

Abstract
Hepatocellular carcinoma (HCC) is a typical inflammation‐induced cancer and displays a complex interaction between the tumor microenvironment and tumor development. Immune cells in the HCC microenvironment play both pro‐ and anti‐tumoral roles in HCC progression. An increasing number of findings indicate that metabolic reprogramming is essential for immune cell differentiation and function. In this review, we discuss the metabolic changes of different immune cells and correlate these findings to HCC progression.
Keywords: hepatocellular carcinoma, immune cells, metabolic reprogramming, tumor immunology, tumor microenvironment
Immune cells in the Hepatocellular carcinoma (HCC) microenvironment play both pro‐and anti‐tumoral roles in HCC progression. An increasing number of findings indicate that metabolic reprogramming is essential for immune cell differentiation and function. In this review, we discuss the metabolic changes of different immune cells and correlate these findings to HCC progression.
1. INTRODUCTION
Hepatocellular carcinoma (HCC) is by far the most common primary liver cancer and also the fifth leading cause of cancer‐related mortality worldwide.1, 2 Despite recent findings with targeted and immune therapies, the prognosis of patients at an advanced HCC stage remains poor with limited options for systemic therapy and high recurrence rates after locoregional therapies.3, 4, 5, 6 The tumor microenvironment (TME) consists of an intensive interplay between tumor cells, stroma, and immune cells with all these interactions being critical in tumorigenesis, metastasis, and drug resistance.3, 7 The initiation and progression of HCC are highly related to chronic inflammation resulting in a complex role of immune cells in cancer‐related inflammation and tumor cell immune evasion with HCC progression, angiogenesis, and eventual metastasis.3, 8, 9, 10, 11, 12 In addition, liver diseases can have varying effects on the TME some of which are not fully understood.13, 14, 15
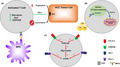
Cellular metabolism and basal metabolic needs are critical to cell growth, development, migration, and tissue‐specific functions.16 Metabolic reprogramming has been shown as a significant sign of cancer progression, not only affecting the survival and proliferation signals within tumor cells themselves but also altering the TME.14, 17, 18, 19, 20, 21 Accumulating evidence indicates that metabolic processes regulate the phenotype, function, and survival of immune cells.14, 17, 21, 22, 23, 24, 25 In this review, we discuss metabolic reprogramming in innate and adaptive immune cells (Figure 1), and their functional effects on HCC progression and metastasis. (Table 1).
FIGURE 1.
Metabolic reprogramming of immune cells in tumor microenvironment. Metabolic alterations in the indicated immune cells (macrophage, neutrophil, NK cells, and T cells) are presented
TABLE 1.
Metabolism and function of immune cells
| Immune cell lineage | Subtypes | Function | Metabolism |
|---|---|---|---|
| Macrophage | |||
| M1 (classical activation) |
Pathogen clearance and antigen presentation Secretes pro‐inflammatory cytokines and express high levels of MHC |
Enhanced glycolysis, PPP, and FA synthesis | |
| M2 (alternate activation) | Produce anti‐inflammatory cytokines to promote immunosuppression and tumor progression |
Increased OXPHOS and FAO decreased glycolysis and PPP |
|
| Neutrophil |
Glycolysis If exposed to a TME deficient in glucose, adapt to mitochondrial FAO |
||
| N1 (anti‐tumor) | Anti‐tumor polarization induced by type 1 IFNs | ||
| N2 (pro‐tumor) | TGF‐β an overexpressed by tumor cells polarize neutrophils to pro‐tumor phenotype | ||
| Neutrophil extracellular traps (NETs) | Fibers of decondensed DNA | Dependent on glucose and to a lesser extent on glutamine | |
| NK cell | Critical in the early immune response regulating the adaptive immune response through the release of IFN‐γ |
Use OXPHOS during their resting state and upon short‐term activation Prolonged stimulation: switch to glycolysis |
|
| T cell | |||
| CD4+ Helper T cells | Mediator of immune function secreting cytokines to heighten immune response | Glycolysis and ACC‐mediated de novo FA synthesis | |
| CD8+ Cytotoxic T cells | Direct cytotoxic killing of cancer cells | Enhanced glycolysis, glutaminolysis, and FAO to exert anti‐tumor cytotoxicity | |
| Regulatory T cells | Dampen the immune response | FAO rather than glycolysis | |
| Memory T cells | Protection against reinfection or tumor re‐emergence | Mitochondrial FAO for development and long‐term survival | |
2. INNATE IMMUNE SYSTEM
2.1. Tumor‐associated macrophages
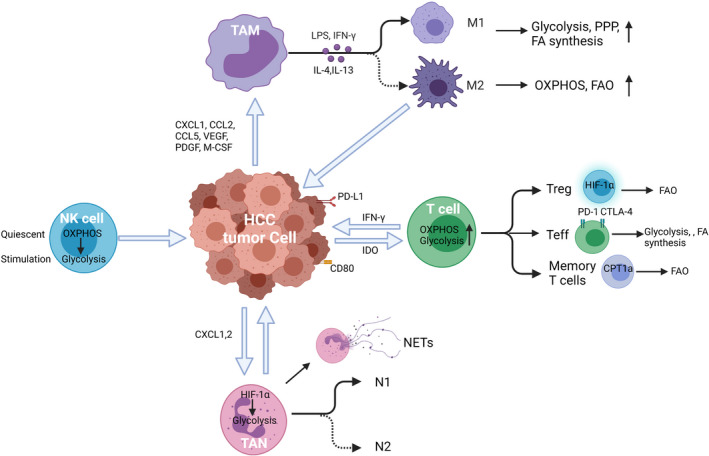
Tumor‐associated macrophages (TAMs) are a prominent component of the TME and are often considered a biomarker of poor clinical prognosis.26, 27 TAMs have long been considered to be recruited to the tumor by chemokines such as CXCL1, CCL2, CCL5, and growth factors (VEGF, PDGF, and M‐CSF).28, 29, 30 TAMs act a critical role in innate and adaptive immunity by recruiting other immune cell types and act as antigen‐presenting cells. However, tumors may be able to alter the normal developmental process of TAMs to express PD‐L1 in the TME of HCC where increased expression of PD‐L1 was linked to disease progression and mortality.27
TAMs have a high functional plasticity with two activation phenotypes: classical (M1) and alternative (M2). M1 polarization can be stimulated by IFN‐γ alone or together with bacterial moieties, such as the lipopolysaccharide (LPS). The M1 phenotype produces several pro‐inflammatory cytokines and effector molecules, such as TNF‐α, IL‐23, IL‐12, and IL‐6, major histocompatibility complex (MHC)‐related molecules, such as class I and II, and iNOS. M1 and M2 subtypes exert contrasting effects in inflammation and tumor development. M1 cells function in pathogen resistance and tumor control. However, M2 macrophages release a great number of immunosuppressive cytokines such as arginase 1 (ARG1), TGF‐β, and IL‐10 to promote immunosuppression, tumor progression, and contribute to resistance to chemotherapies.20, 30 Therefore, the balance between M1 and M2 in TAMs is important for cancer immune therapy.
TAMs are frequently found in the stroma of HCC and are polarized to the M2‐like phenotype.31 M2‐like TAMs in HCC produce immunosuppressive functions with an enhancement of cancer proliferation, invasion, metastasis, extracellular matrix (ECM) remodeling, angiogenesis, epithelial–mesenchymal transition (EMT), and cancer cell stemness maintenance.32, 33, 34 In addition, M2 macrophages have been linked to early tumor recurrence. Krall et al showed that the process of postoperative wound healing leads to myeloid cells mobilization and the shift of TAMs to M2 phenotype, contributing to the immunosuppressive TME and mediating surgery‐induced tumor outgrowth.35
Altered cellular metabolism appears to be responsible for the polarization and function in activated macrophages.36 M1 macrophages display pro‐inflammatory properties defined by an enhanced glycolysis, metabolic flux through pentose phosphate pathway (PPP), fatty acid (FA) synthesis, a decreased oxidative phosphorylation (OXPHOS), and tricarboxylic acid (TCA) cycle. M2 cells are well known as anti‐inflammatory macrophages with enhanced OXPHOS and activated fatty acid oxidation (FAO), reduced glycolysis, and PPP (Figure 2). 37, 38, 39 These metabolic changes in TAMs not only regulate energy production but also mediate the variety of TAM phenotypes. M1 macrophages fail to transform into an M2 phenotype after the stimulation of IL‐4 in vitro and in vivo secondary to M1‐associated inhibition of mitochondrial OXPHOS.40 Metabolic changes promote the development of tumor where M2 macrophages lead to increased proliferation, invasion, and metastasis of HCC cells via FAO pathway by enhancing IL‐1β secretion.41
FIGURE 2.
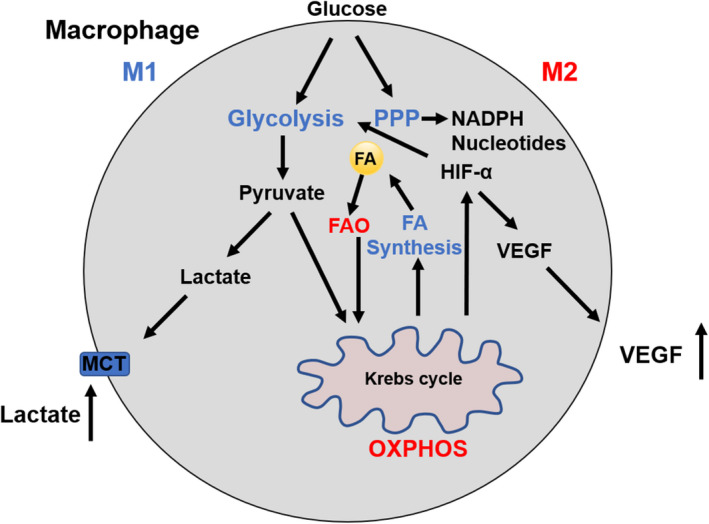
Metabolism in M1 and M2 macrophages. M1 macrophages display an enhanced glycolysis, PPP, FA synthesis, a decreased OXPHOS, and TCA cycle. Activation of glycolysis which results in increased production of lactate, increased FA uptake and synthesis, and increased uptake of glutamine to fuel the TCA cycle. M2 cells are well known as anti‐inflammatory macrophages with enhanced OXPHOS and activated FAO, reduced glycolysis, and PPP
TAMs also have been found to have altered glucose metabolism, and glucose metabolism varies at different stages of carcinogenesis. At the early inflammatory phase of cancer onset, the main energy source of TAMs is through glycolysis; whereas at the later stage of tumor progression TAMs utilize OXPHOS.42, 43 This shift in energy sources is mediated by lactic acid and cytokines. Tumor‐derived lactate also contributes to the polarization of TAMs, which is mediated through hypoxia‐inducible factor‐1α (HIF‐1α). Lactate induces ARG1 expression in TAMs and consequently affects the tumor cell proliferation and collagen synthesis through the production of ornithine and polyamines.44 Lactic acid also induced a malignancy‐promoting phenotype by promoting vascular endothelial growth factor A (VEGFA) expression via triggering HIF‐1α activation.45 In addition, glycolysis inhibition in TAMs was able to reverse TAM‐induced angiogenesis, extravasation, and EMT.46 Furthermore, the increase in glycolysis in the TME secondary to monocytes and macrophages has been linked to PD‐L1 expression through the upregulation of the glycolytic enzyme PFKFB3 in TAMs.47 High levels of PD‐L1 on tumor cells and tumor‐infiltrating immune cells are a negative prognostic factor in tumors and has been found to be a predictor of recurrence.48, 49 Therefore, PFKFB3 can be a potential therapeutic target in both tumor cells and TAMs in HCC.47
2.2. Tumor‐associated neutrophils
Neutrophils has been recognized as the most abundant circulating immune cell type and the first‐line responders to many pathologic processes. However, while providing protection in some scenarios they can cause damage in others.50 It is known that tumors secrete chemokines that support the recruitment of leukocytes including neutrophils from the circulation into the TME thus producing tumor‐infiltrating neutrophils (TANs).51 TANs play a key role in promoting growth, invasion, angiogenesis, and metastasis in HCC.52 Neutrophils are predominantly found in the peritumoral stroma and the levels of TANs can serve as an independent predictor of poor outcome in HCC patients.53
Neutrophils derive most of their energy from glycolysis, which is oxygen‐independent energy generation allows neutrophils to function effectively at low oxygen levels.54, 55 HIF‐1α also known as a crucial regulator of metabolism in myeloid cells.56 HIF‐1α is essential for neutrophil glucose uptake after activation mediated by glucose transporter GLUT1 translocation to the cell surface. Enhanced glucose consumption has been shown to be crucial for enhanced neutrophil survival and function in a hypoxic microenvironment.57, 58 The TCA cycle, OXPHOS, FAO, and PPP play different roles in the energetic, biosynthetic, and functional requirements of neutrophils (Table 2).59, 60, 61, 62, 63, 64 Due to limited glucose, neutrophils utilize mitochondrial FAO to fuel NOX‐2‐dependent reactive oxygen species (ROS) production.65 Tumor‐elicited oxidative neutrophils are an important source of NADPH oxidase‐dependent ROS and are able to maintain immune tolerance in the nutrient‐limited TME. In addition, neutrophils from patients with cancer show increased immaturity, mitochondrial content, and OXPHOS.65
TABLE 2.
Key metabolic pathways involved in different neutrophil functions
| Neutrophil Function | Metabolic pathways |
|---|---|
| Differentiation | OXPHOS, FAO, and TCA |
| Phagocytosis | Glycolysis |
| ROS production | PPP and Glutaminolysis |
| Degranulation | Glycolysis |
| NETs | Glycolysis and PPP |
| Chemotaxis | Glycolysis and mitochondria |
The thought that neutrophils are primarily dependent on the glycolytic pathway is currently being challenged as emerging research is indicating metabolic reprogramming may occur by mediators of inflammation, sepsis, diabetes, and cancer.66 In early stages of cancer, neutrophils display enhances migration with high rates of glycolysis and OXPHOS to produce more ATP.67 However, if exposed to a TME deficient in glucose, neutrophils adapt to mitochondrial FAO.67 Similar to macrophages, TANs can be classified into anti‐tumorigenic phenotype (N1) and pro‐tumorigenic phenotype (N2).68 However, the effect of metabolic reprogramming on the polarization of TANs is still largely unknown.
Recent studies have discovered a new function of neutrophils in cancer progression through the production of neutrophil extracellular traps (NETs). NETs are composed of sticky DNA fibers that are decorated with several proteins like myeloperoxidase and neutrophil elastase released from activated neutrophils.69 NET formation clearly plays an important role in HCC development associated with nonalcoholic steatohepatitis (NASH).70 NETs deletion did not affect liver steatosis and FFA accumulation but ameliorated macrophage infiltration and changed the inflammatory environment to one that is less favorable for the biological behavior of the HCC.70 Metabolically, NET formation is critically dependent on glucose and to a lesser extent on glutamine.61, 62 Future studies will be required to further investigate metabolic reprogramming in neutrophil polarization and its influence on the TME and NETs in tumor formation and progression.
2.3. Natural Killer (NK) cells
NK cells are key regulators of early immune reactions. They also control adaptive immunity through the secretion of cytokines, such as IFN‐γ, that impact dendritic cells, macrophages, and neutrophils.71, 72, 73 NK cells exhibit a superior capacity for expansion to create innate memory in parallel with the adaptive immune system. The number of NK cells was significantly lower in the liver with tumor, when compared with the adjacent normal liver of HCC patients.74, 75 Moreover, the absolute number of NK cells in peripheral blood and liver tumor tissues is positively correlated with the apoptosis rate and patient survival in HCC.76, 77, 78
The interaction of NK cells and tumor cells can induce the expression of CD25, which is identical to the α chain of the high‐affinity interleukin (IL)‐2 receptor. CD25high NK cells are highly sensitive to IL‐2, leading to a robust and prolonged NK cell anti‐tumor response.79 As a result, NK cells show an increase in metabolic signaling pathways, nutrition transport cell growth as well as increased function and survival.79, 80, 81 NK cells require dramatic changes in metabolism upon activation. NK cells utilize OXPHOS during their metabolically quiescent state and after short‐term activation.82 However, upon prolonged stimulation NK cells switch to glycolytic metabolism.83 The mechanistic target of rapamycin (mTOR) exerts a crucial role in attaining a heightened glycolytic state. The mTOR genetic deletion inhibits NK cell effector function, inhibiting IFN‐γ production and granzyme B, which further strengthens the evidence linking cellular metabolism with NK cell function.83, 84, 85 It has been shown that energy metabolism and cell motility defects play pivotal roles in circulating NK cells, which impairs functional NK cell in HCC patients.86 However, the mechanisms that tumor‐derived stimuli regulate NK activation and energy metabolism in anti‐tumor immunity have yet to be established.
3. ADAPTIVE IMMUNE SYSTEM
The effect of T cells in the immunity to cancer, including HCC, is increasingly recognized.87 As immunotherapy is becoming more widespread for malignancies including HCC it is important to further investigate the role of T‐cell metabolism.88 T‐cell metabolic reprogramming processes are closely related to the differentiation and function of T cell (Figure 3). After antigen stimulation, T cells grow, proliferate, and activation occurs releasing cytotoxic factors and cytokines. Therefore, T cells undergo profound metabolic shifts to sustain the biosynthesis of nucleic acids, amino acids, and proteins. In order to meet the requirement for cell proliferation, T cells simultaneously increase glucose and glutamine metabolism.89, 90 Interestingly, T cells, like cancer cells, primarily utilize glycolysis even when oxygen is sufficient for OXPHOS.91, 92, 93, 94, 95 It is hypothesized this is due to provide other advantages to the cell other than mere energy production, such as to promote the acquisition of nutrients to produce daughter cells.91, 96, 97
FIGURE 3.
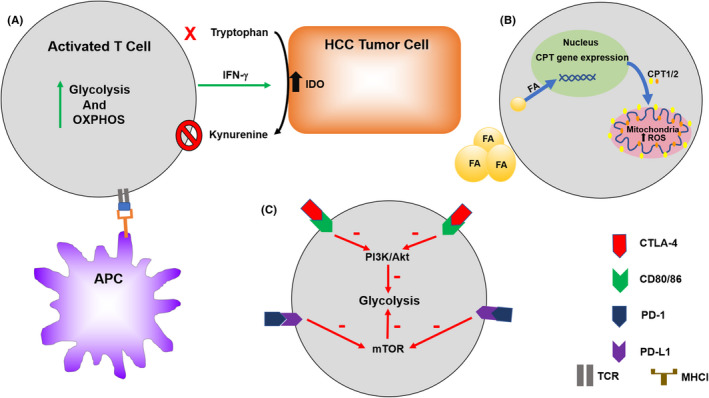
Regulation metabolism of T cells. (A) Activated T cells have increased metabolic processes to secrete activating cytokines such as IFN‐γ. However, tumor cells can have upregulation of alternate immune checkpoints to produce immune escape. (B) Fatty acids can increase the CPT gene expression causing increased ROS and CD4+ T‐cell death produce an immunosuppressive TME. (C) CTLA‐4 and PD‐1 immune checkpoints can alter T‐cell metabolism causing T‐cell anergy, exhaustion, and autophagy
Furthermore, distinct T‐cell subsets possess special metabolic alterations that are associated with their function and may provide a novel therapeutic strategy for immune regulation. Naïve T‐cell activation relies on OXPHOS while IFN‐γ cytokine production depends on glycolysis.91 Glycolysis and acetyl‐CoA carboxylase (ACC)‐mediated de novo fatty acid synthesis are the primary mechanisms to effector CD4+ T‐cell fate.98 GLUT1 knockdown reduces glucose uptake, transportation, and glycolysis, and also impairs the growth, proliferation, and prevented T‐effector (Teff) expansion.99 Cytotoxic CD8+ T cells increase glycolysis, glutaminolysis, and FAO to play effective anti‐tumor cytotoxic activity. mTOR is a central integration site for metabolic switch of CD8+ T cells.100, 101 Upon activation, PI3K/Akt/mTOR, and the transcription factor c‐Myc‐related pathways are activated in CD8+ T cells, as well as Th1, Th2, and Th17 cells, to regulate glycolytic genes such as GLUT1, PDK1, or HK2.89, 90, 102, 103
Regulatory T cells (Tregs), which depend mainly on FAO for energy and minimally utilize glycolysis, can survive under nutrient‐depleted tumor environments and play an immunosuppressive effect.99, 104 The inhibition of T‐cell intrinsic mTORC1 by rapamycin dampens growth and proliferation of Teff cell and reinforces Treg generation.105, 106, 107 HIF‐1α has been suggested to be the main regulator in T‐cell fates, resulting in increased Treg development and suppressed Th17 cells.108, 109, 110 How HIF‐1α mediates Th17 and Treg cell differentiation is unclear, but it has been shown to be involved in both metabolic and nonmetabolic processes. Metabolically, blocking glycolysis by HIF‐1α disturbs the Th17/Treg balance. Nonmetabolically, HIF‐1α activates the transcription factor RORγt and collaborates with RORγt to promote Th17 development. Interestingly, HIF‐1α can directly bind Foxp3 protein and impact its degradation.110 The metabolic switch of hypoxia and HIF‐1α play important roles in T cells and will be crucial for more studies in the future.
Another fundamental ability of the adaptive immune system is to generate a pool of long‐lived, antigen‐specific memory T and B cells. These memory cells protect against reinfection or tumor recurrence. Unlike activated effector T cells, memory T cells have dramatic changes in metabolism which does not require high rates of anabolism to maintain rapid proliferation. However, they still need energy generation to conduct certain basic cellular functions and stay alive. The metabolic switch from aerobic glycolysis to mitochondrial FAO is important for development and long‐term survival.111, 112 In contrast to effector T cells, memory T cells have a quite distinct metabolic state, which reduces glycolysis, maintains greater mitochondrial metabolism, and use fatty acids.111 Memory T cells induce expression of the critical mitochondrial transporter CPT1a, and this protein increases the memory T cells generation and survival.111
As immunotherapy is becoming more widely used in HCC, it is imperative to understand the relationship between tumor immunology and metabolism.113 On CD8+ and CD4+ T cells, isolated from HCC tissues, there is an up‐regulation of the immune checkpoint inhibitory proteins PD‐1, CTLA4, TIM3, and LAG3, compared with T cells from tumor‐free liver tissues.114 Several studies have demonstrated that PD‐1 and CTLA‐4 engagements inhibit glucose metabolism, thereby impairing T‐cell activation and ultimately inducing T‐cell dysfunction (anergy, tolerance, and exhaustion). CTLA‐4 has been shown to block Akt phosphorylation, thereby reducing glycolysis that is supported for cell growth and function.93, 115, 116 Meanwhile, ligand binding to PD‐1 can antagonize mTOR signaling via Akt and PI3K inhibition, thus diminishing glycolysis and glutaminolysis within the target cells.117 Taken together, increasing evidence suggests that metabolic reprogramming exert a key effect on anti‐ and pro‐tumor immunity of tumor‐infiltrating lymphocytes.118
The tumor microenvironment is not only the crucial regulator in HCC development but perhaps also a response to therapy. Nonalcoholic fatty liver disease (NAFLD) and its advanced form nonalcoholic steatohepatitis (NASH) are an increasing problem worldwide as the prevalence of obesity increases. The accumulation of fatty acids stored in liver tissue has been shown to cause selective CD4+ helper T cells loss.13 This is believed to be due to upregulate CPT family genes that induced by fatty acid in the more mitochondria‐rich CD4+ T cells and increase in reactive oxygen species.13, 14 A T cell‐dependent immunotherapy was found to be no longer effective in mice with liver tumors in the setting of fatty liver disease.119 Additionally, tryptophan is an essential amino acid and its deprivation has been shown to cause T cells derrangements.120 Indoleamine 2,3‐dioxygenase (IDO) is an intracellular enzyme that selective cleavage tryptophan and is overexpressed in the TME. IDO can render T cells without the essential amino acid as well as producing an immunosuppressive phenotype with an increase in Tregs.121 IDO production in HCC has been linked to a poor prognosis.121 In mouse models, IDO has been shown to be induced in HCC tumors providing immune escape from immune checkpoint inhibitors. This phenomenon could be substantially reversed by the administration of an IDO inhibitor along with the immune checkpoint inhibitor.21
4. CONCLUSION
In recent years, many advances have been obtained in understanding the effects and mechanisms of tumor‐infiltrating immune cells in the development of HCC. Metabolic reprogramming affects the differentiation stages and functions of tumor‐associated immune cells in the TME and thus plays an important role in the progression of HCC. Immune cells use diverse metabolic pathways including glycolysis, FAO, TCA cycle, PPP, and glutaminolysis to perform distinct functions. The lack of nutrients from the environment suppresses antitumor immune cells, such as M1 macrophages, N1 neutrophils, and CD8+ T cells, promoting the differentiation and activation of pro‐tumor immune cells, including M2 macrophages, N2 neutrophils, and Tregs. All of these features make the immune metabolism in TME a powerful target for cancer therapy. However, numerous findings indicate that the roles of immune cells under different conditions are self‐contradictory, for which many of the mechanisms are still unknown. Future studies are required to unravel the complex interplay between metabolic reprogramming, the immune system, and the TME and increased understanding will prove beneficial in advancing therapeutic options for patients with HCC.
CONFLICT OF INTEREST
The authors made no disclosures.
ETHICAL STATEMENT
This study does not involve any human or animal testing.
ACKNOWLEDGMENTS
This work was supported by the National Institute of Health grants (R01‐GM95566, R01‐CA214865‐02) and by the National Natural Science Foundation of China (No. 81700515).
Xia Y, Brown ZJ, Huang H, Tsung A. Metabolic reprogramming of immune cells: Shaping the tumor microenvironment in hepatocellular carcinoma. Cancer Med. 2021;10:6374–6383. 10.1002/cam4.4177
Yujia Xia and Zachary J. Brown contributed equally.
DATA AVAILABILITY STATEMENT
This is a review article and therefore the data discussed in this study are publicly available. No ethical approval was required.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A, Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. [DOI] [PubMed] [Google Scholar]
- 2.Pang TC, Lam VW. Surgical management of hepatocellular carcinoma. World J Hepatol. 2015;7:245‐252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greten TF, Lai CW, Li G, Staveley‐O'Carroll KF. Targeted and immune‐based therapies for hepatocellular carcinoma. Gastroenterology. 2019;156(2):510–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first‐line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non‐inferiority trial. Lancet. 2018;391:1163‐1173. [DOI] [PubMed] [Google Scholar]
- 5.El‐Khoueiry AB, Sangro B, Yau T, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open‐label, non‐comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389(10088):2492–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown ZJ, Greten TF, Heinrich B. Adjuvant treatment of hepatocellular carcinoma: prospect of immunotherapy. Hepatology. 2019;70:1437‐1442. [DOI] [PubMed] [Google Scholar]
- 7.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309‐322. [DOI] [PubMed] [Google Scholar]
- 8.Tu T, Bühler S, Bartenschlager R. Chronic viral hepatitis and its association with liver cancer. Biol Chem. 2017;398:817‐837. [DOI] [PubMed] [Google Scholar]
- 9.Capece D, Fischietti M, Verzella D, et al. The inflammatory microenvironment in hepatocellular carcinoma: a pivotal role for tumor‐associated macrophages. Biomed Res Int. 2013;2013:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci. 2012;125:5591‐5596. [DOI] [PubMed] [Google Scholar]
- 11.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423‐1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu C, Rong D, Zhang B, et al. Current perspectives on the immunosuppressive tumor microenvironment in hepatocellular carcinoma: challenges and opportunities. Mol Cancer. 2019;18:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma C, Kesarwala AH, Eggert T, et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature. 2016;531:253‐257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown ZJ, Fu Q, Ma C, et al. Carnitine palmitoyltransferase gene upregulation by linoleic acid induces CD4(+) T cell apoptosis promoting HCC development. Cell Death Dis. 2018;9:620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown ZJ, Heinrich B, Greten TF. Mouse models of hepatocellular carcinoma: an overview and highlights for immunotherapy research. Nat Rev Gastroenterol Hepatol. 2018;15:536‐554. [DOI] [PubMed] [Google Scholar]
- 16.Lane AN, Higashi RM, Fan TW. Metabolic reprogramming in tumors: contributions of the tumor microenvironment. Genes & diseases. 2020;7:185‐198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Biswas SK. Metabolic reprogramming of immune cells in cancer progression. Immunity. 2015;43:435‐449. [DOI] [PubMed] [Google Scholar]
- 18.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 19.Robey RB, Weisz J, Kuemmerle NB, et al. Metabolic reprogramming and dysregulated metabolism: cause, consequence and/or enabler of environmental carcinogenesis? Carcinogenesis. 2015;36:S203‐S231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, Wan YY, Zhu B. Immune cell metabolism in tumor microenvironment. Adv Exp Med Biol. 2017;1011:163‐196. [DOI] [PubMed] [Google Scholar]
- 21.Brown ZJ, Yu SJ, Heinrich B, et al. Indoleamine 2,3‐dioxygenase provides adaptive resistance to immune checkpoint inhibitors in hepatocellular carcinoma. Cancer Immunol immunother. 2018;67:1305‐1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biswas SK, Mantovani A. Orchestration of metabolism by macrophages. Cell Metab. 2012;15:432‐437. [DOI] [PubMed] [Google Scholar]
- 23.Ghesquière B, Wong BW, Kuchnio A, Carmeliet P. Metabolism of stromal and immune cells in health and disease. Nature. 2014;511:167‐176. [DOI] [PubMed] [Google Scholar]
- 24.Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38:633‐643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu D. Innate and adaptive immune cell metabolism in tumor microenvironment. Adv Exp Med Biol. 2017;1011:211‐223. [DOI] [PubMed] [Google Scholar]
- 26.Noy R, Pollard JW. Tumor‐associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuang DM, Zhao Q, Peng C, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD‐L1. J Exp Med. 2009;206:1327‐1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Larionova I, Cherdyntseva N, Liu T, Patysheva M, Rakina M, Kzhyshkowska J. Interaction of tumor‐associated macrophages and cancer chemotherapy. Oncoimmunology. 2019;8:1596004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laviron M, Boissonnas A. Ontogeny of Tumor‐Associated Macrophages. Front Immunol. 2019;10:1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677‐686. [DOI] [PubMed] [Google Scholar]
- 31.Sprinzl MF, Reisinger F, Puschnik A, et al. Sorafenib perpetuates cellular anticancer effector functions by modulating the crosstalk between macrophages and natural killer cells. Hepatology. 2013;57:2358‐2368. [DOI] [PubMed] [Google Scholar]
- 32.Clappaert EJ, Murgaski A, Van Damme H, Kiss M, Laoui D. Diamonds in the rough: harnessing tumor‐associated myeloid cells for cancer therapy. Front Immunol. 2018;9:2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petty AJ, Yang Y. Tumor‐associated macrophages: implications in cancer immunotherapy. Immunotherapy. 2017;9:289‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yeung OWH, Lo CM, Ling CC, et al. Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma. J Hepatol. 2015;62:607‐616. [DOI] [PubMed] [Google Scholar]
- 35.Krall JA, Reinhardt F, Mercury OA, et al. The systemic response to surgery triggers the outgrowth of distant immune‐controlled tumors in mouse models of dormancy. Sci Transl Med. 2018;10(436):eaan3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Odegaard JI, Chawla A. Alternative macrophage activation and metabolism. Annu Rev Pathol. 2011;6:275‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Groh L, Keating ST, Joosten LAB, Netea MG, Riksen NP. Monocyte and macrophage immunometabolism in atherosclerosis. Semin Immunopathol. 2018;40:203‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nomura M, Liu J, Rovira II, et al. Fatty acid oxidation in macrophage polarization. Nat Immunol. 2016;17:216‐217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mills EL, O'Neill LA. Reprogramming mitochondrial metabolism in macrophages as an anti‐inflammatory signal. Eur J Immunol. 2016;46:13‐21. [DOI] [PubMed] [Google Scholar]
- 40.Van den Bossche J, Baardman J, Otto N, et al. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep. 2016;17:684‐696. [DOI] [PubMed] [Google Scholar]
- 41.Zhang QI, Wang H, Mao C, et al. Fatty acid oxidation contributes to IL‐1β secretion in M2 macrophages and promotes macrophage‐mediated tumor cell migration. Mol Immunol. 2018;94:27‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baardman J, Licht I, de Winther MP, Van den Bossche J. Metabolic‐epigenetic crosstalk in macrophage activation. Epigenomics. 2015;7:1155‐1164. [DOI] [PubMed] [Google Scholar]
- 43.Boscá L, González‐Ramos S, Prieto P, et al. Metabolic signatures linked to macrophage polarization: from glucose metabolism to oxidative phosphorylation. Biochem Soc Trans. 2015;43:740‐744. [DOI] [PubMed] [Google Scholar]
- 44.Munder M, Eichmann K, Moran JM, Centeno F, Soler G, Modolell M. Th1/Th2‐regulated expression of arginase isoforms in murine macrophages and dendritic cells. J Immunology. 1950;1999(163):3771‐3777. [PubMed] [Google Scholar]
- 45.Colegio OR, Chu NQ, Szabo AL, et al. Functional polarization of tumour‐associated macrophages by tumour‐derived lactic acid. Nature. 2014;513:559‐563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Penny HL, Sieow JL, Adriani G, et al. Warburg metabolism in tumor‐conditioned macrophages promotes metastasis in human pancreatic ductal adenocarcinoma. Oncoimmunology. 2016;5:e1191731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen DP, Ning WR, Jiang ZZ, et al. Glycolytic activation of peritumoral monocytes fosters immune privilege via the PFKFB3‐PD‐L1 axis in human hepatocellular carcinoma. J Hepatol. 2019;71:333‐343. [DOI] [PubMed] [Google Scholar]
- 48.Calderaro J, Rousseau B, Amaddeo G, et al. Programmed death ligand 1 expression in hepatocellular carcinoma: relationship with clinical and pathological features. Hepatology. 2016;64:2038‐2046. [DOI] [PubMed] [Google Scholar]
- 49.Gao Q, Wang XY, Qiu SJ, et al. Overexpression of PD‐L1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin Cancer Res. 2009;15:971‐979. [DOI] [PubMed] [Google Scholar]
- 50.Erpenbeck L, Schön MP. Neutrophil extracellular traps: protagonists of cancer progression? Oncogene. 2017;36:2483‐2490. [DOI] [PubMed] [Google Scholar]
- 51.Coffelt SB, Wellenstein MD, de Visser KE. Neutrophils in cancer: neutral no more. Nat Rev Cancer. 2016;16:431‐446. [DOI] [PubMed] [Google Scholar]
- 52.Granot Z, Jablonska J. Distinct functions of neutrophil in cancer and its regulation. Mediators Inflamm. 2015;2015:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kuang DM, Zhao Q, Wu Y, et al. Peritumoral neutrophils link inflammatory response to disease progression by fostering angiogenesis in hepatocellular carcinoma. J Hepatol. 2011;54:948‐955. [DOI] [PubMed] [Google Scholar]
- 54.Borregaard N, Herlin T. Energy metabolism of human neutrophils during phagocytosis. J Clin Investig. 1982;70:550‐557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fossati G, Moulding DA, Spiller DG, Moots RJ, White MR, Edwards SW. The mitochondrial network of human neutrophils: role in chemotaxis, phagocytosis, respiratory burst activation, and commitment to apoptosis. J Immunol. 1950;2003(170):1964‐1972. [DOI] [PubMed] [Google Scholar]
- 56.Cheng SC, Quintin J, Cramer RA, et al. mTOR‐ and HIF‐1alpha‐mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345:1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schuster DP, Brody SL, Zhou Z, et al. Regulation of lipopolysaccharide‐induced increases in neutrophil glucose uptake. Am J Physiol Lung Cell Mol Physiol. 2007;292:L845‐L851. [DOI] [PubMed] [Google Scholar]
- 58.Stothers CL, Luan L, Fensterheim BA, Bohannon JK. Hypoxia‐inducible factor‐1α regulation of myeloid cells. J Molecular Med. 2018;96:1293‐1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Riffelmacher T, Clarke A, Richter FC, et al. Autophagy‐dependent generation of free fatty acids is critical for normal neutrophil differentiation. Immunity. 2017;47:466‐480.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Six E, Lagresle‐Peyrou C, Susini S, et al. AK2 deficiency compromises the mitochondrial energy metabolism required for differentiation of human neutrophil and lymphoid lineages. Cell Death Dis. 2015;6:e1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rodriguez‐Espinosa O, Rojas‐Espinosa O, Moreno‐Altamirano MM, Lopez‐Villegas EO, Sanchez‐Garcia FJ. Metabolic requirements for neutrophil extracellular traps formation. Immunology. 2015;145:213‐224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Azevedo EP, Rochael NC, Guimarães‐Costa AB, et al. A metabolic shift toward pentose phosphate pathway is necessary for amyloid fibril‐ and phorbol 12‐myristate 13‐acetate‐induced neutrophil extracellular trap (NET) formation. J Biol Chem. 2015;290:22174‐22183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jones R, McDonald KE, Willson JA, et al. Mutations in succinate dehydrogenase B (SDHB) enhance neutrophil survival independent of HIF‐1alpha expression. Blood. 2016;127:2641‐2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Richer BC, Salei N, Laskay T, Seeger K. Changes in neutrophil metabolism upon activation and aging. Inflammation. 2018;41:710‐721. [DOI] [PubMed] [Google Scholar]
- 65.Rice CM, Davies LC, Subleski JJ, et al. Tumour‐elicited neutrophils engage mitochondrial metabolism to circumvent nutrient limitations and maintain immune suppression. Nat Commun. 2018;9:5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kumar S, Dikshit M. Metabolic insight of neutrophils in health and disease. Front Immunol. 2019;10:2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Patel S, Fu S, Mastio J, et al. Unique pattern of neutrophil migration and function during tumor progression. Nat Immunol. 2018;19:1236‐1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Andzinski L, Kasnitz N, Stahnke S, et al. Type I IFNs induce anti‐tumor polarization of tumor associated neutrophils in mice and human. Int J Cancer. 2016;138:1982‐1993. [DOI] [PubMed] [Google Scholar]
- 69.Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532‐1535. [DOI] [PubMed] [Google Scholar]
- 70.van der Windt DJ, Sud V, Zhang H, et al. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology. 2018;68:1347‐1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Orr MT, Lanier LL. Natural killer cell education and tolerance. Cell. 2010;142:847‐856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vivier E, Raulet DH, Moretta A, et al. Innate or adaptive immunity? the example of natural killer cells. Science. 2011;331:44‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Moretta A, Marcenaro E, Sivori S, Della Chiesa M, Vitale M, Moretta L. Early liaisons between cells of the innate immune system in inflamed peripheral tissues. Trends Immunol. 2005;26:668‐675. [DOI] [PubMed] [Google Scholar]
- 74.Cai L, Zhang Z, Zhou L, et al. Functional impairment in circulating and intrahepatic NK cells and relative mechanism in hepatocellular carcinoma patients. Clinical immunology. 2008;129:428‐437. [DOI] [PubMed] [Google Scholar]
- 75.Wu Y, Kuang DM, Pan WD, et al. Monocyte/macrophage‐elicited natural killer cell dysfunction in hepatocellular carcinoma is mediated by CD48/2B4 interactions. Hepatology (Baltimore, MD). 2013;57:1107‐1116. [DOI] [PubMed] [Google Scholar]
- 76.Chew V, Chen J, Lee D, et al. Chemokine‐driven lymphocyte infiltration: an early intratumoural event determining long‐term survival in resectable hepatocellular carcinoma. Gut. 2012;61:427‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chew V, Tow C, Teo M, et al. Inflammatory tumour microenvironment is associated with superior survival in hepatocellular carcinoma patients. J Hepatol. 2010;52:370‐379. [DOI] [PubMed] [Google Scholar]
- 78.Hoechst B, Voigtlaender T, Ormandy L, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology (Baltimore, MD). 2009;50:799‐807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kedia‐Mehta N, Choi C, McCrudden A, et al. Natural killer cells integrate signals received from tumour interactions and IL2 to induce robust and prolonged anti‐tumour and metabolic responses. Immunometabolism. 2019;1:e190014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lee SH, Fragoso MF, Biron CA. Cutting edge: a novel mechanism bridging innate and adaptive immunity: IL‐12 induction of CD25 to form high‐affinity IL‐2 receptors on NK cells. J Immunol (Baltimore, Md. 1950;2012(189):2712‐2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bihl F, Pecheur J, Bréart B, et al. Primed antigen‐specific CD4+ T cells are required for NK cell activation in vivo upon Leishmania major infection. J Immunol (Baltimore, Md). 1950;2010(185):2174‐2181. [DOI] [PubMed] [Google Scholar]
- 82.Keppel MP, Saucier N, Mah AY, Vogel TP, Cooper MA. Activation‐specific metabolic requirements for NK Cell IFN‐γ production. J Immunol (Baltimore, Md). 1950;2015(194):1954‐1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Donnelly RP, Loftus RM, Keating SE, et al. mTORC1‐dependent metabolic reprogramming is a prerequisite for NK cell effector function. Journal of immunology (Baltimore, Md). 1950;2014(193):4477‐4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Marçais A, Cherfils‐Vicini J, Viant C, et al. The metabolic checkpoint kinase mTOR is essential for IL‐15 signaling during the development and activation of NK cells. Nat Immunol. 2014;15:749‐757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Finlay DK. Metabolic regulation of natural killer cells. Biochem Soc Trans. 2015;43:758‐762. [DOI] [PubMed] [Google Scholar]
- 86.Zecca A, Barili V, Canetti D, et al. Energy metabolism and cell motility defect in NK‐cells from patients with hepatocellular carcinoma. Cancer immunology, immunotherapy. CII. 2020;69(8):1589–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kang TW, Yevsa T, Woller N, et al. Senescence surveillance of pre‐malignant hepatocytes limits liver cancer development. Nature. 2011;479:547‐551. [DOI] [PubMed] [Google Scholar]
- 88.Brar G, Greten TF, Brown ZJ. Current frontline approaches in the management of hepatocellular carcinoma: the evolving role of immunotherapy. Therap Adv Gastroenterol. 2018;11:1756284818808086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Siska PJ, Rathmell JC. T cell metabolic fitness in antitumor immunity. Trends Immunol. 2015;36:257‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.MacIver NJ, Michalek RD, Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol. 2013;31:259‐283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chang CH, Curtis J, Maggi L, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153:1239‐1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T‐cell response. Nat Rev Immunol. 2005;5:844‐852. [DOI] [PubMed] [Google Scholar]
- 93.Frauwirth KA, Riley JL, Harris MH, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16:769‐777. [DOI] [PubMed] [Google Scholar]
- 94.Gerriets VA, Rathmell JC. Metabolic pathways in T cell fate and function. Trends Immunol. 2012;33:168‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jones RG, Thompson CB. Revving the engine: signal transduction fuels T cell activation. Immunity. 2007;27:173‐178. [DOI] [PubMed] [Google Scholar]
- 96.Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441‐464. [DOI] [PubMed] [Google Scholar]
- 97.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (New York, NY). 2009;324:1029‐1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Berod L, Friedrich C, Nandan A, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med. 2014;20:1327‐1333. [DOI] [PubMed] [Google Scholar]
- 99.Macintyre AN, Gerriets VA, Nichols AG, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014;20:61‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Araki K, Turner AP, Shaffer VO, et al. mTOR regulates memory CD8 T‐cell differentiation. Nature. 2009;460:108‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pearce EL, Walsh MC, Cejas PJ, et al. Enhancing CD8 T‐cell memory by modulating fatty acid metabolism. Nature. 2009;460:103‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zheng Y, Delgoffe GM, Meyer CF, Chan W, Powell JD. Anergic T cells are metabolically anergic. J Immunol (Baltimore, Md). 1950;2009(183):6095‐6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Maciolek JA, Pasternak JA, Wilson HL. Metabolism of activated T lymphocytes. Curr Opin Immunol. 2014;27:60‐74. [DOI] [PubMed] [Google Scholar]
- 104.Michalek RD, Gerriets VA, Jacobs SR, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. Journal of immunology (Baltimore, Md). 1950;2011(186):3299‐3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zheng Y, Collins SL, Lutz MA, et al. A role for mammalian target of rapamycin in regulating T cell activation versus anergy. J Immunol (Baltimore, Md). 1950;2007(178):2163‐2170. [DOI] [PubMed] [Google Scholar]
- 106.Kopf H, de la Rosa GM, Howard OM, Chen X. Rapamycin inhibits differentiation of Th17 cells and promotes generation of FoxP3+ T regulatory cells. Int Immunopharmacol. 2007;7:1819‐1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Delgoffe GM, Kole TP, Zheng Y, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832‐844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Semenza GL. Hypoxia‐inducible factor 1 (HIF‐1) pathway. Sci STKE. 2007;2007(407):cm8. [DOI] [PubMed] [Google Scholar]
- 109.Shi LZ, Wang R, Huang G, et al. HIF1alpha‐dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208:1367‐1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dang E, Barbi J, Yang HY, et al. Control of T(H)17/T(reg) balance by hypoxia‐inducible factor 1. Cell. 2011;146:772‐784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.van der Windt G, Everts B, Chang CH, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36:68‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pearce EL. Metabolism in T cell activation and differentiation. Curr Opin Immunol. 2010;22:314‐320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Onuma AE, Zhang H, Huang H, Williams TM, Noonan A, Tsung A. Immune checkpoint inhibitors in hepatocellular cancer: current understanding on mechanisms of resistance and biomarkers of response to treatment. Gene Expr. 2020;20(1):53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhou G, Sprengers D, Boor PPC, et al. Antibodies against immune checkpoint molecules restore functions of tumor‐infiltrating T cells in hepatocellular carcinomas. Gastroenterology. 2017;153:1107‐1119.e10. [DOI] [PubMed] [Google Scholar]
- 115.Parry RV, Chemnitz JM, Frauwirth KA, et al. CTLA‐4 and PD‐1 receptors inhibit T‐cell activation by distinct mechanisms. Mol Cell Biol. 2005;25:9543‐9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jacobs SR, Herman CE, MacIver NJ, et al. Glucose uptake is limiting in T cell activation and requires CD28‐mediated Akt‐dependent and independent pathways. J Immunol. 2008;180(7):4476–4486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Francisco LM, Salinas VH, Brown KE, et al. PD‐L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206:3015‐3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang Q, Lou Y, Bai XL, Liang TB. Immunometabolism: A novel perspective of liver cancer microenvironment and its influence on tumor progression. World J Gastroenterol. 2018;24:3500‐3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Heinrich B, Brown ZJ, Diggs LP, et al. Steatohepatitis Impairs T cell‐directed immunotherapies against liver tumors in mice. Gastroenterology. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Munn DH, Mellor AL. Indoleamine 2,3‐dioxygenase and tumor‐induced tolerance. J Clin Investig. 2007;117:1147‐1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Munn DH, Mellor AL. IDO in the Tumor Microenvironment: Inflammation, Counter‐Regulation, and Tolerance. Trends Immunol. 2016;37:193‐207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This is a review article and therefore the data discussed in this study are publicly available. No ethical approval was required.