

Abstract
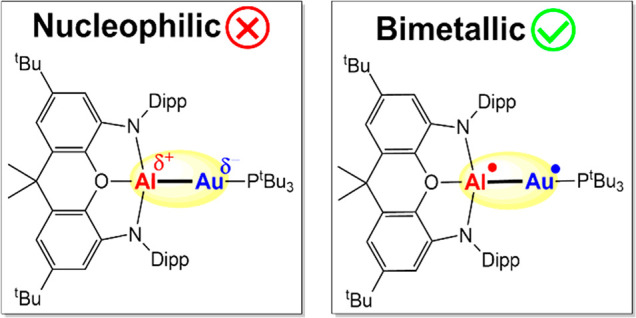
A gold-aluminyl complex has been recently reported to feature an unconventional gold nucleophilic center, which was revealed through reactivity with carbon dioxide leading to the Au-CO2 coordination mode. In this work, we computationally investigate the reaction mechanism, which is found to be cooperative, with the gold–aluminum bond being the actual nucleophile and Al also behaving as electrophile. The Au–Al bond is shown to be mainly of an electron-sharing nature, with the two metal fragments displaying a diradical-like reactivity with CO2.
Recently, a striking reactivity of CO2 with a complex bearing a Au–Al bond has been reported.1 A combination of the new generation aluminyl anion2 [K{Al(NON)}]2 with a phosphine gold tBu3PAuI affords the [tBu3PAuAl(NON)] complex (I) which, in reaction with CO2 (1 atm at room temperature in toluene), leads to the stable [tBu3PAuCO2Al(NON)] complex (II), where Au binds to the CO2 carbon atom (Scheme 1). This CO2 coordination mode has been considered as the revealing of an unconventional nucleophilic behavior of gold, which, in contrast, is well-known to be an extremely powerful electrophile in organic reactions involving unsaturated CC bonds.3 The authors suggested that the aluminyl anion [Al(NON)]− is able to induce an extremely polarized Au(δ-)-Al(δ+) bond, with a significant negative charge at the gold site, which is able to reverse its reactivity. DFT calculations combined with quantum theory of atoms in molecules (QTAIM) charge analysis have shown a substantial electron transfer from [Al(NON)]− to [tBu3PAu]+ (1.56 electrons) and a negative charge at Au (−0.82).1 This picture seems to be consistent with the difference in electronegativity values of the two metals (Au = 2.54, Al = 1.61 on the Pauling scale) and with the relativistic effects on gold which stabilize and contract the 6s orbital,4 resulting in the gold highest electron affinity (2.30 eV) among transition metals (other coinage metals have considerably smaller values, Cu 1.23 eV; Ag 1.30 eV).5 Complex I is not the only complex in which Au would act as a nucleophile toward polar multiple bond.6 Two copper-aluminyl complexes (III and V in Scheme 1) have been reported to insert CO2 into the Cu–Al bond, resulting in complexes IV and VI (Scheme 1), which are very much similar to complex II in terms of structure and kinetic stability.7 A significant covalency of the Al–Cu bond and only slightly negative charges on Cu (e.g., −0.09 in III) have been calculated, revealing here only a small polarization of the M(δ-)-Al(δ+) bond.
Scheme 1. Examples of Nucleophilic Gold (I),1 Copper (III, V),7 and Amido-Digermyne (VII)12 Compounds and Their CO2 Insertion Reaction Products (II, IV, VI, and VIII).

In addition to strongly polarized M(δ-)-Al(δ+) bonds in heterodinuclear complexes,8 CO2 activation by homodinuclear main-group element species,9,10 including diradicals,11 is not uncommon. Frenking and Jones12 demonstrated that the facile reduction of CO2 to CO by a symmetric amido-digermyne compound (VII) proceeds through an asymmetrical intermediate (VIII) that bears a structural analogy with complex II.
The similar coordination modes of CO2 in the Au–Al, Cu–Al, and Ge–Ge bonds is eye-catching, in view of the different degrees of polarization of the insertion metal–metal site, which is expected to determine the effectiveness of the metal nucleophilic behavior. This prompted us to computationally investigate the mechanism of the CO2 insertion into the pioneering nucleophilic [tBu3PAuAl(NON)] complex which has not been explored yet and the actual nucleophilic ability of Au in this “unorthodox reaction “.13 We demonstrate that the nucleophilic attack is actually performed by the Au–Al σ bond, revealing a bimetallic (Au/Al) cooperation in the CO2 binding. The attack is also assisted by a secondary interaction, involving the partially empty 3px atomic orbital of Al, which exploits its Lewis acidity. Transition state and intermediate structures found here suggest a radical-like insertion of CO2 in the Au–Al bond, which has been consistently shown to have an electron-sharing character.
The free energy profile for the CO2 insertion into the Au–Al bond of I was calculated using density functional theory (DFT) with the inclusion of relativistic effects, solvation, and dispersion interactions (see SI for computational details), and it is shown in Figure 1. Complex I was slightly simplified at the NON site (denoted as NON’). The modeling effect is evaluated in Table S1. The optimized geometries of [tBu3PAuAl(NON’)], RC, TSI, INT, TSII, and [tBu3PAuCO2Al(NON’)] (PC) complexes are reported in the SI (Figure S1) and show good agreement with available experimental data (Figure S2 and Table S2).
Figure 1.
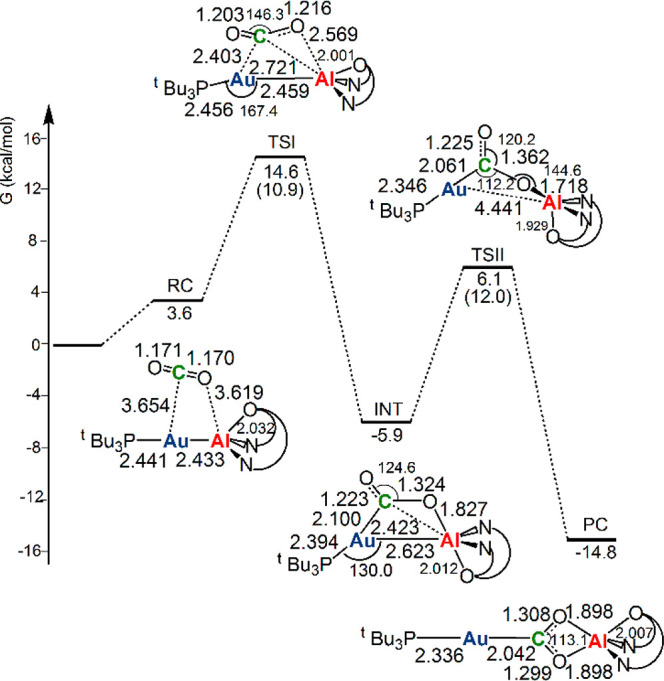
Free energy reaction profile for the CO2 insertion into the Au–Al bond in the [tBu3PAuAl(NON’)] complex. ΔG values refer to the energy of the separated reactants taken as zero. Activation free energy barriers are reported in parentheses. Selected interatomic distances (Å) and bond angles (degrees) are given with the molecular structures.
The nucleophilic attack to the CO2 carbon atom has a relatively low activation free energy barrier (ΔG# = 10.9 kcal/mol). At the TSI, the carbon atom of CO2 is both very close to Au (2.403 Å) and at a relatively short distance from Al (2.721 Å), and a substantial bending of CO2 and asymmetry between the two C–O bonds are also observed (see Table S3 for the evolution of the most relevant Mayer’s bond orders along the reaction path). Subsequent complete bonding of CO2 carbon atom to Au and oxygen atom to Al leads to the formation of intermediate INT, which is stabilized by 20.5 kcal/mol. As a result, the Au–Al bond distance slightly increases and the tBu3PAu moiety coordinates almost linearly with the carbon atom of CO2. A second transition state (TSII) is located with an activation free energy barrier ΔG# of 12.0 kcal/mol, showing a substantial breaking of the Au–Al bond. Finally, the oxygen atom of CO2 attack to the electrophilic Al center leads to the thermodynamically stable product complex PC. The overall CO2 insertion into the Au–Al bond is exergonic by −14.8 kcal/mol.
To get insights into the nature of the CO2 insertion process, we analyze the first activation barrier using the Activation Strain Model (ASM)14−16 (main results in Figure 2, left panel). The ASM formalism is briefly summarized in the SI, and all the results are reported in Table S4.
Figure 2.
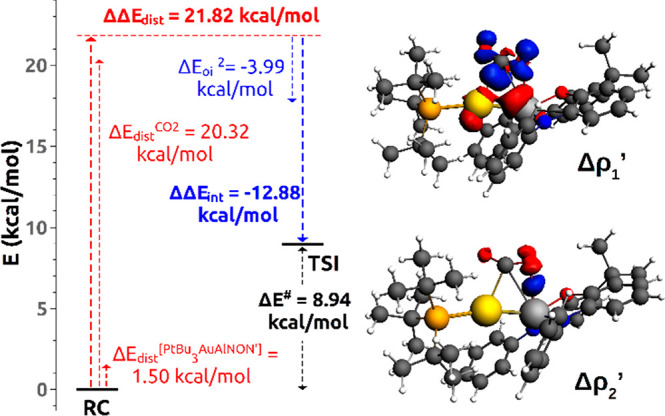
Activation Strain Model (ASM) decomposition of the electronic energy activation barrier ΔE# (left) (see text). Isodensity surfaces (2 me/a03) for the NOCV deformation density maps (charge flux is red → blue) corresponding to the Δρ1’ (top right) and Δρ2’ (bottom right) contributions to the CO2-[tBu3PAuAl(NON’)] fragments interaction in the transition state TSI.
The distortion energy contribution (ΔΔEdist = 21.82 kcal/mol) to the electronic energy activation barrier (ΔE#=8.94 kcal/mol) is almost completely associated with the CO2 bending (20.32 kcal/mol, see Table S5), whereas the stabilizing interaction contribution (ΔΔEint= −12.88 kcal/mol) mainly arises from the orbital interaction energy at TSI (−53.30 kcal/mol) (see Table S6). The results of the ETS-NOCV17 method coupled with the Charge Displacement (CD) Analysis18 (see SI for methodological details) are summarized below. In Figure 2 (right panel), the two most important components (Δρ1’ and Δρ2’) of the total deformation density are shown.
The main interaction component (Δρ1’) is clearly characterized by an electron density depletion localized on both Au and Al atoms and by an electron density accumulation at the CO2 site. This component is associated with a significant energy stabilization (−41.20 kcal/mol) and a charge transfer from the Au–Al bond region to carbon dioxide of 0.326 e. The decomposition into the donor and acceptor NOCV orbitals19 (Figure S3) shows that the electron density accumulation has the main contribution from the LUMO of CO2, while the electron density depletion shows contributions from the HOMO–2 and HOMO of the [tBu3PAuAl(NON’)] fragment, both representing the Au–Al σ bond, where Al 3s3pz–Au 6s6pz type orbitals are involved. Component Δρ2’ reveals an electron density accumulation at the Al center (note that its shape recalls that of an atomic px orbital), coming from one of the oxygen atoms of CO2. Decomposition into donor and acceptor NOCV orbitals (Figure S4) shows that the main contribution to the donor orbital is the HOMO of CO2, whereas a clear characterization of the acceptor orbital is less straightforward, since several delocalized unoccupied MOs, all with small Al 3px orbital mixing, contribute to it. The Δρ2’ contribution is not negligible: 0.047 e are transferred toward Al from CO2, with an associated orbital interaction energy of −3.99 kcal/mol (which notably accounts for one-third of the interaction stabilization to the activation barrier ΔΔEint).
The reaction mechanism in Figure 1 bears surprising analogies with the first steps of the reaction profile for the reduction of CO2 to CO by [LGe-GeL] (see Figure 3 of ref (12)), although complex II does not evolve to CO elimination (the resulting oxide complex [tBu3PAuOAl(NON’)][CO] has been calculated to be highly unstable with ΔG = 29.8 kcal/mol). The high reactivity of digermynes has been often attributed to the nonnegligible biradical character in these systems.11,19,20 A possible diradicaloid character of the Au–Al bond in the [tBu3PAuAl(NON)] complex is certainly very intriguing. Remarkably, the coordination modes of CO2 in the separated neutral open shell doublet [tBu3PAu(CO2)]· and [CO2Al(NON’)]· fragments closely match those found at the TSI and INT (Figures S5, S6 and discussion therein). This prompted us to review the Au–Al bond nature in complex I. We carry out the CD-NOCV analysis on the [tBu3PAuAl(NON’)] complex by choosing the open-shell radical fragments [tBu3PAu]· and [(NON’)Al]· on the basis of EDA analysis21 using different fragmentations (Table S7 and ref (22)). The main results of the CD-NOCV analysis are reported in Figure 3.
Figure 3.
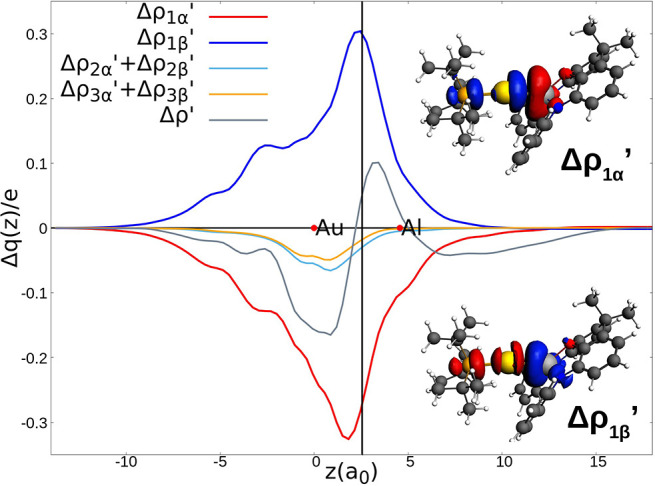
Charge Displacement (CD-NOCV) curves for the interaction between doublet [tBu3PAu]· and [(NON’)Al]· fragments in the [tBu3PAuAl(NON’)] complex. Red dots indicate the position of the nuclei along the z axis. The vertical solid line marks the isodensity boundary between the fragments. Positive (negative) values of the curve indicate right-to-left (left-to-right) charge transfer (see Supporting Information for details). Insets: isodensity surfaces (1 me/a03) of the two NOCV deformation densities Δρ1α’ (top, right) and Δρ1β’ (bottom, right) (charge flux is red → blue).
The CD-NOCV curves clearly exhibit two similar main charge fluxes in opposite directions, which consist of an electron transfer from Au toward Al (Δρ1α’, red curve and inset in Figure 3) and from Al toward Au (Δρ1β’, blue curve and inset in Figure 3). These two charge fluxes have also similar CT absolute values (0.272 and 0.299 e for Δρ1α’ and Δρ1β’, respectively), associated orbital interaction energies (−32.66 and −24.49 kcal/mol for Δρ1α’ and Δρ1β’, respectively), and NOCV eigenvalues (0.45 and 0.42 for Δρ1α’ and Δρ1β’, respectively) as shown in Table S8. Other contributions to the Au–Al bond (CD-NOCV curves labeled as Δρ2α’ + Δρ2β’ and Δρ3α’ + Δρ3β’) describe the π back-donations (see Figure S7) and are definitely smaller in magnitude. The CD curve associated with the total deformation density (Δρ’) shows an almost symmetric charge accumulation at the bonding region with a slightly positive CT (0.05 e) going from the radical [(NON’)Al]· to [tBu3PAu]·, which can be associated with the net polarization of the Au–Al bond. To definitely assess the electron-sharing bond nature, the CD-NOCV analysis for a nonpolar covalent bond system, such as the homonuclear Au2 molecule, is presented in Figures S8, S9 for comparison. The CD-NOCV curves in Figure 3 and Figure S8 are indeed very similar.
We also find that this bonding scheme is not peculiar of the Au–Al bond in [tBu3PAuAl(NON’)]. A qualitatively analogous picture has been also obtained for two complexes with a Cu–Al bond, i.e., a model [tBu3PCuAl(NON’)] (where we substituted gold with copper and reoptimized the structure) and the experimental complex III.7 (see Tables S9, S10 and Figures S10, S11; for a comparative EDA, see Table S11). Before concluding, we comment on the two main theoretical points which suggested in ref (1) the formation of a strongly polarized Au–Al bond with a large negative charge on Au, i.e., (i) the Au/Al difference in the atomic electronegativity values and (ii) the atomic charge on Au. Concerning point (i), although the large Au/Al atomic electronegativity difference (0.93 on the Pauling scale, 2.12 (calculated) and 2.19 (experimental) on the Mulliken scale (see Table S12) seems to be inconsistent with an electron-sharing bond, the calculated Mulliken “molecular electronegativity” is practically identical (2.56 vs 2.53 eV for [tBu3PAu]· and [Al(NON’)]·, respectively) (Table S13). For point (ii), the atomic charges show a huge variability range with the chosen method (consistently with the highly directional and diffuse HOMO of the aluminyl anion, Figure S12): qAu, from −0.83 to +0.22; qAl, from 2.18 to 0.18 (Table S14) which makes an assessment of the bond polarization based on these numbers impossible.
In summary, the reactivity shown here points out that both Au and Al centers act as nucleophiles (radical-like mechanism), with the electrophilic behavior of Al also assisting the interaction with CO2. The Au–Al bonding picture in [tBu3PAuAl(NON)] is consistent with an Au(0) involved in an electron-sharing bond-type. An important general conclusion is that the reactivity of metal-aluminyl complexes with CO2 resulting in M-CO2 coordination mode cannot be considered in itself as a probe for a strongly polarized M(δ-)-Al(δ+) bond and for the metal behaving as a standard nucleophilic center. We believe that the interpretative framework given here may be useful for future experimental investigations on CO2 capture and reduction by these unconventional bimetallic complexes.
Acknowledgments
This work was supported by the Ministero dell’Università e della Ricerca (MUR, project AMIS, through the program “Dipartimenti di Eccellenza −2018-2022″) and the University of Perugia (“Fondo Ricerca di Base 2019″, P.B.).
Glossary
Abbreviations
- NON
4,5-bis(2,6-diisopropylanilido)-2,7-ditert-butyl-9,9-dimethylxanthene
- NHCIPr
N,N′-di-isopropyl-4,5-dimethyl-2-ylidene
- Me2CAAC
1-((2,6-di-isopropylphenyl)-3,3,5,5-tetramethyl-pyrrolidin-2-ylidene)
- SiNDipp
(CH2SiMe2NDipp)2
- Ar*
C6H2{C(H)Ph2}2Me-2,6,4.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c06728.
Methodology and computational details; optimized geometries; Mayer’s bond order along the path; Activation Strain Model and EDA results; donor and acceptor NOCV orbital decomposition; analysis of the [(NON’)Al] and [tBu3PAu] fragments; selection of fragmentations for bonding analysis; CD-NOCV analysis complete results; CD-NOCV and EDA analysis of [tBu3PCuAl(NON’)] and [NHCiPrCuAlSiNDipp] complexes; Au, Al, and “molecular” ionization energy and electron affinity; AIM, Mulliken, Hirshfeld, VDD, Multipole Derived, CM5 calculated atomic charges on Au and Al; [(NON’)Al] HOMO electron density map (PDF)
Author Contributions
$ D.S., L.B., and P.B. contributed equally to this paper
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. All authors contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Hicks J.; Mansikkamäki A.; Vasko P.; Goicoechea J. M.; Aldridge S. A nucleophilic gold complex. Nat. Chem. 2019, 11, 237–241. 10.1038/s41557-018-0198-1. [DOI] [PubMed] [Google Scholar]
- Hicks J.; Vasko P.; Goicoechea J. M.; Aldridge S. The aluminyl anion: a new generation of aluminium nucleophile. Angew. Chem., Int. Ed. 2021, 60, 1702–1713. 10.1002/anie.202007530. [DOI] [PubMed] [Google Scholar]
- a Obradors C.; Echavarren A. M. Intriguing mechanistic labyrinths in gold(I) catalysis. Chem. Commun. 2014, 50, 16–28. 10.1039/C3CC45518A. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wang Y.-M.; Lackner A. D.; Toste F. D. Development of catalysts and ligands for enantioselective gold catalysis. Acc. Chem. Res. 2014, 47, 889–901. 10.1021/ar400188g. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hashmi A. S. K. Dual gold catalysis. Acc. Chem. Res. 2014, 47, 864–876. 10.1021/ar500015k. [DOI] [PubMed] [Google Scholar]; d Zhang L. A non-diazo approach to α-oxo gold carbenes via gold-catalyzed alkyne oxidation. Acc. Chem. Res. 2014, 47, 877–888. 10.1021/ar400181x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwerdtfeger P. Relativistic and electron-correlation contributions in atomic and molecular properties: benchmark calculations on Au and Au2. Chem. Phys. Lett. 1991, 183, 457–463. 10.1016/0009-2614(91)90409-3. [DOI] [Google Scholar]
- Andersen T.; Haugen H. K.; Hotop H. Binding energies in atomic negative ions: III. J. Phys. Chem. Ref. Data 1999, 28, 1511–1533. 10.1063/1.556047. [DOI] [Google Scholar]
- Suzuki A.; Guo X.; Lin Z.; Yamashita M. Nucleophilic reactivity of the gold atom in a diarylborylgold(I) complex toward polar multiple bonds. Chem. Sci. 2021, 12, 917–928. 10.1039/D0SC05478J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.-Y.; Schwamm R. J.; Hill M. S.; Mahon M. F.; McMullin C. L.; Rajabi N. A. Ambiphilic Al-Cu bonding. Angew. Chem., Int. Ed. 2021, 60, 14390–14393. 10.1002/anie.202104658. [DOI] [PMC free article] [PubMed] [Google Scholar]; Note that the Al-Cu complexes reported here are referred as “alumanyl”.
- Escomel L.; Del Rosal I.; Maron L.; Jeanneau E.; Veyre L.; Thieuleux C.; Camp C. Strongly polarized iridium δ- - aluminium δ+ pairs: unconventional reactivity patterns including CO2 cooperative reductive cleavage. J. Am. Chem. Soc. 2021, 143, 4844–4856. 10.1021/jacs.1c01725. [DOI] [PubMed] [Google Scholar]
- Stoy A.; Böhnke J.; Jiménez-Halla J. O. C.; Dewhurst R. D.; Thiess T.; Braunschweig H. CO2 binding and splitting by boron-boron multiple bonds. Angew. Chem., Int. Ed. 2018, 57, 5947–5951. 10.1002/anie.201802117. [DOI] [PubMed] [Google Scholar]
- Weetman C.; Bag P.; Szilvási T.; Jandl C.; Inoue S. CO2 fixation and catalytic reduction by a neutral aluminium double bond. Angew. Chem., Int. Ed. 2019, 58, 10961–10965. 10.1002/anie.201905045. [DOI] [PubMed] [Google Scholar]
- Hinz A.; Schulz A.; Villinger A. Metal-free activation of hydrogen, carbon dioxide, and ammonia by the open-shell singlet biradicaloid [P(μ-NTer)]2. Angew. Chem., Int. Ed. 2016, 55, 12214–12218. 10.1002/anie.201606892. [DOI] [PubMed] [Google Scholar]
- Li J.; Hermann M.; Frenking G.; Jones C. The facile reduction of carbon dioxide to carbon monoxide with an amido-digermyne. Angew. Chem., Int. Ed. 2012, 51, 8611–8614. 10.1002/anie.201203607. [DOI] [PubMed] [Google Scholar]
- Bourissou D. Changing the gold standard. Nat. Chem. 2019, 11, 199–203. 10.1038/s41557-019-0223-z. [DOI] [PubMed] [Google Scholar]; This work also emphasizes the high relevance of a computational study on the gold-aluminyl complex I by an explicit request (″Density functional theory calculations would be very welcome to shed light on the mechanism of this unorthodox reaction “.).
- Fernández I.; Bickelhaupt F. M. The activation strain model and molecular orbital theory: understanding and designing chemical reactions. Chem. Soc. Rev. 2014, 43, 4953–4967. 10.1039/C4CS00055B. [DOI] [PubMed] [Google Scholar]
- Bickelhaupt F. M.; Houk K. N. Analyzing reaction rates with the distortion/interaction-activation strain model. Angew. Chem., Int. Ed. 2017, 56, 10070–10086. 10.1002/anie.201701486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeeren P.; van der Lubbe S. C. C.; Fonseca Guerra C.; Bickelhaupt F. M.; Hamlin T. A. Understanding chemical reactivity using the activation strain model. Nat. Protoc. 2020, 15, 649–667. 10.1038/s41596-019-0265-0. [DOI] [PubMed] [Google Scholar]
- Mitoraj M. P.; Michalak A.; Ziegler T. A combined charge and energy decomposition scheme for bond analysis. J. Chem. Theory Comput. 2009, 5, 962–975. 10.1021/ct800503d. [DOI] [PubMed] [Google Scholar]
- a Belpassi L.; Infante I.; Tarantelli F.; Visscher L. The chemical bond between Au(I) and the noble gases. Comparative study of NgAuF and NgAu+ (Ng = Ar, Kr, Xe) by density functional and coupled cluster methods. J. Am. Chem. Soc. 2008, 130, 1048–1060. 10.1021/ja0772647. [DOI] [PubMed] [Google Scholar]; b Bistoni G.; Rampino S.; Tarantelli F.; Belpassi L. Charge-displacement analysis via natural orbitals for chemical valence: charge transfer effects in coordination chemistry. J. Chem. Phys. 2015, 142, 084112. 10.1063/1.4908537. [DOI] [PubMed] [Google Scholar]; c Bistoni G.; Belpassi L.; Tarantelli F. Advances in charge displacement analysis. J. Chem. Theory Comput. 2016, 12, 1236–1244. 10.1021/acs.jctc.5b01166. [DOI] [PubMed] [Google Scholar]
- The same procedure was successfully employed for studying the H2 activation with a digermyne complexZhao L.; Huang F.; Lu G.; Wang Z.-X.; von Ragué Schleyer P. Why the mechanisms of digermyne and distannyne reactions with H2 differ so greatly. J. Am. Chem. Soc. 2012, 134, 8856–8868. 10.1021/ja300111q. [DOI] [PubMed] [Google Scholar]
- Jung Y.; Brynda M.; Power P. P.; Head-Gordon M. Ab initio quantum chemistry calculations on the electronic structure of heavier alkyne congeners: diradical character and reactivity. J. Am. Chem. Soc. 2006, 128, 7185–7192. 10.1021/ja055374c. [DOI] [PubMed] [Google Scholar]
- a Ziegler T.; Rauk A. On the calculation of bonding energies by the Hartree Fock Slater method. Theor. Chim. Acta 1977, 46, 1–10. 10.1007/BF02401406. [DOI] [Google Scholar]; b Morokuma K. Molecular orbital studies of hydrogen bonds. III. C = O···H-O hydrogen bond in H2CO···H2O and H2CO···2H2O. J. Chem. Phys. 1971, 55, 1236. 10.1063/1.1676210. [DOI] [Google Scholar]
- a Zhao L.; von Hopffgarten M.; Andrada D. M.; Frenking G. Energy decomposition analysis. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2018, 8, e1345 10.1002/wcms.1345. [DOI] [Google Scholar]; b Jerabek P.; Schwerdtfeger P.; Frenking G. Dative and electron-sharing bonding in transition metal compounds. J. Comput. Chem. 2019, 40, 247–264. 10.1002/jcc.25584. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.