

Abstract
The COVID‐19 pandemic has led to a resurgence of the debate on whether host–parasite interactions should evolve towards avirulence. In this review, we first show that SARS‐CoV‐2 virulence is evolving, before explaining why some expect the mortality caused by the epidemic to converge towards that of human seasonal alphacoronaviruses. Leaning on existing theory, we then include viral evolution into the picture and discuss hypotheses explaining why the virulence has increased since the beginning of the pandemic. Finally, we mention some potential scenarios for the future.
Keywords: epidemiology, genomics, immune escape, immunopathology, maladaptation, trade‐off, virus
Why has SARS‐CoV‐2 virulence increased since 2020? Has it reached an evolutionary dead end or will it become a harmless virus? In this review, we particulary highlight three potential scenarios for the future evolution of SARS‐CoV‐2: i) towards avirulence (if tranmission and virulence are independent), ii) towards a higher virulence that optimises its trade‐off with transmission (fully adaptive path), or iii) towards an intermediate virulence level following a path difficult to anticipate, shaped by the yet poorly known adaptive landscape (maladaptive virulence evolution).
![]()
1. INTRODUCTION
The year 2019 witnessed the emergence of one of the largest and fastest spreading pandemics ever, which had caused more than 80 million infections and claimed more than 1.8 million human lives throughout the world by the end of 2020 (World Health Organization, 2021b). In April 2021, the pandemic was still out of control in many countries all over the world. The causative agent was rapidly identified as a novel coronavirus and named SARS‐CoV‐2 (Gorbalenya et al., 2020). Its most likely origin is believed to be from viruses currently circulating in bat populations (World Health Organization, 2021c).
Many reviews have been written on the evolutionary origin of the virus (Andersen et al., 2020; Boni et al. 2020) and on its genetic evolution since then (van Dorp, Acman, et al., 2020; Worobey et al., 2020). Here, we focus on the phenotypic evolution of infection life‐history traits and, in particular, on virulence.
When it comes to the negative effects of infectious diseases on their hosts, a set of related but distinct notions can be found in the literature: mortality, lethality, pathogenicity and virulence. Among the four, mortality is the only aspect that refers to the population (Bonita et al., 2006). The others are defined at the level of an individual. Lethality, or fatality ratio, is the probability for an infected host to die in a given context. For SARS‐CoV‐2, lethality was rapidly shown to be 10 times that of seasonal influenza and also to strongly depend on age (Verity et al., 2020). However, it is important to stress that this trait varies depending on access to healthcare infrastructure or treatment availability. Pathogenicity can be defined at the cellular level and characterizes some of the harmful interactions between the virus and host cells (Isenberg, 1988). Finally, virulence is defined as the decrease in host fitness due to the infection (Read, 1994). Contrarily to the other definitions, it has an evolutionary dimension because it involves the notion of fitness, which is notoriously difficult to measure. Overall, the choice of the most appropriate trait to measure to study virulence evolution should maximize measurement practicality and public health importance, but also, most importantly, trait values should affect infection fitness (Alizon & Michalakis, 2015).
In the following, unless stated otherwise and to simplify the reasoning, we will consider the infection fatality ratio (IFR), which is the proportion of infected hosts that die from the infection, to be a proxy of virulence. At the individual level, it is the metric that has the most direct meaning to a wide audience; however, care must be taken at the population level because IFR strongly varies with age (O’Driscoll et al., 2020; Verity et al., 2020) (Figure 1a). This implies that the age pyramid of a country shapes the IFR, even if the latter is assumed not to depend on nondemographic conditions, for example health care (Figure 1b). Furthermore, host immunity introduces another dimension of variation that needs to be accounted for when estimating the IFR.
FIGURE 1.

Individual SARS‐CoV‐2 infection fatality ratio (IFR) as a function of age (a) and population average IFRs in 8 countries according to their demography since 1950 (b). In panel b, counterfactual mean IFRs were obtained by weighting the age‐stratified IFR data from O’Driscoll et al. (2020) by the relative frequencies of age classes from annual age pyramid data compiled by United Nations (2019). Dots show the median values and shaded areas the 95% confidence intervals
We first present the evidence for the evolution of SARS‐CoV‐2 virulence. We then explain why some predict the virus population will evolve towards avirulence, before explaining why current trends do not follow this expectation. Finally, we present some hypotheses for future evolutionary dynamics.
2. FROM NEUTRAL MUTANTS TO ‘VARIANTS’
For nearly a year, there was little clear evidence of phenotypic variations that could be linked to virus genetics. Of course, as shown by in vitro experiments of deep mutational scanning (Starr et al., 2020), some mutations have strong effects and many decrease the affinity of the receptor‐binding domain (BBD) of the virus spike protein to the human ACE2 receptor so much that they are likely detrimental. An exception that proves the rule is the D614G substitution in the spike protein, which occurred independently in different lineages throughout the world and was shown to increase the affinity of the virus for the ACE2 receptor on human cells (Korber et al., 2020). This was associated with an increased transmission rate in some regions, although teasing apart this effect from genetic drift was difficult in some locations (Volz et al., 2020).
The picture completely shifted at the end of 2020 with the emergence of what are now referred to as ‘variants of concern’ (VOC). To avoid confusion, unless stated otherwise, we will only use the term ‘variant’ to refer to virus strains that cause phenotypically different infections compared with a reference, ‘wild type’, strains. For some strains, this phenotypic effect may be suspected, for instance, if several epidemiological clustered cases are caused by virus lineages bearing some mutations suspected to have strong phenotypic effects. The potential VOCs are referred to as variants of interest (VOI) and closely monitored by health authorities. Less preoccupying variants are referred to as variants under investigation (VUI). We also use the term lineage to refer to all viruses that have a different genotype than the ancestral reference.
As mentioned above, the D614G mutation is associated with an increased transmission rate (Volz et al., 2020) and therefore can be considered as a VOC. A second VOC (see Table 1 for a summary) was detected in September 2020 in Kent (UK) because a high proportion of Thermo Fisher RT‐PCR screening tests were exhibiting an unusual pattern. Indeed, this assay contains probes targeting three areas of the SARS‐CoV‐2 genome (in ORF1, S and N). Although in a small proportion of tests, one of the probes sometimes failed to detect its target, laboratories were seeing rapidly increasing numbers of RT‐PCR tests that seemed positive but where the S‐gene was not detected. This phenomenon was referred to as S‐gene target failure (SGTF). The analysis of sequencing data confirmed that infections with SGTF were caused by viruses belonging to the same lineage (B.1.1.7). The striking surprise was that these bore much more mutations in their genome than expected given the substitution rate estimated so far, with several of them, such as the N501Y mutation and Δ69‐70 deletion in the spike protein, being already under scrutiny (Rambaut et al., 2020). The dense epidemiological survey in the UK was used to track the SGTF in positive RT‐PCR tests, and the statistical analyses identified a significant transmission advantage of what is now known as the α variant over the other lineages (Davies, Abbott, et al., 2021; Volz et al., 2021). The analysis of contact‐tracing data also identified a higher percontact transmission risk (Variant Technical group, 2021). Variant α spread all over the world, and similar transmission advantages were estimated in countries such as Denmark, Switzerland (Davies, Abbott, et al., 2021) or France (Haim‐Boukobza et al., 2021).
TABLE 1.
Taxonomy and properties of the main SARS‐CoV‐2 variants detected to date
| WHO | pango | nextstrain | GISAID | Spike mutations | Phenotypic effect | Detection | Date |
|---|---|---|---|---|---|---|---|
| D614G | Multiple | Multiple | Multiple | D614G | Increased transmissibility | World | Mar 2020 |
| VOC α | B.1.1.7 | 20C/501Y.V1 | GRY | ∆69‐70, ∆144‐145, N501Y, A570G, D614G, P681H, T716I, S982A, D1118H | increased transmissibility, increased virulence | UK | Sept 2020 |
| VOC β | B.1.351 | 20H/501Y.V2 | GH/501Y.V2 | D80A, D215G, ∆242‐244, K417N, E484K, N501Y, D614G, A701V | Immune escape | South Africa | Aug 2020 |
| VOC γ | P.1 | 20J/501Y.V3 | GR/501Y.V3 | L18F, P20N, P26S, D138Y, R190S, K417T, E484K, N501Y, D614G, H655Y, T1027I, V1176F | Increased transmissibility, immune escape | Brazil | Brazil |
| VOC δ | B.1.617.2 | 20J/21A/S:478K | G/452R.V3 | T19R, ∆157‐158, L425R, T478K, D614G, P681R, D950N | Increased transmissibility, immune escape | India | Oct 2020 |
| VOC ϵ |
B.1.427 B.1.429 |
20C/S.452R | GH/452R.V1 | S13I, W152C, L452R, D614G | Increased transmissibility | USA | Jun 2020 |
| VUI | B.1.616 | 20C/H655Y | GH | H66D, G142V, ∆144/145, D215G, V483A, D614G, H655Y, G669S, Q949R, N1187D | Increased virulence | France | Mar 2021 |
Only variants with demonstrated phenotypic differences are shown. ‘Detection’ indicates the first country to report the variant, and ‘date’ indicates the date of the earliest sample known. ‘WHO’ (World Health Organization, 2021a), ‘pango’ (Rambaut et al., 2020), ‘GISAID’ (Elbe & Buckland‐Merrett, 2017) and ‘nextstrain’ (Hadfield et al., 2018) refer to the main nomenclatures. Mutations and deletions (Δ) in the spike protein compared with the first sequence from Wuhan (China) were obtained from outbreak.info (Latif et al., 2021). Mutations of concern are highlighted in italic.
A few months later, two analyses conducted in the UK detected a possible increase in the virulence of the infections caused by the α variant (Challen et al., 2021; Davies, Jarvis, et al., 2021). Care must be taken to interpret these results because many SARS‐CoV‐2 infections are asymptomatic, meaning that the case fatality ratio (CFR) can differ from the infection fatality ratio (IFR) (Verity et al., 2020). Indeed, the former quantifies the fraction of deaths among detected COVID‐19 cases (usually because they are symptomatic), whereas the latter estimates the fraction of deaths among all infected hosts. Logically, CFRs are greater than IFRs. However, as reported by both studies, the fraction of infections caused by 501.V1 is not higher when analysing data from a random sample of the population instead of symptom and contact‐oriented tests. This rules out the possibility that the variant might be causing an increased proportion of asymptomatic infections, which would bias the IFR estimate.
In summary, there is reasonable evidence that variant α, according to the World Health Organization's (WHO) terminology (World Health Organization, 2021a), has an increased transmission rate and an increased virulence compared with the wild‐type strains.
Also at the end of 2020, another lineage (B.1.351) was reported to spread rapidly in South Africa. Sequencing quickly revealed that it too bore a higher number of mutations than expected based on the mean molecular clock value (Tegally et al., 2020). Among these mutations, there was the N501Y substitution found in variant α, but also the E484K mutation, which is associated with immune escape (Cele et al., 2021). Evidence from other countries confirmed that what is now known as the β variant also has a transmission advantage over wild‐type strains. Its virulence remains less known, but its ability to evade natural host immunity and even some vaccine immunity is very likely (Hoffmann et al., 2021).
A third major epidemic took place at the end of 2020 that suggested involvement in virus evolution. This epidemic was unexpected because it took place in Brazil and especially in the region of Manaus, where serological data suggested that, after a major health catastrophe, the cumulative incidence reached the theoretical herd immunity threshold (Buss et al., 2021). As for the other variants, genomic analyses again showed that the new epidemic was associated with a specific lineage bearing several mutations (P.1), including the N501Y and E484K substitutions (Faria et al., 2021). In this case, the analysis was able to detect a significant transmission advantage of what is now known as the γ variant, as well as an ability to reinfect hosts with natural immunity (Hoffmann et al., 2021).
The last WHO VOC, now known as the δ variant, corresponds to the B.1.617.2 lineage. The oldest known sample dates from the end of 2020 and was collected in India, where the variant was associated with a major outbreak in spring 2021. Early analyses in United Kingdom, where it reached the majority of new contaminations early in 2021, suggest that the δ variant is associated with an increased transmissibility and, potentially, an increased risk of hospitalization, compared with the α variant (Public Health England, 2021; Scientific Pandemic Influenza Group on Modelling & Operational sub‐group, 2021). Early epidemiological results also suggest that this new variant may partially evade immunity (Bernal et al., 2021), especially in people with only a single vaccine dose (Bernal et al., 2021). Furthermore, the fact that the δ variant does not bear the N501Y and E484K mutations found in, respectively, all three and two previous VOCs illustrates the limits of SARS‐CoV‐2 convergent evolution (Martin et al., 2021) and challenges the current existence of strong genomic bottlenecks.
Many VOIs have been reported worldwide World Health Organization (2021a) but, by definition, their phenotypic effect on infection life‐history traits remains limited, if not unknown. Note that one of these VOIs, CAL.20C (pango lineages B.1.427 and B.1.429), which was first detected in California (USA), was shown to have a slight transmission advantage (Zhang et al., 2021). In France, variant 20C/H655Y (pango lineage B.1.616) appears to have a pronounced tropism for lower respiratory tracts and seemed difficult to detect using classical nasopharyngeal swabs (Fillatre et al., 2021).
In the context of the ‘test, trace and isolate’ strategy implemented by many countries, this latter VOI raises speculations about a potential transmission‐detection trade‐off. Indeed, specializing in colonizing the epithelium of the upper airways is expected to yield a transmission gain for the virus (Harrison et al., 2020; Wölfel et al., 2020) but it also increases the probability of detection using nasopharyngeal swabs. If case isolation measures are strong (Grassly et al., 2020), such a tropism can greatly affect further virus transmission. Therefore, if the selection pressure exerted by nasopharyngeal mass testing measures is significant enough, the evolution of a virus tropism towards the lower respiratory tract and, hence, an increased virulence, could be favoured if the decrease in detection probability compensates the decrease in transmission rate.
What is likely is the replacement of the ancestral lineages by variants with a significantly different genetic background is likely going to set a new stage for the SARS‐CoV‐2 fitness landscape (Martin et al., 2021). In other words, mutations that had little effect or were deleterious in the ancestral genome may prove to be adaptive. Furthermore, the immunization of the population is introducing a coevolutionary dimension, which makes the effect of specific mutations difficult to anticipate.
3. ON THE ROAD TO AVIRULENCE?
Theory stemming from Theobald Smith's law of declining virulence more than a century ago (Méthot, 2012) postulates that we should expect SARS‐CoV‐2 to evolve to cause avirulent infections in humans. Although this reasoning is frequently invoked for many infectious diseases, in the current case it is reinforced by the fact that the most common, and seasonal, coronaviruses we know cause little mortality (Gorbalenya et al., 2020).
A model by Lavine et al. (2021) elegantly explains why SARS‐CoV‐2 could become a seasonal virus causing little mortality at the population level. The essential ingredients of their reasoning are that (a) the IFR strongly depends on age, and (b) the immunity that prevents severe disease (i.e. anti‐virulence immunity) is long‐lasting. What they show is that such an age‐structured system can converge in a matter of years towards a steady state where individuals are infected as children, which allows them to build an immune response with very low mortality. This immunity prevents severe symptoms (i.e. COVID‐19) when infected as adults. The time to converge towards this state depends on how fast the virus spreads, which itself depends on the transmission rate of the virus and the intensity of transmission‐blocking immunity. The authors also find that this trend is not expected for coronaviruses such as severe acute respiratory syndrome (SARS) and Middle‐East Respiratory Syndrome (MERS) because their virulence in children is so high that it would lead to massive mortality at the population level. Importantly, this reasoning focuses on mortality (i.e. a population‐level variable) and that it does not require any virulence evolution per se (the IFR remains unchanged for a given age).
When virus evolution is allowed, the existence of multiple SARS‐CoV‐2 infection sites can be used to build scenarios consistent with the avirulence hypothesis (Smith, 1887). To simplify, when this virus infects lower respiratory tracts (LRT), it faces a strong immune response, it causes more damage to host tissues, and its transmission to new hosts is limited. Conversely, in the upper respiratory tract (URT), infections face a weak immune response, cause minor damage to the host and achieve high transmission rates (Harrison et al., 2020). The virus can also infect cells in the intestinal tracts (Xiao et al., 2020). This is thought to be associated with diarrhoea, but the transmission potential is unknown (Guo et al., 2021). Therefore, one expects natural selection to favour SARS‐CoV‐2 mutants that specialize in infecting URT because it maximizes transmission and, more importantly, because immunity will build up in the LRT, either through recovery from natural infection or from vaccination.
Given that the ACE2 receptor, which is SARS‐CoV‐2's entry point into the cells, is more expressed in the URT than in the LRT, and given that many mutations in the spike protein increase its affinity for ACE2 (Ziegler et al., 2020), the fixation of the D614G mutation could be interpreted as an evolutionary trend towards avirulence. However, another interpretation is that this increased affinity for the receptor might not be traded‐off for a lower ability to exploit the LRT, therefore combining increased transmission rate and virulence. The latter seems more likely given that variant α represents the majority of the genomes uploaded on the GISAID platform since February 2021, meaning that, so far, the average virulence of the world population of SARS‐CoV‐2 has increased.
4. WHY IS SARS‐CoV‐2 VIRULENT?
When a parasite spills into a new host population, it is often maladapted because there are known trade‐offs relating to host exploitation. This host specialization was actually used to create live‐attenuated vaccines by performing serial passages of a parasite through cell cultures, or chicken eggs, or another host, to generate less virulent strains that could be used for vaccination (Ebert, 1998). As we will see later, when it comes to virulence, there is no rule for maladaptation: the virus can be nearly avirulent if it fails to exploit human cells, but it can also be extremely virulent if it causes massive immunopathology, for example via the activation of cytokine storms. Illustrating these differences in maladaptation is difficult because, ideally, it would require finding different strains of the same parasite species that are maladapted in different ways. In the case of coronaviruses, two extreme examples could be MERS, which is extremely virulent in humans (Lessler et al., 2016), and feline coronavirus, which cannot infect humans (Sykes, 2014). In the case of SARS‐CoV‐2, evidence points more towards maladaptation with high virulence given the physiopathology of the infection (Tay et al., 2020).
Maladaptation to a novel host can account for SARS‐CoV‐2's initially large virulence but it does not explain why it is not decreasing rapidly. A first possibility is that natural selection cannot act on SARS‐CoV‐2 virulence, for instance, because of a lack of genetic variability (Dearlove et al., 2020; van Dorp, Richard, et al., 2020). RNA viruses have to balance large mutation rates and strong genomic constraints, and it could be that there is no viable way for the virus to decrease the immunopathology it causes (Belshaw et al., 2008). Note that, as pointed out earlier, these constraints could be changing and the fixation of many mutations in the variant lineages could have reshaped the fitness landscape (Martin et al., 2021).
Even if we assume that mutants with a comparable transmission ability and a lower virulence than the wild‐type strains could emerge, it is still possible that they would not have a clear advantage in terms of natural selection. Indeed, it is essential to account for the life history of the infection (Figure 2). Although the IFR is approximately 10 times that of seasonal influenza, severe symptoms and hospital admission are a minority and, when they occur, take place on average 14 days after infection (Sofonea et al., 2020). By that time, contagiousness is virtually negligible since 95% of the transmission events seem to occur between 2 and 11 days after infection (He et al., 2020). More generally, the symptoms of an index case appear on average 1.3 days after the mean transmission time to her/his ‘infectee’ (Alene et al., 2021). Therefore, from the virus perspective, harming the host is not very costly as it will have little effect on its epidemiological fitness, which, if we leave aside host immunity, can be approximated through the basic reproduction number (R 0), that is the average number of secondary infections caused by an infected person during her infectious period. As explained by Day (2003), this idea echoes classical results from life‐history theory, especially the evolution of ageing, where traits that have deleterious late in life tend not to be selected against.
FIGURE 2.
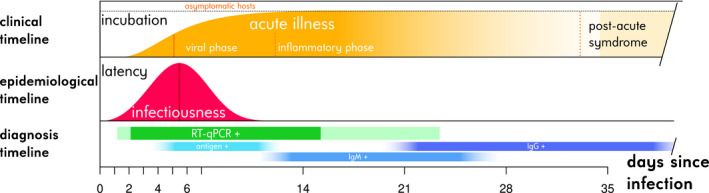
Semi‐quantitative clinical, epidemiological and diagnostic individual history of COVID‐19 infections. The clinical timeline shows the distribution function of the incubation period (time from infection to onset of symptoms), the vertical bar representing the median (McAloon et al., 2020). The breakdowns into clinical phases are exposed are presented with respect to the median symptom onset date (Bouadma et al., 2020; Nalbandian et al., 2021; Polak et al., 2020). The asymptomatic fraction on the top is that estimated by Byambasuren et al. (2020). The epidemiological timeline represents the probability density of the generation time estimated by Ferretti et al. (2020), with the vertical bar showing the median. The latency is the time between infection and the onset of contagiousness. The diagnosis timeline indicates the positivity kinetics of nasopharyngeal RT‐qPCR tests. Following the estimates from Hellewell et al. (2021), more than 50% of cases are positive in the central green band and more than 5% in the peripheral light green bands. Antigenic and serological (immunoglobulin (Ig) M and G) test positivities are shown for qualitative purposes following the estimates from Mercer and Salit (2021)
Since the early 1980s, an explanation for the maintenance of virulence is that the latter can be correlated with traits that are adaptive for the parasite (Anderson & May, 1982; Bremermann & Pickering, 1983; Ewald, 1983; Levin & Pimentel, 1981). For instance, as shown in the case of HIV, strains that cause the most virulent infections tend to be the more transmissible, and the correlation between the two traits could be mediated by the virus load (Fraser et al., 2014). According to such a transmission–virulence trade‐off hypothesis, the exact shape of the relationship between the two traits determines the level of virulence that maximizes the epidemiological fitness of the virus (Alizon et al., 2009). The latter can be seen, in the simplest setting, as the number of secondary infections caused by an infected host. Note that the transmission–virulence trade‐off is restrictive and that other infection life‐history traits should be taken into account to capture the whole life cycle of the virus (Alizon & Michalakis, 2015). In the case of SARS‐CoV‐2, it may be relevant to investigate virulence–recovery trade‐offs (i.e. more virulent strains would have longer infectious periods (Anderson & May, 1982)) or transmission–recovery trade‐offs (if strains that cause shorter infections have higher transmission rates (Alizon, 2008)). Another life‐history trait that could matter is the time until symptom onset because infectiousness of asymptomatic hosts is a key driver of epidemic spread (Fraser et al., 2004). Trade‐offs involving this trait have been less studied but Saad‐Roy et al. (2020) investigated the evolutionary consequence of a trade‐off between the duration of the asymptomatic period and the transmission rate, and, more generally, Sorrell et al. (2009) studied the evolution of asymptomatic infections, which could have implications for SARS‐CoV‐2 given that some individuals appear never to exhibit any symptoms.
In the case of SARS‐CoV‐2, some (limited) evidence suggests that increased virulence can be associated with variations in other life‐history traits that are adaptive for the virus. For instance, a contact‐tracing study showed that individuals with higher viral loads tend to infect a larger proportion of their contacts (Marks et al., 2021). Furthermore, longitudinal follow‐ups show that patients who develop more severe infections tend to have higher virus loads (estimated via the cycle threshold values of RT‐qPCR tests) for a longer time (Néant et al., 2021). Therefore, it could be envisaged that an increased virus load could lead to both increased virulence and contagiousness. Finally, there are data suggesting that the α variant is causing longer infections (Cosentino et al., 2021; Elie et al., 2021).
The adaptive virulence hypothesis is illustrated in Figure 3b, which assumes a transmission–virulence trade‐off. If the virus population is sitting on the trade‐off curve and if the virulence is below the level that maximizes the viral fitness, any increase in transmission rate requires an increase in virulence. However, the trade‐off curve only indicates a constraint and, as we saw above, the virus population could very well be located far from the optimum (Figure 3c). In this case, short‐term evolutionary dynamics are more difficult to anticipate because first, we do not know how far the virus population is from the trade‐off curve (there are many ways to be maladapted) and second, what matters in the short term is the shape of the local fitness landscape. To take a verbal example, one could imagine that a mutant with a virulence and transmission rate could have higher epidemiological fitness (or R 0) than its strain of origin, even if the virulence of this strain is already larger than that of the evolutionary stable strategy (the black dot in Figure 3c). In the other scenarios, such a mutant would most likely have a small R 0 than its strain of origin (Figure 3a) or be a biological aberration (i.e. fall into the nonviable area in Figure 3b).
FIGURE 3.
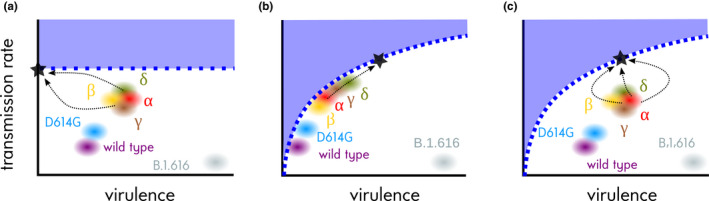
SARS‐CoV‐2 virulence evolution scenarios. (a) More virulent strains are always less fit, (b) virulence and transmission rate are correlated and strains are well‐adapted (i.e. they sit on the trade‐off curve), and (c) same as b but strains are currently maladapted (far from the trade‐off curve). Dashed blue lines show hypothetical transmission–virulence relationships, shaded blue areas the inaccessible state space and black dots the trait combinations maximizing invasion fitness in a naive population (R 0). Dashed arrows show potential evolutionary trajectories. Virulence and transmission rates are in arbitrary units. The virulence and transmission rates of the α and γ variants are currently largely unknown. For the γ variant, the transmission rate appears to be higher (Buss et al., 2021). Viruses can emerge anywhere in the white area, even if they cause virulent and poorly transmissible infections as B.1.616 (Fillatre et al., 2021). For further details about the variants, see Table 1
More generally, nonequilibrium dynamics can explain the persistence of virulent strains, at least transiently. As clearly shown using the Price equation formalism, in the short term, and under the assumption of a correlation between virulence and transmission, more virulent strains tend to be favoured early in an epidemic (Day & Proulx, 2004). The underlying process, which has been shown experimentally using bacteria and phages (Berngruber et al., 2013), is that in an expanding population, an increase in birth rate (or, in the case of an epidemic, transmission) bring more benefits than the same increase in longevity (or infection duration). When the population shrinks, which is the case after an epidemic peak, longevity then becomes more important than birth rates. The essay by Day et al. (2020) discusses these nonequilibrium dynamics in the context of the COVID‐19 pandemic showing, for instance, that the strongest selective pressure in the initial stage of the epidemic is for increased transmission rate. They also expect selection to favour viruses that tend to be less virulent and cause less symptomatic infections.
5. THE FUTURE
The evolution of variants has changed the nature of the SARS‐CoV‐2 pandemic by showing that strong phenotypic evolution is possible for this virus. Interestingly, the difficulty to label these variants is a direct illustration of the ongoing evolutionary dynamics. With the emergence of the D614G mutation in many lineages around the world (Volz et al., 2020), it was tempting to define VOCs through ‘mutations of concern’. This was further supported by the parallel evolution of the N501Y mutation in the α, β and γ variants or of the E484Y mutation in the β and γ variants. However, it is also troubling that VOC all appear to bear a higher number of mutations in their genome than other lineages (Table 1). Furthermore, except for D614G, mutations of concern such as N501Y or E484K seem to be advantageous only in some generic backgrounds. For instance, in the UK, the B.1.1.7 lineage (i.e. the α variant) bearing the E484K mutation appears to have a smaller epidemiological fitness than the β or γ variants. Current trends suggest that epistatic interactions may now dominate SARS‐CoV‐2 evolution and that mutations with effects similar to D614G could be rare. However, it may be hazardous to use past evolutionary trends to anticipate SARS‐CoV‐2 evolution since the fitness landscape that viruses can explore changes as the virus population evolves. For instance, the fixation of the N501Y mutation in the genome has been suggested to deeply affect the fitness landscape compared to that inferred from the original strain detected in Wuhan, China (Martin et al., 2021).
The epidemiological scenario by Lavine et al. (2021), which does not include virus evolution, relies on the assumption that host immunity against symptoms is robust. Unfortunately, field epidemiological data suggest that this might not be the case for the β and γ variants (Abdool Karim & de Oliveira, 2021), and, perhaps, for the δ variant (Bernal et al., 2021). For some vaccines, this immunity appears to be more robust but will this still be the case for future variants? Currently, the greatest unknown appears to reside in immune escape, which can jeopardize the avirulence scenarios. Indeed, with immune escape, absolute fitness (i.e. R 0) should be abandoned in favour or relative fitness measures since the ability of a strain to spread will depend on the immunity of the population, that is on the nature of the strains that are and that have circulated in the past (Lion & Metz, 2018).
With the diversification of SARS‐CoV‐2, the role of multiple infections will likely increase, and these are known to affect virulence evolution (Alizon et al., 2013). Even for short respiratory infections, some infection patterns, in the sense of Sofonea et al. (2017), such as ambinfection, where genotypes are always co‐transmitted, could matter (Lythgoe et al., 2021). Furthermore, the alleviation of nonpharmaceutical interventions implemented to control the pandemic will allow other respiratory infections to spread again. This is particularly true for influenza virus. Here, the evolutionary dynamics are even more difficult to foresee because these involve coevolution between different parasite species and heavily rely on the nature of within‐host interactions (Choisy & de Roode, 2010; Kamiya et al., 2018).
In the long run, immune escape strategies may not be viable for coronaviruses because they impose too many constraints on their genomes (Belshaw et al., 2008). Such reasoning largely rests on our knowledge of the current seasonal coronaviruses, for which large pandemics of immune escape mutants have not been recorded. However, recent results from a time shift experiment conducted using human serum collected from 1985 and 1990 and synthesized spike proteins of the seasonal alphacoronavirus 229E from 1984 to 2016 found that our immune system appears to be less efficient at recognizing ‘future’ coronaviruses (Eguia et al., 2021). This would mean that regular reinfections by seasonal coronaviruses may not just be related to their ability to infect URT, where the immune response is limited, but could also depend on antigenic evolution of the viral spike. Furthermore, an important lesson from this pandemic is that extreme care should be taken before comparing SARS‐CoV‐2 to other viruses, even human coronaviruses. Indeed, this has led to underestimating the transmission before symptoms onset, the airborne transmission and even the magnitude of the pandemic. One of the most recent seasonal coronaviruses is thought to have emerged in the 1950s (Forni et al., 2017). As suggested by Figure 1b, even though half a century ago the age pyramids were different in many countries, the IFR of a coronavirus with a virulence pattern similar to SARS‐CoV‐2 would not have gone unnoticed, although the baseline immunity in the population to viral infections could have been higher at the time due to higher exposition to infectious diseases. This suggests that the virulence of the new virus, which is lower than SARS‐CoV and MERS, with increased transmission before symptoms, but higher than the seasonal coronaviruses, is the worst in terms of population mortality. Again, basing our strategies on immune escape patterns from known coronaviruses can be extremely hazardous.
The high virulence of SARS‐CoV‐2 and its evolution makes it essential to closely monitor this trait. Beyond the definition issues raised in the introduction, a major difficulty for this resides in the proportion of asymptomatic or paucisymptomatic infections, meaning that the IFR is much more difficult to measure than the CFR. To minimize the biases in virulence estimation, the random testing strategy implemented in countries such as the UK seems ideal because it allows controlling for the proportion of variants (Challen et al., 2021; Davies, Jarvis, et al., 2021). International coordination for such random testing appears to be particularly urgent, especially in the context of vaccination (Kennedy & Read, 2020).
On a more positive note, the successful implementation of RNA vaccines does change the dark picture painted by immune escape risk. Indeed, these vaccines theoretically have the potential to follow the coevolutionary race with the virus, at least whereas its genetic diversity remains limited (Dearlove et al., 2020), and this could prove decisive, given the evolutionary rates observed so far. However, we also know that virulence‐blocking vaccines tend to select for strains that are more virulent in nonvaccinated hosts (Gandon et al., 2001). More than ever, we need to monitor virus evolution to avoid an arms race between SARS‐CoV‐2 and public health policies (Kennedy & Read, 2020; Van Baalen, 1998).
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/jeb.13896.
ACKNOWLEDGEMENTS
We thank Yannis Mickalakis and the ETE team for discussion.
Alizon, S. , & Sofonea, M. T. (2021). SARS‐CoV‐2 virulence evolution: Avirulence theory, immunity and trade‐offs. Journal of Evolutionary Biology, 34, 1867–1877. 10.1111/jeb.13896
REFERENCES
- Abdool Karim, S. S. , & de Oliveira, T. (2021). New SARS‐CoV‐2 variants — Clinical, public health, and vaccine implications. New England Journal of Medicine, 384, 1866–1868. 10.1056/NEJMc2100362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alene, M. , Yismaw, L. , Assemie, M. A. , Ketema, D. B. , Gietaneh, W. , & Birhan, T. Y. (2021). Serial interval and incubation period of covid‐19: A systematic review and meta‐analysis. BMC Infectious Diseases, 21, 1–9. 10.1186/s12879-021-05950-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alizon, S. (2008). Transmission‐recovery trade‐offs to study parasite evolution. American Naturalist, 172, E113–E121. 10.1086/589892 [DOI] [PubMed] [Google Scholar]
- Alizon, S. , de Roode, J. C. , & Michalakis, Y. (2013). Multiple infections and the evolution of virulence. Ecology Letters, 344(16), 556–567. 10.1111/ele.12076 [DOI] [PubMed] [Google Scholar]
- Alizon, S. , Hurford, A. , Mideo, N. , & van Baalen, M. (2009). Virulence evolution and the trade‐off hypothesis: History, current state of affairs and the future. Journal of Evolutionary Biology, 22, 245–259. 10.1111/j.1420-9101.2008.01658.x [DOI] [PubMed] [Google Scholar]
- Alizon, S. , & Michalakis, Y. (2015). Adaptive virulence evolution: The good old fitness‐based approach. Trends in Ecology & Evolution, 30, 248–254. 10.1016/j.tree.2015.02.009 [DOI] [PubMed] [Google Scholar]
- Andersen, K. G. , Rambaut, A. , Lipkin, W. I. , Holmes, E. C. , & Garry, R. F. (2020). The proximal origin of SARS‐CoV‐2. Nature Medicine, 26, 450–452. 10.1038/s41591-020-0820-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, R. M. , & May, R. M. (1982). Coevolution of hosts and parasites. Parasitology, 85, 411–426. 10.1017/S0031182000055360 [DOI] [PubMed] [Google Scholar]
- Belshaw, R. , Gardner, A. , Rambaut, A. , & Pybus, O. G. (2008). Pacing a small cage: mutation and RNA viruses. Trends in Ecology & Evolution, 23, 188–193. 10.1016/j.tree.2007.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal, J. L. , Andrews, N. , Gower, C. , Gallagher, E. , Simmons, R. , Thelwall, S. , Stowe, J. , Tessier, E. , Groves, N. , Dabrera, G. , Myers, R. , Campbell, C. , Amirthalingam, G. , Edmunds, M. , Zambon, M. , Brown, K. , Hopkins, S. , Chand, M. , & Ramsay, M. (2021). Effectiveness of COVID‐19 vaccines against the B.1.617.2 variant. medRxiv P. 353 2021.05.22.21257658. [DOI] [PMC free article] [PubMed]
- Berngruber, T. W. , Froissart, R. , Choisy, M. , & Gandon, S. (2013). Evolution of virulence in emerging epidemics. PLoS Path, 9, e1003209. 10.1371/journal.ppat.1003209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boni, M. F. , Lemey, P. , Jiang, X. , Lam, T.‐ T.‐Y. , Perry, B. W. , Castoe, T. A. , Rambaut, A. , & Robertson, D. L. (2020). Evolutionary origins of the SARS‐CoV‐2 sarbecovirus lineage responsible for the COVID‐19 pandemic. Nature Microbiology, 5, 1408–1417. [DOI] [PubMed] [Google Scholar]
- Bonita, R. , Beaglehole, R. , & Kjellström, T. (2006). Basic epidemiology. World Health Organization. [Google Scholar]
- Bouadma, L. , Lescure, F.‐X. , Lucet, J.‐C. , Yazdanpanah, Y. , & Timsit, J.‐F. (2020). Severe SARS‐CoV‐2 infections: Practical considerations and management strategy for intensivists. Intensive Care Medicine, 46, 579–582. 10.1007/s00134-020-05967-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremermann, H. J. , & Pickering, J. (1983). A game‐theoretical model of parasite virulence. Journal of Theoretical Biology, 100, 411–426. 10.1016/0022-5193(83)90438-1 [DOI] [PubMed] [Google Scholar]
- Buss, L. F. , Prete, C. A. , Abrahim, C. M. M. , Mendrone, A. , Salomon, T. , de Almeida‐Neto, C. , França, R. F. O. , Belotti, M. C. , Carvalho, M. P. S. S. , Costa, A. G. , Crispim, M. A. E. , Ferreira, S. C. , Fraiji, N. A. , Gurzenda, S. , Whittaker, C. , Kamaura, L. T. , Takecian, P. L. , da Silva Peixoto, P. , Oikawa, M. K. , … Sabino, E. C. (2021). Three‐quarters attack rate of SARS‐CoV‐2 in the Brazilian Amazon during a largely unmitigated epidemic. Science, 371, 288–292. 10.1126/science.abe9728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byambasuren, O. , Cardona, M. , Bell, K. , Clark, J. , McLaws, M.‐L. , & Glasziou, P. (2020). Estimating the extent of asymptomatic covid‐19 and its potential for community transmission: Systematic review and meta‐analysis. Official Journal of the Association of Medical Microbiology and Infectious Disease Canada, 5, 223–234. 10.3138/jammi-2020-0030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cele, S. , Gazy, I. , Jackson, L. , Hwa, S.‐H. , Tegally, H. , Lustig, G. , Giandhari, J. , Pillay, S. , Wilkinson, E. , Naidoo, Y. , Karim, F. , Ganga, Y. , Khan, K. , Balazs, A. B. , Gosnell, B. I. , Hanekom, W. , Moosa, M.‐Y.‐S. , NGS‐SA, C.‐K. Team , Lessells, R. J. , … Sigal, A. (2021). Escape of SARS‐CoV‐2 501Y.V2 variants from neutralization by convalescent plasma. medRxiv P. 2021.01.26.21250224. [DOI] [PMC free article] [PubMed]
- Challen, R. , Brooks‐Pollock, E. , Read, J. M. , Dyson, L. , Tsaneva‐Atanasova, K. , & Danon, L. (2021). Risk of mortality in patients infected with SARS‐CoV‐2 variant of concern 202012/1: Matched cohort study. BMJ, 372, n579. 10.1136/bmj.n579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choisy, M. , & de Roode, J. C. (2010). Mixed infections and the evolution of virulence: Effects of resource competition, parasite plasticity and impaired host immunity. American Naturalist, 175, E105–E118. 10.1086/651587 [DOI] [PubMed] [Google Scholar]
- Cosentino, G. , Bernard, M. , Ambroise, J. , Giannoli, J.‐M. , Guedj, J. , Débarre, F. , & Blanquart, F. (2021). SARS‐CoV‐2 382 viral dynamics in infections with variants of concern in the French community. https://hal.sorbonne‐universite.fr/hal‐03217231 [DOI] [PubMed]
- Davies, N. G. , Abbott, S. , Barnard, R. C. , Jarvis, C. I. , Kucharski, A. J. , Munday, J. D. , Pearson, C. A. B. , Russell, T. W. , Tully, D. C. , Washburne, A. D. , Wenseleers, T. , Gimma, A. , Waites, W. , Wong, K. L. M. , Zandvoort, K. V. , Silverman, J. D., C. C.‐W. Group , C.‐G. U. C.‐U. Consortium , Diaz‐Ordaz, K. , … Edmunds, W. J. (2021). Estimated transmissibility and impact of SARS‐CoV‐2 lineage B.1.1.7 in England. Science, 372(6538). 10.1126/science.abg3055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, N. G. , Jarvis, C. I. , Edmunds, W. J. , Jewell, N. P. , Diaz‐Ordaz, K. , & Keogh, R. H. (2021). Increased mortality in community‐tested cases of SARS‐CoV‐2 lineage B.1.1.7. Nature, 593, 270–274. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day, T. (2003). Virulence evolution and the timing of disease life‐history events. Trends in Ecology & Evolution, 18, 113–118. 10.1016/S0169-5347(02)00049-6 [DOI] [Google Scholar]
- Day, T. , Gandon, S. , Lion, S. , & Otto, S. P. (2020). On the evolutionary epidemiology of SARS‐CoV‐2. Current Biology, 30(15), R849–R857. 10.1016/j.cub.2020.06.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day, T. , & Proulx, S. R. (2004). A general theory for the evolutionary dynamics of virulence. American Naturalist, 163, E40–E63. 10.1086/382548 [DOI] [PubMed] [Google Scholar]
- Dearlove, B. , Lewitus, E. , Bai, H. , Li, Y. , Reeves, D. B. , Joyce, M. G. , Scott, P. T. , Amare, M. F. , Vasan, S. , Michael, N. L. , Modjarrad, K. , & Rolland, M. (2020). A SARS‐CoV‐2 vaccine candidate would likely match all currently circulating variants. Proceedings of the National Academy of Sciences of the United States of America, 117(38), 23652–23662. https://www.pnas.org/content/117/38/23652.short [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert, D. (1998). Experimental evolution of parasites. Science, 282, 1432–1435. 10.1126/science.282.5393.1432 [DOI] [PubMed] [Google Scholar]
- Eguia, R. T. , Crawford, K. H. D. , Stevens‐Ayers, T. , Kelnhofer‐Millevolte, L. , Greninger, A. L. , Englund, J. A. , Boeckh, M. J. , & Bloom, J. D. (2021). A human coronavirus evolves antigenically to escape antibody immunity. PLoS Path, 17, e1009453. 10.1371/journal.ppat.1009453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbe, S. , & Buckland‐Merrett, G. (2017). Data, disease and diplomacy: GISAID’s innovative contribution to global health. Global Challenges, 1, 33–46. 10.1002/gch2.1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elie, B. , Lecorche, E. , Sofonea, M. T. , Trombert‐Paolantoni, S. , Foulongne, V. , Guedj, J. , Haim‐Boukobza, S. , Roquebert, B. , & Alizon, S. (2021). Inferring SARS‐CoV‐2 variant within‐host kinetics. medRxiv P. 2021.05.26.21257835.
- Ewald, P. W. (1983). Host‐parasite relations, vectors, and the evolution of disease severity. Annual Review of Ecology Evolution and Systematics, 14, 465–485. 10.1146/annurev.es.14.110183.002341 [DOI] [Google Scholar]
- Faria, N. R. , Mellan, T. A. , Whittaker, C. , Claro, I. M. , Candido, D. D. S. , Mishra, S. , Crispim, M. A. E. , Sales, F. C. , Hawryluk, I. , McCrone, J. T. , Hulswit, R. J. G. , Franco, L. A. M. , Ramundo, M. S. , Jesus, J. G. D. , Andrade, P. S. , Coletti, T. M. , Ferreira, G. M. , Silva, C. A. M. , Manuli, E. R. , … Sabino, E. C. (2021). Genomics and epidemiology of a novel SARS‐CoV‐2 lineage in Manaus, Brazil. medRxiv P. 422 2021.02.26.21252554. [DOI] [PMC free article] [PubMed]
- Ferretti, L. , Ledda, A. , Wymant, C. , Zhao, L. , Ledda, V. , Abeler‐Dorner, L. , Kendall, M. , Nurtay, A. , Cheng, H.‐Y. , Ng, T.‐C. , Lin, H.‐H. , Hinch, R. , Masel, J. , Kilpatrick, A.M. , … Fraser, C. (2020). The timing of covid‐19 transmission. medRxiv. [Google Scholar]
- Fillatre, P. , Dufour, M.‐J. , Behillil, S. , Vatan, R. , Reusse, P. , Gabellec, A. , Velmans, N. , Montagne, C. , Geffroy, S. , Droumaguet, E. , Merour, V. , Enouf, V. , Buzele, R. , Valence, M. , Guillotel, E. , Gagniere, B. , Baidaluk, A. , Zhukova, A. , Tourdjman, M. , … Massart, N. (2021). A new SARS‐CoV‐2 variant poorly detected by RT‐PCR on nasopharyngeal samples, with high lethality. medRxiv P. 2021.05.05.21256690. [DOI] [PMC free article] [PubMed]
- Forni, D. , Cagliani, R. , Clerici, M. , & Sironi, M. (2017). Molecular evolution of human coronavirus genomes. Trends in Microbiology, 25, 35–48. 10.1016/j.tim.2016.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser, C. , Lythgoe, K. , Leventhal, G. E. , Shirreff, G. , Hollingsworth, T. D. , Alizon, S. , & Bonhoeffer, S. (2014). Virulence and pathogenesis of HIV‐1 infection: An evolutionary perspective. Science, 343, 1243727. 10.1126/science.1243727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser, C. , Riley, S. , Anderson, R. M. , & Ferguson, N. M. (2004). Factors that make an infectious disease outbreak controllable. Proceedings of the National Academy of Sciences of the United States of America, 101, 6146–6151. 10.1073/pnas.0307506101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandon, S. , Mackinnon, M. J. , Nee, S. , & Read, A. F. (2001). Imperfect vaccines and the evolution of pathogen virulence. Nature, 414, 751–756. 10.1038/414751a [DOI] [PubMed] [Google Scholar]
- Gorbalenya, A. E. , Baker, S. C. , Baric, R. S. , de Groot, R. J. , Drosten, C. , Gulyaeva, A. A. , Haagmans, B. L. , Lauber, C. , Leontovich, A. M. , Neuman, B. W. , Penzar, D. , Perlman, S. , Poon, L. L. M. , Samborskiy, D. V. , Sidorov, I. A. , Sola, I. , Ziebuhr, J. , & Coronaviridae Study Group of the International Committee on Taxonomy of Viruses (2020). The species severe acute respiratory syndrome‐related coronavirus: Classifying 2019‐nCoV and naming it SARS CoV‐2. Nature Microbiology, 5, 536–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassly, N. C. , Pons‐Salort, M. , Parker, E. P. K. , White, P. J. , Ferguson, N. M. , Ainslie, K. , Baguelin, M. , Bhatt, S. , Boonyasiri, A. , Brazeau, N. , Cattarino, L. , Coupland, H. , Cucunuba, Z. , Cuomo‐Dannenburg, G. , Dighe, A. , Donnelly, C. , van Elsland, S. L. , FitzJohn, R. , Flaxman, S. , … Xi, X. (2020). Comparison of molecular testing strategies for covid‐19 control: A mathematical modelling study. The Lancet Infectious Diseases, 20, 1381–1389. 10.1016/S1473-3099(20)30630-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, M. , Tao, W. , Flavell, R. A. , & Zhu, S. (2021). Potential intestinal infection and faecal–oral transmission of SARS‐CoV‐2. Nature Reviews Gastroenterology & Hepatology, 18, 269–283. 10.1038/s41575-021-00416-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadfield, J. , Megill, C. , Bell, S. M. , Huddleston, J. , Potter, B. , Callender, C. , Sagulenko, P. , Bedford, T. , & Neher, R. A. (2018). Nextstrain: Real‐time tracking of pathogen evolution. Bioinformatics, 34, 4121–4123. 10.1093/bioinformatics/bty407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haim‐Boukobza, S. , Roquebert, B. , Trombert‐Paolantoni, S. , Lecorche, E. , Verdurme, L. , Foulongne, V. , Selinger, C. , Michalakis, Y. , Sofonea, M. T. , & Alizon, S. (2021). Detection of Rapid SARS‐CoV‐2 Variant Spread, France, January 26–February 16, 2021. Emerging Infectious Diseases, 372, (6538), eabg3055. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison, A. G. , Lin, T. , & Wang, P. (2020). Mechanisms of SARS‐CoV‐2 transmission and pathogenesis. Trends in Immunology, 41, 1100–1115. 10.1016/j.it.2020.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, X. , Lau, E. H. Y. , Wu, P. , Deng, X. , Wang, J. , Hao, X. , Lau, Y. C. , Wong, J. Y. , Guan, Y. , Tan, X. , Mo, X. , Chen, Y. , Liao, B. , Chen, W. , Hu, F. , Zhang, Q. , Zhong, M. , Wu, Y. , Zhao, L. , … Leung, G. M. (2020). Temporal dynamics in viral shedding and transmissibility of COVID‐19. Nature Medicine, 26, 672–675. 10.1038/s41591-020-0869-5 [DOI] [PubMed] [Google Scholar]
- Hellewell, J. , Russell, T. W. , Beale, R. , Kelly, G. , Houlihan, C. , Nastouli, E. , & Kucharski, A. J. (2021). Estimating the effectiveness of routine asymptomatic PCR testing at different frequencies for the detection of SARS‐CoV‐2 infections. BMC Medicine, 19, 1–10. 10.1186/s12916-021-01982-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann, M. , Arora, P. , Groß, R. , Seidel, A. , Hörnich, B. F. , Hahn, A. S. , Krüger, N. , Graichen, L. , Hofmann‐Winkler, H. , Kempf, A. , Winkler, M. S. , Schulz, S. , Jäck, H.‐M. , Jahrsdörfer, B. , Schrezenmeier, H. , Müller, M. , Kleger, A. , Münch, J. , & Pöhlmann, S. (2021). SARS‐CoV‐2 variants B.1.351 and P.1 escape from neutralizing antibodies. Cell, 184(9), 2384–2393.e12. 10.1016/j.cell.2021.03.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isenberg, H. D. (1988). Pathogenicity and virulence: Another view. Clinical Microbiology Reviews, 1, 40–53. 10.1128/CMR.1.1.40-53.1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya, T. , Mideo, N. , & Alizon, S. (2018). Coevolution of virulence and immunosuppression in multiple infections. Journal of Evolutionary Biology, 31, 995–1005. 10.1111/jeb.13280 [DOI] [PubMed] [Google Scholar]
- Kennedy, D. A. , & Read, A. F. (2020). Monitor for COVID‐19 vaccine resistance evolution during clinical trials. PLOS Biology, 18, e3001000. 10.1371/journal.pbio.3001000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber, B. , Fischer, W. M. , Gnanakaran, S. , Yoon, H. , Theiler, J. , Abfalterer, W. , Hengartner, N. , Giorgi, E. E. , Bhattacharya, T. , Foley, B. , Hastie, K. M. , Parker, M. D. , Partridge, D. G. , Evans, C. M. , Freeman, T. M. , de Silva, T. I. , Angyal, A. , Brown, R. L. , Carrilero, L. , … Montefiori, D. C. (2020). Tracking changes in SARS‐CoV‐2 spike: Evidence that D614G increases infectivity of the COVID‐19 virus. Cell, 182, 812–827.e19. 10.1016/j.cell.2020.06.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latif, A. A. , Mullen, J. L. , Alkuzweny, M. , Tsueng, G. , Cano, M. , Haag, E. , Zhou, J. , Zeller, M. , Matteson, N. , Wu, C. , Andersen, K. G. , Su, A. I. , Gangavarapu, K. , Hughes, L. D. , & the Center for Viral Systems Biology (2021). outbreak.info: Lineage comparison. Retrieved from https://outbreak.info/compare‐lineages/ [Google Scholar]
- Lavine, J. S. , Bjornstad, O. N. , & Antia, R. (2021). Immunological characteristics govern the transition of COVID‐19 to endemicity. Science, 371(6530):741–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessler, J. , Salje, H. , Van Kerkhove, M. D. , Ferguson, N. M. , Cauchemez, S. , Rodriquez‐Barraquer, I. , Hakeem, R. , Jombart, T. , Aguas, R. , Al‐Barrak, A. , Cummings, D. A. T. , for the MERS‐CoV Scenario and Modeling Working Group , Algarni, H. , AlHarbi, K. , Clapham, H. , Collins, C. , Cori, A. , Donnelly, C. , Fraser, C. , … for the MERS‐CoV Scenario and Modeling Working Group (2016). Estimating the severity and subclinical burden of Middle East respiratory syndrome coronavirus infection in the Kingdom of Saudi Arabia. American Journal of Epidemiology, 183, 657–663. 10.1093/aje/kwv452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin, S. , & Pimentel, D. (1981). Selection of intermediate rates of increase in parasite‐host systems. American Naturalist, 117, 308–315. 10.1086/283708 [DOI] [Google Scholar]
- Lion, S. , & Metz, J. A. J. (2018). Beyond R0 maximisation: On pathogen evolution and environmental dimensions. Trends in Ecology & Evolution, 33, 458–473. 10.1016/j.tree.2018.02.004 [DOI] [PubMed] [Google Scholar]
- Lythgoe, K. A. , Hall, M. , Ferretti, L. , Cesare, M. D. , MacIntyre‐Cockett, G. , Trebes, A. , Andersson, M. , Otecko, N. , Wise, E. L. , Moore, N. , Lynch, J. , Kidd, S. , Cortes, N. , Mori, M. , Williams, R. , Vernet, G. , Justice, A. , Green, A. , Nicholls, S. M. , … Golubchik, T. (2021). SARS‐CoV‐2 within‐host diversity and transmission. Science, 372(6539), eabg0821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks, M. , Millat‐Martinez, P. , Ouchi, D. , Roberts, C. H. , Alemany, A. , Corbacho‐Monné, M. , Ubals, M. , Tobias, A. , Tebé, C. , Ballana, E. , Bassat, Q. , Baro, B. , Vall‐Mayans, M. , Beiras, C. G. , Prat, N. , Ara, J. , Clotet, B. , & Mitjà, O. (2021). Transmission of COVID‐19 in 282 clusters in Catalonia, Spain: A cohort study. The Lancet Infectious Diseases, 21(5), 629–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, D. P. , Weaver, S. , Tegally, H. , San, E. J. , Shank, S. D. , Wilkinson, E. , Giandhari, J. , Naidoo, S. , Pillay, Y. , Singh, L. , Lessells, R. J. , N.‐S. Consortium , C.‐. G. U. C.‐U. Consortium , Gupta, R. K. , Wertheim, J. O. , Nekturenko, A. , Murrell, B. , Harkins, G. W. , Lemey, P. , … Pond, S. L. K. (2021). The emergence and ongoing convergent evolution of the N501Y lineages coincides with a major global shift in the SARS‐CoV‐2 selective landscape. medRxiv P. 2021.02.23.21252268.
- McAloon, C. , Collins, Á. , Hunt, K. , Barber, A. , Byrne, A. W. , Butler, F. , Casey, M. , Griffin, J. , Lane, E. , McEvoy, D. , Wall, P. , Green, M. , O'Grady, L. , & More, S. J. (2020). Incubation period of covid‐19: A rapid systematic review and meta‐analysis of observational research. British Medical Journal Open, 10, e039652. 10.1136/bmjopen-2020-039652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer, T. R. , & Salit, M. (2021). Testing at scale during the covid‐19 pandemic. Nature Reviews Genetics, 22(7), 415–426. 10.1038/s41576-021-00360-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méthot, P.‐O. (2012). Why do parasites harm their host? On the origin and legacy of Theobald Smith’s ‘Law of Declining Virulence’— 1900–1980. History and Philosophy of the Life Sciences, 34, 561–601. [PubMed] [Google Scholar]
- Nalbandian, A. , Sehgal, K. , Gupta, A. , Madhavan, M. V. , McGroder, C. , Stevens, J. S. , Cook, J. R. , Nordvig, A. S. , Shalev, D. , Sehrawat, T. S. et al (2021). Post‐acute covid‐19 syndrome. Nature Medicine, 27(4), 601–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Néant, N. , Lingas, G. , Hingrat, Q. L. , Ghosn, J. , Engelmann, I. , Lepiller, Q. , Gaymard, A. , Ferré, V. , Hartard, C. , Plantier, J.‐C. , Thibault, V. , Marlet, J. , Montes, B. , Bouiller, K. , Lescure, F.‐X. , Timsit, J.‐F. , Faure, E. , Poissy, J. , Chidiac, C. , … f. t. F. C. C. I. a. F. C. S. Groups . (2021). Modeling SARS‐CoV‐2 viral kinetics and association with mortality in hospitalized patients from the French COVID cohort. Proceedings of the National Academy of Sciences of the United States of America, 118(8), e2017962118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Driscoll, M. , Dos Santos, G. R. , Wang, L. , Cummings, D. A. T. , Azman, A. S. , Paireau, J. , Fontanet, A. , Cauchemez, S. , & Salje, H. (2020). Age‐specific mortality and immunity patterns of SARS‐CoV‐2. Nature, 590(7844):140–145. [DOI] [PubMed] [Google Scholar]
- Polak, S. B. , Van Gool, I. C. , Cohen, D. , Jan, H. , & van Paassen, J. (2020). A systematic review of pathological findings in covid‐19: A pathophysiological timeline and possible mechanisms of disease progression. Modern Pathology, 33, 2128–2138. 10.1038/s41379-020-0603-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Public Health England (2021). SARS‐CoV‐2 variants of concern and variants under investigation ‐ Technical briefing 15. Tech. Rep. 15. Retrieved from https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/993879/Variants_of_Concern_VOC_Technical_Briefing_15.pdf [Google Scholar]
- Rambaut, A. , Holmes, E. C. , O’Toole, A. , Hill, V. , McCrone, J. T. , Ruis, C. , du Plessis, L. , & Pybus, O. G. (2020). A dynamic nomenclature proposal for SARS‐CoV‐2 lineages to assist genomic epidemiology. Nature Microbiology, 5, 1403–1407. 10.1038/s41564-020-0770-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut, A. , Loman, N. , Pybus, O. , Barclay, W. , Barrett, J. , Carabelli, A. , Connor, T. , Peacock, T. , Robertson, D. L. , & Volz, E. (2020). Preliminary genomic characterisation of an emergent SARS‐CoV‐2 lineage in the UK defined by a novel set of spike mutations. Virological. https://virological.org/t/preliminary‐genomic‐characterisation‐of‐an‐emergent‐sars‐cov‐2‐lineage‐in‐the‐uk‐defined‐by‐a‐novel‐set‐of‐spike‐mutations/563
- Read, A. (1994). The evolution of virulence. Trends in Microbiology, 2, 73–76. 10.1016/0966-842X(94)90537-1 [DOI] [PubMed] [Google Scholar]
- Saad‐Roy, C. M. , Wingreen, N. S. , Levin, S. A. , & Grenfell, B. T. (2020). Dynamics in a simple evolutionary epidemiological model for the evolution of an initial asymptomatic infection stage. Proceedings of the National Academy of Sciences of the United States of America, 117, 11541–11550. 10.1073/pnas.1920761117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scientific Pandemic Influenza Group on Modelling, Operational sub‐group . (2021). SPI‐M‐O: Consensus statement on COVID‐19. Tech. rep. Retrieved from https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/986709/S1237_SPI‐M‐O_Consensus_Statement.pdf [Google Scholar]
- Smith, T. (1887). Parasitic bacteria and their relation to saprophytes. American Naturalist, 21, 1–9. 10.1086/274401 [DOI] [Google Scholar]
- Sofonea, M. , Alizon, S. , & Michalakis, Y. (2017). Exposing the diversity of multiple infection patterns. Journal of Theoretical Biology, 419, 278–289. 10.1016/j.jtbi.2017.02.011 [DOI] [PubMed] [Google Scholar]
- Sofonea, M. T. , Reyné, B. , Elie, B. , Djidjou‐Demasse, R. , Selinger, C. , Michalakis, Y. , & Alizon, S. (2020). Epidemiological monitoring and control perspectives: Application of a parsimonious modelling framework to the COVID‐19 dynamics in France. medRxiv P. 2020.05.22.20110593.
- Sorrell, I. , White, A. , Pedersen, A. B. , Hails, R. S. , & Boots, M. (2009). The evolution of covert, silent infection as a parasite strategy. Proceedings of the Royal Society of London, Series B: Biological Sciences, 276, 2217–2226. 10.1098/rspb.2008.1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr, T. N. , Greaney, A. J. , Hilton, S. K. , Ellis, D. , Crawford, K. H. D. , Dingens, A. S. , Navarro, M. J. , Bowen, J. E. , Tortorici, M. A. , Walls, A. C. , King, N. P. , Veesler, D. , & Bloom, J. D. (2020). Deep mutational scanning of SARS‐CoV‐2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell, 182, 1295–1310.e20. 10.1016/j.cell.2020.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes, J. E. (2014). Feline coronavirus infection. In Sykes J. E. (Ed.), Canine and feline infectious diseases (pp. 195–208). Saint Louis: W.B. Saunders. [Google Scholar]
- Tay, M. Z. , Poh, C. M. , Rénia, L. , MacAry, P. A. , & Ng, L. F. P. (2020). The trinity of COVID‐19: immunity, inflammation and intervention. Nature Reviews Immunology, 20, 363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegally, H. , Wilkinson, E. , Giovanetti, M. , Iranzadeh, A. , Fonseca, V. , Giandhari, J. , Doolabh, D. , Pillay, S. , San, E. J. , Msomi, N. , Mlisana, K. , Gottberg, A. V. , Walaza, S. , Allam, M. , Ismail, A. , Mohale, T. , Glass, A. J. , Engelbrecht, S. , Zyl, G. V. , … Oliveira, T. D. (2020). Emergence and rapid spread of a new severe acute respiratory syndrome‐related coronavirus 2 (SARS‐CoV‐2) lineage with multiple spike mutations in South Africa. medRxiv P. 2020.12.21.20248640.
- United Nations . (2019). World Population Prospects 2019, Online Edition. Rev. 1. Tech. rep., United Nations, Department of Economic and Social Affairs, Population Division. Retrieved from https://population.un.org/wpp/Download/Standard/Population/ [Google Scholar]
- Van Baalen, M. (1998). Coevolution of recovery ability and virulence. Proceedings of the Royal Society of London, Series B: Biological Sciences, 265, 317–325. 10.1098/rspb.1998.0298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dorp, L. , Acman, M. , Richard, D. , Shaw, L. P. , Ford, C. E. , Ormond, L. , Owen, C. J. , Pang, J. , Tan, C. C. S. , Boshier, F. A. T. , Ortiz, A. T. , & Balloux, F. (2020). Emergence of genomic diversity and recurrent mutations in SARS CoV‐2. Infection, Genetics and Evolution, 83, 104351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dorp, L. , Richard, D. , Tan, C. C. S. , Shaw, L. P. , Acman, M. , & Balloux, F. (2020). No evidence for increased transmissibility from recurrent mutations in SARS‐CoV‐2. Nature Communications, 11, 5986. 10.1038/s41467-020-19818-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Variant Technical group (2021). Investigation of novel SARS‐COV‐2 variant: Variant of concern 202012/01. Public Health England. Technical briefing 3.
- Verity, R. , Okell, L. C. , Dorigatti, I. , Winskill, P. , Whittaker, C. , Imai, N. , Cuomo‐Dannenburg, G. , Thompson, H. , Walker, P. G. T. , Fu, H. , Dighe, A. , Griffin, J. T. , Baguelin, M. , Bhatia, S. , Boonyasiri, A. , Cori, A. , Cucunubá, Z. , FitzJohn, R. , Gaythorpe, K. , … Ferguson, N. M. (2020). Estimates of the severity of coronavirus disease 2019: A model‐based analysis. The Lancet Infectious Diseases, 20, 669–677. 10.1016/S1473-3099(20)30243-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volz, E. , Hill, V. , McCrone, J. T. , Price, A. , Jorgensen, D. , O’Toole, A. , Southgate, J. , Johnson, R. , Jackson, B. , Nascimento, F. F. , Rey, S. M. , Nicholls, S. M. , Colquhoun, R. M. , Filipe, A. D. S. , Shepherd, J. , Pascall, D. J. , Shah, R. , Jesudason, N. , Li, K. , … Connor, T. R. (2020). Evaluating the effects of SARS‐CoV‐2 Spike mutation D614G on transmissibility and pathogenicity. Cell, 184(1), 64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volz, E. , Mishra, S. , Chand, M. , Barrett, J. C. , Johnson, R. , Geidelberg, L. , Hinsley, W. R. , Laydon, D. J. , Dabrera, G. , O’Toole, A. , Amato, R. , Ragonnet‐Cronin, M. , Harrison, I. , Jackson, B. , Ariani, C. V. , Boyd, O. , Loman, N. J. , McCrone, J. T. , Goncalves, S. , … Ferguson, N. M. (2021). Transmission of SARS‐CoV‐2 Lineage B.1.1.7 in England: Insights from linking epidemiological and genetic data. medRxiv P. 2020.12.30.20249034.
- Wölfel, R. , Corman, V. M. , Guggemos, W. , Seilmaier, M. , Zange, S. , Müller, M. A. , Niemeyer, D. , Jones, T. C. , Vollmar, P. , Rothe, C. , Hoelscher, M. , Bleicker, T. , Brünink, S. , Schneider, J. , Ehmann, R. , Zwirglmaier, K. , Drosten, C. , & Wendtner, C. (2020). Virological assessment of hospitalized patients with COVID‐2019. Nature, 581(7809), 465–469. [DOI] [PubMed] [Google Scholar]
- World Health Organisation (2021a). Tracking SARS‐CoV‐2 variants. Retrieved from https://www.who.int/en/activities/tracking‐SARS‐CoV‐2‐variants/ [Google Scholar]
- World Health Organisation . (2021b). Weekly epidemiological update on COVID‐19 ‐ 5 January 2021. Tech. rep. Retrieved from https://www.who.int/publications/m/item/weekly‐epidemiological‐update‐‐‐5‐january‐2021 [Google Scholar]
- World Health Organisation . (2021c). WHO‐convened global study of origins of SARS‐CoV‐2: China Part. Tech. rep. Retrieved from https://www.who.int/publications‐detail‐redirect/who‐convened‐global‐study‐of‐origins‐of‐sars‐cov‐2‐china‐part [Google Scholar]
- Worobey, M. , Pekar, J. , Larsen, B. B. , Nelson, M. I. , Hill, V. , Joy, J. B. , Rambaut, A. , Suchard, M. A. , Wertheim, J. O. , & Lemey, P. (2020). The emergence of SARS‐CoV‐2 in Europe and North America. Science, 370, 564–570. 10.1126/science.abc8169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, F. , Tang, M. , Zheng, X. , Liu, Y. , Li, X. , & Shan, H. (2020). Evidence for gastrointestinal infection of sars‐cov‐2. Gastroenterology, 158, 1831–1833. 10.1053/j.gastro.2020.02.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, W. , Davis, B. D. , Chen, S. S. , Sincuir Martinez, J. M. , Plummer, J. T. , & Vail, E. (2021). Emergence of a novel SARS‐CoV‐2 variant in Southern California. JAMA, 325(13), 1324–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler, C. G. K. , Allon, S. J. , Nyquist, S. K. , Mbano, I. M. , Miao, V. N. , Tzouanas, C. N. , Cao, Y. , Yousif, A. S. , Bals, J. , Hauser, B. M. , Feldman, J. , Muus, C. , Wadsworth, M. H. , Kazer, S. W. , Hughes, T. K. , Doran, B. , Gatter, G. J. , Vukovic, M. , Taliaferro, F. , … Zhang, K. (2020). SARS‐CoV‐2 receptor ACE2 Is an interferon‐stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell, 181, 1016–1035.e19. 10.1016/j.cell.2020.04.035 [DOI] [PMC free article] [PubMed] [Google Scholar]