

Abstract
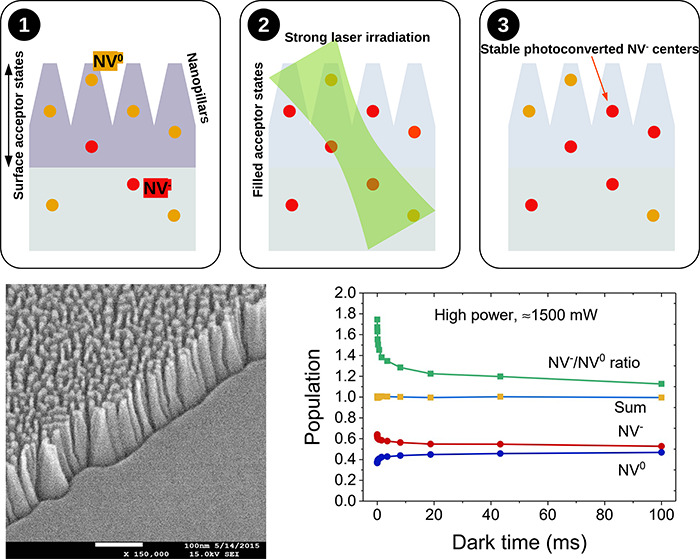
Shallow, negatively charged nitrogen-vacancy centers (NV–) in diamond have been proposed for high-sensitivity magnetometry and spin-polarization transfer applications. However, surface effects tend to favor and stabilize the less useful neutral form, the NV0 centers. Here, we report the effects of green laser irradiation on ensembles of nanometer-shallow NV centers in flat and nanostructured diamond surfaces as a function of laser power in a range not previously explored (up to 150 mW/μm2). Fluorescence spectroscopy, optically detected magnetic resonance (ODMR), and charge-photoconversion detection are applied to characterize the properties and dynamics of NV– and NV0 centers. We demonstrate that high laser power strongly promotes photoconversion of NV0 to NV– centers. Surprisingly, the excess NV– population is stable over a timescale of 100 ms after switching off the laser, resulting in long-lived enrichment of shallow NV–. The beneficial effect of photoconversion is less marked in nanostructured samples. Our results are important to inform the design of samples and experimental procedures for applications relying on ensembles of shallow NV– centers in diamond.
Keywords: diamond, nitrogen-vacancy centers, NV0, photoconversion, nanostructures, surface effects
Introduction
Negatively charged nitrogen-vacancy (NV–) centers are solid-state defects in the diamond lattice whose properties have been exploited to detect temperature gradients,1,2 magnetic3,4 and electric fields5,6 at the nanoscale, and interactions with magnetic molecules and nanoparticles.7−10 Due to their biocompatibility, NV–-enriched fluorescent nanodiamonds represent promising sensors to investigate the cellular microenvironment in living tissues and their use in high-sensitivity bioassays has been proposed.11−13 Furthermore, NV centers can be used in dynamic nuclear polarization (DNP) protocols where the polarization of the NV–s is transferred to 13C nuclei, leading to hyperpolarization of 13C nuclei in the diamond lattice.14−16 Substantial efforts are ongoing to promote polarization transfer from shallow NV centers to molecules absorbed at the diamond surface,17 thus enabling hyperpolarization of high-sensitivity tracers for biomedical magnetic resonance imaging.
For all these applications, the proximity of NV– centers to the diamond surface, where NVs can effectively interact with spins outside the diamond lattice, is of paramount importance, as the coupling strength between magnetic dipoles decreases with increasing distance. To this end, specially engineered layers of shallow NV–s18,19 as well as nanodiamonds20 have been proposed.
Unfortunately, surface states and defects at and close to the diamond surface can affect the charge stability of NV centers, reducing the availability of magnetically active NV– centers in favor of the neutral form (NV0 centers), which do not present the same detection features. The relative stability and interconversion between the neutral and negatively charged states of the NV centers have been the object of investigation in several studies,21−25 and various attempts have been made to increase the stability of NV– centers, for instance, by surface termination,26 by doping of the diamond lattice,27 or by application of an electric field.28
Here, we investigate the effects of laser power and surface structures on charge stability and attainable spin polarization of shallow NV– centers in high-purity diamonds. Specifically, we aim to establish experimental conditions that maximize the availability of magnetically active NV– at the diamond surface. To this end, we apply fluorescence spectroscopy, as well as optically detected magnetic resonance (ODMR), in electronic-grade diamond samples implanted with nitrogen ions at different depths and with flat or nanostructured surfaces.
Importantly, we study the effects of these factors in a regime of high-power laser irradiation not previously explored, and we assess the time dependence of the laser-induced NV–/NV0 distribution during and after irradiation.
Material and Methods
Sample Fabrication
We used four electronic-grade, single-crystal diamond plates, with an initial nitrogen concentration lower than 5 ppb (Element Six Technologies Ltd). The samples were 2.0 × 2.0 mm2, 0.5 mm thick, with {100} face orientation, and they were double-side polished, with roughness Ra < 5 nm on both sides.
Two of these samples were nanostructured to increase the surface-to-volume ratio. Samples nanostructuring was carried out using inductively coupled plasma-reactive ion etching (ICP-RIE) with oxygen gas, with a noncontinuous gold layer as a mask.29 Gold was deposited by DC sputtering (KS500 confocal sputtering system; Kenosistec Srl), using Ar gas (Figure 1a). Etching was performed by means of ICP-RIE (SI500—Sentech Instruments GmbH), using 700 W ICP power, 50 W plate power, and 40 sccm O2, at 1 Pa pressure, for 60 s (Figure 1b). After the ICP-RIE process, gold was removed with a commercial gold etchant (Sigma-Aldrich). Scanning electron microscopy (SEM) shows a tight arrangement of nanostructures (“nanopillars”) (Figure 1e) over the diamond surface, with a height of ≈150 nm. SEM images were acquired before gold removal.
Figure 1.
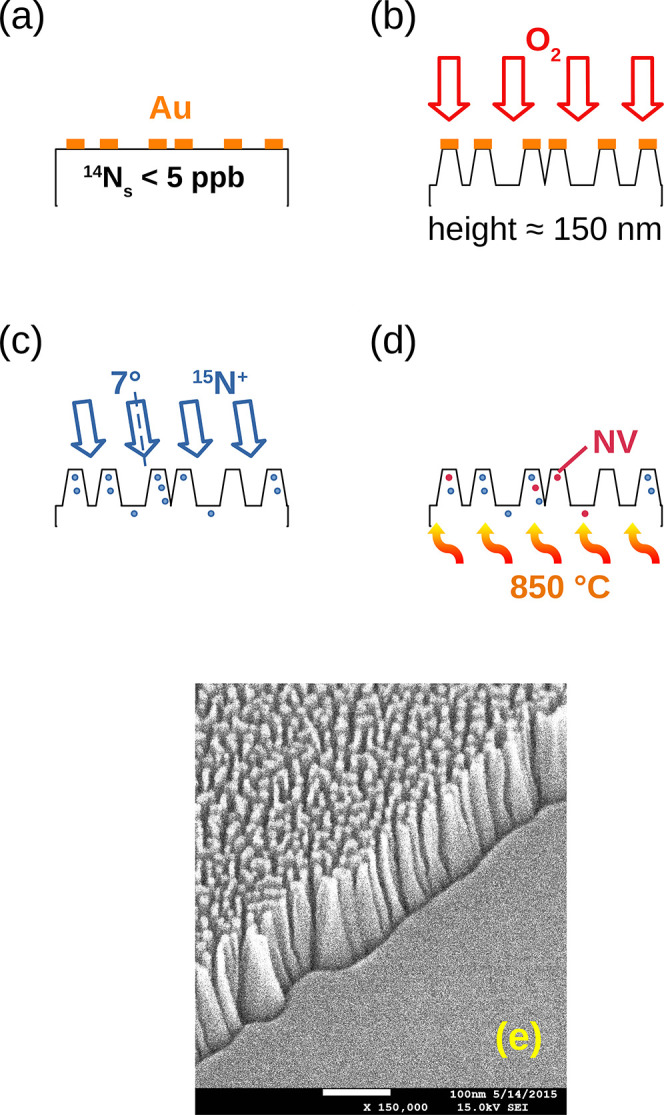
Nanofabrication process and synthesis of shallow NV centers in nanostructured samples. Electronic grade diamond with a very low concentration of nitrogen, less than 5 ppb, was masked with gold by sputtering (a) and then etched using oxygen reactive ion etching (b) to fabricate nanostructures, onto which nitrogen was implanted after gold removal (c). Samples were then annealed (d). SEM images show ≈150 nm high nanopillar-like structures (e). The SEM image was taken between steps (b) and (c), with still some conductive gold on the tips to improve image quality. Flat samples were implanted with 15N ions and then annealed, without application of any etching procedure.
Samples were implanted with 15N (INNOVION Corp., San Jose, CA) at a fluence of 1013 cm–2, at 7° angle from normal to avoid ion-channeling30−32 (Figure 1c), in two different energy conditions: one flat sample and one nanostructured sample were implanted at 10 keV; the other flat and nanostructured ones were implanted at 20 keV (see Table 1). Ion average ranges in the flat samples calculated by SRIM (http://www.srim.org, SRIM-2013.00) are, respectively, ≈15 nm, with straggling of 5 nm, and ≈27 nm with straggling of 8.5 nm.33
Table 1. Type of Surfaces and Implantation Energies of 15N+ for the Four Electronic-Grade Samples Used in This Work.
| sample name | surface type | implantation energy (keV) |
|---|---|---|
| F1 | flat | 20 |
| F2 | flat | 10 |
| N1 | nanostructured | 20 |
| N2 | nanostructured | 10 |
We then annealed the four samples at a temperature of 850 °C for 2 h in high vacuum conditions (10–6 mbar) (Figure 1d). The setup employed consists of a vacuum chamber equipped with a dry pumping system to avoid hydrocarbon contamination and a resistive heater. The heating element is a tantalum box, 5 × 5 × 5 mm3 in size, that keeps the sample in thermal equilibrium with the surrounding blackbody radiation. The temperature was externally monitored through an optical window using a pyrometer, pointed to the tantalum box. The heating and cooling rates were of 10 °C/min to avoid thermal stresses in the diamond structures. At this temperature, vacancies and interstitial nitrogen atoms become mobile but substitutional nitrogen atoms are fixed in their position.34
We expect deeper NV centers for the 20 keV than for 10 keV implantation energies on average. For a rough estimate of the density of NV centers in the flat samples, we consider a conversion efficiency of ≈1% of the substitutional nitrogen after electron irradiation and annealing35 This corresponds to an NV areal density of 103 μm–2. Dividing by the width of the implanted region (10 nm indicatively, the order of magnitude of nitrogen straggling), we find an NV density of 0.57 ppm. In the nanostructured samples, a more inhomogeneous distribution profile is expected, as the implantation occurs on an irregular profile.
The fabrication process for the nanostructured samples is summarized in Figure 1a–d. For flat samples, implantation and annealing procedures were the same as in the nanostructured samples but were performed on a flat surface.
Experimental Setup
Full fluorescence spectra of NVs were taken with a confocal micro-Raman setup (LabRam Aramis, Jobin-Yvon Horiba), equipped with a 532 nm DPSS laser and an air-cooled multichannel CCD detector. Laser power was attenuated by neutral filters with 0, 1, or 2 optical densities (OD), resulting in laser intensities of 41 mW/μm2 (OD0), 4.1 mW/μm2 (OD1), and 0.41 mW/μm2 (OD2). When referring to full fluorescence spectra taken with the micro-Raman, we use interchangeably laser intensities or optical densities.
Time-resolved measurements were performed with a home-built fluorescence microscope equipped with a green 532 nm laser (5 W, Verdi, Coherent) and a 0.25 NA objective (Plan N, Olympus). A focal spot size of 10 μm2 was estimated. We used the same 532 nm laser to initialize and read out the state of the system. Fluorescence was collected by the objective and sent to a photon-counter module (Excelitas SPCM-AQRH-14-FC). Laser power at the sample surface was attenuated with a combination of absorptive filters and ranged from a maximum value of 1500 to 1 mW, depending on the experiment’s purpose. Correspondingly, the laser intensity could be tuned from a maximum of 150 mW/μm2 to 100 μW/μm2. For nanostructured samples, irradiation occurred on an irregular “conical frustum” surface, with flat tops and regions between the structures, and tilted side surfaces. As a result, laser power (and laser intensity) should be downscaled by a factor of 2.5–3. Even if this reduction in intensity may play a role, it cannot explain the large differences between flat and nanostructured samples reported in the Results section (differences are as if the laser power were 2 orders of magnitude less for the nanostructured samples). With the laser spot size calculated above, we estimated 104 NVs to be simultaneously excited by the laser.
We collected fluorescence through two different spectral windows, selected by combinations of filters: 550 to 600 nm (“550–600 nm”) and >750 nm (“750 nm+”). The 550–600 nm window is centered around the zero-phonon-line (ZPL) of the NV0 centers, at 575 nm, and collects exclusively the NV0 signal. Conversely, the >750 nm window is more selective for the NV– centers, even if a tail of the NV0 fluorescence can still leak through (see Section S1 of the Supporting Information).
We used a high-power acousto-optic modulator (ISOMET 523C-6) for pulsed-laser sequences. Typically, a variable preparation pulse was applied to initialize the system, followed by a read-out pulse. A read-out pulse of 5 μs provided good sensitivity while minimally perturbing the distribution of NV charge states even at the highest laser power used (see Section S2 of the Supporting Information).
The microwave lines were composed of a microwave generator (Keysight N5171B), an amplifier (ZHL-16W-43-S+), and a millimeter-sized gold-coated copper loop placed below the sample. A custom three-axis Helmholtz coil (Micro Magnetics, Inc) provided a static magnetic field along any desired direction. In some experiments, a strong magnetic field of 750 G was applied along the z-axis, perpendicularly to the top diamond face, to quench the spin dynamics and enable selective measurement of charge dynamics.36 The z-axis was set parallel to the [100] crystallographic direction so that the applied magnetic field had the same magnitude along the four possible orientations of the NV centers axes in the diamond lattice.
Results
Figure 2 shows the effects of laser power on the fluorescence (FL) spectra from different samples. The schematics of the energy level structures of NV0 and NV– centers are reported in Figure 2a to facilitate the interpretation of the experimental results. In short, NVs are characterized by a ground state (a spin-triplet 3A2 for NV–, and 2E for NV0) and excited states (3E for NV– and 2A1 for NV0) positioned within the diamond band gap,37 each accompanied by a phonon band. Additionally, the NV– has two intermediate states (1A1 and 1E) coupled to the ground and the excited states. The existence of this coupling enables a nonradiative transition that accumulates population in the ms = 0 levels of the ground state under constant laser irradiation (blue arrows). A green laser (532 nm) is used to excite both the NV– and the NV0 centers (green arrows). Laser irradiation can also induce a charge-state conversion, either by promoting an electron from the NV– excited state into the conduction band (NV– → NV0 conversion) or by exciting an electron from the valence band to the NV0 ground state (NV0 → NV– conversion).38 These two routes of photoconversion are represented by green dotted arrows. Experimental observations suggest that, upon switching off the laser, the system goes back to equilibrium through charge conversion in the dark, a process attributed to electron tunneling (black dotted arrows). Indeed, tunneling couples the NV centers to proximal electron donor or acceptor states, such as substitutional nitrogen atoms, vacancy complexes, and surface states.23,25,39
Figure 2.

Fluorescence properties of NV centers. Electronic structures of NV– and NV0 centers are represented in (a). Green arrows indicate optical excitation and photoconversion (continuous and dotted arrows, respectively). Red and yellow arrows denote radiative decay from NV– and NV0 centers. Blue arrows indicate the nonradiative pathway, which leads to polarization of the ms = 0 NV– ground state. Black arrows represent tunneling transitions in the dark between the two charged states. (b) Both NV– and NV0 fluorescence spectra were detected in implanted samples; no signal was detectable in nonimplanted, nonannealed samples (inset). Notably, emission from NV–s increases with laser power, at the expense of NV0s. (c): the FL of nanostructured samples is lower compared to flat samples at all laser power levels (indicated by the filter optical density (OD)), with a larger component of NV0. The ratio of NV– and NV0 FL intensities is plotted in (d), as a function of total integrated fluorescence and laser intensity. The NV–/NV0 ratio increases with laser power in all samples and is systematically larger in flat samples.
Figure 2b shows fluorescence spectra from one sample (F2) at different laser powers. The NV charge states are characterized by different fluorescence spectra, with zero-phonon lines (ZPL) at 575 and 638 nm for the NV0 and NV–, respectively, and a phonon sideband peaked around 620 nm for the NV0 and 700 nm for the NV–, and extending up to ≈800 nm in both cases. Spectral features from both NV– and NV0 centers are apparent, demonstrating the co-occurrence of the two charge states in this sample. The NV centers originate exclusively from implanted nitrogen (pristine diamond samples before implantation and annealing show no detectable fluorescence under a maximum laser intensity excitation of 41 mW/μm2 but only the first- and second-order Raman peaks, shown in the inset). As the laser power intensity increases by 2 orders of magnitude (from 0.41 mW/μm2 for filter optical density OD = 2 to 41 mW/μm2 at OD = 0), the relative intensity of the NV– band increases with respect to that of the NV0 band. This indicates some degree of NV0 → NV– photoconversion at higher laser powers under continuous excitation (curves are normalized to 1 at 638 nm). The spectra in Figure 2c reflect the relative abundances of NV0 and NV– centers in the nanostructured and flat samples for different laser powers. For the two nanostructured samples, the overall FL is lower (not apparent in the figure, where spectra are normalized for comparison), and the FL spectra show a higher component in the NV0 region (blue and red curves compared to yellow and green curves). In all samples, an increase in the NV– component and a decrease in NV0 is observed with increasing laser power. We summarize these observations in Figure 2d, where the x-axis represents the integrated total FL, comprising the signals from both NV types, while the y-axis indicates the ratio of the areas under the NV– and NV0 curves. Fluorescence curves were deconvolved into NV0 and NV– components before calculating the values of Figure 2d, similar to that in Alsid et al.40 (the process of integration and calculating the FL ratio is described in Section S1 of the Supporting Information). For all samples, the NV–/NV0 ratio increases with laser power, suggesting stronger NV0 → NV– photoconversion at higher laser powers. Nanostructured samples (N1 and N2) contain fewer NV centers (reflected by an overall lower FL, integrated over the entire spectrum) and a higher fraction of NV0 compared to flat samples.
To explore the dynamics of laser-induced charge switching and subsequent return to equilibrium, we applied two pulsed experimental schemes (Figure 3). In these experiments, a strong magnetic field (750 G) was applied along the [100] direction to suppress the effects of spin polarization of FL and selectively investigate charge dynamics. In fact, a sufficiently strong magnetic field (>600 G) quenches the polarization of the ms = 0 ground state by spin-state mixing and leads to a reduction of the spin-related component in the FL.36,41,42
Figure 3.

Detection of charge dynamics. (a) Pulse sequence adopted to investigate charge dynamics under laser pumping, for variable irradiation pulse length τL. Laser-induced NV0 → NV– photoconversion results in a decrease of the NV0 signal and an increase of the NV–. As an example, panel (b) shows the decrease in NV0 FL for sample F2, as measured in the 550–600 nm window, for different laser powers. (c) Pulse sequence used to detect the charge recovery in the dark after a preparation pulse. Recovery of NV0 FL for sample F2 at a laser power of 625 mW is shown in panel (d). Various curves in panel (d) correspond to different durations of the initialization pulse.
Figure 3a,c shows the two pulse sequences adopted to assess charge dynamics during laser irradiation and in the following dark time. In the first one (Figure 3a), a laser pulse (532 nm) of variable duration τL (from 10 μs to 100 ms) was focused on the diamond surface to study charge photoconversion during irradiation. This pulse induces photoionization, with an increase in NV– and a decrease in the NV0 signal (blue and orange curves, respectively). FL was recorded by the subsequent short detection pulse of 5 μs. At the end of the sequence, a time interval of 10 ms, during which the laser was off, allowed for charge relaxation before the experiment was repeated for signal averaging. Data points acquired after discrete initialization pulses, for different values of laser power, are shown in Figure 3b (for sample F2). For simplicity, only FL signals acquired in the range 550–600 nm, reflecting predominantly NV0 fluorescence, are reported (the behavior of NV– is symmetrical). The curves reported in this figure show that the degree of photoconversion increases steeply with lasers power, ranging between 50 mW to 1400 mW. Independently of laser power, for pulses longer than 10 ms, the fluorescence level lies within 5% of its equilibrium value, and photoconversion approaches saturation in all samples. Therefore, in the following experiments, we use an initialization pulse of at least 10 ms to maximize the number of NV–.
The second pulse sequence of Figure 3c was used to probe the charge dynamics in the dark. The sequence consists of a laser pulse of duration τL followed by a variable dark time τD (from 1 μs to 10 ms) and a 5 μs readout pulse. In Figure 3d, the FL of the NV0 centers measured through the 550–600 nm spectral window is reported as a function of τD. As discussed above, the first initialization pulse reduces the number of NV0 by photoconversion to NV–. During the dark time, the system goes back to equilibrium and a steady increase in the number of NV0 centers is reflected by increased fluorescence in the spectral region 550–600 nm registered by the readout pulse. A plausible explanation of this recharging-in-the-dark process, previously reported and discussed,23,25 contemplates electrons tunneling from the negative NV– to surface acceptors or vacancies until equilibrium is reached. To exemplify this phenomenon, Figure 3d shows the curves obtained with a laser power of ≈625 mW and initialization times from 10 μs to 100 ms for sample F2. Coherent with the results of Figure 3b, the initial value of fluorescence decreases with longer initialization pulses, meaning that an increasing fraction of NV0 is converted into NV– upon laser irradiation. In all cases, fluorescence from NV0 increases in the dark with longer τD. We also notice that the curves acquired after a τL of 10, 30, and 100 ms are almost identical, consistent with the observation that beyond 10 ms the photoconversion process saturates.
The curves of Figure 3b can be fitted with a stretched exponential law of the type8
| 1 |
where Tr represents a characteristic timescale for the photoconversion process under the laser pulse, n is the stretching factor, Ieq is the FL value at equilibrium, and C is a positive parameter describing the fluorescence drop from the initial value to equilibrium. Curves were normalized by Ieq for easier comparison. Charge recovery curves, as in Figure 3d, can also be fitted with the function of eq 1, even though, in this case, the pre-exponential parameter is negative (the initial value is lower than the equilibrium value). It should be noted that the mechanisms underlying charge conversion in the dark are different from those that induce photoconversion, so the two characteristic times Tr are not related.
The photoconversion data for all samples are reported in Figure 4a, where we plot (1 + C)−1 (eq 1), corresponding to the ratio between the FL equilibrium and initial values, for different laser powers. The largest drop in NV0s due to photoconversion was observed in the flat samples (down to 74% for F2 and 81% for F1) at the largest available laser power, while more limited variation was observed in the nanostructured samples. Note that the large error bars on the x-axis are caused by a slight deterioration in the laser beam’s transmittance at high laser power, an effect that accounts for 5–10% of losses during prolonged irradiation. In Figure 4b, we show the rate of photoconversion (i.e., the inverse of the photoconversion time Tr) for the flat samples (in the case of nanostructured samples, the uncertainties on extracted values are too large to draw conclusions on their laser-power dependence). (Tr)−1 is seen to increase with laser power according to a power law with exponents of 0.95 ± 0.07 and 1 ± 0.07 for samples F1 and F2, respectively (dashed lines). The nearly linear dependence on the laser power is suggestive of a single-photon-mediated NV0 → NV– photoconversion mechanism.
Figure 4.

Parameters describing NV0 → NV– photoconversion under laser irradiation. The quantity (1 + C)−1 in (a) indicates the ratio between the equilibrium value of FL and the initial value, right after the initialization pulse. The decrement is more consistent for the flat samples (down to 75% for sample F2). The photoconversion rate (Tr)−1 displayed in (b) is nearly linear with laser power, following a power law with exponents of 0.95 ± 0.07 for F1 and 1 ± 0.07 for F2 (red and blue dashed curves).
To illustrate the symmetry between the dynamics of the two charge states in the dark, in Figure 5a, we show the FL signal in the 550–600 and >750 nm spectral windows, dominated by NV0 and NV–, respectively, for sample F2 at the highest laser power. The experiment of Figure 3c was performed with or without an applied magnetic field of 750 G (open and solid symbols in Figure 5a, respectively). Indeed, while the evolution of the NV0 signal reflects strictly charge dynamics, the NV– signal is influenced by both charge and spin dynamics. The applied magnetic field suppresses spin dynamics and makes it possible to selectively measure the effects of charge switching for NV–. Under these experimental conditions, the two curves, in red and blue, representing charge state recovery for NV– and NV0, appear symmetrical, thus corroborating the idea that laser light converts one state into the other.
Figure 5.

Spin and charge dynamics after the laser pulse. The effect of a strong magnetic field (750 G) on the fluorescence of sample F2 at a laser power of 1500 mW is shown in (a). FL was collected in the 550–600 nm region (blue curves) and in the >750 nm window (red curves), with and without magnetic field (empty and solid symbols, respectively). When considering only the NV0 centers (550–600 nm window), no change in the FL profile is observed, irrespective of the magnetic field applied. On the contrary, the signal in the >750 nm window is reduced by a strong magnetic field. This difference is the result of quenching NV– spin polarization through spin state mixing. However, beyond ≈10 ms, the difference vanishes, and the evolution at longer times is dominated by charge dynamics. At low (b) and high (c) laser powers, the 10 ms laser pulse creates an excess of NV–s (red curve) that convert to NV0s (blue curve) in the dark. NV– and NV0 populations are normalized such that their sum is 1 (yellow-azure curve). The ratio R is indicated by the black curve. A near-equilibrium ratio (0.15) is reached after 100 ms at low power (50 mW), while a large and sustained R (1.13) value is observed after high power irradiation (1500 mW). Results of (b) and (c) are taken at a magnetic field of 750 G, which enables determining the charge state ratio (for more details, see the text or Section S4 of the Supporting Information).
Performing the experiment of Figure 3c with the two 550–600 and >750 nm spectral windows makes it possible to estimate the charge-state ratio R = [NV–]/[NV0] at a steady state, as proposed by Giri et al.36 (derivation of R is illustrated in Section S4 of the Supporting Information). The two assumptions are (i) all the NV centers are optically active and (ii) the sum of NV– and NV0 is always constant. The latter hypothesis implies that a reduction of NV0 is counterbalanced by an increase of the NV–, and vice versa. Figure 5b,c shows the subdivision of the populations between NV– and NV0 (at B = 750 G) after a 10 ms laser pulse at low and high powers for sample F2, as an example. In both cases, the initialization pulse produces an excess of NV– centers that convert back into NV0 during the dark time. At low laser power (50 mW, ≈5 mW/μm2), the signal is dominated by NV0s, with a ratio R = 0.13/0.87 ≈ 0.15 at the longest time point acquired (100 ms). However, at high power (1500 mW, corresponding to ≈150 mW/μm2) the ratio changes to 0.53/0.47 ≈ 1.13, with the NV– centers now being the dominant state after a sevenfold increase compared to the low power case. Interestingly, after an initial rapid decrease, a large value of R (1.13) is observed after 100 ms, and the curve exhibits a much more slowly decaying component. Thus, the effect of an intense laser pulse is to create a large NV–/NV0 ratio that is sustained over tens or hundreds of milliseconds (see the Discussion section). Evidently, the charge dynamics has fast and slow components, summarized by the stretched exponential behavior, with the fast components in the 10–1000 μs timescale and the slower ones on a 100 ms scale (Figure 5b,c). On the contrary, the timescale of spin dynamics is of the order of ≈1 ms. For this reason, when considering the combined effect of spin and charge dynamics, there will be a substantial overlap in the fluorescence up to 1–10 ms, while at longer times the FL profile will be dominated by charge dynamics. This is also apparent from Figure 5a, where the difference between the two red curves, attributed to spin dynamics, vanishes after ≈10 ms; the evolution at longer times is unaffected by the magnetic field, thus indicating that it can be attributed exclusively to charge dynamics.
Finally, repeating the same experiments with sample N2 gives similar results, but with a modest degree of NV0 → NV– photoconversion and a shorter lifetime of the NV–-photoconverted centers (approximately 10 ms). Even at the highest power of 1500 mW, the ratio NV–/NV0 is 0.1/0.9 ≈ 0.11, much lower compared to the results of sample F2 (as shown in Figure S4 in Section S4).
Finally, we report optically detected magnetic resonance (ODMR) experiments to assess the spin polarization of the ground-state level ms = 0 of NV–. Continuous laser irradiation polarizes the ms = 0 state, while a sweeping microwave field transfers the population to the ms = ± 1 state. However, we note that the detected ODMR contrast is also affected by NV0 centers. Indeed, even though FL is collected in the >750 nm window, where most of the signal comes from the NV– centers, a residual background signal from NV0s is still present. This residual NV0 signal is not modulated by microwave irradiation and cannot be removed entirely by spectral filtering. Hence, we expect the ODMR contrast to be increased at higher laser power due to two independent mechanisms: (i) increased NV– polarization and (ii) reduction in the nuisance background signal from NV0.
In Figure 6a, we show the ODMR spectra from sample F2 for a fixed microwave power and increasing the laser power up to 1500 mW. The contrast (i.e., the depth of the dip) increases rapidly with laser power and makes it possible to resolve the strain-split doublet43 and the 13C sidebands at the highest contrast-to-noise ratio. In Figure 6b, we show the dependence of the ODMR contrast on laser power for all samples. Nanostructured samples exhibit low contrast and limited gain with increasing laser power, consistent with the evidence that NV0 remains the dominant charge state even at the highest laser powers. The ODMR contrast in flat samples is much larger and increases sharply with laser power. Saturation of contrast for the F2 sample at around 600 mW probably reflects laser-induced repolarization related to insufficient microwave power (see the Discussion section). In light of the above results, we can conclude that the increment in the ODMR contrast with laser power does not merely reflect increased ground-state spin polarization, but also a gain in the number density of NV– as a result of photoconversion (see the Discussion section for the consideration on the ODMR contrast).
Figure 6.

Optically detected magnetic resonance. (a) ODMR spectra from sample F2 as a function of laser power. (b) ODMR contrast for all samples. The increase, in contrast, is more pronounced for flat samples.
Discussion
Our results highlight the importance of NV0 to NV– charge conversion in shallow defects, and point to the use of high-power laser irradiation to improve the availability and polarization of shallow NV–. This effect appeared to be more prominent in flat samples, compared to nanostructured samples.
The maximum values of laser intensities used here are larger than those commonly reported in the literature. In fact, NV centers are typically investigated using laser intensities of few mW/μm2 or less21,44,45 and only rarely approach 100 mW/μm2.40 The use of such a high laser power is justified by the lack of saturation of the NV optical transition, as shown in Figure S3 (Section S3 of the Supporting Information). Indeed, we recorded FL emission in the two windows “550–600 nm” and “>750 nm” with increasing laser power: the signals from the >750 nm window barely show any indication of saturation (saturation power estimated at 8.5 W), while the 550–600 nm window shows a flexion only for the flat samples (saturation power estimated at 2.3 W). Thus, we can conclude that the maximum laser power used here was below the onset of saturation. These results are not surprising, as saturation curves depend on whether a single NV center or ensembles are observed and on the sample characteristics (defects type and density). Very different saturation intensities are reported in the literature, ranging from a few mW/μm246−48 to an estimate of thousands of mW/μm2.49
In all four samples investigated, the NV0 centers represent a substantial proportion of NV defects. In the bulk, the presence of electron donors, such as substitutional nitrogen, favors the formation and stability of the NV– center. However, moieties and acceptor states at the diamond surface decrease the stability of the NV–, even at depths of tens of nanometers,50−52 and NV0s may become the dominant charge state53,54 with detrimental effects for sensing and polarization transfer applications. Our samples were engineered to present shallow NV centers (similar to the previous reports55), and it is not surprising to find predominantly NV0 centers. In the case of nanostructured samples, the surface effects are exacerbated by the large surface-to-volume ratio of the nanostructures. Therefore, diamonds with nanopillars have fewer NV–s compared to NV0s, as apparent in the FL and ODMR spectra (Figures 2c, 4a, and 6b, respectively).
Moreover, the integrated fluorescence of Figure 2d suggests that nanostructured samples have a lower number of NV centers altogether, independent of the specific charge state. This could be determined by less efficient ion implantation on the nanostructures or by a loss of nitrogen due to ion straggling.
Laser irradiation has two effects on the NV centers. The first effect is the polarization of the NV–ms = 0 spin state. The second effect is to convert part of the NV0s into NV– and increase the ratio of the negatively charged centers over the total NVs. Upon switching the laser off, a process of recharging in the dark results in an NV– → NV0 back-conversion. An interesting finding of this work is that the excess NV– population is persistent over a relatively long timescale.
The following hypothesis can qualitatively explain the experimental findings. A laser pulse ionizes the substitutional nitrogen atoms,56 and electrons are released into the conduction band and recombine with electron-trapping defects,57 including the NV0. The decrease of NV0 FL in Figure 3b is consistent with this mechanism. The phenomenon of redistribution of charges was recently investigated from a different perspective, namely the Stark shift of optical transition.39 The nearly-linear dependence of (Tr)−1 on laser power (Figure 4b) suggests a single-photon ionization process of substitutional nitrogen as a source of electrons for NV0 → NV– photoconversion. We note that the proposed mechanism for NV0 → NV– charge-state conversion is different from the two-photon excitation of electrons from the valence band (i.e., a photon initializing the NV0 to the excited state and a second photon promoting an electron from the valence band to a trapping NV0 centers),38 which is better described by a quadratic dependence of (Tr)−1 on laser power.21 In the dark, an NV– → NV0 back-conversion is observed. The mechanism proposed for this process is electron tunneling, which critically depends on the NV environment. Tunneling to a neighboring trap can only occur if the trap is empty.25 At weak laser powers, the photoexcited electrons do not saturate the traps in the vicinity of the NV center. At stronger laser powers, increasingly distant traps are populated. Hence, the time the system takes to return to equilibrium depends on laser power, as tunneling to distant traps is a much slower process. This picture is consistent with the results of Figure 5b,c that show, at high laser power, a large [NV–]/[NV0] ratio persistent on a scale of 100 ms. Thus, high-power irradiation increases the availability of NV– and extends the time window in which the NV– spin states can be manipulated and detected, with important implications for sensing and polarization-transfer applications.
The beneficial effects of high-power irradiation are less pronounced in the etched samples, with a modest increase in NV– population. This is consistent with the idea that surface defects compete with NV0 to capture released electrons. Therefore, the NV0 → NV– conversion is more efficient in the flat samples and less efficient in the nanostructured ones, where the amount of surface defects is large compared to the amount of NVs (Figure 4a). In our samples, the advantage of the increased exposed area resulting from nanostructuring appears to be mitigated by the less favorable charge dynamics. Additional treatment of the surface to stabilize NV– (like selective oxidation58 or fluorination26) may improve the performance of nanostructured samples, but we have not explored the effects of different terminations in this paper. It is interesting to compare our results concerning nanopillars with recent work,59 where nanostructures demonstrated higher brightness, larger NV–/NV0 ratio, and better spin properties compared to a bulk reference. We can explain the differences by accounting for the striking different sizes of nanostructures. In the work of McCloskey,59 the nanopillars size was ≈1 μm (as an order of magnitude for both height and width), while our nanostructures were much smaller, with a height of 150 nm and a width narrowing from 50 nm at the base to 10 nm at the top. Therefore, the effects of lateral surfaces are expected to be relevant for small nanostructures but negligible for micrometric pillars, where only a small fraction of the NV centers (the closest to the sidewall) is affected. Additional work would be necessary to trace the transition from nano- to microstructures in terms of NV–/NV0 ratio and to understand if the size and shape can affect the way photoexcited electrons recombine with defects, including NV0.
Interestingly, both routes of photoconversion (NV– → NV0 and NV0 → NV–) have been reported in the literature (see, for instance, refs (40, 44, 57, 60) for NV– → NV0 and refs (25, 55) for NV0 → NV–). These different trends of charge-state conversion are related to the environment of the NV centers, i.e., to the presence of electron donors (mostly substitutional nitrogen) and electron acceptors (like vacancies created by nitrogen implantation or surface acceptors). Thus, the photostability and the routes of photoconversion are in general different for bulk and shallow NV centers, as suggested by the recent literature.36,54 Our results apply to shallow NV centers, which are relevant for sensing and polarization-transfer applications. Importantly, we stress the fact that in our samples a high laser power has a twofold beneficial effect: first, it increases the density of NV–s by photoconverting part of the NV0s, and second, it polarizes the NV– spins. This might not be the case for bulk samples, as improved spin polarization at high laser power could be frustrated by a loss of NV– centers via NV– → NV0 photoionization.
An intermediate scenario between shallow NV centers and native bulk NV centers is represented by high-energy nitrogen implantation (hundreds of keV or MeV). On the one hand, it has been reported that, under these conditions, the NV yield from nitrogen is extremely high (approximately 50% for 2 MeV61 and 18 MeV35 of implantation energy), so a high density of NV centers can be generated at a depth of 1 μm or more, where the effects of surface acceptors on the NV– stability are negligible. On the other hand, high implantation energies and doses are accompanied by the creation of a large number of vacancies, divacancies, and vacancy clusters, which might deteriorate the diamond lattice and compromise the NV– stability.62,63 The effect of nitrogen implantation energy and dose on NV– and NV0 charge dynamics might be the subject of a future analysis.
We used stretched exponentials to fit all the fluorescence curves to account for heterogeneity in the ensemble of NVs, resulting in a superposition of many single exponential curves.8,64 The dynamics of charge tunneling was addressed by Miller65 and Tachiya66 by considering a spatially homogeneous distribution of charge acceptors and recently extended to a discrete distribution.23 In this model, the charge dynamics can be described by a functional dependence of the form
| 2 |
Here, n is the density of trapping defects, and ν and a are parameters linked to the NV– center, defined as the tunneling frequency and the attenuation length of the wavefunction, respectively. Using this expression for the fits, we find a = 0.1 nm, ν = 0.8 ± 0.1 MHz, and n = 2–4 ppm. The density n shows a slight tendency to decrease after long laser pulses, possibly indicating that photoexcited electrons have filled some charge traps. Additional work will be necessary to understand the features of n and some subtleties not captured by the present model, such as nonuniform values of n, ν, and a across a defective sample. However, for our data, we do not observe a substantial improvement of the fit using eq 2 compared with a stretched exponential function (eq 1). We are mostly interested in the initial out-of-equilibrium values of the curves compared to equilibrium. These values are set during the initialization phase by the laser pulse length and power and do not depend critically on the choice of the fitting function. In this respect, the values returned by the two fitting procedures are in good agreement.
The leakage of the NV0 FL component in the NV– spectral region can affect the ODMR contrast.67−69 For NV– centers, the contrast is defined as Cs = (Ioff – Ion)/Ioff, where Ioff and Ion are the collected FL intensities with microwaves off- and on-resonance, respectively. If the intensity contains an NV0-related component I0, then the contrast Cs must be scaled by a factor Ioff/(Ioff + I0). In our case, the intensity Ioff (I0) is the fraction f– (f0) of the total NV– (NV0) intensity filtered by the >750 nm longpass and normalized by photon-counter efficiency. It is possible to relate these intensities to the charge-state ratio R by parameters pertaining to NV– (NV0) centers, such as the absorption cross sections σ– (σ0), and the excited-state lifetime τ– (τ0). The ODMR contrast for the NV ensemble is
| 3 |
where 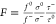 is a numerical factor
that depends on detection
conditions, such as the choice of filters and detectors, and structural
properties of NV centers. Conversely, R can vary
for different samples. For shallow NVs, where the charge state is
dominated by neutral centers, R is small and the
ensemble contrast is substantially reduced. On the contrary, for samples
that mostly contain NV– centers (as seen usually
for bulk NV centers), R is much larger than F and the correction is minimal. For our setup, we estimate f0/f– ≈
0.42 from the shape of fluorescence (see Section S1). Using σ– = 3.1 × 10–17 cm2,70 σ0 = 1.8 × 10–17 cm2,71 τ– = 12 ns, and τ0 = 21 ns,72 we find F ≈ 0.14. Also, the charge state ratio varies dramatically
from 0.15 at low laser power to 1.13 at high power (Figure 5b,c). With an indicative value
of Cs = 10%, we estimate ensemble contrasts
of 5.2 and 8.9% at low and high laser powers, respectively, and a
tendency to increase with R and with laser power,
following the NV0 → NV– photoconversion.
In principle, Cs can be recovered a posteriori from experimental values of the ODMR contrast,
provided that the dependence of R on the laser power
is known. The proposed interpretation explains the trend shown in Figure 6b from low to moderately
high laser powers, although the contrast shows a saturation or even
a slight reduction at the highest power levels (see, for instance,
sample F2). It also explains the striking difference between the flat
and the nanostructured samples: in the latter, NV0s are
by far the dominating species even at high laser power and the extracted
contrast is always poorer than in the flat samples. Concerning the
saturation of the contrast at high powers, we note that an optimal
contrast results from combined laser and microwave spin initialization.
If the laser power is too high, the microwave field cannot compete
in inverting the population levels in the ground state, and some degree
of laser-induced repolarization is expected.49 Despite setting the power of the microwave to a maximum available
value (output of 8 dBm amplified by ≈35 dB), the laser repolarization
effect may prevail, resulting in the saturation and slight reduction
shown in Figure 6b.
is a numerical factor
that depends on detection
conditions, such as the choice of filters and detectors, and structural
properties of NV centers. Conversely, R can vary
for different samples. For shallow NVs, where the charge state is
dominated by neutral centers, R is small and the
ensemble contrast is substantially reduced. On the contrary, for samples
that mostly contain NV– centers (as seen usually
for bulk NV centers), R is much larger than F and the correction is minimal. For our setup, we estimate f0/f– ≈
0.42 from the shape of fluorescence (see Section S1). Using σ– = 3.1 × 10–17 cm2,70 σ0 = 1.8 × 10–17 cm2,71 τ– = 12 ns, and τ0 = 21 ns,72 we find F ≈ 0.14. Also, the charge state ratio varies dramatically
from 0.15 at low laser power to 1.13 at high power (Figure 5b,c). With an indicative value
of Cs = 10%, we estimate ensemble contrasts
of 5.2 and 8.9% at low and high laser powers, respectively, and a
tendency to increase with R and with laser power,
following the NV0 → NV– photoconversion.
In principle, Cs can be recovered a posteriori from experimental values of the ODMR contrast,
provided that the dependence of R on the laser power
is known. The proposed interpretation explains the trend shown in Figure 6b from low to moderately
high laser powers, although the contrast shows a saturation or even
a slight reduction at the highest power levels (see, for instance,
sample F2). It also explains the striking difference between the flat
and the nanostructured samples: in the latter, NV0s are
by far the dominating species even at high laser power and the extracted
contrast is always poorer than in the flat samples. Concerning the
saturation of the contrast at high powers, we note that an optimal
contrast results from combined laser and microwave spin initialization.
If the laser power is too high, the microwave field cannot compete
in inverting the population levels in the ground state, and some degree
of laser-induced repolarization is expected.49 Despite setting the power of the microwave to a maximum available
value (output of 8 dBm amplified by ≈35 dB), the laser repolarization
effect may prevail, resulting in the saturation and slight reduction
shown in Figure 6b.
The resulting increase in ODMR contrast improves sensitivity η
(expressed in 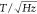 ) and the minimum magnetic field
δB detectable at time Δt, defined
as
) and the minimum magnetic field
δB detectable at time Δt, defined
as 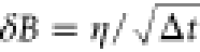 . In continuous-wave ODMR protocols,
sensitivity
is given by the relation73
. In continuous-wave ODMR protocols,
sensitivity
is given by the relation73
| 4 |
where the numerical factor in front is related
to the Lorentzian function used to fit the resonance, h is the Planck constant, μB is the Bohr magneton, g ≈ 2, Γ is the full width at half-maximum
of the Lorentzian, and r is the photodetection rate.
The contrast Cens in eq 4 is the ODMR contrast of the NV ensemble given
by eq 3. At low laser
power (50 mW), we find experimentally r = 0.73 ×
106 s–1, while at high power (1500 mW),
we find r = 17.8 × 106 s–1 (values normalized by limited photon counter efficiency of the detector).
Taking values of Cens and R as before, and a representative Γ = 10 MHz, we find 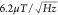 and
and 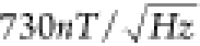 at low and high powers, respectively. Sensitivity
might be further improved with the use of pulsed-wave ODMR73 and extended to the detection of AC magnetic
fields.74,75 Therefore, magnetometry applications benefit
from the use of strong laser power by an increase in the photon detection
rate and improved contrast. We note that the correction factor of eq 3 does not exclusively determine
the ODMR contrast but plays a role in other detection schemes, including
pulsed sequences as those in Figure 3a,c.
at low and high powers, respectively. Sensitivity
might be further improved with the use of pulsed-wave ODMR73 and extended to the detection of AC magnetic
fields.74,75 Therefore, magnetometry applications benefit
from the use of strong laser power by an increase in the photon detection
rate and improved contrast. We note that the correction factor of eq 3 does not exclusively determine
the ODMR contrast but plays a role in other detection schemes, including
pulsed sequences as those in Figure 3a,c.
13C nuclear hyperpolarization is another technique that could benefit from an increased fraction of NV–s. This technique is based on the transfer of spin polarization from highly polarized NV– to first-neighbor 13C nuclei via spin–spin interaction at specific values of the applied magnetic field.76 Polarization can then diffuse through the diamond lattice to the bulk 13C spin reservoir by nuclear spin–spin interactions.77,78 However, this process is much slower compared to the fast hyperpolarization of the first shell 13C nuclei (≈1 ms) in the vicinity of NV– centers, and increasing the amount of NV–s via photoconversion can substantially improve the efficiency of the hyperpolarization process.
The effects reported here may be particularly important for hyperpolarization of nuclei outside the diamond surface. Indeed, it has been proposed that shallow NV– may provide a source of spin polarization of molecular moieties adsorbed at the diamond surface,16,17,79 thus complementing techniques like dynamic nuclear polarization or optical pumping spin-exchange. Increasing their concentration in the vicinity, the diamond surface may enable hyperpolarization of nuclei outside the diamond lattice.
Realistically, laser power is constrained by the available equipment and by the nature of the sample. Some applications of sensing,80 widefield imaging,81 and hyperpolarization17 necessarily address a large number of NVs over broad two-dimensional (2D) or three-dimensional (3D) regions at the expense of a tighter focusing (so, with lower laser intensity). Nevertheless, our results indicate that charge-state photoconversion must be taken into account for shallow NV centers and point to the maximization of laser power for improved NV– concentration and lifetime whenever possible.
In conclusion, we studied the effects of laser power on charge-state photoconversion in ensembles of shallow NV centers in flat and nanostructured diamonds. Surface effects favor the neutral charge state (NV0) over the negative state (NV–). This problem is exacerbated in the nanostructured samples. We show that high-power laser irradiation promotes NV0 to NV– photoconversion, thus significantly increasing the relative abundance of NV–s in flat and in a nanostructured diamond. Interestingly, we find that the excess NV– population is stable over timescales of tens or hundreds of milliseconds. This may provide an important advantage for magnetometry and polarization-transfer applications, where large ensembles of shallow NV– are needed.
Acknowledgments
The authors thank P. Olivero for useful suggestions on sample treatments from the Department of Physics, University of Torino, Italy; L. Basso for fluorescence spectroscopy with the micro-Raman setup from the Department of Physics, Università di Trento, Italy; and Simone Lauciello and Alice Scarpellini of Electron Microscopy Facility of Istituto Italiano di Tecnologia in Genova for SEM characterization. This project has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No 858149 (AlternativesToGd) and from the Marie Skłodowska-Curie program, Grant Agreement No. 766402 (ZULF).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsami.1c09825.
Charge state fluorescence ratio and filtering; effect of the readout pulse; excited state saturation; and calculation of the charged state ratio (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Kucsko G.; Maurer P. C.; Yao N. Y.; Kubo M.; Noh H. J.; Lo P. K.; Park H.; Lukin M. D. Nanometre-Scale Thermometry in a Living Cell. Nature 2013, 500, 54–58. 10.1038/nature12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singam S. K. R.; Nesladek M.; Goovaerts E. Nitrogen-Vacancy Nanodiamond Based Local Thermometry Using Frequency-Jump Modulation. Nanotechnology 2020, 31, 105501 10.1088/1361-6528/ab5a0c. [DOI] [PubMed] [Google Scholar]
- Davis H. C.; Ramesh P.; Bhatnagar A.; Lee-Gosselin A.; Barry J. F.; Glenn D. R.; Walsworth R. L.; Shapiro M. G. Mapping the Microscale Origins of Magnetic Resonance Image Contrast with Subcellular Diamond Magnetometry. Nat. Commun. 2018, 9, 131 10.1038/s41467-017-02471-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss J. M.; Barry J. F.; Turner M. J.; Walsworth R. L. Simultaneous Broadband Vector Magnetometry Using Solid-State Spins. Phys. Rev. Appl. 2018, 10, 034044 10.1103/PhysRevApplied.10.034044. [DOI] [Google Scholar]
- Iwasaki T.; Naruki W.; Tahara K.; Makino T.; Kato H.; Ogura M.; Takeuchi D.; Yamasaki S.; Hatano M. Direct Nanoscale Sensing of the Internal Electric Field in Operating Semiconductor Devices Using Single Electron Spins. ACS Nano 2017, 11, 1238–1245. 10.1021/acsnano.6b04460. [DOI] [PubMed] [Google Scholar]
- Forneris J.; Ditalia Tchernij S.; Traina P.; Moreva E.; Skukan N.; Jakšić M.; Grilj V.; Bosia F.; Enrico E.; Amato G.; Degiovanni I. P.; Naydenov B.; Jelezko F.; Genovese M.; Olivero P. Mapping the Local Spatial Charge in Defective Diamond by Means of NV Sensors – A “Self-Diagnostic” Concept. Phys. Rev. Appl. 2018, 10, 014024 10.1103/PhysRevApplied.10.014024. [DOI] [Google Scholar]
- Tetienne J. P.; Lombard A.; Simpson D. A.; Ritchie C.; Lu J.; Mulvaney P.; Hollenberg L. C. L. Scanning Nanospin Ensemble Microscope for Nanoscale Magnetic and Thermal Imaging. Nano Lett. 2016, 16, 326–333. 10.1021/acs.nanolett.5b03877. [DOI] [PubMed] [Google Scholar]
- Gorrini F.; Giri R.; Avalos C. E.; Tambalo S.; Mannucci S.; Basso L.; Bazzanella N.; Dorigoni C.; Cazzanelli M.; Marzola P.; Miotello A.; Bifone A. Fast and Sensitive Detection of Paramagnetic Species Using Coupled Charge and Spin Dynamics in Strongly Fluorescent Nanodiamonds. ACS Appl. Mater. Interfaces 2019, 11, 24412–24422. 10.1021/acsami.9b05779. [DOI] [PubMed] [Google Scholar]
- Wang P.; Chen S.; Guo M.; Peng S.; Wang M.; Chen M.; Ma W.; Zhang R.; Su J.; Rong X.; Shi F.; Xu T.; Du J. Nanoscale Magnetic Imaging of Ferritins in a Single Cell. Sci. Adv. 2019, 5, eaau8038 10.1126/sciadv.aau8038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radu V.; Price J. C.; Levett S. J.; Narayanasamy K. K.; Bateman-Price T. D.; Wilson P. B.; Mather M. L. Dynamic Quantum Sensing of Paramagnetic Species Using Nitrogen-Vacancy Centers in Diamond. ACS Sens. 2019, 5, 703–710. 10.1021/acssensors.9b01903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian G.; Lazariev A.; Arumugam S. R.; Duan D. wen. Nitrogen-Vacancy Color Center in Diamond-Emerging Nanoscale Applications in Bioimaging and Biosensing. Curr. Opin. Chem. Biol. 2014, 20, 69–77. 10.1016/j.cbpa.2014.04.014. [DOI] [PubMed] [Google Scholar]
- Hsiao W. W. W.; Hui Y. Y.; Tsai P. C.; Chang H. C. Fluorescent Nanodiamond: A Versatile Tool for Long-Term Cell Tracking, Super-Resolution Imaging, and Nanoscale Temperature Sensing. Acc. Chem. Res. 2016, 49, 400–407. 10.1021/acs.accounts.5b00484. [DOI] [PubMed] [Google Scholar]
- Boretti A.; Rosa L.; Blackledge J.; Castelletto S. Nitrogen-Vacancy Centers in Diamond for Nanoscale Magnetic Resonance Imaging Applications. Beilstein J. Nanotechnol. 2019, 10, 2128–2151. 10.3762/bjnano.10.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King J. P.; Jeong K.; Vassiliou C. C.; Shin C. S.; Page R. H.; Avalos C. E.; Wang H. J.; Pines A. Room-Temperature in Situ Nuclear Spin Hyperpolarization from Optically Pumped Nitrogen Vacancy Centres in Diamond. Nat. Commun. 2015, 6, 8965 10.1038/ncomms9965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Álvarez G. A.; Bretschneider C. O.; Fischer R.; London P.; Kanda H.; Onoda S.; Isoya J.; Gershoni D.; Frydman L. Local and Bulk 13 C Hyperpolarization in Nitrogen-Vacancy-Centred Diamonds at Variable Fields and Orientations. Nat. Commun. 2015, 6, 8456 10.1038/ncomms9456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajoy A.; Liu K.; Nazaryan R.; Lv X.; Zangara P. R.; Safvati B.; Wang G.; Arnold D.; Li G.; Lin A.; Raghavan P.; Druga E.; Dhomkar S.; Pagliero D.; Reimer J. A.; Suter D.; Pines A.; Meriles C. A. Orientation-Independent Room Temperature Optical 13C Hyperpolarization in Powdered Diamond. Sci. Adv. 2018, 4, eaar5492 10.1126/sciadv.aar5492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healey A. J.; Hall L. T.; White G. A. L.; Teraji T.; Sani M.-A.; Separovic F.; Tetienne J.-P.; Hollenberg L. C. L. Polarization Transfer to External Nuclear Spins Using Ensembles of Nitrogen-Vacancy Centers. Phys. Rev. Appl. 2021, 15, 054052 10.1103/PhysRevApplied.15.054052. [DOI] [Google Scholar]
- Neu E.; Appel P.; Ganzhorn M.; Miguel-Sánchez J.; Lesik M.; Mille V.; Jacques V.; Tallaire A.; Achard J.; Maletinsky P. Photonic Nano-Structures on (111)-Oriented Diamond. Appl. Phys. Lett. 2014, 104, 153108 10.1063/1.4871580. [DOI] [Google Scholar]
- Alghannam F.; Hemmer P. Engineering of Shallow Layers of Nitrogen Vacancy Colour Centres in Diamond Using Plasma Immersion Ion Implantation. Sci. Rep. 2019, 9, 5870 10.1038/s41598-019-42323-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrich P.; Li J.; Liu X.; Heremans F. J.; Nealey P. F.; Awschalom D. D. Microscale Resolution Thermal Mapping Using a Flexible Platform of Patterned Quantum Sensors. Nano Lett. 2018, 18, 4684–4690. 10.1021/acs.nanolett.8b00895. [DOI] [PubMed] [Google Scholar]
- Aslam N.; Waldherr G.; Neumann P.; Jelezko F.; Wrachtrup J. Photo-Induced Ionization Dynamics of the Nitrogen Vacancy Defect in Diamond Investigated by Single-Shot Charge State Detection. New J. Phys. 2013, 15, 013064 10.1088/1367-2630/15/1/013064. [DOI] [Google Scholar]
- Chen X. D.; Zou C. L.; Sun F. W.; Guo G. C. Optical Manipulation of the Charge State of Nitrogen-Vacancy Center in Diamond. Appl. Phys. Lett. 2013, 103, 013112 10.1063/1.4813120. [DOI] [Google Scholar]
- Dhomkar S.; Jayakumar H.; Zangara P. R.; Meriles C. A. Charge Dynamics in Near-Surface, Variable-Density Ensembles of Nitrogen-Vacancy Centers in Diamond. Nano Lett. 2018, 18, 4046–4052. 10.1021/acs.nanolett.8b01739. [DOI] [PubMed] [Google Scholar]
- Giri R.; Gorrini F.; Dorigoni C.; Avalos C. E.; Cazzanelli M.; Tambalo S.; Bifone A. Coupled Charge and Spin Dynamics in High-Density Ensembles of Nitrogen-Vacancy Centers in Diamond. Phys. Rev. B 2018, 98, 045401 10.1103/PhysRevB.98.045401. [DOI] [Google Scholar]
- Bluvstein D.; Zhang Z.; Jayich A. C. B. Identifying and Mitigating Charge Instabilities in Shallow Diamond Nitrogen-Vacancy Centers. Phys. Rev. Lett. 2019, 122, 076101 10.1103/PhysRevLett.122.076101. [DOI] [PubMed] [Google Scholar]
- Cui S.; Hu E. L. Increased Negatively Charged Nitrogen-Vacancy Centers in Fluorinated Diamond. Appl. Phys. Lett. 2013, 103, 051603 10.1063/1.4817651. [DOI] [Google Scholar]
- Murai T.; Makino T.; Kato H.; Shimizu M.; Murooka T.; Herbschleb E. D.; Doi Y.; Morishita H.; Fujiwara M.; Hatano M.; Yamasaki S.; Mizuochi N. Engineering of Fermi Level by Nin Diamond Junction for Control of Charge States of NV Centers. Appl. Phys. Lett. 2018, 112, 111903 10.1063/1.5010956. [DOI] [Google Scholar]
- Forneris J.; Ditalia Tchernij S.; Tengattini A.; Enrico E.; Grilj V.; Skukan N.; Amato G.; Boarino L.; Jakšić M.; Olivero P. Electrical Control of Deep NV Centers in Diamond by Means of Sub-Superficial Graphitic Micro-Electrodes. Carbon 2017, 113, 76–86. 10.1016/j.carbon.2016.11.031. [DOI] [Google Scholar]
- Trusheim M. E.; Li L.; Laraoui A.; Chen E. H.; Bakhru H.; Schröder T.; Gaathon O.; Meriles C. A.; Englund D. Scalable Fabrication of High Purity Diamond Nanocrystals with Long-Spin-Coherence Nitrogen Vacancy Centers. Nano Lett. 2014, 14, 32–36. 10.1021/nl402799u. [DOI] [PubMed] [Google Scholar]
- Naydenov B.; Reinhard F.; Lämmle A.; Richter V.; Kalish R.; D’Haenens-Johansson U. F. S.; Newton M.; Jelezko F.; Wrachtrup J. Increasing the Coherence Time of Single Electron Spins in Diamond by High Temperature Annealing. Appl. Phys. Lett. 2010, 97, 242511 10.1063/1.3527975. [DOI] [Google Scholar]
- Toyli D. M.; Weis C. D.; Fuchs G. D.; Schenkel T.; Awschalom D. D. Chip-Scale Nanofabrication of Single Spins and Spin Arrays in Diamond. Nano Lett. 2010, 10, 3168–3172. 10.1021/nl102066q. [DOI] [PubMed] [Google Scholar]
- Spinicelli P.; Dréau A.; Rondin L.; Silva F.; Achard J.; Xavier S.; Bansropun S.; Debuisschert T.; Pezzagna S.; Meijer J.; Jacques V.; Roch J. F. Engineered Arrays of Nitrogen-Vacancy Color Centers in Diamond Based on Implantation of Cn? Molecules through Nanoapertures. New J. Phys. 2011, 13, 025014 10.1088/1367-2630/13/2/025014. [DOI] [Google Scholar]
- Ziegler J. F.; Ziegler M. D.; Biersack J. P. The Stopping and Range of Ions in Matter. Nucl. Instrum. Methods Phys. Res., Sect. B 2010, 268, 1818–1823. 10.1016/j.nimb.2010.02.091. [DOI] [Google Scholar]
- Antonov D.; Häuermann T.; Aird A.; Roth J.; Trebin H. R.; Müller C.; McGuinness L.; Jelezko F.; Yamamoto T.; Isoya J.; Pezzagna S.; Meijer J.; Wrachtrup J. Statistical Investigations on Nitrogen-Vacancy Center Creation. Appl. Phys. Lett. 2014, 104, 012105 10.1063/1.4860997. [DOI] [Google Scholar]
- Pezzagna S.; Naydenov B.; Jelezko F.; Wrachtrup J.; Meijer Jjnj. Creation Efficiency of Nitrogen-Vacancy Centres in Diamond. New J. Phys. 2010, 12, 065017 10.1088/1367-2630/12/6/065017. [DOI] [Google Scholar]
- Giri R.; Dorigoni C.; Tambalo S.; Gorrini F.; Bifone A. Selective Measurement of Charge Dynamics in an Ensemble of Nitrogen-Vacancy Centers in Nano- and Bulk Diamonds. Phys. Rev. B 2019, 99, 155426 10.1103/PhysRevB.99.155426. [DOI] [Google Scholar]
- Doherty M. W.; Manson N. B.; Delaney P.; Jelezko F.; Wrachtrup J.; Hollenberg L. C. L. The Nitrogen-Vacancy Colour Centre in Diamond. Phys. Rep. 2013, 528, 1–45. 10.1016/j.physrep.2013.02.001. [DOI] [Google Scholar]
- Bourgeois E.; Gulka M.; Nesladek M. Photoelectric Detection and Quantum Readout of Nitrogen-Vacancy Center Spin States in Diamond. Adv. Opt. Mater. 2020, 8, 1902132 10.1002/adom.201902132. [DOI] [Google Scholar]
- Manson N. B.; Hedges M.; Barson M. S. J.; Ahlefeldt R.; Doherty M. W.; Abe H.; Ohshima T.; Sellars M. J. NV– - N+ Pair Centre in 1b Diamond. New J. Phys. 2018, 20, 113037 10.1088/1367-2630/aaec58. [DOI] [Google Scholar]
- Alsid S. T.; Barry J. F.; Pham L. M.; Schloss J. M.; O’Keeffe M. F.; Cappellaro P.; Braje D. A. Photoluminescence Decomposition Analysis: A Technique to Characterize N-V Creation in Diamond. Phys. Rev. Appl. 2019, 12, 044003 10.1103/PhysRevApplied.12.044003. [DOI] [Google Scholar]
- Tetienne J. P.; Rondin L.; Spinicelli P.; Chipaux M.; Debuisschert T.; Roch J. F.; Jacques V. Magnetic-Field-Dependent Photodynamics of Single NV Defects in Diamond: An Application to Qualitative All-Optical Magnetic Imaging. New J. Phys. 2012, 14, 103033 10.1088/1367-2630/14/10/103033. [DOI] [Google Scholar]
- Capelli M.; Reineck P.; Lau D. W. M.; Orth A.; Jeske J.; Doherty M. W.; Ohshima T.; Greentree A. D.; Gibson B. C. Magnetic Field-Induced Enhancement of the Nitrogen-Vacancy Fluorescence Quantum Yield. Nanoscale 2017, 9, 9299–9304. 10.1039/C7NR02093G. [DOI] [PubMed] [Google Scholar]
- Doherty M. W.; Dolde F.; Fedder H.; Jelezko F.; Wrachtrup J.; Manson N. B.; Hollenberg L. C. L. Theory of the Ground-State Spin of the NV - Center in Diamond. Phys. Rev. B 2012, 85, 205203 10.1103/PhysRevB.85.205203. [DOI] [Google Scholar]
- Manson N. B.; Harrison J. P. Photo-Ionization of the Nitrogen-Vacancy Center in Diamond. Diam. Relat. Mater. 2005, 14, 1705–1710. 10.1016/j.diamond.2005.06.027. [DOI] [Google Scholar]
- Acosta V. M.; Bauch E.; Ledbetter M. P.; Santori C.; Fu K.-M.; Barclay P. E.; Beausoleil R. G.; Linget H.; Roch J. F.; Treussart F.; et al. Diamonds with a High Density of Nitrogen-Vacancy Centers for Magnetometry Applications. Phys. Rev. B 2009, 80, 115202 10.1103/PhysRevB.80.115202. [DOI] [Google Scholar]
- Kurtsiefer C.; Mayer S.; Zarda P.; Weinfurter H. Stable Solid-State Source of Single Photons. Phys. Rev. Lett. 2000, 85, 290 10.1103/PhysRevLett.85.290. [DOI] [PubMed] [Google Scholar]
- Manson N. B.; Harrison J. P.; Sellars M. J. Nitrogen-Vacancy Center in Diamond: Model of the Electronic Structure and Associated Dynamics. Phys. Rev. B 2006, 74, 104303 10.1103/PhysRevB.74.104303. [DOI] [Google Scholar]
- Plakhotnik T.; Gruber D. Luminescence of Nitrogen-Vacancy Centers in Nanodiamonds at Temperatures between 300 and 700 K: Perspectives on Nanothermometry. Phys. Chem. Chem. Phys. 2010, 12, 9751–9756. 10.1039/c001132k. [DOI] [PubMed] [Google Scholar]
- Jensen K.; Acosta V. M.; Jarmola A.; Budker D. Light Narrowing of Magnetic Resonances in Ensembles of Nitrogen-Vacancy Centers in Diamond. Phys. Rev. B 2013, 87, 014115 10.1103/PhysRevB.87.014115. [DOI] [Google Scholar]
- Santori C.; Barclay P. E.; Fu K.-M. C.; Beausoleil R. G. Vertical Distribution of Nitrogen-Vacancy Centers in Diamond Formed by Ion Implantation and Annealing. Phys. Rev. B 2009, 79, 125313 10.1103/PhysRevB.79.125313. [DOI] [Google Scholar]
- Hauf M. V.; Grotz B.; Naydenov B.; Dankerl M.; Pezzagna S.; Meijer J.; Jelezko F.; Wrachtrup J.; Stutzmann M.; Reinhard F.; Garrido J. A. Chemical Control of the Charge State of Nitrogen-Vacancy Centers in Diamond. Phys. Rev. B 2011, 83, 081304 10.1103/PhysRevB.83.081304. [DOI] [Google Scholar]
- Newell A. N.; Dowdell D. A.; Santamore D. H. Surface Effects on Nitrogen Vacancy Centers Neutralization in Diamond. J. Appl. Phys. 2016, 120, 185104 10.1063/1.4967735. [DOI] [Google Scholar]
- Kaviani M.; Deák P.; Aradi B.; Frauenheim T.; Chou J. P.; Gali A. Proper Surface Termination for Luminescent Near-Surface NV Centers in Diamond. Nano Lett. 2014, 14, 4772–4777. 10.1021/nl501927y. [DOI] [PubMed] [Google Scholar]
- Meirzada I.; Hovav Y.; Wolf S. A.; Bar-Gill N. Negative Charge Enhancement of Near-Surface Nitrogen Vacancy Centers by Multicolor Excitation. Phys. Rev. B 2018, 98, 245411 10.1103/PhysRevB.98.245411. [DOI] [Google Scholar]
- Gaebel T.; Domhan M.; Wittmann C.; Popa I.; Jelezko F.; Rabeau J.; Greentree A.; Prawer S.; Trajkov E.; Hemmer P. R.; Wrachtrup J. Photochromism in Single Nitrogen-Vacancy Defect in Diamond. Appl. Phys. B 2006, 82, 243–246. 10.1007/s00340-005-2056-2. [DOI] [Google Scholar]
- Iakoubovskii K.; Adriaenssens G. J. Optical Transitions at the Substitutional Nitrogen Centre in Diamond. J. Phys.: Condens. Matter 2000, 12, L77. 10.1088/0953-8984/12/6/102. [DOI] [Google Scholar]
- Jayakumar H.; Henshaw J.; Dhomkar S.; Pagliero D.; Laraoui A.; Manson N. B.; Albu R.; Doherty M. W.; Meriles C. A. Optical Patterning of Trapped Charge in Nitrogen-Doped Diamond. Nat. Commun. 2016, 7, 12660 10.1038/ncomms12660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu K.-M.; Santori C.; Barclay P. E.; Beausoleil R. G. Conversion of Neutral Nitrogen-Vacancy Centers to Negatively Charged Nitrogen-Vacancy Centers through Selective Oxidation. Appl. Phys. Lett. 2010, 96, 121907 10.1063/1.3364135. [DOI] [Google Scholar]
- McCloskey D. J.; Dontschuk N.; Broadway D. A.; Nadarajah A.; Stacey A.; Tetienne J.-P.; Hollenberg L. C. L.; Prawer S.; Simpson D. A. Enhanced Widefield Quantum Sensing with Nitrogen-Vacancy Ensembles Using Diamond Nanopillar Arrays. ACS Appl. Mater. Interfaces 2020, 12, 13421–13427. 10.1021/acsami.9b19397. [DOI] [PubMed] [Google Scholar]
- Tallaire A.; Mayer L.; Brinza O.; Pinault-Thaury M.-A.; Debuisschert T.; Achard J. Highly Photostable NV Centre Ensembles in CVD Diamond Produced by Using N2O as the Doping Gas. Appl. Phys. Lett. 2017, 111, 143101 10.1063/1.5004106. [DOI] [Google Scholar]
- Meijer J.; Burchard B.; Domhan M.; Wittmann C.; Gaebel T.; Popa I.; Jelezko F.; Wrachtrup J. Generation of Single Color Centers by Focused Nitrogen Implantation. Appl. Phys. Lett. 2005, 87, 261909 10.1063/1.2103389. [DOI] [Google Scholar]
- Waldermann F. C.; Olivero P.; Nunn J.; Surmacz K.; Wang Z. Y.; Jaksch D.; Taylor R. A.; Walmsley I. A.; Draganski M.; Reichart P.; et al. Creating Diamond Color Centers for Quantum Optical Applications. Diam. Relat. Mater. 2007, 16, 1887–1895. 10.1016/j.diamond.2007.09.009. [DOI] [Google Scholar]
- Deák P.; Aradi B.; Kaviani M.; Frauenheim T.; Gali A. Formation of NV Centers in Diamond: A Theoretical Study Based on Calculated Transitions and Migration of Nitrogen and Vacancy Related Defects. Phys. Rev. B 2014, 89, 075203 10.1103/PhysRevB.89.075203. [DOI] [Google Scholar]
- Choi J.; Choi S.; Kucsko G.; Maurer P. C.; Shields B. J.; Sumiya H.; Onoda S.; Isoya J.; Demler E.; Jelezko F.; Yao N. Y.; Lukin M. D. Depolarization Dynamics in a Strongly Interacting Solid-State Spin Ensemble. Phys. Rev. Lett. 2017, 118, 093601 10.1103/PhysRevLett.118.093601. [DOI] [PubMed] [Google Scholar]
- Miller J. R. Scavenging Kinetics of Electrons Produced by Irradiation of Organic Glasses; Experimental Evidence for Long Range Tunneling. J. Chem. Phys. 1972, 56, 5173–5183. 10.1063/1.1677003. [DOI] [Google Scholar]
- Tachiya M.; Mozumder A. Decay of Trapped Electrons by Tunnelling to Scavenger Molecules in Low-Temperature Glasses. Chem. Phys. Lett. 1974, 28, 87–89. 10.1016/0009-2614(74)80022-9. [DOI] [Google Scholar]
- Aude Craik D. P. L.; Kehayias P.; Greenspon A. S.; Zhang X.; Turner M. J.; Schloss J. M.; Bauch E.; Hart C. A.; Hu E. L.; Walsworth R. L. A Microwave-Assisted Spectroscopy Technique for Determining Charge State in Nitrogen-Vacancy Ensembles in Diamond. Phys. Rev. Appl. 2020, 14, 014009 10.1103/PhysRevApplied.14.014009. [DOI] [Google Scholar]
- Yuan Z.; Fitzpatrick M.; Rodgers L. V. H.; Sangtawesin S.; Srinivasan S.; de Leon N. P. Charge State Dynamics and Optically Detected Electron Spin Resonance Contrast of Shallow Nitrogen-Vacancy Centers in Diamond. Phys. Rev. Res. 2020, 2, 033263 10.1103/physrevresearch.2.033263. [DOI] [Google Scholar]
- Hopper D. A.; Lauigan J. D.; Huang T. Y.; Bassett L. C. Real-Time Charge Initialization of Diamond Nitrogen-Vacancy Centers for Enhanced Spin Readout. Phys. Rev. Appl. 2020, 13, 024016 10.1103/PhysRevApplied.13.024016. [DOI] [Google Scholar]
- Wee T. L.; Tzeng Y. K.; Han C. C.; Chang H. C.; Fann W.; Hsu J. H.; Chen K. M.; Yu E. C. Two-Photon Excited Fluorescence of Nitrogen-Vacancy Centers in Proton-Irradiated Type Ib Diamond. J. Phys. Chem. A 2007, 111, 9379–9386. 10.1021/jp073938o. [DOI] [PubMed] [Google Scholar]
- Fraczek E.; Savitski V. G.; Dale M.; Breeze B. G.; Diggle P.; Markham M.; Bennett A.; Dhillon H.; Newton M. E.; Kemp A. J. Laser Spectroscopy of NV- and NV0 Colour Centres in Synthetic Diamond. Opt. Mater. Express 2017, 7, 02571 10.1364/ome.7.002571. [DOI] [Google Scholar]
- Storteboom J.; Dolan P.; Castelletto S.; Li X.; Gu M. Lifetime Investigation of Single Nitrogen Vacancy Centres in Nanodiamonds. Opt. Express 2015, 23, 011327 10.1364/oe.23.011327. [DOI] [PubMed] [Google Scholar]
- Dréau A.; Lesik M.; Rondin L.; Spinicelli P.; Arcizet O.; Roch J. F.; Jacques V. Avoiding Power Broadening in Optically Detected Magnetic Resonance of Single NV Defects for Enhanced Dc Magnetic Field Sensitivity. Phys. Rev. B 2011, 84, 195204 10.1103/PhysRevB.84.195204. [DOI] [Google Scholar]
- Taylor J. M.; Cappellaro P.; Childress L.; Jiang L.; Budker D.; Hemmer P. R.; Yacoby A.; Walsworth R.; Lukin M. D. High-Sensitivity Diamond Magnetometer with Nanoscale Resolution. Nat. Phys. 2008, 4, 810–816. 10.1038/nphys1075. [DOI] [Google Scholar]
- Barry J. F.; Schloss J. M.; Bauch E.; Turner M. J.; Hart C. A.; Pham L. M.; Walsworth R. L. Sensitivity Optimization for NV-Diamond Magnetometry. Rev. Mod. Phys. 2020, 92, 015004 10.1103/RevModPhys.92.015004. [DOI] [Google Scholar]
- Fischer R.; Bretschneider C. O.; London P.; Budker D.; Gershoni D.; Frydman L. Bulk Nuclear Polarization Enhanced at Room Temperature by Optical Pumping. Phys. Rev. Lett. 2013, 111, 057601 10.1103/PhysRevLett.111.057601. [DOI] [PubMed] [Google Scholar]
- Ajoy A.; Safvati B.; Nazaryan R.; Oon J. T.; Han B.; Raghavan P.; Nirodi R.; Aguilar A.; Liu K.; Cai X.; Lv X.; Druga E.; Ramanathan C.; Reimer J. A.; Meriles C. A.; Suter D.; Pines A. Hyperpolarized Relaxometry Based Nuclear T1 Noise Spectroscopy in Hybrid Diamond Quantum Registers. Nat. Commun. 2019, 10, 5160 10.1038/s41467-019-13042-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker A. J.; Jeong K.; Avalos C. E.; Hausmann B. J. M.; Vassiliou C. C.; Pines A.; King J. P. Optically Pumped Dynamic Nuclear Hyperpolarization in C 13 -Enriched Diamond. Phys. Rev. B 2019, 100, 041203 10.1103/PhysRevB.100.041203. [DOI] [Google Scholar]
- Abrams D.; Trusheim M. E.; Englund D. R.; Shattuck M. D.; Meriles C. A. Dynamic Nuclear Spin Polarization of Liquids and Gases in Contact with Nanostructured Diamond. Nano Lett. 2014, 14, 2471–2478. 10.1021/nl500147b. [DOI] [PubMed] [Google Scholar]
- Wolf T.; Neumann P.; Nakamura K.; Sumiya H.; Ohshima T.; Isoya J.; Wrachtrup J. Subpicotesla Diamond Magnetometry. Phys. Rev. X 2015, 5, 041001 10.1103/PhysRevX.5.041001. [DOI] [Google Scholar]
- Tetienne J.-P.; Dontschuk N.; Broadway D. A.; Stacey A.; Simpson D. A.; Hollenberg L. C. L. Quantum Imaging of Current Flow in Graphene. Sci. Adv. 2017, 3, e1602429 10.1126/sciadv.1602429. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.