

Abstract
Intensive gene annotation has revealed many functional and regulatory elements in the human genome. Although eukaryotic protein-coding genes are generally transcribed into monocistronic mRNAs, recent studies have discovered additional short open reading frames (sORFs) in mRNAs. Here, we performed proteogenomic data mining for hidden proteins categorized into sORF-encoded polypeptides (SEPs) in human cancers. We identified a new SEP-encoding overlapping sORF (oORF) on the cell polarity determinant Scribble (SCRIB) that is considered a proto-oncogene with tumor suppressor function in Hippo-YAP/TAZ, MAPK/ERK, and PI3K/Akt/mTOR signaling. Reanalysis of clinical human proteomic data revealed translational dysregulation of both SCRIB and its oORF, oSCRIB, during carcinogenesis. Biochemical analyses suggested that the translatable oSCRIB constitutively limits the capacity of eukaryotic ribosomes to translate the downstream SCRIB. These findings provide a new example of cis-regulatory oORFs that function as a ribosomal roadblock and potentially serve as a fail-safe mechanism to normal cells for non-excessive downstream gene expression, which is hijacked in cancer.
Subject terms: Proteomics, Proteome informatics, Cancer genomics, Open reading frames
Yuhta Nomura and Naoshi Dohmae report the discovery of a small protein-coding gene that overlaps the tumor suppressor gene Scribble. Their data suggest that the overlapping gene, oSCRIB, limits the translation of downstream Scribble and may have important implications in cancer.
Introduction
More accurate and detailed annotation of the human genome has shed light on many functional and regulatory elements, including protein-coding genes and noncoding RNA genes (e.g., ribosomal RNA, transfer RNA, and microRNA genes). Eukaryotic protein-coding genes are generally transcribed into monocistronic messenger RNAs (mRNAs), each with a single protein-coding open-reading frame (ORF) and two untranslated regions (UTRs: 5′-UTR and 3′-UTR) for their subsequent translation1. Contrary to this view, there is growing evidence that eukaryotic mRNAs can code for more than one protein, indicating the existence of exceptions to the dogma2–5. In fact, recent studies have uncovered many classes of additional ORFs (viz., alternative ORFs, altORFs), such as short/small ORFs (sORFs/smORFs), in mRNAs6–10, which are translated on ribosomes and may produce small proteins or polypeptides <150 amino acids, namely sORF-encoded polypeptides (SEPs)6. Based on their relative position against the primary ORF (viz., reference protein-coding sequence, refCDS), which is usually the longest ORF per transcript, they are currently categorized into three major groups: upstream ORFs (uORFs or altORFs5′-UTR), overlapping ORFs (oORFs or altORFsCDS), and downstream ORFs (dORFs or altORFs3′-UTR)3. Interestingly, it has been reported that approximately 50% of mRNAs potentially code for at least one uORF or oORF, and the majority of oORF-containing mRNAs have also at least one potential uORF in humans and mice11,12. In addition, there is emerging evidence that uORFs and oORFs, both of which are altORFs, act as translational cis-regulatory elements of refCDSs12,13 and are closely associated with tumor-initiating and tumor-progressing unconventional translation in cancer13,14. These studies on altORFs are opening new avenues for therapeutic interventions in cancer treatment. Furthermore, the presence of altORFs in some long noncoding RNAs and circular RNAs has also been reported6,8,15–17, termed altORFslnc and altORFscirc, respectively. These striking examples indicate a higher density of genetic information in the human genome and motivate our research on SEP gene discovery.
Mass spectrometry (MS)-based proteomics and ribosome profiling (or Ribo-Seq)-based translatomics are both becoming powerful tools for novel protein/peptide discovery6–10. In particular, the former approach has the capability to conclusively identify peptides and proteins by their direct detection18. This has permitted the simultaneous identification of human peptides and proteins encoded by 17,294 genes, which account for approximately 84% of protein-coding genes previously annotated in the human genome15. On the other hand, there is inherent dependence of current MS-based proteomics on predefined databases of protein sequences and/or translated nucleotide sequences for protein identification. Therefore, a combination of high-quality data on both proteomic mass spectra and genomic/transcriptomic nucleotide sequences (or ORFeomes) is indispensable for the more comprehensive identification of human proteomes19–21. This proteogenomic approach still promises to uncover previously ignored dark proteomes and peptidomes, including novel SEPs.
In this study, we performed proteogenomic data mining for hidden SEP-encoding genes in the human genome through the use of publicly available high-quality datasets of human cancer cell lines, which were obtained from MS-based proteomics and transcriptomic RNA sequencing (RNA-Seq). Our study successfully demonstrated the existence of a new SEP-encoding oORF on the cell polarity-determining scaffold protein gene Scribble (SCRIB), which is considered a proto-oncogene with tumor suppressor function in antitumorigenic Hippo-YAP/TAZ, Ras/Raf/MEK/ERK (MAPK/ERK), and PI3K/Akt/mTOR and proapoptotic c-Myc-induced signaling pathways22–29. Together with the results from our clinical human proteomic data reanalysis and biochemical analysis, we report that the translatable oORF on SCRIB, oSCRIB, is a cis-regulatory oORF potentially providing a fail-safe mechanism to normal cells for nonexcessive downstream SCRIB expression, whereas the mechanism was dysregulated in cancer cells for their survival and proliferation.
Results
Transcriptomic data mining for hidden protein-coding potential of the human genome
We performed proteogenomic data mining in human cancer cell lines, as illustrated in Fig. 1a. High-quality transcriptomic data that can yield high sequence coverage can achieve a more comprehensive estimation of proteomes. In addition, a combination of proteomic and transcriptomic data from the same cell line would maximize the predictive power of proteogenomics (i.e., proteotranscriptomics)-based gene discovery. Therefore, with several publicly available RNA-Seq datasets of the human transcriptome from cancer cell lines (HeLa, MCF-7, A549, and HCT-116; three biological replicates for each cell line), we first performed de novo transcriptome assemblies and searched the resulting contigs for possible ORFs. Additionally, we also extracted potentially translatable ORFs from a nonredundant database of human reference transcript sequences (RefSeq Transcripts30; RefSeq assembly accession GCF_000001405.39), which was prepared from the Genome Reference Consortium Human Build 38 patch release 13 (GRCh38.p13)31 by the National Center for Biotechnology Information (NCBI). Next, we extracted all possible sORFs possibly encoding SEPs of 10–149 amino acids that start and stop with AUG and stop codons, respectively, from the newly constructed datasets of ORFs found in cancer cell lines (HeLa, MCF-7, A549, and HCT-116) and RefSeq Transcripts. The resulting datasets of sORFs from three replicates of each cell line were aggregated into a nonredundant dataset for each cell line. The translated nucleotide sequences of sORFs, i.e., the amino acid sequences of putative SEPs, were further searched using the Basic Local Alignment Search Tool (BLAST)32 against a nonredundant database of human reference protein sequences (RefSeq Proteins30; RefSeq assembly accession GCF_000001405.39), which was also prepared from GRCh38.p13 by NCBI. These preliminary transcriptomic data analyses predicted 3,216,009, 3,704,082, 1,533,942, 2,051,239, and 1,781,513 SEP candidates in HeLa, MCF-7, A549, HCT-116, and RefSeq Transcripts, respectively. These results indicated the presence of a high degree of protein-coding potential of the human genome. Although the retrieved sequences may still include partial protein sequences, such as N-terminally truncated protein sequences, due to incomplete transcriptome assembly, the results indicated that almost two-thirds of these sequences, 2,204,181, 2,506,331, 1,029,024, 1,375,127, and 1,146,023 sequences in HeLa, MCF-7, A549, HCT-116, and RefSeq Transcripts, respectively, were not assigned as genes currently annotated in human RefSeq Proteins and were expected to contain real, previously unidentified SEPs. Therefore, we only retained the unassigned sequences in the SEP datasets. To improve the predictive power of proteogenomics-based gene discovery, we further integrated the SEP dataset of RefSeq Transcripts into that of each cell line and used the integrated nonredundant datasets as the custom SEP sequence databases (for HeLa, MCF-7, A549, and HCT-116) to perform further proteogenomic analyses. These custom SEP sequence databases for each line consisted of 2,852,280, 3,135,500, 1,731,286, and 2,070,511 sequences in HeLa, MCF-7, A549, and HCT-116, respectively (Fig. 1b). Furthermore, we also prepared the complete dataset for which we integrated the SEP datasets of all cell lines (HeLa, MCF-7, A549, and HCT-116) and RefSeq Transcripts into a single nonredundant database; it ultimately consisted of 4,345,441 sequences.
Fig. 1. Proteogenomic data mining for new SEP proteins in human cell lines.

a Proteogenomics workflow for SEP gene discovery in this study. This workflow used a cell-free translation system for authentic protein/peptide production as the final step (yellow highlight) providing conclusive evidence of mass spectrometric protein-identification results, which are normally obtained by probability-based selection in automated search engines (e.g., Mascot). b, c Venn diagrams showing the number of SEP candidates detected in human cell lines at the transcriptome (b) and proteome (c) levels.
Proteomic data mining for new SEPs in humans
High-quality MS-based proteomic data that comprise human proteins with high coverage are also indispensable for a more comprehensive identification of human proteomes. Recently optimized shotgun proteomics with off-line high-pH reversed-phase fractionation of peptides and subsequent multiple injections of each fraction into online low-pH liquid chromatography-tandem MS (LC–MS/MS) accomplished the generation of in-depth human proteomes33,34. The secondary use of these high-quality datasets is also important for mining dark proteomes and peptidomes. Therefore, we also obtained the raw datasets comprising tryptic mass spectra of the human proteome from cancer cell lines (HeLa, MCF-7, A549, and HCT-116; two biological replicates for each cell line) deposited in ProteomeXchange35 (dataset identifier PXD004452) by Bekker-Jensen et al.34 for further proteogenomic analyses.
The raw tryptic mass spectra files were first processed to recalibrate precursor masses and convert the files into Mascot Generic Format (MGF) files, which contain MS/MS peak lists and experimental parameters, with Thermo Proteome Discoverer (Thermo Fisher Scientific). The MGF files were then submitted to an in-house Mascot Server36 through Thermo Proteome Discoverer and searched against the two different sequence databases in parallel, i.e., the combination of human RefSeq Proteins and the custom SEP sequence database (for HeLa, MCF-7, A549, or HCT-116), with target decoy-based false-discovery rate (FDR) filtering37,38 and parameters described in the “Methods” section. The peptide-spectrum matching (PSM) by the Mascot algorithm predicted the inclusion of 258,123, 166,787, 175,353, and 200,071 peptide sequences in the HeLa, MCF-7, A549, and HCT-116 proteomes, respectively, each with an FDR of ≤1% at the peptide level. Among the detected peptides, approximately 0.5–0.7% of peptides (HeLa, 1738; MCF-7, 1,166; A549, 889; and HCT-116, 983) were assigned to SEP candidates (HeLa, 1709; MCF-7, 1120; A549, 861; and HCT-116, 943) derived from the custom SEP sequence databases, whereas the remaining peptides (HeLa, 256,385; MCF-7, 165,621; A549, 174,464; and HCT-116, 199,088) were assigned to known proteins (HeLa, 11,186; MCF-7, 10,939; A549, 11,195; and HCT-116, 11,624) listed in human RefSeq Proteins. On the other hand, when using the single complete database into which we integrated the SEP datasets of all cell lines (HeLa, MCF-7, A549, and HCT-116) and RefSeq Transcripts, Mascot algorithm-assisted PSM predicted the slightly smaller number of peptides (256,228) in the HeLa proteome, among which 2225 and 254,003 peptides were assigned to 2178 SEP candidates and 11,130 known proteins, respectively, each with an FDR of ≤1% at the peptide level. These results indicated the limited advantage of the single complete database as compared with cell-type-matched databases in the detection power of proteomes. Since our SEP discovery was conducted within a commercial proteome informatics pipeline (i.e., Thermo Proteome Discoverer, as described above), a class-specific FDR estimation recommended by Nesvizhskii20 was not taken into consideration. Our results were instead filtered to maintain a low FDR of ≤1% at the peptide level, thereby ensuring high confidence in the detected peptides. In addition, approximately 96–97% and 42–47% of the detected peptides derived from human RefSeq Proteins and the custom SEP sequence database, respectively, had lower FDRs of ≤0.5%.
We next evaluated the results at the protein level. Among the detected SEP candidates (HeLa, 1709; MCF-7, 1120; A549, 861; and HCT-116, 943) described above, 35, 82, 37, and 56 candidates in the HeLa, MCF-7, A549, and HCT-116, respectively, had a relatively low FDR of ≤1% (high confidence) at the protein level; 275, 395, 228, and 231 candidates in the HeLa, MCF-7, A549, and HCT-116, respectively, had a relaxed FDR of >1% but still maintained an FDR of ≤5% (medium confidence); the remaining candidates (HeLa, 1399; MCF-7, 643; A549, 596; and HCT-116, 656) each had a high FDR of >5% (low confidence). The high-to-medium-confidence candidates (HeLa, 310; MCF-7, 477; A549, 265; and HCT-116, 287) included 17 SEP candidates (7 high- and 10 medium-confidence candidates) that were detected in all the human cell lines described above (Fig. 1c). Interestingly, the 7 high-confidence candidates included two SEP candidates with a protein-level FDR of 0%, both of which were detected not only in the HeLa proteome but also in the other human cell lines mentioned above. This suggests the presence of the two SEPs in human cells. One of the two SEP-encoding sORFs overlapped with the cell-polarity protein gene Scribble (SCRIB)28 in the human genome; this overlapping sORF had not previously been annotated. The spectrum-centric PSM approach described above predicted the existence of two peptides unique to this SEP, AGGDLPLQPQPGGAAAR and AAQAFFPAAELAQAGPER, with very low peptide-level FDRs of 0.0018% and 0%, respectively. Furthermore, an alternative peptide-centric approach by PepQuery also retrieved the same unique peptides (PepQuery P-value, 0.001%) from the publicly available proteomic data (MGF format) under a P-value cutoff of 1%, where well-controlled FDRs are produced39. We thus designated this SEP-encoding sORF (120 amino acids in length) as an oORF on SCRIB or oSCRIB (Fig. 2a and Supplementary Table 1). On the other hand, the existence of the other SEP (63 amino acids in length) was also supported by the detection of NDDIPEQDSLGLSNLQK by the Mascot algorithm with very low peptide-level FDR of 0%. The PepQuery also retrieved the same unique peptide from the MGF files (PepQuery P-value, 0.001%; P-value cutoff, 1%). However, this SEP was equivalent to an evolutionarily conserved uORF of the McKusich–Kaufman Syndrome (MKKS) gene, which was previously reported as uMKKS140 (Supplementary Table 1) and whose translation product was also detected by MS-based proteomics41, although it has not been included in human RefSeq Proteins. Accordingly, we focused on the newly discovered oSCRIB protein in this study.
Fig. 2. Investigation of the newly discovered oSCRIB protein.

a The transcript encoding the main SCRIB protein contains additional unidentified sORFs (uSCRIB and oSCRIB). The oSCRIB-coding region overlapped the downstream out-of-frame SCRIB gene in the human genome. Translational start codons (AUGs) of oSCRIB and SCRIB and the surrounding sequences in humans are shown. The most prevalent sequence (GGACU) for the reversible epitranscriptomic m6A modification and the resultant m6A-dependent start codon selection42,43,58,73 are shown in the box. b Survey of publicly available Ribo-Seq data. The aggregated profiles of initiating and elongating ribosomes were obtained from GWIPS-viz74 (https://gwips.ucc.ie). c Translation products of oSCRIB in humans (Homo sapiens), chimpanzees (Pan troglodytes), cattle (Bos taurus), mice (Mus musculus), and rats (Rattus norvegicus). The protein sequences were aligned and colored using the GenomeNet ClustalW 2.1 and EMBnet BoxShade 3.21 servers (https://www.genome.jp/tools-bin/clustalw and https://embnet.vital-it.ch/software/BOX_form.html). Red boxes indicate the amino acid sequences of tryptic peptides detected in LC–MS/MS analyses as shown in Figs. 3 and 4.
Further investigation of the newly discovered oSCRIB protein
It is worth noting that approximately 80% of the oSCRIB-coding region (287 of 363 bp) overlapped with a downstream out-of-frame SCRIB gene (two transcript variants, 1 and 2, 4968 bp and 4893 bp, respectively) in the human genome (Fig. 2a). These two adjacent genes, oSCRIB and SCRIB, correspond to oORF (or altORFCDS) and refCDS, respectively; they share the same genomic region and are transcribed into a single bicistronic mRNA, after which they are translated into proteins with different translational start codons. A survey of the oSCRIB sequence itself and its upstream region in the human genome further indicated the existence of an internal epitranscriptomic N6-methyladenosine (m6A) consensus motif (GGACU)42,43 and an unknown, potentially translatable uORF of SCRIB (uSCRIB), respectively (Fig. 2a). Since our custom SEP sequence databases included the translated uSCRIB sequence, we checked the publicly available MS-based proteomic data again for uSCRIB. Although both oSCRIB and SCRIB were successfully detected in these proteomes with protein-level FDRs of 0%, the translation product of uSCRIB (MRSRRRRRSPRFLRV, 15 amino acids in length) was not detected. This was likely due to its low abundance and/or tryptic digestion of its arginine-rich region during sample preparation. In support of our proteogenomic data mining, the publicly available Ribo-Seq-based translatomic data also implied the translational initiation and elongation of ribosomes at uSCRIB, oSCRIB, and SCRIB in human cancer cell lines (Fig. 2b). Additionally, other mammalian genomes were found to potentially conserve oSCRIB homologous proteins (Fig. 2c).
Conclusive validation of the identification of oSCRIB protein
A Mascot server performs the probability-based protein identification in silico by fitting experimental MS/MS data into theoretical MS/MS models that are constructed by user-selected protein-sequence datasets36. Therefore, it is necessary to validate the identification of SEPs with their authentic proteins. The newly discovered oSCRIB was the target of this study, but the previously reported uMKKS1 was used as a positive control in subsequent investigations.
For the simultaneous synthesis of multiple authentic proteins in small quantities, we employed a commercially available in vitro translation system based on bacterial 70 S ribosomes, called Protein synthesis Using Recombinant Elements (PURE) system44. We first constructed linear DNA fragments that were used as templates for in vitro transcription and translation (Fig. 3a). The DNA fragments contained a bacteriophage T7 RNA polymerase promoter, bacterial ribosome-binding site called a Shine–Dalgarno sequence, and either the sORF encoding oSCRIB or uMKKS1 or the ORF encoding a superfolder green fluorescent protein (sfGFP)45 as a positive control of in vitro translation. Then in vitro translation coupled with transcription was performed with the PURE system in the presence of the DNA fragments, and the synthesized proteins were detected by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). The results showed their successful expression in vitro; oSCRIB and sfGFP proteins were only detected in the soluble fraction, whereas uMKKS1 protein was found in both the soluble and insoluble fractions (Fig. 3b).
Fig. 3. Cell-free translation system-assisted rapid validation of new SEP protein identification.

a Linear DNA fragments (sfGFP, oSCRIB, and uMKKS1) prepared for in vitro production of authentic proteins/peptides were analyzed by agarose gel electrophoresis. The DNA fragments were visualized under ultraviolet light on gels stained with GelRed, and the positions of molecular standards are indicated. Arrowheads indicate the bands of the DNA fragments. b SDS-PAGE analysis of in vitro-synthesized proteins (sfGFP, oSCRIB, and uMKKS1). Coupled in vitro reactions of transcription and translation with and without the DNA fragment encoding sfGFP protein were used as positive and negative controls of the reactions, respectively. The gel was stained with Coomassie Brilliant Blue G-250, and the positions of molecular standards are indicated. Arrowheads indicate the bands of proteins synthesized in vitro, whereas other bands were derived from enzymes for transcription and translation. The following abbreviations are used: S, soluble fractions; and P, insoluble fractions. c–e Mirror plots of MS/MS spectra obtained by LC–MS/MS in DDA mode. The spectra included in publicly available proteomic data on HeLa cells deposited in the ProteomeXchange (dataset identifier PXD004452) by Bekker-Jensen et al.34 are shown above, whereas those of the corresponding authentic peptides obtained by tryptic digestion of in vitro-synthesized proteins are inverted and shown below. The spectra included the profiles of the product ions generated by HCD of divalent precursor ions [M + 2H]2+ that were derived from tryptic peptides: AGGDLPLQPQPGGAAAR (c) and AAQAFFPAAELAQAGPER (d) of oSCRIB protein; and NDDIPEQDSLGLSNLQK (e) of uMKKS1 protein.
Next, we performed in-gel tryptic digestion of the in vitro-synthesized oSCRIB and uMKKS1 proteins excised from the SDS-PAGE gels. The resultant peptides were analyzed as authentic standards by LC–MS/MS under data-dependent acquisition (DDA) control46. These MS/MS spectra included the profiles of the product ions generated by higher-energy collisional dissociation (HCD)46,47 of divalent precursor ions [M + 2H]2+ that were derived from tryptic peptides as follows: AGGDLPLQPQPGGAAAR (m/z 788.41425) and AAQAFFPAAELAQAGPER (m/z 922.96613) of the authentic oSCRIB protein (Fig. 3c, d), and NDDIPEQDSLGLSNLQK (m/z 943.45764) of the authentic uMKKS1 protein (Fig. 3e). Manual comparison of these MS/MS spectra with those of the corresponding peptides obtained from the above-mentioned proteome dataset (PXD004452) confirmed their equivalent fragmentation patterns (Fig. 3c–e). Therefore, this inspection provided evidence for the existence of oSCRIB and uMKKS1 proteins in human cell lines.
Finally, to further validate the newly identified oSCRIB protein, we also extracted HeLa proteins. The extracted HeLa proteins were subjected to SDS-PAGE for protein separation (Fig. 4a), and the enrichment of small proteins approximately 10–15 kDa, which included oSCRIB protein (12 kDa), was carried out by excising the corresponding protein bands from SDS-PAGE gels. These small proteins were then digested in-gel with trypsin and the resultant peptides were analyzed by LC–MS/MS. To obtain high sensitivity and selectivity toward the detection of tryptic peptides of oSCRIB protein, we applied targeted MS/MS acquisition called parallel reaction monitoring (PRM)48. Our results showed the successful detection of two peptides unique to oSCRIB protein, AGGDLPLQPQPGGAAAR and AAQAFFPAAELAQAGPER, and their retention times and fragmentation patterns were identical to those of the corresponding peptides obtained from the authentic oSCRIB protein (Fig. 4b–e). Therefore, our results confirmed the existence of oSCRIB protein in human cells at the protein level.
Fig. 4. Further validation of the newly identified oSCRIB protein.

a SDS-PAGE profile of endogenous HeLa proteins. The gel was stained with Coomassie Brilliant Blue G-250, and the positions of molecular standards are indicated. The broken circle indicates the protein fraction used for LC–MS/MS analysis, where small HeLa proteins of approximately 10–15 kDa, including oSCRIB protein (12 kDa), were enriched. b–e The results of LC–MS/MS analysis in PRM mode. The PRM chromatograms (b, c) and MS/MS spectra (d, e) obtained from the protein fraction in (a) are shown above, whereas those of the corresponding authentic peptides obtained by tryptic digestion of in vitro-synthesized oSCRIB protein are shown below. The lower parts of (d, e) are inverted to make mirror plots. The PRM chromatograms of (b) show the extracted peaks containing the product ions of m/z 824.4100–824.4700 among the precursor ions of m/z 788.4155 for AGGDLPLQPQPGGAAAR and those of (c) show the extracted peaks containing the product ions of m/z 1209.6000–1209.6600 among the precursor ions of m/z 922.9681 for AAQAFFPAAELAQAGPER. The MS/MS spectra (d, e) corresponding to the peaks indicated in (b–c) by arrowheads included the profiles of the product ions generated by HCD of divalent precursor ions [M + 2H]2+ that were derived from tryptic peptides unique to oSCRIB protein, AGGDLPLQPQPGGAAAR (d) and AAQAFFPAAELAQAGPER (e).
The newly discovered oSCRIB is a translational inhibitory element of SCRIB
Recent studies have revealed that the downstream SCRIB is a double-agent gene as a proto-oncogene with tumor suppressor function (POTSF) in normal cells26,27,29. In fact, SCRIB is reported to be overexpressed at the mRNA and protein levels in multiple human cancers, including uterine and breast cancers, and the resultant mislocalization of SCRIB protein promotes tumorigenesis23,26.
To investigate the relationship between oSCRIB and SCRIB expression in carcinogenesis, we reanalyzed publicly available tandem mass tag (TMT) isobaric labeling-based quantitative proteomic data on human primary tumor and adjacent normal tissues from endometria49 and breasts50. The expression levels of oSCRIB and SCRIB proteins were simultaneously increased in human endometrial (Fig. 5a) and breast (Fig. 5b) primary tumors as compared with adjacent normal tissues. This suggests translational dysregulation of both oSCRIB and SCRIB on oSCRIB–SCRIB mRNAs during carcinogenesis.
Fig. 5. The relationship between oSCRIB and SCRIB expression in carcinogenesis.
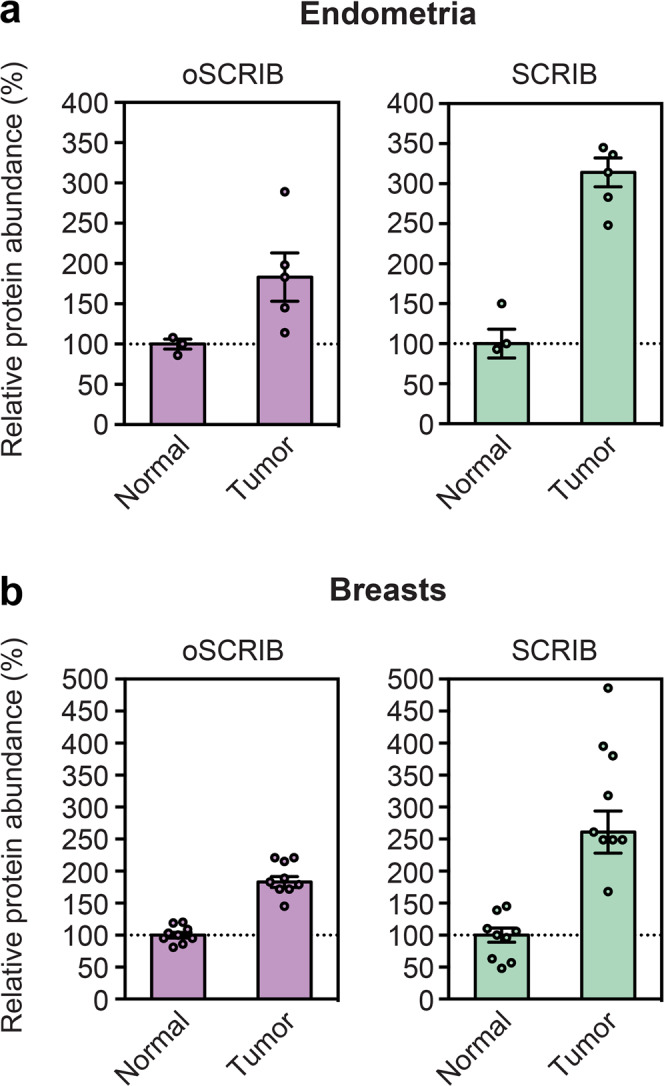
Reanalysis of publicly available quantitative proteomic data on human primary tumor and adjacent normal tissues from endometria (a) and breasts (b). The data calculated by Thermo Proteome Discoverer (Thermo Fisher Scientific) are the median ± SE of independent samples.
To investigate the functional role of oSCRIB in SCRIB translation, we employed an in vitro translation system based on eukaryotic 80 S ribosomes51. We first constructed linear DNA fragments that were used as templates for in vitro transcription and translation. The DNA fragments were constructed to code for a full-length oSCRIB and a partial SCRIB that was C-terminally truncated and fused to the reporter gene sfGFP through a flexible linker (SCRIB-sfGFP) (Fig. 6a). To assess the functionality of oSCRIB, we also mutated the translational start codon (AUG) of oSCRIB into AGG and CCC, both of which are not recognized as the start codons by eukaryotic ribosomes51. The DNA fragment encoding SCRIB–sfGFP alone was used as a positive control of in vitro translation. Then in vitro transcription and the subsequent translation were performed with the DNA fragments. The translational efficiency of SCRIB was quantified by the fluorescence intensity of the reporter protein sfGFP. The oSCRIB–SCRIB–sfGFP and SCRIB–sfGFP constructs showed that existence of the oSCRIB sequence drastically decreased the fluorescence intensity of sfGFP, indicating the inhibitory effect of the oSCRIB sequence on SCRIB translation (Fig. 6b). This was probably due to its extremely high GC base content (87%) (Supplementary Tables 1-2) and potential stem-loop GC base pairing, which leads to reduced translational efficiency52. Furthermore, disrupting the start codon (AUG) of oSCRIB by mutating it to AGG and CCC increased the fluorescence intensity of sfGFP, suggesting that abolishing the translatability of oSCRIB instead increased ribosomal translation of SCRIB (Fig. 6b). Therefore, our results confirmed that the newly discovered oSCRIB and its ribosomal translation constitutively limit the capacity of eukaryotic ribosomes to translate the downstream SCRIB.
Fig. 6. The functional role of oSCRIB in SCRIB translation.

a The design of linear DNA fragments used as templates for in vitro transcription and translation. b The translational efficiency of SCRIB was quantified by the fluorescence intensity of the reporter protein sfGFP. Data are the mean ± SD of three independent reactions.
Discussion
We discovered a new small protein oSCRIB in human cancer proteomes using a proteogenomic approach. Importantly, oSCRIB translation by eukaryotic ribosomes occurred on a polycistronic mRNA encoding two adjacent protein-coding genes, oSCRIB and SCRIB, which correspond to oORF (or altORFCDS) and refCDS, respectively. The generated oSCRIB protein (i.e., an SEP), if bioactive, could directly impact cellular behavior2,7,9.
The next important point to note is that the start codon of the downstream SCRIB is completely embedded in the coding region of oSCRIB (Fig. 2a). Hence, the translation initiation of oSCRIB through 5′ to 3′ scanning of the polycistronic mRNA by eukaryotic ribosomes could hinder that of the downstream out-of-frame SCRIB (Fig. 7), as strongly supported by previous studies on oORFs12,13,53. In other words, SCRIB translation appeared to be dependent on the frequency of leaky scanning2,54, whereby eukaryotic ribosomes bypass the start codon of oSCRIB and reach that of SCRIB (Fig. 7). Our biochemical analysis also supports these views as abolishing the translatability of oSCRIB by disrupting its start codon increased SCRIB translation (Fig. 6). Given that both oSCRIB and SCRIB proteins were detected in human cell and tissue proteomes (as described in the “Results” section), the existence of oSCRIB does not abolish the translatability of SCRIB, likely due to leaky scanning. Eukaryotic ribosomes tend to adopt a start codon surrounded by stronger sequence context or a Kozak sequence (e.g., ACCAUGG in human), and begin translation55,56. Given that oSCRIB lacks a purine (A or G) at position –3 relative to the start codon (AUG), its sequence context is assumed to be weaker than that of SCRIB (Fig. 2a). This disadvantaged sequence context of oSCRIB could permit leaky scanning of the polycistronic mRNA and subsequent translation initiation of SCRIB to some extent.
Fig. 7. Proposed translational switch of the bicistronic oSCRIB–SCRIB gene pair in humans.

Eukaryotic ribosome scanning (5′–3′) of the bicistronic mRNA for a translational start codon (AUG) and the subsequent translation initiation of oSCRIB can hinder that of the downstream out-of-frame tumor suppressor gene SCRIB, as supported by studies of oORFs12,13,53. SCRIB translation seems to be dependent on the frequency of leaky scanning2,54, whereby eukaryotic ribosomes bypass the start codon of oSCRIB and reach that of SCRIB.
Recent work has provided strong evidence that altORF translation, which can arise from unconventional translation with an alternative eukaryotic-initiation factor 2 A in cancer, is an early and important event in oncogene expression and tumor formation14. In addition, translational dysregulation of altORF-dependent refCDS (or cancer driver gene) expression has been demonstrated to be important in tumor development and manipulation of immune-checkpoint proteins13. Similarly, our clinical proteomic data reanalysis also clarified translational dysregulation of the oSCRIB–SCRIB gene pair in human endometrial and breast carcinogenesis.
The translation product of SCRIB is known as a cell-polarity determinant and a large multi-domain scaffold POTSF protein for many key pathways such as antitumorigenic Hippo-YAP/TAZ, Ras/Raf/MEK/ERK (MAPK/ERK), and PI3K/Akt/mTOR and proapoptotic c-Myc-induced signaling pathways22–29. Hence, the regulatory role of oSCRIB in SCRIB translation is likely to be associated with maintaining cellular homeostasis in normal cells. Although further investigations are needed into oSCRIB-dependent SCRIB translation in humans and its biological contribution, our biochemical analysis suggested that the newly discovered oSCRIB and its translation constitutively limit the capacity of eukaryotic ribosomes to translate the downstream SCRIB. Therefore, it is reasonable to assume that the translatable oSCRIB is a cis-regulatory oORF that functions as a ribosomal roadblock and potentially provides a fail-safe mechanism to normal cells for nonexcessive downstream SCRIB expression. This limiting mechanism by oSCRIB region seemed to restrain the overexpression of SCRIB as a cancer-promoting POTSF not only in normal cells but also in cancer cells; however, our clinical proteomic data reanalysis indicated that it was dysregulated in cancer cells for SCRIB overexpression, resulting in their survival and proliferation. These results suggest that cancer cells weakened the effectiveness of the fail-safe mechanism, presumably by increasing the abundance of oSCRIB–SCRIB mRNA and the efficiency of ribosome loading onto the mRNA23. However, since our biochemical analysis clearly indicated that abolishing the translatability of oSCRIB results in SCRIB overexpression (Fig. 6), the existence of oSCRIB region is assumed to partially contribute to limit SCRIB expression in cancer cells and reduce malignant potential of cancer cells to some extent. In other words, lack of the oSCRIB region might further increase malignant potential of cancer cells. Thus, we anticipate that our findings will provide insight into research on cancer and associated biomarkers57. Taken together, the newly identified oORF, oSCRIB, is a promising translational regulatory element for SCRIB translation in the human genome. Additionally, the existence of the most prevalent sequence (GGACU) for the reversible epitranscriptomic m6A modification within oSCRIB-coding region (Fig. 2a) might represent a hidden layer of translational regulation exerted by the dynamic m6A modification58,59.
In this study, we newly constructed the custom sequence databases for SEP discovery. Although our databases consisted of millions of SEP candidates, only a limited number (–0.06%) of SEP candidates were detected in the publicly available high-quality proteomic data. This may imply the existence of SEPs expressed under specific cellular conditions. Thus, together with further validation of the probability-based identification of the remaining high-to-medium-confidence candidates, further proteomic analyses of cells/tissues treated with various stresses might be important in discovering a greater number of SEP candidates in the future.
In conclusion, our proteogenomics-driven gene discovery, which was supported by the integration of publicly available high-quality data from multishot deep proteomics and unbiased transcriptome annotation, proved its capability to mine hidden proteins again. Together with the data-driven multi-omics strategy, cell-free production of authentic proteins facilitated the rapid validation of probability-based SEP identification, thereby providing conclusive evidence of the SEP discovery. This workflow should ultimately lead to complete annotation of the human genome and improve our fundamental understanding of the “blueprint of life” in the future.
Methods
Transcriptomic data processing
Publicly available paired-end RNA-Seq data of polyadenylated mRNAs from several cancer cell lines (three biological replicates for each cell line) were downloaded as a single Sequence Read Archive (SRA) format file or a set of FASTQ format files as follows: the paired-end SRA format data of HeLa (1.57 × 108 read pairs), MCF-7 (1.60 × 108 read pairs), A549 (1.61 × 108 read pairs), and HCT-116 (1.45 × 108 read pairs) cells deposited in the NCBI BioProject (accession PRJNA523380) by the Cancer Cell Line Encyclopedia (CCLE)60,61; the paired-end FASTQ format data of HeLa (1.21 × 108 and 1.18 × 108 read pairs), MCF-7 (1.28 × 108 and 1.32 × 108 read pairs), A549 (7.11 × 107 and 7.72 × 107 read pairs), and HCT-116 (4.59 × 107 and 4.59 × 107 read pairs) cells deposited in the Encyclopedia of DNA Elements (ENCODE)62 database (https://www.encodeproject.org). The paired-end SRA format data were then converted to a set of FASTQ format files using a utility called fasterq-dump in NCBI SRA Toolkit 2.9.6-163. Acquired raw paired-end reads in the FASTQ files were trimmed with Trim Galore! 0.6.264, and the resultant trimmed reads were subjected to de novo assembly using Trinity 2.8.565 with default settings. Possible ORFs within the assembled sequences and, additionally, human RefSeq Transcripts30 (NCBI RefSeq assembly accession GCF_000001405.39) were predicted by TransDecoder.LongOrfs 5.5.065 with a parameter of the minimum codon length of 10, i.e., minimum protein length of nine amino acids. Next, all possible sORFs possibly encoding SEPs of 10–149 amino acids that start and stop with AUG and stop codons, respectively, were extracted using SeqKit 0.12.066. The resulting datasets of sORFs from three replicates of each cell line were aggregated into a nonredundant dataset for each cell line. The translated nucleotide sequences of sORFs, i.e., the amino acid sequences of putative SEPs, were further searched against human RefSeq Proteins30 (NCBI RefSeq assembly accession GCF_000001405.39) using a stand-alone version of the BLAST program, BLAST + 2.9.067. The resulting datasets, SEP sequences and their protein annotations by BLAST+, were combined with SeqKit, and only unannotated sequences were retained in each dataset (HeLa, MCF-7, A549, HCT-116, and RefSeq Transcripts). The dataset of RefSeq Transcripts was further integrated into that of each cell line and the resulting nonredundant datasets were used as the custom SEP sequence databases for HeLa, MCF-7, A549, and HCT-116 cells.
DNA construction
Each DNA fragment that corresponded to oSCRIB, uMKKS1, sfGFP, and oSCRIB–SCRIB–sfGFP was produced and introduced into a pEX-A2J2 vector (Eurofins Genomics, Tokyo, Japan) by Eurofins Genomics (Supplementary Table 2). These DNA sequences were also confirmed by Eurofins Genomics. Then the coding regions with their 3′-UTR derived from the pEX-A2J2 vector were amplified by polymerase chain reaction (PCR) with Tks Gflex DNA polymerase (Takara Bio, Shiga, Japan) and primers listed in Supplementary Table 3 as follows: forward and reverse primers, Fw1-S and Rv1 for oSCRIB and uMKKS1, Fw1-G and Rv1 for sfGFP, Fw1-SG1 and Rv1 for oSCRIB (WT; ATGstart)-SCRIB-sfGFP, Fw1-SG2 and Rv1 for oSCRIB (mt1; ATGstart to AGG)-SCRIB-sfGFP, Fw1-SG3 and Rv1 for oSCRIB (mt2; ATGstart to CCC)-SCRIB-sfGFP, and Fw1-SG4 and Rv1 for SCRIB–sfGFP. Each of the first PCR products was used as a template for a second PCR with primers listed in Supplementary Table 3 as follows: split forward primers, Fw2-E and Fw3-E (3 and 300 nM, respectively), and a nested reverse primer Rv2 (300 nM) for oSCRIB, uMKKS1, and sfGFP, and other split forward primers, Fw2-W and Fw3-W (3 and 300 nM, respectively), and the nested reverse primer Rv2 (300 nM) for oSCRIB (WT, mt1, and mt2)-SCRIB-sfGFP and SCRIB–sfGFP. The second PCR products were purified and concentrated with an Illustra GFX PCR DNA and gel band purification kit (GE Healthcare, Chalfont St. Giles, UK). To confirm the molecular size of the resulting DNA fragments, they were subjected to 1% (w/v) agarose gel electrophoresis and visualized under ultraviolet light on the gel stained with GelRed nucleic acid gel stain (FUJIFILM Wako Chemicals, Osaka, Japan). The DNA fragments were used for in vitro transcription and translation.
In vitro transcription and translation
Bacterial translation coupled with transcription was performed in vitro with the PUREfrex 2.0 system (GeneFrontier, Chiba, Japan). The reactions were initiated with the addition of 5 μL of DNA fragment (oSCRIB, uMKKS1, or sfGFP) into a 15-μL mixture of RNase-free water and PUREfrex 2.0 Solution I, II, and III (GeneFrontier). The reaction mixtures were incubated at 37 °C for 5 h. Then the reaction mixtures were centrifuged at 20,400 × g for 20 min at 4 °C, and the resulting supernatants (soluble fractions) and pellets (insoluble fractions) were applied to SDS-PAGE. The gel was stained with Coomassie Brilliant Blue G-250. Eukaryotic translation was performed in vitro with the WEPRO7240H Expression Kit (CellFree Sciences, Matsuyama, Japan) as previously described51 with minor modifications. The reaction mixtures were then used to quantify the fluorescence intensity of the reporter protein sfGFP at excitation/emission wavelengths of 485/535 nm using a Wallac 1420 Multilabel Counter ARVO MX (PerkinElmer, Waltham, MA, USA).
Protein extraction from HeLa cells
HeLa cells were grown and harvested as previously described68, and the collected cells were frozen at −80 °C until subsequent use. The frozen cells were thawed on ice and lysed by ultrasonic treatment in a solution containing 50 mM sodium phosphate buffer (pH 7.9) and 500 mM NaCl. The cell extracts were applied to SDS-PAGE for protein separation. The gel was stained with Coomassie Brilliant Blue G-250.
Sample preparation for LC–MS/MS analysis
The protein bands that corresponded to in vitro-synthesized proteins (oSCRIB and uMKKS1) and endogenous HeLa proteins of approximately 10–15 kDa, which included oSCRIB protein (12 kDa), were excised from the SDS-PAGE gels. The gel slices were destained and treated with 50 mM dithiothreitol followed by 100 mM sodium iodoacetate for protein reduction and alkylation (carboxymethylation), respectively. Then the gel slices were washed with water. The proteins in the gel were digested with N‐tosyl-l-phenylalanine chloromethyl ketone‐treated trypsin (Worthington Biochemical, Freehold, NJ, USA) in a solution containing 20 mM Tris-HCl buffer (pH 8.0) and 0.05% (w/v) n-dodecyl-β-d-maltoside at 37 °C for 16 h. The resultant peptides were analyzed by LC-MS/MS.
LC–MS/MS analysis
The samples were applied to a packed nanocapillary C18 column (NTCC-360/75-3-105, 0.075 × 105-mm column, particle size: 3 μm; Nikkyo Technos, Tokyo, Japan) for LC–MS/MS analysis with an Easy-nLC 1000 liquid chromatography system (Thermo Fisher Scientific, Waltham, MA, USA). Two types of eluent were used in the column: eluent A consisted of water containing 0.1% (v/v) formic acid and eluent B consisted of acetonitrile containing 0.1% (v/v) formic acid. The column was kept at room temperature, and eluent A was applied as an initial eluent to the column at a flow rate of 300 nL/min. Elution was performed by increasing the proportion of eluent B to eluent A from 0% to 100% over 12 min (35% at 10 min and 100% at 12 min). The last condition was maintained for 8 min. The Q Exactive hybrid Quadrupole-Orbitrap mass spectrometer46 (Thermo Fisher Scientific) was used to analyze the eluate in electrospray-ionization (ESI) positive-ion mode. The MS/MS spectra were obtained by the mass spectrometer that was operated in DDA mode to automatically alternate between a full scan of precursor ions (m/z 300–2000) in the Orbitrap (resolution, 70,000; automatic gain-control target, 3,000,000; maximum injection time, 60 ms) and subsequent HCD–MS/MS scans of the 10 most abundant (top 10) precursor ions (m/z 200–2000) in the Orbitrap (resolution, 17,500; automatic gain-control target, 500,000; maximum injection time, 100 ms; isolation window, 4.0 m/z; normalized collision energy, 30%).
For PRM analysis, sample elution from the column was performed by increasing the proportion of eluent B to eluent A from 0% to 100% over 230 min (1% at 1 s, 2% at 10 min, 10% at 120 min, 20% at 180 min, 40% at 220 min, and 100% at 230 min). The last condition was maintained for 20 min. The mass spectrometer was used to analyze the eluate in ESI positive-ion mode. The MS/MS spectra were obtained by the mass spectrometer that was operated in the PRM mode to perform the HCD–MS/MS scans of selected precursor ions, [M + 3H]3+ of TEPRPPAPSPPSAAAGAR (m/z 577.30419), [M + 3H]3+ and [M + 4H]4+ of AAHPHHAQVHPAVALQPAR (m/z 670.02939 and 502.77386, respectively), [M + 2H]2+ of GVGGQAALFAAGR (m/z 587.82000), [M + 2H]2+ of AGGDLPLQPQPGGAAAR (m/z 788.41553), and [M + 2H]2+ and [M + 3H]3+ of AAQAFFPAAELAQAGPER (m/z 922.96812 and 615.64784, respectively), in the Orbitrap (resolution, 140,000; automatic gain-control target, 3,000,000; maximum injection time, 500 ms; isolation window, 0.8 m/z; normalized collision energy, 30%).
Proteomic data processing
All raw MS/MS files, including the one analyzed above and the publicly available proteome datasets of human cell lines (HeLa, MCF-7, A549, and HCT-116; two biological replicates for each cell line) deposited in the ProteomeXchange35 (dataset identifier PXD00445234) and human primary tumor and adjacent normal tissues (endometria49 and breasts50) deposited in the Clinical Proteomic Tumor Analysis Consortium (CPTAC) Data Portal (https://cptac-data-portal.georgetown.edu/study-summary/S053, https://cptac-data-portal.georgetown.edu/study-summary/S060), were converted into MGF files using Thermo Proteome Discoverer 2.2.0.388 (Thermo Fisher Scientific). The publicly available proteomic data files were first processed to recalibrate precursor masses and then converted into MGF files with Thermo Proteome Discoverer. The MGF files were submitted to an in-house Mascot Server 2.7.0 (Matrix Science, London, UK) and subjected to PSM searches by the Mascot algorithm through Thermo Proteome Discoverer with target- and decoy-sequence databases and the following parameters: variable modifications, acetyl (protein N-term), oxidation (M), and Gln to pyro-Glu conversion (N-term Q); static modification, carboxymethyl (C) for the samples analyzed above or carbamidomethyl (C) for the publicly available proteome datasets and TMT6plex (N-term and K) for the publicly available proteome datasets of human primary tumor/normal adjacent tissues; maximum of three missed cleavages by trypsin digestion; precursor mass tolerance of 15 ppm; fragment mass tolerance of 30 mmu. The following target-sequence databases and their decoy-sequence databases generated by reversing the target sequences with Thermo Proteome Discoverer were each used in separate PSM searches for target decoy-based FDR estimation37,38. For proteogenomic data mining, the two different target databases in parallel, i.e., the combination of human RefSeq Proteins and the custom SEP-sequence database (for HeLa, MCF-7, A549, or HCT-116) as constructed above were used; for the detection of the in vitro-synthesized proteins, the target database that consists of the protein sequences of oSCRIB and uMKKS1 and the whole-protein sequences of Escherichia coli BL21 (DE3) (NCBI GenBank accession CP001509, version CP001509.369) were used; for the detection of endogenous HeLa proteins of approximately 10–15 kDa, the target database that consists of the protein sequences of oSCRIB and uMKKS1 and the previously annotated SEP sequences (–150 amino acids) collected from human RefSeq Proteins was used; and for reanalysis of clinical human proteomic data (TMT-based quantitative proteomic data) on human primary tumor and adjacent normal tissues (endometria and breasts), the target database that consists of the protein sequences of uSCRIB and oSCRIB and human RefSeq Proteins (including SCRIB) was used. The search results were filtered by the Percolator algorithm37,38 to maintain an estimated target decoy-based FDR of 1% at the peptide and protein levels. The identified peptides/proteins were further searched against the universal protein resource (UniProt) database (UniProt Knowledgebase). A peptide-centric search of the oSCRIB-derived peptides (AGGDLPLQPQPGGAAAR and AAQAFFPAAELAQAGPER) and the uMKKS1-derived peptide (NDDIPEQDSLGLSNLQK) from the publicly available proteomic data (MGF format) was executed by a stand-alone version (1.6.0) of the PepQuery program39 with default parameters (reference protein database, human RefSeq Proteins; variable modification, oxidation (M); static modification, carbamidomethyl (C); maximum of two missed cleavages by trypsin digestion; precursor mass tolerance of 10 ppm; fragment mass tolerance of 600 mmu; and scoring algorithm, Hyperscore). Manual inspection of MS/MS spectra was performed with Thermo Proteome Discoverer and Thermo Xcalibur Qual Browser 3.1.66.10 (Thermo Fisher Scientific). The results were then illustrated using an integrative proteomics data viewer PDV 1.6.070. Mapping of DNA sequences corresponding to peptides/proteins of interest onto the human genome was conducted with an integrative genomics viewer IGV 2.8.071.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
The authors thank Dr. Takehiro Suzuki (RIKEN) for his technical support with LC–MS/MS, Dr. Makoto Muroi and Ms. Naoko Ogawa (RIKEN) for their technical assistance with cell culture, and Ms. Tamayo Oishi (RIKEN) for her help during the early days of the study.
Author contributions
Y. N. and N. D. conceived the research; Y. N. designed and conducted the research, Y. N. and N. D. analyzed the results, Y. N. performed the biological interpretation of the results, and Y. N. wrote the paper.
Data availability
All data supporting the findings of this study are available within the paper and its supplementary information file. Uncropped and unedited gel images are included in Supplementary Figure 1. Source data for graphs are included in Supplementary Data 1. The protein-sequence data reported in this paper will appear in the UniProt Knowledgebase under the accession number C0HLS1 for oSCRIB in humans (Homo sapiens). The raw mass spectrometric data and Mascot-related files have been deposited to the ProteomeXchange Consortium35 via the PRIDE partner repository72 with the dataset identifier PXD027841 and 10.6019/PXD027841.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review informationCommunications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Debarka Sengupta, Brooke LaFlamme.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yuhta Nomura, Email: yuta.nomura@riken.jp.
Naoshi Dohmae, Email: dohmae@riken.jp.
Supplementary information
The online version contains supplementary material available at 10.1038/s42003-021-02619-8.
References
- 1.Kozak M. Comparison of initiation of protein synthesis in procaryotes, eucaryotes, and organelles. Microbiol. Rev. 1983;47:1–45. doi: 10.1128/mr.47.1.1-45.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrews SJ, Rothnagel JA. Emerging evidence for functional peptides encoded by short open reading frames. Nat. Rev. Genet. 2014;15:193–204. doi: 10.1038/nrg3520. [DOI] [PubMed] [Google Scholar]
- 3.Mouilleron H, Delcourt V, Roucou X. Death of a dogma: eukaryotic mRNAs can code for more than one protein. Nucleic Acids Res. 2016;44:14–23. doi: 10.1093/nar/gkv1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Couso JP, Patraquim P. Classification and function of small open reading frames. Nat. Rev. Mol. Cell Biol. 2017;18:575–589. doi: 10.1038/nrm.2017.58. [DOI] [PubMed] [Google Scholar]
- 5.Orr MW, Mao Y, Storz G, Qian SB. Alternative ORFs and small ORFs: shedding light on the dark proteome. Nucleic Acids Res. 2020;48:1029–1042. doi: 10.1093/nar/gkz734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Slavoff SA, et al. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nat. Chem. Biol. 2013;9:59–64. doi: 10.1038/nchembio.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Slavoff SA, Heo J, Budnik BA, Hanakahi LA, Saghatelian A. A human short open reading frame (sORF)-encoded polypeptide that stimulates DNA end joining. J. Biol. Chem. 2014;289:10950–10957. doi: 10.1074/jbc.C113.533968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aspden JL, et al. Extensive translation of small Open Reading Frames revealed by Poly-Ribo-Seq. eLife. 2014;3:e03528. doi: 10.7554/eLife.03528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saghatelian A, Couso JP. Discovery and characterization of smORF-encoded bioactive polypeptides. Nat. Chem. Biol. 2015;11:909–916. doi: 10.1038/nchembio.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinez TF, et al. Accurate annotation of human protein-coding small open reading frames. Nat. Chem. Biol. 2020;16:458–468. doi: 10.1038/s41589-019-0425-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calvo SE, Pagliarini DJ, Mootha VK. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc. Natl Acad. Sci. USA. 2009;106:7507–7512. doi: 10.1073/pnas.0810916106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnstone TG, Bazzini AA, Giraldez AJ. Upstream ORFs are prevalent translational repressors in vertebrates. EMBO J. 2016;35:706–723. doi: 10.15252/embj.201592759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu Y, et al. Translation control of the immune checkpoint in cancer and its therapeutic targeting. Nat. Med. 2019;25:301–311. doi: 10.1038/s41591-018-0321-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sendoel A, et al. Translation from unconventional 5’ start sites drives tumour initiation. Nature. 2017;541:494–499. doi: 10.1038/nature21036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim MS, et al. A draft map of the human proteome. Nature. 2014;509:575–581. doi: 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Legnini I, et al. Circ-ZNF609 is a circular RNA that can be translated and functions in myogenesis. Mol. Cell. 2017;66:22–37. doi: 10.1016/j.molcel.2017.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pamudurti NR, et al. Translation of CircRNAs. Mol. Cell. 2017;66:9–21. doi: 10.1016/j.molcel.2017.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eliuk S, Makarov A. Evolution of orbitrap mass spectrometry instrumentation. Annu. Rev. Anal. Chem. 2015;8:61–80. doi: 10.1146/annurev-anchem-071114-040325. [DOI] [PubMed] [Google Scholar]
- 19.Evans VC, et al. De novo derivation of proteomes from transcriptomes for transcript and protein identification. Nat. Methods. 2012;9:1207–1211. doi: 10.1038/nmeth.2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nesvizhskii AI. Proteogenomics: concepts, applications and computational strategies. Nat. Methods. 2014;11:1114–1125. doi: 10.1038/nmeth.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma J, Saghatelian A, Shokhirev MN. The influence of transcript assembly on the proteogenomics discovery of microproteins. PLoS ONE. 2018;13:e0194518. doi: 10.1371/journal.pone.0194518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhan L, et al. Deregulation of scribble promotes mammary tumorigenesis and reveals a role for cell polarity in carcinoma. Cell. 2008;135:865–878. doi: 10.1016/j.cell.2008.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vaira V, et al. Aberrant overexpression of the cell polarity module scribble in human cancer. Am. J. Pathol. 2011;178:2478–2483. doi: 10.1016/j.ajpath.2011.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cordenonsi M, et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell. 2011;147:759–772. doi: 10.1016/j.cell.2011.09.048. [DOI] [PubMed] [Google Scholar]
- 25.Pearson HB, et al. The polarity protein Scrib mediates epidermal development and exerts a tumor suppressive function during skin carcinogenesis. Mol. Cancer. 2015;14:169. doi: 10.1186/s12943-015-0440-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen B, et al. ZDHHC7-mediated S-palmitoylation of Scribble regulates cell polarity. Nat. Chem. Biol. 2016;12:686–693. doi: 10.1038/nchembio.2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kapil S, et al. The cell polarity protein Scrib functions as a tumor suppressor in liver cancer. Oncotarget. 2017;8:26515–26531. doi: 10.18632/oncotarget.15713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bonello TT, Peifer M. Scribble: a master scaffold in polarity, adhesion, synaptogenesis, and proliferation. J. Cell Biol. 2019;218:742–756. doi: 10.1083/jcb.201810103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Santoni MJ, Kashyap R, Camoin L, Borg JP. The Scribble family in cancer: twentieth anniversary. Oncogene. 2020;39:7019–7033. doi: 10.1038/s41388-020-01478-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pruitt KD, et al. RefSeq: an update on mammalian reference sequences. Nucleic Acids Res. 2014;42:D756–D763. doi: 10.1093/nar/gkt1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schneider VA, et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 2017;27:849–864. doi: 10.1101/gr.213611.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 33.Batth TS, Francavilla C, Olsen JV. Off-line high-pH reversed-phase fractionation for in-depth phosphoproteomics. J. Proteome Res. 2014;13:6176–6186. doi: 10.1021/pr500893m. [DOI] [PubMed] [Google Scholar]
- 34.Bekker-Jensen DB, et al. An optimized shotgun strategy for the rapid generation of comprehensive human proteomes. Cell Syst. 2017;4:587–599. doi: 10.1016/j.cels.2017.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vizcaíno JA, et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014;32:223–226. doi: 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 37.Käll L, Canterbury JD, Weston J, Noble WS, MacCoss MJ. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods. 2007;4:923–925. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- 38.Brosch M, Yu L, Hubbard T, Choudhary J. Accurate and sensitive peptide identification with Mascot Percolator. J. Proteome Res. 2009;8:3176–3181. doi: 10.1021/pr800982s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wen B, Wang X, Zhang B. PepQuery enables fast, accurate, and convenient proteomic validation of novel genomic alterations. Genome Res. 2019;29:485–493. doi: 10.1101/gr.235028.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Akimoto C, et al. Translational repression of the McKusick-Kaufman syndrome transcript by unique upstream open reading frames encoding mitochondrial proteins with alternative polyadenylation sites. Biochim. Biophys. Acta. 2013;1830:2728–2738. doi: 10.1016/j.bbagen.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 41.Vanderperre B, et al. Direct detection of alternative open reading frames translation products in human significantly expands the proteome. PLoS ONE. 2013;8:e70698. doi: 10.1371/journal.pone.0070698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dominissini D, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 43.Meyer KD. DART-seq: an antibody-free method for global m6A detection. Nat. Methods. 2019;16:1275–1280. doi: 10.1038/s41592-019-0570-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shimizu Y, et al. Cell-free translation reconstituted with purified components. Nat. Biotechnol. 2001;19:751–755. doi: 10.1038/90802. [DOI] [PubMed] [Google Scholar]
- 45.Pédelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 2006;24:79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- 46.Michalski A, et al. Mass spectrometry-based proteomics using Q Exactive, a high-performance benchtop quadrupole Orbitrap mass spectrometer. Mol. Cell. Proteom. 2011;10:M111.011015. doi: 10.1074/mcp.M111.011015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Olsen JV, et al. Higher-energy C-trap dissociation for peptide modification analysis. Nat. Methods. 2007;4:709–712. doi: 10.1038/nmeth1060. [DOI] [PubMed] [Google Scholar]
- 48.Nguyen CDL, et al. A sensitive and simple targeted proteomics approach to quantify transcription factor and membrane proteins of the unfolded protein response pathway in glioblastoma cells. Sci. Rep. 2019;9:8836. doi: 10.1038/s41598-019-45237-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dou Y, et al. Proteogenomic characterization of endometrial carcinoma. Cell. 2020;180:729–748. doi: 10.1016/j.cell.2020.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krug K, et al. Proteogenomic landscape of breast cancer tumorigenesis and targeted therapy. Cell. 2020;183:1436–1456. doi: 10.1016/j.cell.2020.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ohta T, Matsuoka H, Nomura Y, Tozawa Y. Control of translational initiation in the wheat-embryo cell-free protein expression system for producing homogenous products. Protein Expr. Purif. 2010;73:15–22. doi: 10.1016/j.pep.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 52.Babendure JR, Babendure JL, Ding JH, Tsien RY. Control of mammalian translation by mRNA structure near caps. RNA. 2006;12:851–861. doi: 10.1261/rna.2309906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McGeachy AM, Ingolia NT. Starting too soon: upstream reading frames repress downstream translation. EMBO J. 2016;35:699–700. doi: 10.15252/embj.201693946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sriram, A., Bohlen, J. & Teleman, A. A. Translation acrobatics: how cancer cells exploit alternate modes of translational initiation. EMBO Rep19, e45947 (2018). [DOI] [PMC free article] [PubMed]
- 55.Kozak M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell. 1986;44:283–292. doi: 10.1016/0092-8674(86)90762-2. [DOI] [PubMed] [Google Scholar]
- 56.Lütcke HA, et al. Selection of AUG initiation codons differs in plants and animals. EMBO J. 1987;6:43–48. doi: 10.1002/j.1460-2075.1987.tb04716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang B, et al. Clinical potential of mass spectrometry-based proteogenomics. Nat. Rev. Clin. Oncol. 2019;16:256–268. doi: 10.1038/s41571-018-0135-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou J, et al. N6-Methyladenosine guides mRNA alternative translation during integrated stress response. Mol. Cell. 2018;69:636–647. doi: 10.1016/j.molcel.2018.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dong, L. et al. Relaxed initiation pausing of ribosomes drives oncogenic translation. Sci. Adv.7, eabd6927 (2021). [DOI] [PMC free article] [PubMed]
- 60.Tsherniak A, et al. Defining a cancer dependency map. Cell. 2017;170:564–576. doi: 10.1016/j.cell.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ghandi M, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019;569:503–508. doi: 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Davis CA, et al. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018;46:D794–D801. doi: 10.1093/nar/gkx1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.SRA Toolkit Development Team. NCBI SRA Toolkit. https://github.com/ncbi/sra-tools (2019).
- 64.Krueger, F. Trim Galore! https://github.com/FelixKrueger/TrimGalore (2019).
- 65.Haas BJ, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013;8:1494–1512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shen W, Le S, Li Y, Hu F. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS ONE. 2016;11:e0163962. doi: 10.1371/journal.pone.0163962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Camacho C, et al. BLAST+: architecture and applications. BMC Bioinform. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Muroi M, Osada H. Proteomic profiling for target identification of biologically active small molecules using 2D DIGE. Methods Mol. Biol. 2019;1888:127–139. doi: 10.1007/978-1-4939-8891-4_7. [DOI] [PubMed] [Google Scholar]
- 69.Kim S, et al. Genomic and transcriptomic landscape of Escherichia coli BL21(DE3) Nucleic Acids Res. 2017;45:5285–5293. doi: 10.1093/nar/gkx228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li K, Vaudel M, Zhang B, Ren Y, Wen B. PDV: an integrative proteomics data viewer. Bioinformatics. 2019;35:1249–1251. doi: 10.1093/bioinformatics/bty770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Robinson JT, et al. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vizcaíno JA, et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016;44:D447–D456. doi: 10.1093/nar/gkv1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019;20:608–624. doi: 10.1038/s41580-019-0168-5. [DOI] [PubMed] [Google Scholar]
- 74.Michel AM, Kiniry SJ, O’Connor PBF, Mullan JP, Baranov PV. GWIPS-viz: 2018 update. Nucleic Acids Res. 2018;46:D823–D830. doi: 10.1093/nar/gkx790. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
All data supporting the findings of this study are available within the paper and its supplementary information file. Uncropped and unedited gel images are included in Supplementary Figure 1. Source data for graphs are included in Supplementary Data 1. The protein-sequence data reported in this paper will appear in the UniProt Knowledgebase under the accession number C0HLS1 for oSCRIB in humans (Homo sapiens). The raw mass spectrometric data and Mascot-related files have been deposited to the ProteomeXchange Consortium35 via the PRIDE partner repository72 with the dataset identifier PXD027841 and 10.6019/PXD027841.