

Abstract
Increasingly, research suggests that neurodegenerative diseases and dementias are caused not by unique, solitary cellular mechanisms, but by multiple contributory mechanisms manifesting as heterogeneous clinical presentations. However, diverse neurodegenerative diseases also share common pathological hallmarks and cellular mechanisms. One such mechanism involves the redistribution of the microtubule associated protein tau from the axon into the somatodendritic compartments of neurons, followed by the mislocalization of tau into dendritic spines, resulting in postsynaptic functional deficits. Here we review various signaling pathways that trigger the redistribution of tau to the cell body and dendritic tree, and its mislocalization to dendritic spines. The convergence of multiple pathways in different disease models onto this final common pathway suggests that it may be an attractive pathway to target for developing new treatments for neurodegenerative diseases.
Keywords: Tau, Neuronal Polarity, MAPT, Alzheimer’s disease, Parkinson’s disease, FTDP-17, Huntington’s disease, Dendritic spines, AMPA receptors, Synaptic plasticity
Introduction
Dementia refers to neuronal deterioration in the CNS leading to the inability to care for oneself due to impaired cognition, including deficits in memory acquisition, encoding, or consolidation, as well as behavioral abnormalities. Dementia occurs in a variety of neurodegenerative diseases, Alzheimer’s disease (AD) being the most prevalent disorder (James and Bennett, 2019). AD is characterized by the presence of two histopathological hallmarks, senile plaques and neurofibrillary tangles (NFT). Senile plaques are extracellular deposits composed primarily of amyloid-beta (Aβ), the product of the proteolysis of the C-terminus of the membrane protein, amyloid precursor protein (APP), by beta and gamma secretase enzymes. NFT are intracellular inclusion bodies composed primarily of paired helical filaments of the microtubule-associated protein (MAP) tau which has been “hyperphosphorylated” at multiple residues not observed under normal physiological conditions (Trojanowski and others, 1995). Given the presence of tau aggregates, AD is classified as a member of the tauopathies, a broad family of neurodegenerative diseases including frontotemporal dementia, Pick’s disease, chronic traumatic encephalopathy and, more recently, Parkinson’s disease and Huntington’s disease. Dementia is a feature of all tauopathies. Therefore, it is likely that tau plays a critical role in causing dementia.
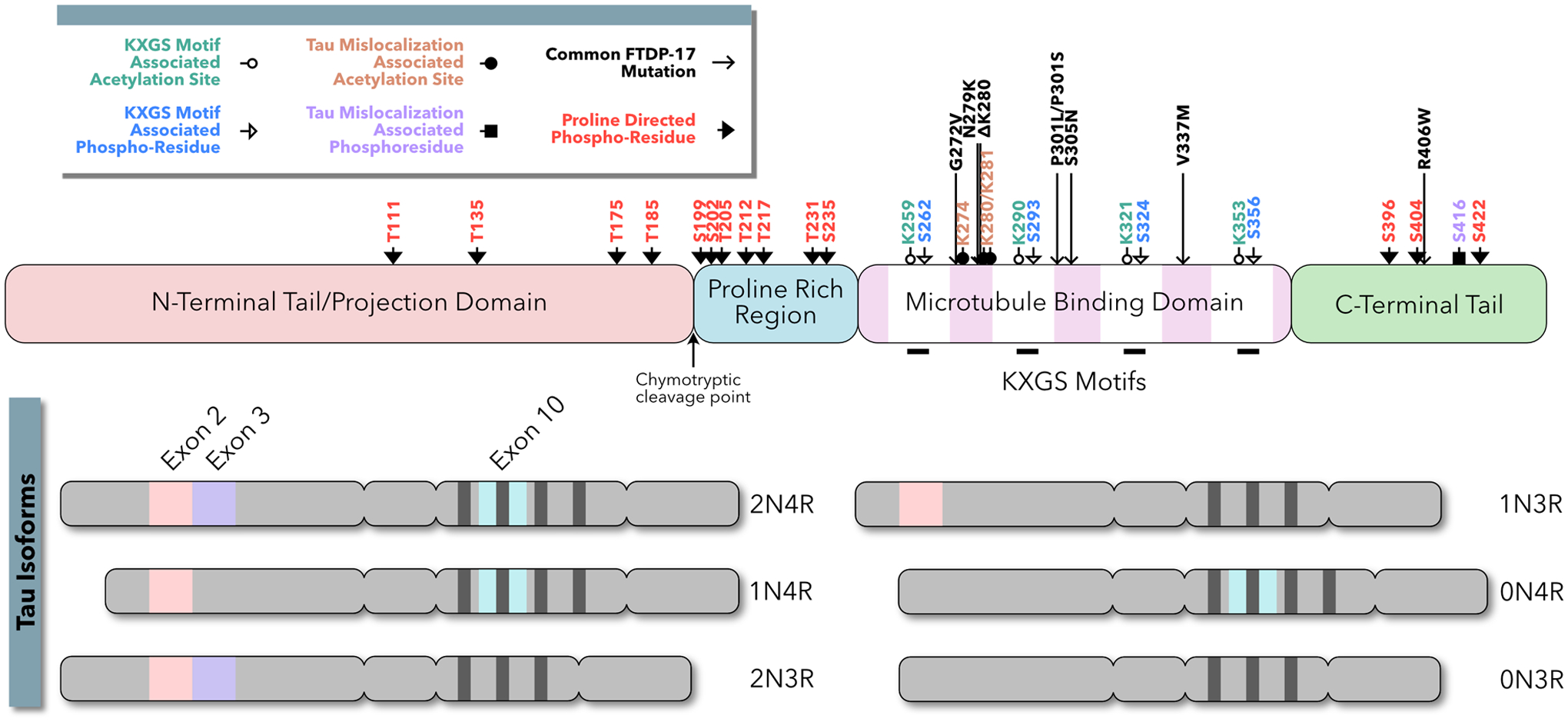
Tau proteins were initially isolated in association with tubulin, purified from porcine brains, and found in microtubule assemblies (Weingarten and others, 1975; Cleveland and others, 1977). Tau binds microtubules through a series of weak interacting sites within the microtubule binding repeat domain; its microtubule interactions are modulated both positively and negatively by amino acid sequences in the adjacent proline rich region (PRR) (Box 1) (Butner and Kischner, 1991; Goode and others, 1997). Tau is responsible, in part, for microtubule stabilization and spacing (reviewed in Barbier and others, 2019; Méphon-Gaspard and others, 2016). Tau consists of six isoforms due to alternative RNA slicing (Box 1). Three isoforms contain four microtubule binding repeats (4R), and three isoforms contain three microtubule binding repeats (3R). As illustrated in Box 1, the primary tauopathy, frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17), is caused by genetic mutations in tau itself. Among secondary tauopathies, including Alzheimer’s disease (AD), Parkinson’s disease dementia (PDD), Lewy body dementia (LBD) and Huntington’s disease (HD), upstream triggers induce posttranslational modifications of tau proteins, such as hyperphosphorylation, acetylation, and truncation (Grundke-Iqbal and others, 1986; Galloway and others, 1989; Vermersch and others, 1993; Spillantini and others, 2000; Kotzbauer and others, 2004; Cohen and others, 2011; Fernández-Nogales and others, 2014; Irwin and others 2017; Liu and others, 2019; reviewed in Tan and others, 2019). At autopsy, the brains of patients with secondary tauopathies often exhibit characteristic pathologies, such as Aβ-containing plaques, tau-containing NFT, α-synuclein-containing Lewy bodies and TAR DNA binding protein 43 (TDP43) inclusions, which appear to interact with each other at the cellular level (reviewed in Spires-Jones and others, 2017). Over the past decade, there has been growing evidence that a myriad of distinct pathological processes converges upon a common signaling pathway, in which the redistribution and mislocalization of tau leads to neurological dysfunction.
The MAPT gene is located on the long arm of chromosome 17 and contains sixteen exons. Exons 0, 1, and 16 are untranslated and three exons (exon 2, 3, and 10) are alternatively spiced to generate mRNA coding for six tau isoforms. The remaining exons are constitutive. The full-length tau isoform contains two N-terminal domains (2N) coded by exons two and three, and four homologous and well-conserved repeat domains (4R). The three tau isoforms lacking exon 10 (2N3R, 1N3R, and 0N3R) are predominant in human fetal development. In adult neurons, all six tau isoforms are expressed including isoforms 2N4R, 1N4R, and 0N4R, which are absent in fetal neurons.
Tau contains little secondary structure and is considered a “natively unfolded” protein. Early protease cleavage studies of tau revealed two functional domains separated by the chymotrypsin cleavage point (Y197–S198): An N-terminal projection domain, which interacts with cytosolic and membrane associated proteins, and a C-terminal assembly domain, which interacts with microtubules. Alternatively tau may be divided into four domains based on its primary structure. First, the acidic N-terminal tail “projects” away from the microtubule and has been implicated in binding membrane associated proteins including apolipoprotein 1A and synaptophysin (a presynaptic protein implicated in presynaptic vesicle release) as well as facilitating a signaling cascade that inhibits microtubule transport. Second, tau contains a proline rich region (PRR), which contains a plurality of putative tau phosphorylation sites. Third, the microtubule binding domain, which encompasses the four repeat domains, associates with microtubules via KGXS sequences contained in each repeat. Finally, the C-terminal tail has been implicated in both microtubule binding and signaling.
The C-terminal and N-terminal tails of tau can curl back upon the repeat domains to form “closed” and “paperclip” structural configurations, potentially modulating microtubule spacing and microtubule binding.
Tau has been shown to be subject to many post-translational modifications including phosphorylation, truncation, acetylation, N-glycocylation, nitrogenation, and ubiquitination The above figure illustrates the phosphorylation sites and acetylation sites associated with tau missorting to dendrites and missorting to dendritic spines.
For a complete review of the structure and function of tau, see Guo and others, 2017.
In this review, we summarize current literature supporting tau redistribution and mislocalization as a common signaling pathway that mediates synaptic and cognitive deficits in neurodegenerative diseases. In disease models and human tissue, the normal gradient of tau is inverted, such that tau becomes “redistributed” to the somatodendritic compartment, where it is scarcely present under healthy conditions. Subsequent modifications of tau cause it to “mislocalize” and accumulate in dendritic spines. Within dendritic spines, tau mediates long-term depression (LTD)-like disruptions to AMPA receptor trafficking, resulting in functional, electrophysiological deficits.
Axonal Localization of Tau in Healthy Neurons
The polarity of tau distribution:
To understand the process of tau redistribution to the somatodendritic compartment in disease, it is helpful first to understand how healthy neurons maintain a gradient of tau. In mature cultured neurons, tau is found along a gradient in which the highest concentrations are in the distal segment of the axon, lower concentrations are found in the proximal segment, and the lowest levels in the soma and dendrites (Black and others, 1996; Mandell and Banker, 1996). While most studies of tau localization have been conducted in cultured neurons, this phenomenon has been confirmed in vivo. There is approximately three times more tau in white matter than gray matter, in rat and bovine brains (Binder and others, 1985). This gradient is fully established concurrent with the elaboration of mature pre- and postsynaptic features (Kosik and Finch, 1987). Even in earlier stages of neuronal development, tau immunostaining is commonly used to indicate axon differentiation (Oliva and others, 2006). Indeed, tau is essential to the stabilization of the growth cone (Black and others, 1996), and isoform-specific tau polarization may be essential for the development of mature postsynaptic structures (Zempel and others, 2017). However, external factors and differential neurite outgrowth are likely upstream of tau polarization (Yamamoto and others, 2012; reviewed in Yogev and Shen, 2017) and, aside from very minor structural and functional abnormalities, tau is not necessary for healthy neuronal development (reviewed in Ke and others, 2012).
Mechanisms underlying tau polarity:
The cellular mechanisms responsible for the axonal tau gradient are hotly debated; current evidence points to multiple overlapping mechanisms (Figure 1). First, in transgenic mouse models, exogenous human tau moves by slow (0.2–0.4 mm/day), anterograde axonal transport (Zhang and others, 2004). This process is facilitated by tau phosphorylation by GSK3β (an axonal kinase) (Cuchillo-Ibanez and others, 2008). Tau transport may result from “piggybacking” on short microtubules in cultured cortical neurons, its rate of transport is retarded by the “stutter-step” motion of this mechanism (Konzack and others, 2007; reviewed in Scholz and Mandelkow, 2014). Tau has also been shown to interact with kinesin short chains (Utton and others, 2005).
Figure 1:
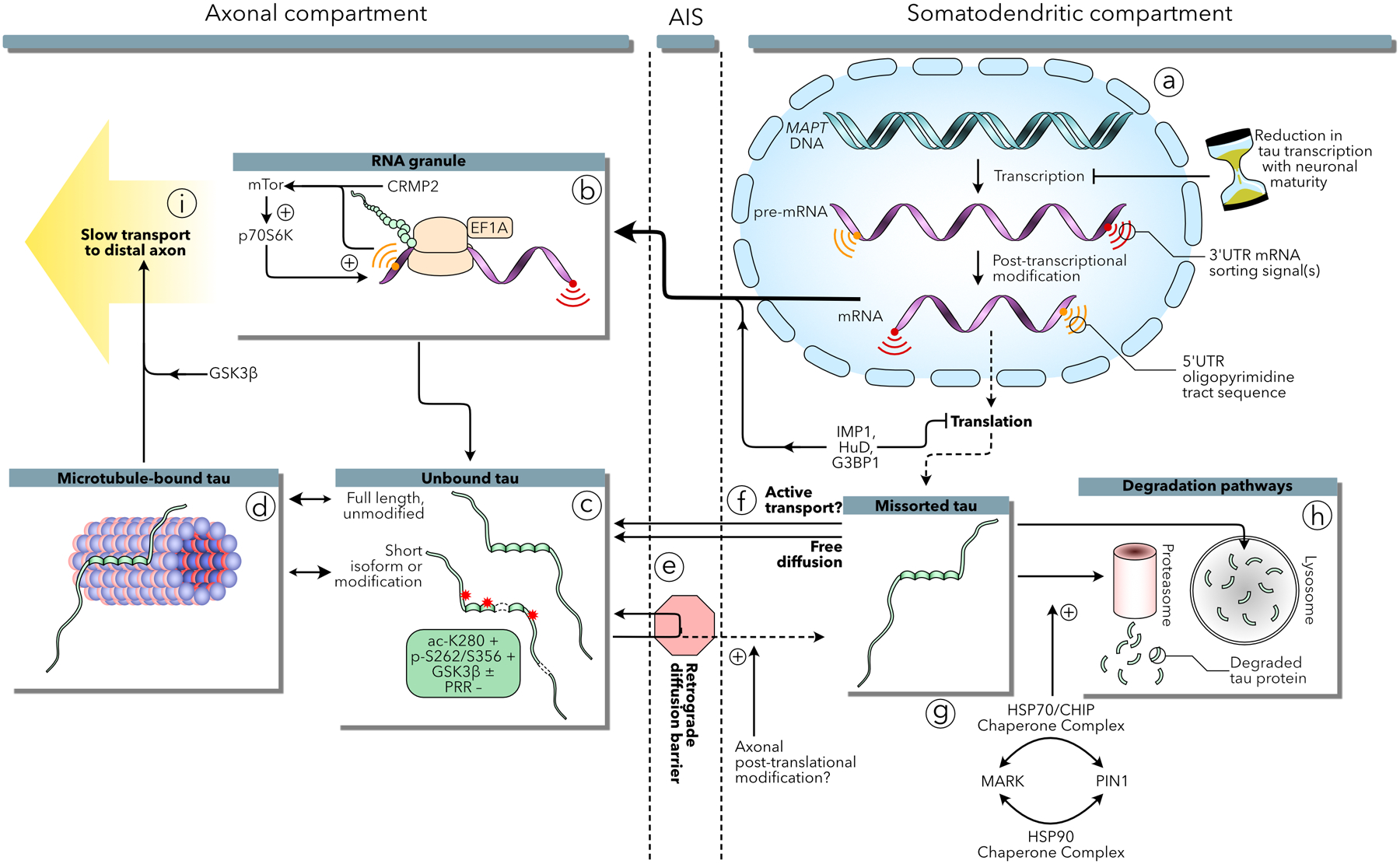
Mechanisms underlying the axonal localization of tau in healthy neurons. (a) The MAPT gene codes for 5’ and 3’UTR signals critical to tau mRNA sorting and selective translation in the axon. After a short burst of expression early in development, tau mRNA transcription is significantly reduced with neuronal maturation by an unknown antenatal mechanism. Tau mRNA may be preserved by the contribution of IMP1, HuD, and G3BP1 during transit to the axon and those proteins may contribute to suppressing expression in the somatodendritic compartment. (b) Tau mRNA undergoes active translation in RNA granules in the axon. The 5’UTR oligopyramidal tract signals of tau and CRMP2 mRNA encourage selective translation via an mTor-p70S6K mechanism. (c) Once translated, tau protein can bind axonal microtubules (d), tau isoform and posttranslational modifications have been shown to have differential effects on microtubule binding that may contribute to efflux from the axon through a retrograde diffusion barrier associated with the axon initial segment (AIS) (e). Tau translated in the somatodendritc compartment can freely diffuse into the axon, or may be involved in an unknown active transport mechanism (f). Alternatively, somatodenritic tau is degraded by proteasome and lysosome proteolytic mechanisms, potentially regulated by the interplay of MARK and Pin1 within a chaperone protein refolding pathway (g–h). Finally, tau may progress into the distal axon via slow transport on microtubule segments or by kinesin motor transport, regulated by GSK3β (i).
Second, tau is selectively degraded in the somatodendritic compartment by autophagy and proteasomes. Pharmacologically inhibiting proteasomes or autophagy leads to a buildup in dendrites of locally synthesized endogenous tau, which is phosphorylated at the Ser262/Ser356 (Balaji and others, 2018) by MARK (Nishimura and others, 2004). Phosphorylation of Ser262/Ser356 prevents tau from being polyubiquitinated and degraded, resulting in its association with chaperone complexes (composed of p23, HOP, HSF1, CHIP, HSP90, HSP70 and PIN1) that refold tau (Dickey and others, 2007). These data suggest that dendritic tau potentially accumulates, in part, due to altered folding and resulted failure of subsequent degradation pathways. Alternatively, we hypothesize that anomalies in the degradation pathway may lead to the retention of a subpopulation of tau via the actions of MARK and chaperone complexes.
Third, tau is physically sequestered in axons in two ways: selective binding to axonal microtubules, and a retrograde diffusion barrier. Biochemical in vitro assays reveal that tau binds more tightly to microtubules prepared from axons than dendrites, potentially due to selective phosphorylation of dendritic tau (Kanai and Hirokawa, 1995). In culture, neurons microinjected with biotinylated tau and imaged by electron microscopy show tau associating with microtubules in axons and cytosolic elements in dendrites (Hirokawa and others, 1996). Additionally, photoconversion studies of cultured neurons demonstrate that exogenous tau is prevented from diffusing from the axon into the soma, whereas somatic tau can freely diffuse into the axon (Li and others, 2011). This “tau diffusion barrier” appears to colocalize with the cellular machinery of the axon initial segment (AIS), and knocking down individual AIS constituent proteins increases retrograde tau diffusion (Zempel and others, 2017).
Active transport, dendritic degradation, and sequestration do not appear to be the sole mechanisms responsible for creating the tau gradient, however. A fourth mechanism involves the distribution of tau mRNA. Expressing exogenous tau cDNA lacking native introns and untranslated regions (UTR) late in neuronal development leads to homogeneous distribution throughout cultured neurons (Hoover and others, 2010; Kanai and Hirokawa, 1995). This effect is not a result of hyperexpression overwhelming tau sorting mechanisms: even extremely low levels of expression (requiring the amplification of immunofluorescence signals to observe) results in the presence of tau in the somatodendritic compartment (Zempel and others, 2017).
Interestingly, endogenous tau retains its axonal gradient in the presence of exogenously expressed tau in the mouse brain (Kubo and others, 2019). In vitro evidence suggests that tau mRNA sorting may contribute to this observation. Early in development, endogenous tau mRNA is localized to the axon hillock (Litman and others, 1993). In the distal axons of cortical neurons, tau mRNA colocalizes within RNA granules with elongation factor 1A, suggesting tau is actively translated in distal axons (Malmquist and others, 2014). This tau mRNA localization mechanism is mediated by an AU-rich cis-acting 3’UTR signal (Aronov and others, 2001; Behar and others, 1995). In addition, an oligopyrimidine tract sequence in the terminal 5’UTR of tau mRNA selectively activates the mTOR-p70S6K pathway, which induces local upregulation of mRNA translation in the axon (Morita and Sobue, 2009). 5’ oligopyrimidine tract sequences have been shown to induce translational control mechanisms in a diverse array of mRNA molecules (Levy and others, 1991). This effect is bolstered by a similar 5’UTR sequence found in CRMP2, another microtubule associated protein localized in the axon (Morita and Sobue, 2009). Finally, the 3’UTR of tau mRNA interacts with polysomal mRNA proteins including IMP1, HuD, and G3BP1, which inhibit translation of tau mRNA in HEK293 cells (Atlas and others, 2007). Thus, inhibiting tau expression in select compartments and facilitating the transport of quiescent tau mRNA to the axon may also help maintain a spatial gradient of tau.
Despite these interesting findings, new research utilizing both cultured neurons and transgenic and knock-in mouse models challenges the role of mRNA sorting in creating the tau gradient. When human tau cDNA sequences lacking 5’ and 3’UTR, and driven by the endogenous mouse tau promotor, were expressed in knock-in mouse models, the exogenous tau was preferentially expressed in axons (Kubo and others, 2019). This effect was accompanied by a dramatic reduction, during the first two postnatal weeks, in both endogenous and knock-in tau expression (Kubo and others, 2019). When exogenous human tau cDNA is transiently expressed using a tetracycline transactivator, rather than constitutively expressed as in the earlier experiments described above, the exogenously expressed tau localized to the axon in day-one cultured neurons, a process that depended on the C-terminal component of the proline-rich region (PRR), and persisted into late neuronal development (Iwata and others, 2019). This PRR-based mechanism operates independently of the retrograde diffusion barrier, mRNA signaling, or microtubule binding and suggests the presence of an active mechanism that biases the transport of exogenous cDNA towards the axon (Iwata and others, 2019).
To reconcile several of the seemingly contradictory results presented above, we propose a hypothetical temporal model (Figure 2). Early in development, tau is highly expressed and homogeneously distributed throughout the nascent neuron. As one neurite begins to take on axonal properties, an active protein transport mechanism emerges and sorts the available, previously expressed tau protein into that neurite. As the axon reaches maturity, this active transport mechanism is attenuated, and the newly formed AIS acts as a retrograde diffusion barrier. Concurrently, tau transcription slows dramatically. Due to mRNA sorting and upregulated axonal translation, the synthesis of new tau proteins occurs preferentially in the axon. Gradually, in the somatodendritic compartment, the remaining tau protein along with the small fraction of tau produced there is degraded, but this process can be overwhelmed by exogenous tau expression, even at relatively low levels.
Figure 2:
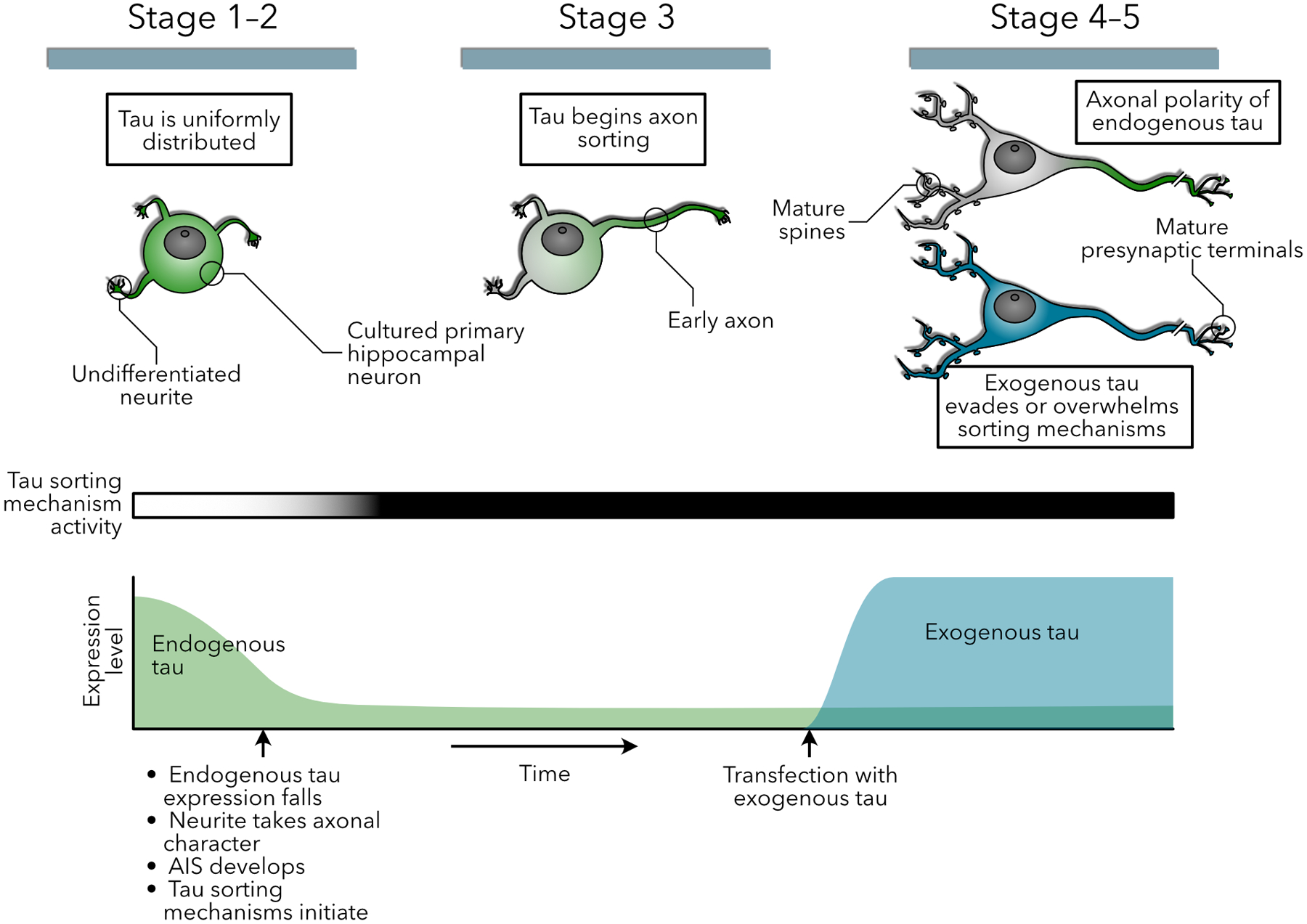
Hypothetical model of the temporal relationship between sorting mechanisms and endogenous and exogenous tau expression. (Top row) Diagrams of endogenous tau (green) and exogenous tau (blue) distribution in a developing cultured hippocampal neuron. While tau is initially uniformly distributed throughout the cellular sphereoid and early, undifferentiated neuronal processes, sorting begins once one of the processes takes on axonal character, and is complete with the elaboration of mature postsynaptic morphology. This is dependent on two mechanisms (1) a drop in mRNA levels and new tau expression shortly after birth, and (2) the initiation of tau sorting mechanisms upon development of axonal characteristics. However, the high expression levels of exogenous tau overcome these sorting mechanisms, especially in the absence of mRNA sorting signals.
Loss of Axonal Polarity: Tau Redistribution to Somatodendritic Compartments
Loss of tau polarity in tauopathies:
The neuropathological hallmark of tauopathies is the accumulation of insoluble tau aggregates in the cell body and dendrites of diseased neurons (Goedert and others, 1989; Pickering-Brown and others, 2004). In Alzheimer’s disease, tau aggregates also appear in dystrophic neurites in the immediate vicinity of senile plaques (Tourtellotte and Van Hoesen, 1991). These observations form the basis for the idea that the redistribution of tau to the somatodendritic compartment is a key step in the pathogenesis of tauopathies. Early research focused on deciphering mechanistic links between tau aggregation and somatodendritic redistribution, but did not specifically address how this process led to neurological dysfunction. Fifteen years ago, it became apparent that neuronal tau inclusions (i.e., neurofibrillary tangles) could be dissociated from impaired memory function and neuron loss (Santa Cruz and others, 2005). Since then, numerous reports have shown that functional deficits can occur in the absence of tau inclusions (Grueninger and others, 2010; Rocher and others, 2010; Sydow and others, 2011; Xu and others, 2014; Maeda and others, 2016). For example, in culture, Aβ-induced calcium influx and tau redistribution has been associated with spine loss, microtubule destruction, dendrite retraction (Zempel and others, 2010), and neuronal cytotoxicity (Tackenberg and Brandt, 2009), in the absence of tau aggregates. The pathways leading to these pathologies are diverse and only now becoming understood; they appear to depend on the presence of tau in dendrites (Figure 3).
Figure 3:
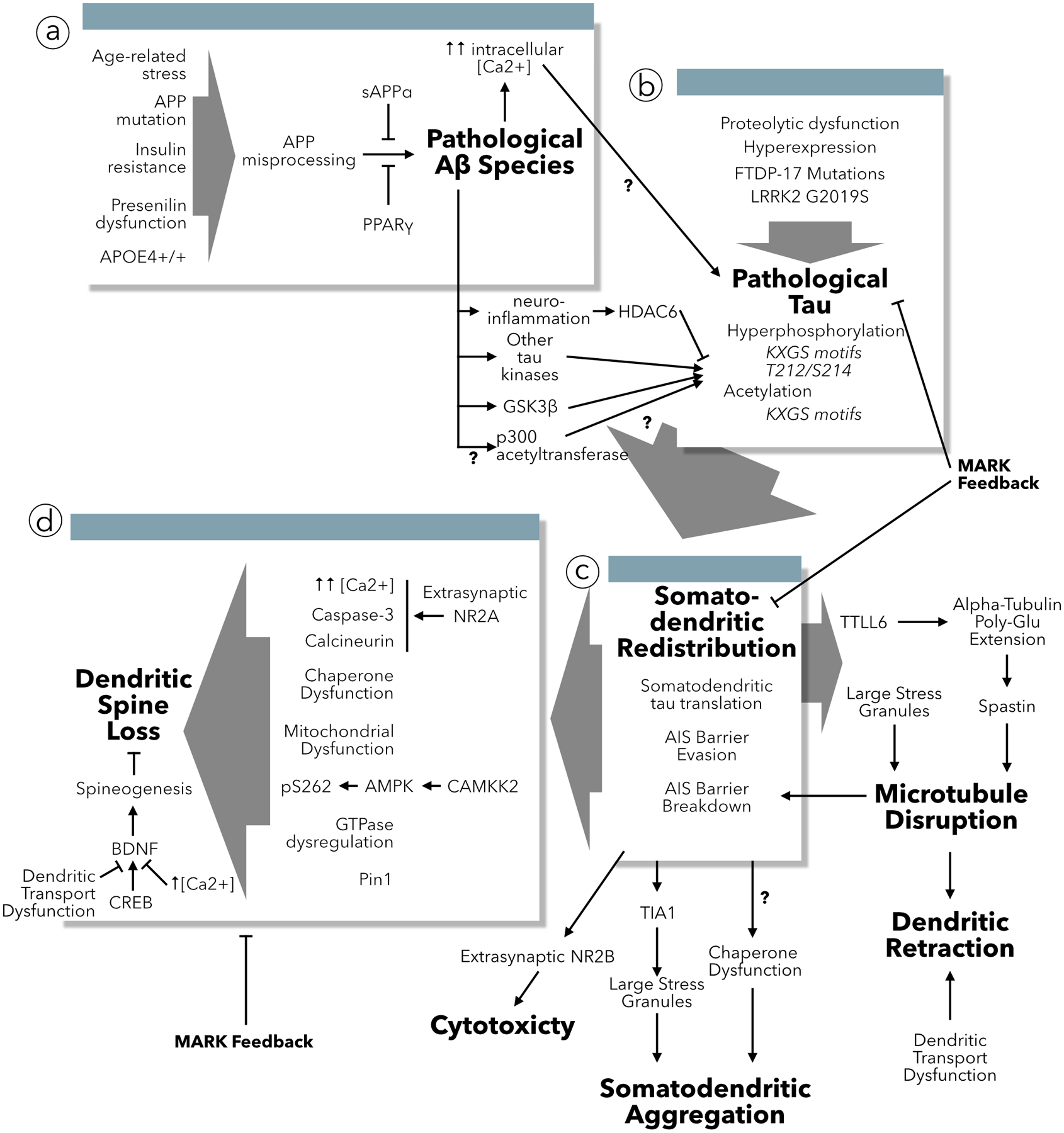
Causes and consequences of tau redistribution to dendrites. (a) Multiple triggers encourage the misprocessing of APP leading to the accumulation of pathogenic Aβ species. Aβ increases intracellular calcium concentration and acts through multiple mediators to change the post-translational modification state of tau. (b) Aside from Aβ, multiple mechanisms can induce changes in tau that lead missorting to dendrites. (c) Once tau has been redistributed into dendrites it effectuates multiple pathologies including disrupting microtubules leading to dendritic retraction and transport dysfunction, undergoing misfolding and aggregation, and mediating cytotoxicity. (d) The primary result of tau redistribution to dendrites is dendritic spine loss, which has been associated with multiple protein signaling cascades as well as potential disruption of BDNF spine stabilization and spinogenesis. This may or may not be associated with tau-induced dendritic microtubule transport dysfunction.
Conversely, tau inclusions do not cause functional deficits or neuron death (Santa Cruz and others, 2005; Calignon and others, 2010; Rocher and others, 2010; Kuchibholtla and others, 2014); and, in multiple models of tauopathic dementia, cell death and functional deficits occur prior to the detection of tau inclusions, or in the absence of tau inclusions entirely (Gueninger and others, 2010; Hampton and others, 2010; Xu and others, 2014; Maeda and others, 2016). Indeed, increasing tau aggregation propensity protects neurons from FTDP-17 mutation-induced cytotoxicity (d’Orange and others, 2018). Further, in postmortem AD brain tissue, while tau inclusion density and extent are closely correlated with disease duration and progression (Fukutani and others, 2000; Theofilas and others, 2018), neuron death cannot be explained by tau inclusion burden alone (Gómez-Isla and others, 1997). Finally, several FTDP-17 tau mutations cause dementia in the absence of appreciable tau inclusion pathology in human tissue (Reed and others, 2001). Current evidence supports the ability of soluble forms of tau to mediate the pathological effects of redistributed tau present in the somatodendritic compartment.
Breakdown of the AIS diffusion barrier leading to the redistribution of tau:
Imaging studies of cultured neurons demonstrate that the AIS plays a critical role in maintaining normal tau polarity by serving as a physical barrier that prevents the retrograde diffusion of tau from the axons to the soma and dendrites (Li and others, 2011; Zempel and others, 2017). Moreover, the AIS may actively transport newly synthesized tau into the axon (Zempel and others, 2013). The AIS barrier depends upon microtubule stability (Li and others, 2011). Tau acetylation destabilizes the cytoskeletal components of the AIS, weakening the barrier, and allowing retrograde redistribution of tau into the soma and dendrites (Sohn and others, 2016). Inhibition of HDAC6 increases the acetylation of tau at Lys280 leading to tau redistribution into somatodendritic structures by weakening the AIS barrier (Tseng and others, 2017; Zempel and others, 2017). Tau phosphorylation, induced by the Pro301→Leu301 mutation linked to FTDP-17, also weakens the AIS barrier by structurally modifying the AIS and shifting its location (Hatch and others, 2017).
Pathological effects on proteins other than tau also erode the integrity of the AIS diffusion barrier. Aβ activates GSK3β, which in turn phosphorylates a non-tau target, destabilizing microtubules and breaking down the AIS diffusion barrier (Zempel and others 2017). Aβ-induced decreases in F-actin expression in the AIS also contribute to the disintegration of the barrier (Zempel and others 2017). Interestingly, the application of Aβ activates the RhoA-mDia1 pathway and decreases HDAC6 activity, which destabilizes microtubules by increasing their tyrosination but decreasing their acetylation (Tsushima and others, 2015; Pianu and others, 2014). Additionally, Aβ-induced integrin signaling and RhoA activation may cooperate to create a positive feedback loop resulting in focal adhesion kinase-dependent cytotoxicity and microtubule glutamylation (Xu and others, 2009; Pianu and others, 2014).
Other factors leading to tau redistribution:
The FTDP-17 Arg406→Trp406 mutation interferes with the slow transport of tau along the axon, which may contribute to a buildup of tau in the somatodendritic compartment (Zhang and others, 2004). Additionally, inhibition of autophagy and proteasome function both induce local tau redistribution to dendrites (Balaji and others, 2018). Other cellular stressors including hydrogen peroxide, serum depravation, glutamate, ATP and glucocorticoids can induce tau to redistribute to the dendritic shaft (Pinheiro and others, 2016; Zempel and others, 2010). The Glu2019→Ser2019 LRKK2 mutation linked to PD induces tau redistribution and GSK3β-mediated dendritic retraction in dopaminergic neurons (Lin and others, 2010). A pathogenic strain of α-synuclein oligomers, correlated with idiopathic PD pathology, induced tau pathology and the redistribution of tau to the somatodendritic compartment (Guo and others, 2013). Tau accumulation in dendrites has also been observed in studies of glaucoma (Chiasseu and others, 2016), suggesting a role for this mechanism in non-dementia neuronal dysfunction, too.
Dendritic tau wreaks havoc:
Microtubule disassembly, simplification of the dendritic tree, retraction of dendritic arbors, and loss of dendritic spines are some of the pathological changes that result from the abnormal accumulation of tau in dendrites. Dendritic tau recruits Tubulin-Tyrosine-Ligase-Like-6 (TTLL6), which polyglutamylates α-tubulin sidechains leading to spastin-induced microtubule disassembly (Zempel and others, 2013), which in turn leads to the simplification and retraction of dendritic arbors (Lin and others, 2010). Dendritic tau also interacts with T-cell intracellular antigen 1 (TIA1) to form large RNA stress granules, which are associated with the retraction of dendrites (Vanderweyde and others, 2016).
Dendritic tau induces the loss of dendritic spines through several mechanisms. First, phosphorylation of serine in KXGS motifs in the microtubule binding domain (detected using an antibody that recognizes pSer262/pSer356) is a regular feature of spine loss associated with dendritic tau, observed in three different experimental models—exogenous tau expression in mature neurons, extracellular application of Aβ, and inhibition of proteasome and autophagic proteolytic pathways (Balaji and others, 2018; Zempel and others, 2013; Thies and Madelkow, 2007). The enzyme that phosphorylates KXGS motifs is MARK (Mandelkow et al 2004). Surprisingly, overexpressing MARK rescues dendritic spines (Zempel and others, 2013; Thies and Madelkow, 2007), and reducing MARK increases Aβ-induced, tau-mediated changes in synaptic morphology (Lee and others, 2012), presumably because phosphorylation of KXGS motifs reduces tau binding to microtubules, thereby removing its interference with microtubule motors. Thus, dysregulation of microtubule dynamics by dendritic tau, although not necessarily by hyperphosphorylated tau, leads to spine loss.
In addition to the MARK pathway, Aβ application and AD-associated mutant APP expression also lead to the activation of a CAMKK2-AMPK pathway, which phosphorylates tau at Ser262 and causes spine loss (Mairet-Coello and others, 2013). Moreover, a decrease in the isomerase activity of Pin1 may also mediate Aβ-induced loss of dendritic spines (Stallings and others, 2018). Given the importance of MARK, Pin1, and phosphorylation at the Ser262/Ser356 epitope; it is likely that HSP90 and HSP70 chaperone complexes (discussed above) are involved modulating the synaptotoxicity of dendritic tau, perhaps allowing aberrant dendritic tau to escape selective proteasome degradation (Dickey and others, 2007).
Second, deprivation of energy and growth factors is a toxic consequence of dysregulated microtubule dynamics. Dendritic tau is associated with reduced concentrations of mitochondria along dendritic shafts (Zempel and others, 2010), which is closely linked to spine loss in mice (Kandimalla and others, 2018). GSK3β expression mimics Aβ-induced spine loss via activation of CREB nuclear signaling and suppression of BDNF (DaRocha-Souto and others, 2012), a growth factor necessary for spine maturation (Kellner and others, 2014).
Finally, small GTPases, including Rho, Cdc42 and Rac1, are key regulators of morphogenesis of dendritic spines (Luo and others, 1996; Nakayama and others, 2000; Wiens and others, 2005). The three GTPases interact with each other and the maintenance of normal actin cytoskeleton of cells requires the balanced regulation of the GTPases (Luo 2000 for review). Therefore, unsurprisingly, GTPases are necessary actors in the Aβ/tau-induced spine loss pathway (Borin and others, 2018). Treatment with Aβ40 increases RhoA activity but decreases Rac1 activity (Petratos et al., 2008). At 6 months-of-age, the GTP activity level of Rac1 is significantly decreased in the brains of mice that expressed the APP Swedish (Lys670→Met670, Asn671→Leu671) mutation (Petratos and others, 2008). However, in another study, Rac1 activity is aberrantly elevated in the hippocampal tissues of AD patients and AD animal models; and pharmacological inhibition of Rac1 activity ameliorated cognitive defects in APP/PS1 mice (Wu et al., 2019). All these results together suggest that the disruption of balanced regulation of small GTPases contributes to spine loss in the pathogenesis of AD and other tauopathies.
Beyond Tau Redistribution: Tau Mislocalization to Dendritic Spines
As discussed above, under pathological conditions tau redistributes to the somatodendritic compartment, thereby providing the spatial proximity necessary for tau to directly modulate dendritic spines. Dendritic spines are postsynaptic structures that mediate excitatory synaptic transmission (Harris and Kater, 1994; Kennedy 2000; Hering and Sheng, 2001). They undergo structural and functional changes underlying some forms of synaptic plasticity, such as long-term potentiation (LTP) and long-term depression (LTD), which play key roles in the most widely accepted model of learning and memory (Bliss and Collingridge, 1993; Malinow and others, 2000; Martin and others, 2000; Malinow and Malenka 2002). Therefore, it was reasonable to examine the effects of tau on dendritic spines in tauopathies. Ten years ago, two independent groups showed that the redistribution of soluble tau to the somatodendritic compartment was not sufficient to disrupt synaptic function and impair memory; tau must also accumulate in dendritic spines (Hoover and others, 2010; Ittner and others, 2010).
Early studies of tau mislocalization to dendritic spines were conducted using mouse and cellular models of FTDP-17 and AD (reviewed in Liao and others, 2014). More recent studies implicate this pathogenic process in two other neurodegenerative conditions: synucleinopathy (Teravskis and others, 2018; Singh and others, 2019; Smith and others, 2019) and Huntington’s disease (Liu and others, 2019) (Figure 4). It will be important to determine whether this pathological mechanism occurs in other forms of dementia, such as those related to traumatic brain injury, vascular, and TDP-43 lesions. If so, then identifying and targeting specific molecules in this signaling pathway could prove beneficial in treating memory loss and dementia in a wide variety of neurodegenerative conditions. Several key signaling molecules have been identified, and are discussed in the following sections.
Figure 4:
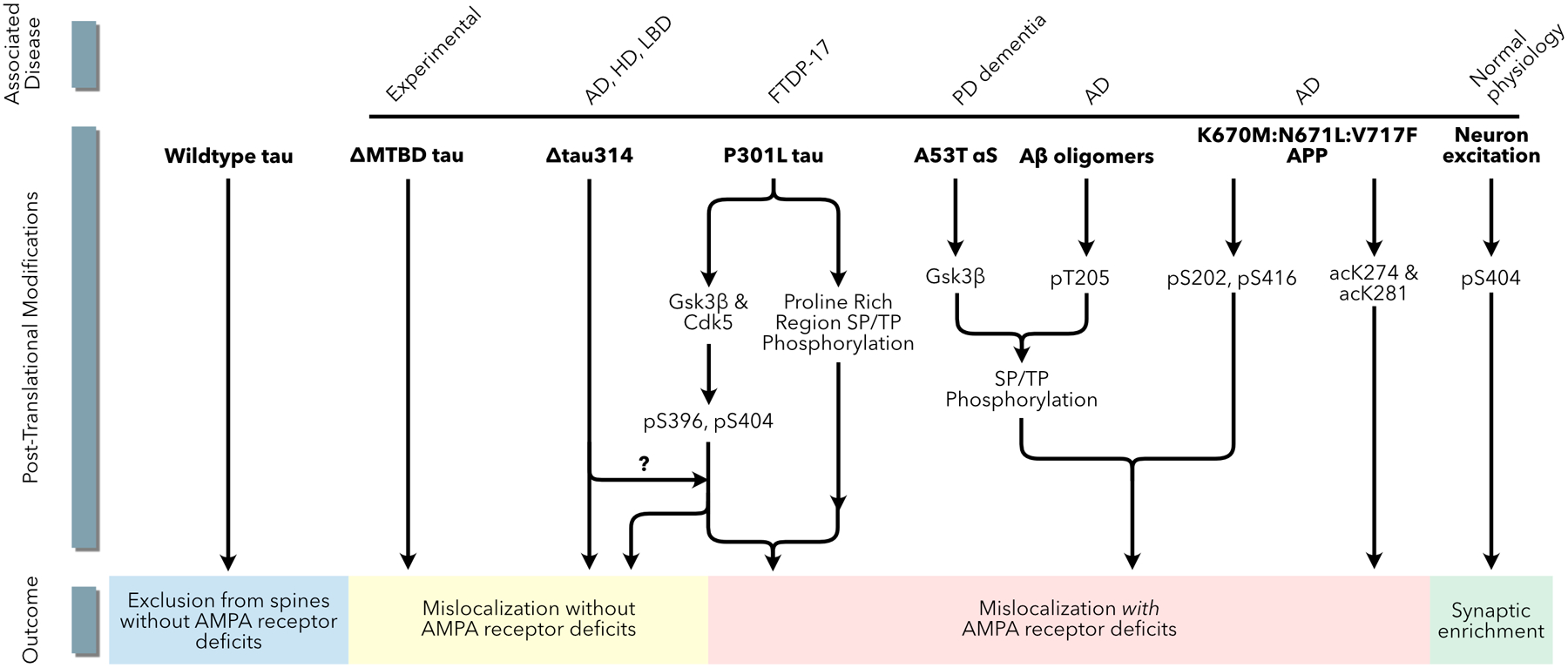
Multiple pathways in multiple disease models lead to tau mislocalization to dendritic spines. Models of AD, HD, PDD, FTDP-17 and LBD all implicate tau mislocalization to dendritic spines via phosphorylation or acetylation posttranslational modifications. The phosphorylation pattern of tau leading to mislocalization may be different in each condition.
FTDP-17:
Using mouse and cellular models of FTDP-17, several steps in the process by which the mislocalization of tau to dendritic spines leads to synaptic dysfunction have been delineated (Figure 5). The FTDP-17-linked tau mutation P301L increases the amount of tau proteins in dendritic spines in cultured rat hippocampal neurons and postsynaptic density (PSD) fractions from mouse brain (Hoover and others, 2010). Under conditions in which tau is expressed at low levels (e.g., in cells transfected using calcium-phosphate), mutant tau mislocalizes to dendritic spines, but wild-type tau generally remains in the dendritic shaft. The mislocalization of tau to dendritic spines depends on its phosphorylation by proline-directed kinases (Hoover and others, 2010; Xia and others, 2015). Specifically, phosphorylation at Ser396 and Ser404 in the C-terminal domain of tau by CDK5 or GSK3β suffices to induce mislocalization to dendritic spines (Teravskis others, 2019).
Figure 5:
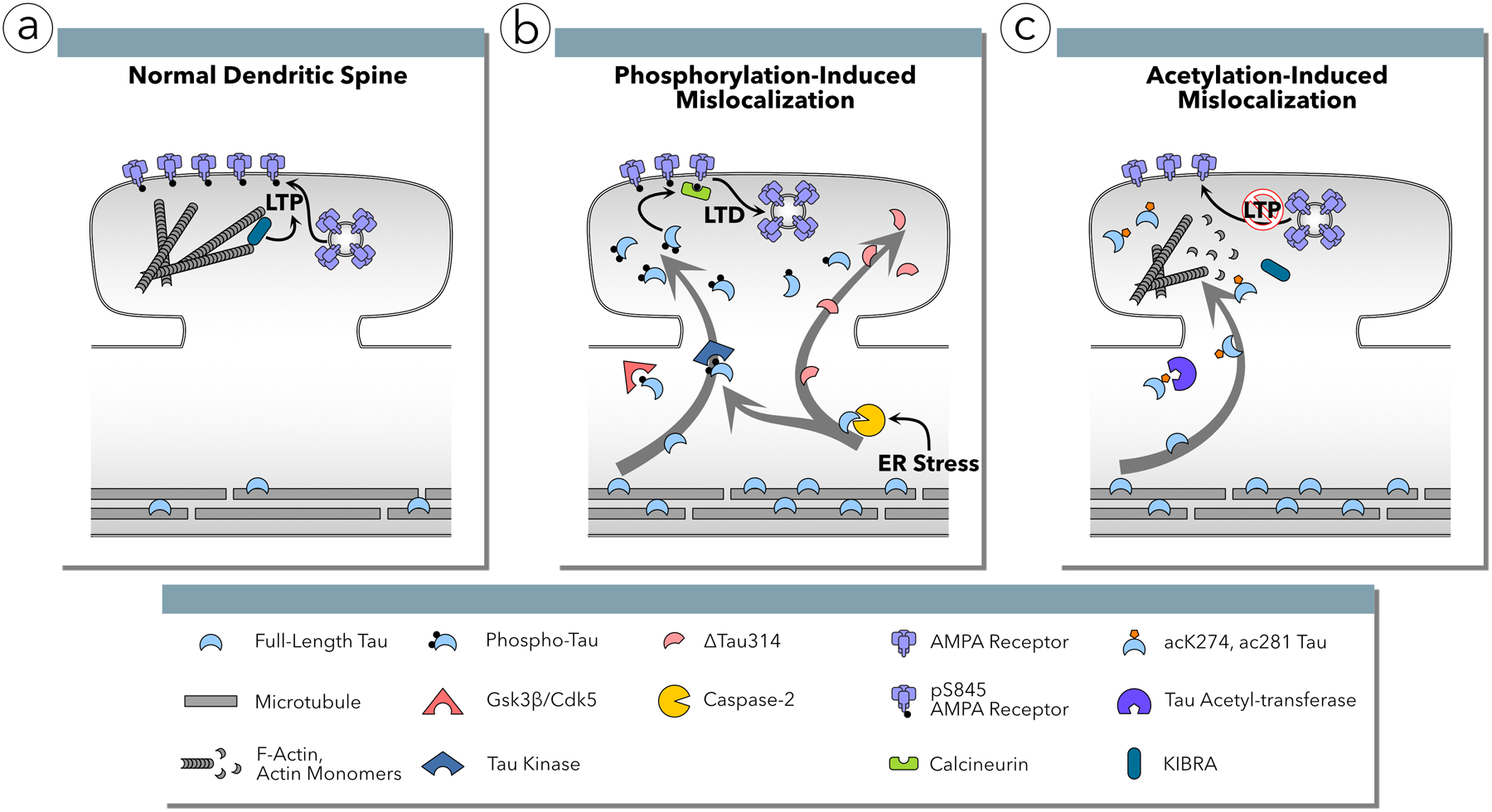
Tau mislocalization to dendritic spines and AMPA receptor functional deficits are mediated by sequential phosphorylation in two domains and acetylation posttranslational modifications. (a) Under normal conditions, tau is excluded from the dendritic spines. The AMPA receptor subunit GluA1 is phosphorylated at Ser845 and the population of AMPA receptors in the postsynaptic membrane is stable. F-actin polymerization supports KIBRA-mediated, activity-induced insertion of AMPA receptors into the postsynaptic membrane, promoting memory formation via long-term potentiation (LTP). (b) In FTDP-17, AD, LBD, PDD and HD, tau is phosphorylated by CDK5 or GSK3β in the C-terminal domain at Ser396 and Ser404 residues resulting in mislocalization to dendritic spines. Mislocalization alone is insufficient to result in functional deficits; separate phosphorylation in the proline rich region (PRR) activates a signaling cascade in which calcineurin dephosphorylates GluA1 at Ser845 leading to AMPA receptor internalization and deficits in excitatory synaptic transmission resembling long-term depression (LTD). This pathway requires the activity of caspase-2 resulting the abnormal cleavage of tau at Asp314 creating Δtau314, which mislocalizes to dendritic spines but cannot induce synaptic deficits without some other cellular stressor. (c) In human AD, tau is abnormally acetylated at residues Lys274 and Lsy281 by an unknown acetyltransferase. Acetylation at these sites results in mislocalization to dendritic spines, actin depolymerization, and reduced KIBRA activity resulting in decreased AMPA receptor insertion into the postsynaptic membrane during LTP induction.
Cleavage at Asp314 (D314) to generate Δtau314 is another necessary step for tau to mislocalize to dendritic spines (Zhao others, 2016). The enzyme that cleaves tau at D314 is caspase-2; mutant tau P301L does not mislocalize to spines in neurons cultured from caspase-2 knock-out mice (Zhao and others, 2016). The upstream activators of caspase-2 are unknown, but may involve endoplasmic reticulum stress (Upton and others, 2008; Bronner and others, 2015). While the activation of caspase-2, GSK3β and CDK5 is required for tau mislocalization to dendritic spines (Hoover and others, 2010; Zhao and others, 2016), they also affect non-tau targets (Xu and others, 2019; Zempel and others 2017). How these targets interact with tau during pathological progression of neurodegenerative diseases remains an open question.
The accumulation of tau in dendritic spines is associated with deficits in spatial memory tasks, a decrease in the strength of AMPA receptor-mediated synaptic responses, and the loss of surface glutamate receptors on dendritic spines (Hoover and others, 2010). The accumulation of full-length tau or Δtau314 in spines is necessary but insufficient to induce synaptic dysfunction and memory deficits (Teravskis and others, 2019; Zhao and others, 2016; Ittner and others, 2010). Additional post-translational modifications of tau are required. The known modifications include phosphorylation of at least one of five residues (Ser202, Thr205, Thr212, Thr217 and Thr231) in the PRR of the N-terminal domain (Teravskis and others, 2019) and acetylation of Lys274 and Lys281 (Tracy and others, 2016).
The final steps by which the presence of hyperphosphorylated tau in dendritic spines results in AMPA receptor internalization are incompletely established. A known step is the activation of calcineurin, which dephosphorylates Ser845 in the GluA1 receptor subunit; AMPA receptor internalization induced by mutant tau P301L does not occur in the presence of the calcineurin inhibitor FK506 (Miller and others, 2014). Calcineurin plays an important role in the normal neurobiology of LTD; the dephosphorylation of Ser845 in GluA1 receptors promotes LTD (Lee and others, 2010). AMPA receptors are also internalized in models of chronic opioid exposure, a process that likewise involves calcineurin (Kam and others, 2010).
The internalization of GluA1, GluA2/3, and NR1 receptor subunits can occur in the absence of spine loss (Hoover and others, 2010), suggesting that the loss of spines commonly observed in models of tauopathy involves pathways other than calcineurin-mediated GluA1 receptor internalization. What those pathways are remains unclear.
Alzheimer’s disease (AD):
The mislocalization of tau to dendritic spines mediates synaptic dysfunction not only in models of primary tauopathy, expressing mutant tau linked to FTDP-17, but also in models of secondary tauopathy, expressing wild-type tau. The best studied are models of familial and sporadic AD relying on the expression of mutant APP or presenilins or the application of amyloid-beta oligomers. Two complementary assessments of the role of tau in dendritic spines have emerged. In one, tau mediates the over-activation of NMDA receptors; in the other, tau mediates the deactivation of NMDA receptors. The reconciliation of these two divergent signaling pathways in Alzheimer’s disease remains unresolved.
Tau mediates the phosphorylation of the NMDA receptor subunit NR2B at Tyr1472 by fyn in APP transgenic mice (Ittner and others, 2010). In synaptosomes from APP23 transgenic mice, which overexpress APP with the Swedish mutation, levels of fyn and Tyr1472-phosphorylated NR2B are significantly higher than in transgene-negative littermates (Ittner and others, 2010). These increases are associated with cognitive deficits and were absent in tau knock-out mice as well as transgenic mice expressing Δtau74 (truncated at aa255, missing the whole microtubule binding domain and the C-terminal tail) (Ittner and others 2010). Δtau74 binds fyn but cannot enter dendritic spines, thereby sequestering fyn in the dendritic shaft (Ittner and others 2010). Although not shown, the authors assumed that full-length tau enters dendritic spines, bringing fyn along with it. Later, imaging experiments in primary hippocampal cultures prepared from mice expressing APP bearing the Swedish mutation and rat hippocampal neurons exposed to oligomeric Aβ provided direct evidence for tau mislocalization to and accumulation in dendritic spines in AD (Frandemiche and others, 2014; Miller and others, 2014; Amar and others, 2017;). Aβ-induced mislocalization of tau to dendritic spines depends on tau phosphorylation (Miller and others, 2014).
Additional studies of fyn have further clarified its role. Fyn is not required for P301L-induced tau mislocalization to spines (Xia and others, 2015). Rather, super-resolution microscopy reveals that tau P301L immobilizes fyn in nanodomains within dendritic spines (Padmanabhan and others, 2019). Furthermore, Aβ activates NMDA receptors, which in turn results in tau phosphorylation and the formation of fyn-PSD95-tau-NMDA receptor complexes in dendritic spines (Mondragón-Rodríguez and others, 2012).
Like AMPA receptors discussed above, NMDA receptor clustering is significantly decreased in an FTDP-17 P301L mouse model, supporting a loss of NMDA receptor function (Hoover and others, 2010); however, the effects of Aβ on NMDA receptor clustering has not been examined. The discrepancy between the findings of the activation of NMDA receptors (Ittner and others, 2010) on the one hand, and the deactivation of NMDA receptors (Hoover and others, 2010) on the other, may arise from additional roles of Aβ beyond inducing phosphorylation-dependent tau mislocalization to dendrites and dendritic spines. In support of this interpretation, NR2A has been linked to Aβ-induced spine loss, whereas NR2B is implicated in Aβ-induced excitotoxicity (Ittner and others, 2010; Tackenberg and Brandt, 2009; Tackenberg and others, 2013).
Acetylation of tau is required for somatodendritic redistribution in AD models (Sohn and others, 2016). It is likely that truncation by caspase-2 at Asp314 is involved in tau mislocalization to dendritic spines because the levels of Δtau314 in the inferior temporal gyrus are elevated in humans with mild cognitive impairment and AD (Zhao and others, 2016). In addition, J20 APP transgenic mice lacking caspase-2 do not exhibit memory deficits or changes in dendritic spine morphology (Pozueta and others, 2013). However, direct evidence is lacking.
Lewy Body Dementia (LBD):
LBD is a member of the family of α-synucleinopathies, which includes PD, and is the second most common form of dementia behind AD. α-synuclein inclusions characterize LBD and PD neuropathologically. Because α-synuclein is highly enriched within presynaptic terminals, it is not surprising that one pathogenic mechanism involves presynaptic dysfunction (Nemani and others, 2010; Burré 2015; Larson and others, 2017).
In recent years, results in human brain specimens and experimental models implicate a role for tau, in addition to α-synuclein. The severity of tau pathology—based on four-level Braak staging (Braak and Braak, 1996), and continuous global assessment of NFT load—was shown in a retrospective study of 213 patients with synucleinopathy to correlate inversely with the age of onset of both motor and cognitive symptoms and a shorter survival period following the onset of symptoms (Irwin and others, 2017). Levels of Δtau314 and caspase-2 are elevated in the superior temporal gyrus of patients with LBD (Smith and others, 2019), suggesting that tau may mislocalize to dendritic spines, since the generation of Δtau314 is associated with this process (Zhao and others, 2016). However, direct evidence for this pathway is currently lacking.
More recent studies have begun to elucidate the signaling pathway mediating α-synuclein-induced post-synaptic deficits (Teravskis and others 2018; Singh and others 2019). Tau and α-synuclein pathologies frequently coexist in the Contursi kindred who carry the A53T α-synuclein point mutation (Duda and others, 2002). Like P301L-induced tau mislocalization to dendritic spines, the postsynaptic deficits induced by A53T α-synuclein depend on tau phosphorylation and mislocalization to dendritic spines (Teravskis and others 2018). As in APP transgenic mice (Roberson and others, 2007), age-dependent spatial learning and memory deficits in transgenic mice expressing A53T α-synuclein are rescued by the deletion of tau (Singh and others 2019). Taken together, these results together support the hypothesis that tau mislocalization to dendritic spines plays an important role in PD dementia and Lewy body dementia.
For reasons that are unclear, transgenic mice expressing mutant A30P or E46K α-synuclein, which are also linked to PD, do not exhibit tau-dependent postsynaptic changes. Therefore, it will be important to examine models of idiopathic Lewy body diseases to understand the extent to which tau-mediated post-synaptic dysfunction contributes to dementia in these disorders.
Huntington’s disease:
Huntington’s disease is a progressive neurodegenerative disease caused by expanded CAG trinucleotide repeats at the N-terminus of IT15 encoding the huntingtin (HTT) protein (The Huntington’s Disease Collaborative Research Group, 1993). Tau proteins colocalize with HTT proteins, and hyperphosphorylated, aggregated tau proteins detected by the AT8 antibody have been found in the somatodendritic compartments of cortex and striatum of Huntington’s disease patients (Vuono and others 2015). The level of Δtau314 is increased in Huntington’s disease patients (Liu and others, 2019), and genetic ablation of either tau (Fernández-Nogales and others, 2014) or caspase-2 (Carroll and others, 2011), which generates Δtau314 (Zhao and others, 2016), ameliorates cognitive and motor deficits of HD mice. These findings support the hypothesis that the accumulation of tau in dendritic spines contributes to the pathogenesis of HD.
Synaptic plasticity:
Interestingly, following electrical and chemical LTP induction, exogenous tau also mislocalizes to dendritic spines, resulting in changes in synaptic strength (Frandemiche and others, 2014). LTD is associated with increased phosphorylation of Ser396/Ser404, and is impaired in tau knock-out mice (Regan and others 2015). LTD is abrogated in mice lacking caspase-2 (Xu and others, 2019). As phosphorylation of tau at Ser396/Ser404 and caspase-2 cleavage of tau at Asp314 play essential roles in tau mislocalization to dendritic spines (Teravskis and others 2019; Zhao and others, 2016), these results suggest that these cellular pathways also participate in synaptic plasticity under physiological conditions. Tau is likely present in the postsynaptic domains of “healthy” neurons, as it is detected in the gray matter of the brain, albeit at levels three times lower than in white matter (Binder and others, 1985), providing the necessary spatial presence for tau to engage in the aforementioned physiological processes. We speculate that neurodegenerative disease processes may exploit and corrupt normal, physiological processes via tau; tau hyperphosphorylation and cleavage may aberrantly suppress excitatory synaptic transmission by over-activating the signaling cascade that leads to LTD, or alternatively, by disrupting an early phase of the LTP pathway.
The chronology of synaptic dysfunction and neuronal loss:
A quantitative morphometric analysis of electron microscopic images of brain tissues from AD patients, who were younger than 65 years old, demonstrated that synapse loss was more substantial than neuronal loss, suggesting that synaptic damages occur before overt neurodegeneration (Davies and others 1987). In an FTDP-17 mouse model, synaptic dysfunction and tau mislocalization preceded loss of dendritic spines and overt brain atrophy (Hoover et al., 2010). Based upon the above studies and other publications, we propose a hypothetical chronology of tau-mediated synaptic dysfunction, spine loss, and neuronal death (Figure 6). Notably, however, the causal relationship between tau-mediated synaptic dysfunction, spine loss, overt dendritic pathology, and neuronal death is not entirely clear. Aβ has been shown to induce spine loss via the activation of caspase-2 (Pozueta and others, 2013), and caspase-3 (D’Amelio and others, 2011). Nevertheless, in vivo imaging has shown that NFT-bearing neurons in transgenic animals expressing human P301L tau have increased caspase activation in the absence of acute neuronal toxicity and cell death (Spires-Jones and others, 2008). Similarly, pathogenic Aβ production in vivo activates caspase-3 in the absence of overt cell death (D’Amelio and others, 2011). These findings accord with studies demonstrating that NFT pathology is not sufficient for behavioral deficits or cell death in inducible models of tauopathy (Santa-Cruz and others, 2005; Calignon and others, 2010; Rocher and others, 2010; Kuchibholtla and others, 2014). Localized mitochondrial stress results in spine loss via the activation caspase-3; however, proteasomal degradation of activated caspase-3 limited its ability to induce cell death (Ertürk and others, 2014). These lines of evidence suggest that caspase-induced dendritic pathology can develop in the absence of neuron death.
Figure 6:
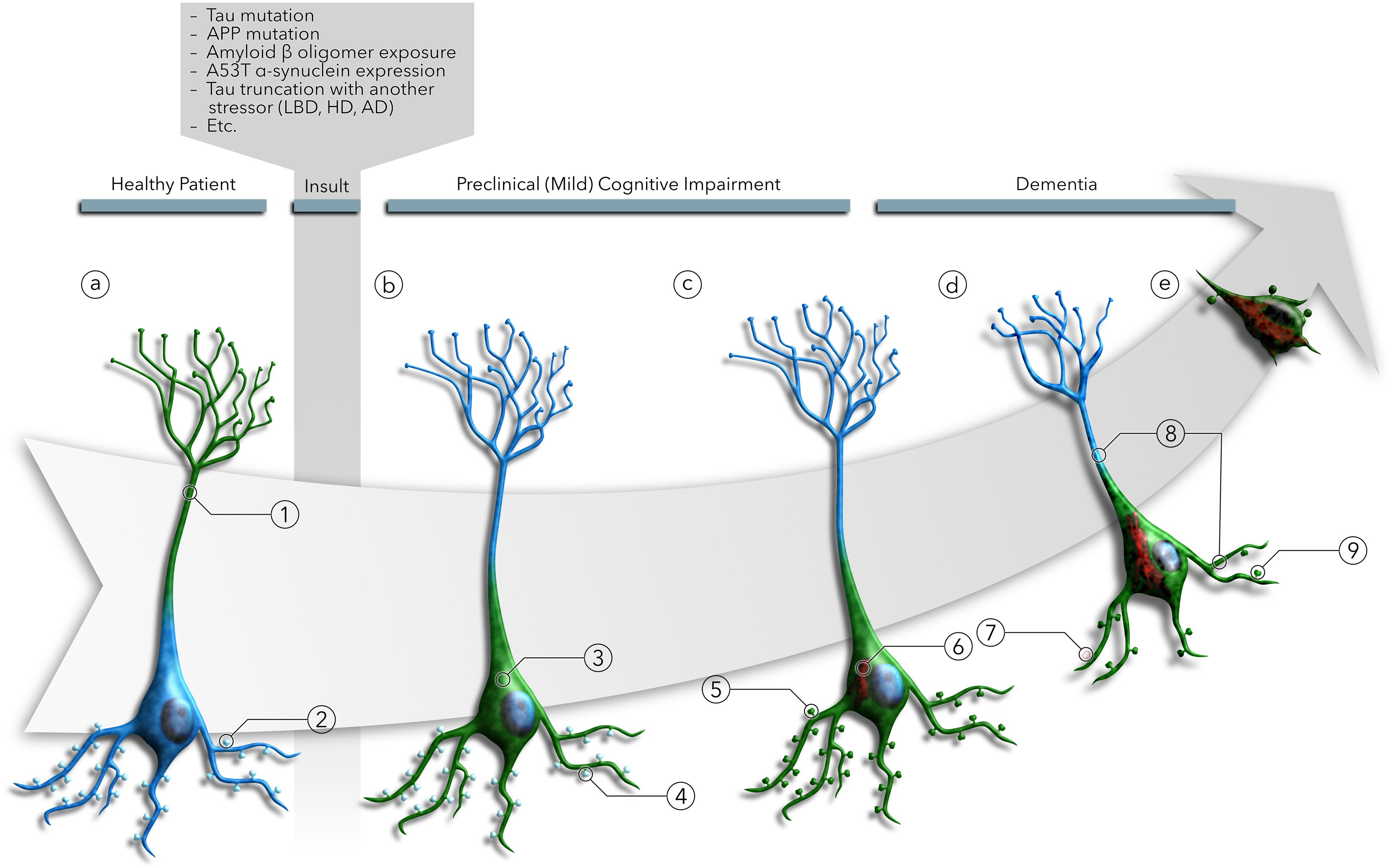
Illustration of the tau missorting sequence. (a) In healthy neurons, tau (green) is (a1) localized to the axon at levels three times greater than the soma and dendrites. (a2) While tau is largely excluded from dendritic spines, the small amount of tau located in the soma and dendrites may enter the spines and facilitate synaptic strengthening during LTP induction. After pathogenic insults from multiple disease mechanisms, (b3) tau is redistributed into the soma and dendrites of neurons due to the breakdown of tau sorting mechanisms or tau modifications leading to evasion of those mechanisms. However, (b4) dendritic tau does not mislocalize to dendritic spines. Once in the dendritic shaft, (c5) phosphorylation, truncation, and acetylation of tau lead to further mislocalization into the dendritic spines. (c6) Tau, redistributed in the soma and dendrites, begins to aggregate to form tau inclusions. Somatodendritic tau redistribution leads to (d7) dendritic spine loss, and (d8) retraction of neuritic processes. (d9) Tau mislocalization to dendritic spines leads to AMPA receptor signaling deficits due to LTD-like AMPA receptor internalization and the failure of LTP induction mechanisms. (e) Ultimately, somatodendritic tau contributes to cytotoxicity and cell death. Notably, overt tau pathology and neuron loss have been shown to be dissociated, even in the presence of pro-apoptotic signals, suggesting that cell death may result from the contribution of an independent pathway.
Conclusion
Neurodegenerative diseases and dementia are complex, multifactorial diseases with multiple etiologies, manifestations, and pathogenic mechanisms. However, there appear to be some shared molecular mechanisms causing neurological dysfunction, one of which we describe here. This common mechanism entails the redistribution of tau to the somatodendritic compartment, followed by the mislocalization of tau to dendritic spines. Evidence for this two-step sequence has been observed in a variety of disease models and diseases, including AD, FTDP-17, PDD, LBD and HD (Figure 6). Future studies will establish the relevance of this pathogenic signaling pathway across a broad range of neurological conditions and cellular stress.
Our current model stipulates a specific sequence of events by which hyperphosphorylated tau leads to synaptic and cognitive deficits. This model differs from models in which hyperphosphorylated tau leads to insoluble aggregates and neurofibrillary tangles. In our model, hyperphosphorylated tau need not form insoluble aggregates, which potentially explains why dementia may occur even in tauopathies lacking classical NFT pathology. Molecules in the signaling pathway such as caspase-2, GSK3β, and CDK5 are promising pharmacological targets for future treatments of neurodegenerative diseases.
Funding Information
NIH Grant Numbers R61 NS115089-01 (D.L.); R21-NS084007-01 (D.L.); R21-NS096437-01 (D.L.); R01-NS79374 (K.H.A.); R01-AG60766 (K.H.A.); Michael J. Fox Foundation Grant (D.L.); UMN-Mayo Partnership Grant (D.L.); Minnesota Higher Education Grant (D.L.).
Footnotes
CONFLICTS: None
References
- Amar F, Sherman M, Rush T, Larson M, Boyle G, Chang L. and et al. 2017. The amyloid-β oligomer Aβ*56 induces specific alterations in neuronal signaling that lead to tau phosphorylation and aggregation. Sci Signal. 10:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Aronov S, Aranda G, Behar L, and Ginzburg I. 2001. Axonal tau mRNA localization coincides with tau protein in living neuronal cells and depends on axonal targeting signal. J Neurosci. 21(17):6577–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asthana J, Kapoor S, Mohan R, and Panda D. 2013. Inhibition of HDAC6 deacetylase activity increases its binding with microtubules and suppresses microtubule dynamic instability in MCF-7 cells. J Biol Chem. 288(31):22516–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atlas R, Behar L, Sapoznik S, and Ginzburg I. 2007. Dynamic association with polysomes during P19 neuronal differentiation and an untranslated-region-dependent translation regulation of the tau mRNA by the tau mRNA-associated proteins IMP1, HuD, and G3BP1. J Neurosci Res. 85(1):173–83. [DOI] [PubMed] [Google Scholar]
- Balaji V, Kaniyappan S, Mandelkow E, Wang Y, and Mandelkow E. 2018. Pathological missorting of endogenous MAPT/Tau in neurons caused by failure of protein degradation systems. Autophagy. 14(12):2169–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbier P, Zejneli O, Martinho M, Lasorsa A, Belle V, Smet-Nocca C and et al. 2019. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front Aging Neurosci. 11:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar L, Marx R, Sadot E, Barg J, and Ginzburg I. 1995. Cis-acting signals and trans-acting proteins are involved in tau mRNA targeting into neurites of differentiating neuronal cells. Int J Dev Neurosci. 13(2):113–27. [DOI] [PubMed] [Google Scholar]
- Binder L, Frankfurter A, and Rebhun L. 1985. The distribution of tau in the mammalian central nervous system. J Cell Biol. 101(4):1371–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black M, Slaughter T, Moshiach S, Obrocka M, and Fischer I. 1996. Tau Is Enriched on Dynamic Microtubules in the Distal Region of Growing Axons. J Neurosci. 16(11):3061–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss T, and Collingridge G. 1993. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 361(6407):31–39. [DOI] [PubMed] [Google Scholar]
- Borin M, Saraceno C, Catania M, Lorenzetto E, Pontelli V, Paterlini A, and et al. 2018. Rac1 activation links tau hyperphosphorylation and Aβ dysmetabolism in Alzheimer’s disease. Acta Neuropatho Comm. 6:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, and Braak E. 1996. Evolution of the neuropathology of Alzheimer’s disease. Acta Neurol Scand Suppl. 165:3–12. [DOI] [PubMed] [Google Scholar]
- Bronner D, Abuaita B, Chen X, Fitzgerald K, Nuñez G, He Y, and et al. 2015. Endoplasmic Reticulum Stress Activates the Inflammasome via NLRP3- and Caspase-2-Driven Mitochondrial Damage. Immunity. 43(3):451–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burré J 2015. The Synaptic Function of α-Synuclein. J Parkinson’s Dis. 5(4): 699–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butner K, and Kirschner M. 1991. Tau protein binds to microtubules through a flexible array of distributed weak sites. J Cell Biol. 115(3):717–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calignon A, Fox L, Pitstick R, Carlson G, Bacskai B, Spires-Jones T, and Hyman B. 2010. Caspase activation precedes and leads to tangles. Nature. 464(7292):1201–04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll J, Southwell A, Graham R, Ehrnhorfer D, Cao L, Zhang W, and et al. 2011. Mice lacking caspase-2 are protected from behavioral changes, but not pathology, in the YAC128 model of Huntington disease. Mol Neurodegener. 6:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiasseu M, Cueva Vargas J, Destroismaisons L, Vande Velde C, Leclerc N, Di Polo A. 2016. Tau Accumulation, Altered Phosphorylation, and Missorting Promote Neurodegeneration in Glaucoma. J Neurosci. 36(21):5785–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland D, Hwo S, and Kirschner M. 1977. Purification of Tau, a Microtubule-Associated Protein that Induces Assembly of Microtubules from Purified Tubulin. J Molec Biol. 116(2):207–25. [DOI] [PubMed] [Google Scholar]
- Cohen T, Guo J, Hurtado D, Kwong L, Mills I, Trojanowski J, and Lee V. 2011. The Acetylation of Tau Inhibits its Function and Promotes Pathological Tau Aggregation. Nat Commun. 2:252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuchillo-Ibanez I, Seereeram A, Byers H, Leung K, Ward M, Anderton B, and Hanger D. 2008. Phosphorylation of tau regulates its axonal transport by controlling its binding to kinesin. FASEB J. 22(9):3186–95. [DOI] [PubMed] [Google Scholar]
- D’Amelio M, Cavallucci V, Middei S, Matchetti C, Pacioni S, and et al. 2011. Caspase-3 Triggers Early Synaptic Dysfunction in a Mouse Model of Alzheimer’s Disease. Nat Neurosci. 14(1):69–76. [DOI] [PubMed] [Google Scholar]
- Davies C, Mann D, Sumpter P, and Yates P. 1987. A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer’s disease. J Neurol Sci. 78(2):151–64. [DOI] [PubMed] [Google Scholar]
- d’Orange M, Aurégan G, Cheramy D, Gaudin-Guérif M, Lieger S, Guillermier M, and et al. 2018. Potentiating tangle formation reduces acute toxicity of soluble tau species in the rat. Brain. 141(2):535–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DaRocha-Souto B, Coma M, Pérez-Nievas B, Scotton S, Siao M, Sánchez-Ferrer P, and et al. 2012. Activation of glycogen synthase kinase-3 beta mediates β-amyloid induced neuritic damage in Alzheimer’s disease. Neurobiol Dis. 45:425–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey C, Kamal A, Lundgen K, Klosak N, Bailey R, Dunmore J, and et al. 2007. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest. 117(3):648–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda J, Giasson B, Mabon M, Miller D, Golbe L, and et al. 2002. Concurrence of alpha-synuclein and tau brain pathology in the Contursi kindred. Acta Neuropathol. 104(1):7–11. [DOI] [PubMed] [Google Scholar]
- Ertürk E, Wang Y, and Sheng M. 2014. Local Pruning of Dendrites and Spines by Caspase-3-Dependent and Proteasome-Limited Mechanisms. J Neurosci. 24(5):1672–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Nogales M, Cabrera J, Santos-Galindo M, Hoozemans J, Ferrer I, Rozemuller A, and et al. 2014. Huntington’s Disease is a Four-Repeat Tauopathy with Tau Nuclear Rods. Nat Med. 20(8):881–85. [DOI] [PubMed] [Google Scholar]
- Frandemiche M, De Seranno S, Rush T, Borel E, Elie A, Arnal I, and et al. 2014. Activity-dependent tau protein translocation to excitatory synapse is disrupted by exposure to amyloid-beta oligomers. J Neurosci. 201434(17):6084–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukutani Y, Cairns N, Shiozawa M, Sasaki K, Sudo S, Isaki K, and et al. 2000. Neuronal loss and neurofibrillary degeneration in the hippocampal cortex in late-onset sporadic Alzheimer’s disease. Psychiatry Clin Neurosci. 54:523–29. [DOI] [PubMed] [Google Scholar]
- Galloway P, Bergeron C, and Perry G. 1989. The Presence of Tau Distinguishes Lewy Bodies of Diffuse Lewy Body Disease from Those of Idiopathic Parkinson Disease. Neurosci Lett. 100(1–3):6–10. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini M, Jakes R, Rutherford D, and Crowther R. 1989. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 3(4):519–26. [DOI] [PubMed] [Google Scholar]
- Goode B, Denis P, Panda D, Radeke M, Miller H, Wilson L, and Feinstein S. 1997. Functional interactions between the proline-rich and repeat regions of tau enhance microtubule binding and assembly. Molec Biol Cell. 8(2):353–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Isla T, Hollister R, West H, Mui S, Growdon J, Petersen R, and et al. 1997. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol. 41(1):17–24. [DOI] [PubMed] [Google Scholar]
- Grueninger F, Bohrmann B, Czech C, Ballard T, Frey J, Weidensteiner C, and et al. 2010. Phosphorylation of Tau at S422 is enhanced by Abeta in TauPS2APP triple transgenic mice. Neurobiol. Dis 37(2):294–306. [DOI] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Tung Y, Quinlan M, Wisniewski H, and Binder L. 1986. Abnormal Phosphorylation Of The Microtubule-Associated Protein τ (Tau) In Alzheimer Cytoskeletal Pathology. Proc Natl Acad Sci. 83(13):4913–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Covell D, Daniels J, Iba M, Stieber A, Zhang B, and et al. 2013. Distinct a-Synuclein Strains Differentially Promote Tau Inclusions in Neurons. Cell. 154:103–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T, Noble W, and Hanger D. 2017. Roles of tau protein in health and disease. Acta Neuropathol. 133(5):665–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton D, Webber D, Bilican B, Goedert M, Spillantini M, and Chandran S. 2010. Cell-mediated neuroprotection in a mouse model of human tauopathy. J Neurosci. 30(30):9973–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris K, and Kater S. 1994. Dendritic spines: cellular specializations imparting both stability and flexibility to synaptic function. Annu Rev Neurosci. 17:341–71. [DOI] [PubMed] [Google Scholar]
- Hatch R, Wei Y, Xia D, and Götz J. 2017. Hyperphosphorylated tau causes reduced hippocampal CA1 excitability by relocating the axon initial segment. Acta Neuropatho. 133:717–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering H, and Sheng M. 2001. Dendritic spines: structure, dynamics and regulation. Nat. Rev. Neurosci 2(12):880–88. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Funakoshi T, Sato-Harada R, and Yoshimitsu K. 1996. Selective Stabilization of Tau in Axons and Microtubule-associated Protein 2C in Cell Bodies and Dendrites Contributes to Polarized Localization of Cytoskeletal Proteins in Mature Neurons. J Cell Biol. 132(4):667–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover B, Reed M, Su J, Penrod R, Kotilinek L, Grant M, and et al. 2010. Tau Missorting to Dendritic Spines Mediates Synaptic Dysfunction Independently of Neurodegeneration. Neuron. 68:1067–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin D, Grossman M, Weintraub D, Hurtig H, Duda J, Xie S, and et al. 2017. Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet Neurol. 16(1):55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner L, Ke Y, Delerue F, Bi M, Gladbach A, Ersel J, and et al. 2010. Dendritic Function of Tau Mediates Amyloid-β Toxicity in Alzheimer’s Disease Mouse Models. Cell. 142:387–97. [DOI] [PubMed] [Google Scholar]
- Iwata M, Watanabe S, Yamane A, Miyasaka T, and Misonou H. 2019. Regulatory mechanisms for the axonal localization of tau protein in neurons. MBoC. 30:2441–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James B and Bennett D. 2019. Causes and Patterns of Dementia: An Update in the Era of Redefining Alzheimer’s Disease. Ann Rev Pub Health. 40:65–84. [DOI] [PubMed] [Google Scholar]
- Kam A, Liao D, Loh H, and Law P. 2010. Morphine Induces AMPA Receptor Internalization in Primary Hippocampal Neurons via Calcineurin-Dependent Dephosphorylation of GluR1 Subunits. J Neurosci. 30(45):15304–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai Y and Hirokawa N. 1995. Sorting Mechanisms of Tau and MAP2 in Neurons:S uppressed Axonal Transit of MAP2 and Locally Regulated Microtubule Binding. Neuron. 14:421–32. [DOI] [PubMed] [Google Scholar]
- Kandimalla R, Manczak M, Yin X, Wang R, and Hemachandra Reddy P. 2018. Hippocampal phosphorylated tau induced cognitive decline, dendritic spine loss and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Human Molec Genet. 27(1):30–40. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ke Y, Suchowerska A, Hoven J, Silva D, Wu C, Eersel J, and et al. 2012. Lessons from Tau-Deficient Mice. Int J Alz Dis. 2012:873270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellner Y, Gödecke N, Dierkes T, Thieme N, Zagrebelsky M, and Korte M. 2014. The BDNF effects on dendritic spines of mature hippocampal neurons depend on neuronal activity. Front Synaptic Neurosci. 6:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy M 2000. Signal-processing machines at the postsynaptic density. Science. 290(5492):750–54. [DOI] [PubMed] [Google Scholar]
- Konzack S, Thies E, Marx A, Mandelkow E, and Mandelkow E. 2007. Swimming against the Tide: Mobility of the Microtubule-Associated Protein Tau in Neurons. J Neurosci. 27(37):9916–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosik and Finch. 1987. MAP2 and tau segregate into dendritic and axonal domains after the elaboration of morphologically distinct neurites: an immunocytochemical study of cultured rat cerebrum. J Neurosci. (10):3142–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotzbauer P, Giasson B, Kravitz A, Golbe L, Mark M, Trojanowski J, Lee V. 2004. Fibrillization of Alpha-Synuclein and Tau in Familial Parkinson’s Disease Caused by the A53T Alpha-Synuclein Mutation. Exp Neurol. 187(2):279–88. [DOI] [PubMed] [Google Scholar]
- Kubo A, Ueda S, Yamane A, Wada-Kakuda S, Marita M, Matsuyama M, and et al. 2019. Ectopic Expression Induces Abnormal Somatodendritic Distribution of Tau in the Mouse Brain. Neurobiol Dis. 39(34):6781–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchibhotla K, Wegmann S, Kopeikina K, Hawkes J, Rudinskiy N, Andermann M, and et al. 2014. Neurofibrillary tangle-bearing neurons are functionally integrated in cortical circuits in vivo. Proc Natl Acad Sci. 111(1):510–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson M, Greimel S, Amar F, LaCroix M, Boyle G, Sherman M, and et al. 2017. Selective lowering of synapsins induced by oligomeric α-synuclein exacerbates memory deficits. Proc Natl Acad Sci. 114(23):E4648–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Wang J, Yu W, and Lu B. 2012. Phospho-dependent ubiquitination and degradation of PAR-1 regulates synaptic morphology and tau-mediated Ab toxicity in Drosophila. Nat Comm 3:1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S, Avni D, Hariharan N, Perry R, and Meyuhas O. 1991. Oligopyrimidine tract at the 5’ end of mammalian ribosomal protein mRNAs is required for their translational control. Proc Natl Acad Sci. 88(8):3316–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Kumar Y, Zempel H, Madnelkow E, Biernat J, and Mandelkow E. 2011. Novel diffusion barrier for axonal retention of Tau in neurons and its failure in neurodegeneration. EMBO J. 30:4825–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Miller E, and Teravskis P. 2014. Tau acts as a mediator for Alzheimer’s disease-related synaptic deficits. Eur J Neurosci. 39(7):1202–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Tsai P, Wu R, and Chien C. 2010. LRRK2 G2019S Mutation Induces Dendrite Degeneration through Missorting and Phosphorylation of Tau by Recruiting Autoactivated GSK3β. J Neurosci. 30(39):13138–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litman P, Barg J, Rindzoonski L, and Ginzburg I. 1993. Subcellular localization of Tau mRNA in Differentiating Neuronal Cell Culture: Implications for Neuronal Polarity. Neuron. 10:627–38. [DOI] [PubMed] [Google Scholar]
- Liu P, Smith B, Huang E, Mahesh A, Vonsattel J, Petersen A, and et al. 2019. A soluble truncated tau species related to cognitive dysfunction and caspase-2 is elevated in the brain of Huntington’s disease patients. Acta Neuropathol Comm. 7(1):111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L 2000. Rho GTPases in neuronal morphogenesis. Nat Rev Neurosci. 1(3):173–80. [DOI] [PubMed] [Google Scholar]
- Luo L, Hensch T, Ackerman L, Barbel S, Jan L, and Jan Y. 1996. Differential effects of the Rac GTPase on Purkinje cell axons and dendritic trunks and spines. Nature. 379(6568):837–40. [DOI] [PubMed] [Google Scholar]
- Maeda S, Djukic B, Taneja P, Yu G, Lo I, Davis A, and et al. 2016. Expression of A152T human tau causes age-dependent neuronal dysfunction and loss in transgenic mice. EMBO Rep. 17(4):530–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mairet-Coello G, Courchet J, Pieraut S, Courchet V, Maximov A, and Polleux F. 2013. The CAMKK2-AMPK Kinase Pathway Mediates the Synaptotoxic Effects of Aβ Oligomers through Tau Phosphorylation. Neuron. 78:94–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R, and Malenka R. 2002. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 25:103–26. [DOI] [PubMed] [Google Scholar]
- Malinow R, Mainen Z, and Hayashi Y. 2000. LTP mechanisms: from silence to four-lane traffic. Curr Opin Neuobiol. 10(3):352–57. [DOI] [PubMed] [Google Scholar]
- Malmquist T, Anthony K, and Gallo J. 2014. Tau mRNA is present in axonal RNA granules and is associated with elongation factor 1A. Brain Res. 1584:22–27. [DOI] [PubMed] [Google Scholar]
- Mandelkow E, Thies E, Trinczek B, Biernat J, and Mandelkow E. 2004. MARK/PAR1 kinase is a regulator of microtubule-dependent transport in axons. J Cell Biol. 167(1):99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell J and Banker G. 1996. A Spatial Gradient of Tau Protein Phosphorylation in Nascent Axons. J Neurosci. 16(18):5727–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S, Grimwood P, and Morris R. 2000. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 23:649–711. [DOI] [PubMed] [Google Scholar]
- Méphon-Gaspard A, Boca M, Pioche-Durieu C, Desforges B, Burgo A, Hamon L, and et al. 2016. Role of Tau in the Spatial Organization of Axonal Microtubules: Keeping Parallel Microtubules Evenly Distributed Despite Macromolecular Crowding. Cell Mol Life Sci. 73:3745–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller E, Teravskis P, Dummer B, Zhao X, Huganir R, and Liao D. 2014. Tau phosphorylation and tau missorting mediate soluble Aβ oligomer-induced AMPA glutamate receptor signaling deficits. Eur J Neurosci. 39(7):1214–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondragón-Rodríguez S, Trillaud-Doppia E, Dudilot A, Bourgeois C, Lauzon M, Leclerc, and Boehm J. 2012. Interaction of Endogenous Tau Protein with Synaptic Proteins Is Regulated by N-Methyl-D-aspartate Receptor-dependent Tau Phosphorylation. J Biol Chem. 287(38):32040–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita T, and Sobue K. 2009. Specification of neuronal polarity regulated by local translation of CRMP2 and Tau via the mTOR-p70S6K pathway. J Biol Chem. 284(40):27734–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama A, Harms M, and Luo L. 2000. Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J Neurosci. 20(14):5329–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemani V, Lu W, Berge V, Nakamura K, Onoa B, Lee M, and Chaudhry F. 2010. Increased Expression of α-Synuclein Reduces Neurotransmitter Release by Inhibiting Synaptic Vesicle Reclustering after Endocytosis. Neuron. 65:66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura I, Yang Y, and Lu B. 2004. PAR-1 kinase plays an initiator role in a temporally ordered phosphorylation process that confers tau toxicity in Drosophila. Cell. 116(5):671–82. [DOI] [PubMed] [Google Scholar]
- Oliva A, Atkins C, Copenagle L, and Banker G. 2006. Activated c-Jun N-Terminal Kinase Is Required for Axon Formation. J Neurosci. 26(37):9462–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan P, Martínez-Mármol R, Xia D, Götz J, and Meunier F. 2019. Frontotemporal dementia mutant Tau promotes aberrant Fyn nanoclustering in hippocampal dendritic spines. Elife. 8: e45040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petratos S, Li Q, George A, Hou X, Kerr M, Unabia S, Hatzinisiriou I, and et al. 2008. The beta-amyloid protein of Alzheimer’s disease increases neuronal CRMP-2 phosphorylation by a Rho-GTP mechanism. Brain. 131(1):90–108. [DOI] [PubMed] [Google Scholar]
- Pianu B, Lefort R, Thuiliere L, Tabourier E, and Bartolini F. 2014. The Aβ1–42 peptide regulates microtubule stability independently of tau. J Cell Sci. 127:1117–27. [DOI] [PubMed] [Google Scholar]
- Pickering-Brown S, Baker M, Nonaka T, Ikeda K, Sharma S, Mackenzie J, and et al. 2004. Frontotemporal dementia with Pick-type histology associated with Q336R mutation in the tau gene. Brain. 127(6):1415–26. [DOI] [PubMed] [Google Scholar]
- Pinheiro S, Joana S, Mota C, Vaz-Silva J, Veloso A, Pinto V, and et al. 2016. Mol Neurobiol. 53:4745–53. [DOI] [PubMed] [Google Scholar]
- Pozueta J, Lefort R, Ribe E, Troy C, Arancio O, and Shelanski M. 2013. Caspase-2 is required for dendritic spine and behavioural alterations in J20 APP transgenic mice. Nat Commun. 4:1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed L, Wszolek Z, and Hutton M. 2001. Phenotypic correlations in FTDP-17. Neurobiol Aging. 22(1):89–107. [DOI] [PubMed] [Google Scholar]
- Regan P, Piers T, Yi J, Kim D, Huh S, Park S, and et al. 2015. Tau phosphorylation at serine 396 residue is required for hippocampal LTD. J Neurosci. 35(12):4804–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson E, Scearce-Levie K, Palop J, Yan F, Cheng I, Wu T, and et al. 2007. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 316(5825):750–54. [DOI] [PubMed] [Google Scholar]
- Rocher A, Crimins J, Amatrudo J, Kinson M, Todd-Brown M, Lewis J, and Luebke J. 2010. Structural and functional changes in tau mutant mice neurons are not linked to the presence of NFTs. Exp Neurol. 223(2):385–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santa Cruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, and et al. 2005. Tau Suppression in a Neurodegenerative Mouse Model Improves Memory Function. Science. 309(5733):476–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz T and Mandelkow E. 2014. Transport and diffusion of Tau protein in neurons. Cell Mol Life Sci. 71:3139–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B, Covelo A, Martell-Martínez H, Nanclares C, Sherman M, Okematti E, and et al. 2019. Tau is required for progressive synaptic and memory deficits in a transgenic mouse model of α-synucleinopathy. Acta Neuropathol. 138(4):551–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B, Nelson K, Kemper L, Leinonen-Wright K, Petersen A, Keene C, and Ashe K. 2019. A soluble tau fragment generated by caspase-2 is associated with dementia in Lewy body disease. Acta Neuropathol Comm. 7:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn P, Tracy T, Son H, Zhou Y, Leite R, Miller B, and et al. 2016. Acetylated tau destabilizes the cytoskeleton in the axon initial segment and is mislocalized to the somatodendritic compartment. Mol Neurodegener. 11(1):47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini M, Van Swieten J, and Goedert M. 2000. Tau Gene Mutations in Frontotemporal Dementia and Parkinsonism Linked to Chromosome 17 (FTDP-17). Neurogenetics. 2(4):193–205. [DOI] [PubMed] [Google Scholar]
- Spires-Jones T, Calignon A, Matsui T, Zehr C, Pitstick R, and et al. 2008. In Vivo Imaging Reveals Dissociation Between Caspase Activation and Acute Neuronal Death in Tangle-Bearing Neurons. J Neurosci. 28(4):862–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires-Jones T, Attems J, and Thal D. 2017. Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol. 134(2):187–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallings N, O’Neal M, Hu J, Kavalali E, Bezprozvanny I, and Malter J. 2018. Pin1 mediates Aβ42-induced dendritic spine loss. Sci Sig. 11:8734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sydow A, Van der Jeugd A, Zheng F, Ahmed T, Balschun D, Petrova O, and et al. 2011. Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic Tau mutant. J Neurosci. 31(7):2511–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tackenberg C, and Brandt R. 2009. Divergent pathways mediate spine alterations and cell death induced by amyloid-beta, wild-type tau, and R406W tau. J Neurosci. 29(46):14439–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tackenberg C, Grinschgl S, Trutzel A, Santuccione A, Frey M, Konietzko U, and et al. 2013. NMDA receptor subunit composition determines beta-amyloid-induced neurodegeneration and synaptic loss. Cell Death Dis. 4:608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S, Karri V, Tay N, Chang K, Ah H, Ng P, and et al. 2019. Emerging pathways to neurodegeneration: Dissecting the critical molecular mechanisms in Alzheimer’s disease, Parkinson’s disease. Biomed Pharm. 111:765–77. [DOI] [PubMed] [Google Scholar]
- Teravskis P, Covelo A, Miller E, Singh B, Martell-Martínez H, Bennyworth M, and et al. 2018. A53T Mutant Alpha-Synuclein Induces Tau-Dependent Postsynaptic Impairment Independently of Neurodegenerative Changes. J Neurosci. 38(45):9754–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teravskis P, Oxnard B, Miller E, Kemper L, Ashe K, Liao D. 2019. Phosphorylation in two discrete tau domains regulates a stepwise process leading to postsynaptic dysfunction. J Physol. doi: 10.1113/JP277459.June13. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 72(6):971–83 [DOI] [PubMed] [Google Scholar]
- Theofilas P, Ehrenberg A, Nguy A, Thackrey J, Dunlop S, Mejia M, and et al. 2018. Probing the correlation of neuronal loss, neurofibrillary tangles, and cell death markers across the Alzheimer’s disease Braak stages: a quantitative study in humans. Neurobiol Aging. 61:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thies E, and Mandelkow E. 2007. Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J Neurosci. 27(11):2896–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourtellotte W and Van Hoesen G. 1991. The axonal origin of a subpopulation of dystrophic neurites in Alzheimer’s disease. Neurosci Lett. 129(1):11–16. [DOI] [PubMed] [Google Scholar]
- Tracy T, Sohn P, Minami S, Wang C, Min S, Li Y, and et al. 2016. Acetylated Tau Obstructs KIBRA-Mediated Signaling in Synaptic Plasticity and Promotes Tauopathy-Related Memory Loss. Neuron. 90:245–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojanowski J, Shin R, Schmidt M, and Lee V. 1995. Relationship Between Plaques, Tangles, and Dystrophic Processes in Alzheimer’s Disease. Neurobiol Ageing. 16(3): 333–45. [DOI] [PubMed] [Google Scholar]
- Tseng J, Xie L, Song S, Xie Y, Allen L, Hong J, and et al. , 2017. The Deacetylase HDAC6 Mediates Endogenous Neuritic Tau Pathology. Cell Rep. 20:2169–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsushima H, Emanuele M, Polenghi A, Esposito A, Vassalli M, Barberis A, Difato F, Chieregatti E. 2015. HDAC6 and RhoA are novel players in Abeta-driven disruption of neuronal polarity. Nat Commun. 6:7781. [DOI] [PubMed] [Google Scholar]
- Upton J, Austgen K, Nishino M, Coakley K, Hagen A, Han D, and et al. 2008. Caspase-2 cleavage of BID is a critical apoptotic signal downstream of endoplasmic reticulum stress. Mol Cell Biol. 28(12):3943–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utton M, Noble W, Hill J, Anderton B, and Hanger D. 2005. Molecular motors implicated in the axonal transport of tau and α-synuclein. J Cell Sci. 118:4645–54. [DOI] [PubMed] [Google Scholar]
- Vanderweyde T, Apicco D, Youmans-Kidder K, Ash P, Cook C, Lummertz da Rocha E, and et al. 2016. Interaction of tau with the RNA-Binding Protein TIA1 Regulates tau Pathophysiology and Toxicity. Cell Rep. 15:1455–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermersch P, Delacourte A, Javoy-Agid F, Hauw J, and Agid Y. 1993. Dementia in Parkinson’s Disease: Biochemical Evidence for Cortical Involvement Using the Immunodetection of Abnormal Tau Proteins. Ann Neurol. 33(5):445–50. [DOI] [PubMed] [Google Scholar]
- Vuono R, Winder-Rhodes S, de Silva R, Cisbani G, Drouin-Ouellet J, and et al. 2015. The role of tau in the pathological process and clinical expression of Huntington’s disease. Brain. 138(7):1907–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiens K, Lin H, and Liao D. 2005. Rac1 induces the clustering of AMPA receptors during spinogenesis. J Neurosci. 25(46):10627–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weingarten M, Lockwood A, Hwo S, Kirschner M. 1975. A Protein Factor Essential for Microtubule Assembly. Proc Natl Acad Sci. 72(5):1858–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Du S, Shi W, Liu Y, Hu Y, Xie Z, and et al. 2019. Inhibition of Rac1-dependent forgetting alleviates memory deficits in animal models of Alzheimer’s disease. Protein Cell. 10(10):745–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia D, Li C, and Götz. 2015. Pseudophosphorylation of Tau at distinct epitopes or the presence of the P301L mutation targets the microtubule-associated protein Tau to dendritic spines. Biochem Biophys Acta. 1852:913–24. [DOI] [PubMed] [Google Scholar]
- Xu H, Rösler T, Carlsson T, de Andrade A, Bruch J, Höllerhage M, and et al. 2014. Memory deficits correlate with tau and spine pathology in P301S MAPT transgenic mice. Neuropathol Appl Neurobiol. 40(7):833–43. [DOI] [PubMed] [Google Scholar]
- Xu Y, Wang H, Yan J, Sun X, Guo J, and Zhu. 2009. Antibody binding to cell surface amyloid precursor protein induces neuronal injury by deregulating the phosphorylation of focal adhesion signaling related proteins. Neurosci Letters. 465:276–81. [DOI] [PubMed] [Google Scholar]
- Xu Z, Tan J, Xu H, Hill C, Ostrovskaya O, Martemyanov K, and Xu B. 2019. Caspase-2 promotes AMPA receptor internalization and cognitive flexibility via mTORC2-AKT-GSK3β signaling. Nat Commun. 10(1):3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Demura T, Morita M, Banker G, Tanii T, and Nakamura S. 2012. Differential neurite outgrowth is required for axon specification by cultured hippocampal neurons. J Neurochem. 123(6):904–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yogev S and Shen K. 2017. Establishing Neuronal Polarity with Environmental and Intrinsic Mechanisms. Neuron. 96:638–50. [DOI] [PubMed] [Google Scholar]
- Zempel H, Dennissen F, Kumar Y, Luedtke J, Biernat J, Mandelkow E, and Mandelkow E. 2017. Axodendritic sorting and pathological missorting of Tau are isoform-specific and determined by axon initial segment architecture. J Biol Chem. 292(29):12192–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zempel H, Luedtke J, Kumar Y, Biernat J, Dawson H, Mandelkow E, and Mandelkow E. 2013. Amyloid-β oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin. EMBO J. 32:2920–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zempel H, Thies E, Madelkow E, and Mandelkow E. 2010. Aβ Oligomers Cause Localized Ca2+ Elevation, Missorting of Endogenous Tau into Dendrites, Tau Phosphorylation, and Destruction of Microtubules and Spines. J Neurosci. 30(36):11938–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Higuchi M, Yoshiyama Y, Ishihara T, Forman M, Martinez D, and et al. 2004. Retarded Axonal Transport of R406W Mutant Tau in Transgenic Mice with a Neurodegenerative Tauopathy. Neurobiol Dis. 24(19):4657–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Kotilinek L, Smith B, Hlynialuk C, Zahs K, Ramsden M, and et al. 2016. Caspase-2 cleavage of tau reversibly impairs memory. Nat Med. 22(11):1268–76. [DOI] [PubMed] [Google Scholar]