

Abstract
Host–virus protein–protein interactions play key roles in the life cycle of severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2). We conducted a comprehensive interactome study between the virus and host cells using tandem affinity purification and proximity‐labeling strategies and identified 437 human proteins as the high‐confidence interacting proteins. Further characterization of these interactions and comparison to other large‐scale study of cellular responses to SARS‐CoV‐2 infection elucidated how distinct SARS‐CoV‐2 viral proteins participate in its life cycle. With these data mining, we discovered potential drug targets for the treatment of COVID‐19. The interactomes of two key SARS‐CoV‐2‐encoded viral proteins, NSP1 and N, were compared with the interactomes of their counterparts in other human coronaviruses. These comparisons not only revealed common host pathways these viruses manipulate for their survival, but also showed divergent protein–protein interactions that may explain differences in disease pathology. This comprehensive interactome of SARS‐CoV‐2 provides valuable resources for the understanding and treating of this disease.
Keywords: BioID2, host–virus protein–protein interaction, interactome, SARS‐CoV‐2, SFB‐TAP
Subject Categories: Microbiology, Virology & Host Pathogen Interaction; Proteomics
Human proteins identified as copurifying with or proximal to SARS‐CoV‐2 proteins suggest cellular response pathways and potential drug targets, as well as divergence of NSP1 and N protein binding specificities compared to related human coronaviruses.

Introduction
The ongoing global pandemic of coronavirus disease 2019 (COVID‐19) was first reported in December 2019. Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) was identified as the virus causing COVID‐19 (Wu et al, 2020). SARS‐CoV‐2 is a highly transmissible and pathogenic coronavirus. It spreads easily through the air when people are physically near each other. As of 29 June 2021, more than 181.5 million cases had been confirmed, resulting in more than 3.9 million deaths.
SARS‐CoV‐2 is a novel beta coronavirus with a genome composed of approximately 30 kb of positive‐strand RNA. It shares 79% genomic sequence identity with SARS‐CoV‐1, which caused the SARS epidemic in 2003 (Lu et al, 2020). The SARS‐CoV‐2 genome contains 14 open reading frames (ORFs), including one large ORF that encodes two large polyproteins (ORF1a and ORF1ab) and 13 small ORFs that encode viral structural proteins and other polypeptides. The polyproteins from the large ORF can be further cleaved into 16 non‐structure proteins (NSP1 to NSP16) (Lu et al, 2020).
Researchers have tried several strategies to develop drugs and vaccines to treat SARS‐CoV‐2 and COVID‐19. Viruses usually encode very limited viral proteins and thus need to recruit many host proteins to complete the viral life cycle. Therefore, identifying protein–protein interactions between viral proteins and their host cellular cofactors is an important and efficient way to understand the virus and uncover potential drug targets. Such host–virus interactome analysis has been reported recently using different strategies, such as affinity purification (AP) (preprint: Davies et al, 2020; Gordon et al, 2020a; Gordon et al, 2020b; preprint: Nabeel‐Shah et al, 2020; Li et al, 2021; Stukalov et al, 2021), proximity labeling‐based strategy (preprint: Laurent et al, 2020; preprint: Samavarchi‐Tehrani et al, 2020; preprint: St‐Germain et al, 2020), and yeast two‐hybrid system (preprint: Kim et al, 2021).
Affinity purification (AP)‐ and proximity labeling‐based strategies followed by mass spectrometry (MS) analysis are two well‐developed ways to study protein‐protein interactome (Smits & Vermeulen, 2016; Chen & Chen, 2021). AP‐MS is a widely used and highly reproducible method that allows identification of physiologically relevant interaction proteins. However, this method may miss weak or transient binding proteins during the pulldown process, even when it is performed in a mild buffer. In addition, AP‐MS may not capture poorly soluble protein partners, such as membrane‐associated proteins, which are critically important for viral life cycle. The proximity labeling‐based strategy solves these problems by labeling nearby proteins before harvesting cells (Roux et al, 2012; Smits & Vermeulen, 2016). This method is based on an enzyme‐substrate reaction with an effective labeling radius of about 10 nm. The drawback of this method is that it may fail to label the binding proteins located outside of this labeling range.
In the current study, we applied these two complementary strategies, i.e., tandem affinity purification (TAP) with SFB (S protein, FLAG epitope, and streptavidin‐binding peptide) tag and proximity labeling with a second‐generation biotin ligase, BioID2, for a comprehensive analysis of host–virus protein–protein interaction network. With an interactome analysis of 29 SARS‐CoV‐2 cDNAs, we uncovered key human proteins that may participate in SARS‐CoV‐2 life cycle of infection, replication, and budding. This interactome dataset not only confirmed some previously reported host–virus interactions but also uncovered numerous new interacting proteins that may be critical for SARS‐CoV‐2 life cycle. This dataset will benefit further investigations of the mechanisms underlying viral infection and life cycle and provide potential new drug targets for the treatment of COVID‐19. Moreover, we compared the interactomes of two critical viral gene products, NSP1 and N proteins, among different human coronaviruses. These analyses will help us to fight SARS‐CoV‐2 and future pandemics caused by new coronaviruses.
Results
Overview of the interactome analysis between host and SARS‐CoV‐2
To comprehensively illustrate the host and SARS‐CoV‐2 protein–protein interaction network, we performed an interactome study using two different strategies, TAP with the SFB tag and proximity labeling with the BioID2 tag. Genome annotation revealed 29 gene products from SARS‐CoV‐2, including 16 NSPs, 4 structure proteins, and 9 accessary factors (Fig 1A). These viral gene products were fused with the SFB or BioID2 tag and stably expressed in HEK293T cells, except NSP1, which was done with transient expression. Viral gene expression in cells was verified by immunoblotting (Fig EV1A and B). Labeling reactions of fused BioID2‐viral genes were confirmed using the streptavidin antibody (Fig EV1C). Immunofluorescence staining was also performed for all the viral gene products (Appendix Fig S1A and B). Several viral proteins, such as NSP3, NSP4, and NSP6, were not detected by Western blotting, likely due to the low expression of these proteins, which were also reported in other studies (Gordon et al, 2020b; preprint: Samavarchi‐Tehrani et al, 2020; Stukalov et al, 2021). We still included the interactome of these genes, since we successfully detected their expression by immunostaining (Appendix Fig S1A and B) and acquired MS signals of these baits in our proteomics analyses. Three biological replicates of the interactome experiments were conducted for each tag and each fused viral gene product, along with controls (vector with tag only or fused with GFP sequence) following the workflow presented in Fig 1B. After analysis by Q Exactive HF MS, the data were searched against a database integrated with all human genes, viral genes, GFP, and the tag sequences (Dataset EV1). All experiments are summarized in Fig 1C. The Pearson correlation coefficient among three independent biological replicates of the SFB‐TAP results and BioID2 labeling experiments was calculated (Fig 1D).
Figure 1. Summary of the SFB‐TAP and BioID2 interactome experiments.
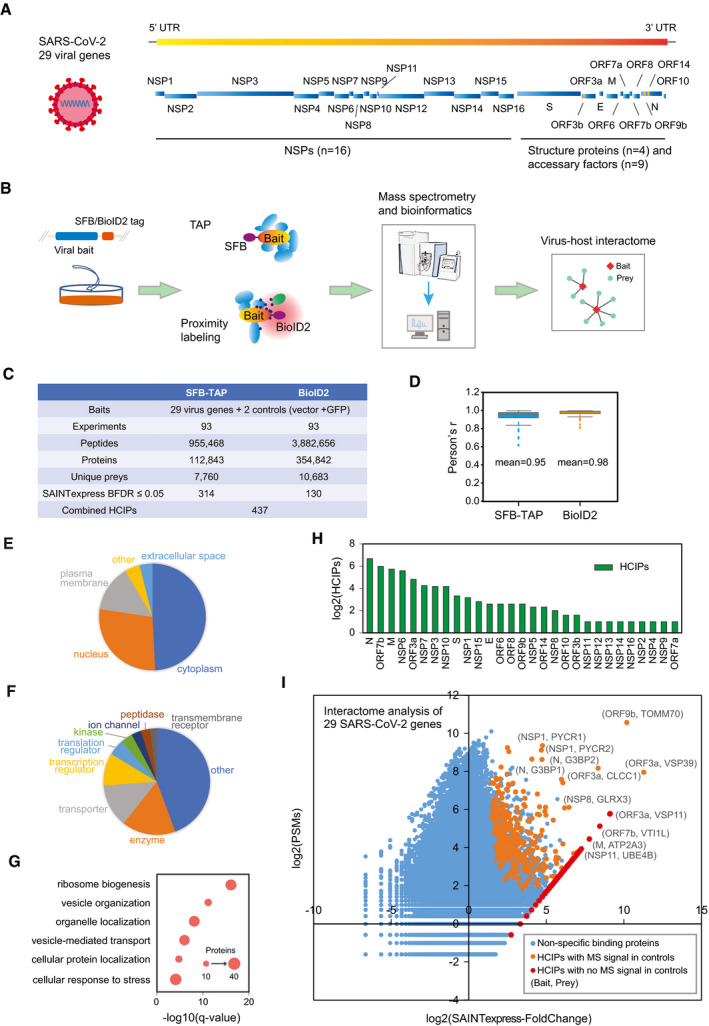
-
ASARS‐CoV‐2 genome annotation, which predicts 29 virus gene products. The 16 non‐structure proteins (NSPs) are cleaved products of the large polyprotein open reading frame (ORF)1ab or ORF1a. These polyproteins are cleaved into small function fragments or NSPs after translation.
-
BWorkflow for the comprehensive virus–host interactome analysis. Two different strategies, SFB‐TAP and BioID2 proximity labeling, were applied in the study. Samples were analyzed by Q Exactive HF mass spectrometry (MS).
-
CSummary of the datasets obtained from SFB‐TAP and BioID2 results, including the number of high‐confidence interacting proteins (HCIPs). BDFR, Bayesian false discovery rate.
-
DPearson correlation coefficient among three independent biological replicates of the SFB‐TAP results and the BioID2 labeling experiments. Box limits represent 25th percentile and 75th percentile; horizontal line represents median. Whiskers display min. to max. values.
-
E–GGO analysis. GO enrichment was performed using Ingenuity Pathway Analysis. Protein localization (E), molecular function (F), and biological function (G) are plotted in a single panel.
-
HHCIPs identified in the purification of each SARS‐CoV‐2 gene.
-
ICorrelation between peptide‐spectrum matches (PSMs) of identified proteins and their fold change calculated by SAINTexpress.
Figure EV1. Analysis of SARS‐CoV‐2 protein expression by Western blotting and interferon (IFN) signaling assays (related to Fig 1).
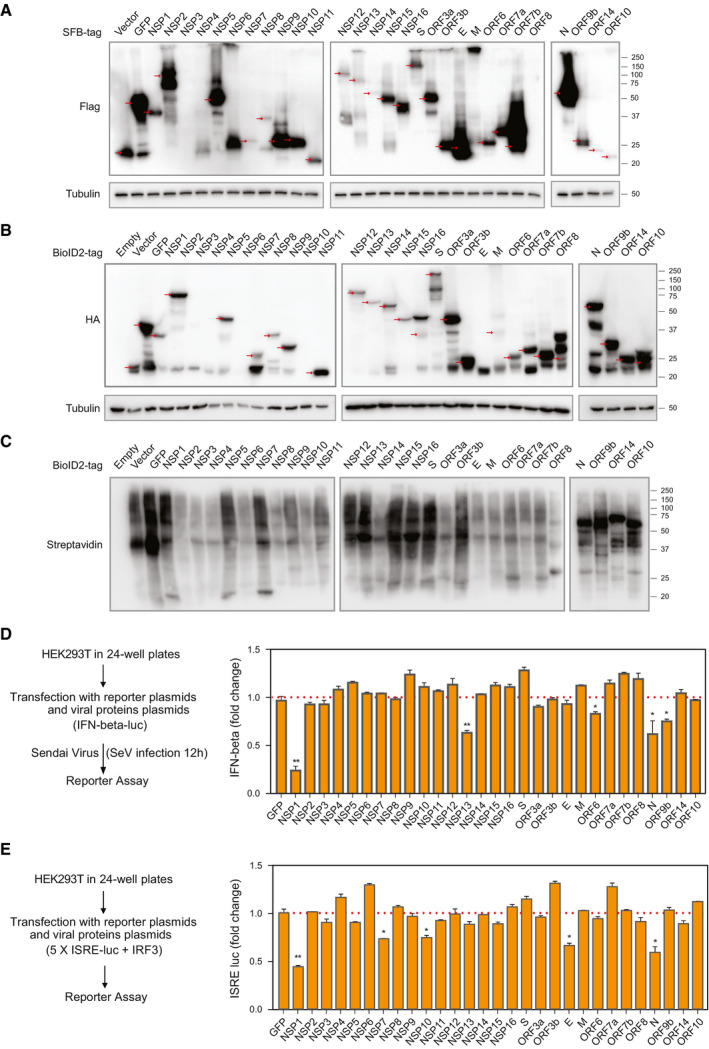
-
ATwenty‐nine SARS‐CoV‐2 genes with the SFB tag were expressed in cells. Cells transfected with vector or construct encoding SFB‐tagged GFP were included as controls. Western blotting was conducted with indicated antibodies. Red arrows indicate the predicted positions of bait gene products.
-
BTwenty‐nine SARS‐CoV‐2 genes with the second‐generation biotin ligase (BioID2) tag were expressed in cells. Untransfected cells, cells transfected with vector, and cells transfected with construct encoding GFP tagged with BioID2 were included as controls. Western blotting was conducted with indicated antibodies. Red arrows indicate the predicted positions of bait gene products.
-
CValidation of BioID2 labeling by blotting with streptavidin antibody.
-
DAnalysis of IFN‐beta‐luciferase reporter 12 h after Sendai virus (SeV) infection with co‐expression of indicated SARS‐CoV‐2 viral proteins.
-
EAnalysis of ISRE‐luciferase reporter when the indicated SARS‐CoV‐2 viral proteins were co‐expressed with IRF3.
Data information: Graphs D and E show mean ± SD, n = 3. **P < 0.01 and *P < 0.05 (Student’s t‐test).Source data are available online for this figure.
The identified proteins were filtered using Significance Analysis of INTeractome (SAINTexpress) (Teo et al, 2014). The SAINTexpress scores were averaged among the three biological replicates to calculate a Bayesian false discovery rate, and preys with a Bayesian false discovery rate of 0.05 or less were considered high‐confidence interacting proteins (HCIPs). In total, we obtained 314 HCIPs from SFB‐TAP and 130 HCIPs from BioID2 experiments (Dataset EV2). We combined these two datasets and generated a list of HCIPs, which included 437 pairs of virus–host interacting proteins (Fig 1C). GO analysis of these 437 viral interacting proteins showed that the preys localize in various subcellular locations (Fig 1E). Many of the preys localize on various plasma membrane structures, such as the endoplasmic reticulum, Golgi membrane, or cell membrane. Protein function analysis also showed that many of the preys have roles in transporter, ion channel, and peptidase processes (Fig 1F). The biological processes enriched included vesicle organization, organelle localization, and vesicle‐mediated transport (Fig 1G). These preys may assist SARS‐CoV‐2 infection, replication, and budding. Another group of enriched proteins is involved in ribosome function, indicating that these preys may be exploited by viral proteins to suppress host‐cell translation and facilitate viral gene expression.
The number of HCIPs for each SARS‐CoV‐2 viral gene product was summarized in Fig 1H. The fold change calculated by SAINTexpress analysis and the averaged peptide‐spectrum matches (PSMs) of the identified preys are plotted in Fig 1I. The orange and red dots in the upper right represent HCIPs. The most significant interaction pair, ORF9b‐TOMM70, was identified with more than 1,000 PSMs of the prey. This interaction was reported recently in a structural study of the complex (Gordon et al, 2020a). We also identified several binding pairs with very high MS signals, including ORF3a‐VSP39, ORF3a‐VSP11, ORF3a‐CLCC1, NSP1‐PYCR1, NSP1‐PYCR2, N‐G3BP1, and N‐G3BP2. These strong interacting partners may have cellular functions important to SARS‐CoV‐2. The functions of these preys may be taken over by the virus to support viral life cycle.
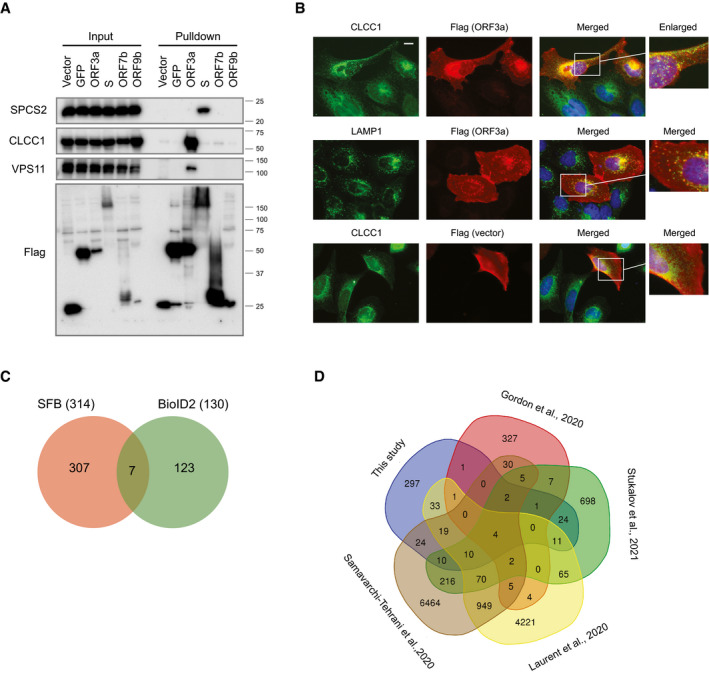
We performed further validation for several binding proteins. In Fig 2A, we confirmed the interaction of S protein with the signal peptidase component SPCS2. Other components of the signal peptidase complex, including SPCS1, SPCS3, and SEC11A, were also identified as HCIPs in our interactome study, which may indicate the involvement of signal peptidase complex in the function of S protein during virus infection cycle.
Figure 2. Validation of selected host–virus protein–protein interactions.
-
APulldown and Western blot analysis validated the interaction between the viral protein ORF3a and its interactors, VPS11 and CLCC1, and the binding of S protein to SPCS2. HEK293T cells with SFB‐tagged bait expression were collected and lysed. Cell lysates were subjected to pulldown assay using S‐protein beads. Western blot analysis was conducted with the indicated antibodies. Cells transfected with vector or construct encoding control genes were included as controls in these experiments.
-
BImmunostaining analysis of protein localization. U2OS cells were transfected with construct encoding ORF3a. Cells were fixed and stained with the indicated antibodies. The green signal is CLCC1 or lysosome marker LAMP1, the red signal is flag (for SFB‐ORF3a), and the blue signal indicates DAPI/nuclei. Scale bar: 10 µm.
-
COverlap of HCIPs identified from SFB‐tandem affinity purification and second‐generation biotin ligase (BioID2) labeling experiments.
-
DOverlap of HCIPs reported previously and those identified in the current study.
Source data are available online for this figure.
We also confirmed the interactions of ORF3a with CLCC1 and VPS11 in Fig 2A. These host proteins bind specifically to ORF3a. CLCC1 is a chloride ion channel protein with limited studies, and its functions are not well known. We obtained it as one of the top SARS‐CoV‐2 and host interaction proteins. Furthermore, we examined the localization of CLCC1 with or without ORF3a overexpression (Fig 2B). When ORF3a was overexpressed, the localization of CLCC1 changed to strong co‐localization with ORF3a in the lysosomes, which happened in higher than 95% of the ORF3a‐expressed cells. Although the detailed mechanisms remain to be elucidated with future functional analysis, the strong interaction between ORF3a and CLCC1 indicates that disrupting this interaction and/or CLCC1 function may affect the SARS‐CoV‐2 life cycle.
Comparison of different datasets of SARS‐CoV‐2 virus–host interaction network
In this study, we chose two distinct approaches, SFB‐TAP and BioID2 proximity‐labeling, to enrich for the viral protein binding partners in host cells. We compared the list of HCIPs obtained from TAP‐MS with that from BioID2‐MS and found only seven common gene products from these results (Fig 2C). We examined the raw list and found 50,523 unique bait–pray pairs identified from TAP approach, and 136,188 unique bait–pray pairs identified from BioID2 method. In the HCIPs from TAP‐MS analysis, 205 out of the 314 HCIPs were also identified in the raw list using BioID2 approach. However, most of these identified HCIPs were filtered out when we performed BioID2 data analysis. This is likely due to the difference in the strategies used, which result in different prey PSM numbers and different background of non‐specific binding proteins. An example of the difference in prey PSMs obtained from two methods in our dataset is the ORF9a–TOMM70 interaction. It was uncovered as an HCIP by both methods. However, we detected an average of 1,518 PSMs of TOMM70 in our TAP‐MS experiments, but only an average of 31 PSMs in the BioID2‐MS study. An example of the different background is the N protein and its binding partners G3BP1 and G3BP2. We identified an average of 403 PSMs for G3BP1 and an average of 397 PSMs for G3BP2 in SFB‐TAP results. With BioID2 method, we also acquired an average of 107 PSMs for G3BP1 and an average of 55 PSMs for G3BP2. However, G3BP1 and G3BP2 are commonly identified in BioID2 controls with about 40 PSMs. Therefore, these two proteins were removed from the list of HCIPs of BioID2 because of low FDR scores in HCIP analysis.
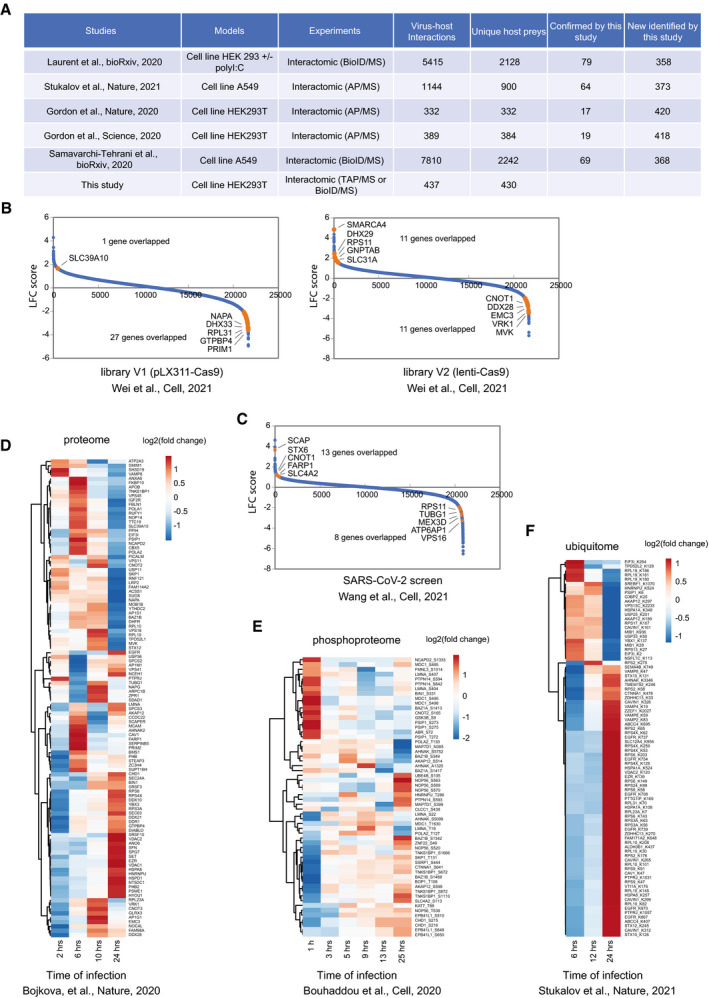
The same issues also show up in other SARS‐CoV‐2 interactome studies. We compared our HCIPs to four other datasets, two generated by AP‐MS (Gordon et al, 2020a; Stukalov et al, 2021), and the other two generated using BioID proximity‐labeling strategy (preprint: Laurent et al, 2020; preprint: Samavarchi‐Tehrani et al, 2020). As shown in Fig 2D, the overlaps between any two datasets are not high. The comparison between our list and each of these datasets is presented in Fig EV2A with detailed list in Dataset EV3. The relative low overlapping between datasets was also noticed by a paper published recently (Stukalov et al, 2021). They compared their SARS‐CoV‐2 host–virus interactome to other studies and found low overlapping between these datasets. This is still an issue in protein–protein interaction studies, which may due to differences in purification strategy, experimental design, and/or data filtration/analysis.
Figure EV2. Comparing HCIPs from this study to other interactome studies with SARS‐CoV‐2 genes or other large‐scale studies analyzing cellular responses upon virus infection (related to Figs 2 and 4).
-
AComparison of HCIPs to other five SARS‐CoV‐2 interactome studies.
-
BComparison of SARS‐CoV‐2 virus–host interaction to the genetic screening with CRISPR technique published by Wei et al in 2021. Two different Cas9 nuclease constructs were used, Lenti‐Cas9‐Blaset (V1) and Plx_311‐Cas9 (V2). Blue dots are the identified genes from the genetic screening. Orange dots are the highlighted overlapped genes between our virus–host interaction study and the results from genetic screening. Up to five top overlapped genes are indicated.
-
CComparison of SARS‐CoV‐2 virus–host interaction to the genetic screening with CRISPR technique published by Wang et al in 2021. Blue dots are the identified genes from the genetic screening. Orange dots are the highlighted overlapped genes between our virus–host interaction study and the results from genetic screening. Top five overlapped genes are indicated.
-
DHeat map of the HCIPs with protein abundance changes during SARS‐CoV‐2 viral infection with the time point of 2, 6, 10, and 24 h.
-
EHeat map of the HCIPs with their phosphorylation levels regulated during SARS‐CoV‐2 viral infection with the monitored time points of 1, 3, 5, 9, 13, and 25 h.
-
FHeat map of the HCIPs with their ubiquitination levels regulated during SARS‐CoV‐2 viral infection with the monitored time points of 6, 12, and 24 h.
Analysis of the interactions between SARS‐CoV‐2 and human proteins
We next built an interaction network using the 437 identified virus–host protein–protein interactions uncovered in our study. Many pathways or function complexes were highly enriched for binding to a specific SARS‐CoV‐2 gene product, as shown in Fig 3. Function enrichment may help us elucidate the biological functions and the underlying mechanisms of these SARS‐CoV‐2 viral gene products and design drugs targeting these virus–host interactions and/or the host complexes/pathways that are most relevant to this virus.
Figure 3. Interactomes between SARS‐CoV‐2 and human proteins.
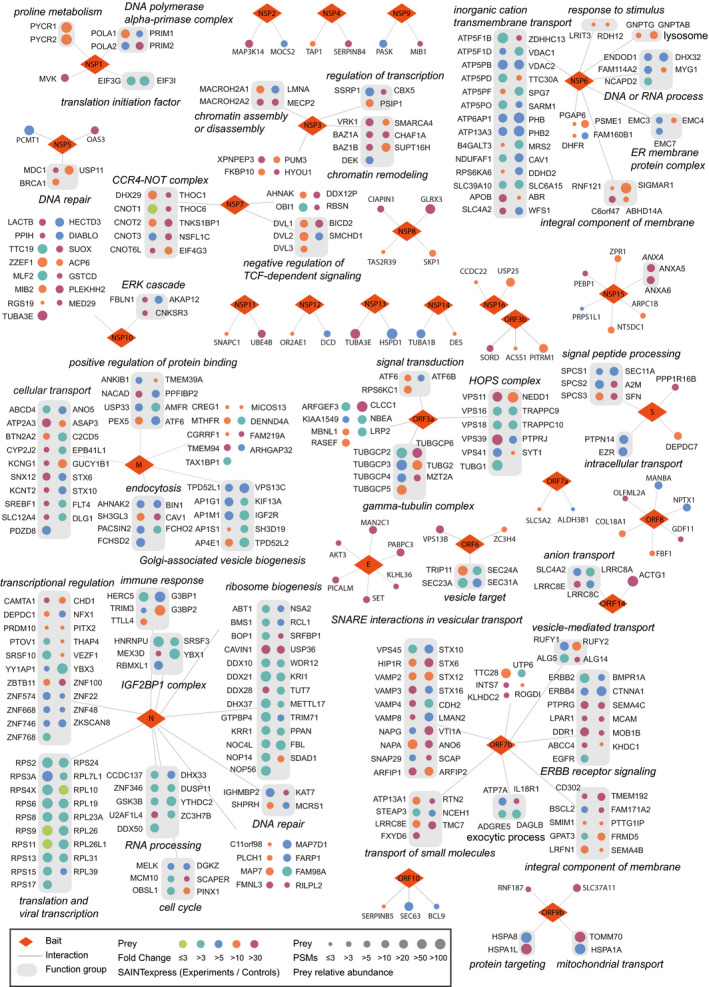
Protein–protein interaction network between 29 viral proteins of SARS‐CoV‐2 and the host proteins is shown for 437 pairs of host–virus interactions. The red diamonds are virus proteins (NSP, non‐structure protein), and gray rounded rectangles are groups of proteins belonging to the same protein complex or from the same pathway. The italic text are the functional characterizations analyzed with Metascape and reference mining, and circles are the high‐confidence interacting proteins (HCIPs) identified from SFB‐tandem affinity purification experiments. The colors of the cycle indicate the fold change of that prey calculated by SAINTexpress comparing to control SFB‐TAP purifications. The size of the cycle are PSMs number identified for that prey.
Most enveloped viruses do not encode their own membrane fission machinery, which is required for viral entry, transport, and budding (Garrus et al, 2001). M protein and ORF7b both interact with several components of the SNARE complex, such as STX6 and STX10 (Fig 3). This is especially the case for ORF7b, since its interactome revealed many additional SNARE complex components (Fig 3), including STX12, STX16, VAMP2, VAMP3, VAMP4, VAMP8, NAPA, NAPG, and SNAP29. ORF3a shows the binding to many components of the HOPS complex (Fig 3), including VPS11, VPS16, VPS18, VPS39, and VPS41, while ORF6 binds to several components of the COPII complex (Fig 3), including SEC23A, SEC24A, and SEC31A. The interactions of different viral proteins with distinct cellular complexes suggest that these associations may be coordinated by the virus to maximize viral protein trafficking between different membrane‐associated compartments.
After a virus infects a host cell, it needs to suppress the host‐cell translation activity to facilitate the life cycle of the virus. NSP1 is a well‐studied coronavirus non‐structural gene product that displays biological functions in suppressing host gene expression (Kamitani et al, 2006; Tohya et al, 2009; Lokugamage et al, 2012). Accordingly, we identified HCIPs of eukaryotic initiation factor 3 (EIF3) complex subunits that bind to NSP1 (Fig 3). We also identified the whole DNA polymerase alpha complex, i.e., POLA1, POLA2, PRIM1, and PRIM2, in the NSP1 interactome (Fig 3), which is similar to a recent report (Gordon et al, 2020b). Our interactome analysis also revealed that NSP5 associates with several well‐studied DNA damage proteins (Fig 3), e.g., MDC1, BRCA1, and USP11. In addition, NSP6 interacts with EMC components (Fig 3), and NSP7 associates with CCR4‐NOT complex and THO/TREX complex (Fig 3), which may inhibit host functions and facilitate viral genome replication and virus production.
N protein is a nucleocapsid protein that binds directly to the virus genome. We identified many ribosome proteins with strong interaction with N protein (Fig 3). This is similar to reports in other virus species showing that viral N proteins bind to host ribosome subunits (Cheng et al, 2011; Li et al, 2018). Our data also indicate the binding of N protein to the IGF2BP1 complex, which has been shown previously to enhance viral translation initiation and stabilize viral RNA (Weinlich et al, 2009). In addition to the abovementioned HCIPs, we identified many other RNA processing, RNA metabolism, and transcriptional regulatory proteins as N protein interacting partners (Fig 3). These data suggest that N protein may be involved in multiple viral RNA processes.
Mitochondria is also a major target manipulated by the virus after infection. By taking control of the mitochondria, the virus disrupts host‐cell functions and forces cells to produce energy and other products needed for the viral life cycle (Chambers et al, 2010; Ripoli et al, 2010). The SARS‐CoV‐2 gene NSP6 may play a role in hijacking mitochondria as it interacts with a few subunits of ATP synthase (Fig 3), e.g., ATP5F1B, ATP5F1D, ATP5PB, ATP5PF, ATP5PO, ATP6AP1, and ATP13A3. It also binds to PHB and PHB2, which stabilizes mitochondrial respiratory enzymes and maintains mitochondrial integrity (Kuadkitkan et al, 2010). These interactions of NSP6 with mitochondrial proteins suggest that NSP6 may help manipulate the host energy production system to serve the life cycle of SARS‐CoV‐2.
SARS‐CoV‐2 interacts with host cell immune and antiviral systems
Host cells have developed efficient antiviral systems to counter viral infections. Similarly, viruses find ways to inhibit host antiviral systems to ensure survival. The interferon (IFN)‐mediated antiviral pathway is one of the major innate immune responses against viral pathogens. We expressed the 29 SARS‐CoV‐2 coding genes in HEK293T cells and then analyzed the activity of IFN‐beta luciferase reporter response to infection with Sendai virus, or the activity of ISRE‐luciferase reporter response when viral proteins were co‐expressed with IRF3 (Fig EV1D and E). We found that two viral proteins, NSP1 and N proteins, showed most significantly suppression of the activation of IFN signaling based on these two assays.
NSP1 acts as the strongest gene product in SARS‐CoV‐2 that helps evade host‐cell antiviral defense. It suppresses global gene expression in the host as we mentioned above. Our host–virus interactome results show that NSP1 interacts with nine components of the EIF3 complex, which may be one of the mechanisms to suppress the translation machinery. Translation of host antiviral genes is also inhibited by this global translation suppression function. Therefore, NSP1 acts as the strongest viral protein that inhibits IFN‐beta signaling when overexpressed. On the other hand, N protein is the first released and most abundant SARS‐CoV‐2 viral protein upon infection. We identified several immune response proteins that are recruited by N protein (Fig 3), which include HERC5 and TTLL4. These proteins may facilitate the viral infection and the later viral life cycle.
In addition, our interactome analysis revealed a potential interaction between NSP5 and an IFN‐induced antiviral enzyme, OAS3 (Fig 3). We also found that both M protein and ORF7 interact with growth factor receptors (Fig 3), with M protein interacts with FLT4 (VEGFR3) and ORF7 binds to ERBB receptors including EGFR (ERBB1), ERBB2, and ERBB4 and proteins involved in related signaling pathways. SARS‐CoV‐2 is suspected to bind to growth factor receptors and regulate the related signaling pathways to suppress host antiviral systems and promote host‐cell survival to increase viral infection and production.
Collectively, the association between several viral proteins and host proteins involved in antiviral defense and other cellular functions may reveal how SARS‐CoV‐2 employs multiple ways to evade host antiviral systems for its survival, reproduction, and release.
Comparison of SARS‐CoV‐2 virus–host interaction network to other cellular responses to SARS‐CoV‐2 infection
Interaction between SARS‐CoV‐2 viral gene products and host proteins play key roles in viral infection (Gordon et al, 2020b; Li et al, 2021). To further mining the key genes involved in SARS‐CoV‐2 infection life cycle, we analyzed other datasets which evaluated cellular responses to SARS‐CoV‐2 infection using different strategies.
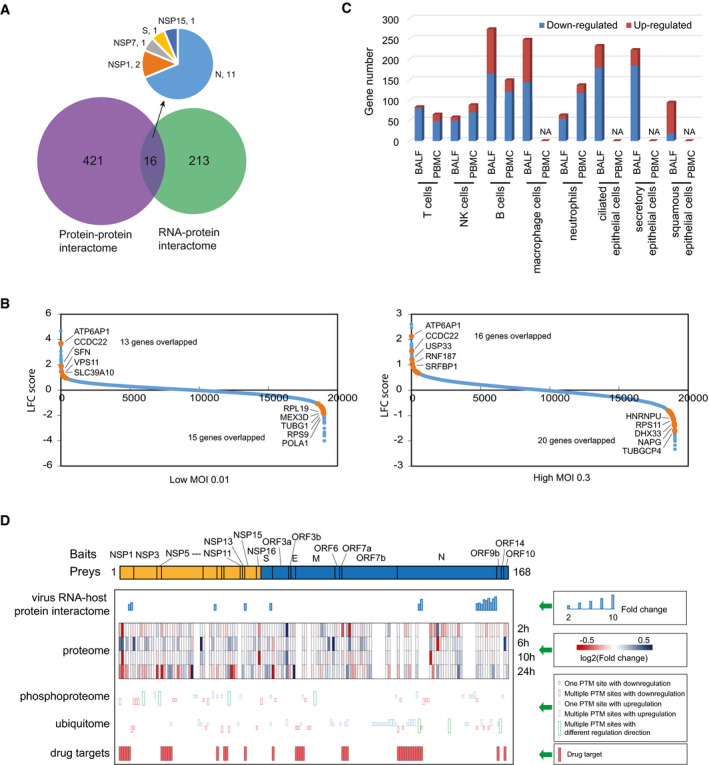
Interactions between virus RNAs and host proteins are also proven to be important in viral infection (Garcia‐Blanco et al, 2016). We compared virus–host protein–protein interactome to the virus RNA–protein interactome (Flynn et al, 2021). In Fig 4A, 16 proteins are common between these two datasets. We also observed that 11 out of these 16 proteins are identified as SARS‐CoV‐2N protein binding partners, which is known as an abundant RNA‐binding protein. Two RNA‐binding proteins were found as NSP1 binding partner, PYCR2 and EIF3I, which imply that these two proteins may work with NSP1 in suppressing host gene expression that requires further investigation.
Figure 4. Comparison of SARS‐CoV‐2 virus–host interaction network to other cellular responses to SARS‐CoV‐2 infection.
-
AOverlapping between SARS‐CoV‐2 virus–host interaction with virus RNA–host protein interaction results.
-
BComparison of SARS‐CoV‐2 virus–host interaction to the genetic screening with CRISPR technique. Two different MOIs (MOI 0.01 and MOI 0.3) were used in these screens, and we compared our results with both of these datasets. Blue dots are the identified genes from the genetic screening. Orange dots are the highlighted overlapped genes between our virus–host interaction study and the results from the genetic screening. Top five overlapped genes are indicated.
-
CAnalyzing the number of HCIPs with RNA expression changes identified by single‐cell RNA sequencing using cells from COVID‐19 patients.
-
DIntegrated analysis of RNA–host protein interaction, proteome or protein profiling, phosphoproteomics, and ubiquitome with our HCIPs. HCIPs overlapped with any of the other omics study were listed, which include a total of 168 proteins. The potential drug targets were also included and presented here.
Genetic screens have been performed to uncover host factors important for SARS‐CoV‐2 life cycle (Daniloski et al, 2021; Wang et al, 2021; Wei et al, 2021). We compared our HCIPs data with all datasets from these screening. An example shown in Fig 4B was the comparison to one dataset (Daniloski et al, 2021).13 genes and 15 genes were identified, respectively, as overlapped genes when knockdown led to increased or decreased viral infection at low MOI of 0.01 (or 16 and 20 genes, respectively, for viral infection at high MOI of 0.3). The top five overlapped genes were highlighted in Fig 4B. ATP6AP1 and CCDC22 were identified as the strongest hits in the screen with different amount of virus (MOI 0.01 or 0.3). The detailed functions of these two genes in SARS‐CoV‐2 virus cell cycle warrant further investigation. The comparison with other two datasets (Wang et al, 2021; Wei et al, 2021) were shown in Fig EV2B and C, and all the comparisons were included in Dataset EV3.
The host may also respond to viral infection by altering host gene transcription. Single‐cell RNA sequencing has been conducted to analyze gene expression in COVID‐19 patients (Ren et al, 2021). The host–virus interactome HCIPs was compared with the single‐cell RNA sequencing data from eight different cell types derived from bronchoalveolar lavage fluid (BALF) or peripheral blood mononuclear (PBMC) cells as shown in Fig 4C. The detailed comparisons were included in Dataset EV3.
It is known that host proteins are also regulated following SARS‐CoV‐2 infection. A number of these proteomics studies, including protein abundance (Bojkova et al, 2020), phosphorylation (Bouhaddou et al, 2020), and ubiquitination (Stukalov et al, 2021), have been conducted. Heat maps of the regulated HCIPs in their abundance, phosphorylation, or ubiquitination with different time point were generated and presented in Fig EV2, EV3, EV4, EV5. In comparison with the proteome dataset (Bojkova et al, 2020), we identified that 110 of the HCIPs changed in their abundance (Fig EV2D), with 67 proteins up‐regulated and 33 proteins down‐regulated (Dataset EV3). When compared with phosphorylation dataset (Bouhaddou et al, 2020), 69 phosphorylation sites belonging to 35 protein were regulated upon viral infection (Fig EV2E). In this list, sixteen proteins have phosphorylation site(s) up‐regulated, with four of them having multiple phosphorylation sites. Another sixteen proteins have phosphorylation site(s) down‐regulated, with seven of them have multiple down‐regulated phosphorylation sites. In addition, we also found that another four proteins have multiple phosphorylation sites but with mixed up‐ or down‐regulation sites (Dataset EV3). Similar comparison was also conducted with the ubiquitination study (Stukalov et al, 2021). In total, 86 ubiquitination sites and 50 HCIPs were regulated via ubiquitination (Fig EV2F). In detail, 33 proteins showed up‐regulated ubiquitination, and 12 of them have multiple ubiquitination sites; 16 proteins showed down‐regulated ubiquitination, and 4 of them have multiple ubiquitination sites; 3 ubiquitination proteins have multiple sites but with mixed results (Dataset EV3).
Figure EV3. Potential drugs targeting SARS‐CoV‐2 and host protein interaction.

The drug‐human target network was analyzed by Metascape and Ingenuity Pathway Analysis/GO analysis. The red diamonds are virus proteins, green circles are the high‐confidence interactive proteins that were identified as drug targets, and red text lists potential drugs. Proteins in the blue rounded rectangles belong to the complex or functional pathway targeted by the same drugs.
Figure EV4. Comparison of interactomes of seven NSP1 proteins from seven human coronaviruses (related to Fig 5).
-
ASequence identity comparison of NSP1 proteins from seven human coronaviruses.
-
B, CInterferon (IFN) signaling assays for NSP1 of seven human coronaviruses. (B) Analysis of IFN‐beta‐luciferase reporter 12 h after Sendai virus infection. (C) Analysis of ISRE‐luciferase reporter when the viral proteins were co‐expressed with IRF3. Graphs show mean ± SD, n = 3. ***P < 0.001, **P < 0.01 and *P < 0.05 (Student’s t‐test).
-
DExpression validation of NSP1 with the SFB tag from different human coronaviruses by Western blotting.
-
EExpression validation of NSP1 with SFB tag from different human coronaviruses by immunofluorescent staining. The green signal is SFB‐tagged bait, and the blue signal indicates DAPI/nuclei. Scale bar: 10 µm.
-
FPSMs identified for the SFB tag in all NSP1 purification experiments which were used as internal control to normalize the interactome list for comparison.
-
GNetwork of high‐confidence interactive proteins (green circles) identified with seven NSP1 proteins (red diamonds).
Source data are available online for this figure.
Figure EV5. Comparison of interactomes of seven N proteins from seven human coronaviruses (related to Fig 6).

-
ASequence identity comparison of N proteins from seven human coronaviruses.
-
BExpression validation of N protein with the SFB tag from different human coronaviruses using Western blotting.
-
CExpression validation of N protein with the SFB tag from different human coronaviruses using immunofluorescent staining. The green signal is SFB‐tagged bait, and the blue signal indicates DAPI/nuclei. Scale bar: 10 µm.
-
D, EInterferon (IFN) signaling assays for N proteins of seven human coronaviruses. (D) Analysis of IFN‐beta‐luciferase reporter 12 h after Sendai virus infection. (E) Analysis of ISRE‐luciferase reporter when the viral proteins were co‐expressed with IRF3. Graphs show mean ± SD, n = 3. ***P < 0.001, **P < 0.01 and *P < 0.05 (Student’s t‐test).
-
FNetwork of high‐confidence interactive proteins (green circles) identified with seven N proteins (red diamonds).
-
GPSMs identified for the SFB tag in all N protein purification experiments which were used as internal control to normalize the interactome list for comparison.
Source data are available online for this figure.
To better understand the regulation of these HCIPs after virus infection, we combined the protein abundance, phosphorylation, and ubiquitination information in Fig 4D and Dataset EV3. We also included the virus RNA–host protein interaction study results. In total, 168 HCIPs have been identified by at least one of the four datasets. Four proteins, i.e., PSIP1, AKAP12, EZR, and HNRNPU, were identified as proteins regulated via their abundance, phosphorylation, and ubiquitination. Furthermore, EZR and HNRNPU were also reported as virus RNA‐binding proteins. These HCIPs are likely to be important during SARS‐CoV‐2 infection. In‐depth functional analysis should be carried out to reveal the key roles of these proteins during viral infection life cycle, which may lead to new strategies to combat this disease.
Potential drug targets revealed by the host–virus interactome study
The interactions between viral and host proteins are believed to play fundamental roles in virus life cycle. It is reasonable to speculate that drugs targeting host proteins may inhibit the functions of host protein complexes and/or disrupt their interactions with viral proteins, which may lead to the collapse of virus life cycle. Therefore, we analyzed potential drug targets and drugs using bioinformatics tools, Metascape (Zhou et al, 2019), or Ingenuity Pathway Analysis. Of the 29 viral proteins and 437 SARS‐CoV‐2 and host HCIPs, we found 18 viral proteins with 60 host interaction proteins that could be targeted by various drugs (Fig EV3 and Table EV1). Of these potentially drugs targets, some well‐studied drugs are available and/or being developed. SIGMAR1, which binds to NSP6, was tested as an important drug target candidate for the treatment of COVID‐19 (Gordon et al, 2020b). Many of our candidate proteins are well studied as drug targets, such as DHFR and VDAC1/VDAC2, which interact with NSP6; GSK3B, which interacts with N protein; FLT4, which interacts with M protein; EGFR, ERBB2, and ERBB4, which interact with ORF7b; A2M, which interacts with S protein; and ANXA5, which interacts with NSP15. Multiple drugs targeting these host proteins have been developed, which should facilitate the development of treatment options for COVID‐19 patients. Indeed, two potential drugs proposed in our study were already tested for COVID‐19 treatment, haloperidol (SIGMAR1 antagonist), and tamoxifer (ERBB2/ERBB4 inhibitor) (Fig EV3) and they both showed effectiveness against COVID‐19 (Gordon et al, 2020a).
Comparison with other proteomics datasets of SARS‐CoV‐2 infection may help us narrow down potential drug targets, since the regulated HCIPs may have additional functions during viral infection beyond host–virus protein–protein interaction. In Fig 4D, the drug targets we analyzed above was integrated with the HCIPs which show changes in their protein level or modifications. For example, the potential drug targets VDAC1/VDAC2 were identified as up‐regulated proteins upon viral infection. Additionally, VDAC2 ubiquitination site K120 was also up‐regulated during SARS‐CoV‐2 infection. Another potential drug target GSK3B was found to be down‐regulated at its phosphorylation site S9 after SARS‐CoV‐2 infection. These alterations underscore the potential roles of these proteins during viral infection, which make them ideal drug targets for COVID‐19.
Comparison of coronavirus–host interaction networks among human coronaviruses
Seven human coronaviruses have been reported since the first one, HCoV‐OC43, was identified in the mid‐1960s. Four of these coronaviruses cause mild to moderate symptoms, including HCoV‐OC43, HCoV‐HKU1, HCoV‐NL63, and HCoV‐229E. The other three coronaviruses, SARS‐CoV‐2, SARS‐CoV‐1, and MERS‐CoV, can cause more serious, even fatal diseases. To understand the overlap and divergence of virus–host interactions among these human coronaviruses, we selected two viral gene products, NSP1 and N protein, for comparison. These two viral proteins also significantly suppress the host antiviral IFN pathway as shown in Fig EV1D and E. These comparisons were meant to identify potential targets that could be used for pan‐viral treatment against currently known coronaviruses and any new coronaviruses identified in the future. The comparisons may also reveal different mechanisms adopted by each coronavirus for its viral life cycle and associated pathologic symptoms.
Comparison of NSP1 interactomes among all seven human coronaviruses
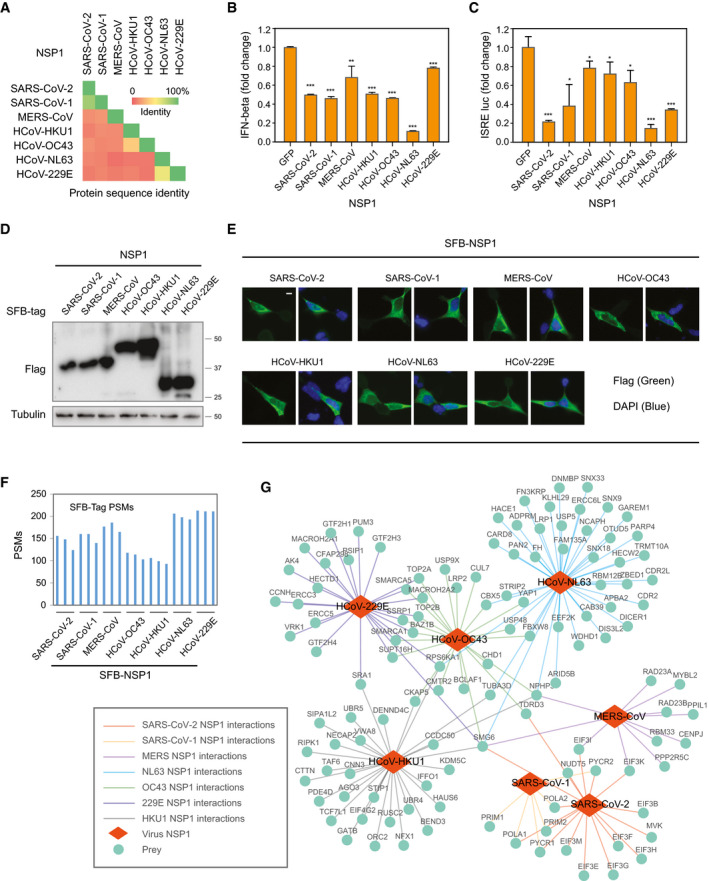
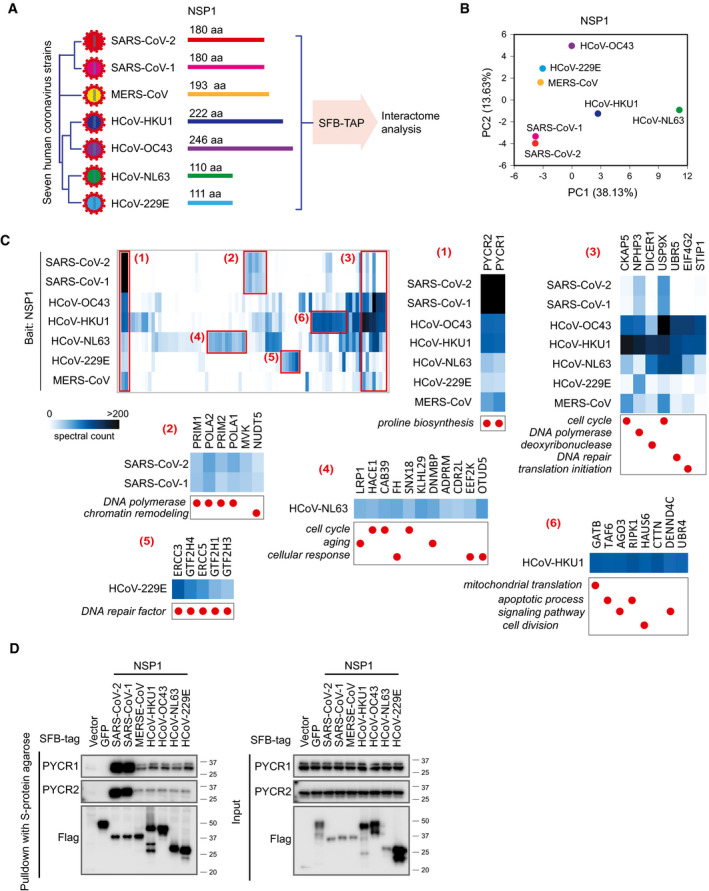
NSP1 is arguably the most important pathogenic determinant of human coronaviruses, because it induces a near‐complete suppression of host gene expression (Benedetti et al, 2020). NSP1 uses a two‐pronged strategy for this host gene suppression, i.e., stalling canonical mRNA translation and triggering the degradation of host mRNAs (Kamitani et al, 2009). NSP1 also suppresses host antiviral gene expression, which inhibits the host antiviral defense systems and therefore facilitates viral replication and immune evasion (Thoms et al, 2020). NSP1 proteins from two alpha coronaviruses, CoV‐229E and CoV‐NL63, are shorter than those from the other five beta coronaviruses (Fig 5A). Most coronavirus NSP1 proteins share low sequence identity, i.e., 10–20%, except for SARS‐CoV‐2 and SARS‐CoV‐1, which have a shared sequence identity of 84.4% (Fig EV4A). However, all of these NSP1 proteins have similar functions in suppressing host gene expression and inhibiting the host antiviral system, such as IFN signaling with varying extend (Fig EV4B and C).
Figure 5. Comparison of interactomes of seven NSP1 proteins from seven human coronaviruses.
-
AComparisons of NSP1 proteins from seven human coronaviruses.
-
BPrincipal Component Analysis of the seven HCIP lists with bait NSP1 from different human coronaviruses.
-
CHeat map of the NSP1 HCIPs obtained using SFB‐TAP. NSP1 HCIPs were compared among seven human coronaviruses using the preys’ spectral counts. Six areas of the heat map are manually selected, enlarged, and labeled as (1), (2), (3), (4), (5), and (6). These groups of proteins are enriched by NSP1 protein specific from one or limited human coronaviruses. Functional characterization of each protein is shown with the red circles below the enlarged images.
-
DPulldown and Western blot validation of the interaction between human coronavirus NSP1 proteins and the human proteins PYCR1/PYCR2. Cells transfected with vector or construct encoding GFP were included as controls in these experiments.
Source data are available online for this figure.
We expressed all seven human coronavirus NSP1 proteins in HEK293T cells, which were validated by immunoblotting and Immunostaining (Fig EV4D and E), and performed SFB‐TAP experiment to uncover the protein–protein interaction networks between NSP1 and host proteins, following a similar strategy as described in the SARS‐CoV‐2 interactome analysis presented in Fig 1. To compare all HCIPs among different human coronavirus strains, we first identified proteins for each coronavirus NSP1 with a SAINTexpress Bayesian false discovery rate ≤ 0.01 in at least one of the seven coronavirus NSP1 interactions. Using these HCIPs, we identified all PSMs captured for that prey in the raw identification data for each coronavirus NSP1 (Dataset EV4). To further compare the NSP1 interacting proteins among different human coronaviruses, we normalized the prey PSMs based on the identified PSMs of the SFB tag which could serve as an internal standard for this comparison (Fig EV4F). Principal component analysis grouped SARS‐CoV‐2 and SARS‐CoV‐1 together (Fig 5B), indicating that these two viruses with high NSP1 sequence identity share similar interactomes and may adopt similar mechanisms to suppress host gene expression. The protein–protein interaction network of NSP1 in the seven human coronaviruses was built and presented in Fig EV4G.
Some HCIPs were unique or showed significantly higher binding signals to NSP1 from one or several coronaviruses. For example, the subunits of DNA polymerase alpha‐primase complex, POLA1, POLA2, PRIM1, and PRIM2, were identified with NSP1 of SARS‐CoV‐1 and SARS‐CoV‐2 (Fig 5C, group 2), which was reported recently (Gordon et al, 2020a). However, there was no or weak signal of this complex in other coronavirus NSP1 interactomes (Fig 5C). We identified more than 500 PSMs for both PYCR1 and PYCR2 in SARS‐CoV‐2 and SARS‐CoV‐1 purifications (Fig 5C, group 1), indicating that these two viruses may have some unique mechanisms via their association with PYCR1/2. Several HCIPs, such as DICER1, UBR5, and EIF4G2, were significantly enriched in HCoV‐OC43 and HCoV‐HKU1 NSP1 interactomes when comparing to the interactomes of other human coronaviruses (Fig 5C, group 3). Additionally, a group of proteins function in several function pathways has been significantly enriched only by HCoV‐NL63 (Fig 5C, group 4), such as LRP1, HACE1, CAB39, and OTUD5. One group of proteins was identified only in HCoV‐229E NSP1 purification (Fig 5C, group 5); these proteins, ERCC3, ERCC5, GTF2H1, GTF2H3, and GTF2H4, are involved in DNA repair pathway. TAF6 was identified only by the NSP1 purification of HCoV‐HKU1, and AGO3, RIPK1, HAUS6, were captured by all seven human coronaviruses NSP1 protein, but only significantly enriched by HCoV‐HKU1 NSP1 (Fig 5C, group 6).
We further confirmed the protein–protein interaction between SARS‐CoV‐1/2 NSP1 with PYCR1/PYCR2 (Fig 5D). PYCR1/PYCR2 proteins catalyze the last step in proline biosynthesis. The functional significance of NSP1‐PYCR1/PYCR2 interaction in viral life cycle warrants further investigation.
Together, these findings suggest that different and unique mechanisms may be utilized by each coronavirus after infection to suppress host gene expression. These differences in NSP1 interactomes may also explain why different coronaviruses have different symptoms in humans. It may indicate potential drug targets against a specific kind or limited number of human coronavirus(es) can be developed with those unique binding host genes.
Comparison of N protein interactomes among human coronaviruses
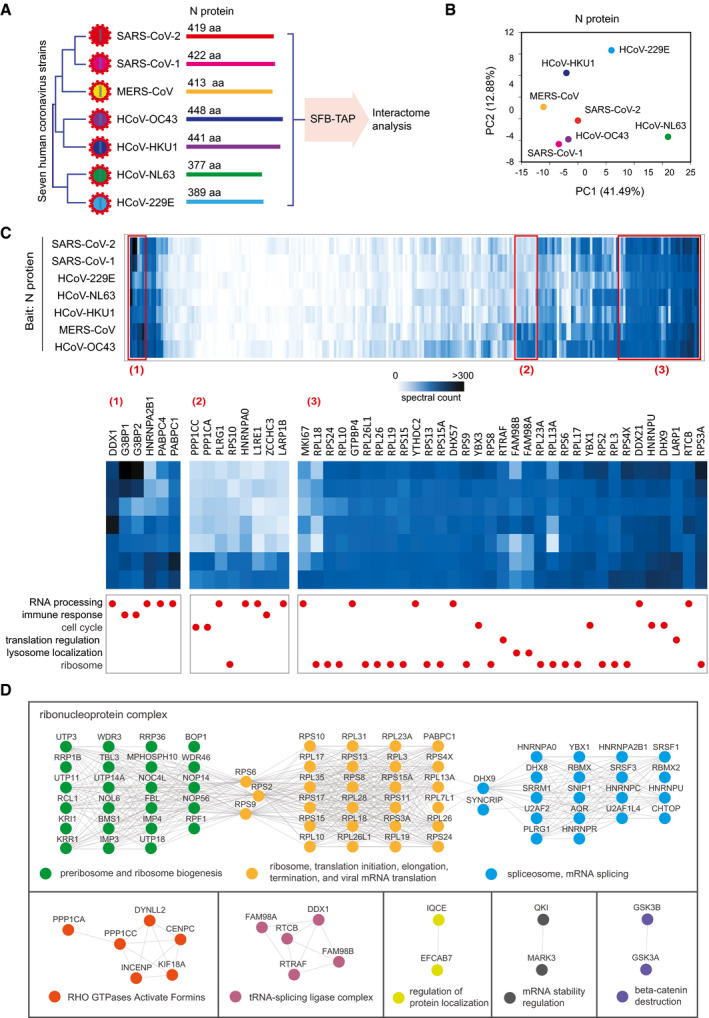
SARS‐CoV‐2N protein is the first released and most abundant protein during viral infection. N protein has several key functions that help viral transcription, translation, genome replication, and packaging before budding. N protein is also one of the key molecules that the virus uses to fight against host antiviral systems. We investigated the interactomes of N protein in different coronaviruses using the same strategy as described above for the study of NSP1 (Fig 6A). Most of the N proteins in different human coronaviruses had sequence identity percentages between 20 and 30%, but the sequence identity was 89.1% between SARS‐CoV‐2 and SARS‐CoV‐1 (Fig EV5A).
Figure 6. Comparison of interactomes of N proteins from different human coronaviruses.
-
AComparison of N proteins from seven human coronaviruses.
-
BPrincipal Component analysis of the seven high‐confidence interacting protein (HCIP) lists with bait N protein from different human coronaviruses.
-
CHeat map of the N protein HCIPs identified by SFB‐tandem affinity purification. N protein HCIPs were compared among seven human coronaviruses using the preys’ spectral counts. Three areas of the heat map are manually selected, enlarged, and labeled as (1), (2), and (3), which are enriched by all N proteins of the seven human coronaviruses. Functional characterization for each protein is shown with the red circles below the enlarged images.
-
DThe human proteins which binding to N proteins of human coronaviruses were integrated and analyzed by STRING to illustrate the protein–protein interaction among the HCIPs. The minimum required interaction score setting in STRING is 0.9. Different function group or complex were colored differently. The filled circles are proteins, and the lines are the interactions.
We expressed all seven human coronavirus N proteins in HEK293T cells, which were validated by immunoblotting and Immunostaining (Fig EV5B and C). We tested IFN signaling suppression by N proteins of all seven human coronaviruses. Our results indicated only SARS‐CoV‐2 and SARS‐CoV‐1N proteins significantly suppressed the activation of IFN signaling (Fig EV5D and E). With the TAP‐MS results, we built the protein–protein interaction network of N proteins in seven human coronaviruses (Fig EV5F). Similar to the analysis of NSP1 interactomes, we normalized the prey PSMs based on the identified PSMs for the SFB tag (Fig EV5G). We performed a principal component analysis (Fig 6B) and generated a heat map of the N protein interactomes in different coronaviruses (Fig 6C and Dataset EV5). Many of the strong binding HCIPs were shared among all coronaviruses, and a large portion of the shared HCIPs play roles in RNA processing, ribosome biogenesis, and translation regulation (Fig 6C and D).
Some strong binding partners were enriched with much higher identified PSMs in one or two coronaviruses comparing to other human coronaviruses. As shown in Fig 7A, G3BP1 and G3BP2 are highly enriched by N proteins from SARS‐CoV‐2 and SARS‐CoV‐1 (please also see Fig 6C); FAM98A, FAM98B, DDX1, and RTRAF were better enriched by HCoV‐OC43 N proteins; PPP1CA, PPP1CC, PABPC1, and PABPC4 have preferred binding to the N proteins belonging to HCoV‐229E and HCoV‐NL63; TOP2A and EXOSC10 were identified as HCIPs only by the N protein of HCoV‐NL63.
Figure 7. Analysis of HCIPs showing specific interaction with one or more N proteins of human coronaviruses.
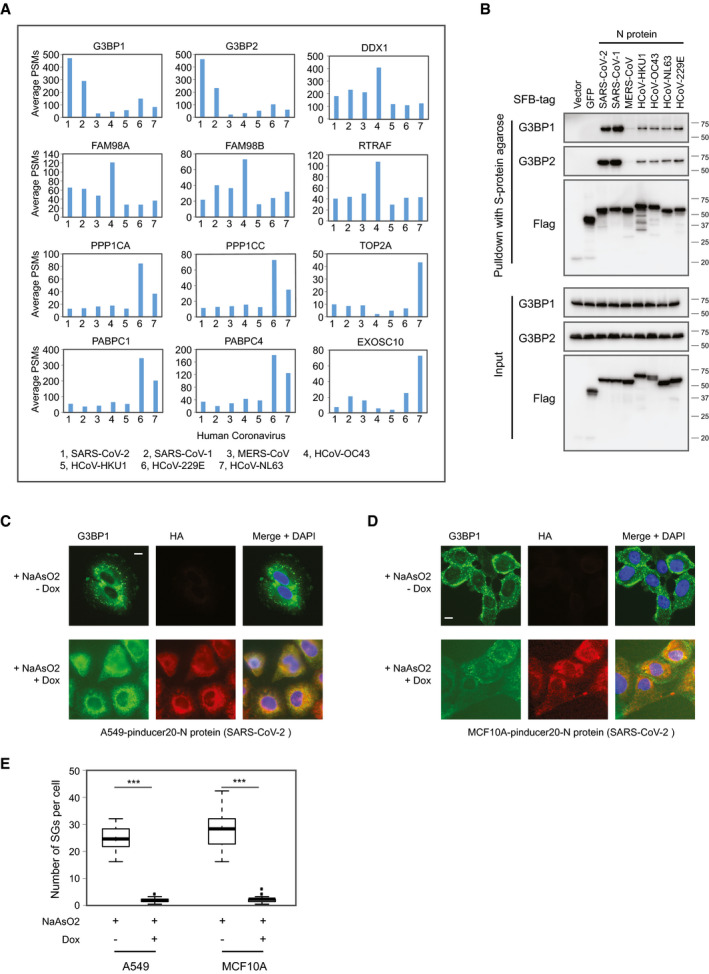
-
APreys identified showing significant enrichment by N proteins specifically from one or more human coronaviruses. PSM, peptide‐spectrum match.
-
BPulldown and Western blot validation of the interaction between N protein and G3BP1/G3BP2. Cells transfected with vector or construct encoding GFP or N protein from different human coronaviruses were compared in these experiments.
-
C, DEffect of SARS‐CoV‐2N protein on stress granule formation. A549 (C) and MCF10A (D) cells were transfected with pinducer20‐N protein of SARS‐CoV‐2. All the cells are treated with sodium arsenite but with or without Dox to induce N protein expression. Cells were fixed and stained with the indicated antibodies. The green signal is G3BP1, the red signal is HA (for N protein), and the blue signal indicates DAPI/nuclei. Scale bar: 10 µm.
-
EQuantification of the stress granule formation in A549 and MCF10A cells. Total of 30 cells were counted and Student’s t‐test was used for the statistical analysis (***P < 0.001). Box limits represent 25th percentile and 75th percentile; horizontal line represents median. Whiskers display min. to max. values.
Source data are available online for this figure.
As shown in Fig 7B, we confirmed the highly enriched G3BP1 and G3BP2 by N proteins. G3BP1 and G3BP2 bind more strongly with the N proteins from SARS‐CoV‐2 and SARS‐CoV‐1 than with the N proteins from other human coronaviruses. Both G3BP1 and G3BP2 are suggested contributing to stress granule (SG) formation. We tested the SG formation before and after inducible expression of SARS‐CoV‐2N protein. A549 and MCF10A cells were transfected with pinduce20‐N protein from SARS‐CoV2. The sodium arsenite treatment was applied to the cell to induced SGs. We found that SG formation under stress were diminished when we induced the expression of SARS‐CoV‐2N protein (Fig 7C–E). These results suggest that SARS‐CoV‐2N protein may affect the function of G3BP1 protein in these SGs.
To further study the biological functions of the interaction between N protein and G3BP1/G3BP2, we tested two other recently reported functions of these two proteins. G3BP1 and G3BP2 have been shown to be important for immunity response (Liu et al, 2019; Yang et al, 2019). We also found that SARS‐CoV‐2N protein significantly suppressed the activation of IFN signaling. However, as shown in Appendix Fig S2A, with or without N protein expression, we did not observe any significant change in SeV infection‐induced luciferase activity in control or G3BP1/2 knockdown HEK293T cells. Another recently reported function of G3BP1 and G3BP2 was their involvement in the regulation of mTORC1 activity (Prentzell et al, 2021). We tested this function in MCF10A cells. However, the results showed some modest upregulation of mTOR pathway activation following SARS‐CoV‐2N protein expression as detected by pS6K signals, but this change was not affected in G3BP1/2 KO cells (Appendix Fig S2B). The biological significance of the strong binding between SARS‐CoV‐2N protein and G3BP1/G3BP2 remains to be elucidated.
Discussion
In the current study, we generated a comprehensive host–virus protein–protein interaction network of SARS‐CoV‐2 by identifying HCIPs using two different interactome approaches, i.e., TAP‐MS and BioID2‐MS. We identified a total of 437 HCIPs that bind to one or several of the SARS‐CoV‐2 gene products. These findings not only validate several known protein–protein interactions but also identify many new protein–protein interactions with potential functions in SARS‐CoV‐2 life cycle. This dataset reveals new mechanistic insights into the diverse functions of SARS‐CoV‐2 genes, which require further investigation. Moreover, this dataset suggests potential drug targets for the treatment of COVID‐19.
We built a host–virus protein–protein interaction network of SARS‐CoV‐2 based on the 437 HCIPs identified in our study. This interaction network provides clues as how these viral proteins participate in virus life cycle. M protein, NSP6, ORF3a, ORF6, and ORF7b help viral infection, trafficking, and maybe the budding of the virion; NSP1, NSP3, NSP5, NSP6, NSP7, and N protein suppress host‐cell replication, transcription, and translation, and at the same time contribute to the same processes in the viral life cycle; S protein facilitates the formation of new viruses after viral replication and translation of structural proteins; NSP1, NSP5, N protein, M protein, and ORF7 may inhibit host‐cell antiviral responses and therefore promote the survival of the infected virus, and they also prolong host‐cell survival to increase viral production. NSP6 and NSP7 may manipulate host‐cell metabolism and signaling transduction pathways. Understanding in detail how the virus and host‐cell communicate will help us to find or design therapeutic strategies to suppress viral infection.
Additionally, we compared NSP1 and N protein interactomes among different human coronaviruses. These comparisons expand our knowledge of these viruses. For the N proteins, there have many common host binding partners, which match their biological functions. The regulation of multiple viral RNA processes in all human coronaviruses may be mediated by the interaction between N protein and host ribosomal proteins or proteins involved in ribosomal biogenesis. Besides common interaction proteins, we also uncovered some host proteins showing distinct binding patterns to a unique or a subset of the seven human coronaviruses, and these specific interactions may explain divergent pathogenesis of these coronaviruses. Indeed, we observed robust interactions between the NSP1 proteins of SARS‐CoV‐2 and SARS‐CoV‐1 with host PYCR1 and PYCR2, two proteins involved in proline biosynthesis. We also validated strong interactions between the N proteins of SARS‐CoV‐2 and SARS‐CoV‐1 with host G3BP1 or G3BP2, two proteins that are known to be involved in SG formation. How these strong protein–protein interactions participate in the pathogenesis of SARS‐CoV‐2 and SARS‐CoV‐1 remain to be determined.
There are limitations in our current study. First, the expression levels across baits are quite different as shown in Fig EV1A and B, and it is a common problem in other interactome studies of SARS‐CoV‐2 (Gordon et al, 2020b; preprint: Samavarchi‐Tehrani et al, 2020; Stukalov et al, 2021). The bait with poor expression level may lead to fewer PSMs identification of that bait in the interactome study. It may lower the interactome data quality of that bait. We tried to use stringent data analysis process to improve the final HCIPs list. However, systematic optimization to improve the expression level of a specific viral gene is preferred if we interested in a specific viral gene interactome. Second, as shown in Figs 2C and D, and EV2, the overlapping between our TAP‐MS and BioID2‐MS is quite low, and also in other SARS‐CoV‐2 interactome datasets. This is still an issue in protein–protein interaction studies, which may due to the difference in detailed experimental setting and data processing procedure tailored to each approach/study. The host–virus protein interaction network may be improved if we integrate all the interactome results with stringent and proper data filtration strategy. Third, this study needs additional validation especially in clinical relevant setting to expand our knowledge how these interactions may be involved in SARS‐CoV‐2 pathogenesis, which will help the fight against COVID‐19. We believe that rapid public access to our findings may help other researchers in their SARS‐CoV2 and COVID‐19 studies.
In conclusion, our systemic study of the SARS‐CoV‐2 protein–protein interaction network provides useful data on viral protein functions and potential underlying mechanisms, which could lead to the identification of new drug targets for the treatment of COVID‐19. The comparison of interactomes among different human coronaviruses elucidated the common and different strategies these human coronaviruses may employ to manipulate host cells and maximize viral infection/production. These findings will benefit our current fight against COVID‐19 and also suggest ways to combat any future coronavirus‐caused diseases.
Materials and Methods
Reagents and Tools table
| Reagent/Resource | Reference or Source | Identifier or Catalog Number |
|---|---|---|
| Experimental Models | ||
| HEK293T | ATCC | Cat#CRL‐3216 |
| HEK293A | ATCC | Cat# CRL‐1573 |
| U2OS | ATCC | Cat#HTB‐96 |
| MCF10A | ATCC | Cat# CRL‐10317 |
| Recombinant DNA | ||
| Gateway pDONR221 vector | ThermoFisher Scientific | Cat#12536017 |
| MCS‐BioID2‐HA | Addgene | Cat#74224 |
| pDEST‐SFB vector | this study | N/A |
| pDEST‐BioID2 vector | this study | N/A |
| Antibodies | ||
| Rabbit anti‐HA | Cell Signaling Technology | Cat#3724S |
| Rabbit anti‐G3BP1 | Cell Signaling Technology | Cat#17798S |
| Rabbit anti‐S6K | Cell Signaling Technology | Cat#2708S |
| Rabbit anti‐pS6K | Cell Signaling Technology | Cat#9208S |
| Rabbit monoclonal anti‐LAMP1 | Cell Signaling Technology | Cat#9091S |
| Rabbit polyclonal anti‐CLCC1 | Atlas Antibodies | Cat# HPA009087 |
| Mouse monoclonal anti‐VPS11 | Santa Cruz Biotechnology | Cat# sc‐100893 |
| Rabbit polyclonal anti‐SPCS2 | ThermoFisher Scientific | Cat# A305‐608A‐M |
| Rabbit polyclonal anti‐G3BP2 | ThermoFisher Scientific | Cat#A302‐040A‐M |
| Rabbit polyclonal anti‐PYCR1 | Proteintech | Cat# 13108‐1‐AP |
| Rabbit polyclonal anti‐PYCR2 | Proteintech | Cat# 17146‐1‐AP |
| Mouse monoclonal anti‐Flag (M2) | Sigma‐Aldrich | Cat#F3165 |
| Mouse monoclonal anti‐Tubulin | Sigma‐Aldrich | Cat#T6199 |
| Mouse monoclonal anti‐Vinculin | Sigma‐Aldrich | Cat#V9264 |
| Streptavidin (HRP) | Abcam | Cat#ab7403 |
| Chemicals, Enzymes and other reagents | ||
| Trypsin, mass spectrometry grade | Promega Corporation | Cat#V5280 |
| Trifluoroacetic acid | Sigma‐Aldrich | Cat#302031 |
| Acetonitrile | Sigma‐Aldrich | Cat#271004 |
| Biotin | Sigma‐Aldrich | Cat#B4501‐25G |
| Streptavidin sepharose beads | ThermoFisher Scientific | Cat# 45‐000‐279 |
| S‐protein agarose | VWR International | Cat#EM69704 |
| Protease inhibitor cocktail | Sigma‐Aldrich | Cat# 11697498001 |
| Polyethyleneimine | ThermoFisher Scientific | Cat# NC1014320 |
| Puromycin | Life Technologies | Cat# A1113803 |
| X‐tremeGENE HP DNA Transfection Reagent | Sigma‐Aldrich | Cat#6366546001 |
| Passive lysis buffer | Promega | Cat# E1941 |
| ECL Plus Western blotting substrate | Bio‐Rad | Cat# 1705061 |
| Dual‐luciferase assay kit | Promega | Cat# E1910 |
| Prolong Gold Reagent with DAPI | Cell Signaling Technology | Cat# 8961S |
| Sodium (meta)arsenite | Sigma‐Aldrich | Cat#S7400‐100G |
| Software | ||
| Proteome Discoverer | Thermo Fisher | V2.2 |
| Mascot | Matrix Science | V2.5 |
| SAINTexpress | Teo et al (2014) | V3.6.3 |
| Metascape | Zhou et al (2019) | https://metascape.org/ |
| Cytoscape | NRNB | V3.8.1 |
| Ingenuity Pathway Analysis | QIAGEN | |
| Other | ||
| Q Exactive HF Orbitrap LC‐MS/MS System | ThermoFisher Scientific | |
Methods and Protocols
Plasmids construction
The cDNAs of viral genes were synthesized using Integrated DNA Technologies (IA, USA) or Twist Bioscience (CA, USA), or ordered from Addgene (MA, USA), and subcloned into pDONOR201 vector as entry clones. Then, the entry clones were recombined into a lentiviral‐gateway‐compatible destination vector to express the fusion proteins with SFB or BioID2 tag.
Cell culture and transfection
HEK293T, HEK293A, U2OS, and MCF10A cell lines were maintained in DMEM supplemented with 10% fetal bovine serum, but MCF10A was maintained in DMEM/F12 medium, and cultured at 37°C in 5% CO2 (v/v). The cell lines were tested to make sure that they were from of mycoplasma contamination.
Recombined destination vector with SFB‐ or BioID2‐fused viral genes were transfected into HEK293T or U2OS cells with polyethyleneimine reagent (Fisher Scientific). After selection with 2 μg/ml puromycin (Life Technologies), the stable clones were picked and validated by Western blot analysis. The exception is the viral gene NSP1: Destination vectors with NSP1 (SFB‐NSP1 and BioID2‐NSP1) were transfected into HEK293T cells 36 h before cell harvest using the reagent X‐tremeGENE HP DNA Transfection Reagent (Sigma‐Aldrich, MO, USA). Cells expressing the BioID2‐tagged genes were treated with 50 mM biotin for 18 h and then harvested. Vector‐transfected cells or cells expressing GFP were used as controls, which were processed along with these experiments. Three biological replicates were performed for each SFB‐ or BioID2‐tagged gene or negative control.
SFB‐TAP and BioID2 assays
Pellets of cells expressing SFB‐ or BioID2‐tagged viral genes were subjected to lysis with NETN buffer (100 mM NaCl, 1 mM EDTA, 20 mM Tris–HCl, and 0.5% Nonidet P‐40) with protease inhibitor cocktail (Sigma‐Aldrich) at 4°C for 20 min. After centrifugation at 16,000 g for 20 min at 4°C, the supernatant was incubated with streptavidin‐conjugated beads (Thermo Fisher Scientific) for 2 h at 4°C. Then, the beads were washed with NETN buffer four times. The BioID2‐tagged samples were subjected to SDS–PAGE, and the gel was fixed and stained with Coomassie brilliant blue. Then, the whole lane of the sample in the gel was excised and subjected to MS analysis. The SFB‐tagged samples were eluted by 2 mg/ml biotin for 1 h at 4°C. The elutes were incubated with S‐protein agarose (VWR International, PA, USA) for 2 h. After four washings with NETN buffer, the beads were subjected to SDS–PAGE and Coomassie brilliant blue staining, as with the BioID2 samples.
Mass spectrometry analysis
The samples in gel were excised and de‐stained completely before digestion. In‐gel digestion was performed with trypsin (V5280, Promega Corporation, WI, USA) in 50 mM NH4HCO3 at 37°C overnight. The extracted peptides were vacuum‐dried and then reconstituted in the MS loading solution (2% acetonitrile and 0.1% formic acid).
The MS sample was loaded onto nano‐reverse‐phase high‐performance liquid chromatography and eluted with acetonitrile gradient from 5 to 35% for 60 min at a flow rate of 300 nl/min. The elute was analyzed by the Q Exactive HF MS system (Thermo Fisher Scientific) with positive ion mode and in a data‐dependent manner, one full scan followed by up to 20 MS/MS scans. The full MS scan was performed with a scanning range of 350–1,200 m/z and resolution at 60,000 at m/z 400.
The raw MS data were submitted to Mascot 2.5 (Matrix Science, MA, USA) using Proteome Discoverer 2.2 (Thermo Fisher Scientific). The database used for searching was Homo sapiens downloaded from Uniprot (July 2020). The sequences of SARS‐CoV‐2 genes, SFB tag, and BioID2 tag were added into the database with 20,414 entries in total. Oxidation for methionine and carboxyamidomethyl for cysteine were set as variable modifications. The mass tolerance for precursor was 10 ppm, and for product ion, 0.02 Da. Tolerance of two missed cleavages of trypsin was applied. Common contaminant proteins were removed from the identification list.
High‐confidence interacting proteins
The identified protein lists were applied to a filtration strategy by comparing with lists obtained from controls to uncover HCIPs. We used SAINTexpress (version 3.6.3) to compare samples with the controls, which included the results from SFB‐TAP or BioID2 experiments using the bait, vector, GFP control, and purification results with other virus genes, except the one we were analyzing. For the SARS‐CoV‐2 HCIPs analysis, we selected the proteins with SAINTexpress Bayesian false discovery rate ≤ 0.05 as our virus interactome HCIPs. The host–virus interactome network was generated by Cytoscape and based on these HCIPs. Functional characterization was carried out by Metascape (Zhou et al, 2019) or Ingenuity Pathway Analysis (Qiagen).
Pulldown and Western blot analysis
To validate the interaction between host and viral gene products, we transfected HEK293T cells with constructs encoding SFB‐tagged viral genes using polyethyleneimine reagent. Cells were collected and subjected to lysis buffer (NETN buffer) on ice for 20 min. The cell lysates were collected and centrifuged. Supernatants were incubated with S‐protein beads for 2 h at 4°C. After three washings with NETN buffer, samples were boiled in 2× Laemmli buffer and the Western blot analysis was conducted with antibodies as indicated in the Figures.
Immunofluorescent staining
Immunofluorescent staining was performed following the method described in our previous study (Chen, Lei et al, 2019). U2OS cells were cultured on coverslips overnight and then transfected with construct encoding SARS‐CoV‐2 viral gene or vector control. Twenty hours after transfection, cells were fixed in 3% paraformaldehyde for 10 min and extracted with a 0.5% Triton X‐100 solution for 5 min. After blocking with 1% bovine serum albumin, cells were incubated with the indicated primary antibodies for 1 h and washed three times before incubation with fluorescein isothiocyanate‐ or rhodamine‐conjugated second primary antibodies (1:3000 dilution; Jackson ImmunoResearch Laboratories, PA, USA) for 1 h. The coverslips were mounted onto glass slides with ProLong gold antifade reagent with DAPI (Cell Signaling Technology) and visualized with a fluorescent microscope.
Luciferase reporter assay
The luciferase reporter assay followed a previous study. Constructs encoding viral proteins were co‐transfected in HEK293T cells along with IFN‐beta‐luciferase or 5× ISRE‐Luc reporter together with construct encoding IRF3, and with the pRL‐Luc with Renilla luciferase as the internal control. 24 h after transfection, for IFN‐beta‐luciferase reporter assay Sendai Virus (Cantell strain; Charles River, SPAFAS) was added directly to the medium to a final concentration of 1 hemagglutinating unit/4.0× 103 cells and incubated for 12 h before harvesting cells. Cells were lysed with passive lysis buffer (Promega), and we performed the luciferase assays using a dual‐luciferase assay kit (Promega) and quantified them with Monolight 3010 (Becton Dickinson).
Statistical analysis
Each experiment was repeated three times or more unless otherwise noted. Differences between groups were analyzed using Student’s t‐test or two‐way analysis of variance with the Tukey multiple comparisons test. P < 0.05 were considered statistically significant.
Author contributions
Conceptualization, ZC and JC; Methodology, ZC, CW, XF, and JC; Investigation, ZC, CW, and XF; Validation, ZC, CW, and XF; Resources, LN, MT, HZ, YX, SKS, and MS; Writing Original Draft, ZC and JC; Review and Editing, all authors; Supervision, JC.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Review Process File
Appendix
Expanded View Figures PDF
Table EV1
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Source Data for Expanded View and Appendix
Source Data for Figure 2
Source Data for Figure 5
Source Data for Figure 7
Acknowledgements
We thank Drs. Peihui Wang and Yuan Wang for their kind help. We thank Erica Goodoff and the Research Medical Library at the University of Texas MD Anderson Cancer Center for editing the manuscript. This work was supported in part by startup and internal funds from MD Anderson Cancer Center to J.C. J.C. also received support from the Pamela and Wayne Garrison Distinguished Chair in Cancer Research. This work was also supported in part by the National Institutes of Health through Cancer Center Support Grant P30CA016672. In addition, JC receives support from Cancer Prevention & Research Institute of Texas award (RP160667 and RP180813) and NIH (P01CA193124, R01CA210929, R01CA216437, and R01 CA216911).
The EMBO Journal (2021) 40: e107776.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD023209 (http://www.ebi.ac.uk/pride/archive/projects/PXD023209).
References
- Benedetti F, Snyder GA, Giovanetti M, Angeletti S, Gallo RC, Ciccozzi M, Zella D (2020) Emerging of a SARS‐CoV‐2 viral strain with a deletion in nsp1. J Transl Med 18: 329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojkova D, Klann K, Koch B, Widera M, Krause D, Ciesek S, Cinatl J, Munch C (2020) Proteomics of SARS‐CoV‐2‐infected host cells reveals therapy targets. Nature 583: 469–472 [DOI] [PubMed] [Google Scholar]
- Bouhaddou M, Memon D, Meyer B, White KM, Rezelj VV, Correa Marrero M, Polacco BJ, Melnyk JE, Ulferts S, Kaake RMet al (2020) The global phosphorylation landscape of SARS‐CoV‐2 infection. Cell 182: 685–712.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers JW, Maguire TG, Alwine JC (2010) Glutamine metabolism is essential for human cytomegalovirus infection. J Virol 84: 1867–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Chen J (2021) Mass spectrometry‐based protein‐protein interaction techniques and their applications in studies of DNA damage repair. J Zhejiang Univ Sci 22: 1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Lei C, Wang C, Li N, Srivastava M, Tang M, Zhang H, Choi JM, Jung SY, Qin Jet al (2019) Global phosphoproteomic analysis reveals ARMC10 as an AMPK substrate that regulates mitochondrial dynamics. Nat Commun 10: 104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng E, Haque A, Rimmer MA, Hussein IT, Sheema S, Little A, Mir MA (2011) Characterization of the Interaction between hantavirus nucleocapsid protein (N) and ribosomal protein S19 (RPS19). J Biol Chem 286: 11814–11824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniloski Z, Jordan TX, Wessels HH, Hoagland DA, Kasela S, Legut M, Maniatis S, Mimitou EP, Lu L, Geller Eet al (2021) Identification of required host factors for SARS‐CoV‐2 infection in human cells. Cell 184: 92–105 e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JP, Almasy KM, McDonald EF, Plate L (2020) Comparative multiplexed interactomics of SARS‐CoV‐2 and homologous coronavirus non‐structural proteins identifies unique and shared host‐cell dependencies. bioRxiv 10.1101/2020.07.13.201517 [PREPRINT] [DOI] [PMC free article] [PubMed]
- Flynn RA, Belk JA, Qi Y, Yasumoto Y, Wei J, Alfajaro MM, Shi Q, Mumbach MR, Limaye A, DeWeirdt PCet al (2021) Discovery and functional interrogation of SARS‐CoV‐2 RNA‐host protein interactions. Cell 184: 2394–2411 e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Blanco MA, Vasudevan SG, Bradrick SS, Nicchitta C (2016) Flavivirus RNA transactions from viral entry to genome replication. Antiviral Res 134: 244–249 [DOI] [PubMed] [Google Scholar]
- Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH, Wang HE, Wettstein DA, Stray KM, Côté M, Rich RLet al (2001) Tsg101 and the vacuolar protein sorting pathway are essential for HIV‐1 budding. Cell 107: 55–65 [DOI] [PubMed] [Google Scholar]
- Gordon DE, Hiatt J, Bouhaddou M, Rezelj VV, Ulferts S, Braberg H, Jureka AS, Obernier K, Guo JZ, Batra Jet al (2020a) Comparative host‐coronavirus protein interaction networks reveal pan‐viral disease mechanisms. Science 370: eabe9403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, O'Meara MJ, Rezelj VV, Guo JZ, Swaney DLet al (2020b) A SARS‐CoV‐2 protein interaction map reveals targets for drug repurposing. Nature 583: 459–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamitani W, Huang C, Narayanan K, Lokugamage KG, Makino S (2009) A two‐pronged strategy to suppress host protein synthesis by SARS coronavirus Nsp1 protein. Nat Struct Mol Biol 16: 1134–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamitani W, Narayanan K, Huang C, Lokugamage K, Ikegami T, Ito N, Kubo H, Makino S (2006) Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proc Natl Acad Sci USA 103: 12885–12890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D‐K, Weller B, Lin C‐W, Sheykhkarimli D, Knapp JJ, Kishore N, Sauer M, Rayhan A, Young V, Marin‐de la Rosa Net al (2021) A map of binary SARS‐CoV‐2 protein interactions implicates host immune regulation and ubiquitination. bioRxiv 10.1101/2021.03.15.433877 [PREPRINT] [DOI]
- Kuadkitkan A, Wikan N, Fongsaran C, Smith DR (2010) Identification and characterization of prohibitin as a receptor protein mediating DENV‐2 entry into insect cells. Virology 406: 149–161 [DOI] [PubMed] [Google Scholar]
- Laurent EMN, Sofianatos Y, Komarova A, Gimeno J‐P, Tehrani PS, Kim D‐K, Abdouni H, Duhamel M, Cassonnet P, Knapp JJet al (2020) Global BioID‐based SARS‐CoV‐2 proteins proximal interactome unveils novel ties between viral polypeptides and host factors involved in multiple COVID19‐associated mechanisms. bioRxiv 10.1101/2020.08.28.272955 [PREPRINT] [DOI]
- Li J, Guo M, Tian X, Wang X, Yang X, Wu P, Liu C, Xiao Z, Qu Y, Yin Yet al (2021) Virus‐host interactome and proteomic survey reveal potential virulence factors influencing SARS‐CoV‐2 pathogenesis. Med 2: 99–112.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Li X, Zhou Y (2018) Ribosomal protein L18 is an essential factor that promote rice stripe virus accumulation in small brown planthopper. Virus Res 247: 15–20 [DOI] [PubMed] [Google Scholar]
- Liu Z‐S, Cai H, Xue W, Wang M, Xia T, Li W‐J, Xing J‐Q, Zhao M, Huang Y‐J, Chen Set al (2019) G3BP1 promotes DNA binding and activation of cGAS. Nat Immunol 20: 18–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokugamage KG, Narayanan K, Huang C, Makino S (2012) Severe acute respiratory syndrome coronavirus protein nsp1 is a novel eukaryotic translation inhibitor that represses multiple steps of translation initiation. J Virol 86: 13598–13608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Zhao X, Li J, Niu P, Yang Bo, Wu H, Wang W, Song H, Huang B, Zhu Naet al (2020) Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395: 565–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabeel‐Shah S, Lee H, Ahmed N, Marcon E, Farhangmehr S, Pu S, Burke GL, Ashraf K, Wei H, Zhong Get al (2020) SARS‐CoV‐2 Nucleocapsid protein attenuates stress granule formation and alters gene expression via direct interaction with host mRNAs. bioRxiv 10.1101/2020.10.23.342113 [PREPRINT] [DOI]
- Prentzell MT, Rehbein U, Cadena Sandoval M, De Meulemeester AS, Baumeister R, Brohee L, Berdel B, Bockwoldt M, Carroll B, Chowdhury SRet al (2021) G3BPs tether the TSC complex to lysosomes and suppress mTORC1 signaling. Cell 184: 655–674 e27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X, Wen W, Fan XR, Hou W, Su B, Cai P, Li J, Liu Y, Tang F, Zhang Fet al (2021) COVID‐19 immune features revealed by a large‐scale single‐cell transcriptome atlas. Cell 184: 1895–1913: e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripoli M, D'Aprile A, Quarato G, Sarasin‐Filipowicz M, Gouttenoire J, Scrima R, Cela O, Boffoli D, Heim MH, Moradpour Det al (2010) Hepatitis C virus‐linked mitochondrial dysfunction promotes hypoxia‐inducible factor 1 alpha‐mediated glycolytic adaptation. J Virol 84: 647–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Raida M, Burke B (2012) A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol 196: 801–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samavarchi‐Tehrani P, Abdouni H, Knight JDR, Astori A, Samson R, Lin Z‐Y, Kim D‐K, Knapp JJ, St‐Germain J, Go CDet al (2020) A SARS‐CoV‐2 – host proximity interactome. bioRxiv 10.1101/2020.09.03.282103 [PREPRINT] [DOI]
- Smits AH, Vermeulen M (2016) Characterizing protein‐protein interactions using mass spectrometry: challenges and opportunities. Trends Biotechnol 34: 825–834 [DOI] [PubMed] [Google Scholar]
- St‐Germain JR, Astori A, Samavarchi‐Tehrani P, Abdouni H, Macwan V, Kim D‐K, Knapp JJ, Roth FP, Gingras A‐C, Raught B (2020) A SARS‐CoV‐2 BioID‐based virus‐host membrane protein interactome and virus peptide compendium: new proteomics resources for COVID‐19 research. bioRxiv 10.1101/2020.08.28.269175 [PREPRINT] [DOI]
- Stukalov A, Girault V, Grass V, Karayel O, Bergant V, Urban C, Haas DA, Huang Y, Oubraham L, Wang Aet al (2021) Multilevel proteomics reveals host perturbations by SARS‐CoV‐2 and SARS‐CoV. Nature 594: 246–252 [DOI] [PubMed] [Google Scholar]
- Teo G, Liu G, Zhang J, Nesvizhskii AI, Gingras AC, Choi H (2014) SAINTexpress: improvements and additional features in significance analysis of INTeractome software. J Proteomics 100: 37–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoms M, Buschauer R, Ameismeier M, Koepke L, Denk T, Hirschenberger M, Kratzat H, Hayn M, Mackens‐Kiani T, Cheng Jet al (2020) Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS‐CoV‐2. Science 369: 1249–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohya Y, Narayanan K, Kamitani W, Huang C, Lokugamage K, Makino S (2009) Suppression of host gene expression by nsp1 proteins of group 2 bat coronaviruses. J Virol 83: 5282–5288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Simoneau CR, Kulsuptrakul J, Bouhaddou M, Travisano KA, Hayashi JM, Carlson‐Stevermer J, Zengel JR, Richards CM, Fozouni Pet al (2021) Genetic screens identify host factors for SARS‐CoV‐2 and common cold coronaviruses. Cell 184: 106–119 e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Alfajaro MM, DeWeirdt PC, Hanna RE, Lu‐Culligan WJ, Cai WL, Strine MS, Zhang SM, Graziano VR, Schmitz COet al (2021) Genome‐wide CRISPR screens reveal host factors critical for SARS‐CoV‐2 infection. Cell 184: 76–91 e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinlich S, Huttelmaier S, Schierhorn A, Behrens SE, Ostareck‐Lederer A, Ostareck DH (2009) IGF2BP1 enhances HCV IRES‐mediated translation initiation via the 3'UTR. RNA 15: 1528–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F, Zhao Su, Yu B, Chen Y‐M, Wang W, Song Z‐G, Hu Yi, Tao Z‐W, Tian J‐H, Pei Y‐Yet al (2020) A new coronavirus associated with human respiratory disease in China. Nature 579: 265–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Ru Y, Ren J, Bai J, Wei J, Fu S, Liu X, Li D, Zheng H (2019) G3BP1 inhibits RNA virus replication by positively regulating RIG‐I‐mediated cellular antiviral response. Cell Death Dis 10: 946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK (2019) Metascape provides a biologist‐oriented resource for the analysis of systems‐level datasets. Nat Commun 10: 1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review Process File
Appendix
Expanded View Figures PDF
Table EV1
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Source Data for Expanded View and Appendix
Source Data for Figure 2
Source Data for Figure 5
Source Data for Figure 7
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD023209 (http://www.ebi.ac.uk/pride/archive/projects/PXD023209).