

Abstract
Mimicking growth factor–ECM interactions for promoting cell migration is a powerful technique to improve tissue integration with biomaterial scaffolds for the regeneration of damaged tissues. This has been attempted by scaffold-mediated controlled delivery of exogenous growth factors; however, the predetermined nature of this delivery can limit the scaffold’s ability to meet each wound’s unique spatiotemporal regenerative needs and presents translational hurdles. To address this limitation, we present a new approach to growth factor presentation by incorporating heparin microislands, which are spatially isolated heparin-containing microparticles that can reorganize and protect endogenous local growth factors via heterogeneous sequestration at the microscale in vitro and result in functional improvements in wound healing. More specifically, we incorporated our heparin microislands within microporous annealed particle (MAP) scaffolds, which allows facile tuning of microenvironment heterogeneity through ratiometric mixing of microparticle sub-populations. In this manuscript, we demonstrate the ability of heparin microislands to heterogeneously sequester applied growth factor and control downstream cell migration in vitro. Further, we present their ability to significantly improve wound healing outcomes (epidermal regeneration and re-vascularization) in a diabetic wound model relative to two clinically relevant controls.
Keywords: porous hydrogel, diabetic wound healing, heparin microislands
1. Introduction
Growth factors are key regulators of each stage of wound repair, including cellular migration, proliferation, angiogenesis, and extracellular matrix remodeling[1]. Their potential for accelerating wound healing has led to a long history of research efforts focused on biomaterial scaffold-mediated delivery of a range of growth factors (e.g. PDGF, VEGF, EGF, FGF)[2–4]. Despite existing FDA approval, applications using growth factor delivery have been limited in clinical translation by safety concerns and cost-effectiveness[2,3,5,6].
Growth factors are an essential component of the instructional microenvironment in uninjured tissue and their spatial organization within that microenvironment is partially shaped by their affinity interactions with the extracellular matrix (ECM)[1,2]. Specifically, the ECM regulates growth factor movement and can create chemotactic gradients for spatio-temporal regulation of cell migration[2]. Heparin, the most negatively charged glycosaminoglycan in human ECM, is an important regulator of growth factor localization and retention due to its high binding affinity with many growth factors via electrostatic interactions, including multiple growth factors that are important for wound repair (e.g. Vascular Endothelial Growth Factor [VEGF] and Platelet-Derived Growth Factor [PDGF])[7–9]. Using its high affinity for many growth factors, heparin has been incorporated homogeneously within biomaterial scaffolds for controlled exogenous growth factor delivery, thus providing chemotactic bioactivity[7,8].
In addition to heparin-based controlled delivery of growth factors, scaffold incorporation of heparin has also been explored for its impact on growth factor signaling. More specifically, these scaffolds are reported to present growth factors biomimetically and release them in response to cells while preserving their bioactivity in protease-rich environments[8,10,11]. However, these techniques do not present heparin heterogeneously on the microscale, which we believe limits the potential impact of heparin’s ability to organize growth factors instructionally (e.g. promoting improved wound healing). By contrast, in this manuscript we present an exciting heterogeneous heparin incorporation technique using spatially isolated hydrogel microspheres with covalently immobilized heparin, or heparin “microislands” (μIslands). This approach offers growth factor-mediated bioactivity and improved healing outcomes that are not reliant on exogenous growth factors. Our heparin μIslands spontaneously create growth factor depots through reorganization of local growth factors.
Importantly, to incorporate heparin μIslands in a porous format that allows cells to freely respond to growth factor depots, we took advantage of an injectable biomaterial platform, microporous annealed particle (MAP) scaffold[12]. MAP scaffolds are composed of micron-scale spherical building blocks (microspheres) that can be mixed ratiometrically with heparin μIslands to achieve controlled heterogeneity. Further, MAP is assembled via covalent inter-microsphere bonding (i.e. annealing) in situ to form structurally stable scaffolds with cell-scale microporosity. In addition to observing the spontaneous reorganization of uniformly applied growth factors by heparin μIslands, we also validated their impact on cell migration and whole tissue regenerative behavior.
Specifically, we chose to focus our application on diabetic wounds, which pathologically suffer from both a lack of organized tissue regeneration and growth factor retention[5,6,13,14]. Diabetic wounds result in approximately 130,000 lower limb amputations each year and affect 15% of diabetic patients in the U.S. (~10% of U.S. population has diabetes)[15]. Despite FDA approval of recombinant growth factor therapies (e.g. PDGF delivery via Regranex™)[16], their clinical use remains limited and even after treatment half of these wounds never heal[6,13,14,17]. We hypothesize that heparin μIslands, with their unique ability to sequester growth factors into spatially-separated depots in vitro (shown in this manuscript) and recognized ability to prolong growth factor bioactivity[18], can improve the healing of these wounds. Therefore, we chose to test heparin μIslands in an established diabetic wound healing animal model[19] using the most common classes of advanced wound treatment (Oasis Wound Matrix™ decellularized ECM) and basic bland emollient wound treatment (Aquaphor™) as our clinically-relevant controls[6,16].
2. Results
2.1. Particle building blocks to create a heterogeneous porous scaffold
Using our MAP scaffolds[12] as a platform technology, we took advantage of the ability to mix and match particle populations to create ratiometrically-controlled heterogenous scaffolds while maintaining an injectable format. To design instructional interaction with chemokines, we chose to focus on heterogeneously distributed heparin particle populations (heparin μIslands). We hypothesized that heparin μIslands would locally sequester growth factors released endogenously in a wound environment. Three particle populations with variable heparin concentrations (HepHigh, HepLow, and no Hep) were produced using a previously published high-throughput microfluidic method[20] to isolate heparin concentration as the only changing variable between particle types by providing uniform geometric (diameter: 90μm, Figure S6) and mechanical (Young’s modulus: 18kPa, Figure S5) properties[21,22]. Particle diameter and particle stiffness were matched between the three formulations because material pore size (directly related to particle diameter) and mechanical properties[21] are influential in determining cellular responses[22]. The particles were composed of a synthetic hydrogel network of 4-arm poly(ethylene glycol) maleimide backbone crosslinked with a peptide sequence optimized for enzymatic resorption by matrix-metalloprotease-2 (MMP-2) and covalently bonded to an RGD cell adhesive peptide ligand (Figure 1a). Different molecular weight PEG-maleimides (10kDa for heparin formulations, 20kDa for no heparin formulations) were used to offset increased microgel swelling following heparin incorporation. A custom heterofunctional 4-arm PEG maleimide/methacrylamide macromer we have previously reported[23] was incorporated to facilitate accelerated scaffold annealing. With consideration for our ultimate application to diabetic wound healing assays, thiolated heparin[24] was incorporated into the heparin μIslands at a concentration chosen to mimic mouse skin (HepHigh) and one-tenth mouse skin (HepLow) (Figure 1b).
Figure 1.
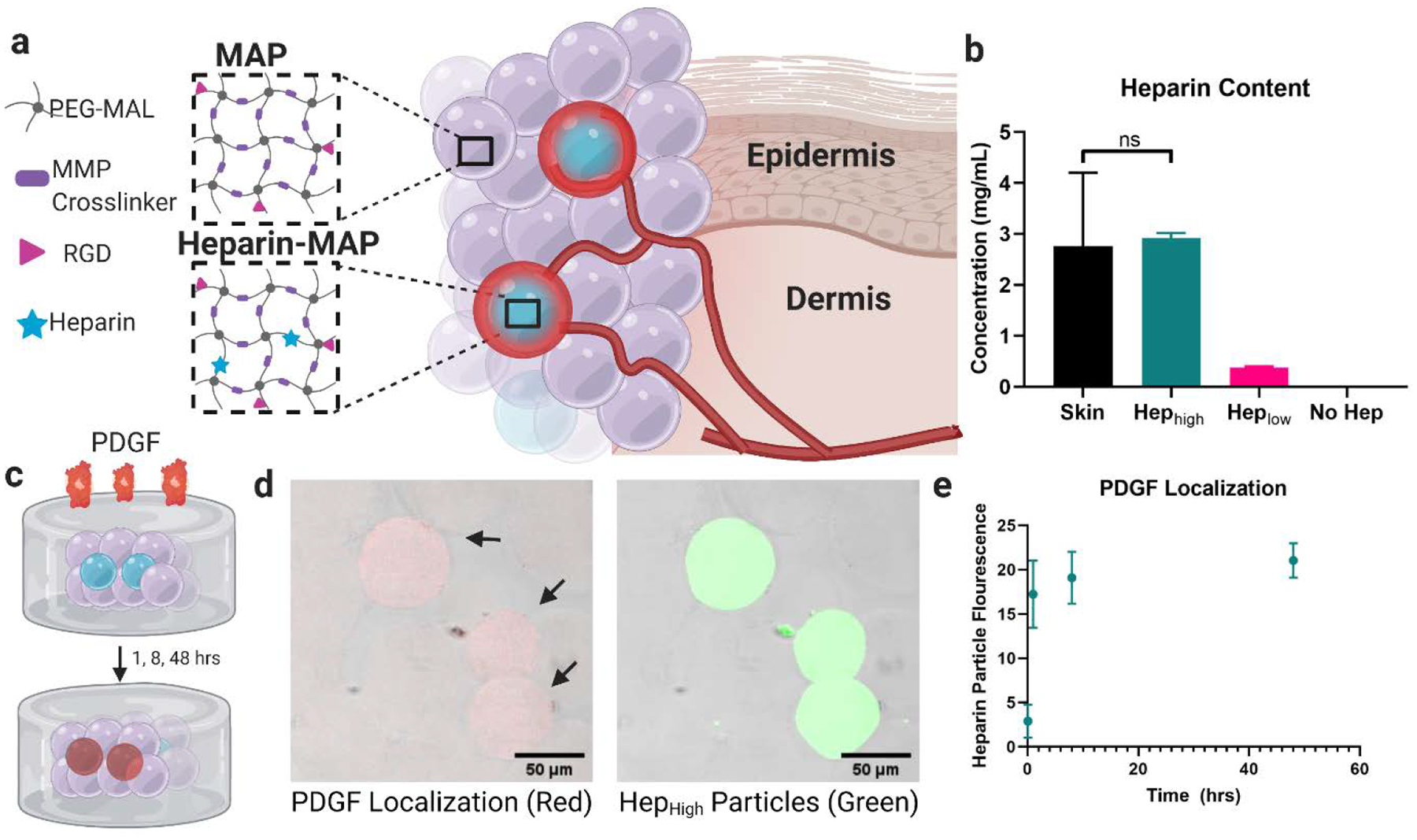
Heparin μIsland Composition and Growth Factor Sequestration. a) Particles were composed of a PEG-maleimide backbone and MMP-2 cleavable crosslinker with an RGD cell adhesive peptide with or without thiolated heparin. Small fractions of heparin particles (μIslands) were mixed with no-heparin particles to generate growth factor depots. b) Heparin concentration within the particles was matched to mouse skin (HepHigh) and one tenth of mouse skin (HepLow). c) To test for PDGF localization around our heparin μislands, we used a well-based assay with heterogenous scaffolds (10% HepHigh) embedded within a collagen-agarose scaffold before introduction of a solution of biotin-labeled PDGF. d) After fixation, fluorescent Streptavidin revealed PDGF localization (red) in the heparin μIslands (green) as early as 1 hour. e) Quantification of fluorescence in the heparin particles across the 48hr time point. Graphs represent mean +/− standard deviation.
2.2. Growth Factor Sequestration in μIslands
To validate our hypothesis that heparin μIslands can sequester and organize uniform growth factor distributions into localized growth factor depots in a “tissue-like” environment and make for more robust handling, we embedded μIsland scaffolds (10% HepHigh) within a collagen-agarose gel to simulate an interpenetrating polymer ECM and incubated them with a solution of platelet-derived growth factor (PDGF) (Figure 1c). We chose PDGF for both its importance in wound healing and its known interactions with heparin[3,9]. After 5 minutes, 1, 8, and 48 hours of incubation with biotinylated PDGF, the gels were fixed with paraformaldehyde and stained with fluorescent streptavidin. Using confocal microscopy, we were able to clearly visualize PDGF sequestered in the heparin particles by 1 hour. This PDGF organization persisted and increased slightly throughout 48 hours. Importantly, in a negative control that was stained with fluorescent streptavidin in the absence of biotinylated PDGF, there were no differences in background fluorescence between heparin and no heparin particles (Figure S8). Thus, with these in vitro assays, we were able to confirm an affinity-based structure-function relationship for heparin μIslands generating growth factor depots. In a separate study observing individual particles embedded within collagen-agarose gels (Figure S9A), we were able to visualize PDGF sequestration within particles and, surprisingly, a clear and quantifiable microscale PDGF gradient surrounding the heparin μIslands. PDGF sequestration and gradient formation were absent around particles without heparin incorporation (Figure S9B). Quantification of relative observable PDGF gradients around μIslands showed a dependence on a) heparin concentration with observable gradients out to 60μm around HepHigh μIslands and 40μm around HepLow μIslands (Figure S9C) and b) presence of a competitive binding agent (e.g. EGF, Figure S9D). In concert, these results prompted further investigation of potential effects on in vitro cell migration.
2.3. Cell migration is dependent on μIsland ratio and heparin concentration
To further investigate the potential functional impact of heparin μIslands, we employed an in vitro spheroid migration assay developed for hydrogel constructs[25] (Figure 2b–c). Due to the potential for subsequent use in a diabetic dermal wound healing model, we chose two primary cell types relevant in dermal wound healing: human dermal fibroblasts and human dermal microvascular endothelial cells. Both ratio (heparin μIslands to no hep particles) and heparin concentration were varied to investigate the ability to tune cell migratory behavior with these predetermined variables in a scaffold using a heparin μIslands approach (Figure 2a) and both variables were found to affect migration behavior for both cell types. Specifically, for ratio impact, we observed a parabolic relationship between cell behavior and scaffold heterogeneity, where 1% HepHigh μIslands had no significant difference in migration compared to 0% heparin gels, but 10% HepHigh had the greatest migration compared to all other groups (Figure 2d–e). Importantly, the cell migration in the 10% HepHigh μIslands was significantly greater than 100% HepLow, which had the same absolute heparin content in a homogenous format (Figure S12 and S13). This result confirmed that heterogeneity and not simple heparin content is responsible for the observed increased in vitro migration. When testing the impact of heparin concentration, we found that using just 10% HepLow particles resulted in an insignificant increase in migration compared to 0% heparin (Figure 2f–g), confirming that heparin concentration remains a critical factor. The enhanced migration seen with 10% HepHigh μIslands in HDFs and HDMVECs prompted the use of this group for the diabetic wound healing studies. Importantly, all migration assays were conducted in manufacturer recommended culture medium for each cell type without additional chemokines beyond those present in medium.
Figure 2.
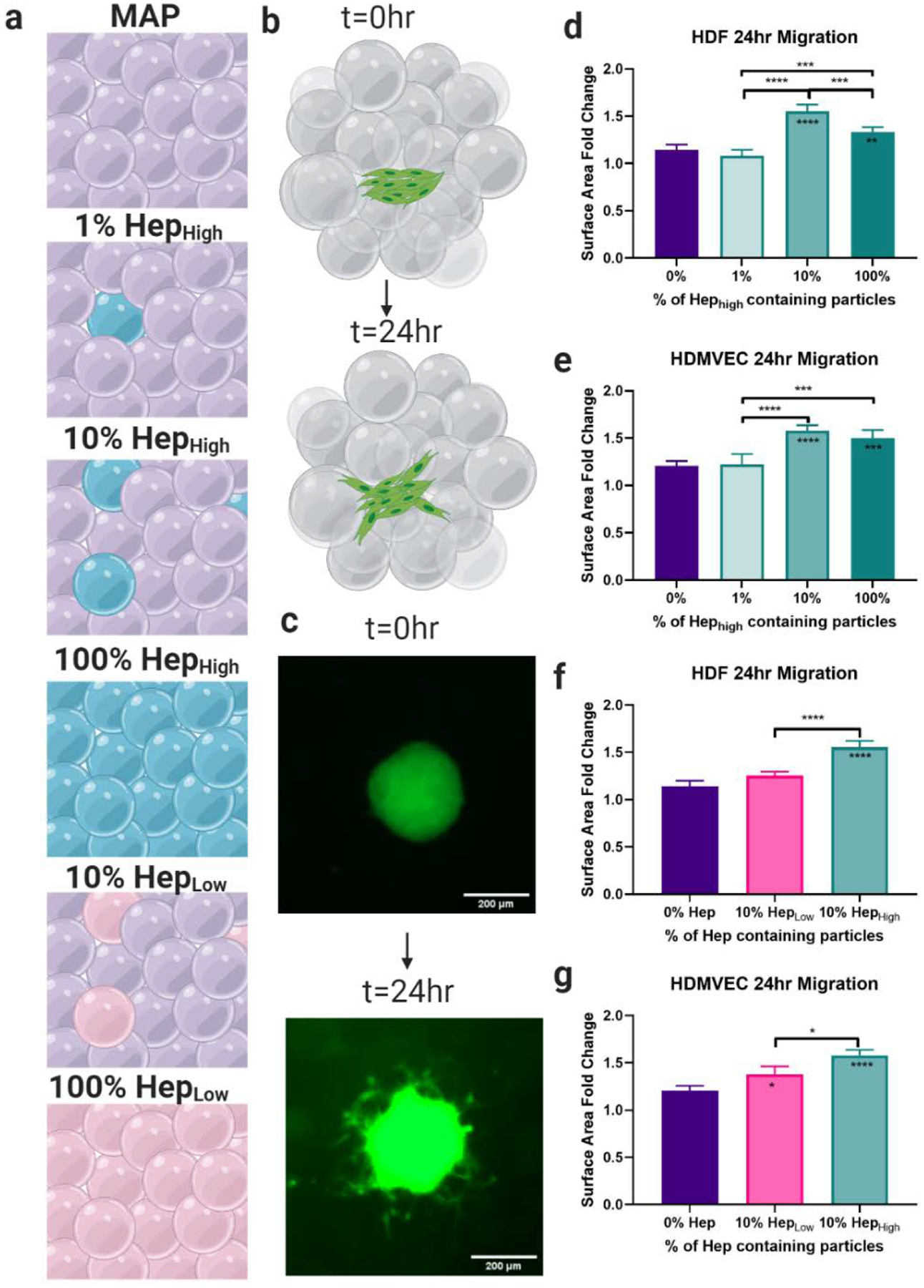
Cellular Migration. a) Six gel conditions were compared with varying concentrations and percentages of heparin particles. b) A spheroid migration assay was used to quantify cell migration over 24 hours. c) Representative images of spheroids at 0 and 24 hrs. d) Quantification of cell migration showed the 10% HepHigh μIslands exhibited the highest migration of Human Dermal Fibroblasts (HDFs) and e) Human Dermal Microvascular Endothelial Cells (HDMVECs) compared to all other groups with the same concentration of heparin, but different spacing. f) The 10% HepHigh μIslands significantly outperformed the 10% HepLow μIslands for HDFs and g) HDMVECs, confirming that both the concentration of heparin and the spacing of heparin particles affect cell migration. All graphs show mean +/− standard deviation. Statistics: ANOVA, Multiple comparisons post-hoc tests (Tukey HSD). N=4. Significance inside of bars represents comparison to no hep gels. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
2.4. Heparin μIslands promote enhanced re-epithelialization in diabetic wounds
Diabetic wounds are characterized by their inability to move from the inflammatory to proliferation phase of wound healing, which is a transition marked by the onset of re-epithelialization and re-vascularization. To characterize the progression of wound healing, we chose two time points for this diabetic wound healing study, Day 3 and Day 7, which allowed observation of the onset and significant progression of the proliferation phase, respectively[26]. The four treatment groups for this study were 10% HepHigh MAP gel (or μIslands), MAP gel without μIslands as a material platform control (or MAP), Oasis wound matrix as an advanced clinical control (currently approved for use in diabetic wounds), and Aquaphor (OTC wound hydration product) as a basic bland emollient clinical control (Figure 3a). Wound re-epithelialization was determined by regenerated epidermal tissue or “tongues” via immunofluorescent staining of keratinocytes (Figure S19). Quantification of re-epithelialization showed the μIslands group resulted in over 60% wound closure by Day 3, which was significantly greater than all other groups (Figure 3b,d). This data aligned with gross observations of wound granulation taken at Days 3 and 7 (Figure S15). By Day 7, the majority of wounds for the μIslands, MAP, and Oasis treated groups had fully re-epithelialized the wound bed (Figure S17), as determined by Keratin-14 (keratinocyte) staining. Epidermal thickness, a clinically relevant marker for healthy wound healing[27], was quantified as a measure of dermal regeneration at Day 7 and μIslands (~90μm thick) were observed to produce at least 39% thicker epidermal layer than all other groups (Figure 3c,d). In a follow-on study to confirm our in vitro observation (Figure 2d–g) of the importance of heterogeneous spacing of heparin as a driver of scaffold impact (rather than simply heparin presentation), we performed a second diabetic wound healing study with four MAP groups: μIslands, 100% HepHigh (uniform matching of heparin-containing microgel formulation), 100% HepLow (matching absolute heparin content of μIslands), and the same no heparin MAP as the previous study (Figure S16). The μIslands group performed similarly to the initial study with an average epidermal thickness of approximately 90μm which was significantly greater than all other groups (Figure 3e,f) indicating a clear benefit to the μislands approach. Interestingly, the 100% HepLow and 100% HepHigh groups were not significantly different (Figure 3e), indicating there may be a threshold to the amount of heparin which has a positive effect. Additionally, the 100% HepHigh group had fewer wounds fully re-epithelialized compared to all other groups (Figure S18). In concert, these results demonstrate that the μIslands treatment provided a more regenerative and accelerated wound re-epithelialization result compared to all controls.
Figure 3.
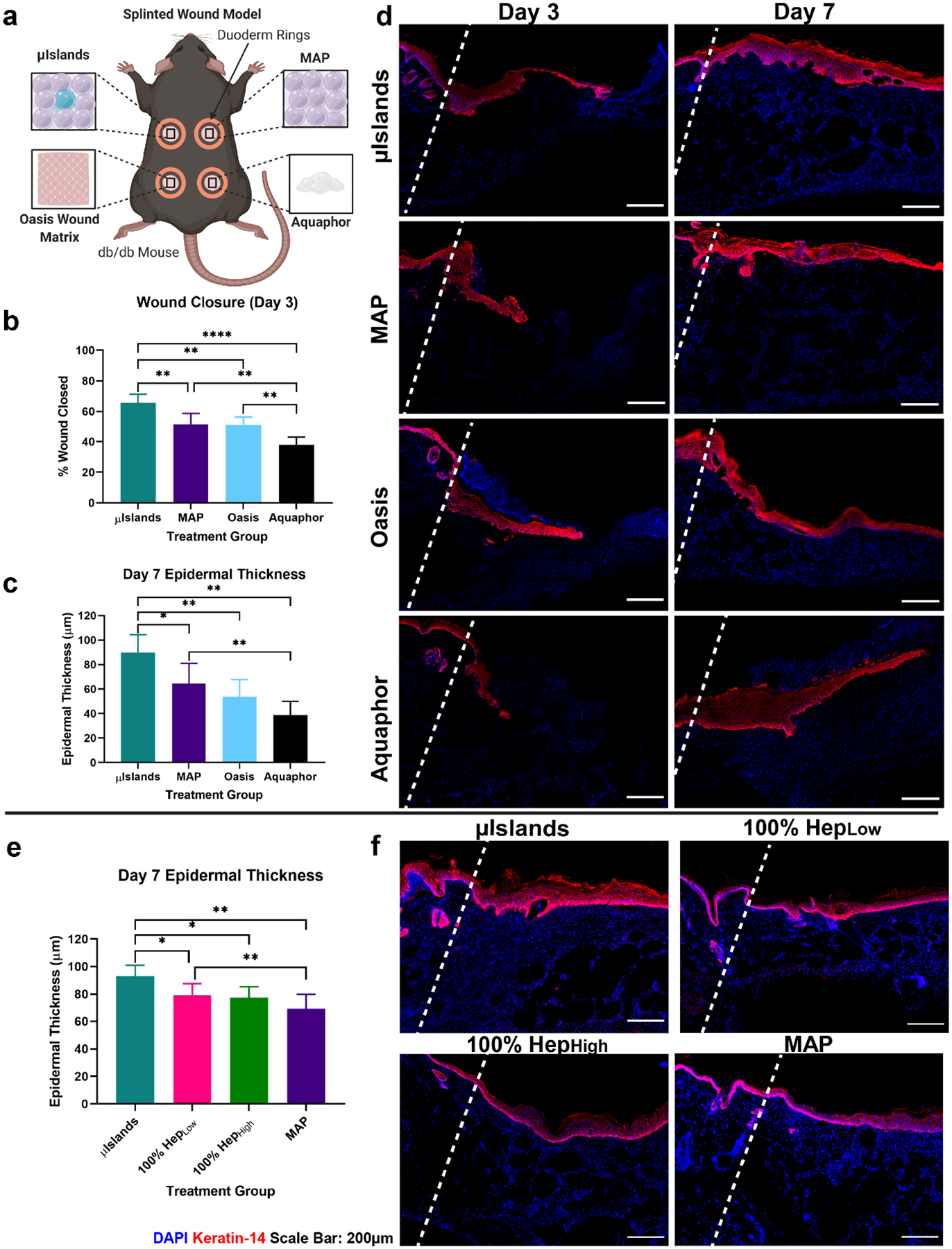
Epidermal Regeneration in a Diabetic Wound Healing Model. a) Four treatment conditions were evaluated in a mouse diabetic wound healing model at Day 3 and Day 7. b) Wound closure at Day 3 was quantified from the epidermal tongue length. c) Epidermal thickness was quantified for all at Day 7 to compare stages of healing. d) Representative images of the four treatment groups keratin-14 staining at Day 3 and Day 7. e) Day 7 epidermal thickness quantification in a separate study confirmed heparin heterogeneity was necessary for improved epidermal thickness. f) Representative keratin-14 staining from the follow-up study at Day 7. Wound edges marked by dashed line. All graphs show mean +/− standard deviation. Statistics: ANOVA, Multiple comparisons post-hoc tests (Tukey HSD). N=6 for a-d, N=7 for e,f. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
2.5. Uniform wound re-vascularization at Day 7
In addition to the investigation of wound re-epithelialization, we used the previously described diabetic wound healing studies to look at the impact of μIslands on wound re-vascularization due to heparin’s known interaction with key growth factors[28,29] in angiogenesis (e.g. VEGF[7] and PDGF) and the importance of re-vascularization to wound healing. At Day 7 for our initial study (comparing μIslands to MAP, Oasis, and Aquaphor), the μIslands group displayed extensive vasculature throughout the entire wound (Figure 4a–d). In contrast, the other three groups had noticeably more of their re-vascularization occurring proximal to the wound edges. Further, our quantification revealed that not only did μIslands provide more total wound re-vascularization than controls (as determined by overall staining for CD31+ endothelial cells), but also the distribution of blood vessels in the μIslands condition was equivalent between the inner 50% and edges of the wound (Figure 4b). Interestingly, when analysis was limited to the edges of the wound, there were no significant differences in re-vascularization among MAP, Oasis, and the μIslands (Figure 4b). By contrast, when analyzing the inner 50% of the wound, only the μIslands show an equivalent re-vascularization as was present in the wound edges. The Aquaphor group had minimal vessels observed within the wound. As an additional metric to characterize the new vasculature, we stained for pericytes by staining for NG2+ cells proximal (<5 μm distant) to endothelial cells (an indicator for vessel maturity[30]). Quantification of pericytes per wound area showed significantly more pericyte staining in the μIslands group, indicating more mature vasculature (Figure 4c,e). In our follow-up study to confirm importance of heterogeneous spacing of heparin, versus simple presentation of heparin alone (comparing μIslands to 100% HepHigh and 100%HepLow group), we observed significantly greater vascularization in the μIslands group compared to both homogenous heparin groups (Fig 4f). 100% HepHigh did promote significantly more vascularization compared to the MAP without heparin control, which we hypothesize is due to its ability to generally sequester VEGF, however further studies are needed to confirm this mechanism. When looking at the vascular (CD31+ staining) localization, the homogenous heparin groups did not significantly differ from the μIslands group on the wound edges but the μIslands group was observed to promote significantly more re-vascularization in the center of the wound (Figure 4f,g). Both homogenous heparin groups had a larger fraction of their vascularization in the wound edges, which further emphasizes the importance of spatial organization of growth factor sequestration in promoting quicker re-vascularization. In concert, these results indicate that μIslands produce a more mature and extensive vascular network throughout the entire diabetic wound compared to controls, including basic (Aquaphor) and advanced clinical controls (Oasis Wound Matrix).
Figure 4.
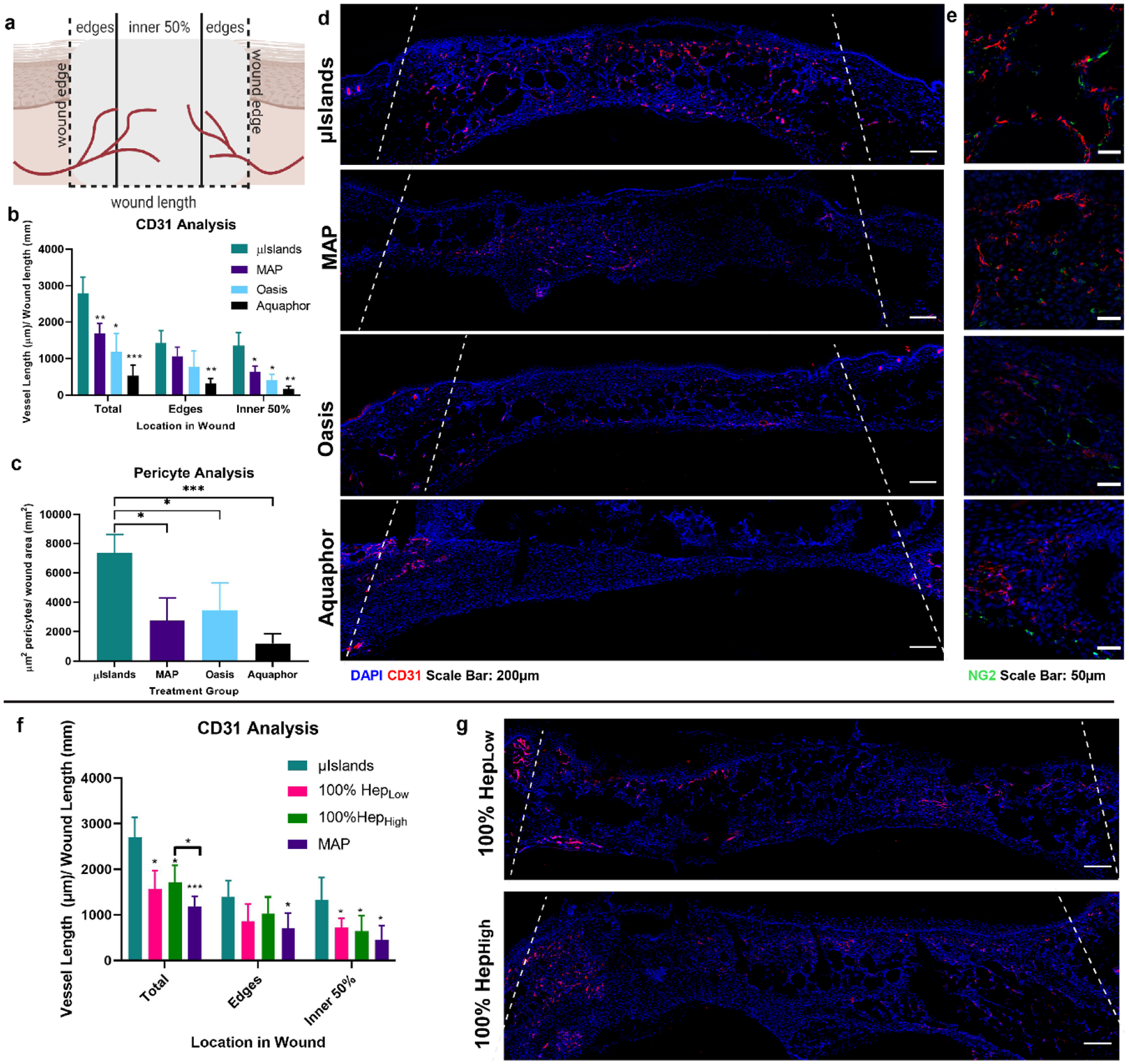
Vascularization at Day 7 in a Diabetic Wound Healing Model. a) Vessel analysis was performed to determine the extent of vascularization throughout the wound. b) Vessel length was quantified for the middle and edges of the wound at Day 7 as determined by CD31 staining. c) Pericyte analysis was performed to assess the presence of mature vasculature as indicated by proximal NG2 staining to a blood vessel. d) Representative images of the four treatment groups CD31 staining Day 7 and e) pericyte staining. f) Vessel length localization at Day 7 in the follow-up wound healing study. g) Representative images of vascularization for the 100% HepLow and 100% HepHigh groups. Wound edges marked by dashed line. All graphs show mean +/− standard deviation. Statistics: ANOVA, Multiple comparisons post-hoc tests (Tukey HSD). N=6 for a-e, N=7 for f,g. *p<0.05, **p<0.01, ***p<0.001.
2.6. No difference in immune modulation between μIslands and MAP
To investigate the potential for altered immune response caused by the presence of heparin μIslands in our initial wound healing study, we stained for macrophage presence and polarization[31] (i.e. M1 versus M2) at Days 3 and 7. There were no significant differences between any of the four groups for total macrophage recruitment within the wounds (Figure S25). While we observed no difference between MAP with or without heparin μIslands, we did observe that MAP groups (with and without heparin μIslands) provided a clear immunomodulatory effect on wound macrophage polarization compared to both the Oasis and Aquaphor treatments (Figure 5). Specifically, both MAP groups promoted more M2 than M1 polarization for both time points compared to Oasis and Aquaphor, and by Day 7 greater than 77% of all macrophages were M2 phenotype for both MAP conditions (Figure 5a,c,d). Notably, these results aligned well with prior macrophage polarization investigations of precast porous hydrogel scaffolds that are the geometric inverse of MAP gel[32,33]. Combined, this data decreases the likelihood that the improved wound closure and re-vascularization effects observed for the heparin μIslands group was due to changes to the local immune response within the wound.
Figure 5.
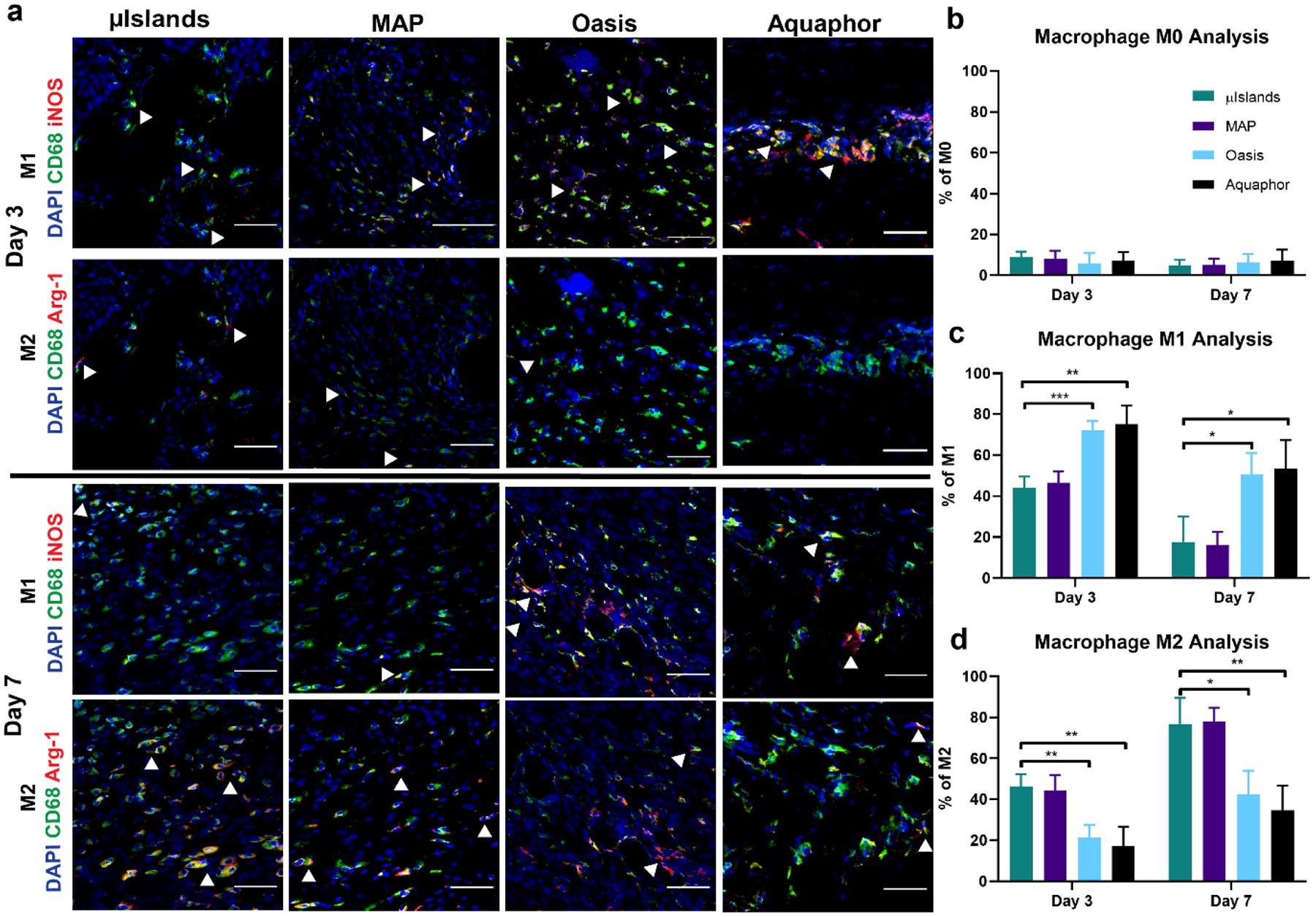
Macrophage Polarization. a) Representative images at Day 3 and Day 7 of macrophage M1 and M2 polarizations. Scale bar represents 50μm. b) Percentage of macrophages in the wounds for each group at Day 3 and 7 that only stained positive for CD68. c) Percentage of macrophages in the wounds that stained positive for the M1 inflammatory phenotype as indicated by CD68+ and iNOS+. d) Percentage of macrophages in the wounds that stained positive for the M2 pro-regenerative phenotype as indicated by CD68+ and Arg-1+. All graphs show mean +/− standard deviation. Statistics: ANOVA, Multiple comparisons post-hoc tests (Tukey HSD). N=6. *p<0.05, **p<0.01, ***p<0.001.
3. Conclusion
Here we present a new class of bioactive scaffold that was observed in vitro to organize local growth factors spatially at the microscale, potentially eliminating the need for exogenous delivery for an instructional microenvironment for in vivo function. Previously, creating heterogenous growth factor distributions within biomaterials has required advanced biofabrication strategies (photolithography[34,35], bioprinting[36,37]) that limits injectability as well as the ability to fill large wounds. In this manuscript, we report that MAP with heparin μIslands significantly improved diabetic wound healing outcomes compared to multiple MAP gel controls (with and without homogeneous heparin) and two clinically relevant control groups. We plan to use future studies to further understand the in vivo mechanism of improved healing by identifying and quantifying the cytokines being organized by the heparin μIslands. Finally, we believe that the presented growth factor free, materials-only approach has high translational potential for biomedical applications requiring accelerated cell or tissue integration.
4. Experimental Section/Methods
Sources/ Storage of Materials:
Four-arm poly(ethylene glycol) maleimide (PEG-MAL, 10kDa and 20kDa) was purchased from Nippon Oil Foundry, Inc (Japan). Using similar peptide components to our previous MAP scaffolds[38], RGD cell adhesive peptide (Ac-RGDSPGGC-NH2) and the MMP-2 degradable crosslinker (Ac-GCGPQGIAGQDGCG-NH2) were purchased from WatsonBio Sciences. All materials were dissolved in either ultrapure water or 0.1% Trifluoracetic Acid solutions (to prevent disulfide bond formation) and aliquoted into specified amounts to ensure precision. MethMal was synthesized as previously reported[23]. The aliquots were lyophilized and stored in −20°C until preparing aqueous gel solutions.
Thiolation of Heparin:
Heparin (MW=15,000Da, Millipore Sigma) was dissolved at 20mg/mL in ultrapure water (300mg reactions, N=3). A 100mM (3-(2-pyridyldithiol) propionyl hydrazide) (PDPH) (CovaChem) solution was prepared in ultrapure water. The 100mM stock PDPH solution was added to each heparin reaction to target a 20% modification. 6.894μL of AlexaFlour hydrazide 555 (1mg/mL) was added to each reaction to fluorescently tag the molecule. 145μLof the 100mM PDPH stock solution was added to each 300mg reaction of heparin. The reactions were mixed well and then the pH was adjusted to 6.5. DMTMM was added to each reaction in a 1:1 molar ratio to the heparin repeat units (estimate 619.49 Da[39]) each day of the reaction. The reaction proceeded 3 days on a rotator at room temperature. Each reaction was dialyzed using 3.5kDa snakeskin dialysis tubing for 3 days in 1M NaCl (4L, changed 2x daily) and then 0.01M NaCl (4L, 6×1hr washes). The product was frozen and lyophilized. The volume before and after dialysis were recorded to determine the percentage of mass after lyophilization that is salt to give a purity value. After quantification of heparin thiolation, the three batches were combined and deprotected with 25mM TCEP (Sigma) at room temperature for 15 minutes. The deprotected solution was dialyzed using 3.5kDa snakeskin dialysis tubing (Fisher) in 0.01% trifluoracetic acid and 1M NaCl (4L, 1×3hrs), then 0.1M NaCl (4L, 2×1hrs), then 0.01M NaCl (4L, 2×1hr). The product was frozen and lyophilized, then stored at −20°C until use.
Quantification of heparin thiolation:
Quantification of thiol concentration was based on a deprotection assay following PDPH manufacturers protocol and the modification was verified by NMR. The Thermofisher Scientific protocol for Pyridine-2-Thione Assay was used to determine the level of sulfhydryl modification. Briefly, the absorbance of the modified heparin was measured using a nanodrop before and after exposure to 1mg/mL DTT for 15 min at 343nm (Figure S1B). HNMR was also used to confirm PDPH modification. A Varian Inova 500 NMR spectrometer located in the UVA Biomolecular Magnetic Resonance Facility was used to acquire the spectra. 25 mg of each reaction was dissolved in deuterium oxide (Sigma). MestReNova was used for analysis, and 3 PDPH peaks (~7.4, 7.95, 8.45ppm) were compared to the acetyl peak of heparin(~2.1ppm) (Figure S1C).
Gel Formulations:
No Hep formulation:
A 3.2wt% (w/v) gel was used for this formulation. The final concentrations in the gel were 45.88mg/mL PEG-MAL (20kDa), 0.82 mg/mL RGD, 8.06 mg/mL MethMal, and 4.62 mg/mL MMP-2 crosslinker. Similar to our previous MAP formulations[40], for macrogel formulations, the PEG-MAL, RGD and MethMal were dissolved in a pH=4.5 10X PBS solution and the MMP-2 crosslinker and heparin were dissolved in pH=7.4 10X PBS. For microgel synthesis the PEG-MAL, RGD, and MethMal were dissolved in pH=1.5 10X PBS and the MMP-2 crosslinker along with 5μM of biotin labeled maleimide was dissolved in pH=7.4 1X PBS.
HepHigh formulation:
A 2.2wt% (w/v) 6 mg/mL heparin gel was used for this formulation. The final concentrations in the gel were 34.83 mg/mL PEG-MAL (10kDa), 0.82 mg/mL RGD, 8.06 mg/mL MethMal, 5.56mg/mL MMP-2 crosslinker, and 6 mg/mL heparin. For macrogel formulations, the PEG-MAL, RGD and MethMal were dissolved in a pH=4.5 10X PBS solution and the MMP-2 crosslinker and heparin were dissolved in pH=7.4 10X PBS. For microgel synthesis the PEG-MAL, RGD, and MethMal were dissolved in pH=1.5 10X PBS and the heparin, 5μM of Alexa Fluor labeled maleimide, and MMP-2 crosslinker was dissolved in pH=7.4 1X PBS.
HepLow formulation:
A 3.0wt% (w/v) 0.6 mg/mL heparin gel was used for this formulation. The final concentrations in the gel were 38.89 mg/mL PEG-MAL (10kDa), 0.82 mg/mL RGD, 8.06 mg/mL MethMal, 7.47 mg/mL MMP-2 crosslinker, and 0.6 mg/mL heparin. For macrogel formulations, the PEG-MAL, RGD and MethMal were dissolved in a pH=4.5 10X PBS solution and the MMP-2 crosslinker and heparin were dissolved in pH=7.4 10X PBS. For microgel synthesis the PEG-MAL, RGD, and MethMal were dissolved in pH=1.5 10X PBS and the heparin, 5μM of Alexa-Fluor labeled maleimide, and MMP-2 crosslinker was dissolved in pH=7.4 1X PBS.
Gelation kinetics:
Macro-scale gels (i.e. macrogels) of 400μL volume were formed on a ThermoScientific Viscometer (20 mm plate attachment). Sufficient gelation time was determined by a minimal change (<10%) in storage modulus over 15 minutes using HAAKE RheoWin (Figure S3).
Macrogel production:
Macro-scale gels (i.e. macrogels) were used to mechanically match hydrogel stiffnesses between groups to be approximately 15–20kPa. 200μL gel pucks were formed between SigmaCote® coated slides for 30 min past total gelation according to the viscometer. The macrogels were collected and swollen to equilibrium in PBS overnight at 37°C prior to testing.
Microgel production:
Microgels were produced using a PDMS mold for 45–55μm particles described in Rutte, et. al[20]. Picosurf surfactant (Sphere Fluidics) was diluted to a 1% solution in NOVEC 7500 (3M). The gel solutions were prepared as described above, however a 10X PBS solution pH=1.5 was used to dissolve the backbone solution to ensure gelation would not occur in the device. Using a syringe pump, the surfactant and aqueous solutions were run through the device at 5 mL/ hour and collected in a 50mL conical tube. The microgels were mixed with a solution of triethylamine (20μL/mL of gel) diluted in NOVEC 7500 to ensure complete gelation before purification.
Microgel purification:
Microgels were washed three times with NOVEC 7500 (1X gel volume). Next, microgels were swelled in PBS (5X volume) and washed 3 more times with NOVEC 7500, allowing separation of the aqueous and oil solutions via settling. The NOVEC oil was removed and microgels were washed with PBS (5X gel volume) and Hexanes (5X gel volume) and centrifuged at 4696gx5min. Particles were incubated with a 100mM N-Acetyl-L-Cysteine (Acros Organics) solution in PBS overnight at 37°C to quench excess maleimides.
Microgel sterilization:
All remaining steps were performed in a biosafety cabinet. After quenching, microgels were washed three times with 70% isopropanol (5X gel volume, 4696gx5min). Microgels were stored at 4°C in 70% isopropanol until being used.
Heparin Quantification in Skin:
Skin of Swiss Webster mice was explanted and digested to isolate heparin using an adaptation of the protocol from Zuo, et. al[41]. Briefly, the tissue specimens are extracted and digested with proteinase K. The amount of total sulfated GAG is quantified using a dimethylmethylene blue (DMMB) assay[42] (Figure S2). Following a protocol by Hahn, et. al[43], the heparin proportion of GAGs was determined by the loss of DMMB signal after sample exposure to heparinase following manufacturer’s protocol (R&D Biosystems).
Heparin Incorporation in Macrogels:
Heparin was tagged with AF555 during the PDPH modification. A standard curve of heparin fluorescence (Figure S4A–B) was produced by making serial dilutions of the heparin dissolved in PBS. 150μL of each solution was placed in a 96 well Greiner SensoPlate and imaged using an ImageXpress MicroConfocal Imaging System (Molecular Devices). A 5 mm biopsy punch of each macrogel containing heparin was placed into a well of a 96 well Greiner SensoPlate and covered with 100μL of PBS. Using the same focus and exposure time as the standard curve the macrogel is acquired. Using a custom ImageXpress module (Molecular Devices) the average intensities for each site of the macrogel are calculated (Figure S4C). The 4 sites with the smallest standard deviation are averaged and the concentration is calculated based on the standard curve (Figure S4D).
Mechanical Analysis of Macrogels:
Macrogels were removed from PBS and excess moisture was wicked from the pucks before testing. An Instron mechanical load device was used to test compressive stiffness (Young’s modulus) at a rate of 0.5 mm/min for 1 mm (Figure S5A) and BlueHill® software analyzed the load (N) and extension (mm) as previously reported by our group[38,44]. Stress-strain curves were produced and the Young’s modulus (Pa) was calculated using the MATLAB SLM package (Figure S5B).
Particle Size Characterization:
Microgel particle spherical diameter was determined similar to our group’s previous work[23,38,44] by using fluorescent images of a dilute solution (1:100) of microspheres and ImageXpress based quantification (Molecular Devices). A minimum of 500 particles (N) were analyzed to determine average diameter (D) and polydispersity index (PDI). The number average was calculated by Dn =∑ NiDi/∑ Ni and weight average was calculated by Dw =∑ NiDi2/∑NiDi. PDI was calculated by Dw/Dn. (Figure S6)
Growth Factor Sequestration Assay:
Platelet Derived Growth Factor (PDGF, R&D Biosystems) was biotinylated (NHS Sulfo EZ Link Biotin) according to manufacturer’s protocol (Fisher) to allow for post-fixation identification. 15μL MAP scaffolds (10% HepHigh) were embedded in 100μL collagen-agarose gel (1 mg/mL collagen, 1% agarose) for structural support. Heparin microgels were labeled with AF555 for post-fixation identification. After gelation at 37°C, the gels were soaked in 10% FBS for 4 hours to mimic in situ conditions. Next, the solutions were removed and gels were incubated with biotinylated PDGF at 1 μg/mL in 1% BSA for either 5 min, 1 hour, 8 hour, or 48 hour (N=3 for each time point) on a shaker plate at 37°C. Following incubation, the gels were fixed with paraformaldehyde for twenty minutes followed by three five-minute PBS washes. Finally, gels were stained with AF647 Streptavidin to visualize growth factor localization for 48 hours, followed by 48 hours of washing in a PBS solution. After PBS washes, the gels were imaged at 20x using a Zeiss 780 Confocal Microscope in the Keck Center for Biologic Imaging.
Quantification of growth factor sequestration:
Using ImageJ the multichannel Zeiss image was used for quantification. First, in the green channel a heparin particle was traced and the fluorescent intensity (ImageJ mean gray value) was measured for the tracing in the red channel which had PDGF (Figure S7). The fluorescence for at least four heparin particles was averaged for each sample (N=3 per time point) at each time point. To account for differences in any autofluorescence the mean fluorescence value was calculated by the mean gray value in the particle minus the mean gray value of the background.
Gradient Formation:
Platelet Derived Growth Factor (PDGF, R&D Biosystems) was biotinylated (NHS Sulfo EZ Link Biotin) according to manufacturer’s protocol (Fisher) to allow for post-fixation identification. MAP particles (no hep, HepLow, and HepHigh) were embedded (1:100) in 100μL collagen-agarose gel (1 mg/mL collagen, 1% agarose) for structural support. Particles were labeled with different Alexa Flour maleimide tags to identify the different types. After gelation at 37°C, the gels were soaked in 10% FBS for 8 hours to mimic in situ conditions. Finally, the solutions were removed and then incubated with the growth factor of interest at 1 μg/mL in 1% BSA for 48 hours on a shaker plate at 37°C. Following incubation, the gels were frozen in OCT and then sectioned into 10μm sections using a cryostat. To visualize the gradients, the slides were rehydrated with PBS and then stained with streptavidin labeled with Cy5 (1:200 dilution) for 1 hour at room temperature. Slides were washed three times with PBS and then coverslipped and imaged at 20x using an ImageXpress Micro-Confocal (Molecular Devices).
Image Analysis:
ImageJ based analysis was used to calculate the gradient formation. The particles were traced using ImageJ in the fluorescence channel that corresponded to the specific particle type. That tracing was overlayed on the Streptavidin fluorescent channel corresponding to the PDGF. 10μm concentric rings were generated surrounding the particle tracing and the mean gray value was measured using ImageJ (Figure S10). All values were subtracted from the background mean gray value in an area without a staining to generate the value above background fluorescence.
Sources of Cells:
Primary adult Human Dermal Fibroblasts (HDFs) and primary Human Dermal Microvascular Endothelial Cells (HDMVECs) were purchased from ATCC and cultured according to manufacturer’s guidelines.
Cell Migration- Spheroid Formation:
Before starting the study, HDFs and HDMVECs were fluorescently labeled using AF488 CellTracker following manufacturer’s guidelines (Fisher). Spheroids were formed at a concentration of 50,000 cells/mL for HDMVECs and 125,000 cells/mL for HDFs. Methylcellulose was prepared following a previously published protocol[45], and it was supplemented as 20% of the media to create circular spheroids. 20μL spheroids were incubated using the hanging drop method for 48 hours, and spheroid formation was confirmed using a brightfield microscope.
Cell Migration Assay:
Cell migration was performed using an assay previously developed to measure migration in hydrogel scaffolds[25]. MAP gel formulations were incubated in complete cell culture media at 37°C overnight before starting the study. After incubation, microgels were spun down at 4696gx5min and suspended in a 2mM LAP (Sigma) solution in media at a 1:1 concentration. Microgels were spun down again at a high speed and the excess solution was poured off. The groups for each study were 1% heparin high, 10% heparin high, 100% heparin high, 10% heparin low, 100% heparin low and no heparin gel. 50μL pucks of gel were annealed using UV light at 14.8mW/cm2 between sterile Sigma-coated slides for 30 seconds and then transferred to the bottom of a 24 well plate. Spheroids were pipetted onto the top of the gel and then imaged to get the 0-hour time point. At 24 hours the spheroids were imaged again using the ImageXpress Micro-Confocal (Molecular Devices). Fold change was measured using ImageJ as previously described[25] using Fiji (Figure S11).
Mouse Excisional Wound Healing Model:
All animal surgeries were performed in accordance with Animal Care and Use Committee guidelines under UVA Animal Protocol 4165. 8-week-old male db/db mice (Jackson stock: 000697 HOM 1) were used for all studies. Mouse excisional wound healing experiments were performed based on a previous protocol for mouse wound healing favoring cutaneous regeneration and preventing wound contraction by splinting[46].
Wound Healing Study 1 Experimental Groups:
The four groups evaluated in the initial diabetic wound healing study were μIslands, no heparin MAP as a materials control, Oasis as an advanced clinical control, and Aquaphor as a basic clinical control. Six mice were evaluated at Day 3 and Day 7.
Wound Healing Study 2 Experimental Groups:
In a follow-on diabetic wound healing study to examine the impact of heterogeneity the four groups were μIslands, 100% HepHigh, 100% HepLow, and no heparin. Eight mice were evaluated at Day 7, however due to one mouse removing all bandages each day it was removed from the study due to observed wound contraction.
Excisional Wound Healing Model Surgery:
Mice were anesthetized using continuous application of isoflurane throughout the duration of the procedure. Hair was removed from the dorsal side of the mouse the day prior to surgery using a combination of electric clippers and Nair, followed by PBS washes to remove the hair. Nails were trimmed to lessen the chance of splint removal. The day of surgery, the mouse backs were first sterilized with iodine and 70% isopropanol. A sterile 5 mm biopsy punch was used to create 4 symmetrical full thickness wounds on the back, two on each side of the midline. Surgical scissors were used to cut out the wound from the biopsy punch indention. Duoderm rings stacked 4 high were used to create the splints. Prior to surgery they were prepared by stacking four layers on top of one another and using a 12 mm biopsy punch to create the outside of the splint and 8 mm biopsy punch to create the inside of the splint. Duoderm splints were superglued (Gorilla Glue) around the wound to prevent contraction. Next, each treatment was applied to the wound in a randomized order which was rotated for each mouse. For MAP conditions, 12μL of gel was applied and smoothed into a flat surface using a positive displacement pipette. Scaffolds were annealed for 30 seconds using 365nm UV light (15.4cm away, 440mA, Thor Labs). Oasis was prepared by using a 5mm biopsy punch to cut out the matrix and then placing it directly on the wound. Aquaphor was spread on the wound with a sterile cotton swab. Pictures of each wound were taken after the treatment was applied. Finally, wounds were covered with small squares of Telfa pads which were superglued on the duoderm rings to protect the wounds. Mice received 0.1cc of Buprinex immediately following surgery and 24 hours later. Mice were singly housed and checked twice a day to confirm they had not removed their bandages. At the end of the wound healing experiments (3 days or 7 days) the wounds were imaged again. Mice were sacrificed by isoflurane overdose and cervical dislocation. The wound samples were retrieved with surgical scissors and immediately embedded in OCT and frozen with dry ice for cryosectioning.
Evaluation of Wound Closure:
Wounds were imaged on day 0 and either day 3 or 7 for each mouse. Closure fraction was determined by comparing the pixel area of the wound to the pixel area within the duoderm ring (Figure S14). Closure fractions were normalized to Day 0 for each condition. Wound closure analysis was performed with investigators blinded to the treatment group identity during analysis. Any wound that had the bandage removed at some point during the study by the mouse was removed from this analysis due to the potential for contraction.
Tissue Section Immunofluorescence:
OCT blocks were sectioned with 20μm sections with 2 sections at different locations in the wound on each slide at least 200μm apart. Slides containing tissue sections were stored at −80°C until staining. Slides containing tissue sections were first fixed in acetone for 10 min. Slides were then washed with PBS (3 × 5min) and then blocked with 1X PBS, 5% goat serum, 0.1% Triton-X and 1% FC block. Following blocking, the slides were incubated with the primary antibody prepared in 5% goat serum listed in Table S1 overnight at 4°C. Secondary antibodies were all prepared in 1X PBS at a 1:1000 dilution. Following incubations slides were washed in PBS (3×5min) to remove any primary antibody and then incubated with the secondary antibody for 1 hour at RT. Following secondary incubation slides were washed again with PBS. Next the slides were stained with DAPI at a 1:1000 dilution in PBS for 20min at RT. Slides were washed again and then mounted with Prolong Gold Antifade mounting medium and stored at 4°C before being imaged.
Keratin-14 Analysis- Epidermal Tongue Length:
Day 3 wound sections were stained with keratin-14 and imaged at 10X using an EVOS microscope. Epidermal tongue length was traced on at least one side of the wound for two sections at different locations within the wound. Tongue length was measured on ImageJ using the line tool and tracing the top of the epidermal tongue from where it starts to ends and measuring the length (Figure S20). The wound edge was determined by the change in morphology from the surrounding tissue typically indicated by an increase in thickness, slope downward, or lack of hair follicles. The wound closure was calculated using the epidermal tongue length as described in Figure S19[40].
Keratin-14 Analysis – Epidermal Thickness:
Epidermal thickness was quantified for wounds at Day 7. If a wound was not fully re-epithelialized, the thickness of the epidermal tongue was quantified. Epidermal thickness was quantified by tracing the area of keratin staining from the wound edges on ImageJ to calculate an area and then dividing it by the length of the wound, which was also measured using the ImageJ (Figure S21). Epidermal thickness was quantified for two sections from each wound.
CD31 Analysis:
Full wounds were imaged at 10X objective using a Molecular Devices Confocal Microscope to visualize CD31 staining and DAPI. Images were cropped to only contain the wound region and not surrounding tissue for quantification (Figure S26). Images were uploaded into the MATLAB REAVER GUI[47] and set to analyze all wounds for vasculature with settings of 400 as the minimum connected component area and vessel thickness set to 1. This batch analysis exported the vessel length and vessel area (Figure S22). These measurements were normalized by tracing the wound length and area in ImageJ. Additionally, a binary image of each quantification was exported. From this image we were able to export X,Y coordinates of each vessel location and determine the percentage of vessels in each quarter of the wound. Vascularization was quantified for two full sections from each wound.
Pericyte Analysis:
Full wounds were imaged at 20X objective using a Molecular Devices Confocal Microscope to visualize CD31, NG2, PDGFRB, and DAPI stains. Images were cropped to only contain the wound region and were analyzed for vessels as described above (Figure S26). The binary image of the vessels was overlayed with the NG2 channel from the original image using ImageJ. NG2 positive cells were cropped out of the image if they were not determined to be proximal to the endothelial cells (<5μm). The area of the NG2 positive staining that was determined to be proximal to vessels was calculated in ImageJ and normalized to the wound area (Figure S23).
Macrophage Analysis:
Full wounds were imaged at 20X objective using a Molecular Devices MicroConfocal Microscope to visualize DAPI, CD68, Arg-1, and iNOS staining. Stitched sections were cropped to only the wound bed and not surrounding skin (Figure S26). After cropping, background removal was performed using ImageJ and each channel was thresholded to obtain positive staining. A MATLAB code was used to determine co-localization (Figure S24). Macrophages were determined to be M1 if there was positive staining for DAPI, CD68, and iNOS. Macrophages were determined to be M2 if there was co-localized positive staining for DAPI, CD68, and Arg-1. Macrophages that only stained positive for DAPI and CD68 were M0. Macrophage density was determined by calculating the number of total macrophages divided by the wound area. Macrophage polarization was quantified for two full sections from each wound.
Statistical Analysis:
All statistical analysis was performed using GraphPad Prism software. Graphs represent mean +- standard deviation.
Supplementary Material
Acknowledgements
We would like to acknowledge Joe de Rutte and the Di Carlo lab for assistance with microfluidic design and device fabrication. We would also like to acknowledge Shayn Peirce-Cottler for providing expertise on angiogenesis analysis. We acknowledge the Keck Center for Cellular Imaging for the usage of the Zeiss 780 microscopy system (PI:AP; NIH-OD016446). Figure schematics created with BioRender.com. L.J.P. was supported by a National Science Foundation Graduate Research Fellowship and by the National Heart, Lung, and Blood Institute of the National Institutes of Health under Award Number F31HL154731. This work was partially supported through the US National Institutes of Health High Priority, Short-Term Project Award (1R56DK126020-01) and The Wallace H. Coulter Translational Partners Program at The University of Virginia.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Competing Interests
D.R.G. has financial interests in Tempo Therapeutics which aims to commercialize MAP technology.
Contributor Information
Lauren Pruett, Department of Biomedical Engineering, University of Virginia, 415 Lane Rd, Charlottesville, VA 22908.
Christian Jenkins, Department of Biomedical Engineering, University of Virginia, 415 Lane Rd, Charlottesville, VA 22908.
Neharika Singh, Department of Biomedical Engineering, University of Virginia, 415 Lane Rd, Charlottesville, VA 22908.
Katarina Catallo, Department of Biomedical Engineering, University of Virginia, 415 Lane Rd, Charlottesville, VA 22908.
Donald Griffin, Department of Biomedical Engineering, University of Virginia, 415 Lane Rd, Charlottesville, VA 22908; Department of Chemical Engineering, University of Virginia, 102 Engineer’s Way Charlottesville, VA 22904.
References
- [1].Greenhalgh DG, Journal of Trauma and Acute Care Surgery 1996, 41, 159. [Google Scholar]
- [2].Briquez PS, Hubbell JA, Martino MM, Advances in Wound Care 2015, 4, 479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Laiva AL, O’Brien FJ, Keogh MB, Journal of Tissue Engineering and Regenerative Medicine 2018, 12, e296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rao SS, Venkatesan J, Prabhu A, Rekha PD, Journal of Drug Delivery Science and Technology 2020, 55, 101385. [Google Scholar]
- [5].Pop MA, Almquist BD, Experimental Dermatology 2017, 26, 760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Futrega K, King M, Lott WB, Doran MR, Trends in Molecular Medicine 2014, 20, 137. [DOI] [PubMed] [Google Scholar]
- [7].Sakiyama-Elbert SE, Acta Biomater 2014, 10, 1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Liang Y, Kiick KL, Acta Biomater. 2014, 10, 1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Capila I, Linhardt RJ, Angewandte Chemie International Edition 2002, 41, 390. [DOI] [PubMed] [Google Scholar]
- [10].Hudalla GA, Murphy WL, Advanced Functional Materials 2011, 21, 1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Belair DG, Nhi Le N, Murphy WL, Chemical Communications 2014, 50, 15651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Griffin DR, Weaver WM, Scumpia PO, Di Carlo D, Segura T, Nature Materials 2015, 14, 737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Turner NJ, Badylak SF, Adv Wound Care (New Rochelle) 2015, 4, 490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kasiewicz LN, Whitehead KA, Biomater Sci 2017, 5, 1962. [DOI] [PubMed] [Google Scholar]
- [15].Singh N, Armstrong DG, Lipsky BA, JAMA 2005, 293, 217. [DOI] [PubMed] [Google Scholar]
- [16].Niezgoda JA, Van Gils CC, Frykberg RG, Hodde JP, Group O. D. U. S., Advances in Skin & Wound Care 2005, 18, 258. [DOI] [PubMed] [Google Scholar]
- [17].Parmaksiz M, Dogan A, Odabas S, Elçin AE, Elçin YM, Biomed. Mater 2016, 11, 022003. [DOI] [PubMed] [Google Scholar]
- [18].Jha AK, Mathur A, Svedlund FL, Ye J, Yeghiazarians Y, Healy KE, Journal of Controlled Release 2015, 209, 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Michaels J, Churgin SS, Blechman KM, Greives MR, Aarabi S, Galiano RD, Gurtner GC, Wound Repair Regen 2007, 15, 665. [DOI] [PubMed] [Google Scholar]
- [20].de Rutte JM, Koh J, Carlo DD, Advanced Functional Materials 2019, 29, 1900071. [Google Scholar]
- [21].Engler AJ, Sen S, Sweeney HL, Discher DE, Cell 2006, 126, 677. [DOI] [PubMed] [Google Scholar]
- [22].Koh J, Griffin DR, Archang MM, Feng A-C, Horn T, Margolis M, Zalazar D, Segura T, Scumpia PO, Carlo DD, Small 2019, 15, 1903147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Pfaff BN, Pruett LJ, Cornell NJ, de Rutte J, Di Carlo D, Highley CB, Griffin DR, ACS Biomater. Sci. Eng 2021, 7, 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rydergren S, Chemical Modifications of Hyaluronan Using DMTMM-Activated Amidation, Uppsala Universitet, 2013. [Google Scholar]
- [25].Nandi S, Brown AC, JoVE (Journal of Visualized Experiments) 2017, e56099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Velnar T, Bailey T, Smrkolj V, Journal of International Medical Research 2009, 37, 1528. [DOI] [PubMed] [Google Scholar]
- [27].Pastar I, Stojadinovic O, Yin NC, Ramirez H, Nusbaum AG, Sawaya A, Patel SB, Khalid L, Isseroff RR, Tomic-Canic M, Advances in Wound Care 2014, 3, 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Johnson KE, Wilgus TA, Advances in Wound Care 2014, 3, 647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic-Canic M, Wound Repair Regen 2008, 16, 585. [DOI] [PubMed] [Google Scholar]
- [30].Gerhardt H, Betsholtz C, Cell Tissue Res 2003, 314, 15. [DOI] [PubMed] [Google Scholar]
- [31].Brown BN, Ratner BD, Goodman SB, Amar S, Badylak SF, Biomaterials 2012, 33, 3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sussman EM, Halpin MC, Muster J, Moon RT, Ratner BD, Ann Biomed Eng 2014, 42, 1508. [DOI] [PubMed] [Google Scholar]
- [33].Ratner BD, Regen Biomater 2016, 3, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Alsop AT, Pence JC, Weisgerber DW, Harley BAC, Bailey RC, Acta Biomaterialia 2014, 10, 4715. [DOI] [PubMed] [Google Scholar]
- [35].Batalov I, Stevens KR, DeForest CA, PNAS 2021, 118, DOI 10.1073/pnas.2014194118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Freeman FE, Pitacco P, van Dommelen LHA, Nulty J, Browe DC, Shin J-Y, Alsberg E, Kelly DJ, Science Advances 2020, 6, eabb5093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bittner SM, Guo JL, Mikos AG, Bioprinting 2018, 12, e00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Pruett L, Koehn H, Martz T, Churnin I, Ferrante S, Salopek L, Cottler P, Griffin DR, Daniero JJ, The Laryngoscope 2020, 130, 2432. [DOI] [PubMed] [Google Scholar]
- [39].Pomin VH, Anal. Chem 2014, 86, 65. [DOI] [PubMed] [Google Scholar]
- [40].Pruett L, Ellis R, McDermott M, Roosa C, Griffin D, Journal of Materials Chemistry B 2021, DOI 10.1039/D1TB00715G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zuo H, Peng D, Zheng B, Liu X, Wang Y, Wang L, Zhou X, Liu J, J Mater Sci: Mater Med 2012, 23, 2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Barbosa I, Garcia S, Barbier-Chassefière V, Caruelle J-P, Martelly I, Papy-García D, Glycobiology 2003, 13, 647. [DOI] [PubMed] [Google Scholar]
- [43].Hahn MS, Jao CY, Faquin W, Grande-Allen KJ, Ann. Otol. Rhinol. Laryngol 2008, 117, 371. [DOI] [PubMed] [Google Scholar]
- [44].Schaeffer C, Pfaff BN, Cornell NJ, Salopek LS, Shan S, Viyar J, Omesiete W, Griffin DR, Cottler PS, DeGeorge BRJ, Annals of Plastic Surgery 2020, 84, S446. [DOI] [PubMed] [Google Scholar]
- [45].Pfisterer L, Korff T, in Angiogenesis Protocols (Eds.: Martin SG, Hewett PW), Springer, New York, NY, 2016, pp. 167–177. [Google Scholar]
- [46].Dunn L, Prosser HCG, Tan JTM, Vanags LZ, Ng MKC, Bursill CA, JoVE (Journal of Visualized Experiments) 2013, e50265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Corliss BA, Doty RW, Mathews C, Yates PA, Zhang T, Peirce SM, Microcirculation 2020, 27, e12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.