

Abstract
Kidney disease affects intestinal structure and function. Although intestinal lymphatics are central in absorption and remodeling of dietary and synthesized lipids/lipoproteins, little is known about how kidney injury impacts the intestinal lymphatic network, or lipoproteins transported therein. To study this, we used puromycin aminoglycoside-treated rats and NEP25 transgenic mice to show that proteinuric injury expanded the intestinal lymphatic network, activated lymphatic endothelial cells and increased mesenteric lymph flow. The lymph was found to contain increased levels of cytokines, immune cells, and isolevuglandin (a highly reactive dicarbonyl) and to have a greater output of apolipoprotein AI. Plasma levels of cytokines and isolevuglandin were not changed. However, isolevuglandin was also increased in the ileum of proteinuric animals, and intestinal epithelial cells exposed to myeloperoxidase produced more isolevuglandin. Apolipoprotein AI modified by isolevuglandin directly increased lymphatic vessel contractions, activated lymphatic endothelial cells, and enhanced the secretion of the lymphaniogenic promoter vascular endothelial growth factor-C by macrophages. Inhibition of isolevuglandin synthesis by a carbonyl scavenger reduced intestinal isolevuglandin adduct level and lymphangiogenesis. Thus, our data reveal a novel mediator, isolevuglandin modified apolipoprotein AI, and uncover intestinal lymphatic network structure and activity as a new pathway in the crosstalk between kidney and intestine that may contribute to the adverse impact of kidney disease on other organs.
Keywords: Kidney, intestine, lymphatics, HDL, reactive dicarbonyls, isolevuglandin
Graphical Abstract
Introduction
Kidney disease is a strong modulator of the composition and metabolism of the intestinal microbiome that produces toxins such as phenols (p-cresyl sulfate), indoles (indoxyl sulfate) and trimethylamine N-oxide1, 2. Kidney injury also disrupts the intestinal barrier promoting translocation of bacterial components and endotoxins into the circulation, which then initiate immune activation and proinflammatory signaling3–6. The primary pathways for mediators in the kidney-gut crosstalk involves blood vessels and nerves2. Little attention has been given to lymphatics which are unique in that in addition to clearing interstitial fluid, macromolecules, and immune/inflammatory cells, are responsible for absorption, transport of dietary lipids as well as intestinally-produced apolipoprotein AI (apoAI)/high density lipoprotein (HDL). Lymphatics are also a primary conduit for transport of ApoAI/HDL from the interstitium to the circulation7. Disruptions in lymph transport and lymphatic vessel integrity are powerful potentiators of disease, including cardiovascular disease (CVD), inflammatory bowel disease, and chronic kidney disease (CKD)8, 9.
Abnormalities plasma lipids/lipoproteins observed in many diseases have been ascribed to changes in production and modifications by the liver10. By contrast, although intestines are also a key source of apoAI/HDL, there is little information about the intestinal contribution to abnormal lipoproteins prevailing in disease. Intestinal microbial variation has recently been shown to modulate plasma lipids, especially metabolism of VLDL and HDL, including the reverse cholesterol transport function of HDL11, 12. This may be pathophysiologically germane since modifications in the structure and composition of apoAI/HDL determine the particle’s beneficial/detrimental effects.
A key mechanism in lipoprotein modification involves adduction by reactive carbonyls including malondialdehyde, 4-hydroxynonenal, 4-oxo-neonenal, and the most reactive among all the carbonyls, isolevuglandin (IsoLG) which impairs the fundamental actions of apoAI: cholesterol efflux, anti-inflammation, and anti-oxidation13, 14. Kidney disease alters the composition and functionality of HDL and increases plasma protein adducts15–19. IsoLG adducts have been detected along the gastrointestinal tract of animals and humans and participate in high salt hypertension linked to changes in the gut microbiome20, 21. These findings raise the possibility that kidney disease-induced intestinal changes include modulation of IsoLG production that adducts intestinal ApoAI/HDL which are then transported by the lymphatic network22, 23. Here we assessed whether proteinuric kidney injury impacts intestinal lymphatics as a novel pathway to deliver gut-generated metabolites and investigated the role of intestinal IsoLG-modified ApoAI/HDL particles in this pathway.
Methods
Animals
Proteinuric kidney injury in adult male Sprague Dawley rats (200–225g, 10 weeks old, Charles River, Wilmington, MA) was induced by a single injection of puromycin aminoglycoside (PAN) (125 mg/kg body weight, i.p.) while saline-injected rats served as controls (Cont)24. Eight days after injection, rats were sacrificed, and blood, urine, and tissues were harvested. Systemic and renal parameters are shown in Table 1. We also studied adult male (12 weeks old) Nphs1-hCD25 transgenic mice (NEP25, C57 bl/6 background), expressing human CD25 on podocytes. These mice then can be selectively injured by recombinant immunotoxin, anti-Tac (Fv)-PE38 (LMB2, 1ng/g BW, i.v. generously provided by Dr. Ira Pastan) resulting in proteinuria25, 26. Two weeks after LMB2, mice were sacrificed, and blood, urine and tissues harvested. Systemic and renal parameters were shown in Table 2. Rats and mice were housed under a 12-h light/dark cycle with free access to regular rodent chow and water. All animal procedures were approved by the Institutional Animal Care and Use Committee at Vanderbilt University.
Table 1:
Systemic and renal parameters in Cont and PAN rats
| Cont | PAN | Statistical significance | |
|---|---|---|---|
|
| |||
| BW (g) | 299.5±24.4 | 291.8±25.9 | pNS |
| SBP (mmHg) | 112.6±8.7 | 115.8±7.5 | pNS |
| BUN (mg/dL) | 29.8±11.0 | 125.7±45.4 | p<0.001 |
| ACR (μg/mg) | 37.2±9.5 | 126652.2±44055.9 | p<0.001 |
| Plasma Albumin (mg/dL) | 523.8±75.67 | 350.7±86.46 | p<0.001 |
| Plasma Cholesterol (mg/dL) | 44.0±12.3 | 236.6±116.4 | p<0.001 |
| Plasma Triglyceride (mg/dL) | 34.9±9.7 | 132.1±60.6 | p<0.001 |
Data expressed as mean±SD
n=12 in each group
Table 2:
Systemic and renal parameters in WT and NEP25 mice
| WT | NEP25 | Statistical significance | |
|---|---|---|---|
|
| |||
| BW (g) | 24.3±1.4 | 24.4±1.1 | pNS |
| SBP (mmHg) | 105.8±8.8 | 112.6±8.8 | pNS |
| BUN (mg/dL) | 52.0±7.4 | 50.5±6.9 | pNS |
| ACR (μg/mg) | 27.3±3.7 | 2114.4±636.9 | p<0.001 |
Data expressed as mean±SD
n=8 in each group
Assessments of lymph, plasma, and urine composition
Mesenteric lymph was collected in a subset of conscious rats following cannulation of the mesenteric lymph duct27. The rats were placed in Bollman cages in a temperature- and humidity-controlled incubator and lymph collected hourly for 3h.
Albumin (Exocell, Philadelphia, PA), apoAI (Mybiosource, San Diego, CA), sphingosine-1 phosphate (S1P) (Mybiosource) and VEGF-C levels (Mybiosource) were measured by ELISA. Albuminuria was measured as urine albumin-to-creatinine ratio (ACR) (Exocell) and QuantiChrom™ Creatinine Assay Kit (Bioassay Systems, Hayward, CA), respectively. Quatichrom™ urea assay kit (Bioassay Systems) was used to measure plasma BUN. Plasma and lymph total cholesterol and triglycerides were measured enzymatically (Cliniqa, CA). HDL and LDL fractions were isolated from plasma and lymph by density-gradient ultracentrifugation28. Lipoprotein particle size was evaluated by NMR (Liposcience, Morrisville, NC). Colorimetric assays were used to measure protein (BCA, ThermoFisher, Rockford, IL). Lipoproteins in plasma and lymph were separated by size-exclusion chromatography (SEC) using an Akta Pure fast-protein liquid chromatography (FPLC) system (GE Healthcare, Chicago, IL). Total IsoLG-protein adduct by LC/MS was measured as IsoLG-lysine after complete proteolytic digestion of lymph samples29. Ileum MPO activity was assessed with 10-acetyl-3,7-dihydroxyphenoxazine (ADHP, ABD Bioquest Inc, Sunnyvale, CA) at an excitation wavelength of 535 nm and an emission wavelength of 590 nm30.
Plasma and lymph levels of interleukin-6 (IL-6), IL-10, IL-17, and IL-1 were determined by Luminex multiplex28. Immune cells in lymph were quantitated by flow cytometry. The samples were incubated with Fc blocking antibody (BD Biosciences, San Jose, CA), then incubated with BV421-conjugated anti-CD3 (BD Biosciences), PE/Cy7-conjugated anti-CD4 (Biolegend, San Diego, CA), Percp-conjugated anti-CD8 (Biolegend), Alexa Flour 488-conjugated anti-CD25 (Biolegend) or PE-conjugated anti-CCR6 (R&D Systems, Minneapolis, MN). Cells were analyzed on a FACSCanto II cytometer with FACSDiva software (BD Biosciences).
Immunostaining on intestinal tissue
Ileal sections were fixed in 4% paraformaldehyde/PBS, paraffin embedded and sections cut. We assessed the ileum because lymph drains into the mesenteric lymph vessels and because the ileum is critical in apoAI synthesis31. For podoplanin staining, rat ileal sections were incubated with mouse anti-podoplanin antibody (1:1000, Novus, Littleton, CO) followed by anti-mouse horseradish peroxidase (HRP) (Vector Laboratories, Burlingame, CA). Mouse ileal sections were incubated with hamster anti-podoplanin antibody (1:2000, ThermoFisher) followed by biotinylated anti-hamster antibody (Vector Laboratories) and ABC reagent. The signal was visualized by diaminobenzidine. Quantitation was done by Image J and represented as percentage of positive area.
Double staining for apoAI and podoplanin used citrate buffer for antigen retrieval followed by primary antibody apoAI (1:200; Novus). ImmPRESS reagent (Vector Laboratories) and Alexa Fluor 488 Tyamide SuperBoost (ThermoFisher) were used as secondary antibodies. Sections were incubated with mouse anti-rat podoplanin followed by anti-mouse HRP and Alexa Fluor 546 Tyamide SuperBoost. Double staining for IsoLG and apoAI used citrate buffer for antigen retrieval, followed by anti-IsoLG (1:10). The second antibody used ImmPACT® Vector® Red Alkaline Phosphatase Substrate kit (Vector Laboratories) as a chromogen. Sections were then incubated with rabbit anti apoAI, followed by anti-rabbit HRP and Alexa Fluor 488 Tyamide SuperBoost. Double-staining for CD68 and VEGF-C used citrate buffer for antigen retrieval, followed by biotinylated primary antibody targeting CD68 (1:10; BioRad, Hercules, CA). The second antibodies were ABC reagent and Alexa Fluor 488 Tyamide SuperBoost. Sections were then incubated with mouse anti-rat VEGF-C (1:200; Abcam, Cambridge, MA), followed by anti-mouse HRP and Alexa Fluor 546 Tyamide SuperBoost. CD68+ cells or CD68+ and VEGFC+ cells were assessed as number per villus. Slides treated with nonspecific antisera instead of primary antibody were used as negative control.
Measurement of lymphatic dynamics ex vivo
Mesenteric lymphatic vessels were collected and mounted in a perfusion chamber32, 33. Chambers were placed on inverted microscopes equipped with a digital image capture system (IonOptix, Westwood, MA) to record intraluminal diameters and frequency of contractions. Vessels were warmed to 37°C, pressurized to 0.5mmHg using a column of Krebs buffer and allowed to equilibrate (20–60 minutes). Viable vessels were then pressurized in a stepwise manner to a constant pressure of 3.5mmHg, then exposed to purified apoAI or modified apoAI using 1 molar equivalent synthetic IsoLG or vehicle (DMSO)14. Fresh Krebs buffer was circulated to facilitate wash out. Vessels were then exposed to IsoLG-modified apoAI. After each challenge, lumen diameters were allowed to plateau (40–60 minutes).
Characterization of intestinal lymphatic endothelial cells
Ilea from PAN and control rats were minced, then incubated with collagenase type D (Roche, Basel, Switzerland), 1mL HBSS medium and 10 μl/mL DNase for 1h and filtered using 70 μm then 40 μm sieves. Cells were resuspended and incubated with podoplanin-selective reagent (Novus). Lymphatic endothelial cells, i.e., podoplanin-positive cells, were isolated using the EasyEights magnetic cell separation system (Stemcell, Cambridge, MA)34.
Total RNA was isolated from ileum lysates and ileal podoplanin-positive cells by RNase Mini kit (QIAGEN, Germantown, MD). Reverse transcription was performed using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Branchburg, NJ). Quantitative real-time PCR was performed in a total reaction volume of 25 μL using 12.5 μL Universal Master Mix II, 1.25 μL forward and reverse primers [podoplanin (PDPN), lymphatic endothelial receptor (LYVE1), vascular endothelial growth factor receptor 3 (VEGFR3, FLT4), sphingosine kinase 2 (SPHK2), sphingolipid transporter 2 (SPNS2), C-C motif chemokine ligand 21 (CCL21) and nitric oxide synthase 3 (eNOS, Nos3)] (ThermoFisher) and 11.25 μL cDNA (10ng/μL). Quantitative real-time PCR used the CFX96™ Real-Time PCR Detection System (RT-PCR, Bio-Rad). Experimental cycle threshold (Ct) values were normalized to 18S measured on the same plate, and fold differences in gene expression were determined by the 2−ΔΔCt method35.
Cell culture
Mouse small intestine epithelial cells (MSIE, generously provided by Dr. Yan Fang)36 were incubated with myeloperoxidase (MPO, 7.5μg/ml, Lee Biosolutions, Maryland Heights, MO) for 24h. Total protein was extracted for quantitation of IsoLG.
Primary adult dermal lymphatic endothelial cells (LECs) (HMVEC-DLyAd, Lonza, Basel, Switzerland) were cultured with conditioned growth medium (Lonza). Cells at passage 5–6 with ~70% confluence were starved in serum-free medium overnight, then incubated with unmodified or IsoLG-modified apoAI (apoAI: 10μg/ml, IsoLG: 1μM/L) for 18h. Previously, we showed that this concentration of IsoLG yields levels of IsoLG-lysine adducts observed in vivo and does not produce unreacted IsoLG14. Quantification of eNOS (Nos3) and β-actin (ACTB) mRNA was performed by RT-PCR and production of superoxide was assessed by high-performance liquid chromatography37.
THP-1 cells were plated and differentiated into macrophages by RPMI 1640 containing 10% FBS and 50 ng/ml phorbol 12-myristate 13-acetate for 3 days38. Cells were incubated with unmodified or IsoLG-modified apoAI (apoAI: 10μg/ml, IsoLG: 1μM/L) for 48h. VEGFC mRNA was assessed by RT-PCR.
Statistical analysis
Data are expressed as mean±SD. The difference between two independent groups was examined with non-parametric Wilcoxon rank sum test, while more than two groups were compared with non-parametric Kruskal-Wallis test. A P-value of 0.05 or less was considered statistically significant.
Results
Proteinuric kidney injury increases mesenteric lymph flow and changes lymph composition
PAN rats developed ascites, proteinuria, hypoalbuminemia, increased plasma cholesterol and triglycerides compared with controls (Table 1). The proteinuric injury caused a striking increase in the mesenteric lymph flow (Fig 1A). Mesenteric lymph in PAN had reduced albumin, and despite increased lymph volume, total mesenteric albumin output was less in PAN vs controls (Fig 1B). Lymphatic concentrations of cholesterol and triglycerides were lower in PAN vs controls. However, total lymphatic output of these lipids increased, paralleling elevated plasma lipids (Fig 1C, Table 1). PAN did not affect lymphatic LDL particle size although triglyceride-containing particles corresponding to VLDL were smaller and HDL particles larger in lymph of PAN vs controls (Fig 1D). Further analysis of HDL particles showed increased total protein, total cholesterol, and phospholipids in fractions coinciding with spherical HDL (Fig 1E). The plasma HDL particle size was not changed (PAN:10.7±0.1 vs Cont:10.5±0.1nm, pNS).
Figure 1. Proteinuric kidney injury increases mesenteric lymph flow and changes lymph composition.
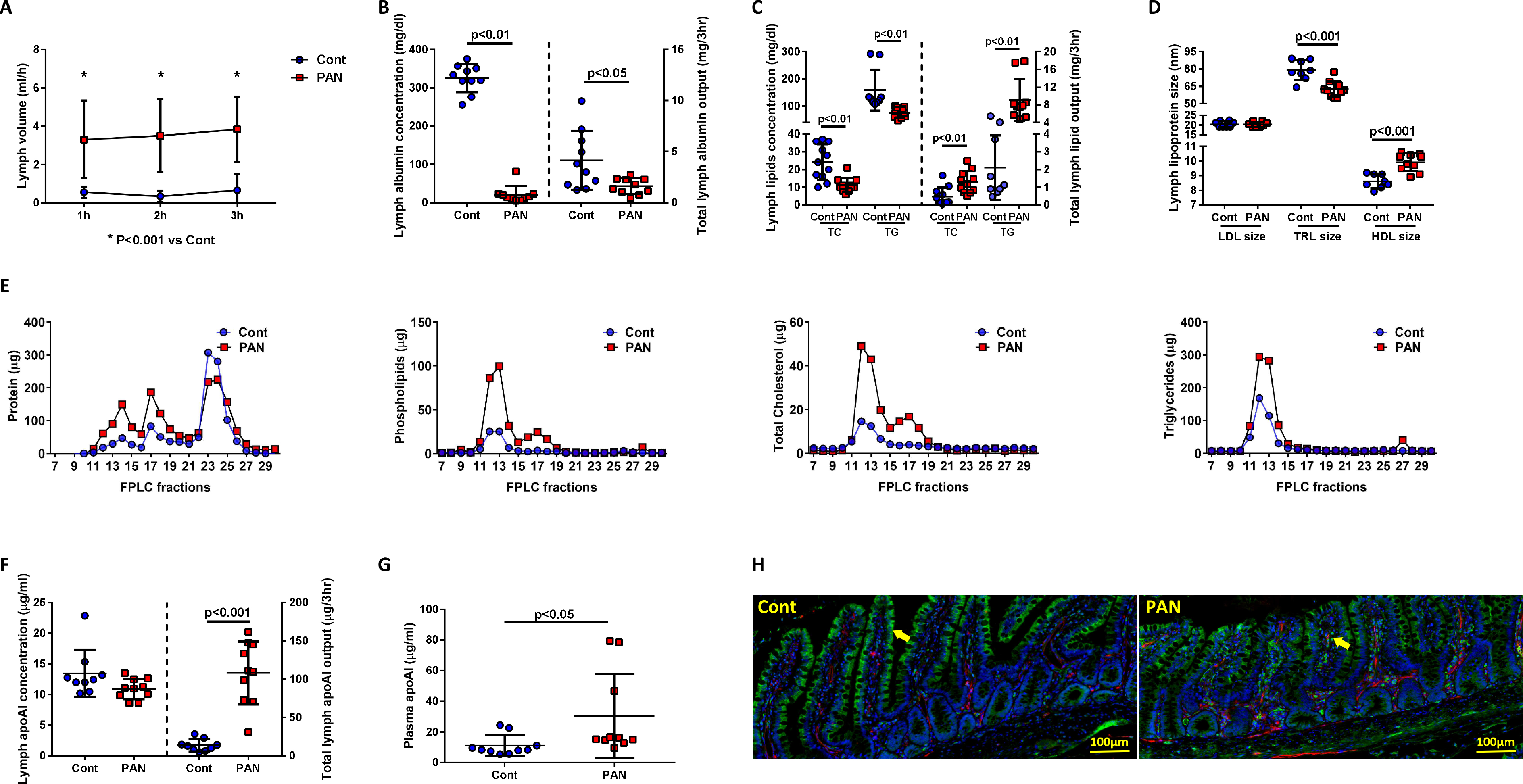
(A) Lymphatic flow rate in mesenteric vessels was consistently higher in PAN-injured animals vs controls. (B) Albumin concentration and output in mesenteric lymph were significantly decreased in PAN vs controls. (C) Cholesterol and triglyceride concentrations in mesenteric lymph were lower in PAN; total output of cholesterol and triglycerides in mesenteric lymph was significantly higher in PAN vs controls. (D) NMR analysis of lipoprotein particles in mesenteric lymph showed similar LDL particles, smaller triglyceride (TRL)-containing particles and larger HDL particles in PAN vs controls. (E) Size-exclusion chromatography (SEC) on the FPLC systems found PAN increased protein, cholesterol, and phospholipids in fractions coinciding with spherical HDL and chylomicrons and increased triglycerides in fractions corresponding to chylomicrons. (F) Lymph apoAI concentration was similar in PAN vs controls; total mesenteric lymph apoAI output was increased in PAN vs controls. (G) Plasma apoAI concentration was increased in PAN vs controls. (H) Double staining of ileal tissue with apoAI (green) and podoplanin (red) showed conspicuous apoAI located in intestinal epithelial cells of controls (arrow), while in PAN injured rats, strong apoAI staining was observed in lacteals (arrow). Results are mean ± SD for 8–10 rats per group.
Since 30% of apoAI in the circulation originates in the ileum39, we assessed the intestinal and lymphatic levels of apoAI. Ileal apoAI protein level was not different (PAN:1.06±0.06 vs Cont:1.16±0.17μg/mg, pNS), although the total mesenteric output of apoAI was significantly higher in PAN vs controls (Fig 1F). This was attended by a higher level of plasma apoAI in PAN vs controls at this early stage of injury (Fig 1G). The ileum of PAN rats showed more prominent apoAI that localized in the lymphatic lacteals, while in the ileum of control rats ApoAI stained strongly in the epithelial cells (Fig 1H).
PAN injury increased the number of Th17 cells (CD3+/CD4+/CCR6+) (Fig 2A, 2B) and cytokines, including IL-6, IL-10 and IL-17 (Fig 2C). Plasma cytokines obtained at the same time were not different in PAN vs controls (Fig 2D). These results demonstrate that proteinuric kidney injury increases mesenteric lymph flow rate, lymph lipids, lipoproteins, immune cells, and cytokines, but that many of these changes are not paralleled by changes in plasma.
Figure 2. Proteinuric kidney injury alters immune cells and cytokines in mesenteric lymph.
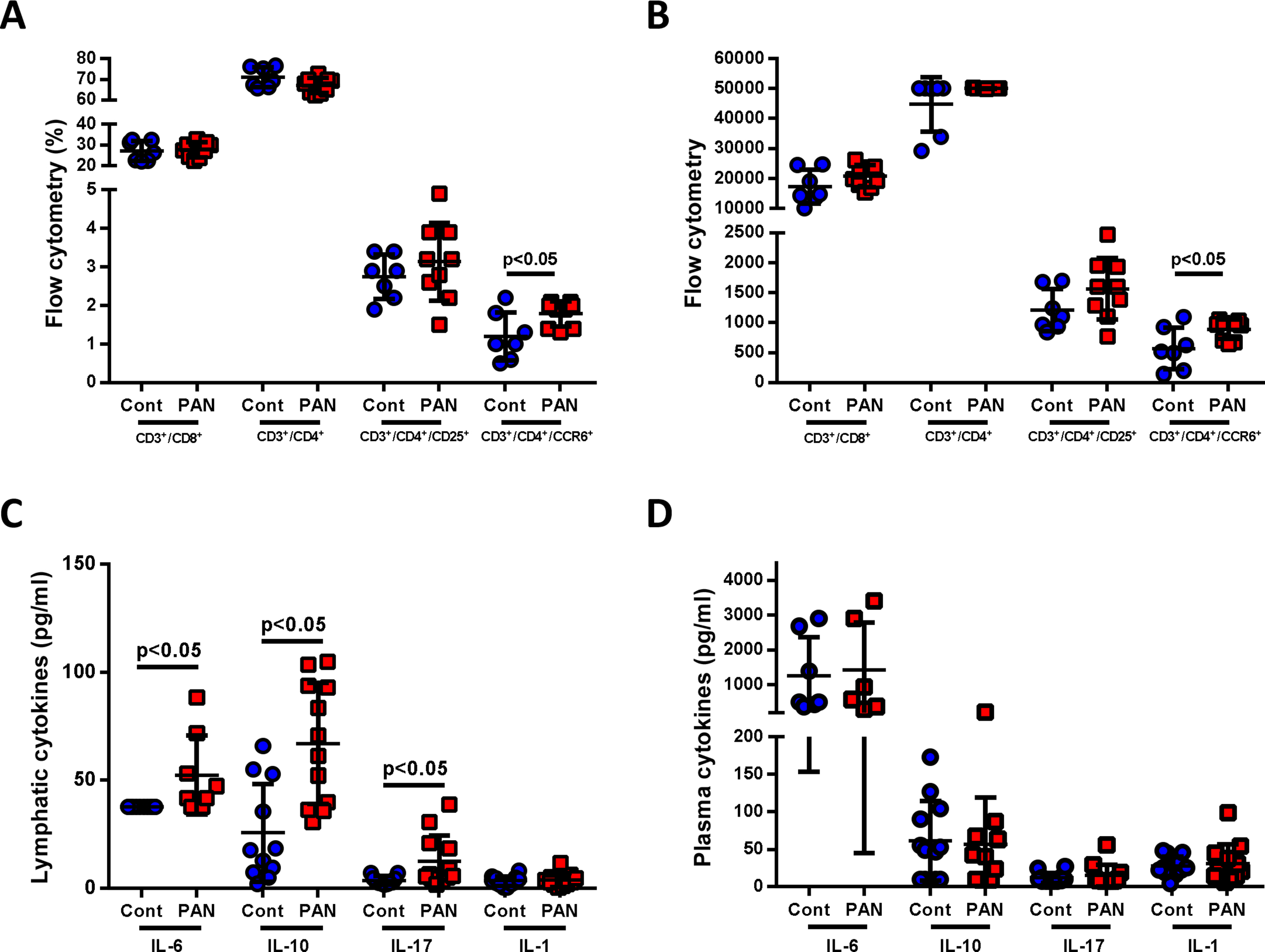
(A, B) Flow cytometry of mesenteric lymph showed more Th17 cells (CD3+/CD4+/CCR6+) in lymph of PAN vs controls. A: percentage of the CD3+ cells, B: absolute number of each cell type. (C) Mesenteric lymph showed more IL-6, IL-10 and IL-17 in PAN vs controls, while IL-1 was not different. (D) Plasma IL-6, IL-10, IL-17 and IL-1 showed no difference between PAN injured rats vs controls. Results are mean ± SD for 6–12 rats per group.
Proteinuric kidney injury expands the intestinal lymphatic vascular network and alters the lymphatic endothelial cell phenotype
Lymphangiogenic markers, podoplanin, LYVE-1 and VEGFR3, were all increased in ileum of PAN vs controls (Fig 3A). Complementing higher mRNAs, PAN ileum showed greater podoplanin-positive lymphatic vessels by IHC vs controls (Fig 3B). These findings were corroborated in the transgenic NEP25 proteinuric mouse, which showed increased ileal gene expression of podoplanin and VEGFR3 (FLT4) vs wild type mice (Fig 3C). Similar to PAN rats, proteinuric mice had increased ileal podoplanin expression compared to uninjured mice (Fig 3D).
Figure 3. Proteinuric kidney injury expands the mesenteric lymphatic vascular network and activates lymphatic endothelial cells (LECs).
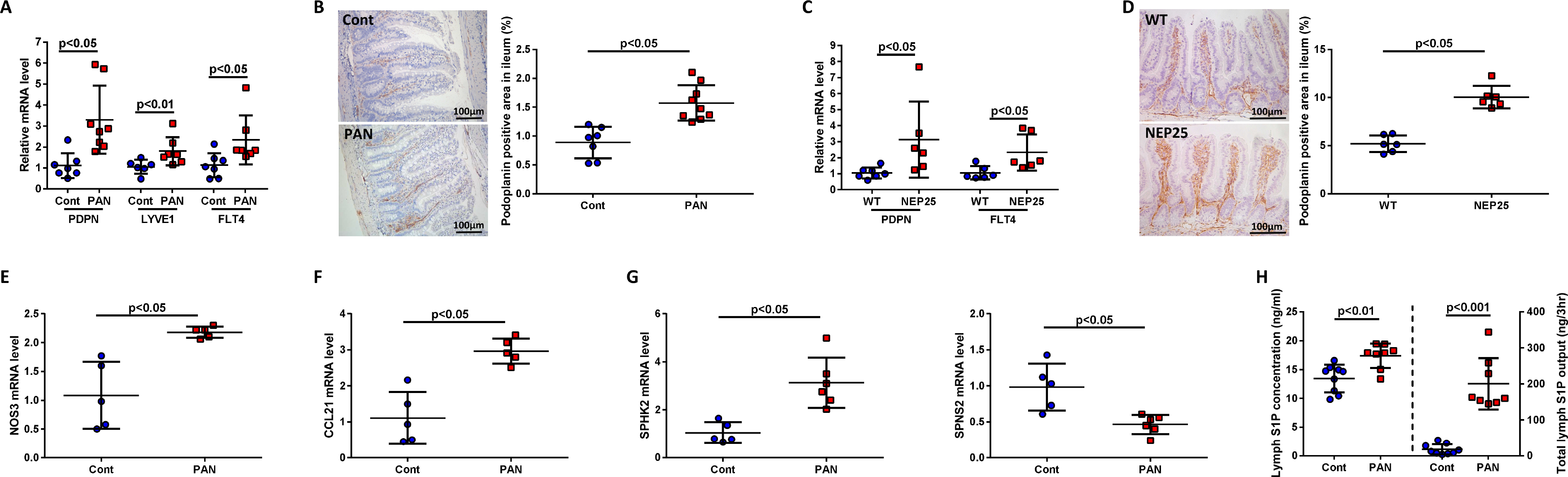
(A) PAN increased ileal expression of lymphangiogenic factors, including podoplanin (PDPN), LYVE-1 (LYVE1) and VEGFR3 (FLT4) mRNA. (B) Staining of PAN injured rats showed increased podoplanin expression vs controls. (C) NEP25 increased ileal gene expression of podoplanin (PDPN) and VEGFR3 (FLT4). (D) Staining of NEP25 ileum showed increased podoplanin expression compared to WT mice. (E) PAN increased eNOS (Nos3) mRNA expression in ileal podoplanin-positive LECs vs controls. (F) PAN kidney injury significantly increased expression of the chemoattractant CCL21 mRNA in ileal podoplanin-positive LECs. (G) Podoplanin-positive LECs isolated from ilea of PAN showed greater SPHK2 mRNA and less SPNS2 mRNA vs ilea of normal controls. (H) Mesenteric lymph from PAN rats had more S1P than control lymph. Results are mean ± SD for 5–9 rats per group and 6–7 mice per group.
To determine if renal injury affected the lymphatic endothelial cells (LECs), expression of key genes was quantified by real-time PCR from LECs isolated from the ileum of PAN and control rats. Endothelial-specific nitric oxide (Nos3), a critical mediator of vasodilation, was significantly increased in intestinal LECs isolated from PAN vs control rats (Fig 3E). PAN also significantly upregulated intestinal expression of the chemokine CCL21, a key mediator of immune cell recruitment (Fig 3F). Podoplanin-positive LECs isolated from PAN ilea showed higher SPHK2 and decreased SPNS2 expression –key regulators of S1P production, which stimulates lymphocyte migration and survival (Fig 3G)40. The mesenteric lymph of PAN contained more S1P vs controls (Fig 3H). Together, these results support the concept that proteinuric kidney injury increases key genes in intestinal LECs involved in lymphangiogenesis, vasodilation and immune cell chemoattraction.
Proteinuric kidney injury increases IsoLG-modified lipoproteins that regulate lymphatic endothelial cells and vessel dynamics
Kidney injury increases oxidative stress and lipid peroxidation that generate a diverse family of lipid aldehydes, including IsoLG, which compromise apoAI functionality14, 41. Both PAN rats and NEP25 mice, had more total IsoLG-lysine content in the ileum vs controls (Fig 4A, 4B) and PAN rats had elevated IsoLG-lysine levels in the mesenteric lymph (Fig 4C), but not in concurrently sampled plasma (Fig 4D). Myeloperoxidase (MPO), a peroxidase enzyme activated at all stages of renal damage42 was elevated in the intestinal wall of proteinuic rats (Fig 4E). Notably, IsoLG production was significantly increased in cultured mouse intestinal cells (MSIE) exposed to MPO compared to vehicle (Fig 4F). ApoAI and IsoLG showed greater co-localization by immunostaining in PAN ilea vs controls (Fig 4G).
Figure 4. Proteinuric kidney injury stimulates ileal production of IsoLG.
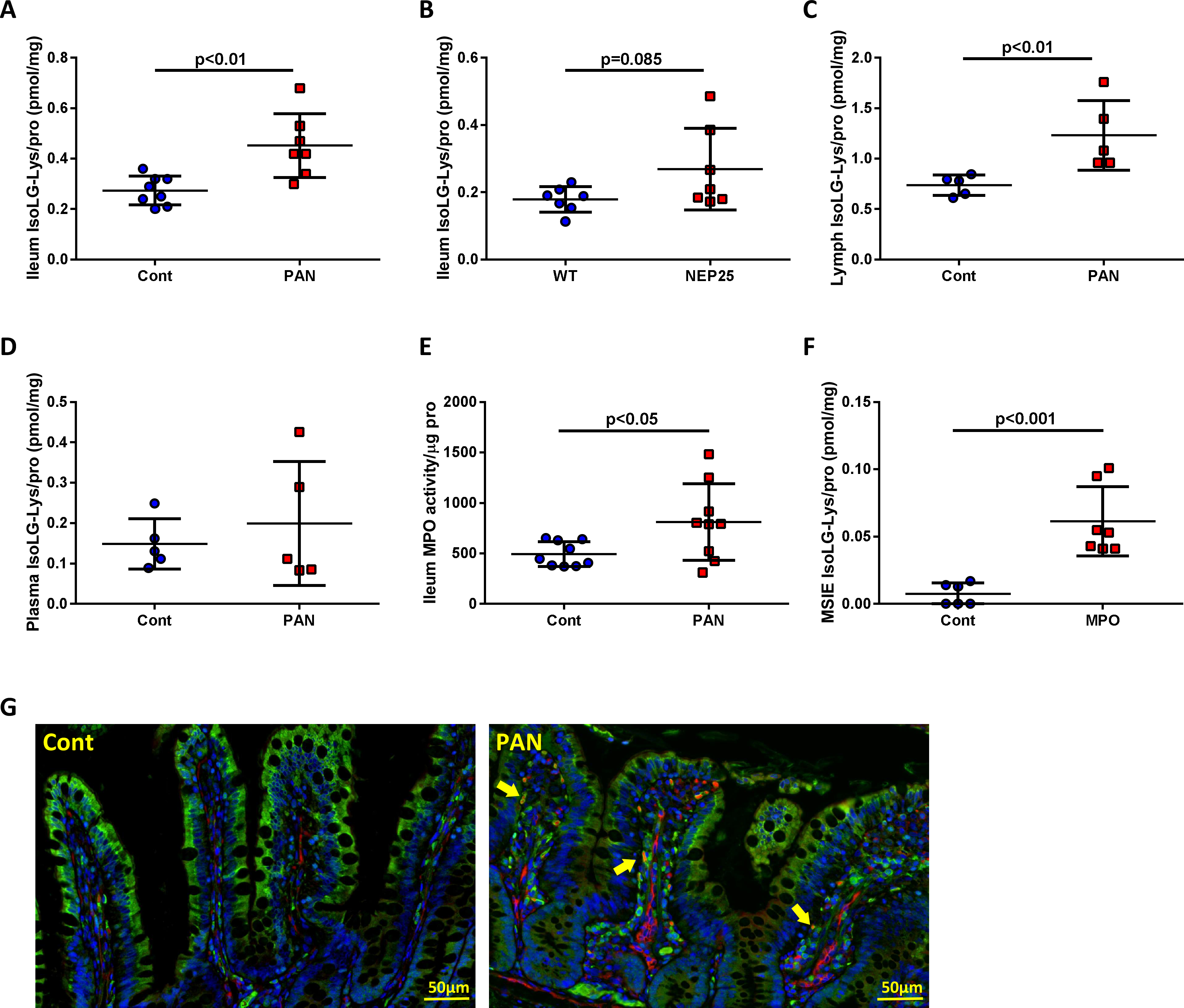
(A) PAN increased IsoLG adducts in ileal tissue vs controls. (B) Ileum IsoLG adducts was numerically increased in NEP25 vs WT. (C) PAN mesenteric lymph contained more IsoLG adducts compared to controls. (D) Plasma IsoLG adducts did not show difference between PAN and controls. (E) PAN increased ileum MPO activity vs controls. (F) In vitro study showed MPO increased MSIE IsoLG adducts vs normal medium exposed cells. (G) Double staining of apoAI (green) and IsoLG (red) in ileum of PAN showed increased IsoLG adducts in lacteals that colocalized with apoAI (arrows) vs controls. In vivo, results are mean ± SD for 5–9 rats per group and 7 mice per group. In vitro, results are mean ± SD for 6–7 wells per group.
To determine if IsoLG-apoAI directly affects lymphatics, cultured LECs were exposed to IsoLG-apoAI. IsoLG-apoAI stimulated production of reactive oxygen species (ROS) compared to unmodified apoAI (Fig 5A). Compared to untreated cells, ApoAI reduced Nos3, while IsoLG-apoAI increased Nos3 (Fig 5B). In ex vivo studies, isolated mesenteric lymphatic vessels exposed to IsoLG-apoAI showed increased frequency of contractions compared to unmodified apoAI (Fig 5C). Although IsoLG-apoAI failed to significantly alter end-systolic diameter (ESD) (Fig 5D), end-diastolic diameter (EDD) was significantly decreased (Fig 5E) together with a significantly reduced amplitude of contraction in IsoLG-apoAI treated lymphatics (Fig 5F). These studies reveal a direct effect of IsoLG-apoAI on lymphatic vessel dynamics which are distinct from native apoAI, suggesting frequency of contraction is a driving force in the increased lymph flow we observed in proteinuric injury in vivo.
Figure 5. IsoLG modified apoAI activates cultured lymphatic endothelial cells (LECs) and alters vasodynamics of isolated mesenteric lymph vessels.
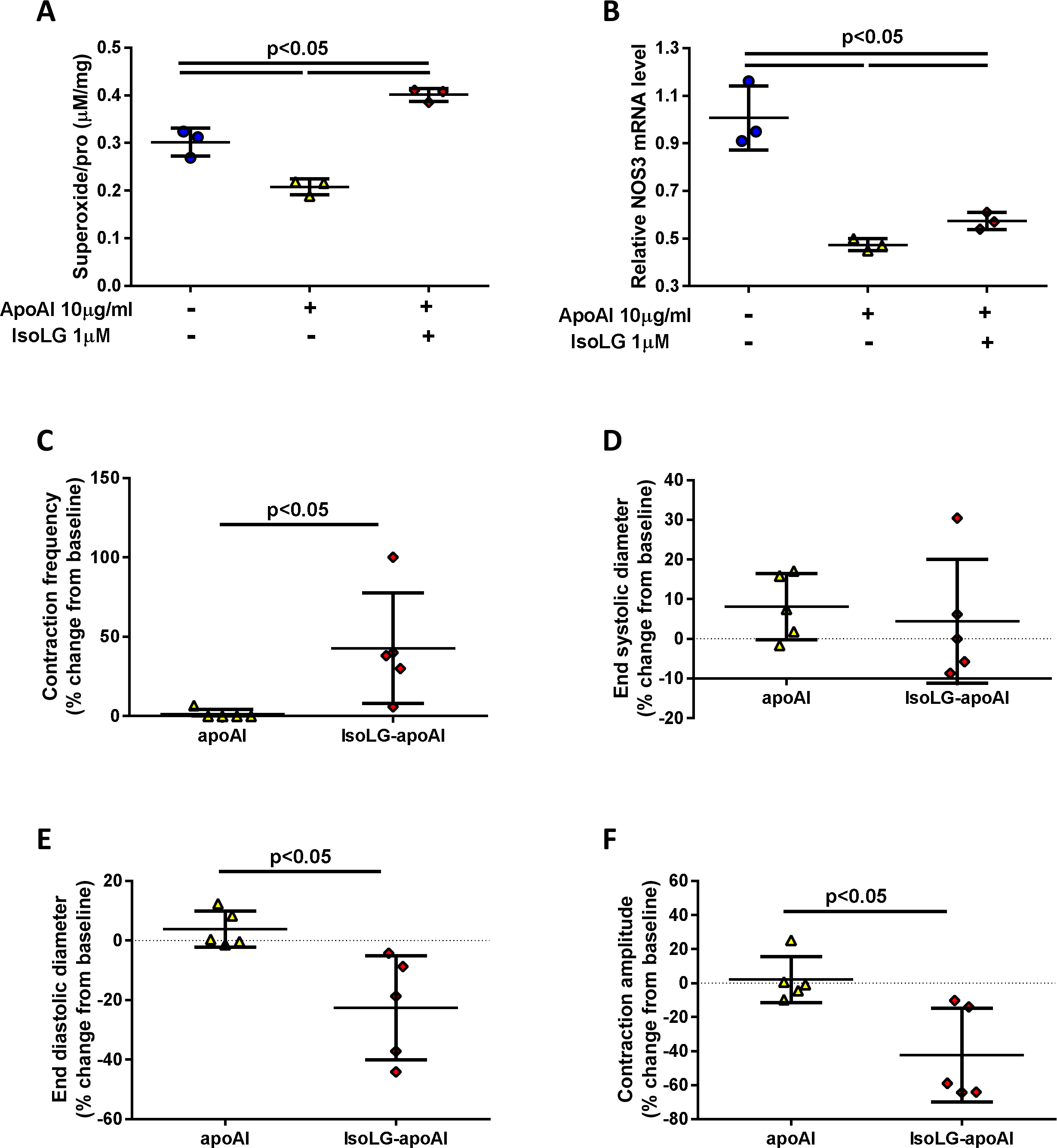
In vitro, cultured LECs exposed IsoLG-apoAI produced (A) more ROS and (B) increased eNOS (Nos3) gene expression vs unmodified apoAI. IsoLG-apoAI (C) increased contraction frequency from baseline, (D) did not change the end systolic diameter, (E) reduced end diastolic diameter from baseline, and (F) reduced contraction amplitude from baseline vs unmodified apoAI. In vitro, experiments were performed independently 3 times with 3 wells per treatment. In vivo, results are mean ± SD for 5 rats per group.
VEGF-C is the major growth factor promoting lymphangiogenesis. VEGF-C protein levels were significantly increased in ilea of PAN vs control rats (Fig 6A). Although VEGF-C concentration was lower in PAN lymph, the higher lymph flow rate increased the total lymphatic VEGF-C mass (Fig 6B), promoting sustained direct exposure of VEGF-C to LECs and thus lymphangiogenesis. The intestinal lacteals in PAN rats had more macrophages (CD68+ cells ), with more co-localization with VEGF-C protein (cells positive for CD68 and VEGF-C) (Fig 6C). Increased VEGF-C protein in lymph is likely linked to IsoLG-modification on lymphatic HDL, as macrophages treated with IsoLG-apoAI increased VEGFC mRNA expression compared to unmodified apoAI (Fig 6D). These data support the hypothesis that the ileal macrophages are a source for increased VEGF-C levels in intestinal lymphatics.
Figure 6. Proteinuric kidney injury stimulates ileal macrophage production of VEGF-C.
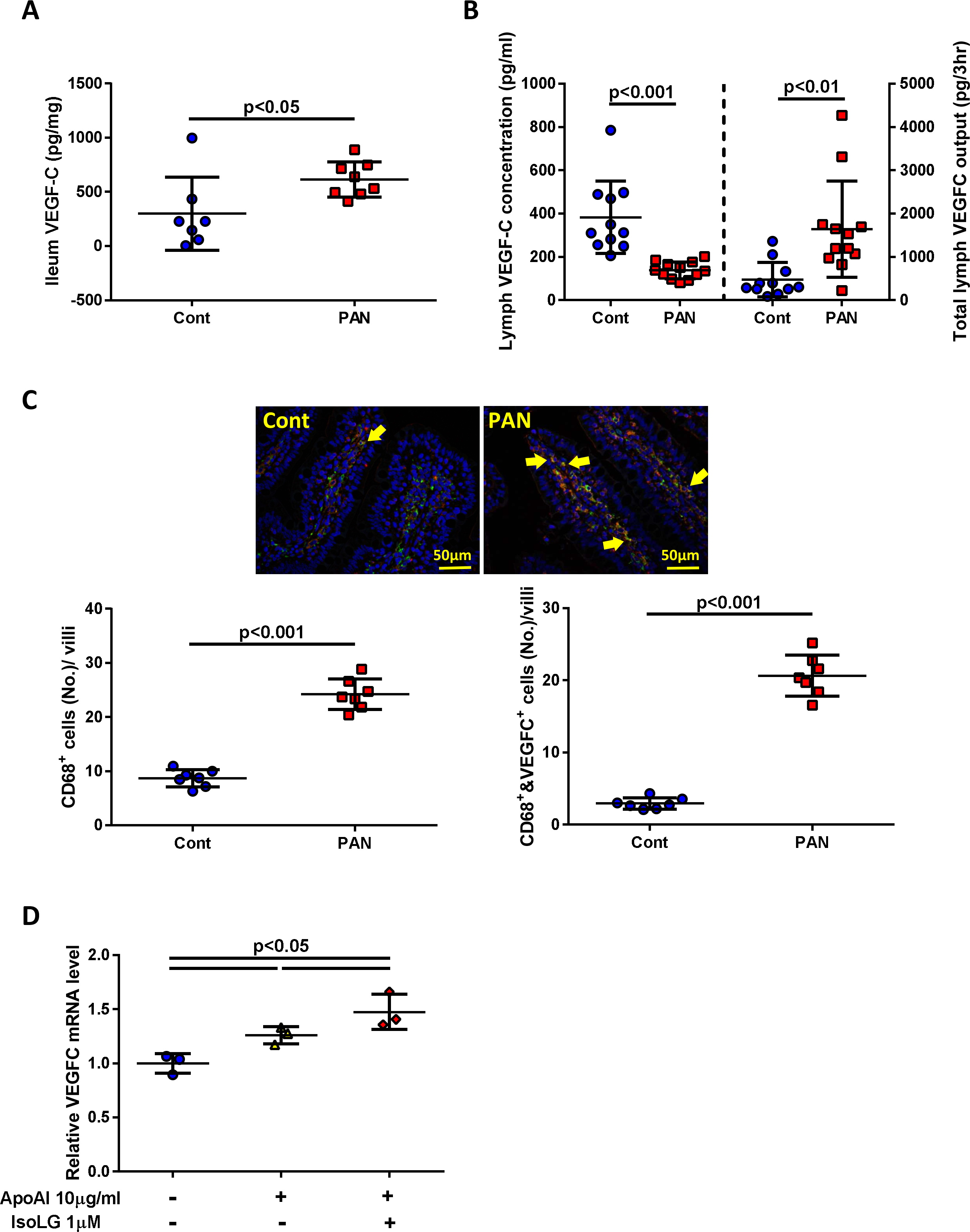
(A) PAN increased ileal VEGF-C vs controls. (B) VEGF-C concentration in PAN lymph was lower but total output of VEGF-C was significantly greater in PAN vs controls. (C) Double staining of ileum with VEGF-C (red) and CD68 (green) showed greater number of CD68-positive cells and CD68 & VEGF-C-positive cells (arrows) in PAN vs controls. (D) Cultured macrophages exposed to IsoLG-apoAI expressed more VEGFC mRNA vs unmodified apoAI. In vivo, results are mean ± SD for 7–12 rats per group. In vitro, experiments were performed independently 3 times with 3 wells per treatment.
Dicarbonyl scavenger, PPM, attenuated IsoLG modified lipoprotein and reduced intestinal lymphangiogenesis
Compared with untreated NEP25 mice, PPM-treated NEP25 mice showed significantly reduced intestinal podoplanin expression (Fig 7A). PPM also decreased the level of IsoLG adducts in the ileum (Fig 7B).
Figure 7. Dicarbonyl scavenger (PPM) treatment decreased ileal lymphangiogenesis and IsoLG adducts.
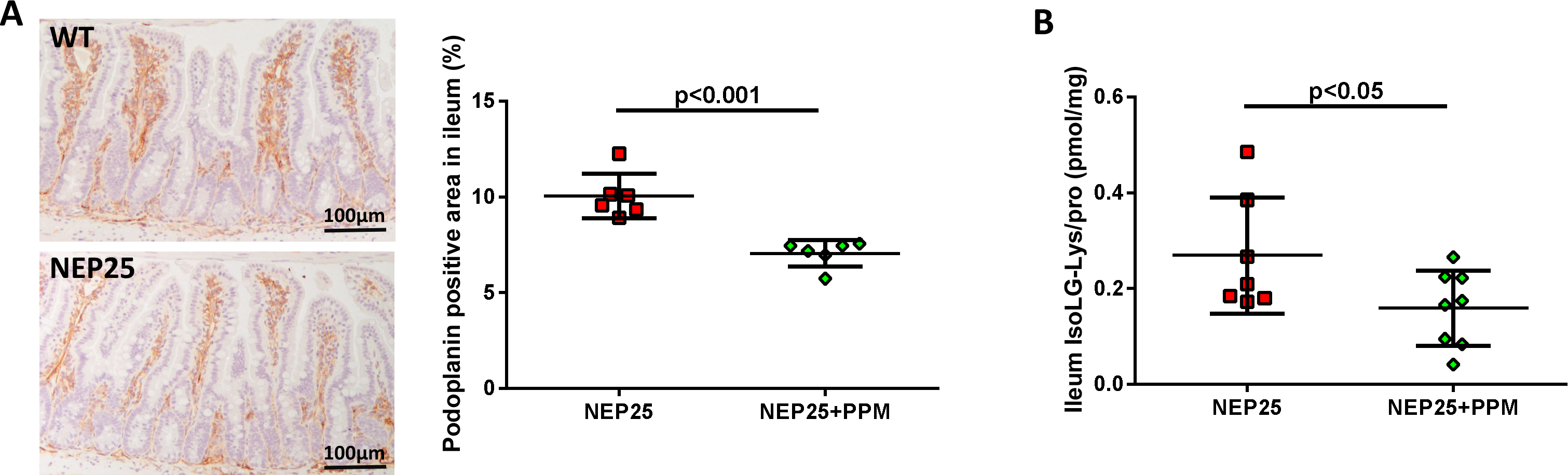
(A) PPM significantly reduced intestinal lymphangiogenesis in proteinuric NEP25 mice. (B) PPM also decreased IsoLG adduct in ileum of NEP25 mice.
Discussion
Experimental and clinical data have established that kidney injury has detrimental consequences on distant organs such as heart, lungs and intestines1, 43–45. Kidney-intestinal crosstalk studies have primarily focused on the adverse effects of kidney disease on the intestinal microbiome8, 9 and barrier dysfunction46. Our study examined the effects of proteinuric kidney injury on intestinal lymphatics, which are central in absorption, metabolism, and transport of lipids and lipoproteins as well as in regulating immunity and inflammation47. Using two models, we demonstrated that proteinuric kidney injury augments intestinal lymphangiogenesis, mesenteric lymph flow, lymphatic vessel contractions, and activation of lymphatic endothelial cells. The composition of the mesenteric lymph was also altered, with increased cytokines and increased enteric production of IsoLG that can adduct local apoAI. Moreover, cytokines and IsoLG adducts were increased in mesenteric lymph but not concurrently collected plasma of kidney injured animals, suggesting intestines are the origin of these potentially harmful particles. These data point to a new pathway in kidney-gut crosstalk that underlies adverse systemic consequences of kidney disease with the intestinal lymphatic network as a conduit and IsoLG-apoAI as a novel mediator of these effects.
Our study demonstrated that proteinuric kidney injury cause intestinal lymphangiogenesis, evidenced by increased mRNA and immunostaining for podoplanin, LYVE-1, and VEGFR3. The expanded lymphatic network transported more than 3-fold higher volume of lymph in the PAN rats vs controls. Thus, while injury is well known to stimulate lymphangiogenesis in the injured organ, our results make the original observation that proteinuric kidney disease activates a lymphatic response in a distant organ. Literature on this subject is limited. Experimental CKD has been shown to expand omental lymphatics48 and an inverse relationship between eGFR and diameter of cisterna chyli draining intestinal lymphatics has been documented in humans49. Our data provides the first direct demonstration that proteinuric damage increases intestinal lymphangiogenesis and increases mesenteric lymph flow. Edema and ascites in our models may have contributed to the expansion of the intestinal lymphatic network. Sodium overload, as well as inflammation, which prevail in kidney disease may have also contributed to lymphangiogenesis and greater lymph flow50, 51. It is possible that kidney disease activates lymphangiogenesis in other organs. Kidney disease is often accompanied by increased levels of VEGF-C and macrophages are an important source for VEGF-C52. Macrophage depletion or blockade of VEGF-C signaling diminishes lymphangiogenesis53. In the current study, macrophage infiltration of intestinal villi colocalized with VEGF-C in PAN. We did not survey other tissues and focused on the intestinal response that revealed kidney injury has major adverse impact on the structure and functions of intestinal lymphatics.
Kidney injury also altered the phenotype of intestinal LECs. Lymphatic endothelial cells isolated from ilea of PAN rats were activated, with significantly increased Nos3 mRNA, results consistent with previous studies showing that elaboration of eNOS by these cells is a major factor in lymphatic dilation54. Our data show that these ileal lymphatic endothelial cells from proteinuric animals upregulate other factors involved in immune cell trafficking, including CCL21, S1P and S1PR1. These factors play key roles and direct immune cells to lymph nodes for antigen presentation and initiation of innate and adaptive immune responses40, 55–57. Proteinuric kidney injury increased levels of potentially toxic immune cells (Th17 lymphocytes) and cytokines.
In addition to lymphangiogenesis and increased lymph flow, kidney injury also modified the composition of mesenteric lymph. Cytokines, including IL-6, IL-10, IL-17 were higher in lymph of PAN rats vs control. Proteinuric injury increased intestinal generation of reactive peroxidation product IsoLG, which is a powerful modifier of apoAI/HDL that degrades many of its beneficial actions, including decreased ability to bind LPS, efflux cellular cholesterol, and inhibit cytokine response14, 58, 59 linked to the pathogenesis of sepsis60, hypertension61 and CVD13, 62. Interestingly, IsoLG adducts have recently been shown to participate in inflammatory gastrointestinal carcinogenesis in animals and humans21 and in high salt hypertension linked to changes in the gut microbiome20. Dicarbonyl compounds reduce anaerobic fermentation processes and strongly depress the microbial community63. Whether the PAN-proteinuric injury altered the intestinal microbiome was not specifically analyzed. Nonetheless, we found increased IsoLG content in the ileal wall and mesenteric lymph of PAN rats vs controls. By immunostaining, we demonstrated that IsoLG-apoAI localized to ileal lymph vessels. Increased levels of IsoLG, as well as cytokines, were detected in the lymph but not in plasma of PAN injured animals, suggesting intestines are the origin of these potentially harmful elements. MPO is activated at all stages of renal damage and binds to apoAI/HDL. We confirmed MPO was increased in ileal walls of our proteinuric animals. Adding MPO increased IsoLG in cultured intestinal epithelial cells. Because intestines synthesize a third of the total apoAI39, we also examined the effects of PAN on intestinal apoAI. Our data failed to show differences in lymphatic apoAI protein concentration between PAN vs controls. Nonetheless, apoAI immunostaining showed strong localization in lymphatic lacteals. It is possible that redistribution and/or enhanced secretion together with increased lymph flow, contributed to increased mesenteric output of apoAI.
Our data indicate that intestinal lymphatics are not only a conduit for lipoprotein transport but present a target for their effects. Previous studies have described that apoAI/HDL regulate lymphangiogenesis and lymphatic integrity22, 23. We examined whether the effects of IsoLG-apoAI on lymphatic vessels or LECs differ from normal apoAI. IsoLG-apoAI increased Nos3 mRNA in cultured LECs compared to unmodified apoAI, paralleling the in vivo effects. IsoLG-apoAI altered functionality of mesenteric lymphatic vessels, including blunted vasoactivity and greater contraction frequency vs unmodified apoAI. Although this ex vivo assessment of lymphatic dynamics does not include contributions from innervation, circulating cytokines, or lymph flow, it revealed a direct impact of IsoLG-apoAI on lymphatic functionality. Together, the results show IsoLG-apoAI can stimulate LEC production of vasodilators and cause the lymphatic vessels to be more flaccid. Nonetheless, the concurrent increase in the contraction frequency may promote greater lymph flow and thus greater delivery of intestinally generated molecules and cells. The pathophysiologic impact of these data was supported by our in vivo studies showing inhibition of IsoLG with PPM significantly lessened injury-induced intestinal lymphangiogenesis in NEP25 proteinuric mice.
We propose that proteinuric kidney injury activates intestinal lymphangiogenesis and increases lymphatic flow via mechanisms involving intestinally generated dicarbonyls carried by lipoproteins. These data identify a new pathway in the kidney-gut axis and present a new target for kidney injury induced intestinal disruptions. Thus, the intestinal/mesenteric lymphatic network serves as both target and perpetrator of IsoLG-HDL’s effects by augmenting lymphangiogenesis, lymphatic vessel contractions, LEC activation, and increased lymph flow. The net effect is greater delivery of intestinally- derived molecules such as IsoLG-modified apoAI/HDL that may contribute to the adverse systemic effects occurring with kidney injury.
Translational Statement.
Kidney disease dysregulates levels, composition and function of lipids and lipoproteins. We know kidney injury stimulates intestinal lymphangiogenesis, activates lymphatic endothelial cells and increases mesenteric lymph flow via mechanisms involving intestinally generated dicarbonyls carried by lipoproteins. These data identify a new pathway in the kidney-gut axis and present a new target for kidney injury-induced intestinal disruptions. Our data suggest kidney injury contributes to increased gut lymphatic network and lymph flow that enhances systemic delivery of gut-originating harmful substances such as IsoLG-modified apoAI/HDL. This gut response to kidney injury may contribute to the adverse systemic effects occurring with kidney injury.
Acknowledgement
We thank Dr. Ira Pastan for LMB2 toxin and Dr. Fang Yan for MSIE.
The sources of support: NIH 1P01HL116263 and APS Vanderbilt Center for Kidney Disease (VCKD) PILOT (Yang), NIDDK RO1 DK056942 (Fogo)
Footnotes
Disclosure
All authors declared no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Meijers B, Jouret F, Evenepoel P. Linking gut microbiota to cardiovascular disease and hypertension: Lessons from chronic kidney disease. Pharmacol Res. 2018;133:101–107. [DOI] [PubMed] [Google Scholar]
- 2.Evenepoel P, Poesen R, Meijers B. The gut-kidney axis. Pediatr Nephrol. 2017;32:2005–2014. [DOI] [PubMed] [Google Scholar]
- 3.Andersen K, Kesper MS, Marschner JA, et al. Intestinal Dysbiosis, Barrier Dysfunction, and Bacterial Translocation Account for CKD-Related Systemic Inflammation. J Am Soc Nephrol. 2017;28:76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wikoff WR, Nagle MA, Kouznetsova VL, et al. Untargeted metabolomics identifies enterobiome metabolites and putative uremic toxins as substrates of organic anion transporter 1 (Oat1). J Proteome Res. 2011;10:2842–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Loor H, Bammens B, Evenepoel P, et al. Gas chromatographic-mass spectrometric analysis for measurement of p-cresol and its conjugated metabolites in uremic and normal serum. Clin Chem. 2005;51:1535–1538. [DOI] [PubMed] [Google Scholar]
- 6.McIntyre CW, Harrison LE, Eldehni MT, et al. Circulating endotoxemia: a novel factor in systemic inflammation and cardiovascular disease in chronic kidney disease. Clin J Am Soc Nephrol. 2011;6:133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Randolph GJ, Miller NE. Lymphatic transport of high-density lipoproteins and chylomicrons. J Clin Invest. 2014;124:929–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramezani A, Massy ZA, Meijers B, et al. Role of the Gut Microbiome in Uremia: A Potential Therapeutic Target. Am J Kidney Dis. 2016;67:483–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Castillo-Rodriguez E, Fernandez-Prado R, Esteras R, et al. Impact of Altered Intestinal Microbiota on Chronic Kidney Disease Progression. Toxins (Basel). 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vaziri ND, Deng G, Liang K. Hepatic HDL receptor, SR-B1 and Apo A-I expression in chronic renal failure. Nephrol Dial Transplant. 1999;14:1462–1466. [DOI] [PubMed] [Google Scholar]
- 11.Fu J, Bonder MJ, Cenit MC, et al. The Gut Microbiome Contributes to a Substantial Proportion of the Variation in Blood Lipids. Circ Res. 2015;117:817–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mistry RH, Verkade HJ, Tietge UJ. Reverse Cholesterol Transport Is Increased in Germ-Free Mice-Brief Report. Arterioscler Thromb Vasc Biol. 2017;37:419–422. [DOI] [PubMed] [Google Scholar]
- 13.Davies SS, May-Zhang LS. Isolevuglandins and cardiovascular disease. Prostaglandins Other Lipid Mediat. 2018;139:29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.May-Zhang LS, Yermalitsky V, Huang J, et al. Modification by isolevuglandins, highly reactive gamma-ketoaldehydes, deleteriously alters high-density lipoprotein structure and function. J Biol Chem. 2018;293:9176–9187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamamoto S, Zhong J, Yancey PG, et al. Atherosclerosis following renal injury is ameliorated by pioglitazone and losartan via macrophage phenotype. Atherosclerosis. 2015;242:56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsuchida Y, Zhong J, Otsuka T, et al. Lipoprotein modulation of proteinuric renal injury. Lab Invest. 2019;99:1107–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Florens N, Calzada C, Lyasko E, et al. Modified Lipids and Lipoproteins in Chronic Kidney Disease: A New Class of Uremic Toxins. Toxins (Basel). 2016;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ikizler TA, Morrow JD, Roberts LJ, et al. Plasma F2-isoprostane levels are elevated in chronic hemodialysis patients. Clin Nephrol. 2002;58:190–197. [DOI] [PubMed] [Google Scholar]
- 19.Kronenberg F HDL in CKD-The Devil Is in the Detail. J Am Soc Nephrol. 2018;29:1356–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferguson JF, Aden LA, Barbaro NR, et al. High dietary salt-induced dendritic cell activation underlies microbial dysbiosis-associated hypertension. JCI Insight. 2019;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gobert AP, Boutaud O, Asim M, et al. Dicarbonyl Electrophiles Mediate Inflammation-Induced Gastrointestinal Carcinogenesis. Gastroenterology. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bisoendial R, Tabet F, Tak PP, et al. Apolipoprotein A-I Limits the Negative Effect of Tumor Necrosis Factor on Lymphangiogenesis. Arterioscler Thromb Vasc Biol. 2015;35:2443–2450. [DOI] [PubMed] [Google Scholar]
- 23.Milasan A, Jean G, Dallaire F, et al. Apolipoprotein A-I Modulates Atherosclerosis Through Lymphatic Vessel-Dependent Mechanisms in Mice. J Am Heart Assoc. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grond J, Weening JJ, van Goor H, et al. Application of puromycin aminonucleoside and adriamycin to induce chronic renal failure in the rat. Contrib Nephrol. 1988;60:83–93. [DOI] [PubMed] [Google Scholar]
- 25.Matsusaka T, Xin J, Niwa S, et al. Genetic engineering of glomerular sclerosis in the mouse via control of onset and severity of podocyte-specific injury. J Am Soc Nephrol. 2005;16:1013–1023. [DOI] [PubMed] [Google Scholar]
- 26.Matsusaka T, Sandgren E, Shintani A, et al. Podocyte injury damages other podocytes. J Am Soc Nephrol. 2011;22:1275–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Albaugh VL, Banan B, Antoun J, et al. Role of Bile Acids and GLP-1 in Mediating the Metabolic Improvements of Bariatric Surgery. Gastroenterology. 2019;156:1041–1051 e1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jerome WG, Cox BE, Griffin EE, et al. Lysosomal cholesterol accumulation inhibits subsequent hydrolysis of lipoprotein cholesteryl ester. Microsc Microanal. 2008;14:138–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yermalitsky VN, Matafonova E, Tallman K, et al. Simplified LC/MS assay for the measurement of isolevuglandin protein adducts in plasma and tissue samples. Anal Biochem. 2019;566:89–101. [DOI] [PubMed] [Google Scholar]
- 30.Huang J, Smith F, Panizzi P. Ordered cleavage of myeloperoxidase ester bonds releases active site heme leading to inactivation of myeloperoxidase by benzoic acid hydrazide analogs. Arch Biochem Biophys. 2014;548:74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Green PH, Tall AR, Glickman RM. Rat intestine secretes discoid high density lipoprotein. J Clin Invest. 1978;61:528–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scallan JP, Hill MA, Davis MJ. Lymphatic vascular integrity is disrupted in type 2 diabetes due to impaired nitric oxide signalling. Cardiovasc Res. 2015;107:89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zawieja SD, Castorena-Gonzalez JA, Dixon B, et al. Experimental Models Used to Assess Lymphatic Contractile Function. Lymphat Res Biol. 2017;15:331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yao L, Wright MF, Farmer BC, et al. Fibroblast-specific plasminogen activator inhibitor-1 depletion ameliorates renal interstitial fibrosis after unilateral ureteral obstruction. Nephrol Dial Transplant. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang HC, Ma LJ, Ma J, et al. Peroxisome proliferator-activated receptor-gamma agonist is protective in podocyte injury-associated sclerosis. Kidney Int. 2006;69:1756–1764. [DOI] [PubMed] [Google Scholar]
- 36.Wang Y, Liu L, Moore DJ, et al. An LGG-derived protein promotes IgA production through upregulation of APRIL expression in intestinal epithelial cells. Mucosal Immunol. 2017;10:373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dikalova A, Clempus R, Lassegue B, et al. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation. 2005;112:2668–2676. [DOI] [PubMed] [Google Scholar]
- 38.Yamamoto S, Yancey PG, Ikizler TA, et al. Dysfunctional high-density lipoprotein in patients on chronic hemodialysis. J Am Coll Cardiol. 2012;60:2372–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arciello A, Piccoli R, Monti DM. Apolipoprotein A-I: the dual face of a protein. FEBS Lett. 2016;590:4171–4179. [DOI] [PubMed] [Google Scholar]
- 40.Stepanovska B, Huwiler A. Targeting the S1P receptor signaling pathways as a promising approach for treatment of autoimmune and inflammatory diseases. Pharmacol Res. 2019;104170. [DOI] [PubMed] [Google Scholar]
- 41.Himmelfarb J, Stenvinkel P, Ikizler TA, et al. The elephant in uremia: oxidant stress as a unifying concept of cardiovascular disease in uremia. Kidney Int. 2002;62:1524–1538. [DOI] [PubMed] [Google Scholar]
- 42.Malle E, Buch T, Grone HJ. Myeloperoxidase in kidney disease. Kidney Int. 2003;64:1956–1967. [DOI] [PubMed] [Google Scholar]
- 43.Husain-Syed F, McCullough PA, Birk HW, et al. Cardio-Pulmonary-Renal Interactions: A Multidisciplinary Approach. J Am Coll Cardiol. 2015;65:2433–2448. [DOI] [PubMed] [Google Scholar]
- 44.Lane K, Dixon JJ, MacPhee IA, et al. Renohepatic crosstalk: does acute kidney injury cause liver dysfunction? Nephrol Dial Transplant. 2013;28:1634–1647. [DOI] [PubMed] [Google Scholar]
- 45.Hoste EAJ, Kellum JA, Selby NM, et al. Global epidemiology and outcomes of acute kidney injury. Nat Rev Nephrol. 2018;14:607–625. [DOI] [PubMed] [Google Scholar]
- 46.Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. [DOI] [PubMed] [Google Scholar]
- 47.Jang JY, Koh YJ, Lee SH, et al. Conditional ablation of LYVE-1+ cells unveils defensive roles of lymphatic vessels in intestine and lymph nodes. Blood. 2013;122:2151–2161. [DOI] [PubMed] [Google Scholar]
- 48.Vlahu CA, de Graaff M, Aten J, et al. Lymphangiogenesis and Lymphatic Absorption Are Related and Increased in Chronic Kidney Failure, Independent of Exposure to Dialysis Solutions. Adv Perit Dial. 2015;31:21–25. [PubMed] [Google Scholar]
- 49.Albayrak E, Ozmen Z, Sahin S, et al. Evaluation of cisterna chyli diameter with MRI in patients with chronic kidney disease. J Magn Reson Imaging. 2016;44:890–896. [DOI] [PubMed] [Google Scholar]
- 50.Machnik A, Neuhofer W, Jantsch J, et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat Med. 2009;15:545–552. [DOI] [PubMed] [Google Scholar]
- 51.Slagman MC, Kwakernaak AJ, Yazdani S, et al. Vascular endothelial growth factor C levels are modulated by dietary salt intake in proteinuric chronic kidney disease patients and in healthy subjects. Nephrol Dial Transplant. 2012;27:978–982. [DOI] [PubMed] [Google Scholar]
- 52.Cursiefen C, Chen L, Borges LP, et al. VEGF-A stimulates lymphangiogenesis and hemangiogenesis in inflammatory neovascularization via macrophage recruitment. J Clin Invest. 2004;113:1040–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim H, Kataru RP, Koh GY. Inflammation-associated lymphangiogenesis: a double-edged sword? J Clin Invest. 2014;124:936–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Datar SA, Gong W, He Y, et al. Disrupted NOS signaling in lymphatic endothelial cells exposed to chronically increased pulmonary lymph flow. Am J Physiol Heart Circ Physiol. 2016;311:H137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Karnezis T, Shayan R, Caesar C, et al. VEGF-D promotes tumor metastasis by regulating prostaglandins produced by the collecting lymphatic endothelium. Cancer Cell. 2012;21:181–195. [DOI] [PubMed] [Google Scholar]
- 56.Weber M, Hauschild R, Schwarz J, et al. Interstitial dendritic cell guidance by haptotactic chemokine gradients. Science. 2013;339:328–332. [DOI] [PubMed] [Google Scholar]
- 57.Card CM, Yu SS, Swartz MA. Emerging roles of lymphatic endothelium in regulating adaptive immunity. J Clin Invest. 2014;124:943–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iyer RS, Ghosh S, Salomon RG. Levuglandin E2 crosslinks proteins. Prostaglandins. 1989;37:471–480. [DOI] [PubMed] [Google Scholar]
- 59.Murthi KK, Friedman LR, Oleinick NL, et al. Formation of DNA-protein cross-links in mammalian cells by levuglandin E2. Biochemistry. 1993;32:4090–4097. [DOI] [PubMed] [Google Scholar]
- 60.Poliakov E, Brennan ML, Macpherson J, et al. Isolevuglandins, a novel class of isoprostenoid derivatives, function as integrated sensors of oxidant stress and are generated by myeloperoxidase in vivo. FASEB J. 2003;17:2209–2220. [DOI] [PubMed] [Google Scholar]
- 61.Wu J, Saleh MA, Kirabo A, et al. Immune activation caused by vascular oxidation promotes fibrosis and hypertension. J Clin Invest. 2016;126:1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Salomon RG, Kaur K, Batyreva E. Isolevuglandin-protein adducts in oxidized low density lipoprotein and human plasma: a strong connection with cardiovascular disease. Trends Cardiovasc Med. 2000;10:53–59. [DOI] [PubMed] [Google Scholar]
- 63.Brighina S, Poveda Turrado C, Restuccia C, et al. Detrimental effect on the gut microbiota of 1,2-dicarbonyl compounds after in vitro gastro-intestinal and fermentative digestion. Food Chem. 2021;341:128237. [DOI] [PubMed] [Google Scholar]