

MEFD2 fusion protein reprograms B-ALL transcriptional circuits, establishing selfreinforcing regulatory network dependent on pre-BCR and SREBF1, which can be exploited to therapeutically target a fusion-driven leukemia.
Abstract
The cellular context that integrates gene expression, signaling, and metabolism dictates the oncogenic behavior and shapes the treatment responses in distinct cancer types. Although chimeric fusion proteins involving transcription factors (TF) are hallmarks of many types of acute lymphoblastic leukemia (ALL), therapeutically targeting the fusion proteins is a challenge. In this work, we characterize the core regulatory circuitry (CRC; interconnected autoregulatory loops of TFs) of B-ALL involving MEF2D-fusions and identify MEF2D-fusion and SREBF1 TFs as crucial CRC components. By gene silencing and pharmacologic perturbation, we reveal that the CRC integrates the pre-B-cell receptor (BCR) and lipid metabolism to maintain itself and govern malignant phenotypes. Small-molecule inhibitors of pre-BCR signaling and lipid biosynthesis disrupt the CRC and silence the MEF2D fusion in cell culture and show therapeutic efficacy in xenografted mice. Therefore, pharmacologic disruption of CRC presents a potential therapeutic strategy to target fusion protein–driven leukemia.
Significance:
Cancer type–specific gene expression is governed by transcription factors involved in a highly interconnected autoregulatory loop called CRC. Here, we characterized fusion protein–driven CRC and identified its pharmacologic vulnerabilities, opening therapeutic avenues to indirectly target fusion-driven leukemia by disrupting its CRC.
See related commentary by Sadras and Müschen, p. 18 .
This article is highlighted in the In This Issue feature, p. 5
Introduction
Cell type–specific gene expression programs in mammals are mainly governed by a small number of transcription factors (TF) that collectively control each other's gene expression to form highly interconnected autoregulatory loops, termed as core regulatory circuitry (CRC; 1–4). Identifying the CRC in cancer can help reveal the cell of origin (5) and critical TFs required to maintain an oncogenic cell state (6–11), thus providing valuable insights into cancer biology. However, such efforts in leukemia have been limited (6, 11). Although fusion proteins involving TF (TF-fusion proteins) are associated with many types of leukemia and are crucial for leukemia development (12, 13), their participation in CRC remains unexplored. Furthermore, CRC may provide the platform for the development of new therapeutics (14, 15), but CRC-targeting therapy is still unestablished in leukemia.
B-cell precursor acute lymphoblastic leukemia (BCP-ALL) is the most common pediatric cancer and its prognosis in adults is generally unfavorable (13). BCP-ALL is characterized by recurrent chromosomal alterations, including chromosomal rearrangements that deregulate oncogenes or result in the formation of chimeric fusion proteins with altered functions of their nonarranged normal counterparts. Such fusion proteins are typically acquired early in leukemogenesis and are supposed to drive tumor formation (12). A systematic analysis of gene expression profiles, gene fusions, and gene mutations recently reported that BCP-ALL was classified into 14 distinct subgroups that are associated with patient survival (16). Most of the subgroups are segregated according to genetic abnormalities, including fusion genes generated from chromosomal rearrangement. A subgroup representing BCP-ALL with MEF2D-fusion genes (hereafter, MEF2D-ALL) is classified as a high-risk group (16), consistent with the results of other studies (17–21).
In mammals, the myocyte enhancer factor 2 (MEF2) family of TFs consists of four members; MEF2A, 2B, 2C, and 2D, which were initially identified as major transcriptional activators of muscle differentiation, but were later shown to be involved in the control of pleiotropic responses, including cardiac morphogenesis, blood vessel formation, neural differentiation, and growth factor responsiveness (22). MEF2 family members have also been implicated in cancer biology. Ectopically activated MEF2C serves as an oncogene in human T-cell ALL (23), and MEF2D is a candidate oncogene in leukemia in mice (24–26). Elevated MEF2D expression in hepatocellular carcinoma and gastric cancer is associated with poor patient prognosis, and MEF2D is critically involved in the proliferation of these cancer cells (27, 28).
In MEF2D-ALL, MEF2D is invariably situated at the 5′ end of the fusion transcripts, whereas 3′ partners are variable, with BCL9 and HNRNPUL1 accounting for the majority (17–19, 21). The close similarity of gene expression profiles of MEF2D-ALL, regardless of 3′ partners, suggests crucial roles of deregulated MEF2D function in leukemogenesis (16–18).
In this study, we examined MEF2D-ALL as an example to investigate the presence of CRC in BCP-ALL and the involvement of TF-fusion protein therein. We then explored the mechanisms involved in controlling the CRC to propose a CRC-targeting therapy.
Results
MEF2D-fusion TF Is Associated with Pre-B-cell Receptor Expression
We first sought to investigate the oncogenic roles of the fusion protein associated with MEF2D TF (hereafter, MEF2D-fusion TF) in MEF2D-ALL, and thus map the genome-wide occupancy of MEF2D-fusion TF in the MEF2D-ALL cell line Kasumi-7. For this purpose, we developed a genome-edited Kasumi-7 cell line in which endogenous MEF2D-HNRNPUL1 fusion TF was tagged at the carboxy-terminal with hemagglutinin (HA) and coexpressed with GFP by virtue of 2A sequence (hereafter, K7-HA-GFP cells; Supplementary Fig. S1A–S1C). Chromatin immunoprecipitation followed by sequencing (ChIP-seq), which used an anti-HA antibody, identified 7,077 unique MEF2D-HNRNPUL1 binding peaks, with a distribution of approximately 20% in the proximity (<3 kb) of transcriptional start sites (TSS) of protein-coding genes, acquiring approximately 40% and 34% in intronic and intergenic regions of the human genome (Supplementary Fig. S1D), respectively, suggesting that MEF2D-fusion TF functions in both promoters and enhancers. The motif for MEF2 family TFs being the most enriched (Supplementary Fig. S1D) in motif searching, and the results of ChIP-quantitative PCR for selected loci (Supplementary Fig. S1E) supported the validity of our ChIP-seq data.
Super-enhancers (SE) are responsible for the robust expression of developmentally regulated genes to specify cell identity (29) and genes required for tumor maintenance in cancer (30, 31). On examining 877 SE regions, which we identified using the H3K27ac ChIP-seq data (29, 32) of Kasumi-7 cells, we observed that 66.3% of the regions (588 regions) were occupied by MEF2D-HNRNPUL1, with prominent assignment of genes associated with pre-B-cell receptor (pre-BCR) signaling (33). Such genes included BLNK, IGLL1, VPREB1, CD79A, and BCL6 (Fig. 1A). MEF2D was also detected. Pre-BCR is composed of immunoglobulin (Ig) μ chain and surrogate light chains, namely, VpreB and lambda 5 (encoded by the IGLL1 gene), along with CD79A/B, and transmits signals via molecules such as the src family kinases (SFK), spleen tyrosine kinase (SYK), and Bruton tyrosine kinase (BTK; ref. 33). Pre-BCR signaling also involves the induced expression of BCL6 (34). Consistently, the “BCR signaling pathway” was the top-ranked Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway (Fig. 1B; Supplementary Fig. S1F) for the genes assigned to SEs involving MEF2D-HNRNPUL1 occupancy. Instead, this result actually showed “pre-BCR signaling” as the top-ranked pathway, considering that; (i) Kasumi-7 cells expressed pre-BCR instead of BCR (description follows), (ii) BCR and pre-BCR signaling pathways share many, if not all, molecules, and (iii) pre-BCR signaling is not included in the KEGG pathways. Indeed, many genes known to be involved in the BCR signaling pathway, such as NFATC1, FOS, PIK3CD, PLCG2, and PPP3CC, were accompanied by SEs involving MEF2D-HNRNPUL1 occupancy near their TSSs, as well as the abovementioned genes, namely, IGLL1, VPREB1, CD79A, BCL6, and MEF2D (Fig. 1C).
Figure 1.

Association of MEF2D-fusion with pre-BCR expression. A, Profile of enhancers in Kasumi-7 cells based on the H3K27ac ChIP-seq signal. Enhancers are ranked by increasing signal level. SEs are represented in the top right quadrant. Genes assigned to selected SEs are indicated. B, KEGG pathway analysis of the genes assigned to SEs involving MEF2D-HNRNPUL1 occupancy. The false discovery rate (FDR) is indicated. C, Occupancy of MEF2D-HNRNPUL1 in genomic regions near the transcription start sites of representative genes involved in pre-BCR signaling. ATAC-seq and H3K27ac ChIP-seq signals are indicated. The MEF2D-HNRNPUL1 signal peaks and SEs are indicated by black lines. The x and y axes indicate the linear sequences of genomic DNA and normalized read densities, respectively. Arrows indicate the locations and directions of the TSSs. D, Flow cytometric analysis of pre-BCR expression on the indicated cells. Cells were labeled using a combination of anti-Igμ and anti-λ5 antibodies (top) and an anti-pre-BCR complex antibody (bottom). E, t-SNE analysis of genes associated with pre-BCR–positive versus -negative BCP ALL in clinical samples from two independent cohorts. Each dot represents one sample. The colors of the dots indicate fusions involving the indicated genes. “Kinase” indicates genes associated with protein kinases. F, Association of enforced MEF2D-BCL9 expression with pre-BCR+ B-cell emergence in mice. Mice were transplanted with mouse pro-B cells infected with retroviruses encoding GFP-only (n = 10) or MEF2D-BCL9 plus GFP (n = 11). Pre-BCR expression in the GFP+ fraction was analyzed by flow cytometry 3 weeks after transplantation. Representative flow-cytometric plots (left) and the % pre-BCR+ cells among total GFP+ cells (right) demonstrate the association of MEF2D-BCL9 expression with the emergence of pre-BCR+ B cells. G, Kaplan–Meier curves of estimated survival among mice transplanted with control (n = 5) and MEF2D-BCL9-expressing pro-B cells (n = 5). This cohort was independent of that described in F. The statistically significant difference in survival was determined using the log-rank test. H, Morphologic (May–Grunwald–Giemsa staining) and flow-cytometric analyses of bone marrow cells of two mice before death revealed many lymphoblasts positive for pre-BCR.
Flow cytometric analysis of cells for the coexpression of Igμ and lambda 5 revealed the presence of pre-BCR on the cell surface of MEF2D-ALL cell lines (Kasumi-7, Kasumi-9, and TS-2), as well as two primary clinical MEF2D-ALL cell samples. The use of an antibody to detect the pre-BCR complex confirmed the results (Fig. 1D). We also examined seven additional MEF2D-ALL cell lines, which were also positive for pre-BCR expression (Supplementary Fig. S1G).
Then, we analyzed the gene expression data of two distinct clinical ALL cohorts (21) for the expression of genes discriminating pre-BCR+ ALL from BCR− ALL (34). This set included genes for pre-BCR signaling components (VPREB1, IGLL1, SYK, PRKCZ, BLNK, BLK, LYN, MERTK, and ZAP70) and TFs involved in B-cell differentiation (BCL6, BACH2, IRF4, TCF3, and POU2AF1), as upregulated in pre-BCR+ ALL. IL2RA (CD25), IL13RA1, CD34, ID2, CD69, CD99, ITGA6, CCND2, SOCS2, PRDM1, TLE3, and EMP1 were included as downregulated genes. Examining the expression of such genes by t-distributed stochastic neighbor embedding (t-SNE) analysis revealed the closest proximity of MEF2D-ALL with PBX1-rearranged ALL, an established pre-BCR+ BCP-ALL (ref. 34; Fig. 1E). Indeed, a comparison of gene expression data in clinical samples between MEF2D-ALL and other B-cell ALLs (PBX1-rearranged ALLs excluded) showed IGLL1, VPREB1, ZAP70, BCL6, IRF4, and BACH2 as overrepresented, and IL13RA1, PRDM1, TLE3, EMP1, CD69, CCND2, CD34, and SOCS2 as underrepresented in MEF2D-ALL. However, BLNK was unexpectedly underrepresented (Supplementary Fig. S1H).
Given the likely association between MEF2D-fusion TF and pre-BCR expression in human MEF2D-ALL cells, as suggested above, we investigated whether MEF2D-fusion TF could invoke pre-BCR expression in normal B cells. To this end, we utilized mouse pro-B cells as the target of retroviral expression of MEF2D-fusion TF. We took advantage of the higher infectivity of retrovirus for MEF2D-BCL9 than for MEF2D-HNRNPUL1; such infectivity was probably due to the smaller cDNA size of the former. Pro-B cells normally differentiate into pre-BCR+, then pre-BCR−, and finally, IgM+ immature B-cell stages in bone marrow (33). Pro-B cells isolated from fetal mouse liver were infected with retrovirus to express MEF2D-BCL9 together with GFP or GFP only (control) and intravenously transplanted into immunodeficient NOD/Scid/IL2Rγnull (NSG) mice. After three weeks, the bone marrow cells in the recipients of GFP-only–transduced cells barely expressed pre-BCR, whereas those in the recipients of MEF2D-BCL9–transduced cells unmistakably expressed pre-BCR (Fig. 1F). Thus, the MEF2D-BCL9 expression was significantly associated with the emergence of pre-BCR+ cells or retardation in differentiation at the pre-BCR+ stage. In a different cohort with more extended observation periods, all 5 mice receiving MEF2D-BCL9–transduced pro-B cells had died or became moribund within 120 days after transplantation, whereas all 5 mice receiving GFP-only–transduced pro-B cells remained healthy (Fig. 1G). We were able to examine 2 mice that received MEF2D-BCL9-transduced pro-B cells before they died and noted an enlarged spleen and many lymphoblasts in the bone marrow, of which >90% were GFP+CD19+ B cells that expressed pre-BCR (Fig. 1H). Therefore, the expression of MEF2D-fusion TF was closely associated with the elevated fraction of pre-BCR+ cells, and leukemia could develop later, probably in some of the pre-BCR+ cells.
Our overall findings, described above, suggested that MEF2D-ALL represented pre-BCR+ ALL. The genes encoding molecules involved in pre-BCR signaling were associated with SEs involving MEF2D-fusion TF occupancy.
MEF2D-ALL Relies on Pre-BCR and MEF2D-fusion TF for Growth and Survival
We next sought to clarify the role of pre-BCR in MEF2D-ALL maintenance. Hence, we utilized lentiviral short hairpin RNA (shRNA) targeting the pre-BCR components, namely, Igμ, CD79A, and CD79B. The utilized lentivirus vectors expressed an extracellular domain of human CD8 (hCD8) along with shRNA, thereby allowing us to identify and isolate the infected cells by using the anti-hCD8 antibody. Pre-BCR expression was downregulated in the hCD8+ but not in the hCD8− fraction, as expected, and the growth of the sorted hCD8+ cells was severely impaired (Fig. 2A). Consistently, CRISPR interference-mediated suppression of the SEs (35) involving MEF2D-HNRNPUL1 binding peaks around the TSSs of either IGLL1 or VPREB1 reduced the expression of pre-BCR in Kasumi-7 cells (Supplementary Fig. S2A) and attenuated cell growth (Supplementary Fig. S2B).
Figure 2.

Roles of pre-BCR expression and MEF2D-fusion in the maintenance of MEF2D-ALL. A, Knockdown of pre-BCR components impaired Kasumi-7 cell growth. Kasumi-7 cells were infected with lentiviruses encoding shRNAs targeting the pre-BCR components Igμ, CD79A, and CD79B while coexpressing an extracellular domain of human CD8 (hCD8). The expression of pre-BCR and hCD8 was then analyzed. Pre-BCR expression was downregulated only in the hCD8+ fraction of cells (top). Purified hCD8+ cells (bottom) exhibited impaired cell growth following pre-BCR knockdown (n = 3). Luciferase shRNA was used as a control. B, Knockdown of GFP reduced the expression of MEF2D-HNRNPUL1 in K7-HA-GFP cells. Cells were infected with two lentiviruses that encoded GFP-specific shRNA while coexpressing hCD8 (i.e., shRNA-GFP_1 and _2). GFP was knocked down only in the hCD8-positive fraction (top). Luciferase shRNA was used as a control. Western blot analysis of flow-sorted hCD8-positive and -negative cells with anti-MEF2D antibody revealed the reduced expression of MEF2D-HNRNPUL1 only in the hCD8-positive fraction of cells infected with shRNA-GFP_1 and _2 viruses (bottom). C, Knockdown of MEF2D-HNRNPUL1 by shRNA-GFP_1 and _2 in K7-HA-GFP cells reduced pre-BCR expression (top) and impaired cell growth (bottom; n = 3). D, MEF2D-HNRNPUL1 knockdown induced apoptosis. K7-HA-GFP cells infected with lentiviruses encoding a control or two shRNAs for GFP, as in B, were subjected to a flow cytometry analysis of apoptosis using Annexin V and 7AAD dual staining. E, Heatmap presentation of gene expression changes associated with differentiation beyond the pre-BCR stage of B-cell development. Six each RNA samples from control- and MEF2D-HNRNPUL1-knocked down (three each infected with shRNA-GFP_1 and shRNA-GFP_2) K7-HA-GFP cells were used. F, Patterns of cell surface expression of the indicated molecules in GFP-positive control (shown in blue) and GFP-low, MEF2D-HNRNPUL1 knocked down cells (in red) suggest differentiation after knockdown. G, Gene-set enrichment analysis of differences in gene expression between control and MEF2D-HNRNPUL1 knocked-down cells. The results are consistent with differentiation after knockdown.
To determine whether the expression of pre-BCR relied on MEF2D-fusion TFs, we used two independent lentiviral shRNA vectors targeting GFP and coexpressing hCD8. GFP expression was reduced only in the CD8+ fraction of shRNA-infected K7-HA-GFP cells as expected, which was accompanied by a reduction in MEF2D-HNRNPUL1 expression (Fig. 2B). The reduction in MEF2D-HNRNPUL1 expression led to impaired pre-BCR expression, attenuated cell growth (Fig. 2C), and cell death (Fig. 2D). Thus, MEF2D-fusion TF was crucial for MEF2D-ALL maintenance, at least partly through the upregulation of pre-BCR expression.
Next, to unravel the transcriptional consequences of MEF2D-HNRNPUL1 binding, we performed RNA-sequencing analysis of gene expression following the MEF2D-HNRNPUL1knockdown. Considering that MEF2D-HNRNPUL1 knockdown induced cell death, we necessarily chose earlier time points before pre-BCR was appreciably downregulated. In normal B-cell development, differentiation beyond the pre-BCR+ stage is associated with the cessation of proliferation and concomitant downregulation of Myc, reactivation of RAG gene expression, and increased cell surface expression of CD19, CD20, and CD45 (33, 36). The gene expression changes observed upon MEF2D-HNRNPUL1 knockdown in K7-HA-GFP cells were generally consistent with differentiation, based on the following findings: (i) by focusing on genes whose expression changes along with differentiation beyond the pre-BCR+ B-cell stage (33, 36); we observed, as in normal differentiation processes, the upregulation of RAG1/RAG2, CD45, CD19, CD20, IKZF3, ELK3, HIVEP3, SPIB, POU2AF1, POU2F1, POU2F2, ELF1, ETS2, and POU4F1 and the downregulation of Myc and BCL6. However, SMARCA5, KLF4, KLF2, and KLF12, which are normally upregulated upon differentiation beyond pre-BCR+ pre-B cells, were instead downregulated in this study (Fig. 2E). (ii) Flow cytometric analysis confirmed the upregulation of CD45, CD19, and CD20 (Fig. 2F). (iii) Gene-set enrichment analysis suggested the consistency of the gene expression changes that we observed after silencing MEF2D-HNRNPUL1 with differentiation beyond the pre-BCR stage (Fig. 2G).
Overall, MEF2D-fusion TF was crucial for MEF2D-ALL maintenance regarding proliferation, differentiation blockade, and cell survival, at least partly through pre-BCR expression.
Self-enforcing Regulatory Loop Involving MEF2D-fusion TF and Pre-BCR Signaling
Tonic pre-BCR signaling involves the constitutive activity of proximal pre-BCR-associated SFKs, such as LYN, FYN, and BLK, as well as SYK, leading to the activation of PI3Ks; this, in turn, activates BTK and PLCγ2 to trigger ERK1/2 MAP kinases (33). We explored the influence of pre-BCR signaling toward MEF2D-fusion TF expression, considering that Erk1/2 activation under pre-BCR signaling leads to phosphorylation of the cAMP response element-binding protein (CREB) TF, which drives the transcription of Mef2d in mice (37). Knockdown of pre-BCR components (CD79A, CD79B, and Igμ) and its signaling molecules, namely, SYK and BTK, in Kasumi-7 cells reduced the phosphorylation of the respective downstream molecules, leading to decreased CREB phosphorylation and a reduction in MEF2D-fusion TF and its transcripts (Fig. 3A and B). Consequently, cell growth was impaired (Fig. 3C). Likewise, CREB knockdown reduced MEF2D-fusion TF expression (Fig. 3D). These findings suggested that pre-BCR signaling positively regulated MEF2D-fusion TF, at least, in part, through CREB-mediated transcription. Similarly, dasatinib (SFK inhibitor), PRT062607 (SYK inhibitor), and ibrutinib (BTK and BLK inhibitor) reduced CREB phosphorylation in Kasumi-7 cells (Fig. 3E) and MEF2D-fusion TF expression in MEF2D-ALL cells (Fig. 3F; Supplementary Fig. S3A). Consistent with MEF2D-fusion TF reduction, the treatment reduced pre-BCR expression on the cell surface in MEF2D-ALL, but not in three non-MEF2D, pre-BCR+ cell lines (Fig. 3G; Supplementary Fig. S3B). FK506 inhibited MEF2D-ALL cell growth like the above tested drugs (description follows), but it did not influence the expression of MEF2D-fusion TF (Supplementary Fig. S3C) and pre-BCR (Supplementary Fig. S3D); hence, the reduction of MEF2D-fusion TF and pre-BCR was not a general consequence of impaired cell growth.
Figure 3.
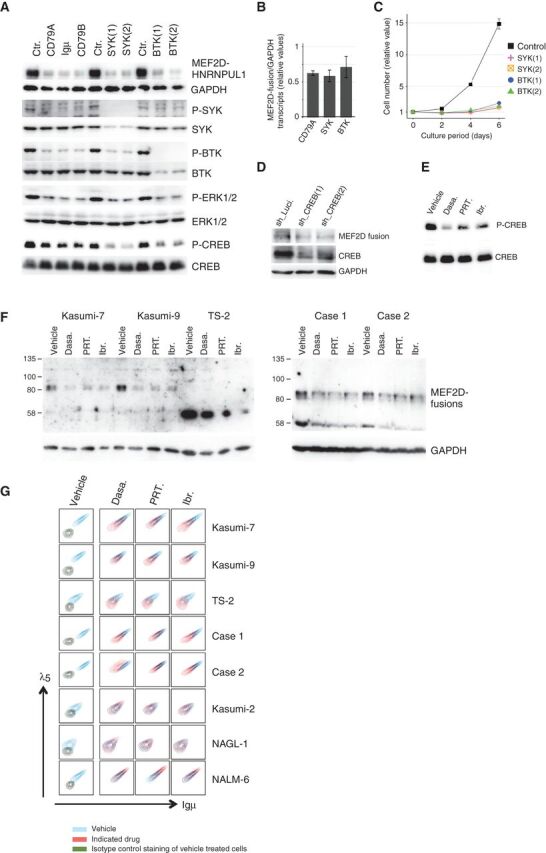
Influence of pre-BCR signaling on MEF2D-fusion expression. A, Knockdown of pre-BCR components (CD79A, Igμ, and CD79B) and downstream signaling molecules (SYK and BTK) reduced the phosphorylation of the respective downstream signaling molecules and CREB. This knockdown was accompanied by the downregulation of MEF2D-HNRNPUL1. Luciferase shRNA was used as a control (Ctr). GAPDH served as a loading control. Results of Western blot analyses are presented. B, Knockdown of the indicated molecules was accompanied by a reduction in MEF2D-HNRNPUL1 transcript levels. Fusion transcript levels were evaluated using a quantitative RT-PCR analysis and are presented as fractions of the control transcript level (means ± SD; n = 3). C, SYK and BTK knockdown impaired Kasumi-7 cell growth (n = 3). D, CREB knockdown reduced MEF2D-HNRNPUL1 protein expression. E, Treating Kasumi-7 cells with the indicated pre-BCR signaling inhibitors reduced CREB phosphorylation in Kasumi-7 cells. Cells were treated with dasatinib (Dasa: 1 μmol/L), PRT062607 (PRT: 10 μmol/L), or ibrutinib (Ibr: 10 μmol/L) for 8 hours. F, Treatment with dasatinib, PRT062607, and ibrutinib reduced MEF2D-fusion protein expression in the indicated MEF2D-ALL cell lines (Kasumi-7, Kasumi-9, and TS-2) and two clinical MEF2D-ALL primary cells. Cells were treated with dasatinib (500 nmol/L), PRT062607 (8 μmol/L), and ibrutinib (8 μmol/L) for 3 days. G, Dasatinib, PRT062607, and ibrutinib reduced pre-BCR expression in the five MEF2D-ALL cell lines, but not in three pre-BCR+ non-MEF2D ALL cell lines. Pre-BCR expression levels in cells treated with the vehicle or the indicated inhibitors as in F are shown in blue and red, respectively, in all panels except those in the leftmost column. Pre-BCR and control staining of vehicle-treated cells in the leftmost column are depicted in blue and dark green, respectively.
Overall, these findings suggested that MEF2D-fusion TF was involved in the expression of pre-BCR, and in turn, the downstream signaling positively influenced the expression of MEF2D-fusion TF, thereby establishing a feed-forward regulatory loop.
CRC of MEF2D-ALL
Next, we sought to unravel the CRC in MEF2D-ALL. CRC has been shown critical in maintaining cancers (6, 11, 38). We used SEs identified in two MEF2D-ALL lines (Kasumi-7 and Kasumi-9) and two non-MEF2D-ALL cell lines (NAGL1 and NALM1) to run the CRC mapper, an algorithm to predict CRC using SE maps (2). Many candidate CRC constituent TFs were shared by the two MEF2D-ALL lines, but not by the two non-MEF2D-ALL cell lines (Supplementary Table S1), thus allowing us to focus on the shared candidate constituent TFs. Given that constituent TFs regulate each other's expression, and that MEF2D-fusion, which is crucial for leukemia maintenance (as described above), was a candidate constituent, we focused on TFs, among candidate CRC constituent TFs, whose expression was downregulated at the transcript level (>1.5-fold) following MEF2D-fusion knockdown in K7-HA-GFP cells (Supplementary Fig. S4A). The reduction of such TFs, namely, EGR1, BCL6, FOS, IRF8, ERG, RUNX1, NFATC1, and SREBF1, was confirmed at the protein level (Fig. 4A). These TFs, along with MEF2D-fusion TF, variably exhibited occupancies in genomic regions near TSSs of genes involved in pre-BCR signaling and probably collectively regulate expression of these genes (Supplementary Fig. S4B); however, IRF8 occupancy data in human immature/lymphoid cells was not available and thus not included in the analysis. Similar observations were made for genomic regions near TSSs of MEF2D and the TFs mentioned above (Supplementary Fig. S4C).
Figure 4.

Identification of TFs comprising the CRC. A, Knockdown of MEF2D-HNRNPUL1 reduces the expression of the indicated TFs. Two GFP-targeting shRNAs were used. PAX5, β-actin, and GAPDH served as controls. B, Knockdown of the indicated putative CRC components reduced MEF2D-HNRNPUL1 expression. Two independent shRNAs were used per component. GAPDH served as a loading control. Relative signal strengths normalized on the basis of GAPDH signals are presented. C, Enforced expression of a truncated form of MEF2D downregulated MEF2D-HNRNPUL1 expression. K7-HA-GFP cells were infected with a lentiviral vector encoding an HA-tagged, truncated form of MEF2D; this form encompassed the first 117 amino acids that encode DNA-binding domains but lacked the C-terminal transcriptional regulatory domains. The vector additionally expressed hCD8 to enable the fractionation of infected (hCD8-positive) from uninfected (hCD8-negative) cells. The expression of MEF2D-HNRNPUL1 and truncated MEF2D in fractionated cells were analyzed using an anti-HA antibody. D, Occupancy of MEF2D-HNRNPUL1 and ATAC-seq and H3K27ac ChIP-seq signals detected around the transcription start sites (TSS) of MEF2D in K7-HA-GFP cells are presented. Also shown are SREBF1, FOS, EGR1, and CREB ChIP-seq signals in GM12878 B lymphoid cells derived from the Encode project, as well as BCL6 ChIP-seq signals detected in pre-B lymphocytes (GSM1438986). SEs and ChIP-seq signal peaks are indicated by black lines (left). ChIP-qPCR analysis of the occupancy of six regions (indicated as 1–6) in the left panel by MEF2D-HNRNPUL1, SREBF1, FOS, EGR1, BCL6, and CREB in K7-HA-GFP cells (right). The fold-increases in the amounts of DNA precipitated by antibodies specific for the indicated TFs were compared with those precipitated by normal IgG and are shown as means ± SDs (n = 3). E, Occupancy of MEF2D-HNRNPUL1 and ATAC-seq and H3K27ac ChIP-seq signals around the TSSs of SREBF1, FOS, EGR1, and BCL6 in K7-HA-GFP cells are presented. SREBF1, FOS, EGR1, BCL6, and CREB ChIP-seq signals are also presented as in D. SEs and ChIP-seq signal peaks are indicated by black lines (left). ChIP-qPCR analysis of occupancy of MEF2D-HNRNPUL1, SREBF1, FOS, EGR1, BCL6, and CREB at the indicated regions (shown in the left) is presented (n = 3) as in D (right). F, Knockdown of either SREBF1, FOS, EGR1, or BCL6 reduced the expression of the other three TFs. G, Treatment of Kasumi-7 cells with the indicated pre-BCR signaling inhibitors as in Fig. 3F reduced the protein expression of SREBF1, EGR1, FOS, and BCL6. H, A putative CRC.
We then narrowed down CRC constituents to TFs, whose knockdown conversely reduced the expression of MEF2D-fusion protein (Fig. 4B). At this point, we deprioritized RUNX1, ERG, and NFATC1 TFs because their knock-down had less impact on the MEF2D-fusion protein level compared with other CRC constituent candidate TFs (Supplementary Fig. S4D), albeit compromising cell growth (Supplementary Fig. S4E). MEF2D-fusion TF was at least partly self-regulatory because the enforced expression of a dominant-negative version of MEF2D (39) reduced MEF2D-fusion protein expression (Fig. 4C). We thus extracted the MEF2D-fusion, SREBF1, FOS, EGR1, BCL6, and IRF8 as constituents of the putative CRC. Knockdown of either one of these TFs or enforced expression of the dominant-negative MEF2D attenuated the growth of K7-2A-GFP cells (Supplementary Fig. S4E), suggesting that these TFs are crucial for leukemia maintenance. Consistently, the MEF2D-fusion TF occupancy data, which we obtained using K7-2A-GFP cells, coupled with ChIP-seq data from GM12878 human lymphoblast cells (40) and human pre-B lymphocytes (34), suggested the cooccupancy of MEF2D-fusion, and four TFs, namely SREBF1, FOS, EGR1, and BCL6 in their respective gene locus; IRF8 was not included (see above; Fig. 4D and E). The occupancy of MEF2D-fusion and the four TFs near the TSSs of their respective gene loci was confirmed by ChIP-quantitative PCR analysis (Fig. 4D and E). Although full elucidation of the CRC was challenging, overall, these findings suggest that MEF2D-fusion TF, SREBF1, FOS, EGR1, and BCL6 constitute the CRC. Congruently, knockdown of SREBF1, FOS, EGR1, and BCL6, respectively, downregulated other constituent TFs (Fig. 4F and B). Interestingly, CREB exhibited occupancy near the TSSs of MEF2D and the four TF genes (Fig. 4D and E), despite not being a CRC constituent. Thus, pre-BCR signaling probably influences the expression of CRC constituent TFs via CREB. Consistently, pre-BCR signaling inhibitors diminished the CRC constituent TFs (Figs. 3F and 4G).
On the basis of these findings, we propose an extended CRC that involves CREB activated through the MEF2D-fusion-pre-BCR signaling axis (Fig. 4H). This network structure involving CRC and pre-BCR signaling may provide the platform by which MEF2D-fusion TF can sustain the entire gene expression program that contributes to the malignant phenotype.
Therapeutic Efficacies of Pre-BCR Signaling Inhibitors in MEF2D-ALL
Next, we evaluated and compared the effects of inhibitors of pre-BCR signaling on the growth of BCP-ALL cell lines, representing MEF2D-ALL (n = 3), pre-BCR+ non-MEF2D-ALL (n = 3; Supplementary Fig. S5A), and pre-BCR− non-MEF2D-ALL (n = 4; Supplementary Fig. S5A). Remarkably, dasatinib, PRT062607, and ibrutinib inhibited the growth of MEF2D-ALL cells, even though these drugs were similarly effective against pre-BCR+ non-MEF2D-ALL cells (Fig. 5A; ref. 34). However, these drugs disrupted the CRC and selectively reduced pre-BCR expression in MEF2D-ALL cells, but not in non-MEF2D-ALL cells (Figs. 3F, G and 4G). Therefore, these drugs most likely provided additional therapeutic benefits beyond the simple inhibition of signaling downstream of the pre-BCR. Two clinical MEF2D-ALL cell samples (Fig. 5B) and seven additional MEF2D-ALL cell lines responded similarly to these inhibitors (Supplementary Fig. S5B). In vivo, dasatinib and ibrutinib effectively reduced the leukemia burden in mice transplanted with Kasumi-7 cells, thereby prolonging survival (Fig. 5C).
Figure 5.

Sensitivity of BCP ALL cells to inhibitors of pre-BCR signaling. A, Ten cell lines (four pre-BCR-negative ALL, three pre-BCR-positive non-MEF2D-ALL, and three MEF2D-ALL lines) were exposed to indicated inhibitors and their sensitivity with respect to growth inhibition was tested. Cells were treated with varying concentrations of the inhibitors as indicated for 3 days. Cell growth is shown relative to that of vehicle-treated control cells (means ± SDs; n = 3). B, Two clinical MEF2D-ALL cell lines were analyzed as in A. C, Immunodeficient mice were transplanted with Kasumi-7 cells engineered to express luciferase, which was monitored by bioluminescence imaging (left). Ibrutinib, dasatinib, and vehicle controls were administered daily from day 1 to day 35 days after transplantation. The survival outcomes were also estimated using the log-rank test (right). Both drugs reduced the leukemia burden and significantly prolonged the survival duration (n = 5 for ibrutinib treatment, otherwise n = 4).
We tested two additional drugs that target probable molecular components in the signaling pathway downstream of the pre-BCR (Supplementary Fig. S5C). Idelalisib, an inhibitor of PI3K delta (PIK3 p110 delta, PIK3CD; ref. 41), had a selective but less potent effect on pre-BCR+ ALL cell growth. Interestingly, FK506 (42), an inhibitor of calcineurin (Supplementary Fig. S1F), exerted more specific growth-inhibitory effects on MEF2D-ALL cells, but it did not affect the expression of MEF2D-fusion and pre-BCR (Supplementary Fig. S3C and S3D). Seven additional MEF2D-ALL cell lines responded similarly to these drugs (Supplementary Fig. S5B).
SREBF1 as a Drug-targetable CRC-constituent TF
Among the identified CRC constituents, we focused on SREBF1 because its activity depends on cellular lipid levels and is susceptible to drug inhibition. SREBF1 is anchored to the endoplasmic reticulum membrane as a precursor protein, and when lipids such as cholesterol and fatty acids are inadequate, it translocates to the Golgi apparatus, where it is cleaved to release a mature functional TF (43). Indeed, the cleaved, active SREBF1 protein amount in MEF2D-ALL cell lines was reduced in the presence of excess cholesterol. The total amount of SREBF1 protein was subsequently reduced, probably because of the disruption of the CRC (Supplementary Fig. S6A), which reflected the reduction of MEF2D-fusion protein (Fig. 6A). Fatty acid supplementation in culture yielded a similar result to that obtained with cholesterol (Supplementary Fig. S6B).
Figure 6.

Effects of lipids and inhibitors of SREBF1 activation on the expression of MEF2D-fusion protein and the therapeutic efficacies of inhibitors. A, Incubation of MEF2D-ALL cells with cholesterol diminished the expression of MEF2D-fusion protein. Cells were cultured in the presence (+) or absence (−) of cholesterol (10 μg/mL cholesterol and 2 μg/mL 25-hydroxycholesterol) for 3 days. Expression of the indicated proteins was analyzed by Western blotting. B, Treatment of five MEF2D-ALL cell lines with fatostatin, FGH10019, and betulin reduced the protein expression of MEF2D-fusion and SREBF1. Cells were treated with fatostatin (3 μmol/L), FGH10019 (3 μmol/L), or betulin (10 μmol/L) for 3 days. All treatments reduced SREBF1 expression. + and – denote treatment with the indicated drug and vehicle, respectively. C, Treatment of Kasumi-7 cells with the indicated inhibitors of SREBF1 activation, as in B, reduced the expression of BCL6, FOS, and EGR1. D, Fatostatin, FGH10019, and betulin selectively inhibited MEF2D-ALL cell growth. Cells were treated with varying concentrations of the drugs for 3 days as indicated. Cell growth was monitored and compared with vehicle treatment as a control. The values relative to the control are presented as means ± SDs (n = 3). E, FGH10019 retards Kasumi-7 cell growth and prolongssurvival in mice. Immunodeficient mice were transplanted with Kasumi-7 cells engineered to express GFP and subsequently treated with FGH10019 (25 mg/kg/day, 6 days per week) or vehicle (n = 5 each). Peripheral blood was collected at 8 and 11 weeks after transplantation and analyzed to determine the %GFP-expressing cells (left). The difference in %GFP between samples from FGH10019- and vehicle-treated mice was statistically significantly different (Wilcoxon rank-sum test; top right). The estimated Kaplan–Meier survival curves of FGH10019- and vehicle-treated mice were also statistically significantly different (log-rank test; bottom right). F, Pre-BCR expression in the indicated cells after treatment with FGH10019 (3 μmol/L) or vehicle for 3 days in culture.
To gain therapeutic insights exploiting this lipid-responsive feature of MEF2D-fusion protein levels, we tested the effects of the inhibitors of SREBF1 activation, such as fatostatin (44), FGH10019 (45), and betulin (46). These drugs reduced the amount of cleaved and total SREBF1, as well as MEF2D-fusion TF protein in Kasumi-7 cells, similar to that observed for cholesterol and fatty acid (Supplementary Fig. S6C; Fig. 6B). Similarly, Kasumi-9 and TS-2 MEF2D-ALL cell lines and two clinical MEF2D-ALL cell samples responded to these drugs by reducing the expression of MEF2D-fusion TF and SREBF1(Fig. 6B). The drug treatment reduced BCL6, FOS, and EGR1 protein levels, probably by disrupting the CRC (Fig. 6C). Seven additional MEF2D-ALL cell lines also responded to fatostatin and FGH10019 by decreasing the expression of MEF2D-fusion TF (Supplementary Fig. S6D). These findings suggest that the CRC of MEF2D-ALL senses cellular lipid levels and shapes its activity in response. The CRC is inhibitable by fatostatin, FGH10019, and betulin. These drugs specifically inhibited MEF2D-ALL cell growth (Fig. 6D; Supplementary Fig. S6E); however, at high concentrations, they exhibited nonspecific toxicity in all BCP-ALL cells tested (Fig. 6D).
Given that FGH10019 is orally administered to mice (45), we focused on this drug and investigated its in vivo effects. Immunodeficient mice transplanted with GFP-expressing Kasumi-7 cells exhibited lower chimerism in peripheral blood and a prolonged survival time following FGH10019 treatment (Fig. 6E). Furthermore, FGH10019 treatment did not affect the survival of mice transplanted with the non-MEF2D BCP-ALL cell line REH; hence, the effects that we observed on Kasumi-7 cells in mice were not the result of nonspecific toxicity (Supplementary Fig. S6F). Consistent with the reduction of MEF2D-fusion TF, FGH10019 reduced pre-BCR expression on the cell surface of MEF2D-ALL cells, but not on pre-BCR+, non-MEF2D cells in culture (Fig. 6F; Supplementary Fig. S6G), further supporting the selectivity of FGH10019 for MEF2D-ALL.
Overall, SREBF1 TF was a critical CRC constituent that integrates cellular lipid levels to leverage CRC. SREBF1 activation inhibitors, such as FGH10019 can target MEF2D-ALL CRC, with therapeutic efficacy in vivo.
Discussion
To our knowledge, this work is the first to characterize B-ALL–specific CRC driven by a fusion TF. In BCP-ALL, fusion genes generated from chromosomal translocations are essential for leukemia biology (12). However, the transcriptional circuits through which fusion proteins establish and maintain ALL, are poorly understood. Specifically, it is unclear whether and how fusion proteins impact CRC. In our study, MEF2D-fusion TF proved critical for MEF2D-ALL maintenance as a constituent of CRC and essential for cell proliferation, differentiation blockade, and cell survival. The roles of MEF2D-fusion TF in leukemia maintenance are at least partly through the upregulation of pre-BCR components Iambda5 and VpreB that emit and relay signals for proliferation and survival (33). Furthermore, we demonstrate that pre-BCR signaling was critical for CRC maintenance; thus, the pre-BCR signaling inhibitors simultaneously disrupt the signaling and CRC with therapeutic efficacy.
The possible involvement of MEF2D in pre-BCR signaling in a normal pre-BCR+ B cell is an interesting research topic. However, the scarcity of these cells in the bone marrow and the lack of available and relevant antibodies for MEF2D have hindered such studies. The elevated gene expression (17, 21, 47) and protein translation of the MEF2D-fusion TF (48), compared with its wild-type counterpart, suggests that the former possesses unique functionalities that distinguish it further from the latter. Further study of this aspect is needed.
Our CRC analysis also identified SREBF1 TF as a crucial component of the CRC. The availability of transcriptionally active SREBF1 depends on the cellular lipid levels (43) and can be reduced by drugs such as FGH10019 (45). Therefore, we propose that the CRC senses the cellular context, including pre-BCR signaling and cellular lipid levels, as inputs, in MEF2D-ALL cells. Subsequently, the CRC integrates these inputs and functions as a rheostat to control an output, such as pre-BCR expression. Pharmacologic or physiologic perturbations of the input can alter the CRC status and thus impair the maintenance of leukemia cells. Although we were unable to fully address the genome-wide cooccupancy of CRC constituent TFs via our mapping project, our results nevertheless revealed a critical regulatory circuit in the maintenance of MEF2D-ALL and clarified how the CRC integrates the cellular context.
Although fusion proteins involving TFs are hallmarks in many types of leukemia (12, 13), ChIP-seq analysis for genes regulated by the fusion TF remains a challenge because of difficulties in discerning genomic regions bound by the fusion and those by normal TF counterparts. In the case of TCF3-PBX1 fusion, fusion TF-bound genomic regions were determined as regions commonly detected by ChIP-seq analyses using anti-TCF3 and anti-PBX2 antibodies (34). Another reason is the frequent unavailability of appropriate antibodies for TFs. The analysis of gene expression changes after the fusion TF is silenced for exploring the fusion TF-regulated genes is also an obstacle, considering that fusion TF-specific knockdown or knockout is technically demanding. In this study, we generated a genome-edited MEF2D-ALL cell line that allowed fusion TF-specific ChIP-seq analysis and knockdown, thereby revealing the central roles of MEF2D-fusion TF in MEF2D-ALL biology.
As a mechanism whereby MEF2D-fusion TF expression relies on pre-BCR signaling, we showed transcriptional upregulation of the fusion TF by CREB TF that was activated through pre-BCR signaling–mediated phosphorylation (37). However, this event may not be the only mechanism at play for this reliance. MEF2D TF is predisposed to various posttranslational modifications, including phosphorylation, acetylation, and sumoylation (22), which may alter the transcription activity of MEF2D. The altered transcription activity of MEF2D-fusion TF might influence the expression of MEF2D-fusion TF itself, considering that MEF2D-fusion TF could be transcriptionally autoregulated (Fig. 4C). In addition, it is likely that the interaction of MEF2D with class II HDACs affects the repression of a subset of genes in cancer (49), and their interaction can be influenced by phosphorylation (22). However, these aspects of MEF2D-fusion TF require further investigation.
Although SEs and CRC are considered therapeutic targets in cancer (31), the availability of drugs targeting them is limited. Inhibitors of bromodomain and extra-terminal proteins, and SE-associated cyclin-dependent kinases, are typically used to inhibit SEs in general with some therapeutic efficacy in experimental models of cancer, but their clinical efficacy and toxicity remain undetermined (50). Drugs targeting specific TFs involved in cancer type–specific CRC would provide additional benefits for cancer treatment. In our study, we presented two distinct types of such drugs for MEF2D-ALL treatment. Pre-BCR signaling inhibitors such as dasatinib, PRT062607, and ibrutinib can disrupt CRC, and FGH10019 directly inhibits the activation of the CRC-constituent SREBF1. As such, seemingly untargetable TF-fusion protein in leukemia can be attacked from behind by disrupting CRC involving the TF-fusion. Mapping CRC and elucidating the drug-targetable pathways influencing the CRC in cancer may provide useful information for the development of new treatment strategies.
Methods
Antibodies, chemicals, shRNA target sequences, and primer sequences for the ChIP-qPCR and RT-PCR analyses are listed in Supplementary Tables S2–S6.
Cell Lines
Kasumi-2, -7, -9, NALL-1, and NAGL-1 cells were purchased from the Japanese Collection of Research Bioresources Cell Bank (Osaka, Japan; http://cellbank.nibiohn.go.jp/english/). NALM-1 and Reh cells were obtained from ATCC (https://www.atcc.org/). NALM6 cells were obtained from DSMZ (https://www.dsmz.de/). AT2 cells were generously provided by J.D. Rowley at the University of Chicago (Chicago, IL). The TS-2 cell line, which was established and deposited by Masue Imaizumi, was kindly provided by Dr. Joji Inazawa (TMDUBR, Tokyo Medical and Dental University, Japan). p30/OHK cells were purchased from the RIKEN BioResource Research Center (Tsukuba, Japan). KOPN41, KOPN61, KOPN70, KOPN71, KOS20, and YAMN96 cell lines were established by Inukai and colleagues; the characterizations of these cell lines and p30/OHK will be published elsewhere. All cells were maintained in Iscove's Modified Dulbecco's Medium (IMDM) supplemented with 10% FCS. Mycoplasma was tested negative using EZ-PCR Mycoplasma Test Kit (Biological Industries; February 2020). Cell line authentication was made by the short tandem repeat (STR) analysis using GenePrint 10 System (Promega; February 2020). For AT2, TS-2, KOPN41, KOPN61, KOPN70, KOPN71, KOS20, and YAMN96 cell lines, STR analysis detected no differences between those frozen within 2 weeks of receipt and those used for the experiments.
Clinical Specimens
This study included cells derived from a 47-year-old male (case 1) and a 16-year-old male (case 2) with primary B-ALL who were treated at Nagoya University Hospital or Japanese Red Cross Nagoya First Hospital. The MEF2D fusion was detected by RT-PCR and subsequent Sanger sequencing. Both patients provided written informed consent to participate in the study. The study protocol was approved by the ethics committees at Nagoya University, Japanese Red Cross Nagoya First Hospital, and Aichi Medical University (Aichi, Japan).
Animals
Fetal liver pro-B cells were isolated from fetal BALB/c mice (SLC). Cells were transplanted into NSG mice (Charles River Laboratories). All animal experiments were performed according to protocols approved by the Institutional Animal Use and Care Committee of Aichi Medical University.
ChIP and Analysis
ChIP was performed using the Simple ChIP-plus kit (Cell Signaling Technology). Control rabbit IgG was included as a control. The DNA library was constructed using the NEBNext Ultra II DNA Library Prep Kit (New England BioLabs) and subjected to massively parallel sequencing on the HiSeq 2500 System platform (Illumina). ChIP-seq reads were aligned to the human genome build hg19 using Bowtie 2 (version 2.3.0). PCR duplicates were removed using the MarkDuplicates function in Picard-tools. Blacklisted genomic regions provided by Encode UCSC were also removed. Chip-seq peaks were called using MACS (1.4.2). The Integrative Genomics Viewer (IGV version 2.3.81) was used to visualize the data. The distributions of ChIP-seq peaks overlapping specific gene features were analyzed using the ChIPseeker with the R package (3.5.1; https://www.r-project.org). Gene annotation and motif discovery were performed using HOMER (v4.10) and i-cisTarget (54), respectively. The precipitated DNA encompassing selected regions was subjected to a qPCR analysis using KOD Fx Neo (Toyobo) with SYBR Green. Reactions were run on the StepOnePlus Real-Time PCR System (Applied Biosystems).
Identification of Enhancer Regions and Extraction of CRC
SE regions were identified by applying the Rank Ordering of Super-Enhancers program to H3K27ac ChiP-seq reads. A stitching distance of 12.5 kb, promoter exclusion zone encompassing ± 2 kb around the TSSs, and default parameters were used as described previously (29, 38). MACS (1.4.2) was used to identify all H3K27ac peaks with a P value of 10−9. The SEs were assigned to the gene for which the TSS was nearest to the center of the stitched enhancer. CRC mapper (2) was used as a default setting during the extraction of candidate CRC constituents.
Assay for Transposase-Accessible Chromatin, Followed by Sequencing
The nuclei of lysed cells were incubated with Tn5 transposase in tagmentation buffer (Nextera, Illumina Inc.). PCR amplification and library purification were performed prior to sequencing.
RNA Extraction and Transcript Quantification
RNA was isolated using the RNeasy Plus Micro Kit (Qiagen). Reverse transcription was performed using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The qPCR analysis was performed as described above.
Gene Expression Analysis
The cDNA library was constructed using the NEBNext Ultra Directional RNA Library Prep Kit (New England BioLabs) and subjected to massively parallel sequencing. The paired-end reads were aligned to the hg19 human genome assembly using TopHat2 (v2.0.9). The expression level of each gene was calculated using HTSeq. The data were normalized using the DESeq2 (1.16.1) with a variance-stabilizing transformation. The differential expression analysis was performed using DESeq2. Genes with P values ≤ 0.01 and fold-change values ≥2 were selected for a WebGestalt pathway analysis (http://www.webgestalt.org/2017/option.php) based on KEGG Pathway (https://www.genome.jp/kegg/pathway.html). A gene set enrichment analysis (GSEA) was performed using the Molecular Signatures Database v6.2.C2 (http://software.broadinstitute.org/gsea/index.jsp). The t-SNE analysis and heatmap generation were performed using the R packages rtsne (version 0.1-3) and pheatmap (version 1.0.12), respectively. The results published in this report are based partly upon data generated by the TARGET Initiative, which is managed by the National Cancer Institute (https://ocg.cancer.gov/programs/target). The RNA-seq dataset used in this analysis and clinical information for the TARGET ALL project are available at the database of Genotypes and Phenotypes (dbGaP) under accession phs000218. We used previously described information regarding fusions (21).
Genome Editing Using the CRISPR-Cas9 System and Enhancer Hindrance Using Crispr-I
To generate K7-HA-GFP cells, we transfected Kasumi-7 cells with the lentiCRISPR v2 (#52961, Addgene), which expresses a guide RNA (GUGUGACCCAGAGGCUCCCGG) that targets a sequence immediately downstream of the stop codon of human HNRNPUL1. We cotransfected cells with a plasmid that contained fragments encompassing a sequence approximately ±1 kbp around the stop codon of human HNRNPUL1, as well as an HA-tag (TACCCATACGATGTTCCAGATTACGCT), 2A sequence (gcaacaaa-cttctctctgctgaaacaagccggagatgtcgaagagaatcctggacc), and cDNA for GFP. The GFP-positive cells were flow-sorted and clones were expanded. To inhibit enhancer activity (35), Kasumi-7 cells were infected with pHR-SFFV-dCas9-BFP-KRAB (Addgene #46911) and lentiGuide-GFP-expressing guide RNA. The lentiGuide-GFP was generated by replacing EF1-puro with PGK-GFP in the lentiGuide-puro construct (Addgene #52963).
Mouse pro-B-Cell Transplantation Assays
This experiment was performed as described previously (21). B220+c-Kit+ pro-B cells were induced from BALB/c fetal liver cells cultured on OP9 cells. The cells were then infected with recombinant retrovirus, sorted for GFP expression (>99% purity), and transplanted (1 × 107 cells) intravenously into sublethally irradiated (2 Gy) female NSG mice (age: 8–10 weeks old). A Kaplan–Meier analysis of survival was performed using the R package survival (version 2.44-1.1).
shRNA-mediated Knockdown
The lentivirus vectors used for shRNA expression were generated by inserting annealed shRNA oligo DNAs into the BamHI/ClaI sites downstream of the hU6 promoter in the CSIIhU6PGKhCD8 vector that enabled coexpression of an extracellular domain of human CD8. Infected cells were isolated using a MACS magnetic cell isolation system (Miltenyi Biotec) in combination with the anti-human CD8 antibody.
Ex Vivo Drug Sensitivity Assays
Cells were cultured in triplicate at a density of 1 × 105/mL in the presence of the indicated concentrations of drugs or vehicle (DMSO) for 3 days. Viable cells were counted using CCK-8 (Dojindo) or by inspection under a microscope. The fraction of surviving cells relative to the control was calculated for each condition. Graphs were generated using the R package ggplot2 (version 3.2.0).
In Vivo Drug Sensitivity Assays
Kasumi-7 cells were infected with a retrovirus engineered to express luciferase-IRES-GFP (Addgene #75021). After cell sorting, 5 × 106 GFP-positive cells were transplanted intravenously into NSG mice that had been preconditioned using two intraperitoneal injections of busulfan (20 mg/kg; B2635, Sigma-Aldrich) at a 24-hour interval. Mice were administered enrofloxacin (170 mg/L; Bayer Pharmaceuticals) in drinking water ad libitum. On posttranslation day 1, the mice were administered daily doses of dasatinib (30 mg/kg/day; Selleck) or ibrutinib (25 mg/kg/day; Selleck) via oral gavage according to the manufacturer's instructions. Bioluminescent imaging was performed using IVIS Lumina (Caliper). In some experiments, mice were administered FGH10019 (MedChemExpress; 25 mg/kg/day, in 3% DMSO/20% PEG300/3% Tween80/saline) via oral gavage 6 days per week, beginning on day 1 posttransplantation. A Kaplan–Meier analysis of survival was performed as described above.
Data Availability
The ChIP-seq, assay for transposase-accessible chromatin, followed by sequencing, and RNA-seq data have been deposited in ArrayExpress under the following accession numbers: E-MTAB-8466, E-MTAB-8463, E-MTAB-8491, and E-MTAB-8480.
Disclosure of Potential Conflicts of Interest
A. Ota reports receiving a commercial research grant from Celgene. H. Mano has ownership interest in a patent. No potential conflicts of interest were disclosed by the other authors.
Authors' Contributions
S. Tsuzuki: Conceptualization, data curation, formal analysis, supervision, funding acquisition, validation, investigation, visualization, methodology, writing-original draft, writing-review and editing. T. Yasuda: Conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology and writing-original draft. S. Kojima: Data curation and methodology. M. Kawazu: Data curation and formal analysis. K. Akahane: Resources. T. Inukai: Resources and funding acquisition. M. Imaizumi: Resources. T. Morishita: Resources. K. Miyamura: Resources. T. Ueno: Data curation and formal analysis. S. Karnan: Methodology. A. Ota: Methodology. T. Hyodo: Methodology. H. Konishi: Methodology. M. Sanada: Resources. H. Nagai: Resources. K. Horibe: Resources. A. Tomita: Resources. K. Suzuki: Resources. H. Muramatsu: Resources. Y. Takahashi: Resources. Y. Miyazaki: Resources. I. Matsumura: Resources. H. Kiyoi: Resources. Y. Hosokawa: Methodology. H. Mano: Data curation and supervision. F. Hayakawa: Conceptualization, resources, funding acquisition and writing-original draft.
Supplementary Material
SupplementaryTables
SupplementaryFigures
Acknowledgments
This work was supported by JSPS KAKENHI grant number 18H02645 (to S. Tsuzuki), 18K16103 (to T. Yasuda), 15K09645 (to T. Inukai), and 18H02835 (to F. Hayakawa); grant (JP19lm0203005) from the Japan Agency for Medical Research and Development (to S. Tsuzuki). Grants for Project for Cancer Research and Therapeutic Evolution (16cm0106525h0001) from the Japan Agency for Medical Research and Development (to T. Yasuda); Grants for Practical Research for Innovative Cancer Control (JP19ck0106331) from the Japan Agency for Medical Research and Development (to F. Hayakawa); Grants for Practical Research for Innovative Cancer Control (JP19ck0106253) from the Japan Agency for Medical Research and Development (to T. Inukai); and a grant from Princess Takamatsu Cancer Research Fund (11-24307; to S. Tsuzuki). This research is partially supported by the Department of Advanced Medicine, Nagoya University Hospital. We thank Keiko Hasegawa for secretary assistance.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Blood Cancer Discovery Online https://bloodcancerdiscov.aacrjournals.org/.
References
- 1. Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 2005;122:947–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Saint-Andre V, Federation AJ, Lin CY, Abraham BJ, Reddy J, Lee TI, et al. Models of human core transcriptional regulatory circuitries. Genome Res 2016;26:385–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Buganim Y, Faddah DA, Jaenisch R. Mechanisms and models of somatic cell reprogramming. Nat Rev Genet 2013;14:427–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Young RA. Control of the embryonic stem cell state. Cell 2011;144:940–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lin CY, Erkek S, Tong Y, Yin L, Federation AJ, Zapatka M, et al. Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature 2016;530:57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sanda T, Lawton LN, Barrasa MI, Fan ZP, Kohlhammer H, Gutierrez A, et al. Core transcriptional regulatory circuit controlled by the TAL1 complex in human T cell acute lymphoblastic leukemia. Cancer Cell 2012;22:209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bradner JE, Hnisz D, Young RA. Transcriptional addiction in cancer. Cell 2017;168:629–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boeva V, Louis-Brennetot C, Peltier A, Durand S, Pierre-Eugene C, Raynal V, et al. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat Genet 2017;49:1408–13. [DOI] [PubMed] [Google Scholar]
- 9. Decaesteker B, Denecker G, Van Neste C, Dolman EM, Van Loocke W, Gartlgruber M, et al. TBX2 is a neuroblastoma core regulatory circuitry component enhancing MYCN/FOXM1 reactivation of DREAM targets. Nat Commun 2018;9:4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Durbin AD, Zimmerman MW, Dharia NV, Abraham BJ, Iniguez AB, Weichert-Leahey N, et al. Selective gene dependencies in MYCN-amplified neuroblastoma include the core transcriptional regulatory circuitry. Nat Genet 2018;50:1240–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ott CJ, Federation AJ, Schwartz LS, Kasar S, Klitgaard JL, Lenci R, et al. Enhancer architecture and essential core regulatory circuitry of chronic lymphocytic leukemia. Cancer Cell 2018;34:982–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hunger SP, Mullighan CG. Redefining ALL classification: toward detecting high-risk ALL and implementing precision medicine. Blood 2015;125:3977–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moorman AV. The clinical relevance of chromosomal and genomic abnormalities in B-cell precursor acute lymphoblastic leukaemia. Blood Rev 2012;26:123–35. [DOI] [PubMed] [Google Scholar]
- 14. Ran L, Sirota I, Cao Z, Murphy D, Chen Y, Shukla S, et al. Combined inhibition of MAP kinase and KIT signaling synergistically destabilizes ETV1 and suppresses GIST tumor growth. Cancer Discov 2015;5:304–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ran L, Chen Y, Sher J, Wong EWP, Murphy D, Zhang JQ, et al. FOXF1 defines the core-regulatory circuitry in gastrointestinal stromal tumor. Cancer Discov 2018;8:234–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li JF, Dai YT, Lilljebjorn H, Shen SH, Cui BW, Bai L, et al. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1,223 cases. Proc Natl Acad Sci U S A 2018;115:E11711–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gu Z, Churchman M, Roberts K, Li Y, Liu Y, Harvey RC, et al. Genomic analyses identify recurrent MEF2D fusions in acute lymphoblastic leukaemia. Nat Commun 2016;7:13331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu YF, Wang BY, Zhang WN, Huang JY, Li BS, Zhang M, et al. Genomic profiling of adult and pediatric B-cell acute lymphoblastic leukemia. EBioMedicine 2016;8:173–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ohki K, Kiyokawa N, Saito Y, Hirabayashi S, Nakabayashi K, Ichikawa H, et al. Clinical and molecular characteristics of MEF2D fusion-positive B-cell precursor acute lymphoblastic leukemia in childhood, including a novel translocation resulting in MEF2D-HNRNPH1 gene fusion. Haematologica 2019;104:128–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Suzuki K, Okuno Y, Kawashima N, Muramatsu H, Okuno T, Wang X, et al. MEF2D-BCL9 fusion gene is associated with high-risk acute B-cell precursor lymphoblastic leukemia in adolescents. J Clin Oncol 2016;34:3451–9. [DOI] [PubMed] [Google Scholar]
- 21. Yasuda T, Tsuzuki S, Kawazu M, Hayakawa F, Kojima S, Ueno T, et al. Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults. Nat Genet 2016;48:569–74. [DOI] [PubMed] [Google Scholar]
- 22. Potthoff MJ, Olson EN. MEF2: a central regulator of diverse developmental programs. Development 2007;134:4131–40. [DOI] [PubMed] [Google Scholar]
- 23. Homminga I, Pieters R, Langerak AW, de Rooi JJ, Stubbs A, Verstegen M, et al. Integrated transcript and genome analyses reveal NKX2-1 and MEF2C as potential oncogenes in T cell acute lymphoblastic leukemia. Cancer Cell 2011;19:484–97. [DOI] [PubMed] [Google Scholar]
- 24. Lund AH, Turner G, Trubetskoy A, Verhoeven E, Wientjens E, Hulsman D, et al. Genome-wide retroviral insertional tagging of genes involved in cancer in Cdkn2a-deficient mice. Nat Genet 2002;32:160–5. [DOI] [PubMed] [Google Scholar]
- 25. Schwieger M, Schuler A, Forster M, Engelmann A, Arnold MA, Delwel R, et al. Homing and invasiveness of MLL/ENL leukemic cells is regulated by MEF2C. Blood 2009;114:2476–88. [DOI] [PubMed] [Google Scholar]
- 26. Suzuki T, Shen H, Akagi K, Morse HC, Malley JD, Naiman DQ, et al. New genes involved in cancer identified by retroviral tagging. Nat Genet 2002;32:166–74. [DOI] [PubMed] [Google Scholar]
- 27. Ma L, Liu J, Liu L, Duan G, Wang Q, Xu Y, et al. Overexpression of the transcription factor MEF2D in hepatocellular carcinoma sustains malignant character by suppressing G2-M transition genes. Cancer Res 2014;74:1452–62. [DOI] [PubMed] [Google Scholar]
- 28. Xu K ZY. MEF2D/Wnt/β-catenin pathway regulates the proliferation of gastric cancer cells and is regulated by microRNA-19. Tumour Biol 2016;37:9059–69. [DOI] [PubMed] [Google Scholar]
- 29. Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013;153:307–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hnisz D, Schuijers J, Lin CY, Weintraub AS, Abraham BJ, Lee TI, et al. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol Cell 2015;58:362–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sengupta S, George RE. Super-enhancer-driven transcriptional dependencies in cancer. Trends Cancer 2017;3:269–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, et al. Super-enhancers in the control of cell identity and disease. Cell 2013;155:934–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Buchner M, Swaminathan S, Chen Z, Müschen M. Mechanisms of pre-B-cell receptor checkpoint control and its oncogenic subversion in acute lymphoblastic leukemia. Immunol Rev 2015;263:192–209. [DOI] [PubMed] [Google Scholar]
- 34. Geng H, Hurtz C, Lenz KB, Chen Z, Baumjohann D, Thompson S, et al. Self-enforcing feedback activation between BCL6 and pre-B cell receptor signaling defines a distinct subtype of acute lymphoblastic leukemia. Cancer Cell 2015;27:409–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, et al. Genome-scale CRISPR-mediated control of gene repression and activation. Cell 2014;159:647–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. van Zelm MC, van der Burg M, de Ridder D, Barendregt BH, de Haas EFE, Reinders MJT, et al. Ig gene rearrangement steps are initiated in early human precursor B cell subsets and correlate with specific transcription factor expression. J Immunol Res 2005;175:5912–22. [DOI] [PubMed] [Google Scholar]
- 37. Yasuda T, Sanjo H, Pagès G, Kawano Y, Karasuyama H, Pouysségur J, et al. Erk kinases link pre-B cell receptor signaling to transcriptional events required for early B cell expansion. Immunity 2008;28:499–508. [DOI] [PubMed] [Google Scholar]
- 38. Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013;153:320–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ornatsky OI, Andreucci JJ, McDermott JC. A dominant-negative form of transcription factor MEF2 inhibits myogenesis. J Biol Chem 1997;272:33271–8. [DOI] [PubMed] [Google Scholar]
- 40. Encode Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012;489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ramadani F, Bolland DJ, Garcon F, Emery JL, Vanhaesebroeck B, Corcoran AE, et al. The PI3K isoforms p110alpha and p110delta are essential for pre-B cell receptor signaling and B cell development. Sci Signal 2010;3:ra60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schreiber SL, Crabtree GR. The mechanism of action of cyclosporin A and FK506. Immunol Today 1992;13:136–42. [DOI] [PubMed] [Google Scholar]
- 43. Shao W, Espenshade PJ. Expanding roles for SREBP in metabolism. Cell Metab 2012;16:414–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kamisuki S, Mao Q, Abu-Elheiga L, Gu Z, Kugimiya A, Kwon Y, et al. A small molecule that blocks fat synthesis by inhibiting the activation of SREBP. Chem Biol 2009;16:882–92. [DOI] [PubMed] [Google Scholar]
- 45. Kamisuki S, Shirakawa T, Kugimiya A, Abu-Elheiga L, Choo HY, Yamada K, et al. Synthesis and evaluation of diarylthiazole derivatives that inhibit activation of sterol regulatory element-binding proteins. J Med Chem 2011;54:4923–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tang JJ, Li JG, Qi W, Qiu WW, Li PS, Li BL, et al. Inhibition of SREBP by a small molecule, betulin, improves hyperlipidemia and insulin resistance and reduces atherosclerotic plaques. Cell Metab 2011;13:44–56. [DOI] [PubMed] [Google Scholar]
- 47. Ying CY, Dominguez-Sola D, Fabi M, Lorenz IC, Hussein S, Bansal M, et al. MEF2B mutations lead to deregulated expression of the oncogene BCL6 in diffuse large B cell lymphoma. Nat Immunol 2013;14:1084–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hirano D, Hayakawa F, Yasuda T, Tange N, Yamamoto H, Kojima Y, et al. Chromosomal translocation-mediated evasion from miRNA induces strong MEF2D fusion protein expression, causing inhibition of PAX5 transcriptional activity. Oncogene 2019;38:2263–74. [DOI] [PubMed] [Google Scholar]
- 49. Di Giorgio E, Franforte E, Cefalu S, Rossi S, Dei Tos AP, Brenca M, et al. The co-existence of transcriptional activator and transcriptional repressor MEF2 complexes influences tumor aggressiveness. PLoS Genet 2017;13:e1006752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stathis A, Bertoni F. BET proteins as targets for anticancer treatment. Cancer Discov 2018;8:24–36. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SupplementaryTables
SupplementaryFigures
Data Availability Statement
The ChIP-seq, assay for transposase-accessible chromatin, followed by sequencing, and RNA-seq data have been deposited in ArrayExpress under the following accession numbers: E-MTAB-8466, E-MTAB-8463, E-MTAB-8491, and E-MTAB-8480.