

Abstract
Current therapies for treating castration resistant prostate cancer (CRPC) include abiraterone and enzalutamide which function by inhibiting androgen signaling by targeting androgen synthesis and antagonizing the androgen receptor (AR) respectively. While these therapies are initially beneficial, resistance inevitably develops. A number of pathways have been identified to contribute to CRPC progression and drug resistance. Among these is aberrant androgen signaling perpetuated by increased expression and activity of androgenic enzymes. While abiraterone inhibits the androgenic enzyme, CYP17A1, androgen synthesis inhibition by abiraterone is incomplete and sustained androgenesis persists, in part due to increased levels of AKR1C3 and steroid sulfatase (STS). Expression of both of these enzymes is increased in CRPC and is associated with resistance to anti-androgens. A number of studies have identified methods for targeting these enzymes. Indomethacin, a non-steroidal anti-inflammatory drug commonly used to treat inflammatory arthritis has been well established as an inhibitor of AKR1C3. Treatment of CRPC cells with indomethacin reduces cell growth and improves the response to enzalutamide and abiraterone. Similarly, STS inhibitors have been shown to reduce intracrine androgens and also reduce CRPC growth and enhance anti-androgen treatment. In this review, we provide an overview of androgen synthesis in CRPC and strategies aimed at inhibiting intracrine androgens.
Keywords: Prostate cancer, intracrine androgens, androgen synthesis inhibition, drug resistance, AKR1C3, STS
Introduction
Prostate cancer (PCa) is the most common cancer in men and is the second leading cause of cancer related death for men in the US [1]. The first line of therapy for these men is often androgen deprivation therapy (ADT) which targets androgen signaling by inhibiting the production of androgens by the testes. In castration-sensitive PCa, this results in tumor shrinkage. Unfortunately, the majority of men experience only transient benefit from these interventions before developing castration-resistant prostate cancer (CRPC), which progresses even in castrate concentrations of androgens. In CRPC, the androgen receptor (AR) is commonly overexpressed, hyper-activated, or both which results in the transcription of downstream AR target genes and promotes tumor progression despite only castrate levels of androgen being present.
Mechanisms resulting in the development of CRPC from castration-sensitive prostate cancer are well studied. The majority of the identified mechanisms involve alterations which result in an increase in AR activation and signaling. This can be the result of increased androgen production, enhanced response to existing androgen, activation of the AR by non-classical ligands and ligand independent AR activation. Specifically identified mechanisms contributing to CRPC include: AR amplification and mutation, AR co-activator and co-repressor modifications, aberrant activation and/or post-translational modification, AR splice variants, and altered steroidogenesis [2-8].
While CRPC is currently considered incurable, the second-generation anti-androgen drugs enzalutamide (Enza), abiraterone (Abi), and more recently apalutamide (Apa) and darolutamide (Daro), are approved for the treatment of CRPC and provide some benefit. Enza, Apa, and Daro function by binding the AR to prevent its activation by androgens. Furthermore, they can block translocation of the AR into the nucleus and inhibit the AR from binding to DNA-binding sites and transcription elements. Abi is a CYP17A1 inhibitor which blocks intracrine androgen synthesis to reduce androgen signaling. Despite the promise of these anti-androgens, it is estimated that one third of patients given Abi and one fourth of patients given Enza will exhibit primary resistance to these therapies [9,10]. Even among those who are initially responsive, resistance occurs frequently, often within a year of therapy initiation, and is the most common cause of treatment failure.
A number of mechanisms by which anti-androgen resistance occurs have been identified. Perhaps the best understood dysregulated pathways in this resistance revolve around irregular androgen signaling, either through expression of mutated AR and expression of AR variants or through an increase in the expression of enzymes involved in androgen synthesis. While Abi inhibits androgen synthesis by blocking CYP17A1 activity, androgen production is not completely abrogated, suggesting other enzymes involved in androgen synthesis may be compensating and promoting tumor progression [11]. This review will focus on dysregulated androgen synthesis and the contribution of intracrine androgen production to CRPC progression and anti-androgen resistance. Furthermore, we will highlight approved and experimental strategies for targeting androgen synthesis (Figure 1).
Figure 1.
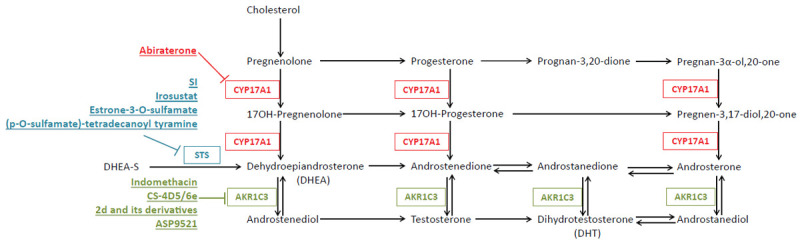
Schematic depicting the androgen synthesis pathway with key enzymes and their experimental and FDA approved inhibitors. Inhibitors block activity of all reactions in their corresponding color.
Androgen synthesis in CRPC
Aberrant androgen synthesis is a hallmark of CRPC progression and is oftentimes further dysregulated in anti-androgen resistant tumors. In healthy individuals, androgen production primarily occurs in the testes and adrenal glands. When this is blocked via ADT, many PCa cells eventually acquire the ability to produce androgens themselves to support their own growth. Androgen synthesis is a complex pathway and the production of intratumoral androgens is hypothesized to arise in multiple ways. De novo androgen synthesis requires cholesterol as a precursor. In the Classic pathway of de novo androgen synthesis, cholesterol is converted into pregnenolone via CYP11A1; CYP17A1 then hydroxylates pregnenolone into 17OH-Pregnenolone which is further converted to dehydroepiandrosterone (DHEA) by the same enzyme. DHEA is converted to androstenedione by HSD3B2 and androstenedione into testosterone by aldo-keto reductase family 1, member C3 (AKR1C3). Dihydrotestosterone (DHT), which is more biologically active than testosterone, is produced from testosterone via activity of SRD5A2 [12].
In addition to the classic de novo androgen synthesis pathway, DHT can be synthesized through a Backdoor pathway. In this pathway, DHT is formed without requiring testosterone as a precursor. Instead, androsterone, which is produced via a number of reactions from progesterone, serves as the main intermediary androgen substrate [12,13]. Interestingly, a study by Deb et al. determined that both peripheral zone and transitional zone prostate tissues have a preference for the Classical pathway, as indicated by increased levels of Classical pathway intermediates compared to those of the Backdoor pathway. However, these same tissues also display increased levels of upstream Backdoor pathway precursors when supplemented with progesterone or 17-OH-pregnenolone [14].
A number of the enzymes involved in de novo androgen synthesis have been demonstrated to be upregulated in CRPC and anti-androgen resistance. Expression levels of CYP17A1, 3βHSD1, and 3βHSD2 were found to be upregulated in metastatic CRPC patient samples compared to primary PCa samples [15]. Liu et al. showed using gene-expression analysis of two independent datasets (Glinsky and Singh Prostate) using the Oncomine database that AKR1C3 expression was correlated with prostate cancer progression and recurrence [16]. They also demonstrated that Enza resistant C4-2B cells (generated via continuous culture of parental C4-2B cells in Enza) display a general upregulation of androgenic enzymes as assessed by microarray gene set enrichment analysis. Specifically, they confirmed via western blotting that significantly increased expression levels of AKR1C3 as well as CYP17A1 and HSD3B were present in Enza resistant cells. This was correlated to an increase in intracellular testosterone and DHT levels [16].
The contribution of the Classic and Backdoor pathways of de novo androgen synthesis to intratumoral androgen concentrations becomes more significant when tied to the fact that cholesterol homeostasis is also dysregulated in CRPC. Leon et al. determined that expression of LDL-r, SR-B1, HMG-CoA reductase, ACAT1,2, and ABCA1 are all altered in CRPC disease progression, resulting in increased influx and synthesis of cholesterol as well as amplified conversion of cholesterol esters into free cholesterol [17]. As both of these pathways utilize cholesterol as a starting substrate, increased levels of cholesterol could promote androgen synthesis. In fact, increased amounts of cholesterol have been linked to PCa tumor growth. Mice maintained on a hypercholesterolemic diet had larger LNCaP xenograft tumors compared to mice fed a low fat/no cholesterol diet [18]. Furthermore, it was observed that the mice on the high cholesterol diet had increased concentrations of intratumoral androgens which may have been the result of upregulated CYP17A expression within these tumors [18].
Another potential source contributing to intracrine androgen synthesis is the presence of the androgen precursor DHEA sulfate (DHEA-S). The adrenal gland in humans is a major source of weak androgens, including DHEA-S. DHEA-S is present at plasma concentrations up to 500 times higher than testosterone and can potentially be delivered into prostate cancer cells via organic anion transporters, and converted via endogenous steroid sulfatase (STS) activity into DHEA and then into testosterone by hydroxysteroid dehydrogenases and into DHT via 5α reductase. STS is present in 85% of prostate cancer specimens and its expression is increased in CRPC; therefore conversion of circulating DHEA-S into DHEA by STS is thought to be an alternate source of androgen [19]. In prostate cancer patients treated with the nonspecific CYP17A1 inhibitor, ketoconazole, or the specific CYP17A1 inhibitor, abiraterone, significant circulating DHEA-S concentrations are still present (~20 µg/dL, about 2000 times higher than castrate levels of testosterone), further suggesting that DHEA-S could act as a depot for downstream androgen formation via desulfatation [11]. A recent study demonstrated that overexpression of STS in vitro results in increased growth of PCa cells and confers resistance to Enza. Furthermore, they found that STS overexpression increased intracellular testosterone levels when cells were supplemented with DHEA-S [20].
Localized androgen production, either de novo or derived from high concentrations of existing precursors, is a major challenge for treating CRPC and anti-androgen resistance. A number of strategies for targeting these pathways have been identified with varying levels of success.
Targeting androgen synthesis
Due to the convoluted pathway resulting in testosterone and DHT production, there are a number of potential targets for inhibiting their synthesis and therefore reducing tumor growth. Abiraterone, an FDA approved treatment for castration-sensitive PCa and CRPC, blocks CYP17A1 activity and provides improved survival in metastatic CRPC patients over placebo (14.8 months vs. 10.9 months) [9]. Unfortunately, inhibition of androgen synthesis by abiraterone is incomplete and resistance occurs frequently, leading to treatment failure. This highlights the need to investigate other enzymes within androgen synthesis as potential targets to reduce androgen concentrations.
AKR1C3
One enzyme that has been under intense investigation is AKR1C3. AKR1C3 is a member of the aldo-keto reductase (AKR) protein family and is highly expressed in the prostate where it catalyzes the formation of testosterone and DHT via NADPH dependent reduction of the weak androgens androstenedione or androstanedione respectively [21,22]. In CRPC, AKR1C3 has been found to be one of the most upregulated enzymes involved in androgen synthesis in patients, being expressed 5.3 fold higher than in primary prostate cancer tissues [23]. Similar observations have been reported in other studies [15,24]. Elevated expression of AKR1C3 has been linked to enhanced PCa progression, aggressiveness, and resistance to anti-androgens and radiation [16,23,25-28]. The importance of AKR1C3 is enhanced by the fact that it is involved in all methods of androgen synthesis, including the Classic and Backdoor pathways, which suggests that targeting this enzyme could inhibit androgen synthesis in multiple ways. Interestingly, AKR1C3 is not only involved in androgen synthesis but also catalyzes the production of prostaglandins which are pro-inflammatory, pro-proliferative, and known to promote cancer growth, progression, and resistance to radiation [26,29]. This suggests that inhibiting AKR1C3 could impair both androgen dependent and androgen independent prostate cancer cell growth. Together, these data make targeting AKR1C3 an attractive strategy for treating CRPC.
To understand the potential mechanisms of resistance to anti-androgens, we have generated and characterized several unique anti-androgen resistant PCa cell lines from C4-2B cells, a widely used castration-resistant prostate cancer cell line generated by Dr. Leland Chung’s laboratory [30]. These anti-androgen resistant cell lines include C4-2B MDVR (enzalutamide resistant), C4-2B AbiR (abiraterone resistant), C4-2B ApalR (apalutamide resistant), and C4-2B DaroR (darolutamide resistant). These resistant cell lines provide us with unique models to study the mechanisms underlying anti-androgen resistance in metastatic PCa.
A study by Liu et al. [16] found that AKR1C3 expression was over 16 fold higher in Enza resistant C4-2B cells (C4-2B MDVR) compared to parental C4-2B cells and this correlated to increased intracrine androgens. Furthermore, they observed that overexpression of AKR1C3 in LNCaP cells significantly increased the cells resistance to treatment with Enza. When they transfected shRNA against AKR1C3 into C4-2B MDVR and the intrinsically Enza resistant CWR22Rv1 cells, both cell lines were sensitized to Enza treatment [16]. This study also investigated the ability of the non-steroidal anti-inflammatory drug, indomethacin, to inhibit AKR1C3 activity and improve the response to Enza. Indomethacin, which is typically used for the treatment of chronic inflammatory arthritis, had been shown to be able to inhibit AKR1C3 activity [31-33]. In their model, when Liu et al. treated C4-2B MDVR and CWR22Rv1 cells with indomethacin in combination with Enza, the response to Enza was greatly enhanced. These results were further confirmed in vivo in mice bearing CWR22Rv1 xenografts [16]. In a separate study, Liu et al. also found that modulation of AKR1C3 activity impacted abiraterone resistance. Similarly as with Enza, they observed that overexpression of AKR1C3 in LNCaP cells promoted abiraterone resistance and that abiraterone resistant C4-2B cells (C4-2B AbiR) had increased AKR1C3 expression over parental C4-2B cells. They further demonstrated that knockdown of AKR1C3 using shRNA or indomethacin enhanced the response to Abi treatment in vitro and in vivo [28].
Other groups have also identified drugs that target AKR1C3. Endo et al. synthesized a number of derivatives of N-(4-fluorophenyl)-8-hydroxy-2-imino-2H-chromene-3-carboxamide (referred to as 2d) with potent AKR1C3 inhibiting capabilities. These inhibitors were observed to suppress proliferation of 22Rv1 and PC3 cells and in vivo tumor xenografts. Additionally, their derivatives improved the response to increase apoptosis in PCa cells treated with Abi and Enza. Importantly, these derivatives were observed to have greater than 220 fold selectivity for AKR1C3 over AKR1C1, AKR1C2, and AKR1C4 [34]. This is important due to the fact that while AKR1C3 is involved in synthesis of testosterone and DHT, other AKR family members can process DHT into pro-apoptotic estrogen receptor β agonists and inactive androgens [22,35,36]. In another study, Zhou et al. combined a mansonone derivative and AKR1C3 inhibitor, 6e, with scFv 4D5-modified chitosan to create a nanodrug-delivery system designed to target HER2 positive CRPC cells which they called CS-4D5/6e. Treatment of AKR1C3 overexpressing LNCaP cells in vitro and in in vivo xenografts with CS-4D5/6e resulted in reduced testosterone levels. Furthermore, CS-4D5/6e reduced the growth rate of 22Rv1 xenografts in vivo [37]. Another novel AKR1C3 inhibitor, ASP9521, was characterized by Kikuchi and colleagues. ASP9521 was observed to selectively inhibit AKR1C3 mediated conversion of androstenedione into testosterone. They further found that ASP9521 reduced androstenedione-dependent PSA production, could suppress prostate cancer cell proliferation, and was orally bioavailable in multiple species [38]. ASP9521 was also tested in Phase I/II clinical trials. It was found to have acceptable safety and tolerability profiles however no significant clinical activity was observed in the patients [39].
Interestingly, in addition to its role in androgen synthesis, AKR1C3 expression has also been implicated in promoting stability of the AR and its variant, ARv7. C4-2B and LNCaP cells overexpressing AKR1C3 have been observed to express higher levels of full length AR and ARv7 protein [40]. AKR1C3 was found to bind to ARv7 as AKR1C3/ARv7 complex and increase the half-life of AR and ARv7 as well as repressing tumor suppressor gene B4GALT1 expression [41]. Inhibition of AKR1C3 with indomethacin suppressed AR and ARv7 signaling as indicated by RNA sequencing data [40]. The AKR1C3/ARv7 axis has been further associated with cross resistance between anti-androgen therapies [42].
Together, these studies highlight the importance of AKR1C3 in CRPC progression and resistance to anti-androgens. Specifically, inhibition of AKR1C3 using shRNA or a variety of drugs was found to successfully inhibit CRPC growth and enhance resistant cell response to treatment with Enza or Abi. The potential of AKR1C3 as a target is enhanced by the fact that it promotes CRPC growth in more than one way.
Steroid sulfatase
In addition to AKR1C3, STS has also garnered interest as a therapeutic target for treating CRPC. Most PCa specimens express STS and in CRPC its expression is even higher [43-46]. This could be due, in part, to production of insulin-like growth factor-II which has been demonstrated to increase STS expression [47]. DHEA is a major source of androgens in prostate cancer tumors and DHEA-S, one of its precursors, is the most abundant adrenal steroid in circulation in males. DHEA-S has been observed to be present at concentrations 400- to 1000-fold higher than DHEA (DHEAS/DHEA; 2-10 µmol/L vs. 10-25 nmol/L) [11,19,48]. Therefore, it is postulated that conversion of circulated adrenal DHEA-S to DHEA by STS may be an alternative source for intracrine androgen production. A number of STS inhibitors have been tested for potential activity both in animal and clinical models in breast cancer, however, to date there are limited studies in PCa despite its potential [49-56].
In LNCaP cells, Selcer et al. demonstrated the ability of estrone-3-O-sulfamate and (p-O-sulfamate)-tetradecanoyl tyramine to reduce STS activity [57]. Similarly, STX-64 (Irosustat), another STS inhibitor, has also been shown to nearly completely inhibit STS activity in LNCaP cells [58]. A more recent study demonstrated that STS expression and activity was low in benign prostate cells and LNCaP cells but increased in C4-2B cells. VCaP cells and the LuCaP35CR PDX model had the highest expression levels of STS [20]. This suggests that STS expression and activity may correlate with disease progression. When STS was knocked down with siRNA, cell growth was reduced in C4-2B and VCaP cells. Overexpression of STS in LNCaP and C4-2B cells resulted in increased STS activity and intracellular androgens. STS overexpression was further observed to promote resistance to Enza and Abi [20]. In this study, novel STS inhibitors (SI) were characterized. These SI were found to significantly inhibit STS activity and suppress cell growth and colony formation. RNAseq analysis determined that treatment with SI reduced expression of both full length and ARv7 associated target genes. In vivo, STS inhibition with SI reduced the growth of relapsed VCaP tumors and improved the response of xenografts to Enza [20]. These data suggest that STS inhibition in PCa could be a viable treatment strategy.
STS inhibitors have been studied in clinical trials with mixed results. However, these studies were primarily conducted in breast and endometrial cancer patients where the goal of STS inhibition is to reduce estradiol levels by blocking the desulfatation of estrone sulphate and estradiol sulfate into estrone and estradiol respectively. Chief among these inhibitors is Irosustat. In a study evaluating Irosustat in endometrial cancer patients, Irosustat was found to be well tolerated with a good safety profile. However, there were no statistically significant differences between Irosustat and the current standard of care on survival rates of patients [59]. Irosustat has also been tested in a Phase I clinical trial in prostate cancer patients where it was found to be well tolerated. Due to the fact that Irosustat was not predicted to reach the primary endpoints of the clinical trial and was shown to have no improvement over the current standard of care, Ipsen, the pharmaceutical company in charge of its development, decided to discontinue development of Irosustat as a monotherapy and instead investigate its potential as a co-treatment with other hormonal therapies. To this end, a Phase II clinical trial using Irosustat in combination with aromatase inhibitors has been launched for patients with estrogen receptor (ER)-positive breast cancer [56].
Overall, these studies hint that STS inhibition is a potential treatment for advanced CRPC. Targeting STS with various inhibitors was observed to reduce STS activity, androgen production, and CRPC growth in vitro and in vivo. Further study is still necessary to discern optimal co-treatment strategies including those combining STS inhibition with Enza.
Conclusion
Androgen synthesis is a complex series of steps involving many enzymes. FDA approved Abi targets CYP17A1 to reduce androgen production however this reduction is incomplete. Other enzymes compensate for the limited CYP17A1 activity in the presence of Abi to drive androgen synthesis regardless. Targeting these additional enzymes, including AKR1C3 and STS, has been demonstrated to effectively reduce androgen levels, impede CRPC growth, and improve the response to Abi and Enza. Continued development of novel inhibitors of AKR1C3 and STS could lead to improvements in the standard of care for CRPC patients.
Acknowledgements
This work was supported in part by grants CA 225836, DOD PC150040, DOD PC180180, and the U.S. Department of Veterans Affairs, Office of Research & Development, BLR&D Research Career Scientist Award IK6BX005222. This review article is dedicated to the memory of Dr. Leland W.K. Chung, who was a mentor and dear friend to us. Dr. Chung fostered our love for science and taught us to pursue it with dedication and curiosity. He inspired us to seek new knowledge and explore better treatments for cancer. His support and kindness have had long lasting impacts on our lives and will always stay with us.
Disclosure of conflict of interest
None.
References
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71:7–33. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 2.Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68:5469–77. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang KH, Ercole CE, Sharifi N. Androgen metabolism in prostate cancer: from molecular mechanisms to clinical consequences. Br J Cancer. 2014;111:1249–54. doi: 10.1038/bjc.2014.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang KH, Li R, Papari-Zareei M, Watumull L, Zhao YD, Auchus RJ, Sharifi N. Dihydrotestosterone synthesis bypasses testosterone to drive castration-resistant prostate cancer. Proc Natl Acad Sci U S A. 2011;108:13728–33. doi: 10.1073/pnas.1107898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shtivelman E, Beer TM, Evans CP. Molecular pathways and targets in prostate cancer. Oncotarget. 2014;5:7217–59. doi: 10.18632/oncotarget.2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steketee K, Timmerman L, Ziel-van der Made AC, Doesburg P, Brinkmann AO, Trapman J. Broadened ligand responsiveness of androgen receptor mutants obtained by random amino acid substitution of H874 and mutation hot spot T877 in prostate cancer. Int J Cancer. 2002;100:309–17. doi: 10.1002/ijc.10495. [DOI] [PubMed] [Google Scholar]
- 7.Uo T, Plymate SR. Clinical significance of AR-V567es in prostate cancer-letter. Clin Cancer Res. 2019;25:6009. doi: 10.1158/1078-0432.CCR-19-1400. [DOI] [PubMed] [Google Scholar]
- 8.Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, Bova GS, Luo J. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB Jr, Saad F, Staffurth JN, Mainwaring P, Harland S, Flaig TW, Hutson TE, Cheng T, Patterson H, Hainsworth JD, Ryan CJ, Sternberg CN, Ellard SL, Fléchon A, Saleh M, Scholz M, Efstathiou E, Zivi A, Bianchini D, Loriot Y, Chieffo N, Kheoh T, Haqq CM, Scher HI COU-AA-301 Investigators. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, de Wit R, Mulders P, Chi KN, Shore ND, Armstrong AJ, Flaig TW, Fléchon A, Mainwaring P, Fleming M, Hainsworth JD, Hirmand M, Selby B, Seely L, de Bono JS AFFIRM Investigators. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–97. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 11.Tamae D, Mostaghel E, Montgomery B, Nelson PS, Balk SP, Kantoff PW, Taplin ME, Penning TM. The DHEA-sulfate depot following P450c17 inhibition supports the case for AKR1C3 inhibition in high risk localized and advanced castration resistant prostate cancer. Chem Biol Interact. 2015;234:332–8. doi: 10.1016/j.cbi.2014.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cai C, Balk SP. Intratumoral androgen biosynthesis in prostate cancer pathogenesis and response to therapy. Endocr Relat Cancer. 2011;18:R175–82. doi: 10.1530/ERC-10-0339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller WL, Auchus RJ. The “backdoor pathway” of androgen synthesis in human male sexual development. PLoS Biol. 2019;17:e3000198. doi: 10.1371/journal.pbio.3000198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deb S, Chin MY, Pham S, Adomat H, Hurtado-Coll A, Gleave ME, Tomlinson Guns ES. Steroidogenesis in peripheral and transition zones of human prostate cancer tissue. Int J Mol Sci. 2021;22:487. doi: 10.3390/ijms22020487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, True LD, Nelson PS. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68:4447–54. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu C, Lou W, Zhu Y, Yang JC, Nadiminty N, Gaikwad NW, Evans CP, Gao AC. Intracrine androgens and AKR1C3 activation confer resistance to enzalutamide in prostate cancer. Cancer Res. 2015;75:1413–22. doi: 10.1158/0008-5472.CAN-14-3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leon CG, Locke JA, Adomat HH, Etinger SL, Twiddy AL, Neumann RD, Nelson CC, Guns ES, Wasan KM. Alterations in cholesterol regulation contribute to the production of intratumoral androgens during progression to castration-resistant prostate cancer in a mouse xenograft model. Prostate. 2010;70:390–400. doi: 10.1002/pros.21072. [DOI] [PubMed] [Google Scholar]
- 18.Mostaghel EA, Solomon KR, Pelton K, Freeman MR, Montgomery RB. Impact of circulating cholesterol levels on growth and intratumoral androgen concentration of prostate tumors. PLoS One. 2012;7:e30062. doi: 10.1371/journal.pone.0030062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakamura Y, Suzuki T, Fukuda T, Ito A, Endo M, Moriya T, Arai Y, Sasano H. Steroid sulfatase and estrogen sulfotransferase in human prostate cancer. Prostate. 2006;66:1005–12. doi: 10.1002/pros.20426. [DOI] [PubMed] [Google Scholar]
- 20.Armstrong CM, Liu C, Liu L, Yang JC, Lou W, Zhao R, Ning S, Lombard AP, Zhao J, D’Abronzo LS, Evans CP, Li PK, Gao AC. Steroid sulfatase stimulates intracrine androgen synthesis and is a therapeutic target for advanced prostate cancer. Clin Cancer Res. 2020;26:6064–74. doi: 10.1158/1078-0432.CCR-20-1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin HK, Jez JM, Schlegel BP, Peehl DM, Pachter JA, Penning TM. Expression and characterization of recombinant type 2 3 alpha-hydroxysteroid dehydrogenase (HSD) from human prostate: demonstration of bifunctional 3 alpha/17 beta-HSD activity and cellular distribution. Mol Endocrinol. 1997;11:1971–84. doi: 10.1210/mend.11.13.0026. [DOI] [PubMed] [Google Scholar]
- 22.Penning TM, Burczynski ME, Jez JM, Hung CF, Lin HK, Ma H, Moore M, Palackal N, Ratnam K. Human 3alpha-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem J. 2000;351:67–77. doi: 10.1042/0264-6021:3510067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, Febbo PG, Balk SP. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815–25. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 24.Pfeiffer MJ, Smit FP, Sedelaar JP, Schalken JA. Steroidogenic enzymes and stem cell markers are upregulated during androgen deprivation in prostate cancer. Mol Med. 2011;17:657–64. doi: 10.2119/molmed.2010.00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wako K, Kawasaki T, Yamana K, Suzuki K, Jiang S, Umezu H, Nishiyama T, Takahashi K, Hamakubo T, Kodama T, Naito M. Expression of androgen receptor through androgen-converting enzymes is associated with biological aggressiveness in prostate cancer. J Clin Pathol. 2008;61:448–54. doi: 10.1136/jcp.2007.050906. [DOI] [PubMed] [Google Scholar]
- 26.Sun SQ, Gu X, Gao XS, Li Y, Yu H, Xiong W, Yu H, Wang W, Li Y, Teng Y, Zhou D. Overexpression of AKR1C3 significantly enhances human prostate cancer cells resistance to radiation. Oncotarget. 2016;7:48050–8. doi: 10.18632/oncotarget.10347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao J, Zhang M, Liu J, Liu Z, Shen P, Nie L, Guo W, Cai D, Liu J, Armstrong CM, Sun G, Chen J, Zhu S, Dai J, Zhang H, Zhao P, Zhang X, Yin X, Zhu X, Ni Y, Chen N, Zeng H. AKR1C3 expression in primary lesion rebiopsy at the time of metastatic castration-resistant prostate cancer is strongly associated with poor efficacy of abiraterone as a first-line therapy. Prostate. 2019;79:1553–62. doi: 10.1002/pros.23875. [DOI] [PubMed] [Google Scholar]
- 28.Liu C, Armstrong CM, Lou W, Lombard A, Evans CP, Gao AC. Inhibition of AKR1C3 activation overcomes resistance to abiraterone in advanced prostate cancer. Mol Cancer Ther. 2017;16:35–44. doi: 10.1158/1535-7163.MCT-16-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsuura K, Shiraishi H, Hara A, Sato K, Deyashiki Y, Ninomiya M, Sakai S. Identification of a principal mRNA species for human 3alpha-hydroxysteroid dehydrogenase isoform (AKR1C3) that exhibits high prostaglandin D2 11-ketoreductase activity. J Biochem. 1998;124:940–6. doi: 10.1093/oxfordjournals.jbchem.a022211. [DOI] [PubMed] [Google Scholar]
- 30.Thalmann GN, Anezinis PE, Chang SM, Zhau HE, Kim EE, Hopwood VL, Pathak S, von Eschenbach AC, Chung LW. Androgen-independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer. Cancer Res. 1994;54:2577–81. [PubMed] [Google Scholar]
- 31.Cai C, Chen S, Ng P, Bubley GJ, Nelson PS, Mostaghel EA, Marck B, Matsumoto AM, Simon NI, Wang H, Chen S, Balk SP. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 2011;71:6503–13. doi: 10.1158/0008-5472.CAN-11-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liedtke AJ, Adeniji AO, Chen M, Byrns MC, Jin Y, Christianson DW, Marnett LJ, Penning TM. Development of potent and selective indomethacin analogues for the inhibition of AKR1C3 (Type 5 17beta-hydroxysteroid dehydrogenase/prostaglandin F synthase) in castrate-resistant prostate cancer. J Med Chem. 2013;56:2429–46. doi: 10.1021/jm3017656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flanagan JU, Yosaatmadja Y, Teague RM, Chai MZ, Turnbull AP, Squire CJ. Crystal structures of three classes of non-steroidal anti-inflammatory drugs in complex with aldo-keto reductase 1C3. PLoS One. 2012;7:e43965. doi: 10.1371/journal.pone.0043965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Endo S, Oguri H, Segawa J, Kawai M, Hu D, Xia S, Okada T, Irie K, Fujii S, Gouda H, Iguchi K, Matsukawa T, Fujimoto N, Nakayama T, Toyooka N, Matsunaga T, Ikari A. Development of novel AKR1C3 inhibitors as new potential treatment for castration-resistant prostate cancer. J Med Chem. 2020;63:10396–411. doi: 10.1021/acs.jmedchem.0c00939. [DOI] [PubMed] [Google Scholar]
- 35.Steckelbroeck S, Jin Y, Gopishetty S, Oyesanmi B, Penning TM. Human cytosolic 3alpha-hydroxysteroid dehydrogenases of the aldo-keto reductase superfamily display significant 3beta-hydroxysteroid dehydrogenase activity: implications for steroid hormone metabolism and action. J Biol Chem. 2004;279:10784–95. doi: 10.1074/jbc.M313308200. [DOI] [PubMed] [Google Scholar]
- 36.Guerini V, Sau D, Scaccianoce E, Rusmini P, Ciana P, Maggi A, Martini PG, Katzenellenbogen BS, Martini L, Motta M, Poletti A. The androgen derivative 5alpha-androstane-3beta,17beta-diol inhibits prostate cancer cell migration through activation of the estrogen receptor beta subtype. Cancer Res. 2005;65:5445–53. doi: 10.1158/0008-5472.CAN-04-1941. [DOI] [PubMed] [Google Scholar]
- 37.Zhou M, Wang X, Xia J, Cheng Y, Xiao L, Bei Y, Tang J, Huang Y, Xiang Q, Huang S. A mansonone derivative coupled with monoclonal antibody 4D5-modified chitosan inhibit AKR1C3 to treat castration-resistant prostate cancer. Int J Nanomedicine. 2020;15:3087–98. doi: 10.2147/IJN.S241324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kikuchi A, Furutani T, Azami H, Watanabe K, Niimi T, Kamiyama Y, Kuromitsu S, Baskin-Bey E, Heeringa M, Ouatas T, Enjo K. In vitro and in vivo characterisation of ASP9521: a novel, selective, orally bioavailable inhibitor of 17beta-hydroxysteroid dehydrogenase type 5 (17betaHSD5; AKR1C3) Invest New Drugs. 2014;32:860–70. doi: 10.1007/s10637-014-0130-5. [DOI] [PubMed] [Google Scholar]
- 39.Loriot Y, Fizazi K, Jones RJ, Van den Brande J, Molife RL, Omlin A, James ND, Baskin-Bey E, Heeringa M, Baron B, Holtkamp GM, Ouatas T, De Bono JS. Safety, tolerability and anti-tumour activity of the androgen biosynthesis inhibitor ASP9521 in patients with metastatic castration-resistant prostate cancer: multi-centre phase I/II study. Invest New Drugs. 2014;32:995–1004. doi: 10.1007/s10637-014-0101-x. [DOI] [PubMed] [Google Scholar]
- 40.Liu C, Yang JC, Armstrong CM, Lou W, Liu L, Qiu X, Zou B, Lombard AP, D’Abronzo LS, Evans CP, Gao AC. AKR1C3 promotes AR-V7 protein stabilization and confers resistance to AR-targeted therapies in advanced prostate cancer. Mol Cancer Ther. 2019;18:1875–86. doi: 10.1158/1535-7163.MCT-18-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang B, Wu S, Fang Y, Sun G, He D, Hsieh JT, Wang X, Zeng H, Wu K. The AKR1C3/AR-V7 complex maintains CRPC tumour growth by repressing B4GALT1 expression. J Cell Mol Med. 2020;24:12032–43. doi: 10.1111/jcmm.15831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao J, Ning S, Lou W, Yang JC, Armstrong CM, Lombard AP, D’Abronzo LS, Evans CP, Gao AC, Liu C. Cross-resistance among next generation anti-androgen drugs through the AKR1C3/AR-V7 axis in advanced prostate cancer. Mol Cancer Ther. 2020;19:1708–1718. doi: 10.1158/1535-7163.MCT-20-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Purohit A, Foster PA. Steroid sulfatase inhibitors for estrogen- and androgen-dependent cancers. J Endocrinol. 2012;212:99–110. doi: 10.1530/JOE-11-0266. [DOI] [PubMed] [Google Scholar]
- 44.James MR, Skaar TC, Lee RY, MacPherson A, Zwiebel JA, Ahluwalia BS, Ampy F, Clarke R. Constitutive expression of the steroid sulfatase gene supports the growth of MCF-7 human breast cancer cells in vitro and in vivo. Endocrinology. 2001;142:1497–505. doi: 10.1210/endo.142.4.8091. [DOI] [PubMed] [Google Scholar]
- 45.Utsumi T, Yoshimura N, Takeuchi S, Ando J, Maruta M, Maeda K, Harada N. Steroid sulfatase expression is an independent predictor of recurrence in human breast cancer. Cancer Res. 1999;59:377–81. [PubMed] [Google Scholar]
- 46.Foster PA, Woo LW, Potter BV, Reed MJ, Purohit A. The use of steroid sulfatase inhibitors as a novel therapeutic strategy against hormone-dependent endometrial cancer. Endocrinology. 2008;149:4035–42. doi: 10.1210/en.2008-0223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sung CH, Im HJ, Park N, Kwon Y, Shin S, Ye DJ, Cho NH, Park YS, Choi HK, Kim D, Chun YJ. Induction of steroid sulfatase expression in PC-3 human prostate cancer cells by insulin-like growth factor II. Toxicol Lett. 2013;223:109–15. doi: 10.1016/j.toxlet.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 48.Mueller JW, Gilligan LC, Idkowiak J, Arlt W, Foster PA. The regulation of steroid action by sulfation and desulfation. Endocr Rev. 2015;36:526–63. doi: 10.1210/er.2015-1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sadozai H. Steroid sulfatase inhibitors: promising new therapy for breast cancer. J Pak Med Assoc. 2013;63:509–15. [PubMed] [Google Scholar]
- 50.Saito T, Kinoshita S, Fujii T, Bandoh K, Fuse S, Yamauchi Y, Koizumi N, Horiuchi T. Development of novel steroid sulfatase inhibitors; II. TZS-8478 potently inhibits the growth of breast tumors in postmenopausal breast cancer model rats. J Steroid Biochem Mol Biol. 2004;88:167–73. doi: 10.1016/j.jsbmb.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 51.Foster PA, Newman SP, Chander SK, Stengel C, Jhalli R, Woo LL, Potter BV, Reed MJ, Purohit A. In vivo efficacy of STX213, a second-generation steroid sulfatase inhibitor, for hormone-dependent breast cancer therapy. Clin Cancer Res. 2006;12:5543–9. doi: 10.1158/1078-0432.CCR-06-0632. [DOI] [PubMed] [Google Scholar]
- 52.Stanway SJ, Delavault P, Purohit A, Woo LW, Thurieau C, Potter BV, Reed MJ. Steroid sulfatase: a new target for the endocrine therapy of breast cancer. Oncologist. 2007;12:370–4. doi: 10.1634/theoncologist.12-4-370. [DOI] [PubMed] [Google Scholar]
- 53.Palmieri C, Januszewski A, Stanway S, Coombes RC. Irosustat: a first-generation steroid sulfatase inhibitor in breast cancer. Expert Rev Anticancer Ther. 2011;11:179–83. doi: 10.1586/era.10.201. [DOI] [PubMed] [Google Scholar]
- 54.Sang X, Han H, Poirier D, Lin SX. Steroid sulfatase inhibition success and limitation in breast cancer clinical assays: an underlying mechanism. J Steroid Biochem Mol Biol. 2018;183:80–93. doi: 10.1016/j.jsbmb.2018.05.009. [DOI] [PubMed] [Google Scholar]
- 55.Coombes RC, Cardoso F, Isambert N, Lesimple T, Soulie P, Peraire C, Fohanno V, Kornowski A, Ali T, Schmid P. A phase I dose escalation study to determine the optimal biological dose of irosustat, an oral steroid sulfatase inhibitor, in postmenopausal women with estrogen receptor-positive breast cancer. Breast Cancer Res Treat. 2013;140:73–82. doi: 10.1007/s10549-013-2597-8. [DOI] [PubMed] [Google Scholar]
- 56.Palmieri C, Stein RC, Liu X, Hudson E, Nicholas H, Sasano H, Guestini F, Holcombe C, Barrett S, Kenny L, Reed S, Lim A, Hayward L, Howell S, Charles Coombes R IRIStrial participants. IRIS study: a phase II study of the steroid sulfatase inhibitor Irosustat when added to an aromatase inhibitor in ER-positive breast cancer patients. Breast Cancer Res Treat. 2017;165:343–53. doi: 10.1007/s10549-017-4328-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Selcer KW, Kabler H, Sarap J, Xiao Z, Li PK. Inhibition of steryl sulfatase activity in LNCaP human prostate cancer cells. Steroids. 2002;67:821–6. doi: 10.1016/s0039-128x(02)00030-2. [DOI] [PubMed] [Google Scholar]
- 58.Day JM, Purohit A, Tutill HJ, Foster PA, Woo LW, Potter BV, Reed MJ. The development of steroid sulfatase inhibitors for hormone-dependent cancer therapy. Ann N Y Acad Sci. 2009;1155:80–7. doi: 10.1111/j.1749-6632.2008.03677.x. [DOI] [PubMed] [Google Scholar]
- 59.Pautier P, Vergote I, Joly F, Melichar B, Kutarska E, Hall G, Lisyanskaya A, Reed N, Oaknin A, Ostapenko V, Zvirbule Z, Chetaille E, Geniaux A, Shoaib M, Green JA. A phase 2, randomized, open-label study of irosustat versus megestrol acetate in advanced endometrial cancer. Int J Gynecol Cancer. 2017;27:258–66. doi: 10.1097/IGC.0000000000000862. [DOI] [PubMed] [Google Scholar]