

Abstract
C-Acyl furanosides are versatile synthetic precursors to a variety of natural products, nucleoside analogues, and pharmaceutical molecules. This report addresses the unmet challenge in preparing C-acyl furanosides by developing a cross-coupling reaction between glycosyl esters and carboxylic acids. A key step is the photoredox activation of the glycosyl ester, which promotes the homolysis of the strong anomeric C–O bond through CO2 evolution to afford glycosyl radicals. This method embraces a large scope of furanoses, pyranoses, and carboxylic acids, and is readily applicable to the synthesis of a thymidine analogue and diplobifuranylone B, as well as the late-stage modification of (+)-sclareolide. The convenient preparation of the redox active glycosyl ester from native sugars and the compatibility with common furanoses exemplifies the potential of this method in medicinal chemistry.
A cross-coupling of glycosyl esters with carboxylic acids to prepare C-acyl furanosides and pyranosides. The reaction proceeds through photoredox activation of the glycosyl ester to afford glycosyl radicals.
Introduction
C-Acyl glycosides are versatile synthetic intermediates to natural products, nucleoside analogues, and pharmaceutical molecules (Scheme 1A).1 Downstream derivatives of C-acyl glycosides, including C-alkyl glycosides,2 diazo derivatives,3 alcohols,4 nucleoside analogues,5 and cyclopentitols,6 display significant potentials in drug discovery and utilities in chemical biology and biochemistry studies. A C-glycoside linkage confers in vivo stability towards hydrolysis and enzymatic degradation.7 Therefore, C-furanosides, including nucleoside analogues in particular, have been extensively explored as antiviral drug candidates. In addition, unnatural nucleosides are essential tools for studying the origins of mutagenicity and the mechanism of replication and evolution.8C-acyl furanosides serve as a convenient intermediate to nucleoside analogues through formation of heterocycles at the anomeric position via condensation and cyclization.5
Scheme 1. Strategies in C-acyl furanoside synthesis.

Synthetic efforts towards C-acyl glycosides have been primarily focused on pyranosides. Conventional methods include the nucleophilic addition of glycosyl lithium9 or samarium10 reagents to carbonyl compounds and the functionalization of C-nitriles,4a,11C-allyls,4bC-allenes3,12 and benzothiazoles (Scheme 1A).13 Recent cross-coupling approaches employ stannane reagents14 and glycosyl bromides15 to deliver a broad range of C-acyl pyranoside products. In contrast to the widespread means to prepare C-acyl pyranosides, access to C-acyl furanosides is limited and lacks a robust scope.16 For example, furanosyl stannane reagents have not yet been reported. Furanosyl bromides, especially 2-deoxy derivatives, are often unstable and have not been applied to C-acyl furanoside synthesis. Currently, the best way to prepare C-acyl furanosides is through the nucleophilic addition of metal reagents to glycosyl nitriles,17 but this method requires multiple steps and precludes base-sensitive functional groups.
The dearth of furanosylation methods may be associated with the lack of furanosyl radical precursors that can be used for cross-coupling. Pyranosyl radicals have been generated from various unnatural glycosyl derivatives, including bromides,18 xanthates,19 glycals,20 stannanes,21 thiol ethers,22 acyl tellurides,23 and trifluoroborates.24 Precursors to furanosyl radicals, however, are limited to unnatural tetrahydrofuran derivatives with a carboxylic acid at the C1 position, restricting the scope of carbohydrate substrates.25 Furthermore, redox auxiliaries based on C–C bond homolysis can only substitute at the C5 of furanosides and the C6 of pyranosides, not allowing for anomeric functionalization.26 The most ideal glycosyl radical precursor is the native carbohydrate, but the BDE of the anomeric C–O bond (99 kcal mol−1) is even higher than that of a typical alcohol (96 kcal mol−1).27 Thus, formation of glycosyl radicals via anomeric C–O bond homolysis represents a significant fundamental challenge.
Herein, we report a solution to the unmet challenge of C-acyl furanoside synthesis by utilizing redox auxiliary 1 to generate furanosyl radicals from furanosyl esters and cross-coupling them to carboxylic acids (Scheme 1B). Inspired by the success of alkyl dihydropyridine (DHP) as a radical precursor28 and its utility in C5- and C6-functionalization of glycosides,26a,b we developed DHP-based redox auxiliary 1 by incorporating a carbonyl group between DHP and the glycoside, which can induce the homolysis of the strong anomeric C–O bond through CO2 evolution.29 The favorable condensation between 1 and furanoses and pyranoses readily furnishes glycosyl esters that are bench stable and compatible with all common native carbohydrates.
Results and discussion
We performed C-acylation of glycosides by cross-coupling glycosyl DHP carboxylate esters with carboxylic acids under modified photoredox-nickel dual catalytic conditions, with diethyl dicarbonate (DEDC) as an activator for the carboxylic acids (Table 1).30 The elevated temperature of 90 °C was necessary to facilitate DHP fragmentation and the subsequent decarboxylation. Lower temperature led to incomplete decarboxylation and the formation of glycosyl ester as a byproduct. We first evaluated the scope with respect to carboxylic acids coupling to diacetonide-protected d-mannofuranosyl DHP carboxylate ester (Table 1A). Aliphatic carboxylic acids containing various functional groups, including amines, alkenes, and ketones, underwent efficient coupling to afford C-acyl d-mannosides 2–13. The scope of the carboxylic acids extends to biologically active substrates, including fenbufen 14, ursodeoxycholic acid 15, l-glutamic acid (Glu) 16, α-d-galactopyranuronic acid 17, d-biotin 18, and mycophenolic acid 19. The high yield of 16 suggests that this method shows promise for glyco-peptide synthesis by allowing conjugation of glutamic acid with a variety of carboxylic acids. The coupling of d-mannofuranosyl DHP ester to benzoic acids utilizes a variant of the standard conditions that employs unsubstituted 2,2′-bi-2-oxazoline (biOx) as the ligand to afford products 20–25. Electron-deficient benzoic acids gave lower yields than electron-rich derivatives. In all reactions, the α-anomer was isolated as the predominant product due to the approach of the incoming catalyst from the α-face, as the β-face was hindered by the C2-substituent. Alternatively, it is possible that the capture of glycosyl radical by Ni is reversible.31 Under the Curtin–Hammett condition, the stereoselectivity is determined by a faster reductive elimination of the α-product relative to the β-product.
Scope of C-acyl glycosylation with DHP-derived glycosyl estersaa.
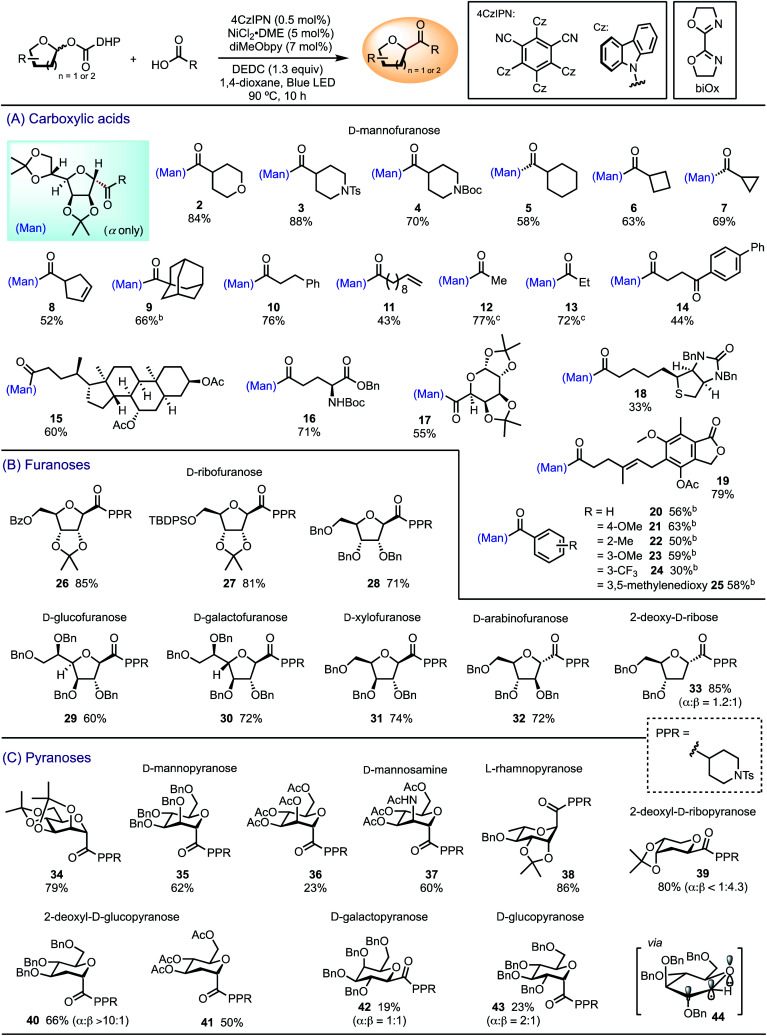
|
Isolated yields. The major anomer was isolated as a pure product, unless specified. Stereochemistry of the products was assigned based on NOESY and COSY experiments. Reaction conditions: carboxylic acid (0.20 mmol), glycosyl ester (0.26 mmol, 1.3 equiv.), 1,4-dioxane (4.8 mL).
2,2′-Bi-2-oxazoline (biOx) as the ligand, NiBr2·diglyme (5 mol%), 4CzIPN (0.25 mol%), glycosyl ester (1.2 equiv.), LiBr (2.5 mol%), 86 °C.
The corresponding anhydride was used as the electrophile without DEDC, 1,4-dioxane (3.2 mL).
We then investigated the scope with respect to furanoses and the compatibility of protecting groups using 4-piperidinecarboxylic acid (PPR–CO2H) as the coupling partner (Table 1B). d-Ribofuranoses bearing common protecting groups, such as benzoyl 26, tert-butyldiphenylsilyl (TBDPS) 27, and benzyl 28, afforded the corresponding β-C-acyl ribofuranosides in good yields. Other furanosides also underwent smooth C-acylation to generate d-glucofuranoside 29, d-galactofuranoside 30, d-xylofuranoside 31, and d-arabinofuranoside 32. The stereoselectivity appears to be governed by the stereochemistry at C2, favoring the entering group from the opposite face of the C2-substituent. While the β-anomer was favored by most products, d-arabinofuranoside 32 was formed as the α-anomer. Benzyl-protected 2-deoxy-d-ribose generated a mixture of α and β anomers of 33 in a ratio of 1.2 : 1, presumably due to the lack of steric hindrance at C2 to distinguish the α from the β face.
A variety of pyranoses were transformed to C-acyl glycosides 34–41 under the standard conditions (Table 1C). α-Anomers were favored for d-mannopyranoses 34–36, d-mannosamine 37, l-rhamnopyranose 38, and 2-deoxy-d-glucopyranosides 40 and 41, as determined by the kinetic anomeric effect, namely the stabilization of the transition state by the donation of the lone electron pair on the ring oxygen to the anti-bonding orbital of the newly-formed σ bond (σ*‡) in the axial position.32 Both anomers were observed for 2-deoxy-d-ribopyranoside 39, with the β-anomer favored due to the steric hindrance remotely imparted by C3 and C4 substituents. C-Acylation of d-galactopyranose and d-glucopyranose was less effective and afforded a mixture of α- and β-anomers 42 and 43 in low yields. We attribute this limitation to the contradictory preferences of the steric and stereoelectronic effects. In the preferred boat conformation 44,32 β-attack of the glycosyl radical intermediate is favored due to the steric hindrance at C2, whereas the transition state of α-attack is stabilized by the kinetic anomeric effect.
The generality of this C-acylation method prompted us to explore its application to derivatize natural products and synthesize biologically relevant molecules (Scheme 2). A (+)-sclareolide derivative 45 can be readily coupled with 4-piperidinecarboxylic acid to afford 46 in 60% yield with a diastereomeric ratio (d.r.) higher than 10 : 1 (Scheme 2A). This late-stage modification can be extended to natural and synthetic molecules containing hemiacetal functionality, in light of a large variety of commercially available carboxylic acids. A thymidine (T) analogue dPzo49 has attracted considerable attention,5a as the corresponding nucleotide can be incorporated into DNA by Klenow fragments and forms a Watson–Crick base pair with adenine (A).5bC-Acylation of d-2-deoxyribosyl ester 47 accomplishes a more efficient formal synthesis of 48 compared to a previous route from 50 (Scheme 2B).5a Alternatively, a better overall yield may be achieved by conducting the C-acylation of d-ribose (74% yield, α only, cf. compound S7 in the ESI†), followed by reduction at C2.5b Finally, we demonstrate that acyl mannofuranose 51, obtained in 68% yield, can lead to natural product diplobifuranylone B 54 33 in a concise synthetic route (Scheme 2C).34 The structure of 55 was assigned to diplobifuranylone A in the original report of these molecules, produced by fungus pathogens,33 but a discrepancy between the spectroscopic data of 55 and that of diplobifuranylone A suggests that the original structural assignment requires reconsideration.
Scheme 2. Synthetic applications of C-acylation of furanosides.

Based on literature precedents and a previous radical trapping experiment,29 we propose that the reaction follows the photoredox-nickel dual catalytic cycles shown in Scheme 3. The key step involves the subsequent fragmentation of the DHP ester to afford the glycosyl radical upon release of Hantzsch pyridine and CO2. The carboxylic acid coupling partner is activated by DEDC by forming the corresponding anhydride.
Scheme 3. Proposed catalytic cycle.

Conclusions
We developed a cross-coupling reaction to prepare C-acyl furanosides and pyranosides from glycosyl esters and carboxylic acids. Upon photoredox activation, the glycosyl ester can fragment to generate a glycosyl radical through CO2 evolution. This method tolerates a broad scope of carboxylic acids, furanoses, and pyranoses to deliver products with excellent diastereoselectivity, governed by the stereochemistry at C2. The reaction is particularly useful for the synthesis of unnatural nucleosides and the late-stage modification of natural products. The convenient preparation of the redox active glycosyl esters, their stability, and their compatibility with common carbohydrates exemplifies the potential of this method in medicinal chemistry.
Data availability
All experimental procedures and spectroscopic data can be found in the ESI.†
Author contributions
Y. W. and T. D. conceived the idea. Y. W. optimized the conditions. Y. W. and J. L. explored the scope. T. D. supervised the project and wrote the manuscript.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Health (R01 GM-127778). T. D. thanks the Alfred P. Sloan Foundation (FG-2018-10354) and the Camille and Henry Dreyfus Foundation (TC-19-019) for providing fellowships to partially support this work.
Electronic supplementary information (ESI) available: Optimisation studies, experimental procedures, spectroscopic data, NMR spectra. See DOI: 10.1039/d1sc03596g
References
- (a) Štambaský J. Hocek M. Kočovský P. Chem. Rev. 2009;109:6729–6764. doi: 10.1021/cr9002165. [DOI] [PubMed] [Google Scholar]; (b) Yang Y. Yu B. Chem. Rev. 2017;117:12281–12356. doi: 10.1021/acs.chemrev.7b00234. [DOI] [PubMed] [Google Scholar]; (c) Levi S. M. Jacobsen E. N. Org. React. 2019:801–852. [Google Scholar]
- (a) Ko K.-S. Kruse J. Pohl N. L. Org. Lett. 2003;5:1781–1783. doi: 10.1021/ol034444m. [DOI] [PubMed] [Google Scholar]; (b) Yang G. Schmieg J. Tsuji M. Franck R. W. Angew. Chem., Int. Ed. 2004;43:3818–3822. doi: 10.1002/anie.200454215. [DOI] [PubMed] [Google Scholar]; (c) Hidaka Y. Kiya N. Yoritate M. Usui K. Hirai G. Chem. Commun. 2020;56:4712–4715. doi: 10.1039/D0CC00785D. [DOI] [PubMed] [Google Scholar]
- Geng Y. Kumar A. Faidallah H. M. Albar H. A. Mhkalid I. A. Schmidt R. R. Bioorg. Med. Chem. 2013;21:4793–4802. doi: 10.1016/j.bmc.2013.05.055. [DOI] [PubMed] [Google Scholar]
- (a) Knapp S. Shieh W.-C. Jaramillo C. Trilles R. V. Nandan S. R. J. Org. Chem. 1994;59:946–948. doi: 10.1021/jo00084a005. [DOI] [Google Scholar]; (b) Wong C.-H. Moris-Varas F. Hung S.-C. Marron T. G. Lin C.-C. Gong K. W. Weitz-Schmidt G. J. Am. Chem. Soc. 1997;119:8152–8158. doi: 10.1021/ja970920q. [DOI] [Google Scholar]
- (a) Maeba I. Iijima T. Matsuda Y. Ito C. J. Chem. Soc., Perkin Trans. 1. 1990;1:73–76. doi: 10.1039/P19900000073. [DOI] [Google Scholar]; (b) Tomori T. Nagaoka K. Takeshita L. Shiozawa T. Miyatake Y. Masaki Y. Sekine M. Seio K. J. Org. Chem. 2018;83:8353–8363. doi: 10.1021/acs.joc.8b00918. [DOI] [PubMed] [Google Scholar]
- Álvarez-Dorta D. León E. I. Kennedy A. R. Riesco-Fagundo C. Suárez E. Angew. Chem., Int. Ed. 2008;47:8917–8919. doi: 10.1002/anie.200803696. [DOI] [PubMed] [Google Scholar]
- (a) Bililign T. Griffith B. R. Thorson J. S. Nat. Prod. Rep. 2005;22:742–760. doi: 10.1039/B407364A. [DOI] [PubMed] [Google Scholar]; (b) De Clercq E. J. Med. Chem. 2016;59:2301–2311. doi: 10.1021/acs.jmedchem.5b01157. [DOI] [PubMed] [Google Scholar]; (c) Seley-Radtke K. L. Yates M. K. Antiviral Res. 2018;154:66–86. doi: 10.1016/j.antiviral.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yates M. K. Seley-Radtke K. L. Antiviral Res. 2019;162:5–21. doi: 10.1016/j.antiviral.2018.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Moran S. Ren R. X.-F. Kool E. T. Proc. Natl. Acad. Sci. U. S. A. 1997;94:10506–10511. doi: 10.1073/pnas.94.20.10506. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lavergne T. Degardin M. Malyshev D. A. Quach H. T. Dhami K. Ordoukhanian P. Romesberg F. E. J. Am. Chem. Soc. 2013;135:5408–5419. doi: 10.1021/ja312148q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Lesimple P. Beau J.-M. Bioorg. Med. Chem. 1994;2:1319–1330. doi: 10.1016/S0968-0896(00)82082-9. [DOI] [PubMed] [Google Scholar]; (b) Beau J.-M., and Gallagher T., Glycoscience Synthesis of Substrate Analogs and Mimetics, ed. H. Driguez and J. Thiem, Springer Berlin Heidelberg: Berlin, Heidelberg, 1998, pp. 1–54 [Google Scholar]
- Mazéas D. Skrydstrup T. Beau J.-M. Angew. Chem., Int. Ed. 1995;34:909–912. doi: 10.1002/anie.199509091. [DOI] [Google Scholar]
- Guisot N. E. S. Ella Obame I. Ireddy P. Nourry A. Saluzzo C. Dujardin G. Dubreuil D. Pipelier M. Guillarme S. J. Org. Chem. 2016;81:2364–2371. doi: 10.1021/acs.joc.5b02853. [DOI] [PubMed] [Google Scholar]
- Kolympadi M. Fontanella M. Venturi C. André S. Gabius H.-J. Jiménez-Barbero J. Vogel P. Chem.–Eur. J. 2009;15:2861–2873. doi: 10.1002/chem.200801394. [DOI] [PubMed] [Google Scholar]
- Dondoni A. Catozzi N. Marra A. J. Org. Chem. 2005;70:9257–9268. doi: 10.1021/jo051377w. [DOI] [PubMed] [Google Scholar]
- (a) Belosludtsev Y. Y. Bhatt R. K. Falck J. R. Tetrahedron Lett. 1995;36:5881–5882. doi: 10.1016/00404-0399(50)1183I-. [DOI] [Google Scholar]; (b) Zhu F. Rodriguez J. O'Neill S. Walczak M. A. ACS Cent. Sci. 2018;4:1652–1662. doi: 10.1021/acscentsci.8b00628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Zhao C. Jia X. Wang X. Gong H. J. Am. Chem. Soc. 2014;136:17645–17651. doi: 10.1021/ja510653n. [DOI] [PubMed] [Google Scholar]; (b) Jia X. Zhang X. Qian Q. Gong H. Chem. Commun. 2015;51:10302–10305. doi: 10.1039/C5CC03113C. [DOI] [PubMed] [Google Scholar]; (c) Boehlich G. J. Schützenmeister N. Angew. Chem., Int. Ed. 2019;58:5110–5113. doi: 10.1002/anie.201900995. [DOI] [PubMed] [Google Scholar]
- DeShong P. Slough G. A. Elango V. Trainor G. L. J. Am. Chem. Soc. 1985;107:7788–7790. doi: 10.1021/ja00311a109. [DOI] [Google Scholar]
- (a) de las Heras F. G. Fernández-Resa P. J. Chem. Soc., Perkin Trans. 1. 1982:903–907. doi: 10.1039/P19820000903. [DOI] [Google Scholar]; (b) Teruaki M. Shu K. Shin-ichiro S. Chem. Lett. 1984;13:1529–1530. doi: 10.1246/cl.1984.1529. [DOI] [Google Scholar]; (c) Kini G. D. Petrie C. R. Hennen W. J. Dalley N. K. Wilson B. E. Robins R. K. Carbohydr. Res. 1987;159:81–94. doi: 10.1016/S0008-6215(00)90007-7. [DOI] [PubMed] [Google Scholar]
- (a) Giese B. Dupuis J. Angew. Chem., Int. Ed. 1983;22:622–623. doi: 10.1002/anie.198306221. [DOI] [Google Scholar]; (b) Giese B. Dupuis J. Tetrahedron Lett. 1984;25:1349–1352. doi: 10.1016/S0040-4039(01)80154-4. [DOI] [Google Scholar]; (c) Andrews R. S. Becker J. J. Gagné M. R. Angew. Chem., Int. Ed. 2010;49:7274–7276. doi: 10.1002/anie.201004311. [DOI] [PubMed] [Google Scholar]; (d) Adak L. Kawamura S. Toma G. Takenaka T. Isozaki K. Takaya H. Orita A. Li H. C. Shing T. K. M. Nakamura M. J. Am. Chem. Soc. 2017;139:10693–10701. doi: 10.1021/jacs.7b03867. [DOI] [PubMed] [Google Scholar]; (e) Liu J. Gong H. Org. Lett. 2018;20:7991–7995. doi: 10.1021/acs.orglett.8b03567. [DOI] [PubMed] [Google Scholar]
- (a) Sakata M. Haga M. Tejima S. Carbohydr. Res. 1970;13:379–390. doi: 10.1016/S0008-6215(00)80595-9. [DOI] [Google Scholar]; (b) Kancharla P. K. Navuluri C. Crich D. Angew. Chem., Int. Ed. 2012;51:11105–11109. doi: 10.1002/anie.201204400. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kiya N. Hidaka Y. Usui K. Hirai G. Org. Lett. 2019;21:1588–1592. doi: 10.1021/acs.orglett.9b00133. [DOI] [PubMed] [Google Scholar]
- (a) Chiara J. L. Sesmilo E. Angew. Chem., Int. Ed. 2002;41:3242–3246. doi: 10.1002/1521-3773(20020902)41:17<3242::AID-ANIE3242>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]; (b) Parrish J. D. Little R. D. Org. Lett. 2002;4:1439–1442. doi: 10.1021/ol025575a. [DOI] [PubMed] [Google Scholar]; (c) Lo J. C. Kim D. Pan C.-M. Edwards J. T. Yabe Y. Gui J. Qin T. Gutiérrez S. Giacoboni J. Smith M. W. Holland P. L. Baran P. S. J. Am. Chem. Soc. 2017;139:2484–2503. doi: 10.1021/jacs.6b13155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F. Zhang S. Q. Chen Z. Rui J. Hong X. Walczak M. A. J. Am. Chem. Soc. 2020;142:11102–11113. doi: 10.1021/jacs.0c03298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang W. Su S.-N. Shi R. Mou Z.-D. Yu G.-Q. Zhang X. Niu D. Angew. Chem., Int. Ed. 2021;60:385–390. doi: 10.1002/anie.202009828. [DOI] [PubMed] [Google Scholar]
- Masuda K. Nagatomo M. Inoue M. Nat. Chem. 2017;9:207–212. doi: 10.1038/nchem.2639. [DOI] [PubMed] [Google Scholar]
- Takeda D. Yoritate M. Yasutomi H. Chiba S. Moriyama T. Yokoo A. Usui K. Hirai G. Org. Lett. 2021;23:1940–1944. doi: 10.1021/acs.orglett.1c00402. [DOI] [PubMed] [Google Scholar]
- (a) Ma Y. Liu S. Xi Y. Li H. Yang K. Cheng Z. Wang W. Zhang Y. Chem. Commun. 2019;55:14657–14660. doi: 10.1039/C9CC07184A. [DOI] [PubMed] [Google Scholar]; (b) Zhu M. Messaoudi S. ACS Catal. 2021;11:6334–6342. doi: 10.1021/acscatal.1c01600. [DOI] [Google Scholar]
- (a) Badir S. O. Dumoulin A. Matsui J. K. Molander G. A. Angew. Chem., Int. Ed. 2018;57:6610–6613. doi: 10.1002/anie.201800701. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dumoulin A. Matsui J. K. Gutiérrez-Bonet Á. Molander G. A. Angew. Chem., Int. Ed. 2018;57:6614–6618. doi: 10.1002/anie.201802282. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ji P. Zhang Y. Wei Y. Huang H. Hu W. Mariano P. A. Wang W. Org. Lett. 2019;21:3086–3092. doi: 10.1021/acs.orglett.9b00724. [DOI] [PubMed] [Google Scholar]; (d) Wan I. C. S. Witte M. D. Minnaard A. J. Org. Lett. 2019;21:7669–7673. doi: 10.1021/acs.orglett.9b03016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALFABET, https://bde.ml.nrel.gov/, retrieved 25 May, 2021
- (a) Gutiérrez-Bonet Á. Tellis J. C. Matsui J. K. Vara B. A. Molander G. A. ACS Catal. 2016;6:8004–8008. doi: 10.1021/acscatal.6b02786. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Buzzetti L. Prieto A. Roy S. R. Melchiorre P. Angew. Chem., Int. Ed. 2017;56:15039–15043. doi: 10.1002/anie.201709571. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Alandini N. Buzzetti L. Favi G. Schulte T. Candish L. Collins K. D. Melchiorre P. Angew. Chem., Int. Ed. 2020;59:5248–5253. doi: 10.1002/anie.202000224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y. Ben-zvi B. Diao T. Angew. Chem., Int. Ed. 2021;60:9433–9438. doi: 10.1002/anie.202014991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Gooßen L. J. Winkel L. Döhring A. Ghosh K. Paetzold J. Synlett. 2002:1237–1240. doi: 10.1055/s-2002-32961. [DOI] [Google Scholar]; (b) Amani J. Molander G. A. Org. Lett. 2017;19:3612–3615. doi: 10.1021/acs.orglett.7b01588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez O. Tellis J. C. Primer D. N. Molander G. A. Kozlowski M. C. J. Am. Chem. Soc. 2015;137:4896–4899. doi: 10.1021/ja513079r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe H. Shuto S. Matsuda A. J. Am. Chem. Soc. 2001;123:11870–11882. doi: 10.1021/ja011321t. [DOI] [PubMed] [Google Scholar]
- Evidente A. Andolfi A. Fiore M. Spanu E. Maddau L. Franceschini A. Marras F. Motta A. J. Nat. Prod. 2006;69:671–674. doi: 10.1021/np050393l. [DOI] [PubMed] [Google Scholar]
- Cheng X. Quintanilla C. D. Zhang L. J. Org. Chem. 2019;84:11054–11060. doi: 10.1021/acs.joc.9b01613. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All experimental procedures and spectroscopic data can be found in the ESI.†