population becomes activated and undergoes proliferation following systemic administration of a model antigen [25]. Furthermore, this antigen‐specific proliferation of aortic T cells was proved to be a local event in situ by blocking lymphocyte trafficking and extravasation. Finally, using confocal microscopy, they have visualized T cells and DCs forming cell contacts in vasculatures following antigen challenge, [25] demonstrating that the aorta can act as a site of naïve CD4+ T cell priming.
THE ROLE OF DC‐DERIVED SOLUBLE FACTORS IN ATHEROSCLEROSIS
In addition to directly interacting with nearby target substances or cells, DCs are involved in the atherosclerotic process by exerting distant immunological effect with soluble factors such as cytokines and chemokines, etc. These soluble factors can be further divided into two different categories, i.e. proinflammatory mediators that stimulate systemic inflammation such as IL‐1 and TNF‐α and counter‐inflammatory mediators involving several immunomodulatory molecules to regulate the proinflammatory cytokine response, such as IL‐35 and IL‐10. Both categories of the DC‐derived factors are implicated in the development of atherosclerosis [6].
IL‐12 family (IL‐12/23/27/35)
When in contact with T cells, DCs can secrete IL‐12 family cytokines in large quantities, stimulate T cell proliferation, induce the generation of specific cytotoxic T lymphocytes (CTLs) for CD8+ T cells and dominate the Th1‐type immune response for CD4+ T cells. The IL‐12 family is evolutionarily associated with the IL‐6 cytokine superfamily and composed of a single group of three possible α chains (p19, p28 or p35) and one of two β chains [p40 or Epstein–Barr virus‐induced gene3 (EBI3)] [76]. In the early stage of atherosclerosis, the IL‐12 cytokine family plays an important role in driving DC‐mediated differentiation of naive T cells [76].
IL‐12
Predominantly expressed by macrophages and monocytes, IL‐12 is also produced by DCs [6]. Recognized as a major regulator of adaptive type 1 cell‐mediated immunity, this phagocyte‐produced cytokine binds to naive CD4+ T cells and promotes the generation of proinflammatory Th1 cells, but antagonizes Th2 responses to have a negative impact upon Treg cells [76]. Significant expression of the IL‐12 transcript and protein has already been identified in the atherosclerotic plaques of human samples [76]. IL‐12 was proved to be a proatherogenic molecule in ApoE−/−/IL‐12−/− mouse models due to the smaller size of atherosclerotic lesions in the absence of this cytokine than in its presence [77]. Moreover, the artificial introduction of IL‐12 by transferring recombinant protein promoted the development of atherosclerosis [77]. Mechanistically, IL‐12 production by DCs works through driving Th1 differentiation, as DCs from IL‐12−/−mice fail to induce Th1 responses: a confirmed pathogenic immune response in atherosclerosis. In addition, IL‐12 was also reported to attract T cell recruitment into the atherosclerotic lesions to accelerate the disease severity, as treatment of IL‐12 resulted in unstable plaque and recurrence of acute coronary events [6]. Consistent with this, vaccination to inhibit the functional endogenous IL‐12 led to a significant mitigation in the atherogenesis of Ldlr−/− mice, [6] reinforcing the pro‐atherogenic role of Th1 polarizing cytokine.
IL‐23
As a newly discovered cytokine of the IL‐12 family, IL‐23 is mainly secreted by activated DCs and macrophages [78]. IL‐23 activates innate and adaptive immune responses through the IL‐23 receptor (IL‐23R) and its shared receptor IL‐12Rb1. The most prominent role of IL‐23 is to regulate the activity of T cells, as IL‐23R is mainly expressed on the surface of T cells [79]. Evidence shows that IL‐23 is essential for the secretion of IL‐17 family cytokines by Th17 T cells and regulates the expression of IFN‐γ and IL‐22 by T cells [78]. Izcue and others have demonstrated that endogenous IL‐23 inhibits the expression of forkhead box protein 3 (FoxP3), which is a key transcription factor in the Treg‐mediated maintenance of immune homeostasis [80]. Thus, L‐23 induces and aggravates the T effector (Teff)/Treg imbalance as well as up‐regulating proinflammatory cytokines and other inflammatory factors to enhance inflammatory responses in atherosclerosis. Interestingly, IL‐23R is expressed not only on T cells but also on DCs, which can enhance its antigen‐presenting ability through the autocrine for lesion infiltration, promote its secretion of a large number of inflammatory factors and facilitate its transformation into inflammatory DCs in a self‐feedback loop [79]. In addition, as a downstream molecule of GM‐CSF, IL‐23 plays an important role in GM‐CSF‐promoted atherosclerosis [79]. The GM‐CSF/IL‐23 axis can promote the rupture of atherosclerotic plaques by enhancing the apoptosis of DCs [81]. Multiple reports have revealed elevated levels of IL‐23 in patients and animals with atherosclerosis, suggesting that IL‐23 may have proinflammatory and proatherogenic effects [79]. However, Koltsova and others have also identified an alternative role for the cytokine IL‐23 in the development of atherosclerosis: IL‐23 may actually act as an immunoregulatory agent in the presence of hypercholesterolaemia, thereby preventing atherosclerosis [77]. Although several animal and clinical studies have shown that IL‐23 is involved in the process of atherosclerosis, research into IL‐23 in the field of atherosclerosis has just begun, its precise effects on and mechanisms underlying the pathogenesis remain unclear.
IL‐27
The major sources of another IL‐12 family cytokine, IL‐27, are from DCs, monocytes and macrophages, [82] which were found to have conflicting outcomes of either pro‐, anti‐inflammatory or immunoregulatory functions in atherosclerosis [83]. CD40–CD40L interactions between DCs and T cells increase the secretion of IL‐27, IL‐23 and IL‐12 [76]. Moreover, DCs incubated with oxLDL can also produce IL‐27. Conversely, incubation of DCs with IL‐27 before lipolysaccharide (LPS) stimulation diminished the expression of CD40, CD86 and MCH‐II compared with DC‐stimulated with LPS alone [82].
The DC‐derived IL‐27 plays a critical role in controlling the differentiation of several T cell subsets in atherosclerosis, [83] because it can not only drive Th1 development but also inhibit Th2 responses, depending on the microenvironment [6]. Circulating protein levels of IL‐27 in serum were remarkably enhanced in patients with carotid atherosclerotic disease with gene expression of IL‐27, and IL‐27R also significantly increased in plaques compared to health controls [84]. IL‐27R limits atherosclerosis in Ldlr−/− mice, which acts to halt the production of proinflammatory cytokines/chemokines and arrest the recruitment of inflammatory myeloid cells into atherosclerotic aortas [17]. Similarly, Apoe−/− IL27ra−/− mice fed with ‘western diet’ for 7 or 18 weeks developed significantly more severe atherosclerosis than Apoe−/−IL27ra+/+ controls, and further study revealed that the accelerated disease was driven by elevated expression of adhesion molecules and chemokines that led to more immune cell accumulation, supporting the notion that IL‐27R signalling controls myeloid cell recruitment and possibly endothelial cells activation during the development of atherosclerosis [85]. This may account for the anti‐atherogenic role of IL‐27 receptor signalling in suppressing the production of proinflammatory cytokines/chemokines [6].
IL‐35
Human tolerogenic DCs produce IL‐35, a heterodimer composed of p35 and Ebi3 subunits, in the absence of other IL‐12 family members [86]. CD8α+ DCs in mice secrete the tolerogenic cytokine IL‐35 at steady states [87] which, in return, induces a tolerogenic phenotype on CD8α+ DCs in terms of up‐regulation of CD11b but down‐regulation of MHC‐II, demonstrating a diminished potential for co‐stimulation but increased production of the immunomodulatory molecule IL‐10 [87].
IL‐35 plays important roles in controlling not only adaptive but also innate immune responses in atherosclerosis [78]. Addition of exogenous IL‐35 in vitro can not only inhibit monocyte‐derived DC (moDC) differentiation and maturation induced by LPS, but also suppresses the moDC‐mediated proliferation of allogeneic T cells, and the differentiation of naive CD4+T cells towards the Th1 phenotype [78]. These results support the conception that IL‐35 is an inhibitory regulator in moDC activation and functions.
Increasing evidence suggests that IL‐35 can serve as a promising target for future anti‐atherosclerotic therapy from its anti‐atherosclerotic properties. First, the immunosuppressive nature of this cytokine might be beneficial against vascular inflammation. In addition, IL‐35 suppresses the differentiation of various T cell subsets, including proinflammatory Th1 and Th17 cells as well as inflammatory moDCs. Thirdly, IL‐35 not only supports the proliferation of conventional Tregs, but also promotes the differentiation of a new subset of Tregs called inducible IL‐35‐producing Tregs (iTr35 cells). Moreover, this cytokine up‐regulates anti‐inflammatory cytokines such as IL‐10 but down‐regulates proinflammatory cytokine such as IL‐17 production. Lastly, IL‐35 can be stimulated by simple compounds such as chemical chaperones, which may help to develop new efficient strategies for the treatment of atherosclerosis [88].
IL‐1 family (IL‐1/37/33)
IL‐1 family members consist of 11 cytokines that constitute complex proinflammatory cytokine network. Of these, IL‐1α and IL‐1β were extensively studied due to their early discovery and strong proinflammatory natures [89]. Innate immune cells such as DCs, macrophages and monocytes produce large amounts of IL‐1 to combat inflammation [90]. First identified as the transferable sterile factor with pyrogen function, IL‐1β was recognized as the mediator of many processes of pathological conditions [91]. During atherosclerosis, many factors such as proinflammatory cytokine, [92] oxidative stress [93] and hypoxia [94] can induce IL‐1β synthesis that finally elicits a strong inflammatory response within the vessel walls and accelerates oxidative stress and cell death. Pro‐atherogenic functions of IL‐1β have been extensively investigated and confirmed in animal experiments: its deficiency or inhibition blunts plaque growth in atherosclerotic‐prone Apoe‐deficient mice, [95] whereas the repeated injection of the recombinant cytokine in the perivascular space increased intima‐media thickness in pigs [96]. To target IL‐1β for effective prevention of cardiovascular disease, canakinumab, a fully human monoclonal antibody specifically inhibiting the function of IL‐1β, was developed in a clinical trial in which 10 061 patients with stable coronary artery disease and high sensitivity C‐reactive protein levels >2 mg/l under optimal CV medical treatment have been randomized to receive either placebo or the monoclonal anti‐IL‐1β antibody. Canakinumab was administered subcutaneously every 3 months at dosages of 50, 150 or 300 mg in three different groups of patients who have been followed‐up for a median period of 3.7 years [97]. While the lowest dose did not show efficacy compared to placebo, the two groups receiving canakinumab at a higher dosage of 150 mg every 3 months led to a significantly lower rate of recurrent cardiovascular events than placebo without reducing the LDL cholesterol levels, indicating the clinical importance of blocking this proinflammatory cytokine in the secondary prevention of cardiovascular disease [97, 98].
Both IL‐37 and IL‐33 belong to IL‐1 family cytokines but with different functions, in that IL‐37 plays anti‐inflammatory but IL‐33 plays conflicting roles in atherosclerosis. Monocytes and DCs are the primary producer of IL‐37 in human immune cells [99]. This IL‐1 family member cytokine is one of few anti‐inflammatory cytokines that can fight against a broad spectrum of proinflammatory assaults. While monocytes and moDCs secrete IL‐37 following LPS stimulation, only moDCs release IL‐37 at steady‐state [99]. Type I IFN‐stimulated pDCs were also the source of the IL‐33, [100] which potently inhibits the uptake of oxLDL to form foam cells.
Because IL‐37‐producing DCs are tolerogenic, the negative cytokine they produced suppresses T cells and also has a negative feedback influence on DCs themselves [101]. On one hand, IL‐37‐expressing DCs demonstrated a diminished ability to stimulate naive T cells but better potential to induce Tregs [102]. On the other hand, the expression of IL‐37 in DCs negatively regulates the maturation as well as the functions of DC, so that semi‐mature tolerogenic DCs are generated to impair the activation of effector T cells and favour the development of Tregs [102]. These IL‐37‐conditioned tolerogenic DCs are associated with the formation of atherosclerosis, as transgenic mice expressing human IL‐37 (IL‐37‐tg) mice have diminished antigen‐specific responses and DCs from these mice exhibit suppressed immunogenicity [103]. Collectively, these experiments demonstrated protective anti‐inflammatory effects of human IL‐37 in various inflammatory mouse models [104]. Moreover, a histopathological examination indicated that IL‐37 was highly expressed in human atherosclerotic plaques, indicating a possible correlation of this IL‐1 family cytokine with the disease, but in an anti‐atherogenic fashion. Furthermore, exogenous IL‐37 mitigates atherosclerosis via Treg induction, [78] suggesting that IL‐37 could serve as a novel therapeutic approach for the prevention and treatment of atherosclerotic diseases.
Belonging to the same IL‐1 family member, IL‐33 regulates Th2 cytokines such as IL‐4, IL‐5 and IL‐13 produced from Th2 T cells, type 2 innate lymphocytes and eosinophils. Mouse bone marrow‐derived DCs (BMDCs) exhibited an enhanced transcription of IL‐33 mRNA upon stimulation with Porphyromonas gingivalis whole cells, whereas prostaglandin E2 (PGE2) dramatically promoted the translation of IL‐33 protein by DCs upon LPS stimulation [105]. IL‐33 was initially found to reduce the onset of atherosclerosis through the induction of IL‐5 and ox‐LDL antibodies [54]. Further study, however, reveals that the IL‐33/ST2 axis can be both pro‐ and anti‐inflammatory under different environmental conditions [106]. IL‐33 and its receptor ST2 play a favourable role during the development of atherosclerosis via promoting a shift of Th1 to Th2 immunity in the established Apoe−/− mouse model of atherosclerosis [106]. Collectively, it can be concluded that IL‐33 is a multi‐functional mediator with double‐edged sword activity, depending on environmental conditions [106].
IL‐6
Many leucocytes, including DCs that produce IL‐6, also express the receptor for this cytokine, whose secretion is limited by self‐signalling in the endosomes of cells, and initially thought to promote activation and maturation of DCs [107]. As a heterodimer composed of IL‐6R and gp130, IL‐6 receptor ligation activates signal transducer and activator of transcription (STAT)‐1 and STAT‐3 transcription factors [108]. Interestingly, IL‐6 knock‐out mice had increased numbers of mature DCs, indicating that IL‐6 plays a major role in blocking DC maturation to maintain the immature state of DCs in vivo [109]. In addition, IL‐6 induces the differentiation of Th17 cells but suppresses the development of Tregs [107].
IL‐6 can play either a pro‐ or anti‐inflammatory role in atherosclerosis depending on the stage of disease [77]. Earlier studies found that administration of recombinant IL‐6 increased the area of atherosclerotic lesions by twofold in Apoe−/− mice, indicating the proinflammatory role of this cytokine [110]. Meanwhile, old Apoe−/− mice in the absence of the IL‐6 gene (IL6−/−Apoe−/−) demonstrated accelerated plaque formation associated with reduced collagen contents, diminished IL‐10 production and decreased accumulation of inflammatory cells in the lesions, suggesting an anti‐inflammatory nature of IL‐6 [111]. In another study, however, younger IL6−/−Apoe−/− mice displayed no such differences when compared to the control group [77]. In general, various researches have highlighted that IL‐6 is an instructive cytokine that plays a central role in promoting the downstream inflammatory response involved in atherosclerosis [112]. Therefore, elevated IL‐6 levels increase both total and cardiovascular mortality over a 5‐year period regardless of the traditional risk factors for atherosclerosis. At cellular levels, IL‐6 mainly contributes to controlling the influx of inflammatory cells in the development of an atherosclerotic lesion [112].
Mechanistically, at molecular levels, recent studies have demonstrated that the receptors for IL‐6 signalling exist not only on the cell membrane as a classical cell surface receptor, but also in cell interstitial as a soluble form (sIL‐6R). Therefore, the different activities of IL‐6 could be achieved through different receptor interactions. The inflammatory response of this cytokine was found to mediate via a process called trans‐signalling in which the IL‐6/sIL‐6R complex reacts directly with gp130 on the surface of almost all cells in an organism [108]. The tissue regeneration and anti‐inflammatory activity of IL‐6, however, are accomplished by the classical IL‐6R signalling pathway, because the introduction of soluble gp130 (sgp130) that specifically blocks the IL‐6/sIL‐6R complex but does not influence the classical IL‐6R‐dependent signalling pathway greatly eases the development of atherosclerosis in Ldlr−/− mice [113]. Collectively, these data indicate that the double‐edged natures of IL‐6 are mediated through two different mechanisms.
IL‐10
IL‐10 is a prototypical anti‐inflammatory cytokine made primarily by the macrophages and Treg. However, DCs and moDCs were also reported to produce this regulatory cytokine [114, 115]. At cellular levels, IL‐10 plays an important role in modulating both innate and adaptive immunity through inhibiting macrophages and Th1 cells but activating B cells for antibody secretion [111].
During the progression of atherosclerosis, the major roles of IL‐10 involve inhibiting the activation of macrophage and matrix metalloproteinase and suppressing the expression of proinflammatory cytokines and cyclo‐oxygenase‐2 in the lipid‐containing foam cells transformed from macrophages, as animal studies have demonstrated that genetic deletion of IL‐10 promotes the development of atherosclerosis by increasing infiltration of inflammatory cells and production of proinflammatory cytokines in lesions [113, 116]. Thus, IL‐10 is an essential regulatory cytokine that effectively counteracts inflammation during the development of atherosclerosis. Chronically, IL‐10 exercises its atheroprotective effects by influencing the local inflammatory process within the atherosclerotic lesion at every stage of atherogenesis. Therefore, predominantly produced within the atherosclerotic plaque by DCs and macrophages, IL‐10 could play a critical role in the modulation of the regional inflammatory reaction on infiltrated innate and adaptive immune cells [117].
Chemokines (CCL‐17/22/19/21)
During the development of atherosclerosis, chemokines play a critical role in attracting monocytes and other immune cells from circulation to the site of lesions where oxLDL are deposited [20]. The chemokines CCL17 and CCL22 are secreted by DCs and macrophages. In IFN‐γR knock‐out mice, CCL17 expression in most splenic DCs is strongly induced as a concerted action of GM‐CSF and IL‐4 [118]. As a ligand of CCR4, the DC‐derived chemokine CCL17 attracts leucocytes and further activates platelets, [23] which accelerates the accumulation of various haematopoietic cells in the vascular walls [119] and facilitates the development of neointima, atherosclerotic plaque and thrombosis [119]. The production of CCL17 from DCs limits Treg numbers by controlling their turnover and promotes atherosclerosis in a T cell‐dependent mechanism. Conversely, a blocking antibody specific for CCL17 enhances Treg expansion and suppresses atherosclerosis progression [23]. Collectively, these data imply that the CCL17 from DCs in the lesions could partly constitute the homeostatic mechanisms of regional inflammation. In other words, the CCL17 in atherosclerotic plaques may also constrain Treg maintenance in a local vascular region to propagate inflammation [2]. Consistent with these assumptions, CCL17, together with high numbers of myeloid DCs, were detected in patients with advanced plaques [2].
CCL17 and CCL22 were previously demonstrated to be capable of activating the chemokine receptor CCR4 to attract not only Th1 and Th2 effector cells but also Tregs [2]. CCL17 compared these CD4+ T cells to an inflammatory air pouch, because CCR4 and CCR8 on the surface of Tregs could interact with CCL22 and CCL17 secreted by DCs, fostering their interaction of Tregs with other T effector cells in the diseased locus [18]. CCL22 induction on tolerogenic DCs leads to the accumulation of Tregs at inflammatory sites, including the atherosclerotic lesion [18]. Serum CCL17 levels are linked to coronary artery disease (CAD) and atherosclerosis severity, regardless of traditional cardiovascular risk factors [83].
With the support of human vulnerable atherosclerotic plaques, CD83+ DCs secrete other CC‐motif chemokines such as CCL19 and CCL21 to accelerate the circulation of naive lymphocytes into atherosclerotic vessels [18]. Similar to CCL17/CCL22, CCL19/CCL21 also plays a critical role during the development of atherosclerosis. The binding receptor for CCL19/CCL21 is CCR7, and mature DCs in the lymphoid organs are potent producers of CCL19 and CCL21 to attract CCR7‐expressing immune cells such as naive and central memory T cells [120, 121]. Interestingly, elevated expression of these homeostatic chemokines CCL19 and CCL21 have increasingly been observed in the plaque regions of clinical and experimental atherosclerosis from both humans and mice to recruit T cells, macrophages [122] and monocytes [123] for vascular inflammation. Interestingly, activated and fully mature human DCs in unstable atherosclerotic plaques were found to produce CCL19 and CCL21 and located in close contact with T cells expressing the activation marker CD40 ligand [124]. Therefore, such plaque‐residing DCs can regulate the T cell traffic via CCL19 and CCL21 as an effective chemoattractant into the atherosclerotic lesion areas and provide optimal T cell priming in situ, as conventional DCs in the organized lymphoid organs do to elicit traditional immune responses.
CONCLUDING REMARKS
DCs distinguish themselves among immune cells in that they link the innate arm of the immune system to that of adaptive immunity and are involved in both immune activation and immunotolerance. The reports of DCs in the murine aorta that were later confirmed in humans have aroused great interest in the role of these cells in the pathogenesis of several inflammatory vascular diseases, including atherosclerosis. Division of labour among different DC subsets certainly exists in the development of atherosclerosis, which is mediated either by direct contact with antigens and target cells or indirectly via secreting soluble factors, especially cytokines. Different soluble factors act on atherosclerosis in different ways (Table 1).
TABLE 1.
Roles of soluble factors in the pathogenesis of atherosclerosis
| Soluble factors | Producers | Target cells | Roles in atherosclerosis | References |
|---|---|---|---|---|
| IL−6 | Macrophages, endothelial cells | Macrophages, Th1 cells | Pro‐atherogenic: promotes the formation of fatty streaks | [107, 125] |
| Anti‐atherogenic: induces IL−1RA and releases soluble TNF‐α that neutralizes proinflammatory molecules | ||||
| IL−10 | Treg cells, myeloid cells | Th1 cells, macrophages, B cells | Anti‐atherogenic: inhibits Th1 cells and macrophages and facilitates B cell survival and their antibody secretion | [126] |
| IL−12 | Macrophages, dendritic cells | Th1 cells, myeloid cells | Pro‐atherogenic: modulates early Th1 differentiation | [76, 127] |
| IL−17 | Th17 cells, γδ T cells, ILCs | Macrophages, neutrophils, T cells | Pro‐atherogenic: advocates the recruitment of monocytes and neutrophils to the intima; modulates t VCAM−1 expression; stimulates proinflammatory cytokines/chemokines (IL−6, TNF‐α and CCL5) secretion | [128, 129] |
| Anti‐atherogenic: presumably enhances IL−5, but inhibits IFN‐γ production | ||||
| IL−23 | Macrophages, dendritic cells | Th17 cells, γδ T cells, ILCs | Probably anti‐atherogenic | [6] |
| IL−27 | Macrophages, dendritic cells | Endothelial cells, all haematopoietic cells | Anti‐atherogenic: inhibits CD4+ T cell activation; suppresses oxidized lipid deposition in macrophages | [130, 131] |
| IL−35 | Treg cells, B cells | Treg cells, Th2 cells, monocytes, endothelial cells, SMCs | Anti‐atherogenic: modulates the expression of the anti‐inflammatory molecule; promotes the differentiation of Tregs; suppresses CD4+ effector T cell responses; inhibits the expression of VCAM−1 | [132, 133] |
| IL−33 | Macrophages, endothelial cells, dendritic cells, epithelial cells, fibroblasts | Th2 cells, B cells, macrophage, ILCs 2 | Anti‐atherogenic: stimulates Th2 cytokines production; inhibits IFN‐γ production; promotes antibody production | [115, 134] |
| CCL−17/22 | Macrophages, dendritic cells | Th1 cells, Th2 cells, Treg cells | Pro‐atherogenic/anti‐atherogenic: interact with the chemokine receptor CCR4 to recruit Th1, Th2 and Treg cells for the dual immunological effects | [18, 83] |
IL = interleukin; CCL5 = chemokine ligand 5; CCR4 = C‐C chemokine receptor type 4; Th1 = T helper type 1; Treg = regulatory T cell; TNF = tumor necrosis factor; VCAM = vascular cell adhesion molecule 1; SMC = smooth muscle cells; ILC = innate lymphoid cells.
A lucid understanding of the direct roles of DCs in atherogenesis is critical in targeting specific cell populations or pathogenic antigens for effective therapeutic approaches. To utilize the medicinal potential of soluble factors derived from DCs, more studies are needed to focus upon the identification of critical mediators in the signalling pathways of the DC‐derived cytokines and chemokines associated with inflammatory cardiovascular disease. Once comprehensive knowledge concerning the detailed roles of DCs in atherogenesis has been obtained, synthetic substitutes or mimetic drugs will be developed as novel therapeutic agents to treat chronic inflammatory atherosclerosis as well as its fetal complications.
CONFLICTS OF INTEREST
The authors declare no financial or commercial competing interests.
AUTHOR CONTRIBUTIONS
Yanfang Zhao performed the literature review, wrote the first draft of the manuscript. Jing Zhang performed the literature review, wrote the revised draft of the manuscript. Wenjie Zhang prepared the figures. Yuekang Xu critically reviewed and wrote the manuscript. All authors approved the final version of the manuscript.
ACKNOWLEDGEMENTS
This work was financially supported by the National Natural Science Foundation of China Major Research Plan Project (91742101); Natural Science Foundation of Anhui Province, China (1608085MH160); Anhui International Science and Technology Collaborative Project, China (1604b0602017); Anhui Provincial Key Laboratory of Molecular Enzymology and Mechanism of Major Diseases, and Key Laboratory of Biomedicine in Gene Diseases and Health of Anhui Higher Education Institutes. Yuekang Xu was supported by the National Natural Science Foundation of China Major Research Plan Project (91742101); Natural Science Foundation of Anhui Province, China (1608085MH160); Anhui International Science and Technology Collaborative Project, China (1604b0602017); Anhui Provincial Key Laboratory of Molecular Enzymology and Mechanism of Major Diseases; and Key Laboratory of Biomedicine in Gene Diseases and Health of Anhui Higher Education Institutes.
Zhao Y, Zhang J, Zhang W, Xu Y. A myriad of roles of dendritic cells in atherosclerosis. Clin Exp Immunol. 2021;206:12–27. 10.1111/cei.13634
Yanfang Zhao and Jing Zhang contributed equally to this work.
DATA AVAILABILITY STATEMENT
Not applicable.
REFERENCES
- 1.Ramji DP, Davies TS. Cytokines in atherosclerosis: key players in all stages of disease and promising therapeutic targets. Cytokine Growth Factor Rev. 2015;26:673–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Busch M, Westhofen TC, Koch M, Lutz MB, Zernecke A. Dendritic cell subset distributions in the aorta in healthy and atherosclerotic mice. PLoS One. 2014;9:e88452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weber C, Zernecke A, Libby P. The multifaceted contributions of leukocyte subsets to atherosclerosis: lessons from mouse models. Nat Rev Immunol. 2008;8:802–15. [DOI] [PubMed] [Google Scholar]
- 4.Niessner A, Weyand CM. Dendritic cells in atherosclerotic disease. Clin Immunol. 2010;134:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu D, Mohanta SK, Yin C, Peng L, Ma Z, Srikakulapu P, et al. Artery tertiary lymphoid organs control aorta immunity and protect against atherosclerosis via vascular smooth muscle cell lymphotoxin beta receptors. Immunity. 2015;42:1100–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abbas A, Gregersen I, Holm S, Daissormont I, Bjerkeli V, Krohg‐Sørensen K, et al. Interleukin 23 levels are increased in carotid atherosclerosis: possible role for the interleukin 23/interleukin 17 axis. Stroke 2015;46:793–9. [DOI] [PubMed] [Google Scholar]
- 7.Bobryshev YV. Dendritic cells in atherosclerosis: current status of the problem and clinical relevance. Eur Heart J. 2005;26:1700–4. [DOI] [PubMed] [Google Scholar]
- 8.Cheong C, Choi JH. Dendritic cells and regulatory T cells in atherosclerosis. Mol Cells. 2012;34:341–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alberts‐Grill N, Denning TL, Rezvan A, Jo H. The role of the vascular dendritic cell network in atherosclerosis. Am J Physiol Cell Physiol. 2013;305:C1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi J‐H, Do Y, Cheong C, Koh H, Boscardin SB, Oh Y‐S, et al. Identification of antigen‐presenting dendritic cells in mouse aorta and cardiac valves. J Exp Med. 2009;206:497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paulson KE, Zhu SN, Chen M, Nurmohamed S, Jongstra‐Bilen J, Cybulsky MI. Resident intimal dendritic cells accumulate lipid and contribute to the initiation of atherosclerosis. Circ Res. 2010;106:383–90. [DOI] [PubMed] [Google Scholar]
- 12.Libby P. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:2045–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rombouts M, Ammi R, Van Brussel I, Roth L, De Winter BY, Vercauteren SR, et al. Linking CD11b (+) dendritic cells and natural killer T cells to plaque inflammation in atherosclerosis. Mediators Inflamm. 2016;2016:6467375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Llodra J, Angeli V, Liu J, Trogan E, Fisher EA, Randolph GJ. Emigration of monocyte‐derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc Natl Acad Sci USA. 2004;101:11779–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fang Z, Deng Q, Hu H, Wang X, Sun X, Ge X, et al. Characteristics of immunogenic and tolerogenic dendritic cells within the arterial wall in atherosclerosis and in vitro. Int J Clin Exp Med. 2014;7:4846–56. [PMC free article] [PubMed] [Google Scholar]
- 16.Choi J‐H, Cheong C, Dandamudi D, Park C, Rodriguez A, Mehandru S, et al. Flt3 signaling‐dependent dendritic cells protect against atherosclerosis. Immunity 2011;35:819–31. [DOI] [PubMed] [Google Scholar]
- 17.Koltsova EK, Ley K. How dendritic cells shape atherosclerosis. Trends Immunol. 2011;32:540–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chistiakov DA, Orekhov AN, Sobenin IA, Bobryshev YV. Plasmacytoid dendritic cells: development, functions, and role in atherosclerotic inflammation. Front Physiol. 2014;5:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takenaka MC, Quintana FJ. Tolerogenic dendritic cells. Semin Immunopathol. 2017;39:113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moss JW, Ramji DP. Cytokines: roles in atherosclerosis disease progression and potential therapeutic targets. Future Med Chem. 2016;8:1317–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goossens P, Gijbels MJJ, Zernecke A, Eijgelaar W, Vergouwe MN, van der Made I, et al. Myeloid type I interferon signaling promotes atherosclerosis by stimulating macrophage recruitment to lesions. Cell Metab. 2010;12:142–53. [DOI] [PubMed] [Google Scholar]
- 22.Gelderblom M, Gallizioli M, Ludewig P, Thom V, Arunachalam P, Rissiek B, et al. IL‐23 (Interleukin‐23)‐producing conventional dendritic cells control the detrimental IL‐17 (interleukin‐17) response in stroke. Stroke 2018;49:155–64. [DOI] [PubMed] [Google Scholar]
- 23.Weber C, Meiler S, Döring Y, Koch M, Drechsler M, Megens RTA, et al. CCL17‐expressing dendritic cells drive atherosclerosis by restraining regulatory T cell homeostasis in mice. J Clin Invest. 2011;121:2898–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zernecke A, Winkels H, Cochain C, Williams JW, Wolf D, Soehnlein O, et al. Meta‐analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ Res. 2020;127:402–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.MacRitchie N, Grassia G, Noonan J, Cole JE, Hughes CE, Schroeder J, et al. The aorta can act as a site of naive CD4+ T‐cell priming. Cardiovasc Res. 2020;116:306–16. [DOI] [PubMed] [Google Scholar]
- 26.Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir E‐A, Amadori L, et al. Single‐cell immune landscape of human atherosclerotic plaques. Nat Med. 2019;25:1576–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wirka RC, Wagh D, Paik DT, Pjanic M, Nguyen T, Miller CL, et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single‐cell analysis. Nat Med. 2019;25:1280–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cole JE, Park I, Ahern DJ, Kassiteridi C, Danso Abeam D, Goddard ME, et al. Immune cell census in murine atherosclerosis: cytometry by time of flight illuminates vascular myeloid cell diversity. Cardiovasc Res. 2018;114:1360–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winkels H, Ehinger E, Vassallo M, Buscher K, Dinh HQ, Kobiyama K, et al. Atlas of the immune cell repertoire in mouse atherosclerosis defined by single‐cell RNA‐sequencing and mass cytometry. Circ Res. 2018;122:1675–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perrins CJ, Bobryshev YV. Current advances in understanding of immunopathology of atherosclerosis. Virchows Arch. 2011;458:117–23. [DOI] [PubMed] [Google Scholar]
- 31.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–95. [DOI] [PubMed] [Google Scholar]
- 32.Nickel T, Schmauss D, Hanssen H, Sicic Z, Krebs B, Jankl S, et al. oxLDL uptake by dendritic cells induces upregulation of scavenger‐receptors, maturation and differentiation. Atherosclerosis 2009;205:442–50. [DOI] [PubMed] [Google Scholar]
- 33.Shashkin P, Dragulev B, Ley K. Macrophage differentiation to foam cells. Curr Pharm Des. 2005;11:3061–72. [DOI] [PubMed] [Google Scholar]
- 34.Cichon N, Lach D, Dziedzic A, Bijak M, Saluk J. The inflammatory processes in atherogenesis. Pol Merkur Lekarski. 2017;42:125–8. [PubMed] [Google Scholar]
- 35.Ishibashi T, Yokoyama K, Shindo J, Hamazaki Y, Endo Y, Sato T, et al. Potent cholesterol‐lowering effect by human granulocyte‐macrophage colony‐stimulating factor in rabbits. Possible implications of enhancement of macrophage functions and an increase in mRNA for VLDL receptor. Arterioscler Thromb. 1994;14:1534–41. [DOI] [PubMed] [Google Scholar]
- 36.Gautier EL, Huby T, Saint‐Charles F, Ouzilleau B, Pirault J, Deswaerte V, et al. Conventional dendritic cells at the crossroads between immunity and cholesterol homeostasis in atherosclerosis. Circulation 2009;119:2367–75. [DOI] [PubMed] [Google Scholar]
- 37.Marvin J, Rhoads JP, Major AS. FcgammaRIIb on CD11c(+) cells modulates serum cholesterol and triglyceride levels and differentially affects atherosclerosis in male and female Ldlr(–/–) mice. Atherosclerosis 2019;285:108–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hansson GK, Nilsson J. Vaccination against atherosclerosis? Induction of atheroprotective immunity. Semin Immunopathol. 2009;31:95–101. [DOI] [PubMed] [Google Scholar]
- 39.Subramanian M, Tabas I. Dendritic cells in atherosclerosis. Semin Immunopathol. 2014;36:93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yilmaz A, Lochno M, Traeg F, Cicha I, Reiss C, Stumpf C, et al. Emergence of dendritic cells in rupture‐prone regions of vulnerable carotid plaques. Atherosclerosis 2004;176:101–10. [DOI] [PubMed] [Google Scholar]
- 41.Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12:204–12. [DOI] [PubMed] [Google Scholar]
- 42.Liu B, Yu H, Sun G, Sun X, Jin H, Zhang C, et al. OX40 promotes obesity‐induced adipose inflammation and insulin resistance. Cell Mol Life Sci. 2017;74:3827–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grabner R, Lotzer K, Dopping S, Hildner M, Radke D, Beer M, et al. Lymphotoxin beta receptor signaling promotes tertiary lymphoid organogenesis in the aorta adventitia of aged ApoE–/– mice. J Exp Med. 2009;206:233–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nishimura T, Kitamura H, Iwakabe K, Yahata T, Ohta A, Sato M, et al. The interface between innate and acquired immunity: glycolipid antigen presentation by CD1d‐expressing dendritic cells to NKT cells induces the differentiation of antigen‐specific cytotoxic T lymphocytes. Int Immunol. 2000;12:987–94. [DOI] [PubMed] [Google Scholar]
- 45.Kyriakakis E, Cavallari M, Andert J, Philippova M, Koella C, Bochkov V, et al. Invariant natural killer T cells: linking inflammation and neovascularization in human atherosclerosis. Eur J Immunol. 2010;40:3268–79. [DOI] [PubMed] [Google Scholar]
- 46.Nakai Y, Iwabuchi K, Fujii S, Ishimori N, Dashtsoodol N, Watano K, et al. Natural killer T cells accelerate atherogenesis in mice. Blood 2004;104:2051–9. [DOI] [PubMed] [Google Scholar]
- 47.Leslie DS, Dascher CC, Cembrola K, Townes MA, Hava DL, Hugendubler LC, et al. Serum lipids regulate dendritic cell CD1 expression and function. Immunology 2008;125:289–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang DH, Osman K, Connolly J, Kukreja A, Krasovsky J, Pack M, et al. Sustained expansion of NKT cells and antigen‐specific T cells after injection of alpha‐galactosyl‐ceramide loaded mature dendritic cells in cancer patients. J Exp Med. 2005;201:1503–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tupin E, Nicoletti A, Elhage R, Rudling M, Ljunggren H‐G, Hansson GK, et al. CD1d‐dependent activation of NKT cells aggravates atherosclerosis. J Exp Med. 2004;199:417–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saez de Guinoa J, Jimeno R, Gaya M, Kipling D, Garzon MJ, Dunn‐Walters D, et al. CD1d‐mediated lipid presentation by CD11c(+) cells regulates intestinal homeostasis. EMBO J. 2018;37:e97537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bondarenko S, Catapano AL, Norata GD. The CD1d‐natural killer T cell axis in atherosclerosis. J Innate Immun. 2014;6:3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.deCathelineau AM, Henson PM. The final step in programmed cell death: phagocytes carry apoptotic cells to the grave. Essays Biochem. 2003;39:105–17. [DOI] [PubMed] [Google Scholar]
- 53.Ravichandran KS, Lorenz U. Engulfment of apoptotic cells: signals for a good meal. Nat Rev Immunol. 2007;7:964–74. [DOI] [PubMed] [Google Scholar]
- 54.Bories GFP, Leitinger N. Macrophage metabolism in atherosclerosis. FEBS Lett. 2017;591:3042–60. [DOI] [PubMed] [Google Scholar]
- 55.Birge RB, Boeltz S, Kumar S, Carlson J, Wanderley J, Calianese D, et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016;23:962–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rosales C, Uribe‐Querol E. Phagocytosis: a fundamental process in immunity. Biomed Res Int. 2017;2017:9042851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Albert ML, Pearce SF, Francisco LM, Sauter B, Roy P, Silverstein RL, et al. Immature dendritic cells phagocytose apoptotic cells via alphavbeta5 and CD36, and cross‐present antigens to cytotoxic T lymphocytes. J Exp Med. 1998;188:1359–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seitz HM, Camenisch TD, Lemke G, Earp HS, Matsushima GK. Macrophages and dendritic cells use different Axl/Mertk/Tyro3 receptors in clearance of apoptotic cells. J Immunol. 2007;178:5635–42. [DOI] [PubMed] [Google Scholar]
- 59.Miksa M, Amin D, Wu R, Jacob A, Zhou M, Dong W, et al. Maturation‐induced down‐regulation of MFG‐E8 impairs apoptotic cell clearance and enhances endotoxin response. Int J Mol Med. 2008;22:743–8. [PMC free article] [PubMed] [Google Scholar]
- 60.Sancho D, Joffre OP, Keller AM, Rogers NC, Martínez D, Hernanz‐Falcón P, et al. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 2009;458:899–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Haddad Y, Lahoute C, Clément M, Laurans L, Metghalchi S, Zeboudj L, et al. The dendritic cell receptor DNGR‐1 promotes the development of atherosclerosis in mice. Circ Res. 2017;121:234–43. [DOI] [PubMed] [Google Scholar]
- 62.Bar‐On L, Jung S. Defining in vivo dendritic cell functions using CD11c‐DTR transgenic mice. Methods Mol Biol. 2010;595:429–42. [DOI] [PubMed] [Google Scholar]
- 63.Beaty SR, Rose CE Jr, Sung SS. Diverse and potent chemokine production by lung CD11bhigh dendritic cells in homeostasis and in allergic lung inflammation. J Immunol. 2007;178:1882–95. [DOI] [PubMed] [Google Scholar]
- 64.Chen L, Flies DB. Molecular mechanisms of T cell co‐stimulation and co‐inhibition. Nat Rev Immunol. 2013;13:227–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Croft M. The role of TNF superfamily members in T‐cell function and diseases. Nat Rev Immunol. 2009;9:271–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gotsman I, Grabie N, Dacosta R, Sukhova G, Sharpe A, Lichtman AH. Proatherogenic immune responses are regulated by the PD‐1/PD‐L pathway in mice. J Clin Invest. 2007;117:2974–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lameijer M, Binderup T, van Leent MMT, Senders ML, Fay F, Malkus J, et al. Efficacy and safety assessment of a TRAF6‐targeted nanoimmunotherapy in atherosclerotic mice and non‐human primates. Nat Biomed Eng. 2018;2:279–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wolf D, Hohmann J‐D, Wiedemann A, Bledzka K, Blankenbach H, Marchini T, et al. Binding of CD40L to Mac‐1’s I‐domain involves the EQLKKSKTL motif and mediates leukocyte recruitment and atherosclerosis – but does not affect immunity and thrombosis in mice. Circ Res. 2011;109:1269–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Buono C, Pang H, Uchida Y, Libby P, Sharpe AH, Lichtman AH. B7–1/B7‐2 costimulation regulates plaque antigen‐specific T‐cell responses and atherogenesis in low‐density lipoprotein receptor‐deficient mice. Circulation 2004;109:2009–15. [DOI] [PubMed] [Google Scholar]
- 70.Matsumoto T, Sasaki N, Yamashita T, Emoto T, Kasahara K, Mizoguchi T, et al. Overexpression of Cytotoxic T‐lymphocyte‐associated antigen‐4 prevents atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2016;36:1141–51. [DOI] [PubMed] [Google Scholar]
- 71.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Foks AC, van Puijvelde GHM, Bot I, ter Borg MND, Habets KLL, Johnson JL, et al. Interruption of the OX40–OX40 ligand pathway in LDL receptor‐deficient mice causes regression of atherosclerosis. J Immunol. 2013;191:4573–80. [DOI] [PubMed] [Google Scholar]
- 73.Jeon HJ, Choi J‐H, Jung I‐H, Park J‐G, Lee M‐R, Lee M‐N, et al. CD137 (4–1BB) deficiency reduces atherosclerosis in hyperlipidemic mice. Circulation 2010;121:1124–33. [DOI] [PubMed] [Google Scholar]
- 74.Fowler AA, Truwit JD, Hite RD, Morris PE, DeWilde C, Priday A, et al. Effect of vitamin C infusion on organ failure and biomarkers of inflammation and vascular injury in patients with sepsis and severe acute respiratory failure: the CITRIS‐ALI randomized clinical trial. JAMA 2019;322:1261–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Niessner A, Sato K, Chaikof EL, Colmegna I, Goronzy JJ, Weyand CM. Pathogen‐sensing plasmacytoid dendritic cells stimulate cytotoxic T‐cell function in the atherosclerotic plaque through interferon‐alpha. Circulation 2006;114:2482–9. [DOI] [PubMed] [Google Scholar]
- 76.Posadas‐Sanchez R, Vargas‐Alarcon G. Innate immunity in coronary disease. The role of interleukin‐12 cytokine family in atherosclerosis. Rev Invest Clin. 2018;70:5–17. [DOI] [PubMed] [Google Scholar]
- 77.Fatkhullina AR, Peshkova IO, Koltsova EK. The role of cytokines in the development of atherosclerosis. Biochemistry 2016;81:1358–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen XI, Hao S, Zhao Z, Liu J, Shao Q, Wang F, et al. Interleukin 35: inhibitory regulator in monocyte‐derived dendritic cell maturation and activation. Cytokine 2018;108:43–52. [DOI] [PubMed] [Google Scholar]
- 79.Zhang S, Yuan J, Yu M, Fan H, Guo Z‐Q, Yang R, et al. IL‐17A facilitates platelet function through the ERK2 signaling pathway in patients with acute coronary syndrome. PLoS One. 2012;7:e40641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schiering C, Krausgruber T, Chomka A, Fröhlich A, Adelmann K, Wohlfert EA, et al. The alarmin IL‐33 promotes regulatory T‐cell function in the intestine. Nature 2014;513:564–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Doring Y. Not growth but death: GM‐CSF/IL‐23 axis drives atherosclerotic plaque vulnerability by enhancing macrophage and DC apoptosis. Circ Res. 2015;116:222–4. [DOI] [PubMed] [Google Scholar]
- 82.Wang Q, Liu J. Regulation and Immune Function of IL‐27. Adv Exp Med Biol. 2016;941:191–211. [DOI] [PubMed] [Google Scholar]
- 83.Aparicio‐Siegmund S, Garbers C. The biology of interleukin‐27 reveals unique pro‐ and anti‐inflammatory functions in immunity. Cytokine Growth Factor Rev. 2015;26:579–86. [DOI] [PubMed] [Google Scholar]
- 84.Gregersen I, Sandanger Ø, Askevold ET, Sagen EL, Yang K, Holm S, et al. Interleukin 27 is increased in carotid atherosclerosis and promotes NLRP3 inflammasome activation. PLoS One. 2017;12:e0188387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Peshkova IO, Fatkhullina AR, Mikulski Z, Ley K, Koltsova EK. IL‐27R signaling controls myeloid cells accumulation and antigen‐presentation in atherosclerosis. Sci Rep. 2017;7:2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dixon KO, van der Kooij SW , Vignali DA, van Kooten C . Human tolerogenic dendritic cells produce IL‐35 in the absence of other IL‐12 family members. Eur J Immunol. 2015;45:1736–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Haller S, Duval A, Migliorini R, Stevanin M, Mack V, Acha‐Orbea H. Interleukin‐35‐producing CD8alpha(+) dendritic cells acquire a tolerogenic state and regulate T cell function. Front Immunol. 2017;8:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sobenin IA, Andrianova IV, Lakunin KY, Karagodin VP, Bobryshev YV, Orekhov AN. Anti‐atherosclerotic effects of garlic preparation in freeze injury model of atherosclerosis in cholesterol‐fed rabbits. Phytomedicine 2016;23:1235–9. [DOI] [PubMed] [Google Scholar]
- 89.Dinarello CA. Interleukin‐1 in the pathogenesis and treatment of inflammatory diseases. Blood 2011;117:3720–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Contassot E, Beer HD, French LE. Interleukin‐1, inflammasomes, autoinflammation and the skin. Swiss Med Wkly. 2012;142:w13590. [DOI] [PubMed] [Google Scholar]
- 91.Libby P. Interleukin‐1 beta as a target for atherosclerosis therapy: biological basis of CANTOS and beyond. J Am Coll Cardiol. 2017;70:2278–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.McGeough MD, Wree A, Inzaugarat ME, Haimovich A, Johnson CD, Peña CA, et al. TNF regulates transcription of NLRP3 inflammasome components and inflammatory molecules in cryopyrinopathies. J Clin Invest. 2017;127:4488–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lavieri R, Rubartelli A, Carta S. Redox stress unbalances the inflammatory cytokine network: role in autoinflammatory patients and healthy subjects. J Leukoc Biol. 2016;99:79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Folco EJ, Sukhova GK, Quillard T, Libby P. Moderate hypoxia potentiates interleukin‐1beta production in activated human macrophages. Circ Res. 2014;115:875–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kirii H, Niwa T, Yamada Y, Wada H, Saito K, Iwakura Y, et al. Lack of interleukin‐1beta decreases the severity of atherosclerosis in ApoE‐deficient mice. Arterioscler Thromb Vasc Biol. 2003;23:656–60. [DOI] [PubMed] [Google Scholar]
- 96.Shimokawa H, Ito A, Fukumoto Y, Kadokami T, Nakaike R, Sakata M, et al. Chronic treatment with interleukin‐1 beta induces coronary intimal lesions and vasospastic responses in pigs in vivo. The role of platelet‐derived growth factor. J Clin Invest. 1996;97:769–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–31. [DOI] [PubMed] [Google Scholar]
- 98.Liberale L, Montecucco F, Schwarz L, Luscher TF, Camici GG. Inflammation and cardiovascular diseases: lessons from seminal clinical trials. Cardiovasc Res. 2021;117:411–22. [DOI] [PubMed] [Google Scholar]
- 99.Rudloff I, Cho SX, Lao JC, Ngo D, McKenzie M, Nold‐Petry CA, et al. Monocytes and dendritic cells are the primary sources of interleukin 37 in human immune cells. J Leukoc Biol. 2017;101:901–11. [DOI] [PubMed] [Google Scholar]
- 100.Kamari Y, Werman‐Venkert R, Shaish A, Werman A, Harari A, Gonen A, et al. Differential role and tissue specificity of interleukin‐1alpha gene expression in atherogenesis and lipid metabolism. Atherosclerosis 2007;195:31–8. [DOI] [PubMed] [Google Scholar]
- 101.Clement M, Guedj K, Andreata F, Morvan M, Bey L, Khallou‐Laschet J, et al. Control of the T follicular helper‐germinal center B‐cell axis by CD8(+) regulatory T cells limits atherosclerosis and tertiary lymphoid organ development. Circulation 2015;131:560–70. [DOI] [PubMed] [Google Scholar]
- 102.Chen HM, Fujita M. IL‐37: a new player in immune tolerance. Cytokine 2015;72:113–4. [DOI] [PubMed] [Google Scholar]
- 103.Dinarello CA, Nold‐Petry C, Nold M, Fujita M, Li S, Kim S, et al. Suppression of innate inflammation and immunity by interleukin‐37. Eur J Immunol. 2016;46:1067–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Palomo J, Dietrich D, Martin P, Palmer G, Gabay C. The interleukin (IL)‐1 cytokine family – balance between agonists and antagonists in inflammatory diseases. Cytokine 2015;76:25–37. [DOI] [PubMed] [Google Scholar]
- 105.Bar‐On L, Jung S. Defining dendritic cells by conditional and constitutive cell ablation. Immunol Rev. 2010;234:76–89. [DOI] [PubMed] [Google Scholar]
- 106.Kunes P, Mandak J, Holubcova Z, Kolackova M, Krejsek J. Actual position of interleukin(IL)‐33 in atherosclerosis and heart failure: great expectations or en attendant Godot? Perfusion 2015;30:356–74. [DOI] [PubMed] [Google Scholar]
- 107.Verboogen DRJ, Revelo NH, Ter Beest M, van den Bogaart G . Interleukin‐6 secretion is limited by self‐signaling in endosomes. J Mol Cell Biol. 2019;11:144–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Garbers C, Hermanns HM, Schaper F, Müller‐Newen G, Grötzinger J, Rose‐John S, et al. Plasticity and cross‐talk of interleukin 6‐type cytokines. Cytokine Growth Factor Rev. 2012;23:85–97. [DOI] [PubMed] [Google Scholar]
- 109.Park S‐J, Nakagawa T, Kitamura H, Atsumi T, Kamon H, Sawa S‐I, et al. IL‐6 regulates in vivo dendritic cell differentiation through STAT3 activation. J Immunol. 2004;173:3844–54. [DOI] [PubMed] [Google Scholar]
- 110.Huber SA, Sakkinen P, Conze D, Hardin N, Tracy R. Interleukin‐6 exacerbates early atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 1999;19:2364–7. [DOI] [PubMed] [Google Scholar]
- 111.Schieffer B, Selle T, Hilfiker A, Hilfiker‐Kleiner D, Grote K, Tietge UJF, et al. Impact of interleukin‐6 on plaque development and morphology in experimental atherosclerosis. Circulation 2004;110:3493–500. [DOI] [PubMed] [Google Scholar]
- 112.Hartman J, Frishman WH. Inflammation and atherosclerosis: a review of the role of interleukin‐6 in the development of atherosclerosis and the potential for targeted drug therapy. Cardiol Rev. 2014;22:147–51. [DOI] [PubMed] [Google Scholar]
- 113.Schuett H, Oestreich R, Waetzig GH, Annema W, Luchtefeld M, Hillmer A, et al. Transsignaling of interleukin‐6 crucially contributes to atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2012;32:281–90. [DOI] [PubMed] [Google Scholar]
- 114.Serbina NV, Salazar‐Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS‐producing dendritic cells mediate innate immune defense against bacterial infection. Immunity 2003;19:59–70. [DOI] [PubMed] [Google Scholar]
- 115.Miller AM, Xu D, Asquith DL, Denby L, Li Y, Sattar N, et al. IL‐33 reduces the development of atherosclerosis. J Exp Med. 2008;205:339–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Elhage R, Clamens S, Besnard S, Mallat Z, Tedgui A, Arnal J, et al. Involvement of interleukin‐6 in atherosclerosis but not in the prevention of fatty streak formation by 17beta‐estradiol in apolipoprotein E‐deficient mice. Atherosclerosis 2001;156:315–20. [DOI] [PubMed] [Google Scholar]
- 117.Mallat Z, Heymes C, Ohan J, Faggin E, Leseche G, Tedgui A. Expression of interleukin‐10 in advanced human atherosclerotic plaques: relation to inducible nitric oxide synthase expression and cell death. Arterioscler Thromb Vasc Biol. 1999;19:611–6. [DOI] [PubMed] [Google Scholar]
- 118.Globisch T, Steiner N, Fülle L, Lukacs‐Kornek V, Degrandi D, Dresing P, et al. Cytokine‐dependent regulation of dendritic cell differentiation in the splenic microenvironment. Eur J Immunol. 2014;44:500–10. [DOI] [PubMed] [Google Scholar]
- 119.Gleissner CA. Platelet‐derived chemokines in atherogenesis: what’s new? Curr Vasc Pharmacol. 2012;10:563–9. [DOI] [PubMed] [Google Scholar]
- 120.Forster R, Schubel A, Breitfeld D, Kremmer E, Renner‐Muller I, Wolf E, et al. Pillars article: CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell 1999;99: 23–33. J Immunol. 2016;196(1):5‐15. [PubMed] [Google Scholar]
- 121.Rossi D, Zlotnik A. The biology of chemokines and their receptors. Annu Rev Immunol. 2000;18:217–42. [DOI] [PubMed] [Google Scholar]
- 122.Damås JK, Smith C, Øie E, Fevang B, Halvorsen B, Wæhre T, et al. Enhanced expression of the homeostatic chemokines CCL19 and CCL21 in clinical and experimental atherosclerosis: possible pathogenic role in plaque destabilization. Arterioscler Thromb Vasc Biol. 2007;27:614–20. [DOI] [PubMed] [Google Scholar]
- 123.Cai W, Tao J, Zhang X, Tian X, Liu T, Feng X, et al. Contribution of homeostatic chemokines CCL19 and CCL21 and their receptor CCR7 to coronary artery disease. Arterioscler Thromb Vasc Biol. 2014;34:1933–41. [DOI] [PubMed] [Google Scholar]
- 124.Erbel C, Sato K, Meyer FB, Kopecky SL, Frye RL, Goronzy JJ, et al. Functional profile of activated dendritic cells in unstable atherosclerotic plaque. Basic Res Cardiol. 2007;102:123–32. [DOI] [PubMed] [Google Scholar]
- 125.Xing Z, Gauldie J, Cox G, Baumann H, Jordana M, Lei XF, et al. IL‐6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J Clin Invest. 1998;101:311–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Moore KW, de Waal MR , Coffman RL, O’Garra A. Interleukin‐10 and the interleukin‐10 receptor. Annu Rev Immunol. 2001;19:683–765. [DOI] [PubMed] [Google Scholar]
- 127.van der Heijden T , Bot I, Kuiper J. The IL‐12 cytokine family in cardiovascular diseases. Cytokine 2019;122:154188. [DOI] [PubMed] [Google Scholar]
- 128.Smith E, Prasad K‐M, Butcher M, Dobrian A, Kolls JK, Ley K, et al. Blockade of interleukin‐17A results in reduced atherosclerosis in apolipoprotein E‐deficient mice. Circulation 2010;121:1746–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Erbel C, Chen L, Bea F, Wangler S, Celik S, Lasitschka F, et al. Inhibition of IL‐17A attenuates atherosclerotic lesion development in apoE‐deficient mice. J Immunol. 2009;183:8167–75. [DOI] [PubMed] [Google Scholar]
- 130.Yoshida H, Hunter CA. The immunobiology of interleukin‐27. Annu Rev Immunol. 2015;33:417–43. [DOI] [PubMed] [Google Scholar]
- 131.Koltsova EK, Kim G, Lloyd KM, Saris CJ, von Vietinghoff S , Kronenberg M, et al. Interleukin‐27 receptor limits atherosclerosis in Ldlr–/– mice. Circ Res. 2012;111:1274–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Collison LW, Delgoffe GM, Guy CS, Vignali KM, Chaturvedi V, Fairweather DeLisa, et al. The composition and signaling of the IL‐35 receptor are unconventional. Nat Immunol. 2012;13:290–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL‐35 contributes to regulatory T‐cell function. Nature 2007;450:566–9. [DOI] [PubMed] [Google Scholar]
- 134.Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL‐33, an interleukin‐1‐like cytokine that signals via the IL‐1 receptor‐related protein ST2 and induces T helper type 2‐associated cytokines. Immunity 2005;23:479–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.