

Abstract
Cardiovascular disease is the number one cause of death globally. Lowering cholesterol levels in plasma is the mainstay therapy; however lifelong treatment and adverse effects call for improved therapeutic interventions. We developed a trivalent vaccine candidate targeting proprotein convertase subtilisin/kexin-9 (PCSK9), apolipoprotein B (ApoB), and cholesteryl ester transfer protein (CETP). Vaccine candidates were developed using bacteriophage Qβ-based virus-like particles (VLPs) displaying antigens of PCKS9, ApoB, and CETP, respectively. Vaccine candidate mixtures were formulated as slow-release PLGA:VLP implants using hot-melt extrusion. The delivery of the trivalent vaccine candidate via the implant produced antibodies against the cholesterol checkpoint proteins at levels comparable to a three-dose injection schedule with soluble mixtures. The reduction in PCSK9 and ApoB levels in plasma, inhibition of CETP (in vitro), and total plasma cholesterol decrease was achieved. All-together, we present a platform technology for a single-dose multi-target vaccination platform targeting cholesterol checkpoint proteins.
Keywords: Cardiovascular disease, trivalent vaccine, virus-like particle (VLP), cholesterol, ApoB, PCSK9, CETP
Graphical Abstract
Statins are the mainstay therapy for cardiovascular disease, but this require requires lifelong treatment. We developed a single-dose, trivalent vaccine candidate using a VLP platform technology delivered via a PLGA slow-release implant. Antibodies raised target cholesterol checkpoint proteins to modulate cholesterol homeostasis. The modular vaccination platform holds great potential for multitarget vaccines for management of cardiovascular disease.
INTRODUCTION
Cardiovascular disease (CVDs) is the number one cause of death globally. An estimated 17.9 million people died from CVD in 2016, representing 31% of all global deaths. Of these deaths, 85% are due to heart attack and stroke and over three quarters of CVD deaths take place in low- and middle-income countries.[1] Atherosclerosis is a chronic inflammatory disease that develops in response to lipid accumulation, immune response, and inflammation.[2,3] The primary events in atherosclerosis are the accumulation and oxidation of lipoproteins such as low-density lipoprotein cholesterol (LDL-C) leading to inflammation of the vascular wall;[2,3] rupture of such plaques can lead to clinical events such as a heart attack and stroke. A primary goal in the treatment of atherosclerosis is to decrease the LDL-C levels in plasma. Statins are the main drugs used to achieve this goal; however, their efficacy is compromised by side effects and the need for lifelong treatments.[3] Immunotherapy holds promise in the management of CVD and the recent US Food and Drug Administration (FDA) approval of monoclonal antibodies (Alirocumab and Evolocumab) targeting the proprotein convertase subtilisin/kexin 9 (PCSK9) pave the way for this development. PCSK9 is a protease that binds and promotes degradation of the LDL receptor. Blocking PCSK9 leads to more LDL receptors on the surface of hepatocyte resulting in lower serum level of LDL-C.[4-6] While this therapy regimen is a promising approach, this passive immunotherapy requires repeated dosing and lifelong treatment; this and the high cost associated with this therapy make it unattainable for a great portion of patients. The costs per dose are approximately $2,300-4,000 per patient for a one-year supply in the United States; these costs are prohibitive for implementation in developing countries. [7]
Keeping in mind the safety and effectiveness of anti-PCSK9 antibodies in lowering LDL-C levels, a potentially more promising approach would be an active immunization as opposed to the repeat treatment with the monoclonal antibody (passive immunization). In modern medicine, vaccines against infectious disease have dramatically improved human health;[8,9] newer vaccination approaches targeting cancer and chronic disease, including CVD are on the horizon and show promise.[9-11] Target antigen selection is crucial for successful vaccine development. In the case of atherosclerosis vaccines progress exists focusing in two main approaches: 1) lowering cholesterol in serum targeting against either the main protein component of LDL (apolipoprotein B100, ApoB100)[12-14] or targeting enzymes related to cholesterol homeostasis, such as the cholesteryl ester transfer protein (CETP) that mediates the transfer of cholesteryl-ester and triglycerides between lipoproteins,[15,16] or the already mentioned protease PCSK9;[17-21] and 2) lowering the inflammatory response through the blockade of proinflammatory molecules (heat shock protein 65, HSP65)[22] or cytokines (interleukin 1α, IL-1α).[23] We opted to design a trivalent vaccine candidate targeting the first approach; specifically, we designed vaccine candidates targeting ApoB, CETP, and PCSK9. These target “cholesterol checkpoints” proteins are validated targets and safety and efficacy in animal models[12-21] or in humans[24-26] has been demonstrated using active and passive immunotherapy approaches.
A successful vaccine requires an antigen, an adjuvant and a delivery strategy. In addition, peptide vaccines require a macromolecular display platform to enhance their stability and enable lymphatic trafficking and processing by antigen presenting cells.[8] To this end, we selected virus-like particles (VLPs) from the bacteriophage Qβ. VLPs can be obtained by expression of the 14 kDa-coat protein in Escherichia coli, resulting in in vivo assembly of VLPs. Scale-up could be achieved by fermentation under current good manufacturing practices (cGMP). Using chemical or molecular biology approaches the VLPs can be engineered to present the target epitopes as coat protein fusions. The VLP is a display platform but also functions as an adjuvant. The repetitive array of the VLP’s coat proteins and its recognition by pattern recognition receptors (PRRs) including toll-like receptors (TLRs) render epitope-displaying VLPs potent B cell immunogens.[9,11] In fact, several Qβ-based VLP vaccine candidates have been produced and entered clinical testing.[11]
Lastly, to address delivery requirements of the vaccine candidates, we formulated slow-release PLGA:VLP implants using hot melt-extrusion.[27-29] We have previously demonstrated that VLPs withstand the rigors of the high-temperature process; VLPs released from hot-melt extruded PLGA:VLP implants maintain their structural and immunogenic properties.[27-29] This manufacturing process is continuous, solvent free, and could lead to the high-throughput production of vaccine delivery devices. In this work, we evaluated the delivery of trivalent VLP vaccine candidates targeting ApoB, CETP, and PCSK9 “cholesterol checkpoint” proteins using the PLGA:VLP implant delivery strategy. VLPs were designed and expressed in E. coli. Vaccination was carried out in Balb/C mice using soluble mixtures vs. slow-release PLGA:VLP implants; monovalent and trivalent vaccine candidates were evaluated and efficacy was determined based on antibody titers against the target proteins, reduction of total cholesterol levels in plasma, lowered abundance of ApoB and PCSK9 proteins, and inhibition of CETP (the latter was tested in vitro using sera from immunized mice). Finally, the immunological and physiological safety of this multitarget method was validated.
MATERIAL AND METHODS
Qβ virus-like particle vaccines production
Bacteriophage Qβ virus-like particles (VLP) were expressed as previously reported.[30] Genes encoding for Qβ coat protein (CP) (NCBI accession: P03615) and Qβ CP-modified with target peptides (ApoB:[12] KTTKQSFDLSVKAQYKKNKH, CETP:[16] FGFPEHLLVDFLQSLS, and PCSK9:[20] NVPEEDGTRFHRQASKC) were codon optimized for E. coli expression and synthesized and cloned by GenScript Biotech Co. into pDUET-1 expression vectors. A linker of GSG was introduced between the C-terminus of Qβ CP and the target peptide. Four vectors were obtained and named corresponding to the carrying genes, pCDF_Qβ (unmodified Qβ CP), pCDF_Qβ_QβApoB (unmodified Qβ CP and QβApoB), pCOLA_Qβ_QβCETP (unmodified Qβ CP and QβCETP), and pRSF_Qβ_QβPCSK9 (unmodified Qβ CP and QβPCSK9). Three different plasmids were used to test whether the trivalent vaccine could be obtained through co-expression in the same cell. This was tested; however, yields of VLP-CETP were low; therefore, we decided to express the vaccine candidates side-by-side through transformation of E. coli Bl21 (DE3) [New England BioLabs] using one plasmid at a time. Positive bacteria colonies were selected and grown for 16 h at 37 °C and 250 rpm in LB medium [Thermo Fisher Scientific] with corresponding antibiotics (pCDF_Qβ and pCDF_Qβ_QβApoB, 25 μg/ml streptomycin [Sigma-Aldrich]; pCOLA_Qβ_QβCETP and pRSF_Qβ_QβPCSK9, 50 μg/ml kanamycin [Sigma-Aldrich]); freezer stocks were prepared using 20% (v/v) sterile glycerol and kept at −80 °C until use. The freezer stock of each transformed bacteria was grown first for 16 h at 37 °C and 250 rpm in 10 ml of MagicMedia™ [Invitrogen] with corresponding antibiotics added; then the culture was scaled up to 200 ml in the same medium and cultured for 20-24 h at 37 °C and 300 rpm. The cell pellet was harvested by centrifugation at 5,000 x g for 20 min at 4 °C and frozen at −80 °C overnight. After that, the pellet was resuspended in 10 ml of lysis buffer [GoldBio] per gram of wet cell mass, a lysis cocktail (1 mg/ml lysozyme [GoldBio], 2 μg/ml of DNase [Promega] and 2 mM MgCl2 [Fisher Scientific]) was added and the reaction mix was incubated at 37 °C for 1 h. To complete the lysis, sonication was performed at Amp 30% and 5-5 sec cycles for 10 min on ice. The lysate was centrifuge at 5,000 x g, 4 °C for 30 min and the clear supernatant was collected. To purify Qβ VLPs and the three vaccine candidates QβApoB, QβCETP, and QβPCSK9, the collected supernatant was precipitated by adding 10% (w/v) PEG8000 (Thermo Fisher Scientific) at 4 °C for 12 h on a rotisserie. The precipitated fraction was pelleted by centrifugation at 5,000 × g and dissolved in 40 ml PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4, pH 7.4) before extraction with a 1:1 (v/v) butanol/chloroform. The aqueous fraction was collected by centrifugation as above (at 5,000 x g) and finally the VLPs were purified on a 10–40% sucrose velocity gradient by ultracentrifugation at 9,6281 × g for 4.5 h. The light-scattering VLP band was collected and pelleted by ultracentrifugation at 16,0326 × g for 3 h. The purified VLPs were resuspended in PBS and stored at 4 °C until further use.
Antigen in silico characterization
Target peptide antigens ApoB = KTTKQSFDLSVKAQYKKNKH,[12] CETP = FGFPEHLLVDFLQSLS,[16] and PCSK9 = NVPEEDGTRFHRQASKC[20] were analyzed using an online peptide calculator (https://pepcalc.com/) to determine their isoelectric point. The sequence identity of the human-specific peptide antigens to corresponding mouse proteins (ApoB = NP_033823.2 and PCSK9= AAP31672.1; mice lack CETP) was determined using protein BLAST software (https://blast.ncbi.nlm.nih.gov/).
VLP characterization
The Qβ-based VLPs (Qβ, QβApoB, QβCETP, and QβPCSK9) were characterized as previously described[29] using fast protein liquid chromatography (FPLC), transmission electron microscopy (TEM), dynamic light scattering (DLS), sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE), and agarose gel electrophoresis. The Qβ VLPs concentration was determined by measuring the total protein using a Pierce BCA assay kit (Thermo Fisher Scientific). FPLC was performed using an AKTA-FPLC 900 system fitted with Superose 6 Increase 10/300 GL columns (GE Healthcare) using PBS as the mobile phase at flow rate 0.5 mL/min. TEM images were acquired on a FEI Tecnai Spirit G2 Bio TWIN transmission electron microscope. Samples were mounted on 400-mesh hexagonal copper grids and stained with 2% (v/v) uranyl acetate. DLS was carried out on a Malvern Instruments Zetasizer Nano at 25 °C in plastic disposal cuvettes. SDS-PAGE was performed under reducing conditions on NuPAGE 12% Bis-Tris protein gels [Thermo Fisher Scientific] at 120 mV for 35 min. and stained with Coomassie Brilliant Blue. Agarose gels (0.8% (w/v) in TAE buffer) were pre-stained with Gelred™ Nucleic Acid Gel Stain [GoldBio] and samples ran under non-reducing conditions at 100 mV for 30min. The gel images were acquired using the ProteinSimple FluorChem R imaging system.
Preparation of PLGA-loaded VLP implants by hot melt-extrusion
The Poly(lactic-co-glycolic acid) PLGA implants were prepared using our previously reported desktop melt-processing system.[27-29] PLGA [Akina Inc. 50:50 LG Ratio, MW 10-15 kDa] was ground in a mechanical blender [Magic Bullet™] for 5 minutes. The resultant powder was progressively passed through fine mesh screens (25, 35, 45, 60, 80, 120, 170, 230 porosity (standard mesh)) [Sigma-Aldrich] and large clumps re-blended. Powder which passed through a −45 mesh was used for implant formulation. The VLP vaccine candidates were individually lyophilized. To obtain the trivalent vaccine candidate the QβApoB, QβCETP, and QβPCSK9 VLPs were mixed at equal ratio before hot melt-extrusion. Implant formulation was as follows: 80% PLGA, 10% VLPs and 10% PEG8000 (by weight). The components were mixed in a 2-mL centrifuge tube by vortexing for 20 min, then loaded into the hot melt-processing system (maximum loading capacity = 60 mg) and heated to 70 °C for 90 s. The air pressure was gradually increased up to 10 psi to extrude the implant bars cylinders, which were dried for 1 h and kept in vacuumed bags with desiccants until use.
Animal immunization
All animal experiments were approved by IACUC of UC San Diego (the Assurance number is D16-00020 and the protocol number is S18021). Six-to-eight weeks old female C57BL/6J mice were purchased from Jackson Laboratory and kept under controlled conditions with chow and water ad libitum. Five animals were assigned per group. For s.c. injection with liquid formulations, the concentration of each VLP vaccine candidate, QβApoB, QβCETP, and QβPCSK9, was adjusted to 1 mg/ml in PBS and 100 μl were injected s.c. (100 μg/dose) every two weeks for a total of three times (prime + 2 boosts). For the mixture of QβApoB, QβCETP, and QβPCSK9 (3 antigens or 3Ag s.c. group) each VLP vaccine candidate was concentrated at 3 mg/ml in PBS, mixed 1:1:1 just before injecting 100 μl s.c. (100 μg/dose/Qβ VLP vaccine) using the same prime-boost administration schedule. For the PLGA:VLP implant (3Ag implant group), the implants were cut into 0.3–0.5 cm lengths according to their weight to yield 300 μg of each VLP vaccine candidate (to match the 300 μg total dose per three injections with liquid format). Implants were administered using a 18G needle [BD Co.] s.c. behind the neck. As control groups, 10 μg/dose of the free peptides [synthesized by GenScript Biotech Co.] were used for prime-boost administration; further a 300 μg/implant of unmodified Qβ VLP served as a control.
Blood collection was done just before injections or implant administration (week 0), and then at weeks 2, 4, 8, and 12. Mice fasted for 4 h prior to retroorbital blood draw and blood was collected in lithium-heparin treated tubes [Thomas Scientific]. Plasma was collected by centrifugation at 2,000 x g for 10 min at room temperature (RT); plasma was kept at −80 °C until use.
ELISA against peptides
Endpoint total IgG titers were determined by enzyme-linked immunosorbent assay (ELISA) against the corresponding peptide displayed in the VLP vaccine candidates (QβApoB, QβCETP, and QβPCSK9). 96-well, maleimide-activated plates [Thermo Fisher Scientific] were prepared following manufacturer directions. In brief, the plates were coated with 100 μl/well of each peptide (containing a N-terminal cysteine) at 25 μg/ml in coating buffer (0.1 M sodium phosphate, 0.15 M sodium chloride, 10 mM EDTA, pH 7.2) overnight at 4 °C. Three washes with 200 μl/well of PBST (PBS + 0.5% (v/v) Tween-20 [Thermo Scientific]) were done between every step. Then, plates were blocked for 1 h at RT using 200 μl/well of 10 μg/ml L-cysteine [Sigma-Aldrich]. After washing, 2-fold serial dilutions of plasma samples from immunized animals in coating buffer were added and incubated for 1 h at RT. After washing, an HRP-labeled goat anti-mouse IgG secondary antibody [Thermo Fisher Scientific] diluted 1:5,000 in PBST was added (100 μl/well) and incubated for 1 h at RT. After a final washing step, 1-Step Ultra TMB substrate [Thermo Fisher Scientific] was added (100 μl/well) and developed for 10 min; the reaction was then stopped using 2N H2SO4 (100 μl/well) [Thermo Scientific]. The absorbance was read at 450 nm on a Tecan microplate reader. The endpoint antibody titers were defined as the reciprocal serum dilution at which the absorbance exceeded two times the background value (blank wells without plasma sample).
Immunoglobulin (Ig) isotyping
The ELISA was adapted from the protocol described above; instead of serial plasma dilutions, here samples a 1:1,000 dilutions were tested using sera collected 8 post-immunization. The secondary antibodies used were an HRP-labeled goat anti-mouse IgG1 (1:5,000) or IgG2b (1:5000) secondary antibody [Abcam]. The IgG2b/IgG1 ratio was reported for each group and a ratio lower than 1 was considered as Th2 response.
ELISA against target proteins
To determine whether the antibodies raised were indeed specific against the target protein, ELISA against intact, full-length target proteins (PCKS9 or LDL) was performed. First, 96-well PolySorp plates [Thermo Fisher Scientific] were coated using 100 μl of recombinant mouse PCKS9 [Abcam] (10 μg/ml in PBS) or 100μl of human LDL [Thermo Fisher Scientific] (10μg/ml in PBS) or 100μl human oxidized LDL [Thermo Fisher Scientific] (10μg/ml in PBS) and incubated at 4 °C overnight. After washing with PBST, the plates were blocked with 200 μL PBST + 2% (w/v) BSA for 1 h at RT. The ELISA protocols described above were used for detection and quantification of PCKS9 or (oxidized) LDL-specific antibody titers. Plasma samples collected 8 weeks post immunization were analyzed. The plasma samples were evaluated per animal or pooled by group and tested in triplicate.
ApoB and PCSK9 plasma levels
The ApoB and PCSK9 plasma levels were determined using blood draws from week 0 and week 8 post first immunization. The ApoB and PCSK9 protein plasma concentration was determined using commercial ELISA kits [Abcam] following the manufacturer’s protocols. The plasma samples were evaluated per animal or pooled by group and tested in triplicate.
CETP inhibition assay in vitro
To determine whether antibodies produced would bind and block the activity of CETP, a commercial CETP Activity Assay Kit II [Abcam] was utilized. Plasma samples collected 4 weeks post immunization were tested following the manufacturer’s protocols. Results were reported as a kinetic curve of relative fluorescent units (Ex480/Em511) over the 90 min time course and as CETP percentage activity at 90 min. The plasma samples were evaluated per animal or pooled by group and tested in triplicate.
Measurement of total cholesterol levels
Total cholesterol from plasma samples was determined using a commercial Amplex™ Red Cholesterol Assay [Thermo Fisher Scientific] following the manufacturer’s protocols. In brief, plasma samples collected at week 0, 8 and 12 post t immunization were diluted 1:200 and analyzed. The plasma samples were evaluated per animal or pooled by group and tested in triplicate.
Measurement of liver and kidney biomarkers
To determine the safety of the VLP vaccine candidates and their implant formulations, especially focusing on the trivalent formulation, toxicity plasma biomarkers were assessed. For liver damage the concentrations of the enzymes aspartate transaminase (AST) and alanine transaminase (ALT) were established by Aspartate Aminotransferase Activity kit [Abcam] and Alanine Transaminase Activity Assay Kit [Abcam], respectively. For kidney damage the concentration of kidney injury molecule-1 (KIM-1) by Mouse KIM-1 ELISA Kit [Abcam] was determined. Here, plasma samples collected at week 0 and 12 weeks post-immunization were tested. The plasma samples were evaluated per animal or pooled by group and tested in triplicate.
ELISPOT assay
Briefly, 96-well ELISPOT plates [Cellular Technology Ltd] were coated with an 1:166 anti-mouse interferon-gamma (IFN-γ) capture antibody and anti-mouse interleukin-4 (IL-4) capture antibody overnight at 4 °C. Splenocyte suspensions collected 2 weeks post immunization using the VLP 3Ag s.c. group were analyzed and added to the plates (5 x 105 cells/well) following stimulation with medium alone (negative control), a mixture of free-peptides (20μg/ml) of each epitope, a mixture of the target proteins (5μg/ml each; recombinant mouse PCSK9 protein [Abcam], human LDL [Thermo Fisher Scientific], and recombinant human CETP [MyBioSource]), unmodified Qβ VLP (10μg/ml), or 50 ng/ml phorbol 12-myristate 13-acetate (PMA)/1 μg/ml Ionomycin [Sigma-Aldrich] (positive control) at 37 °C and 5% CO2 for 24 h. The plates were washed with PBST and the incubated with 1:1000 anti-murine IFN-γ (FITC-labeled) and 1:666 anti-murine IL-4 (Biotin-labeled) antibodies for 2 h at RT. Plates were washed, and 1:1000 streptavidin-alkaline phosphatase (AP) and anti-FITC-HRP secondary antibodies were added to each well and incubated for 1h at RT. Plates were washed with PBST and distilled water, then incubated with AP substrate for 15 min at RT, washed with distilled water and incubated with HRP substrate for 10 min at RT. Plates were rinsed with water and air-dried at RT overnight. Colored spots were quantified using a Immunospot S6 ENTRY Analyzer. The splenocytes were evaluated per animal and tested in triplicate for each stimulant.
Statistical analysis
Data are presented as the mean ±SD. Single comparisons were made with unpaired, two-tailed t-tests using SPSS Statistics software or GraphPad Prism 6. P values lower than 0.5 were considered as statistically significant. Replicates per experiment are detailed in each method section and figure.
RESULTS AND DISCUSSIONS
Characterization of VLP vaccine candidates.
We selected the following peptide antigens; these antigens have been validated as B cell epitopes:
Table 1.
Peptide antigen properties.
The peptide epitopes were cloned into VLP expression vectors for display at the C-terminus of the Qβ coat protein (CP) via a GSG linker. Unmodified Qβ CP and the epitope-displaying Qβ CPs (QβApoB, QβCETP, and QβPCSK9, respectively) were cloned in the same vector to produce hybrid Qβ VLP containing both, unmodified and modified Qβ CP (Figure 1). We chose the hybrid assembly by expressing free and modified CP, because it has been previously established that hybrid assembly is required for the genetic display strategy; free coat protein is required for assembly yielding intact VLPs.[30] The VLP vaccine candidates were expressed in E. coli and purified over sucrose gradients via ultracentrifugation. Purified VLPs were characterized using a combination of size exclusion chromatography (fast protein liquid chromatography, FPLC), dynamic light scattering (DLS), transmission electron microscopy (TEM), and gel electrophoresis (native agarose gels and denaturing SDS-PAGE) to confirm their structural integrity and degree of antigen incorporation (Figure 2).
Figure 1.

Qβ VLP vaccine design. Top) Genes encoding unmodified and modified Qβ coat proteins (CP) with the corresponding peptides (QβApoB, QβCETP, and QβPCSK9); genes are not drawn to scale; middle) Vectors used for expression in E. coli BL21 (DE3): unmodified Qβ CP gene was cloned into pCDF_Qβ and served as a control. Modified and unmodified Qβ CP genes were cloned resulting in the following expression vectors: pCDF_Qβ_QβApoB, pCOLA_Qβ_QβCETP, and pRSF_Qβ_QβPCSK9; the peptide-displaying CP was put under control of a second T7 promoter; bottom) hybrid Qβ VLP vaccine candidates; inset shows the number of peptides displayed per VLP as determined by SDS-PAGE analysis. lacI= lactose repressor protein gene; CloDF13 ori= CloDF13 origin of replication; ColA ori= ColA origin of replication; RSF ori= RSF origin of replication; SmR= Streptomycin resistance gene; AmpR= Ampicillin resistance gene promoter; KnR= Kanamycin resistance gene.
Figure 2.

Characterization of Qβ VLP vaccines. A) FPLC chromatogram of unmodified Qβ VLP and Qβ VLP vaccine candidates QβApoB, QβCETP, or QβPCSK9. B) DLS of unmodified Qβ VLPs and Qβ VLP vaccine candidates. C) TEM images of negatively-stained unmodified Qβ VLPs and Qβ VLP vaccine candidates (scale bar = 100 nm). D) Electrophoretic mobility of unmodified Qβ VLPs and Qβ VLP vaccine candidates on native 0.8% (w/v) agarose gels stained with Gelred™ Nucleic Acid Gel Stain (VLPs package random host RNA enabling detection by nucleic acid staining). E) SDS-PAGE analysis of unmodified Qβ VLP and Qβ VLP vaccine candidate under reducing conditions. Unmodified Qβ coat protein (CP, white triangle, ~14 kDa) and modified Qβ CPs (yellow dot, QβApoB= ~16.4 kDa, red dot, QβCETP= ~15.9 kDa, and blue dot, QβPCSK9= ~16 kDa) were visualized after Coomassie Brilliant Blue staining. All samples were tested in triplicate (n= 3) for all the characterization methods, and the pictures and data shown are representative from those triplicates.
FPLC analysis revealed a single peak for each of the Qβ VLP vaccine candidates, indicating that the VLPs were intact (no free CPs were detected) and free of aggregation (Figure 2A). DLS measurement revealed the hydrodynamic diameter of the VLPs being close to 30 nm for QβApoB and QβCETP VLPs; QβPCSK9 VLPs showed in increase in size with a diameter of 38 nm (Figure 2B); this increase in size may be attributed to interparticle interactions promoted by formation of disulfide bonds between the C-terminal Cys side chain on the PCSK9 antigen (Table 1, Figure 1). Extensive particle aggregation, however, was not apparent by any method (FPLC, DLS, or TEM). TEM imaging of negatively-stained VLPs further indicated that particle preparation yielded monodisperse and intact VLPs (Figure 2C).
Agarose gels further confirmed the presence of intact VLPs (Figure 2D); the electrophoretic mobility of ApoB- and PCSK9-epitope displaying VLPs was slightly reduced compared to native Qβ or QβCETP VLPs, which can be explained by the higher IP of these antigens (Table 1). Lastly, SDS-PAGE analysis confirmed the presence of unmodified Qβ CP (~14 kDa) and modified Qβ CPs (QβApoB= ~16.4k Da, QβCETP= ~15.9 kDa, and QβPCSK9= ~16.0 kDa) for each of the preparation (Figure 2E), and according to the relative intensity of the bands the percentage of modified Qβ CPs (QβApoB= ~38%, QβCETP= 26%, and QβPCSK9= 35%). Therefore, the number of peptide epitopes per VLP lies between 50-70 peptides per VLP (each VLP is comprised of 180 copies of the CPs). Overall, these results indicate that intact Qβ VLPs displaying the antigens ApoB, CETP, and PCSK9 were successfully produced.
Immunogenicity of Qβ VLP vaccines.
After characterization of the Qβ VLP vaccine candidates, we determined whether s.c. immunization using single antigen VLPs (QβApoB, QβCETP, or QβPCSK9) versus a mixture of the three Qβ VLP vaccine candidates (the trivalent vaccine candidate), either injected or delivered via a slow-release implant, could produce antibody titers against the target proteins (Figure 3A). The implants of the following composition 80% PLGA, 10% VLP, and 10% PEG8000, were produced as previously reported; the VLPs are released from the implants over a time course of 30 days.[27-29] We found the monovalent vs. trivalent vaccinations resulted in comparable antibody endpoint titers; VLP display of the peptide antigens was effective to yield high antibody titers without the need of additional adjuvants. Furthermore, immunization using the slow-release implants versus prime-boost administration schedules using soluble VLPs was equally effective. Antibody titers were detectable as early as 2-weeks post immunization with maximum titers achieved at week 4 post immunization. The soluble peptides were, as anticipated, not effective to raise target-specific antibodies (Figure 3B).
Figure 3.

Immunization and antibody response. A) Vaccination schedules for the Qβ VLP vaccine candidates QβApoB, QβCETP or QβPCSK9. Mice in the single antigen group were immunized s.c. using a 100 μg-dose at weeks 0, 2, and 4 (black arrows) of each Qβ, QβApoB, QβCETP or QβPCSK9, respectively. The 3Ag s.c. group received a mixture of the three Qβ VLP vaccine candidates at 100 μg-per VLP dose. Lastly, the 3Ag implant group was immunized using PLGA:VLP implants containing a mixture of the three Qβ VLP vaccine candidates, loaded into the PLGA implant at a 100 μg-per VLP dose (300 μg protein total). B) Endpoint antibody titers at different weeks after first immunization. Week 0 corresponds to plasma collected prior to the first immunization. Plasma samples from free-peptides (-ApoB, -CETP or -PCSK9) and unmodified Qβ VLP (Qβ) were used as control groups. Plasma samples from five mice (n= 5) were tested individually in duplicate. Data is presented as mean ± SD. C) IgG isotype profile (IgG2b/IgG1 ratio), a ratio lower than 1 was considered Th2-biased. Plasma samples from five mice (n= 5) from the same group were pooled and tested five times for each Ig isotype. Data is presented as mean ± SD.
For the ApoB peptide groups, the titers were slightly higher at week two comparing the monovalent versus trivalent vaccine 52 candidates; however, at later time points the 3Ag groups resulted in slightly higher titers (Figure 3B). For CETP peptide, titers remained the same between groups and slightly decreased only for the monovalent vaccine candidate during weeks 8 and 12 (Figure 3B). For PCSK9 peptide, antibody titers overall were consistent between groups; with the 3Ag implant resulting in slightly reduced antibody titers over longer time periods (Figure 3B). Overall though, the various vaccination groups, monovalent versus trivalent administered as soluble vaccine versus slow-release implant, were consistent. The subtle variations may be explained by differences and bias of antigen processing by antigen processing cells (APC).
Our next goal was to establish what IgG isotypes (IgG1 and IgG2b) were elicited and whether the ratio would be altered when Qβ VLPs was injected as a single antigen or in a mixture (3Ag) either s.c. (prime-boost) or implant (slow-release). Data indicate that IgG2b/IgG1 ratios remained essentially the same between groups; and IgG2b/IgG1 ratios were lower than 1. IgG1 antibodies are primarily induced via Th2-type cytokines (e.g. IL-4), and the production of IgG2b antibodies reflects the involvement of Th1-type cytokines (e.g. IFN-γ);31 therefore, the IgG2b/IgG1 ratio of < 1 can be interpreted as a T helper cells 2 (Th2) biased response (Figure 3C). This is consistent with previous reports on cardiovascular vaccine formulations incorporating these peptide epitopes.12,16,20
Interaction and specificity of antibodies raised from the VLP vaccinated mice against the target proteins, ApoEB, CETP, and PCSK9.
Once we confirmed that antibodies against the peptide epitopes were raised, we also confirmed that these antibodies indeed recognize the naïve target protein. First, plasma was isolated from animals receiving the monovalent QβApoB vaccine candidates versus the trivalent 3Ag vaccine candidates and sera were tested against human LDL and oxidized LDL (oxLDL). ELISA confirmed recognition of LDL and oxLDL (Figure 4A and B). ApoB is the primary apolipoprotein of cholesterol and the anti-atherosclerotic effect of anti-ApoB antibodies is related to preferential inhibition of inflammation and reversion of cholesterol transportation by altering the pathway by which macrophages phagocytize ox-LDL.[32] We also confirmed that vaccination using the monovalent QβApoB or trivalent 3Ag vaccine candidates lowered plasma levels of ApoB protein (Figure 4C-E). It is important to note that the difference was more profound when analyzing sera obtained from the trivalent vaccine groups (3Ag s.c. and 3Ag implant) versus sera obtained from mice immunized with the monovalent ApoB vaccine candidate. This effect could be attributed to the synergy of simultaneous PCSK9 neutralization in the trivalent vaccine candidate group (Figure 4J and K). Binding of the sera to PCSK9 protein was validated by direct ELISA; sera from all vaccinated groups but not control groups were found to recognize PCSK9 (Figure 4H). Then, the PCSK9 plasma concentration was examined at week 8 post first vaccination; data indicate increased levels of PCSK9 and this phenomenon was observed across all vaccine groups; others have made similar observations and the apparent increase can be explained by detection of IgG-PCSK9 immunocomplexes.[17-21] Therefore, total IgG was depleted prior to assessing the plasma levels of PCSK9 again. Now, vaccinated groups (using any formulation or administration schedule) showed a significant reduction in PCSK9 levels, indicting effectiveness of the vaccination approach (Figure 4I-K). Finally, we evaluated the neutralizing effect of the various vaccine candidates on CETP activity; because mice do not express CETP, an in vitro assay was established. Results showed that plasma from the three groups were effective to inhibit the CETP activity by up to 15% (Figure 4F and G). Together, these findings demonstrate that antibodies are generated from vaccination using the monovalent or trivalent vaccine candidates administered as prime-boost or slow-release implant are effective in that they recognize and neutralize their target proteins and reduce target protein concentration in plasma.
Figure 4.

Plasma levels of ApoB and PCSK9, and CETP in vitro. A and B) ELISA against LDL and oxidized LDL (oxLDL), respectively. Plasma samples from five mice were pooled and five technical replicas were analyzed (n=5). C, D, and E) Plasma ApoB protein concentration for single antigen, 3Ag s.c., and 3Ag implant groups, respectively. Plasma was evaluated at week 0 (pre-immunization) and week 8 post first immunization. Plasma samples from five mice were pooled and five technical replicas were analyzed twice (n=10). F and G) CETP activity kinetic curve after incubation with plasma samples and CETP percentage activity after 90min. Plasma samples from five mice were pooled and three technical replicas were analyzed twice (n=6). H) ELISA of sera from vaccinated groups against mouse PCSK9 protein. Plasma samples from five mice were pooled and three technical replicas were analyzed twice (n=6). I, J, and K) PCSK9 protein concentration in plasma from mice receiving the various vaccine candidates at week 0 (pre-immunization) and week 8 post first immunization. Plasma samples from five mice were pooled and five technical replicas were analyzed (n=5). Depleted IgG = plasma after depletion of IgGs to remove IgG-PCSK9 immunocomplexes. Single antigen group = QβApoB, QβCETP or QβPCSK9 VLPs; 3Ag s.c.= mixture of QβApoB, QβCETP, and QβPCSK9 injected s.c.; 3Ag implant = mixture of QβApoB, QβCETP, and QβPCSK9 delivered via PLGA:VLP implants. Plasma samples from free-peptides (-ApoB, -CETP, and -PCSK9) and unmodified Qβ VLP (Qβ) were used as control groups. Data is shown as mean ±SD. All comparison between means of vaccinated and control group were done using unpaired t test (two-tailed, 95% confidence value). P values < 0.05 were considered as statistically significant.
Total cholesterol plasma levels in naïve versus vaccinated mice.
After we corroborated that antibodies generated from the VLP vaccine candidates bind, neutralize, and lower their target proteins (Figure 4), we set out to determine plasma cholesterol levels comparing the various vaccine candidates. We first measured the effect of monovalent vaccine candidates and determined that immunization with QβApoB and QβPCSK9 led to a significant reduction on total cholesterol at week 12 post first vaccination, and only QβPCSK9 group showed a lower concentration at week 8 as well (Figure 5A). QβCETP did not indicate a significant difference compared to control (Figure 5A), although plasma samples from this group effectively neutralized recombinant CETP in vitro (Figure 4F and G). This data is as expected, because mice lack CETP,[33] therefore further experiments with transgenic mice expressing CETP[19] or other animal models such as rabbit[16] could help to elucidate how important the effect on CETP inhibition using our QβCETP or trivalent vaccine candidate. Data also indicate that the trivalent vaccine candidates, whether administered as prime-boost or slow-release implant, were effective in decreasing cholesterol levels in mice (Figure 5B and C). These results correlate with antibody titers (Figure 3) and reduction of PCSK9 and ApoB proteins levels in plasma (Figure 4). Data are also consistent with previous reports indicating a key role of these proteins on regulation of plasma cholesterol levels.[12-14,17-21]
Figure 5.

Total cholesterol concentration in plasma. Total cholesterol was determined at different time points after, pre- and post-immunization (week 0, 8, and 12) for A) single antigen groups, B) 3Ag s.c. group, and C) 3Ag implant group. Single antigen group= QβApoB, QβCETP or QβPCSK9 VLP vaccine; 3Ag s.c.= mixture of QβApoB, QβCETP, and QβPCSK9 injected subcutaneously; 3Ag implant= mixture of QβApoB, QβCETP, and QβPCSK9 prepared as loaded PLGA implants; ns= no significance (p>0.05). Plasma samples from free-peptides (-ApoB, -CETP, and -PCSK9) and unmodified Qβ VLP (Qβ) were used as control groups. Plasma samples from five mice were pooled and four technical replicas were analyzed twice (n=8). Data is shown as mean ±SD. All comparison between means of vaccinated and control group were done using unpaired t test (two-tailed, 95% confidence value). P values < 0.05 were considered as statistically significant.
Qβ VLP vaccine candidate safety in healthy mice.
Evaluation of safety is critical, because self-antigens are the therapeutic target. Safety parameters such as the absence of a target-specific T-cell response (Th and cytotoxic T-cells) and blood biomarkers were measured. While safety of the monovalent vaccine candidates has been demonstrated,[12-21] a trivalent vaccination strategy has not yet been reported. First, we examined the concentration of Kidney Injury Molecule-1 (KIM-1), a transmembrane glycoprotein and blood biomarker of kidney injury,[34] pre- and post-immunization (week 0 and 12). There was not difference in KIM-1 levels comparing mice receiving the trivalent vaccine candidate versus the control group, i.e. age-matched mice (Figure 6A), demonstrating that blocking multiple self-proteins (i.e. PCSK9 and ApoB; CETP is not expressed in mice) did not alter kidney physiology in healthy animals. Second, we measured the activity of the enzymes aspartate transaminase (AST) and alanine transaminase (ALT) in plasma as a biomarker for liver injury.[35] ALT levels were comparable in the vaccinated versus the age-matched mice control group; some elevation of the AST levels were observed in mice receiving the trivalent vaccine candidate (Figure 6B and C) – however, levels were in the normal range.[36] The data indicate safety of the trivalent vaccine candidate with no apparent kidney or liver damage.
Figure 6.
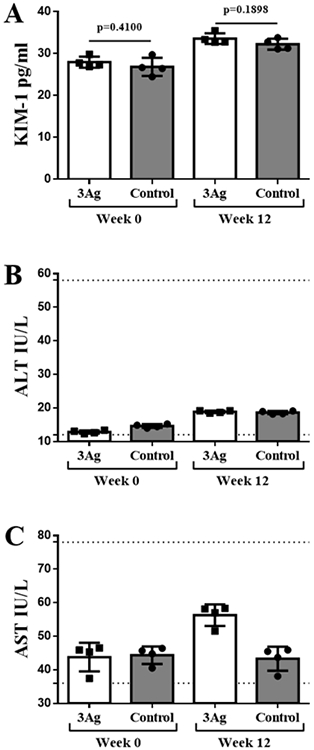
Kidney and liver plasma biomarkers. A) Kidney Injury Molecule-1 (KIM-1); B) aspartate transaminase (AST); and C) alanine transaminase (ALT) levels in plasma samples collected from mice receiving the trivalent vaccine candidate (3Ag s.c. group) at week 0 and 12 after first vaccination compared to age-matched mice. 3Ag s.c.= mixture of QβApoB, QβCETP, and QβPCSK9 injected subcutaneously. Dotted lines= threshold for normal values in healthy mice. Plasma samples from five mice were pooled and four technical replicas were analyzed twice (n= 4). Data is shown as mean ±SD. All comparison between means of vaccinated and control group were done using unpaired t test (two-tailed, 95% confidence value). P values < 0.05 were considered as statistically significant.
Finally, we assessed whether T cell activation was primed upon vaccination using the trivalent vaccine candidate; this was assayed using splenocytes and ELISpot assay. We monitored the production of IFN-γ and IL-4 cytokines, which are linked to a Th1 versus Th2 profile, respectively, and compared splenocytes isolated from mice vaccinated using the mixture of QβApoB, QβCETP, and QβPCSK9 (3Ag s.c. group). Stimulation of splenocytes with Qβ VLPs yielded comparable levels of IFN-γ and IL-4, respectively, when comparing the Qβ versus trivalent vaccine candidate groups; PMA/Ionomycin served as a positive control (Figure 7A). T cell responses against the vaccination platform, the Qβ VLPs, is apparent and expected, because the VLPs contain a mixture of epitopes. Importantly, stimulation with free-peptides or a mixture of the target proteins, ApoB, CETP, and PCSK9, did not elicit a significant IFN-γ production compared to the medium only-control group (Figure 7A). Splenocytes stimulated with a mixture of whole targeted proteins resulted in a reduction of IL-4 levels (Figure 7A). These data indicate that T cell activation against the target proteins ApoB, CETP, and PCSK9 is absent, thus laying a foundation that the trivalent vaccine candidate is safe and does not elicit T cell mediated immunotoxicity directed against the self-proteins; this is consistent with the safety parameters established with others works on self-antigen vaccines.[17,18,21] Data are also in agreement with the IgG isotype, which we also found to have a Th2 bias (see Figure 3B).
Figure 7.

T-cell activation using ELISpot assay and splenocytes from immunized mice. Splenocytes (5 x 105 cells per well) from 3Ag s.c. immunized mice were stimulated with medium only (no stimulation control), a mixture of free-peptides (-ApoB, -CETP, and -PCSK9), a mixture of targeted proteins (ApoB, CETP, and PCSK9), unmodified Qβ VLP or PMA/Ionomycin (positive control). A) Cytokine (IFN-γ and IL-4) producing cells were counted as splenocytes forming colonies (SFC). B) Representative images of blue (IL-4) and red (IFN-γ) spots formed by stimulated splenocytes. Splenocytes from three mice (n= 3) were tested individually in triplicate. Data is shown as mean ±SD. All comparison between means of vaccinated and control group were done using unpaired t test (two-tailed, 95% confidence value). P values < 0.05 were considered as statistically significant. *= p<0.05 compared to medium.
CONCLUSION
In summary, we developed and tested a trivalent VLP vaccine candidate targeting the cholesterol checkpoints proteins (ApoB, CETP, and PCSK9); the vaccine candidates were effective when administered as soluble mixtures or delivered via a PLGA:VLP polymer implant. Data indicate that high levels of functional antibodies are produced that recognize, bind, and inhibit their target proteins leading to reduced cholesterol levels in mice. Immunotoxicity or other adverse effects on liver and kidney function were not apparent. The proposed active immunotherapy approach could provide a cost-effective alternative and efficacious alternative to contemporary monoclonal antibody therapy (e.g. Alirocumab and Evolocumab). In particular, melt-manufacturing could lead to mass production of implants or microneedles – the latter would be self-administered; and this could be a potentially transformative technology for the developing world. This multi-target vaccination approach combined with the innovative vaccine delivery technology could be a vaccination platform approach for alleviating the burden of hypercholesteremia and cardiovascular diseases, especially for developing countries where the cold-chain requirements and high costs are prohibitive for implementation of therapeutic biologics. Finally, given the high degree of flexibility of the VLP platform technology, additional and alternate targets involved in lipid homeostasis could be added in a plug-and-play manner.
ACKNOWLEDGMENTS
This work was supported in part by a grant from the National Institute of Health (R01 HL137674 to N.F.S.) and O.A.O.-R. acknowledges the UC MEXUS-CONACYT Postdoctoral Fellowship 2019-2020 number FE-19-58.
ABBREVIATIONS
- FPLC
fast protein liquid chromatography
- TEM
transmission electron microscopy
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- IFN-γ
interferon
- IL-4
interleukin
- IgG
immunoglobulin G
- FITC
fluorescein isothiocyanate
- HRP
horseradish peroxidase
Footnotes
CONFLICT OF INTEREST STATEMENT
Drs. Steinmetz and Pokorski are co-founders of Mosaic ImmunoEngineering Inc. Dr. Ortega-Rivera declares no potential conflict of interest.
REFERENCES
- [1].World Health Organization (2016) Descriptive note 3 17 May/2017, http://www.who.int/mediacentre/factsheets/fs317/en/ [accessed: 11/01/2020]
- [2].Gisterå A, Hansson GK, Nature Reviews Nephrology 2017, 13, 368. [DOI] [PubMed] [Google Scholar]
- [3].Robinson JG, Heistad DD, Fox KAA, Atherosclerosis 2015, 243, 593. [DOI] [PubMed] [Google Scholar]
- [4].Sabatine MS, Giugliano RP, Wiviott SD, Raal FJ, Blom DJ, Robinson J, Ballantyne CM, Somaratne R, Legg J, Wasserman SM, Scott R, Koren MJ, Stein EA, New England Journal of Medicine 2015, 372, 1500. [DOI] [PubMed] [Google Scholar]
- [5].Robinson JG, Farnier M, Krempf M, Bergeron J, Luc G, Averna M, Stroes ES, Langslet G, Raal FJ, El Shahawy M, Koren MJ, Lepor NE, Lorenzato C, Pordy R, Chaudhari U, Kastelein JJP, New England Journal of Medicine 2015, 372, 1489. [Google Scholar]
- [6].Nissen SE, Stroes E, Dent-Acosta RE, Rosenson RS, Lehman SJ, Sattar N, Preiss D, Bruckert E, Češka R, Lepor N, Ballantyne CM, Gouni-Berthold I, Elliott M, Brennan DM, Wasserman SM, Somaratne R, Scott R, Stein EA, Sullivan D, Kostner K, Don Wauchope A, Hartleib M, Baass A, Bergeron J, Joy T, Adamkova V, Blaha V, Jeppesen J, Jensen HK, Krempf M, Bully C, Bosiljanoff P, Steinhagen-Thiessen E, Pintus P, Borghi C, Sampietro T, Vigna GB, Mannarino E, Pozzi C, Liem A, Van Leendert R, Scott R, Kjaernli T, Langslet G, Jacovides A, Klug E, Blom D, Neely D, Dawson C, Miller M, McCullum K, Rocco M, Shah P, Rosenblit P, Pokrywka G, Blazing M, Toth P, Moriarty P, Rubenfire M, Morris P, Quyyumi A, Kopecky S, JAMA - Journal of the American Medical Association 2016, 315, 1580. [DOI] [PubMed] [Google Scholar]
- [7].ICER, Institute for Clinical and Economic Review. PCSK9 Inhibitors for Treatment of High Cholesterol: Effectiveness, Value, and Value-Based Price Benchmarks Final Report. Published February24th, 2019.
- [8].Shin MD, Shukla S, Chung YH, Beiss V, Chan SK, Ortega-Rivera OA, Wirth DM, Chen A, Sack M, Pokorski JK, Steinmetz NF, Nature Nanotechnology 2020, 15, 646. [DOI] [PubMed] [Google Scholar]
- [9].Chackerian B, Frietze KM, Expert Review of Vaccines 2016, 15, 561. [DOI] [PubMed] [Google Scholar]
- [10].Shukla S, Wang C, Beiss V, Cai H, Washington T, Murray AA, Gong X, Zhao Z, Masarapu H, Zlotnick A, Fiering S, Steinmetz NF, Biomaterials Science 2020, 8, 5489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bachmann MF, Jennings GT, Philosophical Transactions of the Royal Society B: Biological Sciences 2011, 366, 2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fredrikson GN, Söderberg I, Lindholm M, Dimayuga P, Chyu KY, Shah PK, Nilsson J, Arteriosclerosis, Thrombosis, and Vascular Biology 2003, 23, 879. [DOI] [PubMed] [Google Scholar]
- [13].Klingenberg R, Lebens M, Hermansson A, Fredrikson GN, Strodthoff D, Rudling M, Ketelhuth DFJ, Gerdes N, Holmgren J, Nilsson J, Hansson GK, Arteriosclerosis, Thrombosis, and Vascular Biology 2010, 30, 946. [DOI] [PubMed] [Google Scholar]
- [14].Kobiyama K, Vassallo M, Mitzi J, Winkels H, Pei H, Kimura T, Miller J, Wolf D, Ley K, European Journal of Immunology 2018, 48, 1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Qi G, Li J, Wang S, Xin S, Du P, Zhang Q, Zhao X, Peptides 2011, 32, 790. [DOI] [PubMed] [Google Scholar]
- [16].Rittershaus CW, Miller DP, Thomas LJ, Picard MD, Honan CM, Emmett CD, Pettey CL, Adari H, Hammond RA, Beattie DT, Callow AD, Marsh HC, Ryan US, Arteriosclerosis, Thrombosis, and Vascular Biology 2000, 20, 2106. [DOI] [PubMed] [Google Scholar]
- [17].Rittershaus CW, Miller DP, Thomas LJ, Picard MD, Honan CM, Emmett CD, Pettey CL, Adari H, Hammond RA, Beattie DT, Callow AD, Marsh HC, Ryan US, Arteriosclerosis, Thrombosis, and Vascular Biology 2000, 20, 2106. [DOI] [PubMed] [Google Scholar]
- [18].Pan Y, Zhou Y, Wu H, Chen X, Hu X, Zhang H, Zhou Z, Qiu Z, Liao Y, Scientific Reports 2017, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Landlinger C, Pouwer MG, Juno C, Van Der Hoorn JWA, Pieterman EJ, Jukema JW, Staffler G, Princen HMG, Galabova G, European Heart Journal 2017, 38, 2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Crossey E, Amar MJA, Sampson M, Peabody J, Schiller JT, Chackerian B, Remaley AT, Vaccine 2015, 33, 5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kawakami R, Nozato Y, Nakagami H, Ikeda Y, Shimamura M, Yoshida S, Sun J, Kawano T, Takami Y, Noma T, Rakugi H, Minamino T, Morishita R, PLoS ONE 2018, 13, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Jing H, Yong L, Haiyan L, Yanjun M, Yun X, Yu Z, Taiming L, Rongyue C, Liang J, Jie W, Li Z, Jingjing L, Vaccine 2011, 29, 4102. [DOI] [PubMed] [Google Scholar]
- [23].Tissot AC, Spohn G, Jennings GT, Shamshiev A, Kurrer MO, Windak R, Meier M, Viesti M, Hersberger M, Kündig TM, Ricci R, Bachmann MF, European Journal of Immunology 2013, 43, 716. [DOI] [PubMed] [Google Scholar]
- [24].Davidson MH, Maki K, Umporowicz D, Wheeler A, Rittershaus C, Ryan U, Atherosclerosis 2003, 169, 113. [DOI] [PubMed] [Google Scholar]
- [25].Clinical trial NCT01284582 https://clinicaltrials.gov/ [accessed: 11/01/2020]
- [26].Clinical trial NCT02508896 https://clinicaltrials.gov/ [accessed: 11/01/2020]
- [27].Lee PW, Shukla S, Wallat JD, Danda C, Steinmetz NF, Maia J, Pokorski JK, ACS Nano 2017, 11, 8777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wirth DM, Pokorski JK, Polymer 2019, 181, 121802. [Google Scholar]
- [29].Shao S, Ortega-Rivera OA, Ray S, Pokorski JK, Steinmetz NF, Vaccines 2021, 9 (1), 6633478147 [Google Scholar]
- [30].Brown SD, Fiedler JD, Finn MG, Biochemistry 2009, 48, 47, 11155–11157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Finkelman FD, Holmes J, Katona IM, Urban JF, Beckmann MP, Park LS, Schooley KA, Coffman RL, Mosmann TR, Paul WE, Annual Review of Immunology 1990, 8, 303. [DOI] [PubMed] [Google Scholar]
- [32].Zeng Z, Cao B, Guo X, Li W, Li S, Chen J, Zhou W, Zheng C, Wei Y, American Journal of Translational Research 2018, 10, 1817. [PMC free article] [PubMed] [Google Scholar]
- [33].Tanigawa H, Billheimer JT, Tohyama JI, Zhang YZ, Rothblat G, Rader DJ, Circulation 2007, 116, 1267. [DOI] [PubMed] [Google Scholar]
- [34].Sabbisetti VS, Waikar SS, Antoine DJ, Smiles A, Wang C, Ravisankar A, Ito K, Sharma S, Ramadesikan S, Lee M, Briskin R, De Jager PL, Ngo TT, Radlinski M, Dear JW, Park KB, Betensky R, Krolewski AS, Bonventre JV, Journal of the American Society of Nephrology 2014, 25, 2177 LP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].McGill MR, EXCLI Journal 2016, 15, 817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kollmus H, Fuchs H, Lengger C, Haselimashhadi H, Bogue MA, Östereicher MA, Horsch M, Adler T, Aguilar-Pimentel JA, Amarie OV, Becker L, Beckers J, Calzada-Wack J, Garrett L, Hans W, Hölter SM, Klein-Rodewald T, Maier H, Mayer-Kuckuk P, Miller G, Moreth K, Neff F, Rathkolb B, Rácz I, Rozman J, Spielmann N, Treise I, Busch D, Graw J, Klopstock T, Wolf E, Wurst W, Yildirim AO, Mason J, Torres A, Balling R, Mehaan T, Gailus-Durner V, Schughart K, Hrabě de Angelis M, Mammalian Genome 2020, 31, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]