

Abstract
Type 1 diabetes (T1D) is a disease of insulin deficiency that results from autoimmune destruction of pancreatic islet β cells. The exact cause of T1D remains unknown, although asymptomatic islet autoimmunity lasting from weeks to years before diagnosis raises the possibility of intervention before the onset of clinical disease. The number, type, and titer of islet autoantibodies are associated with long-term disease risk but do not cause disease, and robust early predictors of individual progression to T1D onset remain elusive. The Environmental Determinants of Diabetes in the Young (TEDDY) consortium is a prospective cohort study aiming to determine genetic and environmental interactions causing T1D. Here, we analyzed longitudinal blood transcriptomes of 2013 samples from 400 individuals in the TEDDY study before both T1D and islet autoimmunity. We identified and interpreted age-associated gene expression changes in healthy infancy and age-independent changes tracking with progression to both T1D and islet autoimmunity, beginning before other evidence of islet autoimmunity was present. We combined multivariate longitudinal data in a Bayesian joint model to predict individual risk of T1D onset and validated the association of a natural killer cell signature with progression and the model’s predictive performance on an additional 356 samples from 56 individuals in the independent Type 1 Diabetes Prediction and Prevention study. Together, our results indicate that T1D is characterized by early and longitudinal changes in gene expression, informing the immunopathology of disease progression and facilitating prediction of its course.
INTRODUCTION
An autoimmune pathogenesis for type 1 diabetes (T1D) is indicated by strong genetic association of Human leukocyte antigen (HLA) and other immune variants (1) along with progressive development of pancreatic islet β cell autoantibodies (IAbs) (2), although metabolic, microbial, and dietary factors also contribute (3). Genetic risk scores can identify those at highest risk of developing disease (4) in whom peak onset occurs in early childhood, around 1 to 2 years of age (5). Studying early events associated with T1D progression is challenging given the need to identify at-risk individuals and to sample before evidence of autoreactivity begins. Previous studies have typically been cross-sectional investigations of small numbers of individuals after the onset of islet autoimmunity or T1D. Expansions of islet antigen-reactive T cells have been documented in the blood of both T1D cases and healthy controls (6), whereas age-dependent changes in immune phenotype at diagnosis (7) have underlined the complexity of studying disease in the context of a developing immune system. Within the pancreas, longitudinal single-cell RNA sequencing analysis has indicated complex, dynamic changes in infiltrating immune cell populations (8). Although comparable study of human tissue is not possible, pseudotime mass cytometry of pancreatic tissue from T1D-diagnosed donors has illustrated an influx of cytotoxic and T helper cells associated with reductions in β cell mass (9).
Longitudinal metabolomic (10) and microbiomic (11, 12) analyses before onset of autoimmunity or T1D diagnosis have indicated marked age-dependent effects but not a clean association with autoimmunity or disease progression. A viral trigger for pancreatic autoimmunity has long been proposed (13) and has been supported by both early transcriptional signatures of type 1 interferon (IFN1) response (14) and prolonged enteroviral shedding (15) before islet autoimmunity. Enteroviruses can directly infect pancreatic β cells and have been linked to a natural killer (NK) cell insulitis (16). Early changes in blood gene expression (17, 18) before islet autoimmunity are likely to be driven by changes in relative proportions of constituent blood cells (19), although their interpretation is confounded by a lack of data describing how such changes develop in healthy infancy.
T1D onset occurs in the dynamic context of a maturing immune system: Changes observed in children developing autoimmunity must be carefully related to healthy developmental changes to focus our attention on factors that initiate and propagate autoreactive responses. Recently, systems immunology studies of immune development during infancy have indicated early, stereotyped changes in blood cell and protein composition (20) and dynamic changes in the gut microbiome related to breastfeeding patterns, islet autoimmunity (11, 12), and onset of childhood inflammatory disease (21). Such high-throughput analyses create the potential for identification of previously unsuspected pathways associated with disease initiation and progression to improve understanding of disease biology and aid the building of predictive models to suggest targeted therapies (22).
The Environmental Determinants of Diabetes in the Young (TEDDY) study aims to identify gene-environment interactions causing T1D in high-risk infants participating in serial prospective sample collection and monitoring every 3 to 6 months from birth to age 15 years (23). By combining genetic risk stratification and prospective follow-up, it has been possible to collect samples and data from infants before development of both islet autoimmunity and T1D onset, facilitating systematic analysis of early and longitudinal changes associated with the later development of disease. This can facilitate both an improved understanding of the pathogenetic mechanism of human islet autoimmunity and early prediction of its subsequent course. Although the presence of IAbs indicates the development of islet autoimmunity and hence long-term risk of T1D development (2), validated predictive markers have not been able to report serial, individual risk over a near-term horizon such as is necessary to inform clinical decision-making (24).
Here, we undertook transcriptional network analysis of gene expression microarray data using a nested case:control cohort (25) from the TEDDY study to identify early longitudinal changes in whole blood gene expression in healthy infancy or tracking with progression to both islet autoimmunity and T1D. We also built a predictive model incorporating multivariate longitudinal features including gene expression and islet autoantibodies to estimate individual risk of T1D progression.
RESULTS
A dynamic landscape of whole-blood gene expression during infancy
We analyzed data from nested, matched case control cohorts (25) comprising 2013 whole blood transcriptomes sampled longitudinally from 401 individuals, divided into those developing islet autoimmunity or T1D (Fig. 1, A and B, and figs. S1 to S2). We applied transcriptional network analysis (26) to identify groups of coexpressed genes (modules) based solely on patterns of transcription in the data (Fig. 1, C and D, and fig. S1B). We first constructed independent transcriptional networks for both cases and controls, observing closely matched coexpression patterns in each with no evidence of disease-specific modules (fig. S1B): Genes coexpressed in cases were found to be similarly coexpressed in the control cohort and vice versa (Fig. 1, C and D). As genes in a module are by definition coexpressed, they can be summarized by a single profile known as an eigengene (27). As coexpression patterns were preserved in disease and control groups, we constructed a coexpression network based on all samples considered together (fig. S1B) and applied linear mixed modeling (lmm) to whole blood modular eigengenes from this combined network (table S1). We found that a substantial proportion of modules (23 of 85, 27%) demonstrated significant temporal changes during infancy [false discovery rate (FDR) < 5%; Fig. 1E]. Patterns of gene coexpression are highly conserved (28), and modular signatures can be interpreted by screening their composite genes for enrichment of well-defined gene expression signatures in external relational databanks and public repositories (fig. S1) (29, 30). In this way, the biological meaning of the identified patterns of gene coexpression can be interpreted, with the caveat that repositories typically include samples and data from adults rather than infants. We compared each age-associated module (Fig. 1E) to the largest compendium of cell- and tissue-specific transcriptomic signatures [ARCHS4 repository (30)], identifying frequent modular enrichment for cell type–specific transcripts (17 of 23, 74%), upstream kinases (22 of 23, 96%), and transcription factors (22 of 23, 96%) (fig. S3) including progressive reductions in stem cell–specific transcripts (Fig. 1F) along with B cell– and neutrophil-specific transcripts and associated transcription factors (fig. S3).
Fig. 1. Dynamic changes in the infant blood transcriptome.
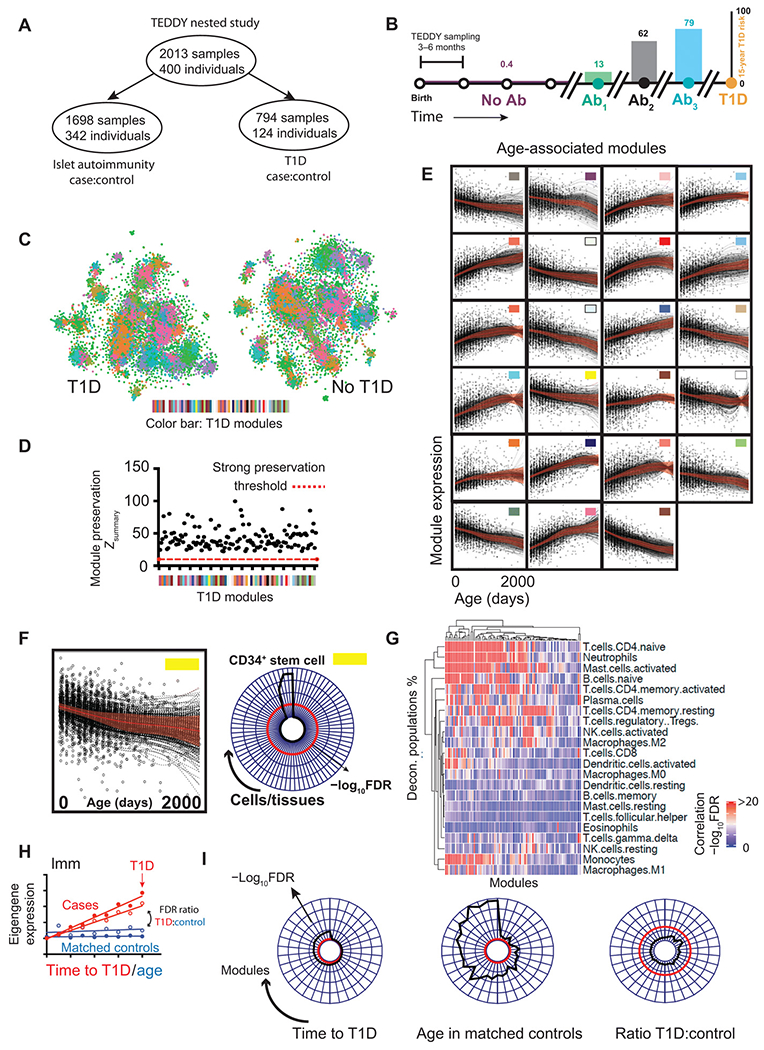
(A and B) Schematic illustration of the (A) TEDDY cohort (B) sampling from birth, through IAbs seroconversion to T1D diagnosis illustrating population-level risFk of T1D. IAb+ samples may appear in both case:control cohorts hence subgroups do not add up to the total. Ab, antibody. (C) tSNE plot illustrating the dissimilarity matrix of gene coexpression networks in T1D (left) and matched controls (right). Each dot represents a distinct gene (n = 15,000). Genes in both plots are colored by modular assignment in the T1D coexpression network. (D) Scatterplot showing strength of T1D module preservation (y axis, Zsummary score) in matched control data (red dashed line = strong preservation threshold, Zsummary = 10). (E) Line and scatterplots showing lmm effects [red, ±95% confidence interval (CI)] and gene expression eigenvalues (black dots) for 23 modular eigengenes showing significant (FDR < 5%) age association in infancy. Colors are matched to (C to E). (F) Example module enrichment: Line and scatterplot (left) showing lmm effects (red line, ±95% CI) for the “yellow” module alongside radar plot (right) showing module enrichment (radial axis, −log10FDR) for cell type–specific transcripts. (G) Clustered heatmap illustrating significance (−log10FDR) of correlation (Pearson) of deconvolved cell subset proportions (y axis) against modular eigengene values (x axis). (H) Schematic line and scatterplot illustrating the use of lmm to compare modular gene expression signatures in matched cases and controls. (I) Radar plots showing all modules (arranged around plot circumference) associated with time to T1D onset (FDR < 5%, left), association of the same modules with sampling age in matched controls (center) and the ratio of observed significance in each (FDRT1D:FDRcontrol, right). For radar plots, radial distance from the center = −log10FDR, red line = threshold FDR < 5%.
We also undertook “digital cytometry” using a deconvolution approach comparing whole blood transcriptional profiles to immune cell–specific transcripts to estimate cell type frequencies (Fig. 1G) (31). This confirmed the association of the majority of modular signatures identified with the frequency of circulating immune cell phenotypes (Fig. 1G). Individual modular signatures correlated with multiple deconvoluted cell frequencies as expected, as cell percentages often vary together during an immune response.
Multiple modular signatures mapped to the same cell type despite showing distinct longitudinal trajectories, suggesting that they cannot be explained by changes in the proportion of circulating cell types in blood alone and likely reflect more subtle phenotypic differences than can currently be mapped by deconvolution or enrichment strategies. Together, these data show marked gene expression changes during healthy infancy, highlighting the dynamic context in which autoimmune diseases such as T1D occur.
Distinct disease-specific transcriptional signatures in T1D subgroups
We next asked whether longitudinal changes in modular patterns of gene expression tracked with progression toward T1D onset. As children get older, they inevitably approach the time of disease onset. Consequently, it is necessary to ensure that the longitudinal changes seen in cases are not simply age-associated changes expected to occur in healthy children. To overcome this, we identified modular eigengenes correlating with time to T1D in cases and, as the study design included age-matched healthy samples, compared these to the longitudinal changes seen in infants who did not progress to disease (Fig. 1, H and I). Although 35 modular signatures significantly correlated with T1D progression (FDR < 5%), none were specific to T1D cases and all showed stronger association with healthy aging in matched controls (Fig. 1I), highlighting the importance of placing longitudinal changes into the dynamic immune context of early infancy.
T1D is thought to be a heterogeneous condition; however, evidence supporting the existence of disease subgroups has proven elusive (32, 33). Those developing autoantibodies to insulin (IAAs) show earlier and more rapid progression to both additional IAbs and to T1D than those initially developing IAbs to the other major pancreatic autoantigen, GAD (glutamic acid decarboxylase) (2). However, it remains unclear whether this observation reflects a distinct immunopathology, or simply autoimmunity occurring at younger age. We next stratified the T1D cohort by target of first appearing autoantibody, comparing modular gene expression changes in IAAs-first and GADA (GAD antibody)–first subgroups with their matched controls as before. Whereas disease-specific longitudinal changes were not apparent in T1D on the whole (Fig. 1I), distinct gene expression signatures showed clear age-independent association with time to T1D onset in IAbs subgroups (Fig. 2, A and B). Among IAAs cases, one dominant signature (IAAsig) showed an early increase in expression with a later secondary increase before diagnosis (Fig. 2C), a pattern not seen in most matched controls. The nested study design allowed for IAbs seroconversion to occur in the T1D control group (25), provided that T1D progression did not occur. Closer analysis of the control group demonstrated that IAAsig expression showed a similar early rise in seroconverting controls, falling off with advancing age where they were maintained in those progressing to T1D (IAbposT1Dneg versus IAbnegT1Dneg; Fig. 2C, inset). By contrast, seronegative controls showed no comparable rise in early IAAsig expression (Fig. 2C, inset). In GADA cases, a group of four closely correlated signatures termed together as GADAsig (Fig. 2B) showed an age-independent decrease toward T1D onset, a pattern absent from matched controls and occurring closer to diagnosis in contrast to the earlier increases in the IAAsig (Fig. 2, C and D). We compared mixed models to ensure that observed gene expression changes were independent of additional clinical covariates including sex, ethnicity, and HLA risk group (table S2) alongside mode of birth delivery and patterns of maternal breastfeeding (fig. S4, C to F). This analysis indicated an independent and significant association of both gene expression signatures and sex, but not the other variables, to the models (table S3). We therefore took these covariates forward into predictive model building as described below.
Fig. 2. Age-independent changes in gene expression accompany T1D progression in subgroups defined by first IAbs specificity.
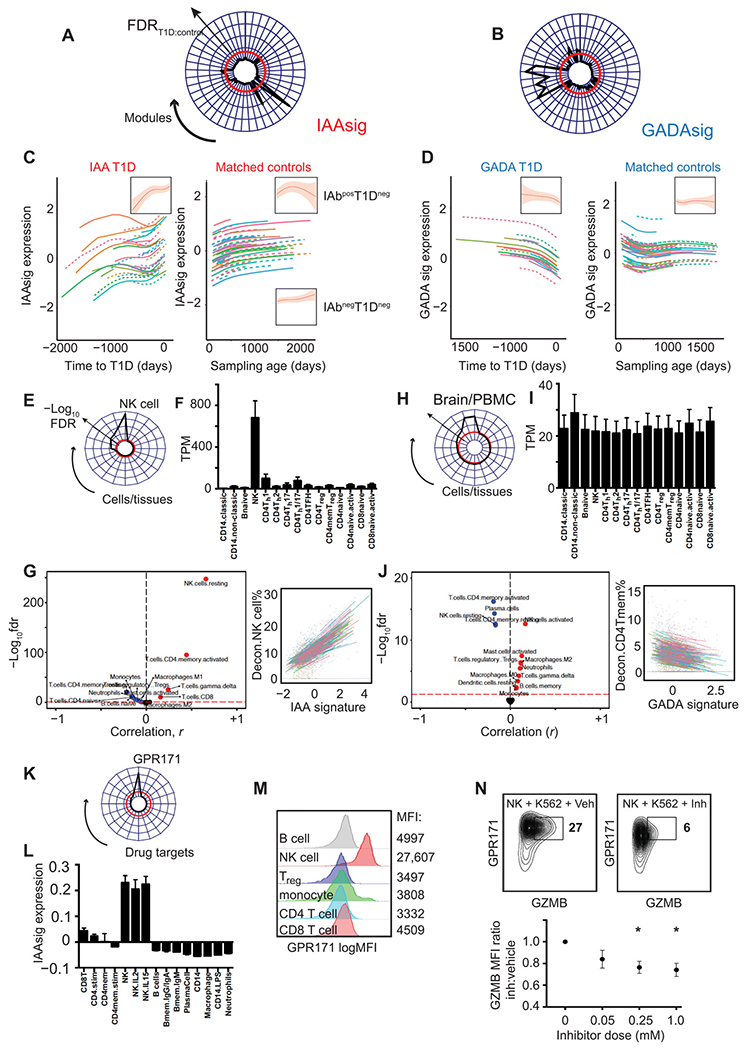
(A and B) Radar plots showing age-corrected associations (Imm FDRT1D:FDRcontrol) of all module eigengenes (shown around plot circumference) with time to T1D onset in IAAs-first (A) and GADA-first (B) cases. Disease specificity indicated by a T1D:control significance ratio > 1 (−log10FDR) = red line. (C and D) Line plots showing individual and summary (inset) effects of natural cubic spline–fitted lmm (±95% CI) fitted to IAAsig (C) or GADAsig (D) eigenvalues in cases (left) and matched controls (right). (E and F) Radar plot (E) and barplot (F) showing IAAs module enrichment for cell type–specific transcripts compared to ARCHS4 (E) and DICE (F) relational databanks. (G) Volcano plot (left) and line plots (right) showing all (left) and selected (right) correlations (Pearson) of IAAsig eigengenes against deconvoluted cell proportions. Significance threshold (FDR < 5%) = red dashed line. (H to J) Radar plot (H) and barplot (I) showing IAAs module enrichment for cell type–specific transcripts compared to ARCHS4 (H) and DICE (I) relational databanks. (J) Volcano plot (left) and line plots (right) showing all (left) and selected (right) correlations (Pearson) of GADAsig eigengenes against deconvoluted cell proportions. Significance threshold (FDR < 5%) = red dashed line. For barplots (F and I), TPM, transcripts per million reads; expression (means ± SEM) of IAAsig (F) and GADAsig (I) per cell type is shown. (K) Radar plot showing enrichment (−log10FDR) of IAAsig genes against 352 druggable targets linked to 20,883 genes from the IDG repository. Drug targets showing any overlap with IAAsig genes are included around the radar plot circumference. (L) Barplot (means ± SEM, n = 3 to 12 per group) showing IAAsig eigengene expression and (M) representative histograms showing GPR171 surface protein expression in circulating immune cell subsets (GSE22886). (N) Representative contour plot (top) and scatterplot (bottom) showing GPR171 surface protein expression (y axis, log10 MFI) and Granzyme B (GZMB) protein expression (x axis) after coculture of purified primary human NK cells with K562 target cells along with vehicle (left) or a titrated dose range of specific GPR171 inhibitor (Inh; right). *P < 0.05, MFI, median fluorescence index; Treg, regulatory T cell.
Biological interpretation of T1D-specific signatures
We next performed enrichment analysis of both IAAsig and GADAsig (table S4) against relational databases [ARCHS4 (30) and Database of Immune Cell eQTLs (DICE) (34)] providing the largest and most granular coverage of immune cell–specific gene expression and also correlated each module’s eigengene against cell subset frequencies estimated through deconvolution analysis (31). IAAsig genes showed strong, specific enrichment for NK cell–specific transcripts (Fig. 2, E and F), transcription factors, and kinases (fig. S5) and correlated with deconvoluted percentage of NK cells, and to a lesser but still significant extent with CD4+ memory T cells (Fig. 2G). Genes comprising GADA-specific modules were enriched for transcripts shared by both blood and brain tissue but not clearly with a single cell type (Fig. 2, H and I, and fig. S5). However, comparison with deconvolved cell type frequencies indicated strongest associations with reduced percentage of CD4+ memory T cells and NK cells, with a relative increase in an activated NK phenotype (Fig. 2J). This observation suggests that the early stages of T1D pathogenesis are associated with different immune cell trajectories that involve similar immune cell types, namely, NK and CD4+ memory T cells, depending on the pattern of initial IAbs seroconversion.
To investigate potential means of therapeutically modulating IAAsigs and GADAsigs, we compared each to an integrated repository of drug response data [the Harmonizome (35, 36)] that links functional associations between genes and proteins based on collated genomic data including physical associations, knockout or knockdown phenotypes, and response to drug treatment. We screened IAAsig genes against all 352 “druggable” targets (30) linked to 20,883 genes and identified a single candidate G protein-coupled receptor (GPR171; Fig. 2K) as a potential controller of IAAsig genes. That is, GPR171 was not itself part of the modular signature but instead predicted to be functionally associated with genes comprising it. We confirmed NK cell expression of IAAsig and GPR171 at both mRNA (Fig. 2L) and protein abundance (Fig. 2M) and demonstrated that a specific inhibitor of its signaling (Inh) could attenuate both GPR171 expression and NK cytotoxicity in an in vitro killing assay (Fig. 2N). Together, these data demonstrate that distinct cell-specific gene expression changes characterize progression to disease onset in subgroups of patients with T1D defined by their sequence of IAbs seroconversion.
Transcriptional signatures associated with islet autoimmunity
Next, we asked whether specific changes in gene expression occur around the onset of islet autoimmunity (IAbs seroconversion), rather than tracking with progression to disease onset (Fig. 3A). Among 50 modular signatures showing significant association with islet autoimmunity onset (Fig. 3, B and C), a dominant signature is associated with seroconversion in both subgroups. This signature (IAsig) was common to both GADA and IAAs subgroups and, although it showed significant dynamic changes in matched healthy controls, there were more marked and sustained reductions in infants progressing to IAbs seroconversion (Fig. 3, D and E). Interrogation of this islet autoimmunity signature (table S5) revealed strong enrichment for B cell–specific transcripts, kinases, and transcription factors (Fig. 3F). Six additional signatures (Fig. 3, B and C) showed a weaker but still significant age-independent association with islet autoimmunity onset, again in both GADA and IAAs subgroups. The second most strongly associated signature was the same NK-enriched signature that associated with T1D onset in IAAs, which here increased toward islet autoimmunity onset but showed no change over time in the control group (Fig. 3, G and H). Together, these data indicate that longitudinal changes in NK- and B cell–associated gene expression track with progression toward the onset of islet autoimmunity, with NK-associated changes also tracking with progression to disease in the IAAs subgroup.
Fig. 3. Age-independent changes in longitudinal gene expression accompany islet autoimmunity.
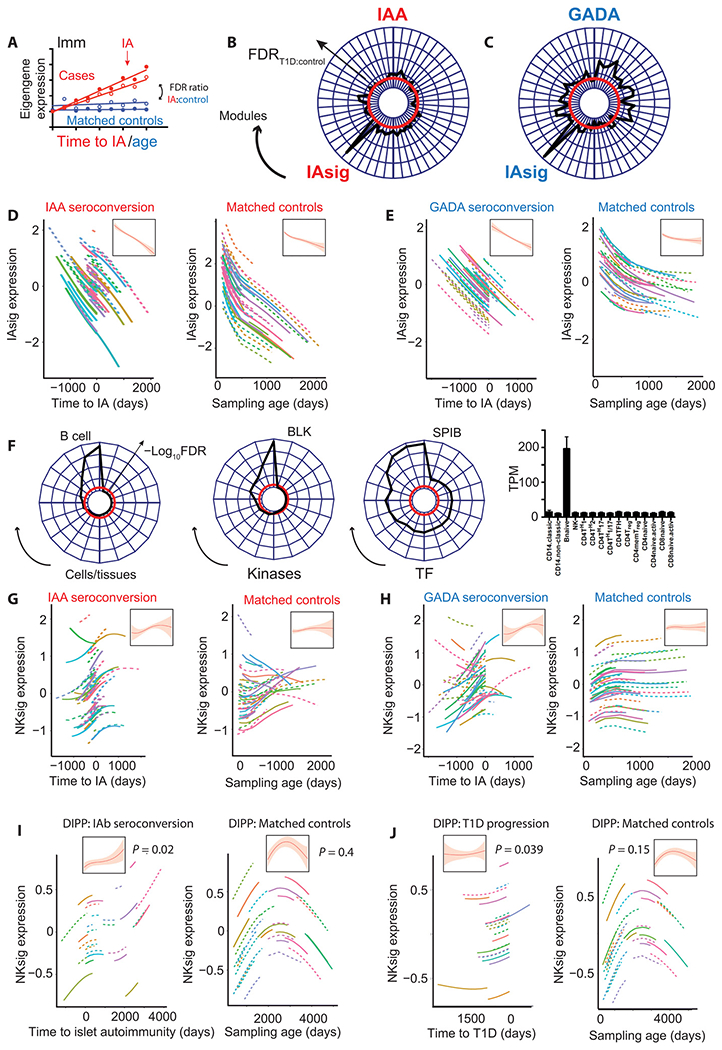
(A) Schematic line and scatterplot illustrating the use of a lmm to compare modular gene expression signatures in matched islet autoimmunity cases and healthy controls. (B and C) Radar plots showing age-independent associations (FDRIA:FDRcontrol) of all gene expression modules significantly associated (FDR < 5%) with time to islet autoimmunity onset in either IAAs-first (B) or GADA-first (C) cases. Disease specificity indicated by a T1D:control significance ratio > 1 (−log10FDR) = red line. (D) Line plots showing individual and summary (inset) effects of a natural cubic spline–fitted lmm (±95% CI) fitted to IAsig eigenvalues in IAAs-first individuals (left, n = 82 versus time to islet autoimmunity) and matched controls (right, n = 63 versus age). (E) Line plots showing individual and summary (inset) effects of a natural cubic spline–fitted lmm (±95% CI) fitted to IAsig eigenvalues in GADA-first cases (left, n = 54 versus time to islet autoimmunity) and matched controls (right, n = 49 versus age). (F) Radar plots showing IAsig module enrichment for cell type–specific transcripts, kinase targets, and transcription factor targets from the ARCHS4 dataset and barplot (right) showing cell specific expression of IAsig genes in the DICE dataset. TPM with expression (means ± SEM) per cell type is shown. (G) Line plots showing individual and summary (inset) effects of natural cubic spline–fitted lmm (±95% CI) fitted to the NK cell–enriched module eigenvalues (from Fig. 2B) in IAAs first cases (left, n = 82 versus time to islet autoimmunity) and matched controls (right, n = 63 versus age). (H) Line plots showing individual and summary (inset) effects of natural cubic spline–fitted lmm (±95% CI) fitted to the NK cell-enriched module eigenvalues (from Fig. 2B) in GADA first cases (left, n = 54 versus time to islet autoimmunity) and matched controls (right, n = 49 versus age). (I and J) Line plots showing individual and summary (inset) effects of natural cubic spline–fitted lmm (±95% CI) fitted to the DIPP NK module in IApos cases [(I, left) n = 26 versus time to islet autoimmunity] and matched IAneg controls [(I, right) n = 32 versus sampling age] and in T1D cases [(J, left) n = 24 versus time to T1D] and matched controls [(J), right, n = 34 versus sampling age]. For radar plots (F and J), radial distance = −log10FDR with threshold FDR (5%) in red.
Validation of T1D-specific longitudinal gene expression changes
We next sought to validate our findings in an independent cohort of IAbs and T1D cases. The Type 1 Diabetes Prediction and Prevention (DIPP) cohort (37) is a prospective, population study of incident islet autoimmunity and T1D with a comparable nested case:control design, although with sampling commencing in slightly older children (18). We undertook an independent network transcriptomic analysis of 356 DIPP samples from 58 individuals, again comparing dynamic changes in modular gene expression to both islet autoimmunity (Fig. 3I) and T1D onset (Fig. 3J). In this smaller cohort, an NK cell–enriched signature (directly comparable to that identified in TEDDY; fig. S6 and table S6) was the only gene expression pattern that showed a significant association with both IAbs seroconversion and T1D progression, which did not similarly change in matched controls (Fig. 3, I and J). A B cell signature comparable to the IAsig seen in TEDDY (Fig. 3, G and H) was significantly associated with both clinical end points but, in the DIPP cohort, showed a comparable association with sampling age. Similar to what was observed in the TEDDY cohort, the NK signature increased toward T1D onset with a later decline in matched controls who did not go on to develop disease (Fig. 3, I and J).
Together, these data confirm an independent association of an NK cell–enriched transcriptional signature with both IAbs seroconversion and rate of progression to T1D, validating the finding in the larger TEDDY discovery cohort.
Gene expression in early infancy associates with rate of disease progression
We next sought to investigate whether whole blood gene expression changes in early infancy, before demonstrable evidence of islet autoimmunity, were related to later risk of progression toward T1D. For this “snapshot” of early risk, we identified the earliest samples available within the case:control cohorts, comprising 288 samples from 288 individuals, all taken before seroconversion and with >85% taken within the first 12 months of life (Fig. 4, A and B). In this cross-sectional analysis, we constructed a gene coexpression network and looked for evidence of association with both disease risk (outcome T1D+ versus T1D−) and rate of subsequent progression toward T1D. As longitudinal changes were not being considered, we used regression analysis to adjust for variable sampling age and sex. At this early time point, four modules were associated with the rate of subsequent progression toward T1D (Fig. 4, C and D). Both enrichment and deconvolution correlation analyses of these modules indicated specific excess of B lymphoblast and monocyte expressed transcripts respectively with the latter also enriched for tumor necrosis factor (TNF) and complement pathway signaling (Fig. 4E, table S7, and fig. S6, H and I). These data indicate that early high expression of a TNF-enriched monocyte signature and early low expression of a B lymphoblast signature were associated with slower progression to T1D onset (Fig. 4, C and D). No modular signatures were significantly different between T1D and healthy control groups, although the TNF-enriched monocyte signature that associated with protection against T1D was markedly higher in infants who later seroconverted without developing T1D (IA+T1D−; Fig. 4F). We also observed that the same pattern of monocyte/TNF-associated transcripts showed significantly lower expression in children within a window of 12 months before diagnosis of T1D (Fig. 4G), a finding that was also validated in the DIPP cohort (Fig. 4H).
Fig. 4. Preseroconversion gene expression changes associate with rate of T1D progression.
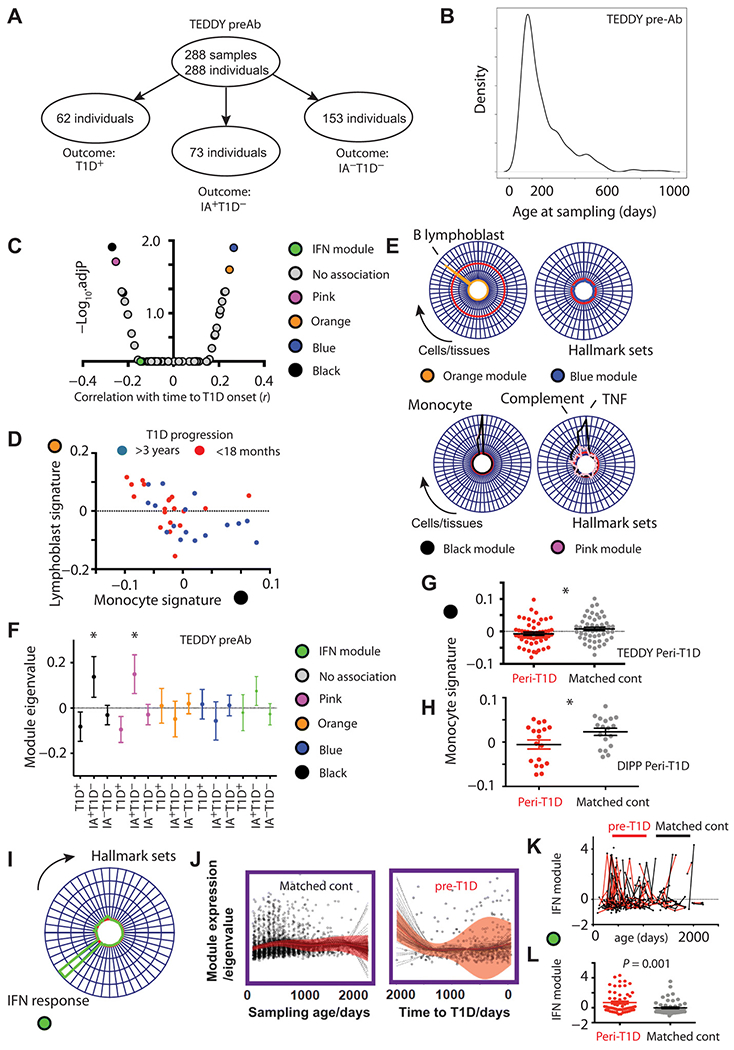
(A) Schematic illustration and (B) density plot showing age distribution of earliest preseroconversion samples taken in the TEDDY transcriptomic study (TEDDY preAb). “Outcome” indicates later progression rather than state at time of sampling. (C) Volcano plot showing correlation of all network modules (x axis, r) against adjusted significance (−log10FDR, y axis). (D) Scatterplot illustrating inverse correlation of B lymphoblast (orange) and monocyte (black) modular signatures and their association with rate of T1D progression. (E) Radar plots illustrating enrichment of the orange and blue (top) and black and pink (bottom) modules against the human cell atlas (left) and hallmark signature sets (right). Radial axis = −log10FDR, red line = significance threshold FDR (5%). (F) Scatterplot illustrating expression (means ± SEM) of black, pink, orange, and blue, and IFN response signatures by outcome group in the TEDDY preAb cohort, * = Mann Whitney P < 0.05. Outcome group reflects final clinical status rather than status at the time of sampling. (G) Scatterplot showing age-corrected eigengene expression of the “black” monocyte/TNF-enriched signature in pre-T1D samples from TEDDY (n = 54 samples within 12 months before diagnosis). (H) Scatterplot showing age-corrected eigengene expression of the black monocyte/TNF-enriched signature in pre-T1D samples from the DIPP cohort (n = 18 samples within 3 months before diagnosis) and their matched controls (n = 18). (I) Radar plot showing modular enrichment for interferon (IFN) response transcripts. (J) Scatterplot and line plot showing lmm summary of longitudinal type1 IFN module expression (red line, ± 95% CI shaded) in pre-T1D children (right, n = 62) and matched controls (left, n = 62). (K) Scatter and line plot illustrating spikes of type 1 IFN response module and (L) age-matched peak expression in pre-T1D (red, n = 57) and age-matched control samples (black, n = 57) in the 12-month preceding T1D onset.
Previous analyses have identified a IFN1 response signature expressed in at-risk children before antibody seroconversion and associated with previous respiratory infections (14). Such a signature was clearly visible in the TEDDY cohort (Fig. 4I), although it was not associated with risk of T1D (Fig. 4C) or rate of progression to T1D (Fig. 4J). However, the IFN response signature conformed to a pattern of transient “spikes” of expression, likely after infectious triggers, most frequently observed in the 12 months before disease onset (Fig. 4, J and K) and to an extent that exceeded those seen in age-matched control samples (Fig. 4L).
Prediction of individual T1D risk using longitudinal data
Recently, statistical learning methods have improved our ability to integrate baseline covariates, longitudinal data, and clinical end points to estimate instantaneous event hazards (38). Current T1D prediction methods stratify disease risk by number of IAbs present, indicating cohort level risk over a horizon of many years, making it difficult to incorporate this information into treatment pathways or to enable recruitment into clinical trials of targeted therapy (Fig. 1B). Individual risk prediction over a short time horizon is necessary to guide clinical decisions and preventive therapy trials (24). We therefore aimed to build a predictive model that could estimate, with an indication of uncertainty, the near-term hazard of T1D for an individual, dynamically updating that prediction as additional data become available. We used a multivariate Bayesian joint model (39) to combine baseline stratification (using a Cox proportional hazards model) with longitudinal variables such as IAbs type, status, timing, and gene expression, returning an event hazard with associated confidence bounds. As our earlier analysis of comparative mixed models had demonstrated independent association of both gene expression signatures and sex (table S3) with gene expression signatures depending on the sequence of serial IAbs seroconversion, we incorporated these covariates into a joint model (Fig. 5A). We chose to build and test the performance of three distinct models in each of two clinical scenarios. Predictive performance [area under the receiver operator characteristic curve (AUC ROC) and prediction error (PE)] was estimated using 10-fold cross-validation on the discovery cohort (TEDDY; Fig. 5B) and on an independent validation dataset (DIPP). The three models each included baseline stratification by sex and (i) IAbs status over time (IAb+/−), (ii) maximal IAbs information (IAbs specificity [IAAs, GADA, IA-2A], timing of IAbs seroconversion, sequence of IAbs seroconversion specificity and associated interaction effects), or (iii) maximal IAbs information along with longitudinal gene expression data (the eigenvalues of the IAAs or GADA signatures). The two clinical scenarios tested were serial prediction over a fixed future horizon of 12 months using all cumulative data available at each time point (mimicking a child being followed up over time; Fig. 5, C to F) and prediction at 1.5 years of age over a serially increasing future horizon (mimicking prediction with early limited data; Fig. 5, C to F).
Fig. 5. Validated prediction of T1D hazard in at-risk infants.
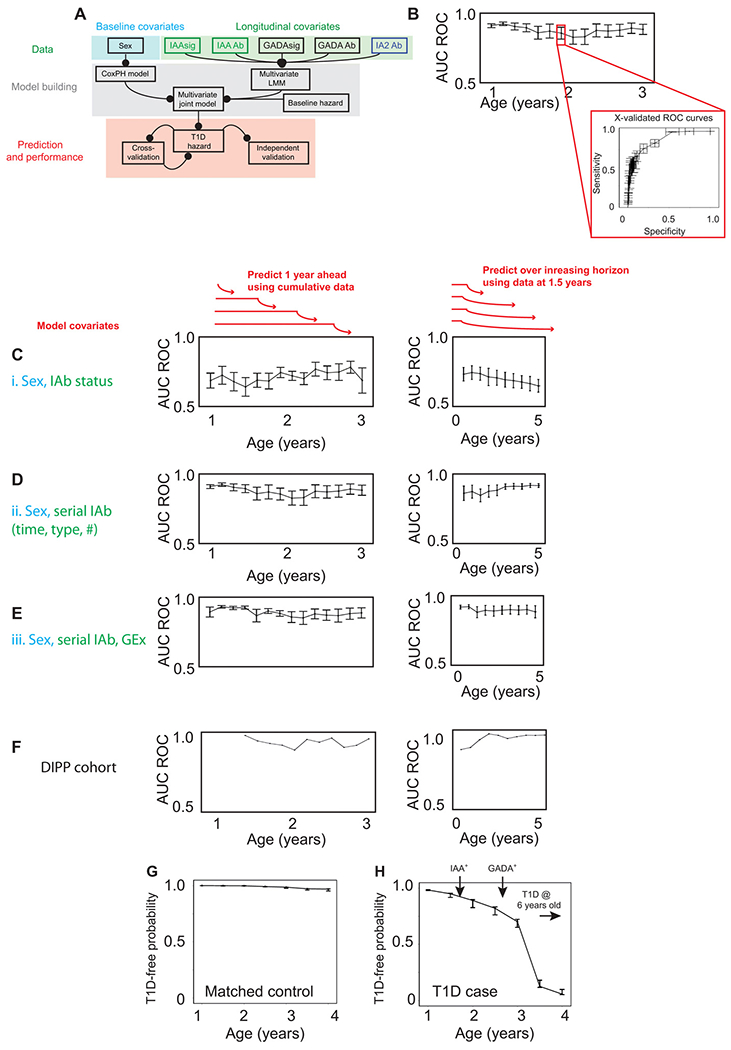
(A) Schematic of multivariate Bayesian joint model data input (top), model building (middle), and model performance assessment (bottom). (B) Illustration of cross-validation method used on discovery TEDDY cohort. (C to F) Line and scatterplots showing predictive accuracy of models applied to each of two clinical scenarios (schematically illustrated above plots in red). Left: Serial prediction over future 12-month horizon using cumulative data. Right: Serial prediction over extended future horizon using fixed data. (C) Model i incorporating IAbs status (IAb+/−) only. (D) Model ii incorporating longitudinal IAbs type, status, timing, and interaction effects. (E) Model iii incorporating model ii plus IAAsig/GADAsig and interaction effects. Predictive accuracy (AUC ROC) determined through 10-fold cross-validation on the discovery TEDDY dataset as illustrated in (B). (F) Predictive accuracy (AUC ROC) of independent validation of model iii on the DIPP dataset. (G and H) Representative line and scatterplots illustrating serial prediction of individual T1D risk for a T1D case (G) and matched control (H) with predictions every 6 months, made over a horizon of 1 year. Error bars represent means ± SEM. Arrows indicate timing of seroconversion events.
Using islet autoimmunity status alone—the scenario most comparable to current methods (24)—had modest predictive accuracy in both clinical scenarios (model i, Fig. 5C). The inclusion of maximal IAbs information (model ii) allowed for robust prediction in the first clinical scenario (serial prediction over a fixed horizon of 12 months; Fig. 5D, left: AUC ROC > 0.9, PE < 10%) with only modest improvement by inclusion of longitudinal gene expression signatures (model iii, Fig. 5E). Although the performance of serial IAbs in this scenario is an improvement in T1D prediction, it is also apparent that making predictions close to diagnosis (in this scenario predicting 12 months ahead) is supported by using IAbs data. By contrast, and consistent with the importance of early gene expression measures, gene expression signatures supported model performance (model iii) more strongly in the second clinical scenario, where predictions were made early over a serially extending time horizon. This was particularly apparent with prediction over the first few years of life, when the majority of infant T1D cases occurred (Fig. 5, D and E). These data show that, although the presence of islet autoantibodies is associated with disease risk (40), incorporating information on serial changes in the type, number, and timing of seroconversion can facilitate T1D risk prediction at an individual level over a time horizon short enough to facilitate changes in clinical monitoring or therapeutic trials. Gene expression measures provided greatest support for prediction when measured early (up to 18 months) to predict T1D risk over a longer time horizon (up to 5 years in this dataset).
DISCUSSION
Together, our data describe dynamic changes in the infant blood transcriptome and show that patterns of islet antibody seroconversion define subgroups of T1D with both distinct rates of progression and distinct age-independent gene expression signatures associated with time to disease onset. Among healthy infants, we observed extensive longitudinal changes in gene expression over the first 5 years of life, highlighting the dynamic immune context in which early islet autoimmunity develops. This observation reinforces the importance of taking such changes into account when seeking to differentiate disease-specific changes from those reflecting “healthy” immune development.
On taking age-associated changes into account, we observed specific, longitudinal changes in gene expression tracking with progression toward both islet autoimmunity and T1D onset. Distinct changes were associated with T1D progression in subgroups defined by the target of initial seroconversion. The two dominant serospecificities at onset of islet autoimmunity (IAAs and GADA) have been proposed as distinct disease “endotypes,” with the former developing earlier and showing faster progression toward T1D onset (41). Consistent with a distinct pathogenetic mechanism underpinning this stratification, we observed that distinct transcriptional signatures tracked with progression to T1D onset in subgroups defined by the specificity of the first appearing islet autoantibody (IAAs or GADA). Earlier changes tracking progression to onset of islet autoimmunity were similar in both groups, however. We identified an NK cell–based signature that increased in expression with progression toward both islet autoimmunity and T1D in IAAs-first individuals. The same signature was similarly seen to associate with time to islet autoimmunity but not T1D onset in GADA-first individuals. Association of a very similar NK cell signature was validated in an independent analysis of longitudinal samples from the DIPP study. However, further work is required to understand the mechanism underlying this association. NK cells have a complex relationship to autoimmunity and may function as either effector cells contributing to tissue damage, or as regulators of immunopathology (42). Differences in NK cell phenotype have been described after T1D diagnosis (43)—although accompanied by many other late differences (44)—whereas changes in their number and phenotype have been variably linked to either aggressive insulitis (45) or protection from it (46) in animal models. Our data indicate a prominent and specific role for NK cells in the development and progression of autoimmunity and T1D in humans, beginning at the earliest stages and tracking longitudinally with rate of progression rather than simply differentiating those who already have disease from those who do not. Although it is difficult to further refine the source of the NK-specific transcriptional signature using whole blood data, it is unlikely that it simply reflects a relative expansion of peripheral NK cells, as evidenced by our identification of other NK cell–enriched modular signatures that did not associate with disease progression. Perhaps the most likely explanation for the observed NK association with T1D progression is that a viral trigger results in altered NK cell phenotype that tracks with progressive insulitis. Persisting enteroviral infection has been associated with T1D progression (15) and enteroviruses are known to infect pancreatic β cells, inducing early NK infiltration and cytolysis in animal models (47, 48). An NK-predominant insulitis has also been observed in pancreas from diabetic organ donors with Coxsackie B4 enteroviral infection (16) and is consistent with an autoreactive effector role for NK in T1D pathogenesis. However, NK cells may also function to regulate T cell–mediated immunity during persistent viral infection (49). A limitation of the current study is that, while the association of an NK transcriptional signature is validated and may be used for predicting progression, the mechanism linking NK cells to insulitis requires further investigation.
Despite these limitations, we also demonstrate that a disease-relevant transcriptional signature can serve as the starting point for further mechanistic understanding and previously unidentified therapeutic approaches. By screening the NK-enriched T1D progression–associated signature against collated genomic information from many sources, we predicted and confirmed in vitro that inhibition of a poorly characterized G protein–coupled receptor [GPR171, previously known for its role in controlling satiety signaling in the hypothalamus (50)] was capable of suppressing NK cytolytic protein expression.
We also undertook a systematic network analysis of age-independent blood transcription, before IAbs emergence with the large majority of samples taken during the first year of life. In these earliest samples, we observed reciprocal association of B lymphoblastic and TNF-enriched monocytic signatures that associated with the subsequent rate of progression to T1D. Although these signatures were not different between those later developing T1D and matched controls, they were specifically increased in individuals who progressed later to islet autoimmunity without developing T1D (IAb+T1D−). Although it is tempting to speculate that this evidence supports a protective role for early inflammatory signals—such as proposed by proponents of the “hygiene hypothesis” (51)—studies in animal models have highlighted the complexity of altered TNF signaling with evidence for distinct roles at different disease stages (52, 53). However, the validated association of increased expression of this signature in T1D-protected individuals despite islet autoimmunity may help inform interventional study design (54).
Previous hypothesis-driven analyses of early gene expression changes identified an increase in IFN1 signaling in pre-T1D children linked to history of recent infection (14). A comparable signature was apparent in the TEDDY cohort and showed transient elevation in spikes consistent with response to an infectious stimulus (and quite different to the chronic, progressive increase seen in the NK signature). Greater IFN1-induced gene expression was observed during the 12-month preceding T1D onset compared with age-matched controls but was not associated with the rate of progression to islet autoimmunity or T1D. This is consistent with a role for IFN1 signaling—and perhaps viral infections that provoke transient IFN1 elevations—in modifying disease progression. However, as with NK cells, evidence from animal models shows that IFN signaling may play either a role in promoting T cell–mediated insulitis (55) or in protecting β cells from NK cell–mediated attack (48).
Last, we sought to incorporate the longitudinal measurement of immune traits—both gene expression and IAbs—into a predictive model that could provide an estimate of an individual’s T1D risk and the confidence of that estimate. Long-term risk of T1D (over the subsequent 10 to 15 years) can currently be informed by the extent of IAbs seropositivity. However, for a predictive model to affect on clinical decision-making—whether by altering the frequency of clinical review to monitor for severe complications such as diabetic ketoacidosis (56) or by facilitating early intervention studies (24)—it is necessary to obtain a robust estimate of near-term risk of T1D onset. We therefore sought to build a predictive model that could estimate individual T1D risk in two specific scenarios: either by making an early prediction (at 18 months) over a longer horizon (5 years) or by using cumulative data to make serial predictions over the subsequent 12 months. To test the ability of both baseline and longitudinal measures to inform this prediction, we built a Bayesian joint model incorporating either Ab status alone, or with more extensive IAbs features (serospecificity, timing, and interaction of IAbs development) with or without gene expression signatures. We included stratification by sex (as this was the only other covariate demonstrating independent association with progression rate) but intentionally excluded HLA stratification (despite a demonstrated association with progression (57)) to facilitate extrapolation between global populations with distinct HLA distributions. This approach allowed direct comparison between both simple and more complex models, aiming to establish optimal prediction with the simplest approach requiring as few measurements as possible. With predictions made over a short horizon of 12 months, the model with extensive IAbs features outperformed standard prediction using IAbs status alone and gained little support from including gene expression data: This is consistent with observations that IAbs are often positive within 12 months of diagnosis (58) and that our model supported robust prediction of T1D progression in this scenario. However, it is an onerous task to repeatedly sample children at such an early age to obtain longitudinal data on timing and sequence of seroconversion specificities. We therefore tested a second scenario using data only from the first 18 months and making predictions progressively further ahead. Predicting from this earlier time point—arguably a more feasible clinical scenario given the reduced sampling requirement—showed a benefit of gene expression signatures in addition to IAbs measures with robust performance on both cross-validation and independent validation cohort testing.
The current study identified extensive, longitudinal changes in the whole blood transcriptome occurring during both healthy infancy and progression to T1D. This finding has been made possible through assiduous prospective collection of samples by the TEDDY consortium. We show here that these changes can be both interpreted and used to inform prediction of T1D risk from an early age. Extensive sampling at an early age is facilitated by the simplicity of whole blood collection. However, this method also limits the biological interpretation of modular signatures identified. The modular signatures identified here are dominated by cell-subset specific transcripts, with both module enrichment and deconvolution methods in broad agreement. Each method can identify the likely cellular source of a transcriptional signature but, when that signal is derived from a mixed cell population like peripheral blood, it is more difficult to pin down the cell-intrinsic pathways responsible for that change in gene expression. Improved methods for deconvolution may help to address this problem (59) but require robust validation against concurrently sampled cell-intrinsic transcriptomes. Transcriptional profiling of sorted cell populations (60) or single-cell profiling (61) methods can similarly overcome this limitation, but they inevitably result in sampling of a much smaller cohorts. It is clear from our analyses that enrichment and deconvolution approaches can be complementary. As deconvoluted cell subset proportions may vary together, for example, increasing together during an inflammatory response, it is expected that a transcriptional signature may correlate with multiple cell subset proportions, making it harder to define the source of that signal through deconvolution alone. Enrichment is not similarly encumbered by this problem, relying instead on coexpressed features within the module itself for interpretation, although it is inevitably constrained by the availability of external signatures for enrichment analysis.
We have demonstrated and validated an association of NK cell gene expression signature with T1D progression. It remains to be determined whether this change reflects a causal contribution to T1D related immunopathology, a host response to an infectious trigger, or both. An answer to this fundamental question will require further analyses and more detailed investigation of prospective data and samples.
Longitudinal measurement of gene expression patterns in infancy is dynamic, but accounting for these changes allows identification of an age-independent NK gene expression signature that tracks with rate of progression to T1D. Incorporating gene expression signatures alongside patterns of islet autoimmunity seroconversion facilitates robust prediction of individual risk, validated in an independent cohort. This creates the potential for early monitoring of at-risk infants for T1D onset, facilitating the prevention of severe complications such as ketoacidosis (62), effective trialing of preventive therapies, or the identification of targets for immunomodulation (63).
MATERIALS AND METHODS
Study design
The current study was designed to identify transcriptional coexpression networks in longitudinal whole blood transcriptomes in the TEDDY nested case:control study. Independent transcriptional networks were identified in and compared between individuals progressing to T1D or islet autoimmunity and age-matched controls (fig. S1). Eigengenes summarizing coexpressed gene modules were then generated and modeled against the principal end points of the TEDDY study, namely, the onset of islet autoimmunity and diagnosis of T1D. Association of early coexpression networks (measured in the earliest sampling time point for each individual) with later progression to either T1D or islet autoimmunity was also undertaken. For validation purposes, independent network analysis was undertaken of whole blood gene expression data from the publicly available DIPP cohort (GSE30211).
TEDDY and nested case:control study design
Enrolment to the TEDDY study and design of the nested case-control biomarker discovery study is described in full elsewhere (25) and summarized here (fig. S2). In brief, the TEDDY study enrolled children <4.5 months of age from December 2004 to July 2010 through newborn screening for high-risk HLA-DR-DQ genotypes at six international centers (three in United States and three in European Union). Written consent was obtained from primary carers for all participants, ethical approval was obtained from local institutional review boards, and the study is monitored by an external evaluation committee formed by the National Institutes of Health. Blood samples were prospectively collected from 3 months of age, continuing at 3 monthly intervals until age 4, and then every 6 months until age 15 unless seroconversion to persistent islet autoimmunity has occurred when they continued every 3 months until age 15. The primary end points of the TEDDY study are (i) the appearance of persistent, confirmed islet autoimmunity, defined as the presence of one confirmed islet autoantibody (IAAs, GAD65A, or IA-2A) on at least two consecutive samples. Islet autoimmunity result confirmation was obtained through reciprocal sample testing at two laboratories with the date of persistent seroconversion being the date of first detection of islet autoimmunity that was subsequently shown to be persistent and (ii) the clinical appearance of T1D, as defined by the American Diabetes Association diagnostic criteria (64).
Samples used for genomic analysis within the nested case:control study design used here were identified by risk set sampling in which islet autoimmunity and T1D controls were randomly selected from individuals who were free of the relevant event within 45 days of the case’s event time using best available sample matching for clinical center, sex, family history of T1D, and age (fig. S7). This identified two separate nested, matched cohorts each relating to one of the primary end points of the TEDDY study, namely, T1D onset, and onset of islet autoimmunity (Fig. 1A, fig. S2, and table S2) (25).
RNA extraction and microarray hybridization
The TEDDY study collected 2.5 ml of peripheral blood to extract total RNA from enrolled children. Total RNA was extracted using a high throughput 96-well format extraction protocol using magnetic (MagMax) beads technology at the TEDDY RNA Laboratory, Jinfiniti Biosciences. Purified RNA (200 ng) was further used for complementary RNA (cRNA) amplification and labeling with biotin using the Target Amp cDNA Synthesis Kit (Epicenter). About 750 ng of labeled cRNA was hybridized to the Illumina HumanHT-12 Expression BeadChips as per the manufacturer’s instructions. The HumanHT-12 Expression BeadChip provides coverage for more than 47,000 transcripts and known splice variants across the human transcriptome. After hybridization, arrays were washed, stained with Cy3-conjugated streptavidin, and scanned.
Microarray data preprocessing and normalization
The BeadArray and lumi Bioconductor packages were used for preprocessing microarray data including image analysis, quality control (QC), variance stabilization transformation, normalization, and gene annotation. The MedianBackground method was used for local background correction. In addition, the BeadArray Subversion of Harshlight (BASH) method was used for beads artifact detection, which takes local spatial information into account when determining outliers. Each probe is replicated a varying number of times on each array; the summarization procedure produces a bead summary data in the form of a single signal intensity value for each probe. Illumina’s default outlier function and modified mean and SD were used to obtain a bead summary data. Variance-stabilizing transformation (65) and robust spline normalization (66) method which combines the features of quantile and loess normalization were used for generating between-array normalization data. QC was performed by excluding arrays from further analysis with the corrupted image files, high gradient effects on the probe intensities, high percentage of beads that were masked by the BASH method (67), low mean or median number of beads used to create the summary values for each probe on each array after outliers removal, low proportion of detected probes, low percentage of housekeeping genes expressed above the background level of the array, gender discrepancies using massiR package, and poor pairwise array correlations. Transcriptional data from the DIPP cohort (GSE302011) was accessed from the National Center for Biotechnology Information Gene Expression Omnibus (GEO) repository using the GEOquery package from Bioconductor in RStudio (version 3.5.1).
Transcriptomic QC and batch correction
All the nested case-control pairs for the longitudinal transcriptome data were assigned to the same batch to constrain batchwise variation. In total, 2013 TEDDY samples were processed in 31 batches with a median batch size of 74 samples per batch (range: 18 to 86 samples per batch). In addition, two external QC samples (donor 1 and donor 2) were included in each batch to estimation of batch-to-batch variations. The MedianBackground method was used for local background correction. In addition, the BASH method was used for beads artifact detection (67), which takes local spatial information into account when determining outliers. The first two principal components of the gene expression data before and after normalization, respectively, are shown in fig. S8. The mean pairwise Pearson correlation coefficients after normalization were 0.97 (SD = 0.04) for donor 1 and 0.99 (SD = 0.01) for donor 2.
Statistical analysis
Transcriptional network analysis
After data processing and QC, 2013 samples from 401 individuals were included in the current analysis, representing 1698 samples from 342 individuals in the islet autoimmunity case:control study and 795 samples from 125 individuals in the T1D case:control study (fig. S2). For islet autoimmunity analyses, samples taken before onset of islet autoimmunity from both T1D and IA case:control cohorts were included along with their respective matched controls, stratified by the specificity of the first seroconversion as indicated. Transcriptional data were variance filtered (using the inflection point of cumulative median absolute deviation distribution) with data from 15,000 probes included in modular network analyses. The weighted gene coexpression networks (WGCNA) Bioconductor package in RStudio (version 3.5.1) was used to identify networks of coexpressed transcripts with scaled eigenvalues taken forward for lmm modeling. Scale-free topology was confirmed, and a soft thresholding power was selected by serial modeling of mean connectivity and adjacency functions. The network was constructed with a specified minimum module size of n = 30 and medium sensitivity to cluster splitting (deepsplit = 2). Independent networks were generated on cases and controls with comparison of network structure undertaken using WGCNA in RStudio applying a composite preservation statistic as described (fig. S1B) (68). Modular structure in selected subgroups was visualized using t-distributed stochastic neighbor embedding (t-SNE) plots using the Rtsne package from CRAN. As equivalent modular structure was identified in cases and controls, network analysis was repeated using the full cohort of 2013 samples to identify “universal” modular eigengenes applicable to the entire cohort (rather than define them separately; fig. S1B).
For the DIPP cohort, the public dataset (GSE30211) was downloaded from GEO into R followed by filtering to retain unique genes, selecting those with the largest interquartile range per gene resulting in n = 18,469 features. This was mapped against the Refseq identifiers in the TEDDY dataset to identify a matching set of n = 9313 unique features that were used for modular network analyses as for the TEDDY dataset.
Longitudinal modeling
Longitudinal changes in gene expression were modeled by applying lmm to scaled modular eigenvalues using the lme4 package from CRAN in RStudio (version 3.5.1). To identify changes in gene expression of cases that were not seen in matched controls, models were fitted for each modular eigenvector against either time to event (for cases, T1D diagnosis, or islet autoimmunity onset) or to chronological age (matched controls) and the observed fit compared between cases and matched controls. Significance of effects was determined using a likelihood ratio test against a null model in the absence of that effect. This was repeated for additional covariates to test their independent association with progression rate including HLA subgroup, ethnicity, and sex. For effects deemed significant (FDR < 5%), specificity of association was determined by comparing observed significance in cases to that in controls in the form of a ratio of FDR values (FDRT1D/IA:FDRcontrol). lmm was fitted including fixed terms (modular eigengene values and sex) and both random intercept and random slope terms for individuals. All identified modular signatures were iteratively tested with the extracted significance corrected for multiple testing using the Benjamini-Hochberg FDR method with a threshold for significance set at FDR of 5%. Where indicated, for modular signatures of interest, modeling was repeated incorporating a natural cubic spline, implemented using the splines package in RStudio (version 3.5.1). Case:control lmm FDR ratios were visualized as radarcharts including all modules significantly associated with time to event, using the package radarchart from CRAN in RStudio (version 3.5.1). Individual fits from lmm models were visualized using the ggplot2, sme, and effects packages from CRAN in RStudio (version 3.5.1).
Early and pre-T1D cohorts
For the TEDDY cohort, earliest available samples were identified from each individual and these were filtered for those obtained before IAbs seroconversion (TEDDY preAb cohort, Fig. 4A). For the peri-T1D cohort, individuals were identified from whom a sample was taken within 365 days of diagnosis (serial IFN analysis) or the sample closest to diagnosis used (peri-T1D). The closest matched sample from the paired, matched control subject was used for comparative purposes. Gene expression modular signatures in these cross-sectional analyses were adjusted for sampling age, taking the residuals of a linear model including the relevant eigenvector and sampling age.
Module enrichment analysis
Module interpretation was performed using enrichment analysis against public repositories of defined transcriptional signatures as described in the text. Genes comprising selected modules were compared to reference signature repositories as indicated including ARCHS4, DICE, and Gene Ontology (GO) with a corrected Fisher’s exact test computed using Enrichr and visualized as the −log10 transformed adjusted value in a radar chart. Deconvolution analysis was undertaken using the CIBERSORT method against the LM22 dataset (31), with imputed cell proportions being correlated against module-specific eigenvectors. GPR171 was identified through a systematic screen against a relational database (36)) linking candidate druggable targets (35) to associated transcriptional changes and other genomic data. T1D-associated signatures were screened against the existing Illuminating the Druggable Genome (IDG) library in the ARCHS4 dataset comprising 352 druggable targets linked to 20,883 genes. All targets showing any overlap with T1D signature genes were included in the radar plot visualization (Fig. 2I), with only GPR171 achieving significant overlap.
NK cell analysis
Primary human NK cells obtained from healthy volunteers and stained with an excess of recombinantly engineered FcR-defective antibodies (CD3 clone REA641 and CD56 clone REA196, Miltenyi Biosciences) to avoid preactivation. Flow sorting of NK cells (CD3−CD56+) was performed using an AriaIII sorter (BD) in the Cambridge BRC flow phenotyping hub. Purified NK cells were cultured for 48 hours in complete RPMI 1640 in the presence of target K562 cells and either a GPR171 inhibitor (MS21570, Tocris Bio-Techne) or vehicle (phosphate-buffered saline) and stained with an excess of antibodies against GPR171 (polyclonal rabbit anti-human GPR171, Abcam), CD71 (clone CY1G4, BioLegend), and Granzyme B (clone GB11, BioLegend).
Bayesian joint modeling
For prediction, we sought a method that could incorporate both baseline risk stratification and multiple longitudinal covariates to provide an estimate of event hazard with associated uncertainty. Joint models applied to longitudinal and survival data allow modeling of the error-free biomarker trajectories and disease process simultaneously and have several advantages over similar alternatives. Joint models have been shown to provide unbiased estimates of hazard ratios, unlike models using time-dependent covariates with increased performance compared to either baseline-only or time-dependent Cox models (39, 69). Joint modeling (jm) was performed using the mvjmbayes, jmbayes, and coxph packages (70) from Bioconductor and CRAN in RStudio (version 3.5.1) to estimate the probability of getting disease at a given point in time given the data available. Let S(t) denotes the survival function, which we define to be , where is the true time of getting disease for the jth patient. S(t) is estimated using the hazard function h(t), the instantaneous risk of getting disease
Whereas standard approaches to survival analysis model and estimate the hazard function given the survival data at hand, Bayesian joint models allow hazard function estimation using both baseline covariates and longitudinal data in a proportional hazard model, using the predicted value from the lmm in the hazard model. This at once aims to reduce noise inherent in sparse biological data while not relying on assumptions that observations remain unchanged between measurements. The Bayesian methodology to compute the parameters allows for uncertainty estimates on predictions, achieved through Monte Carlo Markov chain sampling. We used the JMbayes package (70) from CRAN in RStudio (version 3.5.1).
The complete model including all covariates considered is given below
Not all covariates were included in all model scenarios as an underlying goal was a sparse model incorporating covariates that can be measured in a simple, robust, and cost-effective manner and which are likely to withstand later extension of the model into additional populations. Covariates used were factors that are known or suspected to correlate with progression to disease onset (IA) or progression (T1D diagnosis) and included whole blood transcriptional signatures and serial IAbs data with time-varying effects (the hazard ratio was allowed to vary with time) and interaction effects between covariates (the type, number, and sequence of IAbs seroconversion were accounted for). Sex is included in our model as it has shown to correlate with T1D progression (table S3) (71) and is simple to obtain. HLA risk category is collected in the TEDDY study but was excluded to facilitate extension of the model between populations and ethnicities and because HLA risk groups also did not contribute to model performance on testing in the TEDDY discovery cohort (table S3). Longitudinal data were fitted with a natural cubic spline–fitted lmm from the JMbayes library using the mvglmer function, and survival predictions were made using survFitJM function. The input features to predict longitudinal outcome include the natural spline with three degrees of freedom fitted to time.
Predictive model performance estimates
When building any predictive model, it is imperative to balance predictive performance against the risk of “overfitting,” whereby the model performs well on a training dataset but fails to predict on unseen data. Predictive performance was first estimated using 10-fold cross-validation on the TEDDY discovery dataset. Application in a clinical context was simulated by first making predictions on data collected up to 1 year of age and then serially increasing the amount of data available in steps of 0.15 years (mimicking clinical follow up), making disease predictions at each step over a constant time horizon of 1 year ahead. Model performance was evaluated using metrics addressing two key parameters, again using the JMbayes package: model discrimination (how well the model differentiates between individuals who do/do not reach an end point) and model calibration (how well the model predicts the observed data).
For discrimination, AUC ROC was selected to reflect both sensitivity and specificity of predictive accuracy. For calibration, PE was used as defined below. Each metric was applied to both cross-validated performance estimates on the discovery TEDDY cohort and after application of a “fixed,” optimal model from the discovery set to the independent validation DIPP cohort (which played no part in model training).
AUC is defined for a prediction horizon of Δt as follows
where πj(u | t) is the probability that patient j will survive up to time u given that they are alive at time t and is the true event time (T1D onset or IAbs seroconversion). PE is defined as the expected loss given the difference between the predicted Ni(u) and the true value πj(u | t) as given below
Supplementary Material
Fig. S1. Graphical abstract and analysis overview.
Fig. S2. CONSORT diagram of TEDDY nested case:control study design.
Fig. S3. Age-associated module enrichment.
Fig. S4. Sex and breast-feeding associations of modular expression changes.
Fig. S5. IAAs and GADA signature enrichment.
Fig. S6. DIPP module enrichment.
Fig. S7. Transcriptomic QC and batch correction.
Data file S1. Coexpression network modules in TEDDY.
Data file S2. TEDDY cohort characteristics.
Data file S3. Extended association analysis of T1D-associated eigenvectors.
Data file S4. IAAs and GADA modules.
Data file S5. IAb-associated B cell modular signature.
Data file S6. DIPP cohort NK-enriched modular signature.
Data file S7. TEDDY preAb-modular signatures.
Data file S8. Source data.
Acknowledgments:
We thank A. Wood (Department of Public Health and Primary Care, University of Cambridge and Cambridge Centre for Artificial Intelligence in Medicine, CCAIM) for helpful discussions and all patients and families for participation.
Funding:
The TEDDY study is funded by U01 DK63829, U01 DK63861, U01 DK63821, U01 DK63865, U01 DK63863, U01 DK63836, U01 DK63790, UC4 DK63829, UC4 DK63861, UC4 DK63821, UC4 DK63865, UC4 DK63863, UC4 DK63836, UC4 DK95300, UC4 DK100238, UC4 DK106955, UC4 DK112243, UC4 DK117483, and contract no. HHSN267200700014C from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), National Institute of Allergy and Infectious Diseases (NIAID), Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), National Institute of Environmental Health Sciences (NIEHS), Centers for Disease Control and Prevention (CDC), and JDRF. This work supported in part by the NIH/NCATS Clinical and Translational Science Awards to University of Florida (UL1 TR000064) and University of Colorado (UL1 TR001082). K.G.C.S. is a Lister Prize fellow and is supported by a Wellcome Trust Senior Investigator award (200871/Z/16/Z). E.F.M. is a Wellcome-Beit prize fellow (10406/Z/14/A) supported by the Wellcome Trust and Beit Foundation (10406/Z/14/Z) and by the National Institutes for Health Research Biomedical Research Centre (Cambridge). L.-P.X.’s affiliation changed after completion of the manuscript and is now Département d’Informatique et de Recherche Opérationnelle, Université de Montréal, Montréal, Canada and Mila, Quebec Institute for Learning Algorithms, Montréal, Canada.
Footnotes
Competing interests: A.L. is a member of the scientific advisory board of Diamyd Medical AB; A.-G.Z. is a member of the Data Safety and Monitoring Board of the Protect study for PreventionBio. J.P.K. provides advice to ActoBio. K.G.C.S. is a cofounder of PredictImmune and Rheos Medicines and provides advice to Kymab, GSK, and Elstar. E.F.M. is a cofounder of PredictImmune and provides advice to Kymab. The other authors declare that they have no competing interests.
Data and materials availability:
The TEDDY study gene expression data that support the findings of this study have been deposited in NCBI’s database of Genotypes and Phenotypes (dbGaP) with the primary accession code phs001562.v1.p1.
REFERENCES AND NOTES
- 1.Todd JA, Etiology of type 1 diabetes. Immunity 32, 457–467 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Ziegler AG, Rewers M, Simell O, Simell T, Lempainen J, Steck A, Winkler C, Ilonen J, Veijola R, Knip M, Bonifacio E, Eisenbarth GS, Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA 309, 2473–2479 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Atkinson MA, von Herrath M, Powers AC, Clare-Salzler M, Current concepts on the pathogenesis of type 1 diabetes–considerations for attempts to prevent and reverse the disease. Diabetes Care 38, 979–988 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharp SA, Rich SS, Wood AR, Jones SE, Beaumont RN, Harrison JW, Schneider DA, Locke JM, Tyrrell J, Weedon MN, Hagopian WA, Oram RA, Development and standardization of an improved type 1 diabetes genetic risk score for use in newborn screening and incident diagnosis. Diabetes Care 42, 200–207 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ziegler AG, Nepom GT, Prediction and pathogenesis in type 1 diabetes. Immunity 32, 468–478 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cerosaletti K, Barahmand-Pour-Whitman F, Yang J, DeBerg HA, Dufort MJ, Murray SA, Israelsson E, Speake C, Gersuk VH, Eddy JA, Reijonen H, Greenbaum CJ, Kwok WW, Wambre E, Prlic M, Gottardo R, Nepom GT, Linsley PS, Single-cell RNA sequencing reveals expanded clones of islet antigen-reactive CD4+ T cells in peripheral blood of subjects with type 1 diabetes. J. Immunol 199, 323–335 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dufort MJ, Greenbaum CJ, Speake C, Linsley PS, Cell type-specific immune phenotypes predict loss of insulin secretion in new-onset type 1 diabetes. JCI Insight 4, e125556 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zakharov PN, Hu H, Wan X, Unanue ER, Single-cell RNA sequencing of murine islets shows high cellular complexity at all stages of autoimmune diabetes. J. Exp. Med 217, e20192362 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Damond N, Engler S, Zanotelli VRT, Schapiro D, Wasserfall CH, Kusmartseva I, Nick HS, Thorel F, Herrera PL, Atkinson MA, Bodenmiller B, A map of human type 1 diabetes progression by imaging mass cytometry. Cell Metab. 29, 755–768.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Q, Parikh H, Butterworth MD, Lernmark A, Hagopian W, Rewers M, She JX, Toppari J, Ziegler AG, Akolkar B, Fiehn O, Fan S, Krischer JP; TEDDY Study Group, Longitudinal metabolome-wide signals prior to the appearance of a first islet autoantibody in children participating in the TEDDY Study. Diabetes 69, 465–476 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vatanen T, Franzosa EA, Schwager R, Tripathi S, Arthur TD, Vehik K, Lernmark A, Hagopian WA, Rewers MJ, She JX, Toppari J, Ziegler AG, Akolkar B, Krischer JP, Stewart CJ, Ajami NJ, Petrosino JF, Gevers D, Lahdesmaki H, Vlamakis H, Huttenhower C, Xavier RJ, The human gut microbiome in early-onset type 1 diabetes from the TEDDY study. Nature 562, 589–594 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stewart CJ, Ajami NJ, O’Brien JL, Hutchinson DS, Smith DP, Wong MC, Ross MC, Lloyd RE, Doddapaneni H, Metcalf GA, Muzny D, Gibbs RA, Vatanen T, Huttenhower C, Xavier RJ, Rewers M, Hagopian W, Toppari J, Ziegler A-G, She J-X, Akolkar B, Lernmark A, Hyoty H, Vehik K, Krischer JP, Petrosino JF, Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature 562, 583–588 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christen U, von Herrath MG, Do viral infections protect from or enhance type 1 diabetes and how can we tell the difference? Cell. Mol. Immunol 8, 193–198 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferreira RC, Guo H, Coulson RM, Smyth DJ, Pekalski ML, Burren OS, Cutler AJ, Doecke JD, Flint S, McKinney EF, Lyons PA, Smith KG, Achenbach P, Beyerlein A, Dunger DB, Clayton DG, Wicker LS, Todd JA, Bonifacio E, Wallace C, Ziegler A-G, A type I interferon transcriptional signature precedes autoimmunity in children genetically at risk for type 1 diabetes. Diabetes 63, 2538–2550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vehik K, Lynch KF, Wong MC, Tian X, Ross MC, Gibbs RA, Ajami NJ, Petrosino JF, Rewers M, Toppari J, Ziegler A-G, She J-X, Lernmark A, Akolkar B, Hagopian WA, Schatz DA, Krischer JP, Hyöty H, Lloyd RE; TEDDY Study Group, Prospective virome analyses in young children at increased genetic risk for type 1 diabetes. Nat. Med 25, 1865–1872 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dotta F, Censini S, van Halteren AG, Marselli L, Masini M, Dionisi S, Mosca F, Boggi U, Muda AO, Del Prato S, Elliott JF, Covacci A, Rappuoli R, Roep BO, Marchetti P, Coxsackie B4 virus infection of β cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc. Natl. Acad. Sci. U.S.A 104, 5115–5120 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mehdi AM, Hamilton-Williams EE, Cristino A, Ziegler A, Bonifacio E, Le Cao KA, Harris M, Thomas R, A peripheral blood transcriptomic signature predicts autoantibody development in infants at risk of type 1 diabetes. JCI Insight 3, e98212 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kallionpaa H, Elo LL, Laajala E, Mykkanen J, Ricano-Ponce I, Vaarma M, Laajala TD, Hyoty H, Ilonen J, Veijola R, Simell T, Wijmenga C, Knip M, Lahdesmaki H, Simell O, Lahesmaa R, Innate immune activity is detected prior to seroconversion in children with HLA-conferred type 1 diabetes susceptibility. Diabetes 63, 2402–2414 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Cabrera SM, Chen YG, Hagopian WA, Hessner MJ, Blood-based signatures in type 1 diabetes. Diabetologia 59, 414–425 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olin A, Henckel E, Chen Y, Lakshmikanth T, Pou C, Mikes J, Gustafsson A, Bernhardsson AK, Zhang C, Bohlin K, Brodin P, Stereotypic immune system development in newborn children. Cell 174, 1277–1292.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arrieta MC, Stiemsma LT, Dimitriu PA, Thorson L, Russell S, Yurist-Doutsch S, Kuzeljevic B, Gold MJ, Britton HM, Lefebvre DL, Subbarao P, Mandhane P, Becker A, McNagny KM, Sears MR, Kollmann T; CHILD Study Investigators, Mohn WW, Turvey SE, Finlay BB, Early infancy microbial and metabolic alterations affect risk of childhood asthma. Sci. Transl. Med 7, 307ra152 (2015). [DOI] [PubMed] [Google Scholar]
- 22.McKinney EF, Lee JC, Jayne DRW, Lyons PA, Smith KGC, T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature 523, 612–616 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hagopian WA, Lernmark A, Rewers MJ, Simell OG, She JX, Ziegler AG, Krischer JP, Akolkar B, TEDDY–The environmental determinants of diabetes in the young: An observational clinical trial. Ann. N. Y. Acad. Sci 1079, 320–326 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Bonifacio E, Predicting type 1 diabetes using biomarkers. Diabetes Care 38, 989–996 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Lee H-S, Burkhardt BR, McLeod W, Smith S, Eberhard C, Lynch K, Hadley D, Rewers M, Simell O, She J-X, Hagopian B, Lernmark A, Akolkar B, Ziegler AG, Krischer JP; TEDDY study group, Biomarker discovery study design for type 1 diabetes in The Environmental Determinants of Diabetes in the Young (TEDDY) study. Diabetes Metab. Res. Rev 30, 424–434 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Langfelder P, Horvath S, WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langfelder P, Horvath S, Eigengene networks for studying the relationships between co-expression modules. BMC Syst. Biol 1,54 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stuart JM, Segal E, Koller D, Kim SK, A gene-coexpression network for global discovery of conserved genetic modules. Science 302, 249–255 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, Yefanov A, Lee H, Zhang N, Robertson CL, Serova N, Davis S, Soboleva A, NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 41, D991–D995 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lachmann A, Torre D, Keenan AB, Jagodnik KM, Lee HJ, Wang L, Silverstein MC, Ma’ayan A, Massive mining of publicly available RNA-seq data from human and mouse. Nat. Commun 9, 1366 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, Khodadoust MS, Esfahani MS, Luca BA, Steiner D, Diehn M, Alizadeh AA, Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol 37, 773–782 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leslie RD, Jerram ST, Stratifying diabetes: Desperately seeking specificity. Diabetes 66, 801–803 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Krischer JP, Lynch KF, Schatz DA, Ilonen J, Lernmark A, Hagopian WA, Rewers MJ, She JX, Simell OG, Toppari J, Ziegler A-G, Akolkar B, Bonifacio E; TEDDY Study Group, The 6 year incidence of diabetes-associated autoantibodies in genetically at-risk children: The TEDDY study. Diabetologia 58, 980–987 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmiedel BJ, Singh D, Madrigal A, Valdovino-Gonzalez AG, White BM, Zapardiel-Gonzalo J, Ha B, Altay G, Greenbaum JA, McVicker G, Seumois G, Rao A, Kronenberg M, Peters B, Vijayanand P, Impact of genetic polymorphisms on human immune cell gene expression. Cell 175, 1701–1715.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodgers G, Austin C, Anderson J, Pawlyk A, Colvis C, Margolis R, Baker J, Glimmers in illuminating the druggable genome. Nat. Rev. Drug Discov 17, 301–302 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rouillard AD, Gundersen GW, Fernandez NF, Wang Z, Monteiro CD, McDermott MG, Ma’ayan A, The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016, baw100 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haller MJ, Schatz DA, The DIPP project: 20 years of discovery in type 1 diabetes. Pediatr. Diabetes 17 (Suppl 22), 5–7 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Chen JH, Asch SM, Machine learning and prediction in medicine - Beyond the peak of inflated expectations. N. Engl. J. Med 376, 2507–2509 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rizopoulos D, Joint Models for Longitudinal and Time-To-Event Data: With Applications in R (Chapman & Hall/CRC biostatistics series, CRC Press, Boca Raton, 2012), pp. xiv, 261 pages. [Google Scholar]
- 40.Endesfelder D, Castell WZ, Bonifacio E, Rewers M, Hagopian WA, She JX, Lernmark A, Toppari J, Vehik K, Williams AJK, Yu L, Akolkar B, Krischer JP, Ziegler AG, Achenbach P; TEDDY Study Group, Time-resolved autoantibody profiling facilitates stratification of preclinical type 1 diabetes in children. Diabetes 68, 119–130 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Battaglia M, Ahmed S, Anderson MS, Atkinson MA, Becker D, Bingley PJ, Bosi E, Brusko TM, DiMeglio LA, Evans-Molina C, Gitelman SE, Greenbaum CJ, Gottlieb PA, Herold KC, Hessner MJ, Knip M, Jacobsen L, Krischer JP, Long SA, Lundgren M, McKinney EF, Morgan NG, Oram RA, Pastinen T, Peters MC, Petrelli A, Qian X, Redondo MJ, Roep BO, Schatz D, Skibinski D, Peakman M, Introducing the endotype concept to address the challenge of disease heterogeneity in type 1 diabetes. Diabetes Care 43, 5–12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Flodstrom-Tullberg M, Bryceson YT, Shi FD, Hoglund P, Ljunggren HG, Natural killer cells in human autoimmunity. Curr. Opin. Immunol 21,634–640 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Rodacki M, Svoren B, Butty V, Besse W, Laffel L, Benoist C, Mathis D, Altered natural killer cells in type 1 diabetic patients. Diabetes 56, 177–185 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Oras A, Peet A, Giese T, Tillmann V, Uibo R, A study of 51 subtypes of peripheral blood immune cells in newly diagnosed young type 1 diabetes patients. Clin. Exp. Immunol 198, 57–70 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Poirot L, Benoist C, Mathis D, Natural killer cells distinguish innocuous and destructive forms of pancreatic islet autoimmunity. Proc. Natl. Acad. Sci. U.S.A 101,8102–8107 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee IF, Qin H, Trudeau J, Dutz J, Tan R, Regulation of autoimmune diabetes by complete Freund’s adjuvant is mediated by NK cells. J. Immunol 172, 937–942 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Nekoua MP, Bertin A, Sane F, Alidjinou EK, Lobert D, Trauet J, Hober C, Engelmann I, Moutairou K, Yessoufou A, Hober D, Pancreatic beta cells persistently infected with coxsackievirus B4 are targets of NK cell-mediated cytolytic activity. Cell. Mol. Life Sci 77, 179–194 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Flodstrom M, Maday A, Balakrishna D, Cleary MM, Yoshimura A, Sarvetnick N, Target cell defense prevents the development of diabetes after viral infection. Nat. Immunol 3, 373–382 (2002). [DOI] [PubMed] [Google Scholar]
- 49.Waggoner SN, Cornberg M, Selin LK, Welsh RM, Natural killer cells act as rheostats modulating antiviral T cells. Nature 481,394–398 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gomes I, Aryal DK, Wardman JH, Gupta A, Gagnidze K, Rodriguiz RM, Kumar S, Wetsel WC, Pintar JE, Fricker LD, Devi LA, GPR171 is a hypothalamic G protein-coupled receptor for BigLEN, a neuropeptide involved in feeding. Proc. Natl. Acad. Sci. U.S.A 110, 16211–16216 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bach J-F, Chatenoud L, The hygiene hypothesis: An explanation for the increased frequency of insulin-dependent diabetes. Cold Spring Harb. Perspect. Med 2, a007799 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Christen U, Wolfe T, Mohrle U, Hughes AC, Rodrigo E, Green EA, Flavell RA, von Herrath MG, A dual role for TNF-alpha in type 1 diabetes: Islet-specific expression abrogates the ongoing autoimmune process when induced late but not early during pathogenesis. J. Immunol 166, 7023–7032 (2001). [DOI] [PubMed] [Google Scholar]
- 53.Lee LF, Xu B, Michie SA, Beilhack GF, Warganich T, Turley S, McDevitt HO, The role of TNF-alpha in the pathogenesis of type 1 diabetes in the nonobese diabetic mouse: Analysis of dendritic cell maturation. Proc. Natl. Acad. Sci. U.S.A 102, 15995–16000 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nepom GT, Ehlers M, Mandrup-Poulsen T, Anti-cytokine therapies in T1D: Concepts and strategies. Clin. Immunol 149, 279–285 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marro BS, Ware BC, Zak J, de la Torre JC, Rosen H, Oldstone MB, Progression of type 1 diabetes from the prediabetic stage is controlled by interferon-α signaling. Proc. Natl. Acad. Sci. U.S.A 114, 3708–3713 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Duca LM, Wang B, Rewers M, Rewers A, diabetic ketoacidosis at diagnosis of type 1 diabetes predicts poor long-term glycemic control. Diabetes Care 40, 1249–1255 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Ferrat LA, Vehik K, Sharp SA, Lernmark Å, Rewers MJ, She J-X, Ziegler A-G, Toppari J, Akolkar B, Krischer JP, Weedon MN, Oram RA, Hagopian WA; TEDDY Study Group; Committees, A combined risk score enhances prediction of type 1 diabetes among susceptible children. Nat. Med 26, 1247–1255 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bonifacio E, Achenbach P, Birth and coming of age of islet autoantibodies. Clin. Exp. Immunol 198, 294–305 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kang K, Meng Q, Shats I, Umbach DM, Li M, Li Y, Li X, Li L, CDSeq: A novel complete deconvolution method for dissecting heterogeneous samples using gene expression data. PLOS Comput. Biol 15, e1007510 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lyons PA, Koukoulaki M, Hatton A, Doggett K, Woffendin HB, Chaudhry AN, Smith KGC, Microarray analysis of human leucocyte subsets: The advantages of positive selection and rapid purification. BMC Genomics 8, 64 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aldridge S, Teichmann SA, Single cell transcriptomics comes of age. Nat. Commun 11, 4307 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hekkala AM, Ilonen J, Toppari J, Knip M, Veijola R, Ketoacidosis at diagnosis of type 1 diabetes: Effect of prospective studies with newborn genetic screening and follow up of risk children. Pediatr. Diabetes 19, 314–319 (2018). [DOI] [PubMed] [Google Scholar]
- 63.Bresson D, von Herrath M, Immunotherapy for the prevention and treatment of type 1 diabetes: Optimizing the path from bench to bedside. Diabetes Care 32, 1753–1768 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Puavilai G, Chanprasertyotin S, Sriphrapradaeng A, Diagnostic criteria for diabetes mellitus and other categories of glucose intolerance: 1997 criteria by the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus (ADA), 1998 WHO consultation criteria, and 1985 WHO criteria. World Health Organization. Diabetes Res. Clin. Pract 44, 21–26 (1999). [DOI] [PubMed] [Google Scholar]
- 65.Lin SM, Du P, Huber W, Kibbe WA, Model-based variance-stabilizing transformation for Illumina microarray data. Nucleic Acids Res. 36, e11 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Du P, Kibbe WA, Lin SM, lumi: A pipeline for processing Illumina microarray. Bioinformatics 24, 1547–1548 (2008). [DOI] [PubMed] [Google Scholar]
- 67.Cairns JM, Dunning MJ, Ritchie ME, Russell R, Lynch AG, BASH: A tool for managing BeadArray spatial artefacts. Bioinformatics 24, 2921–2922 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Langfelder P, Luo R, Oldham MC, Horvath S, Is my network module preserved and reproducible? PLoS Comput. Biol 7, e1001057 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Henderson R, Diggle P, Dobson A, Joint modelling of longitudinal measurements and event time data. Biostatistics 1 , 465–480 (2000). [DOI] [PubMed] [Google Scholar]
- 70.Rizopoulos D, The R Package JMbayes for fitting joint models for longitudinal and time-to-event data using MCMC. J. Stat. Softw 72, (2016). [Google Scholar]
- 71.Krischer JP, Liu X, Lernmark A, Hagopian WA, Rewers MJ, She JX, Toppari J, Ziegler AG, Akolkar B; TEDDY Study Group, The influence of type 1 diabetes genetic susceptibility regions, age, sex, and family history on the progression from multiple autoantibodies to type 1 diabetes: A TEDDY study report. Diabetes 66, 3122–3129 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Graphical abstract and analysis overview.
Fig. S2. CONSORT diagram of TEDDY nested case:control study design.
Fig. S3. Age-associated module enrichment.
Fig. S4. Sex and breast-feeding associations of modular expression changes.
Fig. S5. IAAs and GADA signature enrichment.
Fig. S6. DIPP module enrichment.
Fig. S7. Transcriptomic QC and batch correction.
Data file S1. Coexpression network modules in TEDDY.
Data file S2. TEDDY cohort characteristics.
Data file S3. Extended association analysis of T1D-associated eigenvectors.
Data file S4. IAAs and GADA modules.
Data file S5. IAb-associated B cell modular signature.
Data file S6. DIPP cohort NK-enriched modular signature.
Data file S7. TEDDY preAb-modular signatures.
Data file S8. Source data.
Data Availability Statement
The TEDDY study gene expression data that support the findings of this study have been deposited in NCBI’s database of Genotypes and Phenotypes (dbGaP) with the primary accession code phs001562.v1.p1.