

Abstract
Background and aims
Advances in genomic technologies have led to increasing reports of monogenic inflammatory bowel disease (IBD). However, there has not been a detailed evaluation of the characteristics of the published cases of monogenic IBD. Here we systematically review the literature to determine the clinical features, genetic profile, and previously used treatments strategies in monogenic IBD.
Methods
A systematic review of MEDLINE articles published between January 2000 and December 2020 was conducted. Seven hundred and fifty individual monogenic IBD cases were identified from 303 eligible articles.
Results
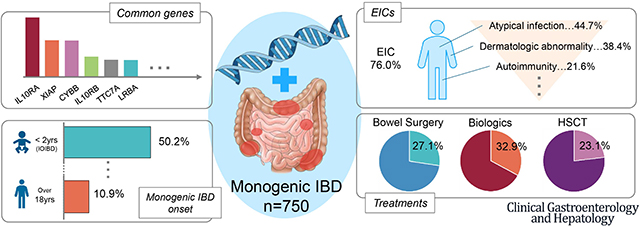
The most frequently reported monogenic IBD genes were IL10RA/B, XIAP, CYBB, LRBA and TTC7A. In total, 63.4% of patients developed IBD before six years of age, 17.4% between ages 10 and 17.9 years, and 10.9% after 18 years of age. There was substantial difference between these age groups and the underlying monogenic disorders. Only 31.7% had any history of extra-intestinal comorbidity (EIC) before IBD onset but, 76.0% developed at least one EIC during their clinical course. The most common EICs were atypical infection (44.7%), dermatologic abnormality (38.4%), and autoimmunity (21.9%). Bowel surgery, biologic therapy, and hematopoietic stem cell transplantation (HSCT) were performed in 27.1%, 32.9%, and 23.1% of patients, respectively.
Conclusion
Monogenic IBD cases while rare have varied extra-intestinal comorbidities and limited treatments options including surgery and transplant. Early identification and improved understanding of the characteristics of the genes and underlying disease processes in monogenic IBD is important for effective management.
Keywords: IBD, VEOIBD, monogenic disorder, whole exome sequence, pediatric
Graphical Abstract
Introduction
Inflammatory bowel disease (IBD) is a disease of intractable chronic intestinal inflammation that is classified based on clinical characteristics as Crohn’s disease (CD), ulcerative colitis (UC), and IBD-unclassified (1). Genetic factors, abnormal immune responses associated with environmental predisposition, and microbial dysbiosis in the gut are thought to cause IBD (2). Advances in next-generation DNA sequencing (NGS) technology have made it possible to genetically diagnose patients with IBD and IBD-like disease. These monogenic forms of IBD are typically rare, severe, and refractory to conventional therapies (3, 4). IBD and IBD-like diseases that are caused by monogenic variants with Mendelian inheritance patterns have been described as “monogenic IBD” in contrast to typical idiopathic (or “polygenic”) IBD. Age of onset, family history, atypical endoscopic findings, severity, atypical infection history, and extra-intestinal comorbidity (EIC) are factors that may help distinguishing monogenic from polygenic IBD (5). To date, between 80 and 90 causative genes associated with monogenic IBD have been reported. Recently, the Pediatric IBD Porto group of ESPGHAN agreed on a consensus list of 75 monogenic IBD genes (6).
The number of reported cases of monogenic IBD has increased in recent years. There are several high quality reviews of very early onset IBD (VEOIBD) that help guide clinical practice (7–9). However, these reviews focus on IBD diagnosis prior to 6 years of age, rather than on monogenic IBD cases. Although there is overlap, with a high frequency of monogenic IBD in this population, the monogenic IBD population is likely unique with separate clinical characteristics and management strategies. Also, there are no detailed reports containing a comprehensive picture of monogenic IBD with specific numbers on age distribution, proportion of EICs, relationship of EICs to age of onset, or detailing the clinical course of monogenic IBD. Since each gene-specific cause of monogenic IBD is rare, it is difficult to collect cases in a single study even with international collaborative cohort studies such as COLORS, Care-for-Rare, NEOPICS.org and VEOIBD.org. To this end, we conducted a systematic review of all published cases of monogenic IBD to review these clinical questions.
Methods
Search Strategy
A comprehensive search of MEDLINE limited to English language was performed using PubMed (http://pubmed.ncbi.nlm.gov/). The following search terms were used: gene names, protein names, protein abbreviations, disease names or disease abbreviations from 75 genes respectively, and the combination of “IBD”, “inflammatory bowel disease”, “colitis”, “Crohn” or “enteropathy” (see Supplemental Table 1 for a full list of the search strings used).
Study selection
The inclusion criteria were: (1) peer-reviewed article, (2) articles including clinical information of monogenic IBD case or cases, (3) articles published between January 2000 and December 2020, (4) cases in which genetic testing was definitively performed, and (5) cases in which GI manifestations were directly marked as IBD or IBD-like by the authors (including, “ulcerative colitis”, “Crohn disease”, “intestinal Bechet” and “granulomatous colitis”). The exclusion criteria were: (1) articles not in English, (2) review articles with previously published cases, (3) articles not containing the details of individual cases (e.g., a study showing only number of patients with IBD in a large cohort of one monogenic disorder or intestinal disease was not described as IBD), and (4) interventional clinical trials. Monogenic IBD was defined as (1) caused by a gene variant that were (2) damaging and rare, (3) known to cause a monogenic disorder from previous reports and/or (4) consistent with the patient’s clinical phenotype. Articles were initially screened according to the title and abstract to exclude irrelevant studies. Full-text manuscripts were then evaluated for the inclusion and exclusion criteria. Simultaneously, the reference lists of all full-text articles were manually reviewed for further eligible articles. This process was performed for each gene. Finally, we removed duplicated articles.
Data Extraction and Quality Assessment
Data were collected and organized in a spreadsheet. The following data were collected for each selected article: name of the first author, article year, country of the corresponding author, journal name, sex, family history of inflammatory bowel disease (within first or second degree relatives), consanguinity, age of onset of first manifestation, age of IBD onset, IBD subtype (UC, CD or IBD-U), if IBD or EICs developed first, GI complications, atypical infection history (including recurrent or severe infection), EICs during clinical course (including atypical infection), modality that first identified the genetic mutation, genetic variant, inheritance pattern, identified variant type and treatment history (including biologics, surgery, and hematopoietic stem cell transplantation [HSCT]). “Atypical infection” included recurrent infection, severe infection, and atypical infection type (such as fungal infection). Finally, cases that appeared in more than one article were excluded based on the genetic variant, onset age of IBD and name of authors. Two authors independently evaluated this procedure.
Statistical Analysis
Descriptive data were calculated as percentages for discrete findings. Chi-square or Fisher exact tests were used for group comparison. We considered P values less than 0.05 as statistically significant. Statistical analyses were performed using PRISM version 8.0 (GraphPad Software, San Diego, USA) and EZR (Jichi Medical University, Saitama, Japan).
Results
Article Selection through Systematic Review
The initial electronic database search identified 11,702 articles, of which 10,930 were excluded after screening their titles and abstracts (Supplemental Figure 1). Subsequently, 772 articles were identified. Additionally, 415 articles were selected from the reference list of the initial 772 studies. From those 1,187 articles, 448 were excluded for not including or demonstrating IBD, 87 for review not showing new cases, 74 for basic research not including clinical cases, 74 for not showing monogenic IBD, 63 for not performing genetic tests, and 53 for not containing sufficient individual clinical information after full-text screening. After screening, 388 articles remained. Of these, 85 articles were excluded because they summarized data from cases of two or more genes causative of monogenic IBD. Overall, 303 articles matched the inclusion criteria for this systematic review. These articles covered 68 of the 75 genes known to be associated with monogenic IBD. For the remaining 7 described monogenic genes, the articles did not meet the inclusion criteria. Regarding 6 of these genes (ADA, IL2RA, IL2RB, ITCH, POLA1, and RAG2), several articles described GI symptoms as “enteropathy” or “chronic colitis” (10–12), but the cases were not described definitively as IBD through explanations of endoscopy or pathology. Also, there were no published full reports of STXBP3 causing monogenic IBD during the review period, only a published abstract (13).
There has been a dramatic increase in the number of articles reporting monogenic IBD cases since 2012 (Supplementary Figure 2). This is presumably because of increased awareness and widespread use of NGS. The use of whole-exome sequencing (WES) increased after approximately 2012, and the utilization of targeted gene-panel sequencing (TGPS) increased after approximately 2017. The use of whole genome sequencing (WGS) in this population remains uncommon but appears to be increasing.
Basic Case Characteristics
The 303 articles included a total of 955 cases of monogenic IBD, originating from 31 different countries (Supplemental Figure 3). However, 205 cases appeared in two or more articles. Thus, after removing repeated cases, 750 individual patients with monogenic IBD were analyzed (472 males, 259 females, 19 with gender not reported). 173 of 750 cases (23.1%) were shown to have family history of IBD in first or second degree relatives, and 163 cases (21.7%) reported known consanguinity (Supplemental Table 2). Within each article, 34% were labeled as Crohn’s disease, 6% as UC, and 49.5% as IBD (see Supplemental Table 3 for details on the clinical subtype frequency for each monogenic disorder).
Figure 1A shows the number of reported monogenic IBD cases and articles per causative gene for the 17 most frequently reported genes (see Supplemental Figure 4 for details on the number of all 68 genes reported). The most commonly reported gene was IL10RA (124 cases in 34articles), followed by XIAP (69 in 33 articles), CYBB (68 in 24 articles), IL10RB (33 in 21 articles), LRBA (31 cases in 16 articles) and TTC7A (31 in 11 articles).
Figure 1. Common genes and age of onset of monogenic IBD.

(A) Number of reported monogenic IBD cases and articles stratified by gene. Only genes that have ten or more cases are listed. Dark blue; number of cases, Light blue; number of articles.
(B) Distribution of IBD onset age. Orange bar (left Y axis) is the number of cases in each age group. The line graph (right Y axis) is the proportion of patients with extraintestinal manifestations before onset of IBD.
Figure 1B shows the distribution of age of IBD onset and the proportion with a history of EICs prior to the onset of IBD in each age group. Infantile onset IBD (IOIBD; from 28 days to less than 2 years of age) was the most common group (n = 181, 28.2%), and 431 of 679 cases (63.4%) had developed IBD prior to the age of six (for 71 cases, onset age was not provided). On the other hand, 17.4% and 10.9% of patients developed IBD between 10 and 17.9 years and after 18 years of age, respectively. Of 606 cases, 192 (31.7%) had EICs before IBD onset (clear information about EICs onset was not provided for 144 cases). Almost all patients in the neonatal IBD group (98.6%) and the IOIBD group (82.6%) had no EICs before the onset of IBD. However, more than half of the patients diagnosed with monogenic IBD over two years of age had EICs before the onset of IBD. There was no significant difference between primary EIC onset rate and any of the groups over the age of two.
Underlying Monogenic Disorders
The most often reported underlying monogenic disorder was IL10-signalling colitis (164 cases; involving the IL10, IL10RA and IL10RB genes), followed by chronic granulomatous disease (CGD; 99 cases; involving the CYBA, CYBB, NCF1, NCF2 and NCF4 genes) and XIAP deficiency (69 cases; involving the XIAP gene) (Supplemental Figure 5). The age of onset of IBD for each monogenic disorder is shown in Figure 2. Some underlying monogenic disorders had a significant difference in the age of presentation (Supplemental Table 4). Most cases of IL10-signalling colitis, TTC7A deficiency, and RIPK1 deficiency developed IBD within the first six months of life, whereas most cases of Hermansky-Pudlak syndrome (HPS) and familial GUCY2C diarrhea syndrome developed IBD in adulthood. Patients with the XIAP, CGD and haploinsufficiency of A20 (HA20) subtypes developed IBD at varying ages, from infancy to their third decade of life.
Figure 2. Distribution of age of IBD onset stratified by underlying monogenic disorder.

The blue shaded region; 0–1.9 years, the red shaded region; 2–5.9 years. Each blue dot is a separate patient, and the vertical red lines indicates the mean with the horizontal line showing the standard error. GUCY2C, Familial GUCY2C diarrhea syndrome (GUCY2C); HPS, Hermansky-Pudlak syndrome (HPS1, HPS4, HPS6,); CEAS, Chronic enteropathy associated with SLCO2A1 gene (SLCO2A1); Agammaglobulinemia, (BTK, PIK3CD, PIK3R1); GSD1b, Glycogen storage disease type 1B (SLC37A4); NP-C, Niemann-Pick disease type C (NPC1); G6PC3, G6PC3 deficiency (G6PC3); XIAP, XIAP deficiency (XIAP); Loeys-Dietz, Loeys-Dietz syndrome (TGFBR1, TGFBR2); CGD, Chronic granulomatous disease (CYBA, CYBB, NCF1, NCF2, NCF4); HA20, haploinsufficiency of A20 (TNFAIP3); WAS/WAS-like, Wiskott-Aldrich syndrome/Wiskott-Aldrich -like syndrome (WAS, ARPC1B); IPEX-like immunodysregulation polyendocrinopathy enteropathy X-linked -like syndrome (STAT1, STAT3, CTLA4, LRBA, IL21, MALT1); Kindler, Kindler syndrome (FERMT1); IPEX, immunodysregulation polyendocrinopathy enteropathy X-linked syndrome (FOXP3); NEMO, Nuclear factor-kappa B Essential MOdulator deficiency (IKBKG); H-HS, Hoyeraal-Hreidarsson Syndrome (DKC1, RTEL1); THE-S, Trichohepatoenteric syndrome (SKIV2L, TTC37); RIPK1, RIPK1 deficiency (RIPK1); SCID, severe combined immunodeficiency (CD3G, DCLRE1C, IL2RG, LIG4, RAG1); IL10/R, IL-10 and IL-10-receptor associated colitis (IL10, IL10RA, IL10RB); TTC7A, TTC7A deficiency (TTC7A).
The proportion of patients with a history of EICs prior to the onset of IBD varies widely depending on the underlying monogenic disorder (Supplemental Figure 5). In most cases with glycogen storage disease type 1B (GSD-1b) and Niemann-Pick disease type C (NP-C), EICs specific to each monogenic disorder were often observed prior to the development of IBD. However, GI symptoms were the primary manifestation of disease in most cases with IL10-signalling colitis, TTC7A deficiency and chronic enteropathy associated with SLCO2A1 gene (CEAS). CGD and XIAP has lower rates of EICs before the onset of IBD (36.8% and 19.0%, respectively), despite the high frequency of these diseases among monogenic IBD.
Extraintestinal Comorbidity and GI Complication
In total, 76.0% (570 out of 750) of all patients had EICs. 62.7% (470 out of 750) had at least one EIC besides atypical infection. The number of reported individual EIC and proportion seen prior to IBD diagnosis is listed in Figure 3A (also see Supplemental Table 5 and Supplementary Figure 6 for further subset data). The most common EICs were atypical infection (n = 335, 44.7%), skin lesion/nail dystrophy/hair abnormality (n = 288. 38.4%), and autoimmunity (e.g., arthritis, type 1 diabetes mellitus, thyroiditis, and autoimmune hemolytic anemia; n = 162, 21.6%, see Supplementary Table 6 for further subset data). Hemophagocytic lymphohistiocytosis (HLH)/macrophage activation syndrome (MAS)-like symptoms were seen in 25 cases across 4 genes (XIAP, STXBP2, CD40LG and CYBB), most of which were in XIAP cases (Figure 5B). Malignancies were seen in 18 cases involving 8 genes in this review (Figure 5C). The frequencies of oral ulcer and perianal disease, which are GI complications commonly associated with non-monogenic IBD, were identified as 15.1% and 35.9%, respectively.
Figure 3. Number of extraintestinal comorbidities across monogenic IBD cases.

(A) Number of EICs in all monogenic IBD cases, where reported (red highlights show GI complications of perianal disease and oral ulcers). Autoimmunity includes autoimmune hepatitis, arthritis, arthralgia, type 1 diabetes mellitus, hypothyroiditis, psoriasis, autoimmune hemolytic anemia, autoimmune neutropenia, immune thrombocytopenic purpura, uveitis, primary sclerosing cholangitis, vasculitis, autoimmune pancreatitis, autoimmune growth hormone deficiency, glomerular nephropathy, nephrotic syndrome and autoimmune lymphoproliferative syndrome. Chronic lung disease includes interstitial lung disease, bronchiectasis, and pulmonary fibrosis. Perianal disease includes fistula, abscess, rectovaginal fistula and ulcer (not including fissure or skin tags).
(B) Number of monogenic IBD cases with HLH/MAS by causative genes. HLH, hemophagocytic lymphohistiocytosis; MAS, macrophage activation syndrome.
(C) Number of monogenic IBD cases associated with malignancy by causative genes. LCH, Langerhans cell histiocytosis; AC, adenocarcinoma.
Figure 5.

Executive flowchart for the management of monogenic IBD.
HLH, hemophagocytic lymphohistiocytosis; MAS, macrophage activation syndrome; CBC, complete blood count; TRECs, T-cell receptor excision circles; DHR-123, dihydrorhodamine 123; HSCT, hematopoietic stem cell transplantation.
Inheritance Pattern and Variant Type
Table 1 shows the inheritance pattern and variant types for the monogenic IBD cases. Autosomal recessive was the most common inheritance pattern (AR, 62.0%). Fifteen cases were described as monoallelic inheritance pattern including nine cases of heterozygous female carriers, while eight cases had only single allele mutations despite the fact that they were in genes usually described with AR inheritance (Supplemental Table 7). Regarding variant type, there were 534 unique variants across 694 patients (no detailed variant description was provided for the remaining 56 cases). Missense was the most common variant type (45.7%), followed by frameshift (20.4%), and stop-gain (16.5%). Deletions and intronic variants (which are sometimes challenging to identify with WES or TGPS) were found in some cases (4.1% and 1.7%), and only seven (1.0%) cases showed copy number variants (CNVs).
Table 1.
Inheritance pattern and variant types identified in this study
| Inheritance pattern | (N) | % | Variant types identified | (N) | % |
|---|---|---|---|---|---|
|
| |||||
| Autosomal recessive | 465 | 62.0 | Missense | 244 | 45.7 |
| X-linked recessive | 204 | 27.2 | Frameshift | 109 | 20.4 |
| Autosomal dominant | 64 | 8.5 | Stop gained | 88 | 16.5 |
| Heterozygous-like form | 17 | 2.3 | Splice site | 42 | 7.9 |
| Deletion | 22 | 4.1 | |||
| Intronic | 9 | 1.7 | |||
| Inframe | 7 | 1.3 | |||
| Insertion | 4 | 0.7 | |||
| Start/Stop loss | 4 | 0.7 | |||
| Inversion | 2 | 0.4 | |||
| Synonymous | 2 | 0.4 | |||
| Duplication | 1 | 0.2 | |||
| Total | 750 | 100.0 | 534 | 100 | |
Therapeutic Management of Monogenic IBD
We collected information on previously attempted treatment options for severe monogenic IBD cases including surgery, biologic therapy and HSCT. Bowel surgery was noted in 203 cases (27.1%), of which bowel resection was performed in 63.1% (Figure 4A). The number and rate of bowel surgery and bowel resection per gene are shown in Supplemental Table 8A. In IL10-signalling colitis and TTC7A deficiency, surgery was required early in disease course (Supplemental Figure 7A). The rates of surgery were similar between the groups with an age of onset less than six years and those with an age of onset greater than six years (29.9% vs. 30.7%) (Supplemental Figure 7B).
Figure 4. Therapeutic management across monogenic IBD cases.

(A) Proportion of monogenic IBD cases shown requiring bowel surgery and for those cases requiring surgery, proportions of the types of surgery.
(B) Proportion of monogenic IBD cases receiving biologic therapies (including infliximab, adalimumab, golimumab, ustekinumab and vedolizumab, abatacept, anakinra, basiliximab, canakinumab, cerotilizumab, etanercept, natalizumab, rituximab, tocilizumab, tofacitinib* and Ruxolitinib*), and efficacy. *a small molecule drug
(C) Proportion of monogenic IBD cases that underwent hematopoietic stem cell transplant (HSCT) and, of those, the response rate. The rightmost image illustrates the details of the cases that HSCT that did not improve.
Biologic therapy was used in 247 cases (32.9%) (Figure 4B, and number and rate of biologics used per gene are shown in Supplemental Table 8B and Supplemental Figure 8). In addition to conventional biologics used for pediatric IBD (infliximab, adalimumab, golimumab, ustekinumab and vedolizumab), other specific therapies have been used, including abatacept, anakinra, basiliximab, canakinumab, cerotilizumab, etanercept, natalizumab, rituximab, tocilizumab, and tofacitinib and ruxolitinib (small molecule drugs) were also used (Supplemental Table 9). Biologics were shown to be effective in 25.5% of 247 cases (not including partial effectiveness). Notably, the efficacy of anti-tumor necrosis factor α (TNF-α) agents for monogenic IBD with HPS was 100% in the seven reported cases. However, anti-TNFα agents were not shown to be effective for IL10-signalling colitis and LRBA defects. Overall, there was insufficient data regarding biologic use in monogenic IBD.
HSCT was performed in 173 cases (23.1%) (Figure 4C) with resolution of intestinal inflammation in 124 cases with no improvement in 42 cases including death in 30 cases. The number of HSCT and rate of IBD improvement per gene are shown in Supplemental Table 8C and Supplemental Figure 8). In this systematic review, HSCT in patients with CGD, LRBA deficiency, XIAP deficiency, and IL10-signalling colitis had a high success rate (96.2%, 90.0%, 81.8%, and 78.8%, respectively). In contrast, the success rates were low for cases with TTC7A deficiency or nuclear factor-kappa B essential modulator (NEMO) deficiency. Three of the five TTC7A deficiency cases did not improve and two died after transplant. Four of the 18 cases with NEMO deficiency did not improve, five died and three cases developed IBD following HSCT, which was performed for severe immunodeficiency and/or autoimmune skin disease.
Discussion
Monogenic forms of IBD are rare making it challenging to collect and analyze large patient cohorts. This study aimed to improve the understanding of monogenic IBD through a systematic review of the monogenic IBD literature. We reviewed published cases of monogenic IBD genes described in patients from across six continents (31 countries) representing a diverse ethnic background. This study provides a comprehensive picture of the heterogeneity of the genetic mechanisms underlying these diseases, clinical presentation and response to therapy. Another strength of this systematic review is that we focused not only on children with VEO-IBD (defined as those with disease onset before age six years of age) but also on monogenic IBD occurring in adolescence and adulthood, although the majority of studies thus far on monogenic IBD focused on those diagnosed before six years of age (14–16).
Diseases such as IL10-associated colitis, CGD, XIAP, IPEX-like disease, and TTC7A deficiency, which have been widely reported - should be considered based on clinical presentation enabling early diagnosis using targeted sequencing (such as Sanger sequencing or TGPS). On the other hand, more than half of all monogenic IBD genes that met criteria for inclusion in this review had fewer than five cases reported.
In terms of the utility of age of diagnosis of monogenic IBD, patients presenting before six years of age (labelled VEOIBD), account for the majority of cases (63.4%), and adolescent- and adult-onset monogenic IBD cases comprised over one third of the cases. This finding differs from previous reports that estimate the prevalence of monogenic IBD in children younger than six years of age with IBD as an even larger majority of cases (17–19). On the other hand, there are no reports demonstrating the proportion of monogenic IBD in adult-onset IBD. Charbit-Henrion et al. performed genetic testing on severe, treatment-resistant cases of IBD between the ages of 6 and 18 years and found that 18.2% (4 of 22) were monogenic IBD, while Uchida et al. similarly found monogenic IBD in 25.9% (7 of 27) of cases in the same age category (17, 18). Additionally, in our local Toronto based cohort we have previously shown that 2.3% (20 of 863) of 6 to 18 year old IBD patients have monogenic IBD (19). Given that genetic testing is generally less aggressively pursued for adult-onset and older pediatric cases than for those under six years of age, clinicians should suspect monogenic IBD more than previously thought especially if a patient has EIC or shows refractory responses to conventional therapy, even if the patient is not under six years of age.
Many of the reported monogenic IBD disorders including CGD, XIAP, CEAS and haploinsufficiency A20 (HA20) are difficult to differentiate from polygenic IBD. These disorders present across a wide range of age and there is infrequently a history of disease specific EICs before IBD onset. Early diagnosis is particularly beneficial for CGD and XIAP patients because HSCT can be performed prior to the development of HLH for XIAP deficiency (20), while the use of anti-TNF drugs can lead to severe infections and even death in CGD (21). These findings further highlight the importance of obtaining timely genetic testing in patients where there is a high suspicion for monogenic disorders (Figure 5).
Further variant types, such as CNVs, intronic variants, synonymous variants, and inversions, are often missed by WES (22, 23), and poor coverage of some regions of known monogenic IBD genes by WES is well described (19, 24) It is likely that eight cases with heterozygous-like inheritance in AR monogenic disorder had these variants (Supplementary Table 7). The advantages and disadvantages for each genetic detection method should be considered when investigating a patient for monogenic IBD. Given that there are not yet many reports of monogenic IBD diagnosis using WGS (Supplementary Figure 1), we expect that the number of reports WGS will increase as the use of WGS becomes more widespread. It is also likely that monogenic IBD variants that are more amenable to discovery by WGS, such as CNVs and intronic variants, will similarly increase over time. The method of detection is also in evolution as the cost of NGS technology in general continues to decrease as does the time to reporting.
In terms of therapy, of the 46 genes responsible for monogenic IBD for which biologics were used, many genes had only a few reported cases, and monogenic cases linked to 15 genes showed 0% efficacy from biologic therapy (see Supplementary Table 8B). Some treatments, such as CTLA4-immunoglobulin, have been reported to be effective therapies in LRBA and CTLA4 deficiency (25), and anti-IL-1β biologics for MVK and NLRC4 (26); however, the use of specific biologics in most monogenic IBDs has not been established and should be used with caution. In our study, HSCT improved IBD in approximately 70% of cases. This number is likely an overestimate due to reporting bias where successful HSCT is likely reported more often than unsuccessful HSCT. These results particularly emphasize the poor transplant outcomes in non-hematopoietic forms of monogenic IBD where the gene expression is known to be present in the intestinal epithelium, such as IKBKG, TTC7A and TTC37 (27, 28). Interestingly, a preclinical study identified leflunomide as a potential therapy for TTC7A deficiency (29). Thus, the discovery of new therapeutic agents and the establishment of treatment modalities though bedside to bench research are required.
This systematic review has some limitations. Firstly, the reason why genetic testing was pursued was not clearly reported for many cases. It is probable that the study population includes a selection bias in which severe cases with treatment resistance or extraintestinal comorbidity are preferentially tested. Secondly, outcomes regarding complications and treatment might be underestimated because the actual events are not sufficiently described or occurred after publication. Thirdly, we excluded cases in which genetic testing was not performed. As such, monogenic IBD with CGD, GSD-type1b, and WAS (which can be diagnosed by molecular testing), and with dystrophic epidermolysis bullosa (which can be diagnosed with physical examination findings) are likely to be more common in clinical practice than in the present study.
Overall, monogenic IBD diagnosis and management is a challenging clinical problem across many age groups. The EICs of monogenic IBD are highly diverse, and the management of monogenic IBD is difficult. Many monogenic IBD genes have only a few reported cases and therefore generalizations about clinical course must be limited. However, we now have an improved understanding of the underlying genetic basis of these diseases, especially those with relatively high frequencies. In recent years, the number of novel genes causing monogenic IBD has been increasing, but in parallel with these discoveries, there is an urgent need to better understand this group of diseases to enable prompt diagnosis, improve prognosis, predict the clinical course, and to establish new treatment strategies. From this systematic review, we now have an improved understanding of the underlying genetic basis of these diseases, especially those with relatively high frequencies, paving the way for prognostication and effective management.
Supplementary Material
What You Need to Know:
Background:
Monogenic IBD has been extensively studied in very young children and infants, but there is limited data regarding older patients, clinical presentation, and treatment options.
Findings:
While monogenic IBD is more common in young children, nearly one-third of all patients were diagnosed after 6 years of age and over 10% as adults. The non-gastrointestinal characteristics of monogenic IBD are diverse with 76% having developed at least one EIC during clinical course. Treatments including bowel surgery and HSCT were performed in 27.1% and 23.1% of patients.
Implications for Patient Care:
Monogenic IBD is rare but can be identified in adolescents and adults with IBD. Early identification and improved understanding of the characteristics monogenic IBD is critical to caring for this patient group.
Acknowledgments
Grant support
RN is supported by a Uehara Memorial Foundation fellowship. DJM is supported by funded by a Canadian Institutes of Health Research (CIHR)-Canadian Association of Gastroenterology (CAG) Fellowship Award and a Southeastern Ontario Academic Medical Organization Clinical Research Fellowship Program Award. CK and DK are funded by German Research Foundation CRC1054 and the Care-for-Rare Foundation. AMM, CK, SBS are funded by the Leona M. and Harry B. Helmsley Charitable Trust. AMM, CK, JC, DM and SBS are funded by NIH (RC2DK122532) Grant. AMM is funded by a Canada Research Chair (Tier 1) in Pediatric IBD, CIHR Foundation Grant and NIDDK NIH (RC2DK118640).
Abbreviations
- AD
autosomal dominant
- AIH
autoimmune hepatitis
- AR
autosomal recessive
- CD
Crohn disease
- CGD
chronic granulomatous disease
- CNVs
copy number variants
- EIC
extraintestinal comorbidity
- GI
gastrointestinal
- GSD-1b
glycogen storage disease type 1B
- HA20
haploinsufficiency A20
- HLH
hemophagocytic lymphohistiocytosis
- HPS
Hermansky-Pudlak syndrome
- HSCT
hematopoietic stem cell transplantation
- IBD
inflammatory bowel disease
- IOIBD
infantile onset inflammatory bowel disease
- IPEX
immunodysregulation polyendocrinopathy enteropathy X-linked syndrome
- MAS
macrophage activation syndrome
- NEMO
nuclear factor-kappa B Essential Modulator
- NGS
next-generation DNA sequencing
- NP-C
Niemann-Pick disease type C
- PSC
primary sclerosing cholangitis
- TGPS
targeted gene-panel sequencing
- UC
ulcerative colitis
- VEOIBD
very early onset inflammatory bowel disease
- WES
whole-exome sequencing
- WGS
whole-genome sequencing
- XR
X-linked recessive
Footnotes
Disclosures:
None declared
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Levine A, Koletzko S, Turner D, et al. ESPGHAN revised porto criteria for the diagnosis of inflammatory bowel disease in children and adolescents. J Pediatr Gastroenterol Nutr. 2014;58:795–806. [DOI] [PubMed] [Google Scholar]
- 2.Sartor RB. Mechanisms of disease: pathogenesis of Crohn’s disease and ulcerative colitis. Nat Clin Pract Gastroenterol Hepatol 2006;3:390–407. [DOI] [PubMed] [Google Scholar]
- 3.Muise AM, Snapper SB, Kugathasan S. The age of gene discovery in very early onset inflammatory bowel disease. Gastroenterology 2012;143:285–8. [DOI] [PubMed] [Google Scholar]
- 4.Uhlig HH, Muise AM. Clinical Genomics in Inflammatory Bowel Disease. Trends Genet. 2017;33:629–641. [DOI] [PubMed] [Google Scholar]
- 5.Uhlig HH, Schwerd T, Koletzko S, et al. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology 2014;147:990–1007.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Uhlig HH, Charbit-Henrion F, Kotlarz D, et al. Clinical Genomics for the Diagnosis of Monogenic forms of Inflammatory Bowel Disease: A Position Paper from The Paediatric IBD Porto Group of ESPGHAN. J Pediatr Gastroenterol Nutr 2020. Dec 16;Publish Ahead of Print. doi: 10.1097/MPG.0000000000003017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ouahed J, Spencer E, Kotlarz D, et al. Very Early Onset Inflammatory Bowel Disease: A Clinical Approach With a Focus on the Role of Genetics and Underlying Immune Deficiencies. Inflamm Bowel Dis 2020;26:820–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pazmandi J, Kalinichenko A, Ardy RC, et al. Early-onset inflammatory bowel disease as a model disease to identify key regulators of immune homeostasis mechanisms. Immunol Rev 2019;287:162–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kelsen JR, Sullivan KE, Rabizadeh S, et al. North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition Position Paper on the Evaluation and Management for Patients With Very Early-onset Inflammatory Bowel Disease. J Pediatr Gastroenterol Nutr 2020;70:389–403. [DOI] [PubMed] [Google Scholar]
- 10.Fernandez IZ, Baxter RM, Garcia-Perez JE, et al. A novel human IL2RB mutation results in T and NK cell-driven immune dysregulation J Exp Med 2019;216:1255–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Starokadomskyy P, Gemelli T, Rios JJ, et al. DNA polymerase-α regulates the activation of type I interferons through cytosolic RNA:DNA synthesis. Nat Immunol 2016;17:495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lohr NJ, Molleston JP, Strauss KA, et al. Human ITCH E3 ubiquitin ligase deficiency causes syndromic multisystem autoimmune disease. Am J Hum Genet 2010;86:447–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelsen JR, Ouahed J, Spessott WA, et al. Mutations in STXBP3 contribute to very early onset of IBD, immunodeficiency and hearing loss. www.gastrojournal.org/article/S0016-5085(17)36478-8/fulltext?referrer=https%3A%2F%2Fwww.gastrojournal.org%2F (accessed 13 Oct 2020).
- 14.Kammermeier J, Drury S, James CT, et al. Targeted gene panel sequencing in children with very early onset inflammatory bowel disease--evaluation and prospective analysis. J Med Genet 2014;51:748–55 [DOI] [PubMed] [Google Scholar]
- 15.Fang YH, Luo YY, Yu JD, et al. Phenotypic and genotypic characterization of inflammatory bowel disease in children under six years of age in China. World J Gastroenterol 2018;24:1035–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ye Z, Zhou Y, Huang Y, et al. Phenotype and Management of Infantile-onset Inflammatory Bowel Disease: Experience from a Tertiary Care Center in China. Inflamm Bowel Dis 2017;23:2154–2164. [DOI] [PubMed] [Google Scholar]
- 17.Charbit-Henrion F, Parlato M, Hanein S, et al. Diagnostic Yield of Next-generation Sequencing in Very Early-onset Inflammatory Bowel Diseases: A Multicentre Study. J Crohns Colitis 2018;12:1104–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uchida T, Suzuki T, Kikuchi A, et al. Comprehensive Targeted Sequencing Identifies Monogenic Disorders in Patients With Early-onset Refractory Diarrhea. J Pediatr Gastroenterol Nutr 2020;71:333–339 [DOI] [PubMed] [Google Scholar]
- 19.Crowley E, Warner N, Pan J, et al. Prevalence and Clinical Features of Inflammatory Bowel Diseases Associated With Monogenic Variants, Identified by Whole-Exome Sequencing in 1000 Children at a Single Center. Gastroenterology 2020;158:2208–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ono S, Okano T, Hoshino A, et al. Hematopoietic Stem Cell Transplantation for XIAP Deficiency in Japan. J Clin Immunol 2017;37:85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uzel G, Orange JS, Poliak N, et al. Complications of tumor necrosis factor-α blockade in chronic granulomatous disease-related colitis. Clin Infect Dis 2010;51:1429–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murugan D, Albert MH, Langemeier J, et al. Very early onset inflammatory bowel disease associated with aberrant trafficking of IL-10R1 and cure by T cell replete haploidentical bone marrow transplantation. J Clin Immunol 2014;34:331–9. [DOI] [PubMed] [Google Scholar]
- 23.Charbit-Henrion F, Bègue B, Sierra A, et al. Copy number variations and founder effect underlying complete IL-10Rβ deficiency in Portuguese kindreds. PLoS One 2018;13:e0205826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kammermeier J, Drury S, James CT, et al. Targeted gene panel sequencing in children with very early onset inflammatory bowel disease--evaluation and prospective analysis. J Med Genet 2014;51:748–55. [DOI] [PubMed] [Google Scholar]
- 25.Lo B, Zhang K, Lu W, et al. AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science 2015;349:436–40. [DOI] [PubMed] [Google Scholar]
- 26.Levy M, Arion A, Berrebi D, et al. Severe early-onset colitis revealing mevalonate kinase deficiency. Pediatrics 2013;132:e779–83. [DOI] [PubMed] [Google Scholar]
- 27.Jardine S, Dhingani N, Muise AM, et al. TTC7A: Steward of Intestinal Health. Cell Mol Gastroenterol Hepatol 2019;7:555–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miot C, Imai K, Imai C, et al. Hematopoietic stem cell transplantation in 29 patients hemizygous for hypomorphic IKBKG/NEMO mutations. Blood 2017;130:1456–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jardine S, Anderson S, Babcock S, et al. Drug Screen Identifies Leflunomide for Treatment of Inflammatory Bowel Disease Caused by TTC7A Deficiency. Gastroenterology 2020;158:1000–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.