

Abstract
The central role of iron in tumor progression and metastasis motivates the development of iron-binding approaches in cancer chemotherapy. Disulfide-based prochelators are reductively activated upon cellular uptake to liberate thiol chelators responsible for iron sequestration. Herein, a trimethyl thiosemicarbazone moiety and the imidazole-2-thione heterocycle are incorporated in this prochelator design. Iron binding of the corresponding tridentate chelators leads to the stabilization of a low-spin ferric center in 2:1 ligand-to-metal complexes. Native mass spectrometry experiments show that the prochelators form stable disulfide conjugates with bovine serum albumin, thus affording novel bioconjugate prochelator systems. Antiproliferative activities at submicromolar levels are recorded in a panel of breast, ovarian and colorectal cancer cells, along with significantly lower activity in normal fibroblasts.
Keywords: iron chelation, cancer, thiosemicarbazone, imidazole-2-thione, albumin
Entry for the Table of Contents
Targeting the altered iron metabolism of cancer cells, disulfide-based prochelators are activated for metal coordination upon intracellular reduction. Novel substituted thiosemicarbazone and imidazole-2-thione systems covalently modify bovine serum albumin forming stable bioconjugate prochelators. Antiproliferative activities at submicromolar levels are recorded in breast, ovarian, and colon cell lines.
((Twitter:)) @elisatomat
((Sonderheft Text:))
This article belongs to the joint Special Collection with the European Journal of Inorganic Chemistry, “Metals in Medicine”.
((Achtung: VIP))
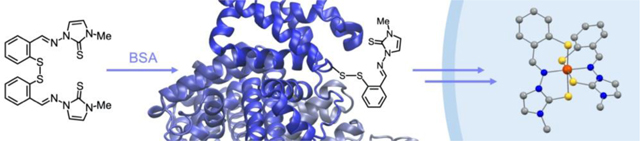
An altered iron metabolism characterizes malignant cells, in which enhanced iron uptake and retention support rapid proliferation rates.[1] This “iron addiction” affects multiple aspects of cancer progression, including not only proliferation but also metastasis and the reprogramming of tumor microenvironments to facilitate growth.[2] Critically, observations at the molecular and cellular level are reflected by the prognostic value of the expression levels of several proteins involved in iron handling (e.g., lipocalin 2,[3] ferroportin[4]). Iron-binding small molecules (chelators), either alone or as adjuvants, are therefore viewed as promising candidates for therapeutic interventions in cancer treatment.[5, 6] Several studies have focused on clinically approved chelators for iron overload disorders (e.g., desferrioxamine, DFO, Figure 1); however, antiproliferative thiosemicarbazones (e.g., Dp44mT, Figure 1) have emerged as a major class of drug candidates for anticancer indications.[7, 8, 9] For instance, thiosemicarbazones Triapine, DpC, and more recently COTI-2 have been investigated in several clinical trials for cancer chemotherapy.[7, 8]
Figure 1.

Structures of reported iron chelators and disulfide-based prochelators.
With the goal of improving the intracellular metal-binding selectivity of thiosemicarbazone and hydrazone chelators, we have developed a class of prochelators (e.g., (TC1-S)2, (AH1-S)2, Figure 1) in which a disulfide linkage functions as a switch to activate iron binding upon intracellular reduction.[10] The resulting tridentate chelators, which feature (S–,N,S) or (S–,N,O) binding units, lead to intracellular iron deprivation.[11, 12] These compounds have antiproliferative effects at low micromolar concentrations in cultured cancer cells (including neuroepithelioma, breast, and colon cancer cell lines).[10, 12, 13] Higher concentrations of intracellular reductants (e.g., reduced glutathione, GSH) enhance the toxicity of these disulfide-masked prochelators,[11] therefore this approach could benefit from preferential activation in the more reducing environment of rapidly proliferating malignant cells relative to normal cells.[14] Additionally, these thiosemicarbazone prochelators affect the iron-releasing phenotype of alternatively activated (M2) macrophages,[15] which play a key role in tumor promotion.[2] Indeed iron-binding strategies are poised to impact not only cancer cell proliferation but also iron availability in the tumor microenvironment.[16]
Herein, we examine the effect of N-alkylation of the thiosemicarbazone unit on lipophilicity, iron coordination, and antiproliferative activity of the disulfide prochelators. In particular, we describe the synthesis and biological activity evaluation of fully methylated thiosemicarbazone (244mTC-S)2 and imidazole-2-thiones (IT1-S)2 and (IT2-S)2 (Figure 2), thus introducing a heterocyclic moiety in this prochelator design. Furthermore, we sought to investigate the reactivity of these compounds with serum albumin, thereby revealing a new class of antiproliferative bioconjugate prochelators.
Figure 2.

Disulfide-based prochelators characterized in this study.
In -N-pyridine thiosemicarbazones such as Triapine and Dp44mT (Figure 1), methylation of the hydrazonic nitrogen prevents formation of the deprotonated thioenolate within the typical (N,N,S–) donor unit thus impacting metal-binding affinities and generally decreasing toxicity.[17, 18] Conversely, tridentate thiosemicarbazones deriving from salicylaldehyde and mercaptobenzaldehyde (such as the reduction products of prochelators (TC1-S)2 and (TC2-S)2) coordinate as thioketones with (O–,N,S) or (S–,N,S) binding units upon deprotonation of the phenolic (or thiophenolic) donor.[7, 10] As we have recently shown in the case of the AH1 system,[19] however, deprotonation of the iron-bound chelators at the hydrazonic nitrogen affects the charge and speciation of the iron complexes in solution and possibly their distribution in cells. In this study, we investigate (244mTC-S)2 as a methylated analog of previously reported disulfide-based prochelators of the TC series, as well as (IT1-S)2 and (IT2-S)2, which feature a different sulfur donor associated to the π system of a heterocyclic ring. In particular, the imidazole-2-thione moiety, which is found in naturally occurring histidine derivative ergothioneine and in clinically approved antithyroid drug methimazole,[20] was chosen for its well-established metal-binding properties.[21]
The thiosemicarbazone prochelator (244mTC-S)2 was synthesized in two steps (Scheme S1) via formation of 2,4,4-trimethyl thiosemicarbazide and then, as in the preparation of other disulfide-masked prochelators of this class,[12] condensation with 2,2’-dithiodibenzaldehyde in ethanol. For the synthesis of (IT1-S)2 (Scheme S2), 1-methylimidazole was aminated using O-(2,4-dinitrophenyl)hydroxylamine (DNPH)[22] and the resulting heterocycle was then treated with elemental sulfur to give 1-amino-3-methylimidazoline-2-thione[23] as a precursor for the final condensation step. (IT2-S)2 was prepared through an analogous procedure (Scheme S3).
The lipophilicity of iron chelators affects their membrane permeability and ultimately their ability to mobilize intracellular iron.[24, 25, 26] For a comparative assessment of the lipophilicity of the new compounds, the octanol/water partition coefficients (log Po/w values) were determined using the stir-flask method and analytical HPLC (see ESI for details). The log Po/w values (Table S1) of (244mTC-S)2 and (IT1-S)2 are 4.0 ± 0.1 and 3.3 ± 0.2, respectively, whereas the concentration of (IT2-S)2 in the aqueous phase was not sufficient for reliable measurements. Indeed, using five different predictive models, the SwissADME web tool[27] predicts a log Po/w of 6.6 for this compound and therefore a low solubility in water. As expected, the additional N-substitution on the new compounds led to higher lipophilicity relative to the first-generation prochelator (TC2-S)2 (Figure 1), which has a measured log Po/w of 2.0 ± 0.2. Additionally, in our experimental conditions, the log Po/w of highly antiproliferative thiosemicarbazone Dp44mT was also found to be 2.0 ± 0.2, in accordance with previously reported data.[28] Overall, the log Po/w values of (244mTC-S)2 and (IT1-S)2 fall within the range (between ~2.5 and ~4.0) which was found to be optimal for antiproliferative activity in a series of thiosemicarbazone chelators.[29]
Because the new functionalization has the potential to affect the donor ability of the thiocarbonyl donor in the metal-binding unit, we sought to characterize the iron complexes of the new chelators and their electronic structure. The disulfide prochelators were reduced quantitatively using dithiothreitol (DTT) under an inert atmosphere and the thiol chelators were isolated by precipitation (see ESI for characterization data). The iron complexes were then prepared by combining the thiol-based ligands with Fe(BF4)2•6H2O (0.5 equiv) in THF under aerobic conditions. Crystallization from acetone/pentane solutions afforded single crystals suitable for X-ray diffraction analysis (see below). These complexes also formed promptly in neutral aqueous solutions as confirmed by electrospray ionization mass spectrometry (Figures S1–S2).
The 244mTC chelator forms a 2:1 complex presenting a pseudo-octahedral geometry and a [BF4]– counterion (Figures 3, S3, Table S2). With two tridentate monoanionic ligands, the iron center is ferric as in previously reported thiosemicarbazone chelators of this series, which stabilize the ferric oxidation state even in the presence of trace oxygen.[10] Unlike iron-bound TC2, the tridentate ligand platform of 244mTC departs significantly from planarity: specifically, the methyl substitution at N2 disrupts the π delocalization of the thiosemicarbazone moiety, in which the N2-C8 distance (1.362(4)-1.383(4) Å) has a more pronounced single-bond character relative to those in N2-unsubstituted analogs (1.325(4)-1.348(3) Å).[10] The iron-thiolate and iron-imine bond distances in the primary coordination sphere (Table S2) are consistent with those in other low-spin ferric complexes of aromatic thiolates,[30] hydrazones,[19] and thiosemicarbazones,[7, 10] thus suggesting an S=½ electronic configuration (vide infra).
Figure 3.

Crystal structures of [Fe(244mTC−H)2]+ (left) and [Fe(IT1−H)2]+ (right) showing a partial atom labeling scheme. Thermal ellipsoids are scaled to the 50% probability level. Carbon-bound hydrogen atoms in calculated positions and [BF4]– counter ions are not shown (CCDC, left: 2060902, right: 2060901)
The crystal structure of the iron complex of imidazole-2-thione chelator IT1 (Figures 3, S4, Table S2) also presents Fe–S and Fe–N distances consistent with a ferric center as well as an overall cationic charge balanced by a tetrafluoroborate ion. Whereas several imidazole-2-thione ligands were found to stabilize ferrous ions,[31, 32] the incorporation of this moiety in a tridentate mercaptobenzaldehyde hydrazone leads to a stable ferric complex. The two ligands coordinate in meridional fashion but again lack the planarity of a fully delocalized π system. Notably, the thione bonds (C10–S2, 1.6869(17)-1.6917(17) Å) are slightly contracted relative to those in the 244mTC complex (C8–S2, 1.699(3)-1.706(3) Å), consistent with a heterocyclic thiocarbonyl and indicating that the ligand maintains an imidazole-2-thione structure as opposed to adopting an imidazolium-2-thiolate donor structure upon iron binding.[21] As such, the introduction of a heterocycle within the ligand framework in IT1 does not alter significantly the iron-binding unit when compared to the thiosemicarbazones of this class. These binding units remain distinct from those of the pyridyl thiosemicarbazone systems (e.g., Triapine, Dp44mT, DpC) that coordinate as ene-thiolates.[7, 18, 29]
The continuous-wave EPR spectra of complexes [Fe(244mTC−H)2][BF4] and [Fe(IT1−H)2][BF4] confirmed a low-spin ferric configuration (Figure 4). The rhombic signals, with turning points at g = (2.129, ~2.110, 2.010) and g = (2.132, 2.096, 2.015) respectively, are similar to the rather narrow spectra of other cationic low-spin Fe(III) complexes deriving from prochelators of the same class, such as [Fe(TC1−H)2]+ and [Fe(AH1−H)2]+.[11, 19] The new complexes, however, exhibit a simpler speciation in aqueous solution: whereas the AH1 complex is present in two protonation states at pH 7.0,[19] [Fe(244mTC−H)2]+ and [Fe(IT1−H)2]+ cannot be deprotonated at the hydrazonic nitrogen and their EPR spectra are indicative of a single species at neutral pH.
Figure 4.

EPR spectra of [Fe(244mTC−H)2]+ (top) and [Fe(IT1−H)2]+ (bottom) in frozen DMSO/water solution (70/30%, v/v). The pH of the solution was estimated at 7.0 using colorimetric indicator strips with a sensitivity of 0.5 pH units. Experimental conditions: microwave frequency, 9.4 GHz; microwave power, 2 mW; magnetic field modulation amplitude, 0.2 mT; temperature, 77 K.
Prior to testing the antiproliferative activity of the new prochelators in cultured cells, we sought to determine their stability and reactivity in the presence of serum albumin. Human serum albumin (HSA) has been widely recognized as an effective drug-delivery carrier owing to its abundance in human blood, non-immunogenicity, good biodegradability, and long lifetime in circulation.[33, 34] Its tendency to accumulate in tumors, both through passive processes and active internalization, make albumin an appealing carrier for anticancer drugs.[35] For instance, clinically approved Abraxane is an albumin-paclitaxel nanoparticle preparation that capitalizes on the ability of albumin to encapsulate small molecules via non-covalent interactions.[34] Notably, non-covalent HSA binding increases the cellular uptake and antiproliferative activity of thiosemicarbazone Dp44mT.[36]
Our in-vitro experiments probed the interaction with bovine serum albumin (BSA), which is abundant in cell growth media and has 78% sequence homology with HSA.[37] Prochelators (244mTC-S)2 and (IT1-S)2 are not prone to hydrolytic degradation in phosphate-buffered saline solutions (PBS, pH 7.40, 0.15% DMSO) and their optical absorption spectra remain unchanged over a period of 8 h (Figure S5). Conversely, in the presence of BSA (40 μM), the absorption profile of both compounds (in the 300 to 400 nm range) undergoes a gradual change that is complete within the first 0.5 h for (244mTC-S)2 and the first 2 h for (IT1-S)2 (Figure S6). Possibly owing to low solubility and aggregation, the spectrum of prochelator (IT2-S)2 presented low absorbance in the tested conditions and no conclusions could be drawn. For (244mTC-S)2 and (IT1-S)2, the putative albumin adducts remain stable in solution for at least 8 h. Overall, these data indicated that the prochelators are stable in neutral aqueous conditions, and that the potential for interaction with serum albumin warranted further investigation.
The accessible Cys34 residue of albumin, which is present both in HSA and BSA and remains mostly reduced in blood circulation, has been long recognized as a reactive handle to connect maleimide-containing molecules[38], such as doxorubicin in Aldoxorubicin[39] and several platinum compounds.[40, 41] Because Cys34 is also accessible for disulfide exchange reactions,[42] we sought to investigate the interaction between BSA and our disulfide-based prochelators using native mass spectrometry methods.
Solutions of BSA (1.0 μM in 0.2 M aqueous ammonium acetate, pH 6.8) were incubated at room temperature for 24 hours in the presence of the disulfide prochelators (10 μM) or of thiosemicarbazone thioether TE1 (10 μM), which was employed as a negative control lacking the disulfide linkage.[12] For prochelators (244mTC-S)2 and (IT1-S)2, mass spectrometric data indicated modification of the protein and formation of a conjugate consistent with disulfide exchange (Figure 5, panels a-d). Conversely, (IT2-S)2 and the thioether control failed to modify the protein, which was detected unchanged (Figure 5, panels e-h). As such, the facile conversion of the symmetric disulfide prochelators (244mTC-S)2 and (IT1-S)2 into BSA conjugates is expected to generate new prochelator systems in the cell growth media. In addition to higher solubility and bioavailability,[43] and of interest for future applications in vivo, these albumin-based prochelators may also elicit enhanced tumor accumulation and longer lifetimes in blood circulation.
Figure 5.

Covalent modifications of BSA by disulfide prochelators as monitored by native mass spectrometry. Spectra are shown on the left with annotated charge states. Corresponding deconvolved mass distributions are shown on the right. Solutions of BSA (1.0 μM) in aqueous ammonium acetate (0.2 M, pH 6.8) were incubated in the presence of prochelators (244mTC-S)2 (panels a, b), (IT1-S)2 (c, d), (IT2-S)2 (e, f), or control thioether TE1 (g, h). The concentration of all tested compounds was 10 μM, and the incubations were conducted at room temperature for 24 h.
The antiproliferative activity of the compounds was investigated in a panel of malignant cells, featuring two breast (MDA-MB-231, MCF-7), one ovarian (A2780), and two colorectal (Caco-2, HT-29) cancer cell lines. These cell lines were chosen because of the established connections between iron dyshomeostasis and breast,[4] ovarian,[44] and colorectal[45] cancers. In addition, a normal lung fibroblast cell line (MRC-5) was included as a comparison. Because of its generally low aqueous solubility, which was not ameliorated by albumin conjugation, (IT2-S)2 was not included in the cell-based assays of antiproliferative activity. After incubation periods of 72 h, the IC50 values of (244mTC-S)2 and (IT1-S)2 were below 0.4 μM in four cancer cell lines and consistently below 1 μM (Table 1), thus presenting the highest antiproliferative activities in their class of disulfide-based prochelators. For instance, the most toxic compounds of this series, thiosemicarbazone (TC1-S)2 and hydrazone (AH1-S)2, have significantly higher IC50 values in breast cancer cells, ranging from 4 to 12 μM (Table 1). Siderophore DFO was included in this screen as a reference iron-sequestering compound and its IC50 values in the low micromolar range (2–20 μM) were consistent with previous reports in various cell lines.[10, 17]
Table 1.
Antiproliferative activity of prochelators and comparison compounds in cancer cell lines and normal lung fibroblasts.
| IC50 values (μM)[a] | ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| MDA-MB-231 (Breast) | MCF-7 (Breast) | A2780 (Ovary) | Caco-2 (Colon) | HT-29 (Colon) | MRC-5 (Normal lung) | |
|
| ||||||
| (244mTC-S) 2 | 0.33 ± 0.06 | 0.12 ± 0.04 | 0.14 ± 0.04 | 0.38 ± 0.06 | 0.9 ± 0.2 | 17 ± 1 |
| (IT1-S) 2 | 0.35 ± 0.02 | 0.36 ± 0.02 | 0.21 ± 0.06 | 0.37 ± 0.02 | 0.85 ± 0.15 | 24 ± 2 |
| (TC1-S) 2 | 4.4 ± 0.7[b] | 12.3 ± 0.8[b] | n.d. | n.d. | n.d. | 30 ± 5[b] |
| (AH1-S) 2 | 6.7 ± 0.5[b] | 4.6 ± 0.9[b] | n.d. | n.d. | n.d. | 20 ± 1[b] |
| DFO | 5.8 ± 0.4 | 21 ± 2 | 3.83 ± 0.02 | 3.7 ± 0.3 | 2.4 ± 0.1 | 9 ± 1 |
IC50 values from MTT assays in the indicated cell lines after exposure to tested compounds for 72 h; values are presented as mean ± SDM, n = 3; n.d.: not determined.
As reported in Ref. 12.
The antiproliferative activity of iron chelators is generally lower in normal cells, reflecting a lower susceptibility to iron deprivation when compared to malignant cells. In addition, the reductive activation switch of disulfide prochelators often leads to higher antiproliferative effects in the more reducing environments of cancer cells, as shown for instance in the case of (TC1-S)2 and (AH1-S)2 (Table 1).[11, 12] The difference in growth inhibition for normal cells relative to malignant cells, however, is far more pronounced for (244mTC-S)2 and (IT1-S)2, with IC50 values in normal vs cancer cells that differ by two orders of magnitude. As such, the prochelators showing the highest antiproliferative activity in cancer cells are also presenting the highest therapeutic indexes in the selected panel of cultured cell lines.
In summary, the N2 substituted prochelators (244mTC-S)2 and (IT1-S)2, which cannot be deprotonated at the hydrazonic nitrogen atom, maintain an optimal overall lipophilicity and lead to the stabilization of ferric complexes of simple speciation in aqueous solutions. These disulfide-masked prochelators promptly modify serum albumin via disulfide exchange, with reduced Cys34 being the most likely candidate for this reactivity. In a panel of breast, ovarian and colon cancer cells, these systems present antiproliferative activities at submicromolar levels, almost two orders of magnitudes from those in normal fibroblasts. Investigations of the structural requirements for albumin modification and its specific impact on toxicity are underway along with studies of intracellular effects, including for instance generation of oxidative stress, which will delineate the mechanism of action for these compounds. Overall, with high antiproliferative activities and the ability to form albumin conjugates, these prochelators are of potential interest for further testing of their lifetime in blood circulation, tumor accumulation, and anticancer activity in vivo.
Supplementary Material
Acknowledgements
This work was supported by the US National Institute of General Medical Sciences of the National Institutes of Health (awards R35 GM128624 to M.T.M. and R01 GM127646 to E.T.), which also supported a fellowship to M.A.E. through the Biological Chemistry Program (T32 GM008804). The Bruker NEO-500 spectrometer in the UArizona Dept. of Chemistry and Biochemistry NMR Facility was purchased thanks to support from the National Science Foundation (MRI award CHE-1920234). The UArizona Cancer Center Flow Cytometry Shared Resource is supported by the National Cancer Institute of the National Institutes of Health (award P30 CA023074). We thank Dr. Andrei Astashkin for assistance in the acquisition and analysis of X-ray diffraction and EPR data.
Footnotes
Supporting information for this article can be found under: https://doi.org/10.1002/cmdc.202100278.
References
- [1].Torti SV, Torti FM, Cancer Res. 2020, 80, 5435–5448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Jung M, Mertens C, Tomat E, Brüne B, Int. J. Mol. Sci 2019, 20, 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bauer M, Eickhoff JC, Gould MN, Mundhenke C, Maass N, Friedl A, Breast Cancer Res. Treat 2008, 108, 389–397. [DOI] [PubMed] [Google Scholar]
- [4].Pinnix ZK, Miller LD, Wang W, D’Agostino R, Kute T, Willingham MC, Hatcher H, Tesfay L, Sui G, Di X, Torti SV, Torti FM, Sci. Transl. Med 2010, 2, 43ra56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Crielaard BJ, Lammers T, Rivella S, Nat. Rev. Drug Discov 2017, 16, 400–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Utterback RD, Tomat E, in Encycl. Inorg. Bioinorg. Chem, 2019, pp. 1–19. [Google Scholar]
- [7].Yu Y, Kalinowski DS, Kovacevic Z, Siafakas AR, Jansson PJ, Stefani C, Lovejoy DB, Sharpe PC, Bernhardt PV, Richardson DR, J. Med. Chem 2009, 52, 5271–5294. [DOI] [PubMed] [Google Scholar]
- [8].Heffeter P, Pape VFS, Enyedy ÉA, Keppler BK, Szakacs G, Kowol CR, Antioxid. Redox Signal 2019, 30, 1062–1082. [DOI] [PubMed] [Google Scholar]
- [9].Matesanz AI, Herrero JM, Quiroga AG, Curr. Top. Med. Chem 2021, 21, 59–72. [DOI] [PubMed] [Google Scholar]
- [10].Chang TM, Tomat E, Dalton Trans. 2013, 42, 7846–7849. [DOI] [PubMed] [Google Scholar]
- [11].Akam EA, Chang TM, Astashkin AV, Tomat E, Metallomics 2014, 6, 1905–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Akam EA, Utterback RD, Marcero JR, Dailey HA, Tomat E, J. Inorg. Biochem 2018, 180, 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Akam EA, Tomat E, Bioconjugate Chem. 2016, 27, 1807–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gamcsik MP, Kasibhatla MS, Teeter SD, Colvin OM, Biomarkers 2012, 17, 671–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mertens C, Akam EA, Rehwald C, Brüne B, Tomat E, Jung M, PLOS ONE 2016, 11, e0166164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Schnetz M, Meier JK, Rehwald C, Mertens C, Urbschat A, Tomat E, Akam EA, Baer P, Roos FC, Brüne B, Jung M, Cancers 2020, 12, 530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yuan J, Lovejoy DB, Richardson DR, Blood 2004, 104, 1450–1458. [DOI] [PubMed] [Google Scholar]
- [18].Enyedy ÉA, May NV, Pape VFS, Heffeter P, Szakács G, Keppler BK, Kowol CR, Dalton Trans. 2020, 49, 16887–16902. [DOI] [PubMed] [Google Scholar]
- [19].Astashkin AV, Utterback RD, Sung Y-S, Tomat E, Inorg. Chem 2020, 59, 11377–11384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cooper DS, Engl N. J. Med 2005, 352, 905–917. [DOI] [PubMed] [Google Scholar]
- [21].Akrivos PD, Coord. Chem. Rev 2001, 213, 181–210. [Google Scholar]
- [22].Legault C, Charette AB, J. Org. Chem 2003, 68, 7119–7122. [DOI] [PubMed] [Google Scholar]
- [23].Laus G, Kahlenberg V, Wurst K, Müller T, Kopacka H, Schottenberger H, Naturforsch Z. B 2013, 68, 1239–1252. [Google Scholar]
- [24].Edward JT, Chubb FL, Sangster J, Can. J. Physiol. Pharmacol 1997, 75, 1362–1368. [PubMed] [Google Scholar]
- [25].Bergeron RJ, Wiegand J, McManis JS, Weimar WR, Park J-H, Eiler-McManis E, Bergeron J, Brittenham GM, J. Med. Chem 2005, 48, 821–831. [DOI] [PubMed] [Google Scholar]
- [26].Edward JT, Ponka P, Richardson DR, BioMetals 1995, 8, 209–217. [Google Scholar]
- [27].Daina A, Michielin O, Zoete V, Sci. Rep 2017, 7, 42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Richardson DR, Sharpe PC, Lovejoy DB, Senaratne D, Kalinowski DS, Islam M, Bernhardt PV, J. Med. Chem 2006, 49, 6510–6521. [DOI] [PubMed] [Google Scholar]
- [29].Lovejoy DB, Sharp DM, Seebacher N, Obeidy P, Prichard T, Stefani C, Basha MT, Sharpe PC, Jansson PJ, Kalinowski DS, Bernhardt PV, Richardson DR, J. Med. Chem 2012, 55, 7230–7244. [DOI] [PubMed] [Google Scholar]
- [30].Noveron JC, Olmstead MM, Mascharak PK, Inorg. Chem 1998, 37, 1138–1139. [DOI] [PubMed] [Google Scholar]
- [31].Stadelman BS, Kimani MM, Bayse CA, McMillen CD, Brumaghim JL, Dalton Trans. 2016, 45, 4697–4711. [DOI] [PubMed] [Google Scholar]
- [32].Plaza-Lozano D, Morales-Martínez D, González FJ, Olguín J, Eur. J. Inorg. Chem 2020, 2020, 1562–1573. [Google Scholar]
- [33].Liu Z, Chen X, Chem. Soc. Rev 2016, 45, 1432–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kratz F, Control J. Release 2014, 190, 331–336. [DOI] [PubMed] [Google Scholar]
- [35].Hoogenboezem EN, Duvall CL, Adv. Drug Deliv. Rev 2018, 130, 73–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Merlot AM, Sahni S, Lane DJR, Fordham AM, Pantarat N, Hibbs DE, Richardson V, Doddareddy MR, Ong JA, Huang MLH, Richardson DR, Kalinowski DS, Oncotarget 2015, 6, 10374–10398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Carter DC, Ho JX, in Adv. Protein Chem, Vol. 45 (Eds.: Anfinsen CB, Edsall JT, Richards FM, Eisenberg DS), Academic Press, 1994, pp. 153–203. [DOI] [PubMed] [Google Scholar]
- [38].Kratz F, Müller-Driver R, Hofmann I, Drevs J, Unger C, J. Med. Chem 2000, 43, 1253–1256. [DOI] [PubMed] [Google Scholar]
- [39].Unger C, Häring B, Medinger M, Drevs J, Steinbild S, Kratz F, Mross K, Clin. Cancer Res 2007, 13, 4858–4866. [DOI] [PubMed] [Google Scholar]
- [40].Warnecke A, Fichtner I, Garmann D, Jaehde U, Kratz F, Bioconjugate Chem. 2004, 15, 1349–1359. [DOI] [PubMed] [Google Scholar]
- [41].Mayr J, Heffeter P, Groza D, Galvez L, Koellensperger G, Roller A, Alte B, Haider M, Berger W, Kowol CR, Keppler BK, Chem. Sci 2017, 8, 2241–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bocedi A, Cattani G, Stella L, Massoud R, Ricci G, FEBS J 2018, 285, 3225–3237. [DOI] [PubMed] [Google Scholar]
- [43].Gou Y, Yang F, Liang H, Curr. Top. Med. Chem 2016, 16, 996–1008. [DOI] [PubMed] [Google Scholar]
- [44].Basuli D, Tesfay L, Deng Z, Paul B, Yamamoto Y, Ning G, Xian W, McKeon F, Lynch M, Crum CP, Hegde P, Brewer M, Wang X, Miller LD, Dyment N, Torti FM, Torti SV, Oncogene 2017, 36, 4089–4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Padmanabhan H, Brookes MJ, Iqbal T, Nutr. Rev 2015, 73, 308–317. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.