

Abstract
BACKGROUND
Safe and effective long-acting injectable agents for preexposure prophylaxis (PrEP) for human immunodeficiency virus (HIV) infection are needed to increase the options for preventing HIV infection.
METHODS
We conducted a randomized, double-blind, double-dummy, noninferiority trial to compare long-acting injectable cabotegravir (CAB-LA, an integrase strand-transfer inhibitor [INSTI]) at a dose of 600 mg, given intramuscularly every 8 weeks, with daily oral tenofovir disoproxil fumarate–emtricitabine (TDF–FTC) for the prevention of HIV infection in at-risk cisgender men who have sex with men (MSM) and in at-risk transgender women who have sex with men. Participants were randomly assigned (1:1) to receive one of the two regimens and were followed for 153 weeks. HIV testing and safety evaluations were performed. The primary end point was incident HIV infection.
RESULTS
The intention-to-treat population included 4566 participants who underwent randomization; 570 (12.5%) identified as transgender women, and the median age was 26 years (interquartile range, 22 to 32). The trial was stopped early for efficacy on review of the results of the first preplanned interim end-point analysis. Among 1698 participants from the United States, 845 (49.8%) identified as Black. Incident HIV infection occurred in 52 participants: 13 in the cabotegravir group (incidence, 0.41 per 100 person-years) and 39 in the TDF–FTC group (incidence, 1.22 per 100 person-years) (hazard ratio, 0.34; 95% confidence interval, 0.18 to 0.62). The effect was consistent across prespecified subgroups. Injection-site reactions were reported in 81.4% of the participants in the cabotegravir group and in 31.3% of those in the TDF–FTC group. In the participants in whom HIV infection was diagnosed after exposure to CAB-LA, INSTI resistance and delays in the detection of HIV infection were noted. No safety concerns were identified.
CONCLUSIONS
CAB-LA was superior to daily oral TDF–FTC in preventing HIV infection among MSM and transgender women. Strategies are needed to prevent INSTI resistance in cases of CAB-LA PrEP failure. (Funded by the National Institute of Allergy and Infectious Diseases and others; HPTN 083 ClinicalTrials.gov number, NCT02720094.)
Despite the availability of highly effective strategies for the prevention of human immunodeficiency virus (HIV) infection, the number of new infections worldwide continues to exceed 5000 per day.1 Daily oral tenofovir disoproxil fumarate–emtricitabine (TDF–FTC) has been reported to provide protection against HIV infection across various populations.2–7 The efficacy of oral preexposure prophylaxis (PrEP) agents is directly correlated with adherence to prescribed dosing.4–9 PrEP agents that do not require regular or planned oral dosing may increase acceptability and protection during periods of risk, thereby reducing the risk of HIV acquisition.
A Quick Take is available at NEJM.org
Long-acting injectable cabotegravir (CAB-LA) is an integrase strand-transfer inhibitor (INSTI) that is administered as an intramuscular injection into the gluteus muscle. CAB-LA has potent anti-HIV activity, protects nonhuman primates from a broad range of HIV exposure types, and is generally safe, without limiting toxic effects, in humans.10–15 (See the Supplementary Appendix, available with the full text of this article at NEJM.org.)
We report the primary results of the HIV Prevention Trials Network (HPTN) 083 trial, a phase 2b–3, multicenter, randomized, double-blind, double-dummy, active-controlled trial. We compared the safety and efficacy of CAB-LA, administered intramuscularly every 8 weeks, with daily oral TDF–FTC in cisgender men who have sex with men (MSM) and in transgender women who have sex with men in the United States, Latin America, Asia, and Africa.
METHODS
TRIAL OVERSIGHT AND PARTICIPANTS
The HPTN 083 trial protocol, which is available at NEJM.org, was approved by the institutional review board, ethics committee, ministry of health, or a combination of these entities at each participating site. All the participants provided written informed consent. Full details of the trial design can be found in the trial protocol. The Division of AIDS of the National Institute of Allergy and Infectious Diseases provided regulatory sponsorship of the trial. The Division of AIDS was responsible for clinical monitoring of the trial. ViiV Healthcare and Gilead Sciences donated trial medications and matching placebos. ViiV Healthcare also provided additional funding and contributed to the design of the trial.
Eligible participants were adults (≥18 years of age) who were in general good health as determined by clinical and laboratory assessments and who had a negative HIV serologic test at enrollment, had an undetectable blood HIV RNA viral load within 14 days before trial entry, and had a creatinine clearance of 60 ml or more per minute.16 Cisgender MSM and transgender women who have sex with men who were recruited for the trial were at high risk for HIV infection, as defined in the protocol. Key exclusion criteria were the use of illicit intravenous drugs within 90 days before enrollment, previous participation in the active treatment group of an HIV vaccine trial, coagulopathy, buttock implants or fillers, a seizure disorder, or a corrected QT interval of greater than 500 msec. Participants who had positive results on a hepatitis B virus surface antigen test or hepatitis C virus antibody test were also excluded.
RANDOMIZATION AND TRIAL PROCEDURES
Randomization was stratified according to site and was performed with the use of permuted blocks of 8, 10, and 12; trial-group assignments occurred electronically at enrollment. Eligible participants were assigned, in a 1:1 ratio, to receive either active cabotegravir with TDF–FTC placebo (cabotegravir group) or active TDF–FTC with cabotegravir placebo (TDF–FTC group).
The trial included three phases: an oral-tablet lead-in phase, an injection phase, and a “tail phase,” the time period beginning 8 weeks after the final injection and continuing for approximately 48 weeks, when plasma drug concentrations are in terminal decline. During the lead-in phase, all the participants received two oral tablets (one active and one placebo) daily for 5 weeks to verify the safety of the tablets. Active cabotegravir was given as a 30-mg tablet, and active TDF–FTC was given as a fixed-dose combination of 300 mg of TDF plus 200 mg of FTC. Participants who had at least 50% adherence to the oral tablets, as determined by pill count, and had acceptable safety laboratory results were permitted to progress to injections. During the injection phase, participants received a supply of daily oral tablets and a 3-ml intramuscular injection on inception of this phase, 4 weeks after the beginning of the phase, and every 8 weeks thereafter. CAB-LA was administered as a single 3-ml injection containing 600 mg of cabotegravir. Placebo for CAB-LA was an injectable fat emulsion (20% intralipid solution) that was visually similar to CAB-LA. Active TDF–FTC was given as described above. Masked oral tablets were dispensed at enrollment, and masked tablets were dispensed and injections administered at weeks 5, 9, and 17 and every 8 weeks thereafter through week 153, thus resulting in approximately 3 years of exposure to CAB-LA. If a planned injection visit was delayed by 8 weeks or more, a 4-week interval was used for the next two injections. Participants who discontinued injections received open-label TDF–FTC for 48 weeks to provide ongoing HIV PrEP during the tail phase.17
Trial visits were scheduled at weeks 2 and 4 during the oral-tablet lead-in phase. Visits during which no injections were administered and only safety was assessed were scheduled at weeks 6 and 10, and then 2 weeks after each injection. Beginning with the last scheduled visit of the injection phase (week 153), participants were provided with open-label TDF–FTC daily over 48 weeks, with four quarterly trial visits (Fig. 1A).
Figure 1. Trial Design, Screening, Randomization, and Follow-up.

CAB denotes cabotegravir, CAB-LA long-acting injectable CAB, HIV human immunodeficiency virus, PK pharmacokinetic, and TDF–FTC tenofovir disoproxil fumarate–emtricitabine.
All the visits included an HIV rapid test (cleared by the Food and Drug Administration) and a laboratory-based HIV antigen and antibody test, assessment of adverse events, collection and storage of plasma samples, and adherence and risk-reduction counseling. (Further details are provided in the Supplementary Appendix.)18 Routine laboratory testing was performed at all visits except the week 5 visit. Injection-site reactions were assessed at visits that occurred 1 or 2 weeks after each injection. Testing for syphilis and nucleic acid amplification testing for rectal and urethral gonorrhea and chlamydia were conducted at least every 6 months. Testing for hepatitis C virus antibodies and measurement of fasting glucose and lipid levels were performed annually. Interviewer-led and computer-assisted structured interviews were conducted approximately every second injection cycle for evaluation of adherence, sexual behaviors, alcohol and drug use, and acceptability of the oral tablets and injections. Among participants in whom HIV infection had been confirmed at the trial site, measurement of the CD4+ T-cell count and the HIV viral load for clinical use was performed at quarterly visits for 1 year after diagnosis. Additional HIV testing was performed retrospectively at the HPTN Laboratory Center to confirm HIV status and determine the timing of HIV infection; this testing included assessment of HIV resistance for selected samples with a viral load of 500 copies or more per milliliter.18
PRIMARY END POINTS
The primary efficacy end point of the trial was incident HIV infection. The methods used for determination of HIV status and for end-point adjudication are described in the Supplementary Appendix; additional information is provided in a separate report.18 An independent committee, whose members were unaware of the trial-group assignments, adjudicated HIV infections and determined the dates of first evidence of infection.18 The primary safety end point was the occurrence of an adverse event of grade 2 or higher. The severity of all adverse events was graded according to the Division of AIDS Table for Grading the Severity of Adult and Pediatric Adverse Events, corrected version 2.1.19
ANALYSIS OF DRUG CONCENTRATIONS
A cohort of 390 randomly selected participants in the TDF–FTC group was assessed for measurement of the tenofovir concentration in plasma and the intraerythrocytic tenofovir–diphosphate concentration in dried blood spots.18 Plasma cabotegravir concentrations were measured in all samples obtained from all the participants in the cabotegravir group who had HIV infection (either incident or prevalent).18 For participants in the TDF–FTC group who had incident HIV infection, tenofovir and tenofovir diphosphate concentrations were measured in samples obtained at the first visit at which the participant was determined to be HIV positive and at selected previous visits.17
STATISTICAL ANALYSIS
The noninferiority margin was a hazard ratio of 1.23, which was chosen on the basis of previous placebo-controlled trials. (Details are provided in the protocol.) To achieve 90% power to detect an alternative hazard ratio of 0.75 (at a one-sided type I error rate of 0.025), we estimated that 172 incident HIV infections (events) would need to occur. Assuming an incidence of 1.75 events per 100 person-years, we enrolled approximately 4500 participants. Interim analyses were planned to be performed with the use of O’Brien–Fleming group-sequential monitoring boundaries after 43, 86, and 129 events had occurred. The primary end point was evaluated in a modified intention-to-treat analysis, which excluded participants who were found to have HIV infection at enrollment. All incident infections were included in the analysis, regardless of when the infection occurred and regardless of whether the participant received an injection. Cox regression, stratified according to geographic region, was used to estimate the hazard ratio for incident HIV infection in the cabotegravir group as compared with the TDF–FTC group; 95% confidence intervals and P values were based on the Wald statistic. The primary hazard ratio was adjusted for interim monitoring.20 A test for proportional hazards was performed with the use of Schoenfeld residuals, and a log-rank test, stratified according to geographic region, was performed as a sensitivity analysis.
Trial data were reviewed approximately every 6 months by an independent data and safety monitoring board. On May 14, 2020, on review of the results of the first preplanned interim end-point analysis, the data and safety monitoring board concluded that the results met the prespecified criteria for stopping the trial on the basis of efficacy (Supplementary Appendix). The analyses in this report include data collected during the blinded phase of the trial up to May 14, 2020. The trial is currently ongoing with an open-label design.
RESULTS
TRIAL PARTICIPANTS
Between December 6, 2016, and March 16, 2020, a total of 6333 participants were screened at 43 sites in the United States, Latin America, Asia, and Africa (Fig. 1B and the Supplementary Appendix). Of the 4570 participants who underwent randomization and were enrolled, 4 were ineligible, resulting in an intention-to-treat population of 4566 participants. Demographic and clinical characteristics of the two groups were similar at baseline (Table 1) and met or exceeded prespecified metrics for enrollment of certain subgroups and populations. Five participants who acquired HIV infection before enrollment (2 in the cabotegravir group and 3 in the TDF–FTC group) and 71 participants who had no follow-up HIV testing after enrollment were excluded from the primary efficacy analysis.
Table 1.
Demographic and Clinical Characteristics of the Participants at Baseline.*
| Characteristic | Overall (N = 4566) | Cabotegravir Group (N = 2282) | TDF-FTC Group (N = 2284) |
|---|---|---|---|
| Cohort — no. (%) | |||
| MSM | 3992 (87.4) | 2013 (88.2) | 1979 (86.6) |
| Transgender women who have sex with men | 570 (12.5) | 266 (11.7) | 304 (13.3) |
| Participant preferred not to answer | 4 (0.1) | 3 (0.1) | 1 (<0.1) |
| Age category — no. (%) | |||
| 18–29yr | 3080 (67.5) | 1572 (68.9) | 1508 (66.0) |
| 30–39yr | 1048 (23.0) | 498 (21.8) | 550 (24.1) |
| 40–49 yr | 315 (6.9) | 145 (6.4) | 170 (7.4) |
| 50–59yr | 110 (2.4) | 60 (2.6) | 50 (2.2) |
| ≥60 yr | 13 (0.3) | 7 (0.3) | 6 (0.3) |
| Median age (IQR) — yr | 26 (22–32) | 26 (22–32) | 26 (22–32) |
| Latinx or Hispanic ethnic group, according to geographic region — no./total no. (%)† | |||
| United States | |||
| Yes | 303/1698 (17.8) | 149/849 (17.6) | 154/849 (18.1) |
| No | 1394/1698 (82.1) | 700/849 (82.4) | 694/849 (81.7) |
| Data missing | 1/1698 (<0.1) | 0 | 1/849 (0.1) |
| Latin America | |||
| Yes | 1806/1964 (92.0) | 894/980 (91.2) | 912/984 (92.7) |
| No | 158/1964 (8.0) | 86/980 (8.8) | 72/984 (7.3) |
| SexPro score, according to geographic region — no./total no. (%)‡ | |||
| United States | |||
| ≤16 | 1447/1698 (85.2) | 729/849 (85.9) | 718/849 (84.6) |
| >16 | 251/1698 (14.8) | 120/849 (14.1) | 131/849 (15.4) |
| Latin America | |||
| ≤16 | 1675/1964 (85.3) | 825/980 (84.2) | 850/984 (86.4) |
| >16 | 289/1964 (14.7) | 155/980 (15.8) | 134/984 (13.6) |
| Geographic region — no. (%) | |||
| United States | 1698 (37.2) | 849 (37.2) | 849 (37.2) |
| Latin America | |||
| Argentina | 337 (7.4) | 169 (7.4) | 168 (7.4) |
| Brazil | 796 (17.4) | 395 (17.3) | 401 (17.6) |
| Peru | 831 (18.2) | 416 (18.2) | 415 (18.2) |
| Asia | |||
| Thailand | 553 (12.1) | 275 (12.1) | 278 (12.2) |
| Vietnam | 199 (4.4) | 100 (4.4) | 99 (4.3) |
| Africa | 152 (3.3) | 78 (3.4) | 74 (3.2) |
| Race, according to geographic region — no./total no. (%)† | |||
| United States | |||
| Black | 845/1698 (49.8) | 411/849 (48.4) | 434/849 (51.1) |
| Non-Black | 851/1698 (50.1) | 437/849 (51.5) | 414/849 (48.8) |
| Data missing | 2/1698 (0.1) | 1/849 (0.1) | 1/849 (0.1) |
| Latin America | |||
| Black or mixed | 392/1964 (20.0) | 198/980 (20.2) | 194/984 (19.7) |
| Indigenous | 862/1964 (43.9) | 435/980 (44.4) | 427/984 (43.4) |
| Asian | 8/1964 (0.4) | 6/980 (0.6) | 2/984 (0.2) |
| White | 659/1964 (33.6) | 319/980 (32.6) | 340/984 (34.6) |
| Other | 43/1964 (2.2) | 22/980 (2.2) | 21/984 (2.1) |
| Asia | |||
| Asian | 749/752 (99.6) | 374/375 (99.7) | 375/377 (99.5) |
| Other | 3/752 (0.4) | 1/375 (0.3) | 2/377 (0.5) |
| Africa | |||
| Black | 119/152 (78.3) | 62/78 (79.5) | 57/74 (77.0) |
| Other | 5/152 (3.3) | 2/78 (2.6) | 3/74 (4.1) |
| Mixed | 28/152 (18.4) | 14/78 (17.9) | 14/74 (18.9) |
| Marital status — no. (%) | |||
| Married, civil union, or legal partnership | 177 (3.9) | 79 (3.5) | 98 (4.3) |
| Living with primary or main partner | 292 (6.4) | 138 (6) | 154 (6.7) |
| Have primary or main partner, not living together | 335 (7.3) | 171 (7.5) | 164 (7.2) |
| Single, divorced, or widowed | 3751 (82.2) | 1888 (82.7) | 1863 (81.6) |
| Other | 11 (0.2) | 6 (0.3) | 5 (0.2) |
| Educational level — no. (%) | |||
| No schooling | 8 (0.2) | 2 (0.1) | 6 (0.3) |
| Primary school | 70 (1.5) | 28 (1.2) | 42 (1.8) |
| Secondary school | 1012 (22.2) | 490 (21.5) | 522 (22.9) |
| Technical training | 375 (8.2) | 187 (8.2) | 188 (8.2) |
| College or university or higher | 3101 (67.9) | 1575 (69.0) | 1526 (66.8) |
Percentages may not sum to 100 because of rounding. IQR denotes interquartile range, MSM men who have sex with men, and TDF–FTC tenofovir disoproxil fumarate–emtricitabine.
Race and ethnic group were reported by the participant.
SexPro is a Web-based tool that provides a sexual health promotion score. It is validated to predict the 6-month risk of HIV acquisition on the basis of sexual behaviors, sexual networks, substance use, history of sexually transmitted infections, race or ethnic group (United States only), and age. Additional background on the SexPro tool is provided in the protocol. Scores range from 1 to 20, with higher scores indicating a lower risk of acquiring HIV infection.
Participant retention was 86.5% at 1 year, with a median follow-up of 1.4 years (interquartile range [IQR], 0.8 to 1.9). Masked oral tablets and injections were permanently discontinued in 908 participants (19.9%), including 445 participants in the cabotegravir group and 463 in the TDF–FTC group (Table S1 in the Supplementary Appendix).
ADHERENCE TO ORAL TABLETS AND INJECTIONS
The median adherence during the oral-tablet lead-in phase, as determined by pill count, was 96.6% (IQR, 89.7 to 100.0) across the trial groups. During the course of the trial, in a randomly selected subgroup of 390 participants in the TDF–FTC group, 74.2% had tenofovir concentrations of greater than 40 ng per milliliter, which is consistent with the receipt of daily TDF–FTC doses in the previous week; 86.0% had concentrations above the lower limit of quantitation (0.31 ng per milliliter). Tenofovir–diphosphate concentrations (measured in dried blood spots) that were consistent with receipt of at least four TDF–FTC doses per week21 over the previous 1 to 2 months were detected in 72.3% of the samples overall (Fig. S1); 91.5% of person-years were considered to have been “covered” by injectable CAB-LA placebo, which was defined as injections having been received with a delay of less than 2 weeks.
HIV INFECTION
Using a prespecified testing algorithm,17 we identified HIV infection in 57 participants, including 5 (2 in the cabotegravir group and 3 in the TDF–FTC group) who had undetected HIV infection at enrollment (“baseline” infections). A total of 52 participants who acquired HIV infection after enrollment were included in the prespecified primary efficacy analysis: 13 in the cabotegravir group (incidence, 0.41 per 100 person-years) and 39 in the TDF–FTC group (incidence, 1.22 per 100 person-years). The hazard ratio for incident HIV infection in the cabotegravir group as compared with the TDF–FTC group was 0.34 (95% confidence interval [CI], 0.18 to 0.62; P<0.001). The test for proportional hazards had modest evidence of nonproportionality (P = 0.07); a stratified log-rank test showed a significant difference between the groups (P<0.001). The direction and overall magnitude of the effect were consistent across prespecified subgroups and populations (Fig. 2) and in a per-protocol analysis (Fig. S2).
Figure 2. Incident HIV infection.
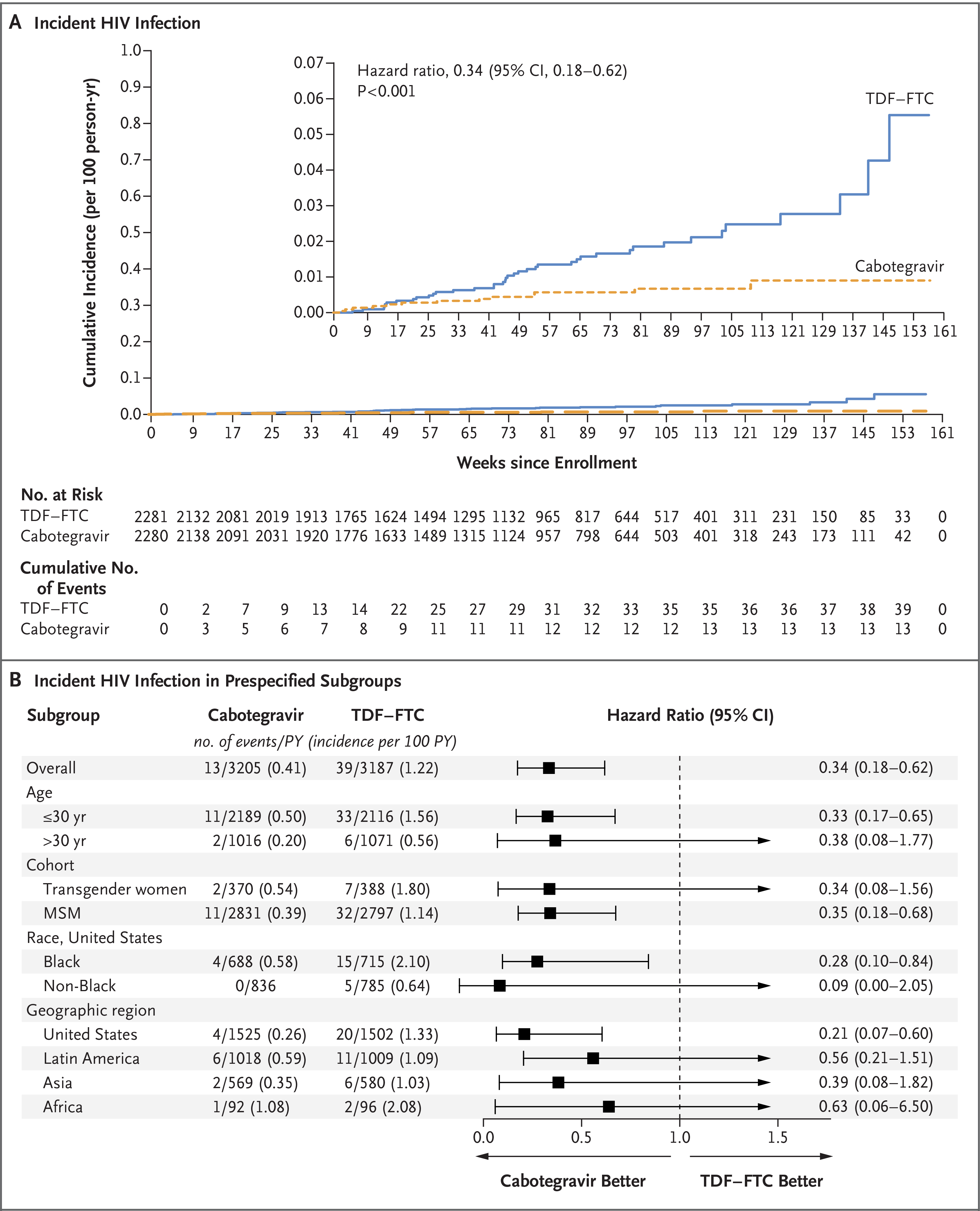
Panel A shows Kaplan–Meier estimates of incident HIV infection. The inset shows the same data on an enlarged y axis. Panel B shows hazard ratios for incident HIV infection in the prespecified subgroups. Race was reported by the participant. MSM denotes men who have sex with men, and PY person-years.
Post hoc centralized testing of stored plasma samples resulted in readjudication of the timing of the first visit at which an HIV test was positive for 2 participants in the cabotegravir group; 1 of the original 13 incident HIV infections in the cabotegravir group was readjudicated as a base line infection (case A3) (Fig. 3). No incident infections in the TDF–FTC group were readjudicated as baseline infections. An analysis that included the post hoc readjudication data provided a revised estimate of incident HIV infection in the cabotegravir group of 0.37 (95% CI, 0.19 to 0.65; hazard ratio, 0.32 [95% CI, 0.16 to 0.58]). One additional case (A4) (Fig. 3) in the cabotegravir group was also identified post hoc as a baseline infection; thus, the final number of observed infections was 58 (16 in the cabotegravir group and 42 in the TDF–FTC group). Detailed descriptions of each case, including virologic and pharmacologic assessments, are presented in a separate report.17 The timing of the first visit at which an HIV test was positive for each of the 16 infections in the cabotegravir group is shown in Figure 3.
Figure 3. Pharmacologic and Virologic Data for HIV Infections in the Cabotegravir Group.
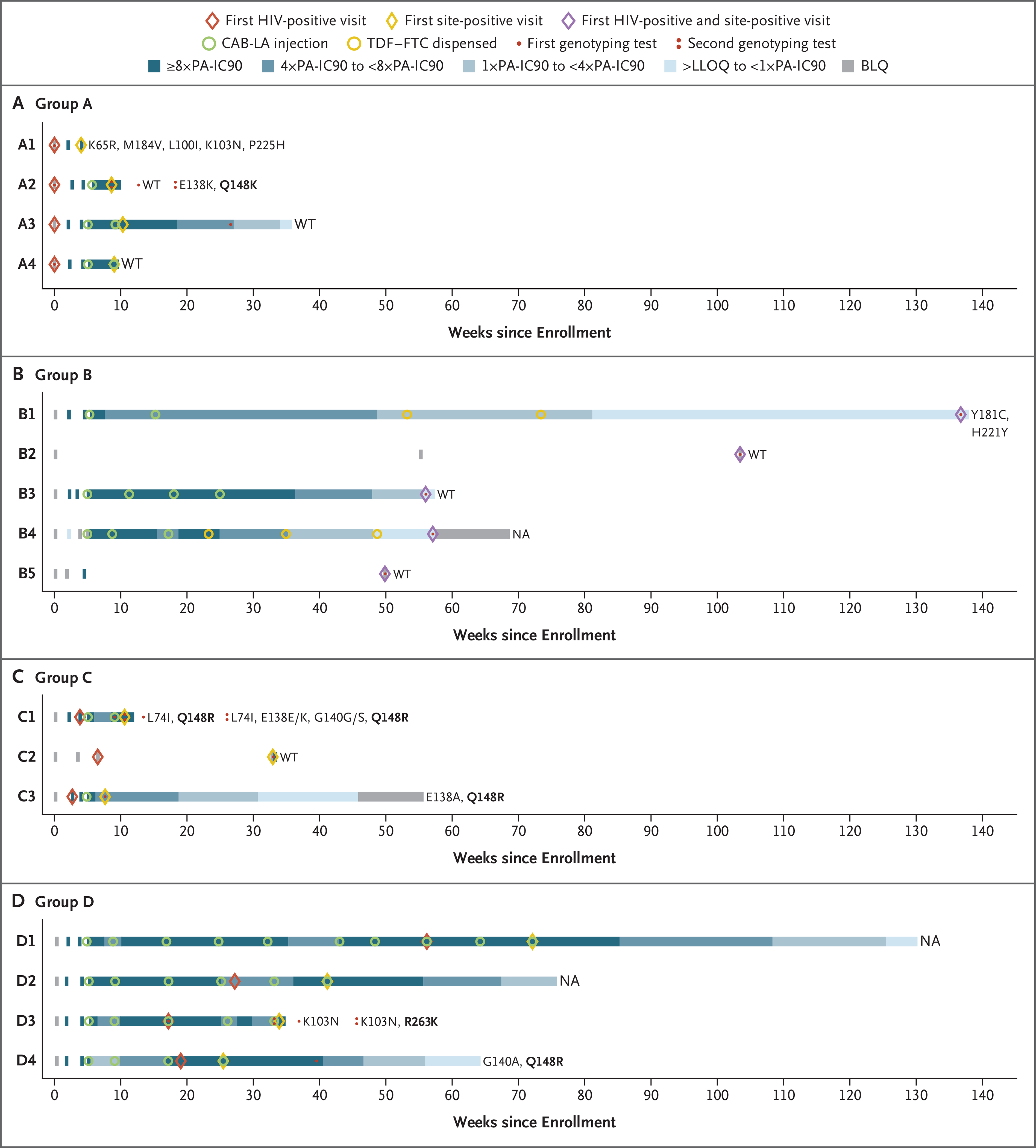
Panels A through D show the timing of key events for the 16 infections that occurred in the cabotegravir group. These infections were classified into four groups: group A (Panel A) includes infections that occurred before enrollment; group B (Panel B) includes infections that occurred with no recent exposure to cabotegravir; group C (Panel C) includes infections that occurred before cabotegravir injection; and group D (Panel D) includes infections that occurred in participants with appropriately timed CAB-LA doses and expected plasma cabotegravir concentrations. The “first HIV-positive visit” refers to the first visit at which the participant was determined to be HIV positive. The “first site-positive visit” refers to the first visit at which evidence of HIV infection was identified at the trial site. HIV genotyping results are shown to the right of each horizontal bar. Major resistance mutations are shown for nucleoside or nucleotide reverse-transcriptase inhibitors (K65R and M184V) and nonnucleoside reverse-transcriptase inhibitors (L100I, K103N, Y181C, H221Y, and P225H). All integrase strand-transfer inhibitor (INSTI) resistance mutations are shown (L74I, E138K or E138E/K, E138A, G140A, G140G/S, Q148R or Q148K, and R263K); major INSTI mutations are shown in bold text. Genotyping results are shown for the first visit at which the viral load was 500 copies or more per milliliter and for follow-up visits at which such a viral load occurred, denoted as one dot or two dots, respectively. The number of days between the first HIV-positive visit and the visit with additional mutations was 60 days for case A2, 10 days for case C1, and 112 days for case D3. NA indicates that the genotyping result was not available (either no visit occurred at which viremia was found to be sufficient for performance of the genotyping assay or the assay failed to produce a genotyping result). BLQ denotes below the limit of quantification, LLOQ lower limit of quantification, PA-IC90 protein-adjusted 90% cabotegravir inhib itory concentration, and WT wild type.
The 16 infections in the cabotegravir group were classified into four groups (A through D) (Fig. 3). Four infections occurred before enrollment (cases A1 through A4). Five infections occurred with no recent exposure to cabotegravir (cases B1 through B5); in 2 of these cases (B1 and B4), open-label TDF–FTC was initiated after the participant had discontinued CAB-LA. Three infections occurred before cabotegravir injection (cases C1 through C3); in 1 of these cases (C2), the participant was nonadherent to oral cabotegravir, and in the other 2 cases (C1 and C3), infection occurred during the oral-tablet lead-in phase. The remaining 4 infections occurred in participants with appropriately timed CAB-LA doses and expected plasma cabotegravir concentrations (cases D1 through D4).
DRUG RESISTANCE MUTATIONS
INSTI resistance mutations were detected in 1 of the 4 cases identified as a baseline infection (case A2) and in 4 of 9 incident cases that had a resistance test result (cases C1, C3, D3, and D4). No resistance was detected when viral escape or HIV acquisition occurred during the period of cabotegravir decay after the last injection (tail phase; cases A3, B1, B3, and B4) (Fig. 3), although varying intervals of follow-up before initiation of antiretroviral therapy (ART) do not allow direct comparison. In the TDF–FTC group, 2 of the 39 incident HIV infections occurred in cases in which the drug concentrations that were measured were consistent with good PrEP adherence (cases E16 and E34). Four incident infections and 2 baseline infections had nucleoside or nucleotide reverse-transcriptase inhibitor mutations (K65R, M184V, M184I, or a mixture of M184V and M184I with or without nonnucleoside reverse-transcriptase inhibitor [NNRTI] mutations; cases E3, E13, E16, E25, E41, and E42) (Figs. S3 and S4).18
SAFETY
The primary safety population (which comprised participants who had received at least one dose of any of the oral tablets or injections) included 4562 participants (2280 in the cabotegravir group and 2282 in the TDF–FTC group). Adverse events of grade 2 or higher were reported in 92.5% of the participants overall, with no marked difference in the overall frequency between the trial groups (Table 2). Adverse events of grade 3 or higher were reported in 1494 participants (32.7%) overall; the overall frequency of these events was similar in the two trial groups. Permanent discontinuation of the oral tablets or both oral tablets and injections owing to adverse events other than injection-site reactions occurred in 172 participants (3.8%) overall, with a similar overall frequency in the two groups.
Table 2.
Adverse Events.*
| Event | Overall (N = 4562) | Cabotegravir Group (N = 2280) | TDF-FTC Group (N = 2282) |
|---|---|---|---|
| number of participants (percent) | |||
| Any adverse event of grade 2 or higher | 4222 (92.5) | 2106 (92.4) | 2116 (92.7) |
| Most common adverse events of grade 2 or higher† | |||
| Decreased creatinine clearance | 3257 (71.4) | 1588 (69.6) | 1669 (73.1) |
| Increased creatine kinase | 953 (20.9) | 484 (21.2) | 469 (20.6) |
| Nasopharyngitis‡ | 844 (18.5) | 447 (19.6) | 397 (17.4) |
| Increased serum creatinine | 810 (17.8) | 382 (16.8) | 428 (18.8) |
| Upper respiratory infection‡ | 516 (11.3) | 257 (11.3) | 259 (11.3) |
| Musculoskeletal discomfort‡ | 513 (11.2) | 254 (11.1) | 259 (11.3) |
| Increased lipase | 509 (11.2) | 249 (10.9) | 260 (11.4) |
| Headache‡ | 458 (10.0) | 237 (10.4) | 221 (9.7) |
| Increased aspartate aminotransferase | 384 (8.4) | 188 (8.2) | 196 (8.6) |
| Increased alanine aminotransferase | 351 (7.7) | 159 (7.0) | 192 (8.4) |
| Increased blood glucose | 322 (7.1) | 199 (8.7) | 123 (5.4) |
| Increased amylase | 317 (6.9) | 148 (6.5) | 169 (7.4) |
| Diarrhea‡ | 313 (6.9) | 152 (6.7) | 161 (7.1) |
| Rash‡ | 260 (5.7) | 116 (5.1) | 144 (6.3) |
| Hypoglycemia‡ | 246 (5.4) | 120 (5.3) | 126 (5.5) |
| Pyrexia | 185 (4.1) | 122 (5.4) | 63 (2.8) |
| Any adverse event of grade 3 or higher | 1494 (32.7) | 727 (31.9) | 767 (33.6) |
| Most common adverse events of grade 3 or higher§ | |||
| Increased creatine kinase | 633 (13.9) | 324 (14.2) | 309 (13.5) |
| Decreased creatinine clearance | 349 (7.7) | 159 (7.0) | 190 (8.3) |
| Increased serum creatinine | 156 (3.4) | 80 (3.5) | 76 (3.3) |
| Increased lipase | 152 (3.3) | 76 (3.3) | 76 (3.3) |
| Increased aspartate aminotransferase | 122 (2.7) | 53 (2.3) | 69 (3.0) |
| Increased alanine aminotransferase | 55 (1.2) | 23 (1.0) | 32 (1.4) |
| Serious adverse event | 241 (5.3) | 120 (5.3) | 121 (5.3) |
| Adverse events of special interest | |||
| Seizure | 7 (0.2) | 2 (0.1) | 5 (0.2) |
| Liver-related adverse event resulting in discontinuation of oral tablets or both oral tablets and injections | 95 (2.1) | 47 (2.1) | 48 (2.1) |
Included are only adverse events that were assigned Medical Dictionary for Regulatory Activities, version 23.1 (MedDRA) terms by clinical staff. Injection-site reactions and sexually transmitted infections are not included. Inappropriately enrolled participants and participants who did not receive any oral trial drug are excluded. In cases in which a participant had multiple events with the same MedDRA term, only one event is counted.
Only adverse events that were reported in at least 5% of the participants in either trial group are shown.
This adverse event category combines multiple MedDRA terms that were too similar to report individually.
Only adverse events that were reported in at least 1% of the participants in either trial group are shown.
Serious adverse events were reported in 241 participants (5.3%) overall and were balanced between the two groups. Additional adverse events of interest included seizures and liver-related adverse events resulting in discontinuation of the oral tablets or both oral tablets and injections; both were uncommon, and the overall frequency of such events was similar in the two groups. A total of 11 participants died (7 in the TDF–FTC group and 4 in the cabotegravir group; hazard ratio, 0.57 [95% CI, 0.17 to 1.96]) (Table S2); 1 death in the TDF–FTC group that resulted from cardiovascular disease was considered to be related to the oral tablets or injections.
INJECTION-SITE REACTIONS
Injection-site reactions were reported in 1724 participants (81.4%) in the cabotegravir group who received at least one injection and in 652 participants (31.3%) in the TDF–FTC group who received at least one placebo injection. Injection-site reactions were mostly mild or moderate in severity and decreased in frequency over time (Fig. S5). Of the 2117 participants who received at least one active CAB-LA injection, 50 (2.4%) permanently discontinued the injections owing to an injection-related adverse event; discontinuation was strongly associated with increased severity of the injection-site reactions (Table S3). Of 10,666 injection-site reactions in the cabote gravir group, 6486 (60.8%) were pain and 2530 (23.7%) were tenderness; the events began a median of 1 day (IQR, 0 to 2) after injection and lasted a median of 3 days (IQR, 2 to 6) (Fig. S6).
SEXUALLY TRANSMITTED INFECTIONS
The overall incidence of new rectal or urethral gonorrhea was 13.49 per 100 person-years, and the incidence of new rectal or urethral chlamydia was 21.36 per 100 person-years. The incidence of new syphilis infections, which were centrally adjudicated (details provided in the Supplementary Appendix), was 16.69 per 100 person-years. The occurrence of each of these infections was similar in the two groups (Tables S4 and S5).
CHANGES IN BODY WEIGHT AND METABOLIC VARIABLES
In a post hoc analysis, a mean annualized increase in body weight of 1.23 kg per year (95% CI, 1.05 to 1.42) was noted in the cabotegravir group, as compared with an increase of 0.37 kg (95% CI, 0.18 to 0.55) in the TDF–FTC group. Differences in weight change between the groups were observed primarily in the first 40 weeks of participation and were similar in the two groups later in the trial (Fig. S12). Changes in fasting glucose and fasting lipid levels are shown in Figures S7 through S11.
DISCUSSION
In the HPTN 083 trial, we compared the safety and efficacy of CAB-LA with that of TDF–FTC for the prevention of HIV infection in MSM and transgender women who have sex with men. Although TDF–FTC is known to be effective in preventing HIV infection when adherence is high, we found that the risk of HIV infection was lower by 66% in the cabotegravir group than in the TDF–FTC group in the prespecified analysis, a result that showed the superiority of CAB-LA to TDF–FTC. This finding was similar in magnitude and direction in key populations that have lower reported rates of adherence to daily oral PrEP agents, including Black MSM in the United States, transgender women, and participants younger than 30 years of age.
Although most of the participants in the cabotegravir group reported injection-site reactions, only 2.4% chose not to receive further injections as a result. Increases in body weight associated with the use of INSTIs in the treatment of HIV infection have been reported in the literature.22–24 We previously observed no significant difference in weight gain between HIV-uninfected participants who received CAB-LA and those who received placebo; participants in both groups gained approximately 1 kg over the course of 41 weeks. In the current trial, differences in weight change between the two groups were driven by weight loss in the TDF–FTC group in year 1; thereafter, the weight changes were similar (approximately 1 kg per year in both groups).
CAB-LA provided protection against HIV acquisition; however, 4 incident infections were observed despite on-time injections and expected cabotegravir concentrations in plasma. The risk of PrEP failure may be influenced by low plasma cabotegravir concentrations between initial injections, low cabotegravir concentrations in rectal tissue,25 rectal inflammation related to sexually transmitted infection, or a combination of these factors.26–30 INSTI resistance was detected in 5 participants in the cabotegravir group (1 with baseline infection and 4 with incident infection). NNRTI-based or boosted protease inhibitor–based ART should suppress viral replication in such cases, absent transmitted resistance; viral replication in some such participants may be suppressed by a regimen that includes twice-daily dolutegravir. To reduce the risk of resistance, HIV assays with increased sensitivity are being evaluated to minimize the interval between HIV infection and diagnosis; ART should be started promptly when long-acting PrEP agents have been used.
CAB-LA provided greater protection against HIV infection than TDF–FTC among MSM and transgender women who have sex with men. A similarly designed randomized, controlled trial evaluating the efficacy of CAB-LA in cisgender women in Africa was recently unblinded early after showing superiority of CAB-LA over daily oral TDF–FTC (ClinicalTrials.gov number, NCT03164564); the safety and acceptability of CAB-LA in adolescents is under evaluation (NCT04692077 and NCT04824131). Although overall adherence to daily oral TDF–FTC was higher than anticipated in the current trial, inadequate TDF–FTC adherence among some participants appeared to drive the overall finding of HIV incidence that was three times the incidence in the cabotegravir group18; daily oral TDF–FTC, when used as prescribed, has been estimated to be more than 99% protective.8
This trial showed that CAB-LA was superior to TDF–FTC in preventing HIV acquisition among MSM and transgender women who have sex with men. The logistics involved in implementation of the use of CAB-LA for PrEP will require new consideration. CAB-LA is an effective strategy for the prevention of HIV infection that will expand PrEP options.
Supplementary Material
Acknowledgments
Supported by the National Institute of Allergy and Infectious Diseases, Office of the Director, National Institutes of Health, the National Institute of Mental Health, the National Institute on Drug Abuse, and the Eunice Kennedy Shriver Institute for Child Health and Human Development, under award numbers UM1AI068619 (HIV Prevention Trials Network [HPTN] Leadership and Operations Center), UM1AI068617 (HPTN Statistical and Data Management Center), and UM1AI068613 (HPTN Laboratory Center). ViiV Healthcare and Gilead Sciences donated trial medications and matching placebos, and ViiV Healthcare provided additional funding.
We thank the participants in the HPTN 083 trial, without whom this work would not have been possible; Dr. Sheryl Zwerski for her expertise, leadership, support, and guidance with respect to the National Institutes of Health–sponsored cabotegravir prevention development initiative; Dr. Wafaa El-Sadr for her leadership and continued support; Drs. Grace Aldrovandi and Daniel Kuritzkes for their independent review of the virologic data for the HPTN 083 trial; Ms. Kathy Hinson and Ms. Sarah Stone for their financial and budgetary support, oversight, and guidance; Ms. Jackie Talley for her administrative assistance; the HPTN Black Caucus for preparatory training, engagement, and support; and our partners and colleagues at ViiV Healthcare and Gilead Sciences. (See the Supplementary Appendix.)
Footnotes
The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Centers for Disease Control and Prevention.
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
A data sharing statement provided by the authors is available with the full text of this article at NEJM.org.
Contributor Information
R.J. Landovitz, Center for Clinical AIDS Research and Education, David Geffen School of Medicine, University of California, Los Angeles, California
D. Donnell, Fred Hutchinson Cancer Research Center, Seattle, Brazil
M.E. Clement, Louisiana State University Health Sciences Center, New Orleans, Brazil
B. Hanscom, Fred Hutchinson Cancer Research Center, Seattle, Brazil
L. Cottle, Fred Hutchinson Cancer Research Center, Seattle, Brazil
L. Coelho, Instituto Nacional de Infectologia Evandro Chagas-Fiocruz, Rio de Janeiro, Brazil
R. Cabello, Via Libre, Peru
S. Chariyalertsak, Research Institute for Health Sciences, Chiang Mai University, Chiang Mai, Thailand
E.F. Dunne, Division of HIV–AIDS Prevention, Centers for Disease Control and Prevention
I. Frank, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania
J.A. Gallardo-Cartagena, Universidad Nacional Mayor de San Marcos, Peru
A.H. Gaur, St. Jude Children's Research Hospital, Memphis, TN, North Carolina
P. Gonzales, Asociacion Civil Impacta Salud y Educacion, Lima, Peru
H.V. Tran, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina
J.C. Hinojosa, Asociacion Civil Selva Amazonica, Iquitos, Peru
E.G. Kallas, University of São Paulo, Brazil
C.F. Kelley, School of Medicine, Emory University, Atlanta
M.H. Losso, Hospital General de Agudos José María Ramos Mejia, Buenos Aires
J.V. Madruga, Centro de Referência e Treinamento DST–AIDS-SP, São Paulo, Brazil
K. Middelkoop, Institute of Infectious Disease and Molecular Medicine, University of Cape Town, Cape Town, South Africa
N. Phanuphak, Institute of HIV Research and Innovation, Bangkok, Thailand
B. Santos, Hospital Nossa Senhora da Conceição, Porto Alegre, Brazil
O. Sued, Fundación Huésped, Buenos Aires
J. Valencia Huamaní, Asociacion Civil Impacta Salud y Educacion, Lima, Peru.
E.T. Overton, University of Alabama at Birmingham, Birmingham
S. Swaminathan, Rutgers New Jersey Medical School, Newark
C. del Rio, School of Medicine, Emory University, Atlanta Rollins School of Public Health, Emory University, Atlanta.
R.M. Gulick, Weill Cornell Medicine, New York
P. Richardson, Johns Hopkins University, Baltimore
P. Sullivan, Johns Hopkins University, Baltimore
E. Piwowar-Manning, Johns Hopkins University, Baltimore
M. Marzinke, Johns Hopkins University, Baltimore
C. Hendrix, Johns Hopkins University, Baltimore
M. Li, Fred Hutchinson Cancer Research Center, Seattle, Brazil
Z. Wang, Fred Hutchinson Cancer Research Center, Seattle, Brazil
J. Marrazzo, University of Alabama at Birmingham, Birmingham
E. Daar, Lundquist Institute at Harbor–UCLA Medical Center, Torrance, California
A. Asmelash, FHI 360, Durham, North Carolina
T.T. Brown, Johns Hopkins University, Baltimore
P. Anderson, University of Colorado Anschutz Medical Campus, Aurora
S.H. Eshleman, Johns Hopkins University, Baltimore
M. Bryan, FHI 360, Durham, North Carolina
C. Blanchette, FHI 360, Durham, North Carolina
J. Lucas, FHI 360, Durham, North Carolina
C. Psaros, Massachusetts General Hospital, Boston
S. Safren, University of Miami, Coral Gables, FL
J. Sugarman, Johns Hopkins University, Baltimore
H. Scott, San Francisco Department of Public Health, San Francisco, California
J.J. Eron, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina
S.D. Fields, Pennsylvania State University, State College, Pennsylvania
N.D. Sista, FHI 360, Durham, North Carolina
K. Gomez-Feliciano, FHI 360, Durham, North Carolina
A. Jennings, FHI 360, Durham, North Carolina
R.M. Kofron, Center for Clinical AIDS Research and Education, David Geffen School of Medicine, University of California, Los Angeles, California
T.H. Holtz, Office of AIDS Research, Rockville, MD
K. Shin, Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Rockville, MD
J.F. Rooney, Gilead Sciences, Foster City, California
K.Y. Smith, ViiV Healthcare, Research Triangle, North Carolina
W. Spreen, ViiV Healthcare, Research Triangle, North Carolina
D. Margolis, ViiV Healthcare, Research Triangle, North Carolina
A. Rinehart, ViiV Healthcare, Research Triangle, North Carolina
A. Adeyeye, Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Rockville, MD
M.S. Cohen, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina
M. McCauley, FHI 360, Durham, North Carolina
B. Grinsztejn, Instituto Nacional de Infectologia Evandro Chagas-Fiocruz, Rio de Janeiro, Brazil
REFERENCES
- 1.2020 global AIDS update — seizing the moment — tackling entrenched inequalities to end epidemics. Geneva: Joint United Nations Programme on HIV/AIDS, July6, 2020. (https://www.unaids.org/en/resources/documents/2020/global-aids-report). [Google Scholar]
- 2.Mayer KH, Molina J-M, Thompson MA, et al. Emtricitabine and tenofovir alafenamide vs emtricitabine and tenofovir disoproxil fumarate for HIV pre-exposure prophylaxis (DISCOVER): primary results from a randomised, double-blind, multicentre, active-controlled, phase 3, non-inferiority trial. Lancet 2020;396239–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Molina JM, Capitant C, Spire B, et al. On-demand preexposure prophylaxis in men at high risk for HIV-1 infection. N Engl J Med 2015;373:2237–46. [DOI] [PubMed] [Google Scholar]
- 4.Grant RM, Lama JR, Anderson PL, et al. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med 2010;363:2587–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baeten JM, Donnell D, Ndase P, et al. Antiretroviral prophylaxis for HIV prevention in heterosexual men and women. N Engl J Med 2012;367:399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thigpen MC, Kebaabetswe PM, Paxton LA, et al. Antiretroviral preexposure prophylaxis for heterosexual HIV transmission in Botswana. N Engl J Med 2012;367: 423–34. [DOI] [PubMed] [Google Scholar]
- 7.Choopanya K, Martin M, Suntharasamai P, et al. Antiretroviral prophylaxis for HIV infection in injecting drug users in Bangkok, Thailand (the Bangkok Tenofovir study): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2013;381:2083–90. [DOI] [PubMed] [Google Scholar]
- 8.Grant RM, Anderson PL, McMahan V, et al. Uptake of pre-exposure prophylaxis, sexual practices, and HIV incidence in men and transgender women who have sex with men: a cohort study. Lancet Infect Dis 2014;14:820–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marrazzo JM, Ramjee G, Richardson BA, et al. Tenofovir-based preexposure prophylaxis for HIV infection among African women. N Engl J Med 2015;372:509–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Müller RH, Gohla S, Keck CM. State of the art of nanocrystals — special features, production, nanotoxicology aspects and intracellular delivery. Eur J Pharm Biopharm 2011;78:1–9. [DOI] [PubMed] [Google Scholar]
- 11.Andrews CD, Spreen WR, Mohri H, et al. Long-acting integrase inhibitor protects macaques from intrarectal simian/ human immunodeficiency virus. Science 2014;343:1151–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andrews CD, Bernard LS, Poon AY, et al. Cabotegravir long acting injection protects macaques against intravenous challenge with SIVmac251. AIDS 2017;31: 461–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dobard C, Makarova N, Nishiura K, et al. Long-acting cabotegravir protects macaques against repeated penile simian-human immunodeficiency virus exposures. J Infect Dis 2020;222:391–5. [DOI] [PubMed] [Google Scholar]
- 14.Markowitz M, Frank I, Grant RM, et al. Safety and tolerability of long-acting cabotegravir injections in HIV-uninfected men (ECLAIR): a multicentre, double-blind, randomised, placebo-controlled, phase 2a trial. Lancet HIV 2017;4(8):e31–e340. [DOI] [PubMed] [Google Scholar]
- 15.andovitz RJ, Li S, Grinsztejn B, et al. Safety, tolerability, and pharmacokinetics of long-acting injectable cabotegravir in low-risk HIV-uninfected individuals: HPTN 077, a phase 2a randomized controlled trial. PLoS Med 2018;1(11):e002690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron 1976;1:3–41. [DOI] [PubMed] [Google Scholar]
- 17.Landovitz RJ, Li S, Eron JJ Jr, et al. Tail-phase safety, tolerability, and pharmacokinetics of long-acting injectable cabotegravir in HIV-uninfected adults: a secondary analysis of the HPTN 077 trial. Lancet HIV 2020;7(7):e472–e481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marzinke MA, Grinsztejn B, Fogel JM, et al. Characterization of HIV infection in cisgender men and transgender women who have sex with men receiving injectable cabotegravir for HIV prevention: HPTN 083. J Infect Dis 2021March19 (Epub ahead of print). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Department of Health and Human Services, National Institutes of Health, National Institute of Allergy and Infectious Diseases. Division of AIDS (DAIDS) table for grading the severity of adult and pediatric adverse events, corrected version 2.1. National Institutes of Health, July2017. (https://rsc.niaid.nih.gv/sites/default/files/daidsgradingcorrectedv21.pdf). [Google Scholar]
- 20.Emerson SS, Fleming TR. Parameter estimation following group sequential hypothesis testing. Biometrika 1990;77: 875–92. [Google Scholar]
- 21.Castillo-Mancilla JR, Zheng J-H, Rower JE, et al. Tenofovir, emtricitabine, and tenofovir diphosphate in dried blood spots for determining recent and cumulative drug exposure. AIDS Res Hum Retroviruses 2013;29:384–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sax PE, Erlandson KM, Lake JE, et al. Weight gain following initiation of antiretroviral therapy: risk factors in randomized comparative clinical trials. Clin Infect Dis 2020;71:1379–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bourgi K, Rebeiro PF, Turner M, et al. Greater weight gain in treatment-naive persons starting dolutegravir-based antiretroviral therapy. Clin Infect Dis 2020;70: 1267–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Venter WDF, Moorhouse M, Sokhela S, et al. Dolutegravir plus two different prodrugs of tenofovir to treat HIV. N Engl J Med 2019;381:803–15. [DOI] [PubMed] [Google Scholar]
- 25.Spreen W, Ford SL, Chen S, et al. GSK1265744 pharmacokinetics in plasma and tissue after single-dose long-acting injectable administration in healthy subjects. J Acquir Immune Defic Syndr 2014; 67:481–6. [DOI] [PubMed] [Google Scholar]
- 26.Hoornenborg E, Prins M, Achterbergh RCA, et al. Acquisition of wild-type HIV-1 infection in a patient on pre-exposure prophylaxis with high intracellular concentrations of tenofovir diphosphate: a case report. Lancet HIV 2017;4(11):e522–e528. [DOI] [PubMed] [Google Scholar]
- 27.Tansey C, Zhao C, Hopkins A, et al. A nonhuman primate model for rectally transmitted syphilis. J Infect Dis 2018; 217:1139–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Solomon MM, Mayer KH, Glidden DV, et al. Syphilis predicts HIV incidence among men and transgender women who have sex with men in a preexposure prophylaxis trial. Clin Infect Dis 2014;59: 1020–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murnane PM, Celum C, Mugo N, et al. Efficacy of preexposure prophylaxis for HIV-1 prevention among high-risk heterosexuals: subgroup analyses from a randomized trial. AIDS 2013;27:2155–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barbee LA, Khosropour CM, Dombrowksi JC, Golden MR. New human immunodeficiency virus diagnosis independently associated with rectal gonorrhea and chlamydia in men who have sex with men. Sex Transm Dis 2017;44: 385–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.