

Abstract
Background and Objective
To determine whether human anti-LRP4/agrin antibodies are pathogenic in mice and to investigate underpinning pathogenic mechanisms.
Methods
Immunoglobulin (Ig) was purified from a patient with myasthenia gravis (MG) with anti-LRP4/agrin antibodies and transferred to mice. Mice were characterized for body weight, muscle strength, twitch and tetanic force, neuromuscular junction (NMJ) functions including compound muscle action potential (CMAP) and endplate potentials, and NMJ structure. Effects of the antibodies on agrin-elicited muscle-specific tyrosine kinase (MuSK) activation and AChR clustering were studied and the epitopes of these antibodies were identified.
Results
Patient Ig-injected mice had MG symptoms, including weight loss and muscle weakness. Decreased CMAPs, reduced twitch and tetanus force, compromised neuromuscular transmission, and NMJ fragmentation and distortion were detected in patient Ig-injected mice. Patient Ig inhibited agrin-elicited MuSK activation and AChR clustering. The patient Ig recognized the β3 domain of LRP4 and the C-terminus of agrin and reduced agrin-enhanced LRP4–MuSK interaction.
Discussion
Anti-LRP4/agrin antibodies in the patient with MG is pathogenic. It impairs the NMJ by interrupting agrin-dependent LRP4–MuSK interaction.
Myasthenia gravis (MG) is an autoimmune disorder of the neuromuscular junction (NMJ),1,2 a cholinergic synapse that rapidly conveys action potentials from motoneuron terminals by releasing acetylcholine (ACh) to depolarize the muscle cell and initiate muscle contraction by activating ACh receptors (AChRs) of muscle fibers.3 In patients with MG, autoantibodies bind to the components of postsynaptic muscle endplates and destroy the function and structure of NMJ, thus leading to impaired neuromuscular transmission and characteristic fatigable skeletal muscle weakness.4-6 AChR is the most common target protein,7 which is accounted for in ∼80% of total MG cases. The agrin/low-density lipoprotein receptor-related protein 4 (LRP4)/muscle-specific tyrosine kinase (MuSK) pathway is critical for NMJ formation, maintenance, and regeneration.3,8 Mutations in agrin, LRP4, and MuSK have been reported in congenital myasthenic syndromes.9,10 Autoantibodies against these proteins have been detected in seronegative MG (anti-AChR antibody–negative MG)11-15 as well as in patients with anti-AChR antibody.16-18 Immunizing animals with LRP4 or agrin induces MG-like deficits presumably by generating relevant antibodies in active experimental autoimmune MG (EAMG) animals.19,20 However, it remains unclear whether human LRP4 or agrin antibodies are pathogenic and if so, what the underlying mechanisms could be—a glaring gap in the understanding of the pathology of these antibodies. To address these questions, we purified immunoglobulin (Ig) from a patient with MG with antibodies against LRP4 and agrin and investigated their effects on NMJ function and structure in passive EAMG mice. The results demonstrate that human anti-LRP4 and anti-agrin antibodies are causal to MG, likely via blocking the agrin-LRP4-MuSK signaling.
Methods
Standard Protocol Approvals, Registrations, and Patient Consents
This study was approved by the Ethics Committee of Georgia Regents University. The project title of the institutional review board (IRB) that approved the study is “(713,470-4) Characterization of Agrin/LRP4 Antibody-Positive Myasthenia Gravis, Georgia Regents University Institutional Review Board.” The patient was given written informed consent. The healthy control plasma sample was approved by the same IRB with waiver of consent.
Patient Information and Plasmapheresis Fluid Collection
One patient (female, age 47, non-Hispanic, Caucasian) had severe MG, Myasthenia Gravis Foundation of America class IVb, and underwent therapeutic plasmapheresis. Her sera tested positive for anti-LRP4 and anti-agrin antibodies, but negative for anti-AChR and MuSK antibodies. The clinic information of this patient can be seen in supplemental clinic information on Zenodo at doi.org/10.5281/zenodo.5143859. Healthy control plasma was obtained from a healthy blood donor from a blood bank. Control plasma was screened negative for anti-AChR, MuSK, LRP4, and agrin antibodies.
Purification of Patient/Healthy Control Ig
Ig was purified from plasmapheresis fluid/healthy control plasma by rivanol and saturated ammonium sulfate (SAS) as previously described with slight modification.21 After adjusting the pH to 8.0, the plasma was added to 0.4% rivanol (D16606; Sigma-Aldrich) (vol/vol, 3.5/1), stirred for 30 minutes, and incubated at room temperature (RT) overnight. Tenacious yellow albumin precipitates were removed by filtering with Whatman No. 1 paper. Rivanol was removed by incubating with activated charcoal (8 g/100 mL) overnight at 4°C and centrifugation at 3,000 g for 30 minutes at 4°C. The supernatant was collected and passed through 0.22 μm Millex-GP filter (Millipore) and was added slowly with equal volume of SAS. After incubation overnight, the sample was centrifuged at 3,000 g for 30 minutes at 4°C; pellets were dissolved in saline (∼10 mL) and dialyzed (Spectra/Por MWCO 50,000; Spectrum Laboratories) at 4°C for 3 hours against 1 L of saline, 2 hours against 1 L of phosphate-buffered saline (PBS), and finally in 1 L of PBS overnight. After centrifugation at 3,000 g for 30 minutes at 4°C, the supernatant was sterilized by passing through a 0.22 µm Millex-GP filter and concentrated by Amicon Ultra 50K (Millipore). A total of 280 nanometers absorbance was used to determine Ig concentration.
Passive Experimental Autoimmune MG
All experiments were performed in a blinder manner where the investigators were blinded to Ig treatment. C57/B6 mice (6–8 weeks old, female) were used. Sample size of mice was estimated by following previous similar studies and by using a sample size calculator at ClinCalc.com (clincalc.com/Stats/SampleSize.aspx). In general, 3 or more mice were used in each group. Mice were housed in ventilated cages in a 21–23°C room with a 12-hour light/dark cycle, with no more than 5 mice per cage. Water and rodent chow diet were provided ad libitum. Experimental procedures were approved by the Institutional Animal Care and Use Committee of Case Western Reserve University.
Passive EAMG was created as previously described.19 Briefly, mice were injected IP with 10 mg purified patient or healthy Ig in 200 μL sterile PBS per day for 24 days; the control group of mice were injected with same volume of PBS. To minimize pain associated with multiple injections, injections were given in different abdominal locations on alternate days. A total of 24 hours after the first Ig injection, mice were injected IP with cyclophosphamide monohydrate (300 mg/kg; Sigma-Aldrich) to suppress potential immune reactions. A total of 24 hours after the last injection, mice were characterized for muscle strength, compound muscle action potential (CMAP), and twitch and tetanic force before being killed for electrophysiologic and histologic studies.
Measurement of Muscle Strength and Contractile Force
Muscle strength was characterized by hanging scale, rotarod, and 2 limb hanging tests as previously described.19,22,23 Torque muscle tension analysis was performed as previously reported.22 Details can be seen in the eMethods (Zenodo, doi.org/10.5281/zenodo.5143859).
Characterization of Neuromuscular Transmission
CMAP and endplate potentials were characterized as previously reported.19,22 Details can be seen in the eMethods (Zenodo, doi.org/10.5281/zenodo.5143859).
NMJ Morphology Characterization
Muscles were stained with whole-mount staining as described previously.19 Muscles were fixed in 4% paraformaldehyde (PFA) for 1 hour at RT, washed 3 times with PBS, and teased to individual fibers that were incubated with the blocking buffer (0.5% Triton X-100, 2% bovine serum albumin, 5% donkey serum in PBS) for 3 hours at RT. The samples were then incubated with primary antibodies in the blocking buffer overnight at 4°C, washing 3 times with PBS, and incubated with α-bungarotoxin (α-BTX)–Alexa Fluor 594 (1: 1,000; B13423; Invitrogen) and goat anti-rabbit/human IgG–Alexa Fluor 488 (1:1,000; A-11034, A-11013; Invitrogen) in the blocking buffer overnight at 4°C. After washing with PBS, muscle fibers were mounted on slides and imaged with a confocal microscope (Zeiss). Antibodies used were rabbit anti-Neurofilament-L (1: 1,000; 2837s; Cell Signaling Technology) and rabbit anti-synapsin-1 (1:500; 5297s; Cell Signaling Technology) or purified Ig (1:500).
Construct Generation
pCNDA3.1-LRP4-ECD was generated by subcloning C-terminal Myc-tagged human LRP4 extracellular domain (ECD) (1–1679) at EcoRV and EcoRI sites in pCDNA3.1. pN1-LRP4-ECD was generated by subcloning C-terminal Flag-tagged human LRP4-ECD (1–1679) at EcoRI and SalI sites in pFlag-N1, replacing enhanced green fluorescent protein. pCMV6-XL4-Flag-LRP4-AP constructs were generated by subcloning N-terminal Flag-tagged and C-terminal alkaline phosphatase (AP)–tagged rat LRP4-ECD and individual domains at NotI and XbaI sites in pCMV6-XL4: extracellular domain (ECD) (23–1,651), LDLa (26–351), β1 (460–693), β2 (765–998), β3 (1,070–1,306), and β4 (1,377–1,610). pFlag-CMV1-agrin constructs were generated by subcloning N-terminal Flag-tagged rat agrin LG123 (1,137–1,940) or agrin laminin G-like 3 domain (LG3) (1,759–1,940) at BamHI and XhoI in pFlag-CMV1. (This backbone has been described by Zhang et al.24). pJZ-152-agrin constructs were generated by subcloning N-terminal hemagglutinin (HA)–tagged agrin-LG23 (1,481–1,940) or agrin-LG3 (1,759–1,940) at StuI and AgeI in pJZ-152 (gift from Rongsheng Jin). pCMV6-XL4-Flag-MuSK-ECD-IgG was generated by subcloning N-terminal Flag-tagged and C-terminal IgG-tagged mouse MuSK ECD (22–487) at NotI and XbaI sites in pCMV6-XL4.
Biochemical Analysis
To determine whether Ig binds to LPR4, condition media were collected from HEK293 cells transfected with pCDNA3.1-based LRP4 constructs, incubated with anti-Myc antibody (positive control), healthy Ig or patient Ig at 4°C overnight, and with protein A/G-immobilized beads. Immobilized proteins were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis and Western blot analysis. To analyze MuSK phosphorylation, C2C12 myotubes were pretreated with purified Ig (50 µg/mL) for 3 hours, then with agrin LG3 for 30 minutes, lysed in the lysis buffer as described previously.19 Lysates were incubated with protein A/G beads (Roche) for 1 hour at 4°C on a rotator and centrifuged at 2,500 g for 5 minutes at 4°C. Supernatants were incubated with anti-MuSK antibody19 at 4°C overnight and with protein A/G beads for 2 hours. Immobilized proteins were subjected to Western blot analysis with mouse 4G10 (anti-phospho-tyrosine; 1:1,000; 05-1050; Millipore). Other antibodies used included rabbit anti-MuSK (1:1,000),19 mouse anti-GAPDH (1:5,000; sc-137179; Santa Cruz), and mouse anti-Flag (1:1,000; 1084; Sigma-Aldrich). Immunoreactive bands were visualized by HRP-conjugated goat anti-mouse/rabbit IgG (1:5,000; PI-31430, PI-31460) and enhanced chemiluminescence (Pierce) and analyzed by Image J.
Solid Phase Binding Assay
To determine the epitopes of patient Ig, 96-well ELISA plate (NUNC maxisorp Apogent) were incubated with purified patient Ig, healthy Ig, or commercial anti-LRP4 antibody (clone N207/27; UC Davis/NIH NeuroMab Facility) in the carbonate buffer (500 mM, pH 9.6) overnight at 4°C. After washing with 0.1% TBST, samples were incubated with the blocking buffer (5% milk in 0.3% TBST) for 2 hours at RT and with C-terminal AP-tagged LRP4-ECD or individual domains that were purified from condition medium of transfected HEK293 cells transfected with Flag and AP-tagged LRP4-ECD and individual domains overnight at 4°C. After washing, AP activity was assayed by p-nitrophenyl phosphate as described previously.24
Effects of Patient Ig on Agrin-Induced AChR Clustering in C2C12 Cells
AChR cluster analysis was performed as previously described.19 C2C12 myotubes were stimulated with agrin LG3 together with purified healthy/patient Ig (50 µg/mL) for 16 hours at 37°C. Myotubes were fixed in 4% PFA, washed by PBS, and incubated with α-BTX-Alexa Fluor 594 (1: 1,000) for 1 hour at RT. After washing 3 times by PBS, AChR clusters were examined by a BZX fluorescence microscope and scored when diameter or axis was ≥4 μm.
Statistical Analysis
Data are shown as mean ± SEM; 2-tailed paired or unpaired Student t test was used to analyze data. Statistical difference was considered when p < 0.05.
Data Availability
Anonymized data not published within the article are available on request. Supplemental data are available from Zenodo at doi.org/10.5281/zenodo.5143859.
Results
Recognizing LRP4 and Agrin Proteins at NMJ by Patient Ig
Igs were purified from the plasmapheresis fluid of the patient with MG and plasma from a healthy control by rivanol and SAS (see Methods for details). Albumin was present in the plasma, but not in purified Ig samples; Ig dimer (heavy and light chains) ran at ∼150 kDa under nonreducing conditions (eFigure 1A, Zenodo doi.org/10.5281/zenodo.5143859), at 50 kDa and 25 kDa under reducing condition (eFigure 1B), but most albumin and other proteins were moved in the purified Ig line (eFigure 1A and 1B). Quantitative interleukin-6 data showed that interleukin-6 was removed from heathy/patient purified Ig (eFigure 1C). These results showed that the preparation for purity is good. Note that the purified Igs contain IgG, IgA, and IgM (eFigure 1D) and are referred as Ig unless otherwise indicated. To determine whether the patient Ig was able to recognize human LRP4, Myc-tagged human LRP4-ECD was produced (as described in Methods) and incubated with patient Ig, healthy Ig, or mouse anti-Myc antibody and subsequently with protein A/G-immobilized beads. Bound proteins were subjected to Western blot analysis with anti-Myc antibody. LRP4-ECD-Myc was detected at ∼250 kDa in the complex with patient Ig, but not with healthy Ig, indicating that patient Ig recognized human LRP4 (Figure 1A). Furthermore, patient Ig, but not healthy Ig, was also able to recognize LRP4 of mouse C2C12 muscle cells (Figure 1B). Next, we determined whether patient Ig recognizes agrin. Flag-agrin-LG123 containing C-terminal 1,137–1,940 amino acid residues (generated in HEK293 cells transfected with pFlag-CMV1-agrin-LG123) was incubated with patient Ig, healthy Ig, or anti-Flag antibody and subsequently with protein A/G-immobilized beads. Bound proteins were subjected to Western blot analysis with anti-Flag antibody. Flag-agrin was detected at ∼130 kDa by patient Ig or anti-Flag antibody, but not healthy Ig (Figure 1C), indicating that patient Ig was able to recognize agrin.
Figure 1. Patient Immunoglobulin (Ig) Recognized Lipoprotein Receptor-Related Protein 4 (LRP4) and Agrin and Labeled the Neuromuscular Junction (NMJ).

(A) The patient Ig recognized human LRP4-ECD-Myc protein. (B) The patient Ig recognized the endogenous Lrp4 protein in C2C12. (C) The patient Ig recognized Flag-agrin-LG123 protein (a C-terminal 110-KDa fragment). (D) Staining the NMJ with the patient Ig, but not healthy Ig. Scale bars:10 μm. IP = immunoprecipitation.
To detect whether the patient Ig can interact with endogenous LRP4 and agrin at NMJ in mice, tibialis anterior muscles were subjected to whole-mount staining with α-BTX to label AChR and with the patient or healthy Ig, which were visualized by Alexa Fluor 488–conjugated anti-human IgG antibody. The NMJs presented characteristic pretzel-like morphology, with complex continuous branches. Staining with patient Ig, but not healthy Ig, co-localized with α-BTX (Figure 1D). These results suggested that the patient Ig recognized endogenous LRP4 or agrin at NMJ in mice. Together, these results indicated that the Ig of the patient with MG recognized human LRP4 and agrin in solution and at the NMJ.
Weight Loss and Muscle Weakness in Passive EAMG Mice
To determine whether the patient's Ig is causally pathogenic, we established a passive EAMG mouse model, as described previously.19,25 C57BL/6 female mice were injected with patient or healthy Ig for 24 days (Figure 2A). Anti-LRP4 antibodies and anti-agrin antibodies were detected in the sera of mice injected with patient Ig, but not with healthy Ig (eFigure 2, available from Dryad at doi.org/10.5061/dryad.44j0zpcdh). Mice injected with patient Ig showed time-dependent weight loss during injection, compared with mice injected with PBS or healthy Ig (Figure 2B). Muscle strength was evaluated by forelimb grip, forelimb hanging, and rotarod test. As shown in Figure 2, C and D, muscle weakness and increased fatigue were noted in patient Ig-injected mice, compared to mice injected with PBS (p < 0.01). Patient Ig-injected mice also displayed a reduction in hanging time in forelimb hanging test (p < 0.01; Figure 2E) and in running time in rotarod test (p < 0.01; Figure 2F). Mice injected with healthy Ig showed no difference in muscle strength or fatigue compared to PBS-injected mice. These results indicated that the patient IgG reduced muscle strength in injected mice.
Figure 2. Weight Loss, Muscle Weakness, and Twitch and Tetanus Force Decreased in Patient Immunoglobulin (Ig)–Injected Mice.

(A) Scheme of passive experimental autoimmune myasthenia gravis. (B) Decreased body weight in patient Ig-injected mice, not in healthy Ig-injected mice, compared with phosphate-buffered saline (PBS)–injected control mice. (C) Declined forelimb grip strength in patient Ig-injected mice, not in healthy Ig-injected mice, compared with PBS-injected control mice. (D) Increased forelimb grip fatigue in patient Ig-injected mice, not in healthy Ig-injected mice, compared with PBS-injected control mice. (E) Reduced forelimbs hanging time in patient Ig-injected mice, not in healthy Ig-injected mice, compared with PBS-injected control mice. (F) Decreased running time in patient Ig-injected mice, not in healthy Ig-injected mice, compared with PBS-injected control mice. (G) Reduced single twitch force by muscle stimulation in patient Ig-injected mice, not in healthy IgG-injected mice, compared with PBS-injected control mice. (H) Reduced single twitch force by sciatic nerve stimulation in patient Ig-injected mice, not in healthy Ig-injected mice, compared with PBS-injected control mice. (I) Representative tetanic curves at stimulation frequency 50 and 100 Hz by muscle stimulation. (J) Reduced tetanic force by muscle stimulation at different stimulation frequencies in patient Ig-injected mice, not in healthy Ig-injected mice, compared with PBS-injected control mice. (K) Representative tetanic curves at stimulation frequency 50 and 100 Hz by sciatic nerve stimulation. (L) Reduced tetanic force muscle stimulation at different stimulation frequencies in patient Ig-injected mice, not healthy Ig-injected mice, compared with PBS-injected control mice. n = 3 mice per group, **p < 0.01.
Frequency-Dependent Reduction in Stimulation-Induced Muscle Contraction
To investigate the underlying mechanism of muscle weakness, first, we studied muscle contractions in response to nerve stimulation and direct muscle stimulation. As shown in Figure 2G, twitch force by muscle stimulation was reduced in patient Ig-injected mice compared with PBS-injected mice. There was also a reduction in twitch force by nerve stimulation in patient Ig-injected mice (Figure 2H). When tetanic contraction was induced by nerve and muscle stimulation (300 ms stimuli at different frequencies), it was reduced in patient Ig-injected mice beginning at 50 Hz, compared to PBS-injected mice, and the peak reduction occurred at 100 Hz (Figure 2, I–L). In contrast, twitch and tetanic contractions were similar between mice injected with healthy Ig and PBS. These observations indicate that patient Ig reduces muscle strength in passive EAMG mice. Interestingly, the reduction in nerve stimulation–induced twitch and tetanic contraction was more than that in muscle stimulation–induced (70% vs 40%, p < 0.01), suggesting an impairment in neuromuscular transmission.
Impaired Neuromuscular Transmission
To test this hypothesis further, we measured CMAPs in response to repetitive nerve stimuli.19 Supramaximal stimulation was applied to the sciatic nerve with trains of 10 stimuli at different frequencies (with a 30-second pause between trains). There was no difference in CMAPs between PBS- and healthy Ig-injected mice (Figure 3C). However, CMAPs by the 10th stimuli (compared with those by the first stimuli) were reduced in patient Ig-injected mice at 10 Hz (by ∼15%) (Figure 3C). At 40 Hz, CMAP reduction was observed even at the second stimulus (Figure 3A, B, D). The frequency-dependent reduction suggests a progressive loss of effective neuromuscular transmission after repeated stimulations, revealing a mechanism of fatigable muscle weakness in the passive EAMG mice.
Figure 3. Decreased Compound Muscle Action Potentials (CMAPs) and Presynaptic and Postsynaptic Deficits in Patient Immunoglobulin (Ig)–Injected Mice.

(A) Stacked 10 succession CMAP traces at 40 Hz. (B) CMAP traces in response to the first, second, and 10th stimuli. (C) Reduced CMAP amplitudes of the 10th stimulation at different stimulation frequencies in patient Ig-injected mice, not in healthy Ig-injected mice, compared with phosphate-buffered saline (PBS)–injected control mice. (D) Reduced CMAP amplitudes at 40 Hz. (E) Representative miniature end plate potential (mEPP) traces. (F, H) Cumulative plots of mEPP events against interval (F) or amplitude (H). (G, I) Reduced mEPP frequency (G) and amplitude (I) in patient Ig-injected mice, not in healthy Ig-injected mice, compared with PBS-injected control mice. (J) Reduced end plate potential (EPP) amplitude in patient Ig-injected mice, not in healthy Ig-injected mice, compared with PBS-injected control mice. (K) Decreased PPR in patient Ig-injected mice, not in healthy Ig-injected mice, compared with PBS-injected control mice. n = 3 mice per group. *p < 0.05, **p < 0.01.
To further characterize the neuromuscular transmission, we measured miniature end plate potentials (mEPPs), which were generated by spontaneous vesicle release. Both amplitudes and frequencies of mEPPs were reduced in patient Ig-injected mice compared with PBS- and healthy Ig-injected mice (Figure 3, E–I). These results suggest that in patient Ig-injected mice, AChR density at the postjunctional membrane or ACh concentration in individual synaptic vesicles was reduced and presynaptic spontaneous ACh release was decreased. Consistently, the amplitudes of end plate potentials (EPPs), events that are elicited by nerve stimulation, were reduced in patient Ig-injected mice compared with PBS- or healthy Ig-injected mice (Figure 3J). In light of reduced mEPP frequency, we measured paired-pulse ratio (PPR) at different stimulus intervals, which is an indicator of presynaptic vesicle release probability (Figure 3K). At 10-ms intervals, PPR was lower in patient Ig-injected mice than PBS-injected mice. With increased intervals, the difference was reduced and diminished at 120-ms intervals. These electrophysiologic results indicated that passive EMAG mice showed both postsynaptic deficits (reduced AChR density) and presynaptic deficits (reduced probability of ACh vesicle release).
Increased NMJ Fragment, Reduced AChR Density, and Increased Denervation
To determine whether patient Ig changes structural NMJ, tibialis anterior muscles were stained whole mount with α-BTX to label AChR and antibodies against neurofilament-L and synapsin-1 to label nerve branches and terminals. PBS- and healthy Ig-injected mice displayed NMJs with characteristic pretzel-like morphology with complex continuous branches (Figure 4A). However, NMJs in patient Ig-injected mice were fragmented; AChR cluster fragment numbers in patient Ig-injected mice were 3- to 4-fold higher than control mice (6.4 ± 0.66 vs 1.7 ± 0.13; p < 0.01; Figure 4B). In addition, α-BTX staining area per NMJ was reduced (332 ± 41.9 vs 749 ± 73.7 μm2) (p < 0.01; Figure 4C) and AChR intensity decreased by 34% in patient Ig-injected mice (p < 0.01; Figure 4D), in agreement with reduced mEPP amplitudes. Moreover, total AChR area covered by axon terminals was 94.0% ± 1.3% in PBS-injected control mice, but reduced to 48.8% ± 6.1% in patient Ig-injected mice (p < 0.01; Figure 4E), indicating presynaptic deficits, in agreement with reduced mEPP frequency. Together, these results from structural and functional studies demonstrate that patient Ig impairs both pre- and postsynaptic structure of the NMJ.
Figure 4. Increased Neuromuscular Junction (NMJ) Fragment, Reduced Acetylcholine Receptor (AChR) Density, and Increased Denervation in the NMJ of Patient Immunoglobulin (Ig)–Injected Mice.

(A) z-Stack images of NMJs from phosphate-buffered saline (PBS)–injected control mice, healthy Ig-injected mice, and patient Ig-injected mice. Scale bars 10 μm. (B–E) Quantitative analysis of data. (B) Increased fragments of AChR clusters in patient Ig-injected mice, not in healthy Ig-injected mice, compared with PBS-injected control mice. (C) Reduced AChR area per NMJ in patient Ig-injected mice, not in healthy Ig-injected mice, compared with PBS-injected control mice. (D) Decreased AChR intensity in patient Ig-injected mice, not in healthy Ig-injected mice, compared with PBS-injected control mice. (E) Reduced overlap area of neurofilament-L/synapsin-1 and AChR staining in patient Ig-injected mice, not in healthy Ig-injected mice, compared with PBS-injected control mice. n = 3 mice per group. **p < 0.01.
Inhibition of AChR Cluster Formation by Patient Ig
To investigate molecular mechanisms of patient Ig, we determined the IgG subclass. As shown in Figure 5, A and B, the major isotype for anti-LRP4 and anti-agrin antibodies was IgG2, an isotype that is less able to activate the complement system.26 We next determined whether patient Ig interferes with the agrin/LRP4/MuSK signaling. Preincubation with patient Ig reduced phosphorylated MuSK in agrin-stimulated C2C12 myotubes compared with healthy Ig (Figure 5, C and D), indicating that patient Ig prevented agrin from activating MuSK (Figure 5, A and B). In accord, patient Ig blocked agrin-induced AChR cluster formation (Figure 5, E and F) compared with clusters in untreated myotubes or those pretreated with healthy IgG. These results suggested that patient Ig inhibited agrin/LRP4/MuSK signaling. Basal MuSK phosphorylation and AChR clusters (i.e., in the absence of agrin) were not changed by patient Ig (Figure 5, E and G), suggesting it may not alter basal MuSK activity or related AChR clustering.
Figure 5. Patient Immunoglobulin (Ig) Impaired Agrin-Elicited Muscle-Specific Tyrosine Kinase (MuSK) Activity and Acetylcholine Receptor (AChR) Clustering.

(A, B) IgG2 as the major isotype of anti-LRP4 and anti-Agrin antibodies in patient Ig. (C) Inhibition of MuSK activation by patient Ig. Phospho-MuSK elicited by agrin (C-terminal 22-KDa fragment) in myotubes treated without or with patient Ig or healthy Ig. (D) Quantitative analysis of data in C. (E–G) Inhibition of agrin-induced AChR clustering by patient Ig. Myotubes were treated without or with agrin LG3 in the presence or absence of patient or healthy Ig and assayed for AChR clusters. (E) Representative images. Scale bars: 50 μm. (F, G) Quantification of data in E. AChR clusters >4 μm in length were scored. n = 3, **p < 0.01.
β3 Domain of LRP4 and LG1/2 Domain of Agrin as Epitopes
Next, we attempted to identify the epitopes that patient Ig recognizes. LRP4-ECD and individual domains were tagged by Flag at N-terminus and by AP at C-terminus (Figure 6A). They were transfected in HEK293 cells, purified from condition medium, and incubated with commercial anti-LRP4 antibody or patient or healthy Ig that was immobilized on 96-well ELISA plates. After wash, AP activity was measured using p-nitrophenyl phosphate as a substrate. High AP activity was detected in wells with full-length LRP4-ECD and β3 domain, but not LDLa, β1, β2, or β4 domains, identifying the β3 domain as an epitope (Figure 6B). As control, healthy Ig did not recognize the β3 domain. To test this notion further, lysates from transfected HEK293 cells were incubated with patient or healthy Ig and then with protein A/G-immobilized beads. Bound proteins were probed with anti-Flag antibody. As shown in Figure 6C, β3 as well as full-length ECD was detected in the precipitates, again indicating β3 as the epitope.
Figure 6. Patient Immunoglobulin (Ig) Recognized β3 Domain of Low-Density Lipoprotein Receptor-Related Protein 4 (LRP4) and LG1 or LG2 Domain of Agrin.

(A) Schematic diagrams of LRP4 extracellular domain (ECD) and individual domains. (B, C) Identification of β3 domain as the epitope by ELISA (B) and by coprecipitation (C). (B) Healthy or patient Ig was immobilized on wells and incubated with alkaline phosphatase (AP)–tagged LRP4 proteins, and AP activity was assayed by ELISA. n = 3, **p < 0.01. (C) Lysates of HEK293 cells expressing respective LRP4 proteins were incubated with indicated Ig or anti-Flag antibody and bound proteins were detected by Western blotting. (D) Schematic diagrams of agrin constructs. (E) Agrin-LG123 and LG23, not agrin-LG3, bound patient Ig. n = 3.
The LG3 domain of agrin is required and sufficient to bind to the β1 domain of LRP4 whereas LRP4 via β3 domain interacts with and activates MuSK.27,28 To identify the epitope in agrin, we generated Flag-agrin-LG123, HA-agrin-LG23, and HA-agrin-LG3 in HEK293 cells (Figure 6D). The recombinant proteins were purified from condition medium and incubated with patient or healthy Ig, mouse anti-Flag, or anti-HA antibody. Antibody-bound agrin proteins were precipitated with protein A/G-immobilized beads and probed with anti-Flag or HA antibodies. As shown in Figure 6E, Flag-agrin-LG123 and HA-agrin-LG23, but not HA-agrin-LG3, were detected in the complex of patient Ig. Note that the expression of the 3 agrin proteins was evident as they were detected in precipitates by anti-Flag or HA antibodies (Figure 6E). These results indicate that the LG1 or LG2 domain, but not LG3 domain, are the epitopes of patient Ig.
Disruption of Agrin-Enhanced LRP4–MuSK Interaction by Patient Ig
Patient Ig did not recognize the β1 domain in LRP4 or LG3 of agrin (Figure 6, C and E, respectively), and thus was not expected to disrupt the interaction between LRP4 and agrin. To test this hypothesis, we incubated Flag-LRP4-ECD first with anti-Flag antibody–immobilized beads and then with HA-agrin LG3. As shown in eFigure 3 (available from Zenodo at doi.org/10.5281/zenodo.5143859), HA-agrin LG3 was detected in precipitates with beads, indicating that LG3 interacts with LRP4-ECD. The interaction was not altered by the addition of patient Ig, indicating that patient Ig had little effect on the LRP4–agrin interaction.
Upon agrin stimulation, the β3 domain of LRP4 was shown to interact with MuSK and thus activate MuSK.28 Knowing that patient IgG recognizes β3 (Figure 6, B and C), we determined whether it was able to disrupt LRP4–MuSK interaction, following a previous protocol.24 Flag-MuSK-ECD-IgG or protein A/G immobilized beads were incubated with LRP4-ECD-Myc without (control) or with agrin. The precipitate was probed with anti-Myc antibody for LRP4-ECD-Myc. As shown in Figure 7, A and B, agrin increases the amount of LRP4-ECD-Myc, indicative of increased LRP4–MuSK interaction. Interestingly, patient Ig reduced LRP4-ECD-Myc in the complex (Figure 7, C and D), indicating that patient Ig blocks the LRP4–MuSK interaction. This effect was not observed with healthy IgG (eFigure 4, A and B). Patient or healthy Ig had little effect on the LRP4–MuSK interaction in the absence of agrin (Figure 7, E and F, and eFigure 4, C and D). Together with the observation that β3 domain binds to patient Ig, these results support a working model that patient Ig acts by disrupting the LRP4–MuSK interaction and thus reducing agrin signaling.
Figure 7. Patient Immunoglobulin (Ig) Inhibited Agrin-Enhanced Low-Density Lipoprotein Receptor-Related Protein 4 (LRP4)–Muscle-Specific Tyrosine Kinase (MuSK) Interaction.
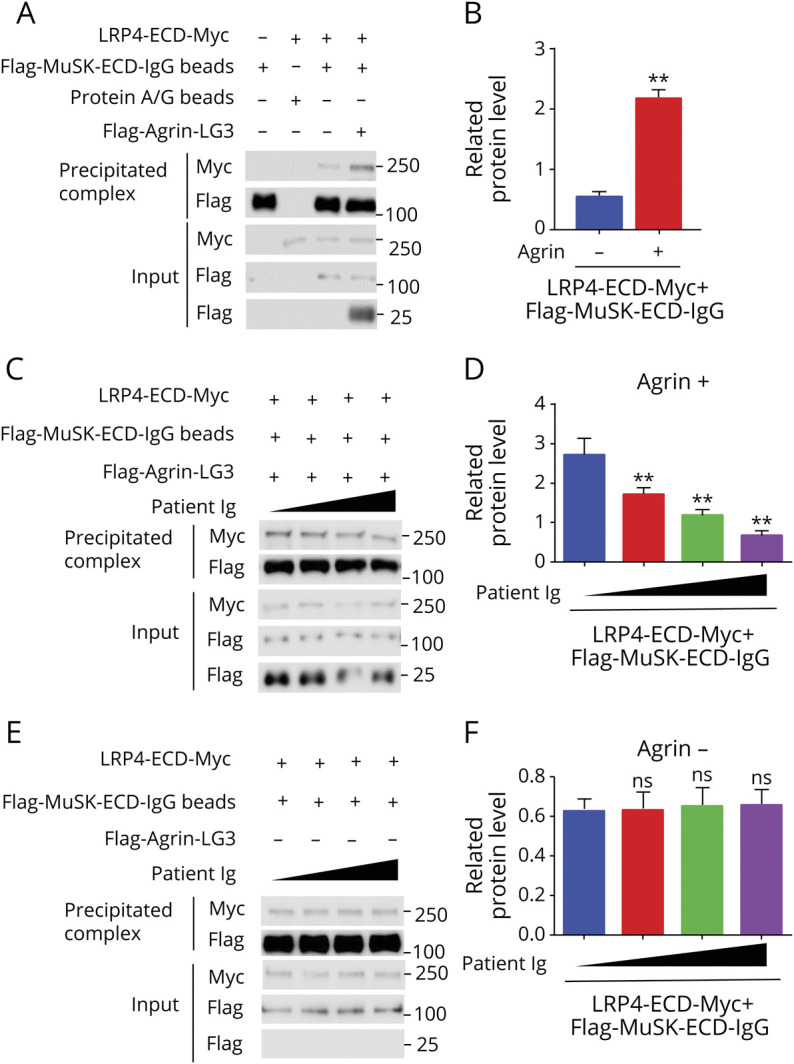
(A) Increased LRP4-ECD-MuSK interaction by agrin. Condition medium of HEK293 cells transfected with LRP4-ECD-Myc was incubated with Flag-MuSK-ECD IgG beads in the presence or absence of agrin LG3. LRP4-ECD-Myc in the precipitates with Flag-MuSK-ECD-IgG beads was revealed by Western blotting. (B) Quantification of data in A. (C) Inhibition of agrin-enhanced LRP4–MuSK interaction by patient Ig. (D) Quantitative analysis of data in C. (E) Little effect of patient Ig on basal LRP4–MuSK interaction. (F) Quantitative data in E. n = 3, *p < 0.05, **p < 0.01.
Discussion
LRP4 and agrin autoantibodies are detected in the sera of patients with double-seronegative (anti-AChR and anti-MUSK negative) MG (DNMG).12,13,15,29 Studies of active EAMG demonstrated that anti-LRP4 or agrin autoantibodies are pathogenic and able to impair NMJ structure or function.19,20,30 When anti-LRP4 antibodies generated in rabbits were transferred to mice, they caused MG-like symptoms. However, whether anti-LRP4 or agrin autoantibodies from patients with MG are pathogenic remains unclear. Here, we generated a passive EAMG mouse model by injecting Ig purified from a patient with DNMG who was positive for anti-LRP4/agrin antibodies. Our results demonstrated that patient Ig induced MG-like deficits by impairing the NMJ. In particular, patient Ig-injected mice exhibited signs of muscle weakness, fatigue, and weight loss, and reduced twitch and tetanus force (Figure 2). NMJs became fragmented, disorganized, and poorly innervated (Figure 4). Concomitantly, the neuromuscular transmission was compromised with reduced CMAPs, mEPPs, and EPPs (Figure 3). Mechanistically, patient Ig was able to interact with LRP4 and agrin protein (Figure 1, A–C), and recognized the NMJ in mice (Figure 1D) and inhibited agrin-elicited MuSK activation and AChR clustering (Figure 5, C–G). Epitope mapping studies demonstrated that the patient Ig recognized LRP4's β3 domain and agrin's LG1 or LG2 domain (Figure 6, B, C, and E). They inhibit the agrin signaling likely by preventing agrin-enhanced LRP4–MuSK interaction (Figure 7, C and D). Together, these observations demonstrated that anti-LRP4/agrin antibodies from the patient with MG are pathogenically causal to MG by inhibiting the agrin-LRP4-MuSK signaling.
Pathogenic autoimmune antibodies produce postsynaptic abnormalities by 3 mechanisms of action: complement activation, antigenic modulation, and functional binding inhibition.31,32 AChR antibody-positive MG may involve complement activation that causes membrane destruction at NMJ33 and antigenic modulation or functional binding inhibition.34,35 On the other hand, anti-MuSK antibodies, mostly of IgG4, damage the NMJ by blocking MuSK binding to ColQ (decreasing AChE in MNJ) and to LRP4 (decreasing AChR aggregation in NMJ).21,36-40 Anti-LRP4 antibodies from patients with MG were shown to block the agrin–LRP4 interaction,12 which decreases agrin-induced MuSK activity and AChR clustering in vitro.12,15,29 LRP4 antibodies in active EAMG mouse models diminished AChR clustering by decreasing agrin-induced MuSK activity and LRP4 surface expression and by mediating cell lysis by activating complement.19,41 These observations suggest that the pathologic mechanisms of anti-LRP4 antibodies could be complex and may involve not only functional binding inhibition, but also antigenic modulation and complement activation. Anti-agrin antibodies from patients with MG inhibit agrin-elicited MuSK phosphorylation and AChR clustering in myotubes.13,20 However, whether they are able to activate complement remains unclear. Here, the isotope of this patient with MG under study is mainly IgG2 (Figure 5, A and B), a subtype whose complement activation ability is less than that of IgG1 and IgG3, suggesting that complement activation may not be a major mechanism. Consistently, we showed that patient Ig impairs agrin-LPR4-MuSK signaling.
LRP4 has multiple domains in the ECD, an LDLa domain and 4 β-propeller domains, and several EGF-like motifs, 1 or 2 of which separate LDLα and the β1 propeller domain or between β-propeller domains.3,42 The first β-propeller domain interacts with the C-terminus of agrin to form a heterodimer; 2 such heterodimers form a tetramer that is critical to agrin signaling.27 On the other hand, the β3 propeller domain was shown to interact with and activate MuSK.28 Interestingly, the epitopes of the patient Ig are the β3 domain of LRP4 and a C-terminal 90-KDa fragment of agrin (Figure 6). Furthermore, patient Ig blocked the enhancement of the LRP4–MuSK interaction by C-terminal 22-KDa fragment of agrin (Figure 7, C and D), but had little effect on the LRP4–MuSK interaction in the absence of agrin (Figure 7, E and F). These observations suggest that the patient Ig impairs the NMJ by inhibiting agrin-induced AChR clustering by preventing the LRP4–MuSK interaction. Patient Ig was able to recognize the 90-KDa fragment, but not the LG3 domain (i.e., the C-terminal 22-KDa fragment) (Figure 6E), suggesting that the epitope in agrin resides outside of the minimal binding domain necessary for interaction with LRP4.27 In accord, patient Ig had little effect on agrin–LRP4 interaction (eFigure 3, A and B, available from Zenodo at doi.org/10.5281/zenodo.5143859). A parsimonious explanation of these results is that agrin-LRP4-MuSK signaling is disrupted by anti-LRP4 antibodies, but not anti-agrin antibodies, in MG serum. Intriguingly, an active EAMG model with neural agrin–LG3 induced anti-agrin antibodies that block the LG3–LRP4 interaction,20 suggesting a complex pathologic mechanism.
The passive EAMG mice displayed presynaptic deficits including reduced axon terminal staining at the NMJ, CMAP amplitudes at higher frequency, mEPP frequency, and PPR. The causes of these presynaptic deficits could be complex. Presynaptic deficits are observed in many mutant mice where critical NMJ genes are mutated such as rapsyn, LRP4, MuSK, and Dok7.3,43 They are also displayed in mutant mice when critical genes are specifically mutated in muscles, such as LRP4 and β-catenin. These deficits could occur as a mechanism to compensate postsynaptic deficits. Alternatively, they may also be caused by deficient retrograde signaling. For example, a stop signal is thought to be encoded by the agrin pathway so that motor nerve terminals could form the NMJ in the middle region of muscle fibers. Finally, LRP4 may be expressed in motor neurons at low levels,43 which may be a target of patient Ig.
Acknowledgment
The authors thank Guanglin Xing, PhD (Department of Neurosciences, Case Western Reserve University) for guidance on methods and Heath Robison, PhD (Department of Neurosciences, Case Western Reserve University) for discussion.
Glossary
- α-BTX
α-bungarotoxin
- ACh
acetylcholine
- AChR
acetylcholine receptor
- AP
alkaline phosphatase
- CMAP
compound muscle action potential
- DNMG
double seronegative (anti-AChR and anti-MUSK negative) myasthenia gravis
- EAMG
experimental autoimmune myasthenia gravis
- ECD
extracellular domain
- EPP
end plate potential
- HA
hemagglutinin
- Ig
immunoglobulin
- IRB
institutional review board
- LG3
laminin G-like 3 domain
- LRP4
low-density lipoprotein receptor-related protein 4
- MEPP
miniature end plate potential
- MG
myasthenia gravis
- MuSK
muscle-specific tyrosine kinase
- NMJ
neuromuscular junction
- PBS
phosphate-buffered saline
- PFA
paraformaldehyde
- PPR
paired-pulse ratio
- RT
room temperature
- SAS
saturated ammonium sulfate
Appendix. Authors
Footnotes
Editorial, page 463
Study Funding
Supported by NIH (5R01NS090083).
Disclosure
Dr. Z. Yu, Dr. Zhang, Dr. Jing, Dr. Chen, Dr. Cao, Dr. Pan, Dr. Luo, Y. Yu, B.M. Quarles, and Dr. Xiong report no disclosures relevant to the manuscript. Dr. Rivner has served as a consultant and received research support from Alexion and Allergan and received research support from UCB, Momenta, Shire Takeda, Orion, Biohaven, Catalyst, Seikagaku, Viela Bio, Apellis Pharmaceuticals, MediciNova, Millennium Pharmaceuticals, Anelixis Therapeutics, and RA Pharmaceutical. Dr. Mei reports no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Gilhus NE, Tzartos S, Evoli A, Palace J, Burns TM, Verschuuren J. Myasthenia gravis. Nat Rev Dis Primers. 2019;5(1):30. [DOI] [PubMed] [Google Scholar]
- 2.Conti-Fine BM, Milani M, Kaminski HJ. Myasthenia gravis: past, present, and future. J Clin Invest. 2006;116(11):2843-2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li L, Xiong WC, Mei L. Neuromuscular junction formation, aging, and disorders. Annu Rev Physiol. 2018;80:159-188. [DOI] [PubMed] [Google Scholar]
- 4.Lazaridis K, Tzartos SJ. Autoantibody specificities in myasthenia gravis: implications for improved diagnostics and therapeutics. Front Immunol 2020;11:212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gilhus NE, Verschuuren JJ. Myasthenia gravis: subgroup classification and therapeutic strategies. Lancet Neurol. 2015;14(10):1023-1036. [DOI] [PubMed] [Google Scholar]
- 6.Tzartos SJ, Barkas T, Cung MT, et al. Anatomy of the antigenic structure of a large membrane autoantigen, the muscle-type nicotinic acetylcholine receptor. Immunol Rev. 1998;163:89-120. [DOI] [PubMed] [Google Scholar]
- 7.Patrick J, Lindstrom J. Autoimmune response to acetylcholine receptor. Science. 1973;180(4088):871-872. [DOI] [PubMed] [Google Scholar]
- 8.Barik A, Lu Y, Sathyamurthy A, et al. LRP4 is critical for neuromuscular junction maintenance. J Neurosci. 2014;34(42):13892-13905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohkawara B, Shen X, Selcen D, et al. Congenital myasthenic syndrome-associated agrin variants affect clustering of acetylcholine receptors in a domain-specific manner. JCI Insight. 2020;5(7):e132023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ohkawara B, Cabrera-Serrano M, Nakata T, et al. LRP4 third beta-propeller domain mutations cause novel congenital myasthenia by compromising agrin-mediated MuSK signaling in a position-specific manner. Hum Mol Genet. 2014;23(10):1856-1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoch W, McConville J, Helms S, Newsom-Davis J, Melms A, Vincent A. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med. 2001;7(3):365-368. [DOI] [PubMed] [Google Scholar]
- 12.Zhang B, Tzartos JS, Belimezi M, et al. Autoantibodies to lipoprotein-related protein 4 in patients with double-seronegative myasthenia gravis. Arch Neurol. 2012;69(4):445-451. [DOI] [PubMed] [Google Scholar]
- 13.Zhang B, Shen C, Bealmear B, et al. Autoantibodies to agrin in myasthenia gravis patients. PLoS One. 2014;9(3):e91816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gasperi C, Melms A, Schoser B, et al. Anti-agrin autoantibodies in myasthenia gravis. Neurology. 2014;82(22):1976-1983. [DOI] [PubMed] [Google Scholar]
- 15.Rivner MH, Quarles BM, Pan JX, et al. Clinical features of LRP4/agrin-antibody-positive myasthenia gravis: a multicenter study. Muscle Nerve. 2020;62(3):333-343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bokoliya SC, Kumar VP, Nashi S, Polavarapu K, Nalini A, Patil SA. Anti-AChR, MuSK, and LRP4 antibodies coexistence: a rare and distinct subtype of myasthenia gravis from Indian subcontinent. Clin Chim Acta. 2018;486:34-35. [DOI] [PubMed] [Google Scholar]
- 17.Tsivgoulis G, Dervenoulas G, Tzartos SJ, Zompola C, Papageorgiou SG, Voumvourakis K. Double seropositive myasthenia gravis with acetylcholine receptor and lipoprotein receptor-related protein 4 antibodies. Muscle Nerve. 2014;49(6):930-931. [DOI] [PubMed] [Google Scholar]
- 18.Inoue H, Yamada K, Fujii A, et al. A patient with fulminant myasthenia gravis is seropositive for both AChR and LRP4 antibodies, complicated by autoimmune polyglandular syndrome type 3. Intern Med. 2020;59(17):2177-2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen C, Lu Y, Zhang B, et al. Antibodies against low-density lipoprotein receptor-related protein 4 induce myasthenia gravis. J Clin Invest. 2013;123(12):5190-5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan M, Liu Z, Fei E, et al. Induction of anti-agrin antibodies causes myasthenia gravis in mice. Neuroscience. 2018;373:113-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawakami Y, Ito M, Hirayama M, et al. Anti-MuSK autoantibodies block binding of collagen Q to MuSK. Neurology. 2011;77(20):1819-1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao K, Shen C, Lu Y, et al. Muscle yap is a regulator of neuromuscular junction formation and regeneration. J Neurosci. 2017;37(13):3465-3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aartsma-Rus A, van Putten M. Assessing functional performance in the mdx mouse model. J Vis Exp. 2014(85):51303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang B, Luo SW, Wang Q, Suzuki T, Xiong WC, Mei L. LRP4 serves as a coreceptor of agrin. Neuron. 2008;60(2):285-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Toyka KV, Brachman DB, Pestronk A, Kao I. Myasthenia gravis: passive transfer from man to mouse. Science. 1975;190(4212):397-399. [DOI] [PubMed] [Google Scholar]
- 26.Tao MH, Smith RI, Morrison SL. Structural features of human immunoglobulin G that determine isotype-specific differences in complement activation. J Exp Med. 1993;178(2):661-667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zong Y, Zhang B, Gu S, et al. Structural basis of agrin-LRP4-MuSK signaling. Genes Dev. 2012;26(3):247-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang W, Coldefy AS, Hubbard SR, Burden SJ. Agrin binds to the N-terminal region of Lrp4 protein and stimulates association between Lrp4 and the first immunoglobulin-like domain in muscle-specific kinase (MuSK). J Biol Chem. 2011;286(47):40624-40630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pevzner A, Schoser B, Peters K, et al. Anti-LRP4 autoantibodies in AChR- and MuSK-antibody-negative myasthenia gravis. J Neurol. 2012;259(3):427-435. [DOI] [PubMed] [Google Scholar]
- 30.Yan M, Xing GL, Xiong WC, Mei L. Agrin and LRP4 antibodies as new biomarkers of myasthenia gravis. Ann NY Acad Sci. 2018;1413(1):126-135. [DOI] [PubMed] [Google Scholar]
- 31.Gilhus NE, Skeie GO, Romi F, Lazaridis K, Zisimopoulou P, Tzartos S. Myasthenia gravis: autoantibody characteristics and their implications for therapy. Nat Rev Neurol. 2016;12(5):259-268. [DOI] [PubMed] [Google Scholar]
- 32.Hughes BW, Moro De Casillas ML, Kaminski HJ. Pathophysiology of myasthenia gravis. Semin Neurol. 2004;24(1):21-30. [DOI] [PubMed] [Google Scholar]
- 33.Lennon VA, Seybold ME, Lindstrom JM, Cochrane C, Ulevitch R. Role of complement in the pathogenesis of experimental autoimmune myasthenia gravis. J Exp Med. 1978;147(4):973-983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Drachman DB, Angus CW, Adams RN, Michelson JD, Hoffman GJ. Myasthenic antibodies cross-link acetylcholine receptors to accelerate degradation. N Engl J Med. 1978;298(20):1116-1122. [DOI] [PubMed] [Google Scholar]
- 35.Gomez CM, Richman DP. Anti-acetylcholine receptor antibodies directed against the alpha-bungarotoxin binding site induce a unique form of experimental myasthenia. Proc Natl Acad Sci USA. 1983;80(13):4089-4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McConville J, Farrugia ME, Beeson D, et al. Detection and characterization of MuSK antibodies in seronegative myasthenia gravis. Ann Neurol. 2004;55(4):580-584. [DOI] [PubMed] [Google Scholar]
- 37.Tsiamalos P, Kordas G, Kokla A, Poulas K, Tzartos SJ. Epidemiological and immunological profile of muscle-specific kinase myasthenia gravis in Greece. Eur J Neurol. 2009;16(8):925-930. [DOI] [PubMed] [Google Scholar]
- 38.Klooster R, Plomp JJ, Huijbers MG, et al. Muscle-specific kinase myasthenia gravis IgG4 autoantibodies cause severe neuromuscular junction dysfunction in mice. Brain. 2012;135(Pt 4):1081-1101. [DOI] [PubMed] [Google Scholar]
- 39.Niks EH, van Leeuwen Y, Leite MI, et al. Clinical fluctuations in MuSK myasthenia gravis are related to antigen-specific IgG4 instead of IgG1. J Neuroimmunol. 2008;195(1-2):151-156. [DOI] [PubMed] [Google Scholar]
- 40.Huijbers MG, Zhang W, Klooster R, et al. MuSK IgG4 autoantibodies cause myasthenia gravis by inhibiting binding between MuSK and Lrp4. Proc Natl Acad Sci USA. 2013;110(51):20783-20788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mori S, Motohashi N, Takashima R, Kishi M, Nishimune H, Shigemoto K. Immunization of mice with LRP4 induces myasthenia similar to MuSK-associated myasthenia gravis. Exp Neurol. 2017;297:158-167. [DOI] [PubMed] [Google Scholar]
- 42.Shen C, Xiong WC, Mei L. LRP4 in neuromuscular junction and bone development and diseases. Bone. 2015;80:101-108. [DOI] [PubMed] [Google Scholar]
- 43.Wu H, Lu Y, Shen C, et al. Distinct roles of muscle and motoneuron LRP4 in neuromuscular junction formation. Neuron. 2012;75(1):94-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data not published within the article are available on request. Supplemental data are available from Zenodo at doi.org/10.5281/zenodo.5143859.