

Abstract
Metabolic syndrome prevalence has increased among US adults, particularly among non-hispanic white and black women. Sedentary behavior often leads to chronic inflammation, a triggering factor of metabolic syndrome. Given that intrinsic exercise capacity is genetically inherited, we questioned if low-grade chronic inflammation would be present in a female rat model of low intrinsic exercise capacity-induced metabolic syndrome, while beneficial increase of resolution of inflammation would be present in a female rat model of high intrinsic exercise capacity. In the vascular system, two primary markers for inflammation and resolution of inflammation are cyclooxygenase (COX) and lipoxygenase (LOX), respectively. Our study focused on the novel hypothesis that untrained, inherited exercise capacity induces divergent vascular plasticity via changes in the delicate balance between COX and LOX inflammatory mediators.
We used divergent rat strains with low (LCR) and high (HCR) aerobic running capacity. By using animals with contrasting intrinsic exercise capacities, it is possible to determine the exact triggers that lead to inherited vascular plasticity in female rats. We observed that female LCR displayed increased periovarian fat pad and body weight, which is congruent with their obesity-presenting phenotype. Furthermore, LCR presented with vascular hypocontractility and increased COX and LOX-derived pro-inflammatory factors. On the other hand, HCR presented with a “shutdown” of COX-induced vasoconstriction and enhanced resolution of inflammation to maintain vascular tone and homeostasis. In conclusion, LCR display low-grade chronic inflammation via increased COX activity. These results provide mechanistic clues as to why lower intrinsic aerobic capacity correlates with a predisposition to risk of vascular disease. Conversely, being born with higher intrinsic aerobic capacity is a significant factor for improved vascular physiology in female rats.
Keywords: Test
1. Introduction
Physical inactivity is responsible for 3.2 million deaths per year worldwide and is defined as a cause of chronic diseases by the Centers for Disease Control and Prevention1,2. Physical inactivity leads to cardiovascular diseases3, and one of the major pathophysiological characteristics of cardiovascular diseases is the presence of endothelial dysfunction and inflammation4,5. Furthermore, endothelial dysfunction and vascular remodeling are signatures for physical inactivity, obesity, and metabolic syndrome6–11. Specifically, human studies have demonstrated that even acute exposure to sedentarism results in decreased endothelial function, increased arterial stiffening, and increased presence of pro-inflammatory arachidonic acid metabolites10.
Inflammation is a beneficial process in the cardiovascular system and is necessary for a proper innate and adaptive immune response; however, during chronic and/or dysregulated inflammation, this process is often pathological. For this reason, resolution of acute inflammation is an important process to limit persistent chronic inflammation and maintain homeostasis12,13. Resolution of inflammation is our body’s natural response to inflammation; however, deficiency and/or an imbalance in any of its components may lead to uncontrolled and chronic inflammation12.
In the vasculature, endothelial cells utilize the arachidonic acid (AA) metabolism pathway to regulate vascular tone, induce acute inflammation, and resolve acute inflammation13. Accordingly, pro-inflammation occurs mainly through cyclooxygenases (COX-1 and -2), while 5-lipoxygenase (5-LOX) is the key enzyme for the biosynthesis of leukotrienes and specialized pro-resolving lipid mediators13–16. In physiological conditions, the activity of the COX and LOX enzymes oscillate to maintain vascular tone and homeostasis; however, in disease states, this balance is disrupted, resulting in increased production of COX-2 derived metabolites, such as thromboxane (TXA2), and 5-LOX derived leukotrienes, such as leukotriene B4 (LTB4)13,16 Conversely, lipoxin A4 (LXA4), an endogenous 15-LOX-derived eicosanoid mediator, has potent pro-resolving and anti-inflammatory properties13,17. In fact, studies have shown that aspirin activates production of LXA4 through acetylation of COX-2, creating 15-epi LXA4, or “aspirin-triggered” LXA4, a well-known treatment for heart diseases17.
Given that intrinsic exercise capacity, in addition to cardiovascular disease, is genetically inheritable18–19, we questioned if AA-induced low-grade chronic inflammation would be present in a rat model of low intrinsic exercise capacity-induced metabolic syndrome, while a beneficial increase of AA-induced resolution of inflammation would be present in a rat model of high intrinsic exercise capacity. We used divergent rat strains with low (LCR) and high (HCR) intrinsic aerobic running capacities to answer these questions18. Using animals with contrasting intrinsic exercise capacities, it is possible to determine the exact triggers that lead to inherited vascular plasticity in female rats. We used female rats for three reasons (1) cardiovascular disease is the most prevalent cause of death in both men and women, yet much less is known about its effects in women20; (2) metabolic syndrome prevalence has increased among US adults, particularly among non-hispanic white women, non-hispanic black women, and individuals of low socioeconomic status21; and (3) women are more likely to lead inactive lifestyles22. Our study focuses on the novel hypothesis that untrained, inherited exercise capacity alters the delicate balance between COX and LOX inflammatory mediators. This leads to an increased COX activity in arteries from female LCR and increased resolution of inflammation in arteries from female HCR.
2. Materials and Methods
2.1. Animal Models for Low and High Intrinsic Aerobic Capacity
High‐ and low‐capacity runner rats were generated from genetically heterogenous N:NIH stock18. The development of HCR and LCR displaying high and low intrinsic exercise capacity has been described previously18 and these strains have been cataloged in the rat genome database (https://rgd.mcw.edu/rgdweb/report/strain/main.html?id=10402167; https://rgd.mcw.edu/rgdweb/report/strain/main.html?id=10402163). In the present study, we used untrained, female LCR and HCR from generation 44 (University of Toledo, Toledo, OH, USA). The rats were 28–30 weeks old at the time of use. The rats were all fed the same and kept at a 12-hour light cycle, ensuring that the changes in body weight were due to the genetic strain, and not external factors. The rats were fed Laboratory Rodent Diet ad libitum with access to autoclaved water at all times. The rats were pair-housed with their own models to allow for motility. Please note that the sample size indicated per experiment is the number of independent rats used. All procedures were performed according to NIH guidelines, and University of Toledo’s Institutional Animal Care and Use Committee (IACUC) guidelines. Tissue harvesting and protocols used to measure vascular function are all described below. The following flowchart (Fig. 1) describes the methodology used to conduct the experiments in this study.
Figure 1.

Methodological procedure of the steps taken to study the function of inflammatory mediators in the vascular system of divergent exercise capacity rat strains. Low (LCR) and High (HCR) capacity runner; Mesenteric Resistance arteries (MRA); Potassium Chloride (KCl); Acetylcholine (ACh); Phenylephrine (PE); Arachidonic acid (AA); Cyclooxygenase (COX).
2.2. Harvested Tissues
After the rats were weighed, blood was collected directly from the abdominal aorta. The rats were killed via thoracotomy and exsanguination via cardiac puncture while anesthetized under isoflurane anesthesia (5% in 100% O2 administered via nose cone). Serum was extracted from blood through centrifugation (15 min at 2000 rcf). Ovarian fat pad was dissected and weighed. The mesenteric bed, whole hearts, and tibias were harvested from all rats. Third-order mesenteric resistance arteries were isolated to evaluate vascular function, as described in the section 2.3.
The right and left ventricles were dissected from the whole hearts and weighed. Tibia length was measured for normalization and quantification of cardiac mass, according to a novel method determined in a 2019 study specific for small animal models23. Accordingly, it has been shown that body weight correlated nonlinearly (cubically) with tibia length (TL), but linearly with TL3 (cubed)23. The linear relation with left ventricle (LV) and TL3 showed that LV weight crossed the x-axis at TL3 − 26.73 (95% CI −29.83; −23.43). Therefore, TL and LV weight were indexed by dividing the weights by 26.73 + TL3. TL and right ventricle (RV) weight were indexed by dividing the weights by 26.53 + TL3.
2.3. Vascular Function Protocols
Third-order mesenteric resistance arteries (MRA) were extracted from the mesenteric bed of individual rats and mounted onto DMT wire myographs (Danish MyoTech, Aarhus, Denmark). MRA segments were normalized to the optimal lumen diameter for active tension development5,24. The MRA were oxygenated, heated (37°C), and submerged in Krebs solution in mM: (NaCl 118; KCl 4.7; NaHCO3 25; CaCl2.2H2O 2.5; KH2PO4 1.2; MgSO4.7H2O 1.2; EDTA 0.01; glucose 11) to mimic an environment for optimal function. To test for vascular smooth muscle cell integrity and for maximum contractile response, MRA were contracted with 120 mM of potassium chloride (KCl). MRA were then tested for endothelial integrity through contraction with phenylephrine (PE; 3*10−6 mol/L), an α1 adrenergic receptor agonist, and relaxation with acetylcholine (ACh; 3*10−6 mol/L), a muscarinic receptor agonist. Subsequently, concentration responses curves to ACh or PE were performed (10−9 mol/L to 3*10−5 mol/L). Concentration response curves were also conducted to a thromboxane (TXA2) mimetic, U46619 (10−10 mol/L to 10−5 mol/L) and LXA4 (10−10 mol/L to 3*10−7 mol/L, after contraction with PE 3*10−6 mol/L). To evaluate endothelium-independent relaxation, concentration response curves to sodium nitroprusside (SNP) were performed (10−9 mol/L to 3*10−5 mol/L). Relaxation responses are shown as a percentage of the initial contraction by PE. Contraction responses are shown as a percentage of the maximum contractile response to KCl.
To evaluate the contribution of AA-derived metabolites, concentration response curves to ACh or PE were also performed in the presence or absence of indomethacin (Indo; 10−5 mol/L), a non-selective COX inhibitor; 17-Octadecynoic acid (17-ODYA; 10−6 mol/L), an irreversible cytochrome P450 inhibitor, or Cinnamyl-3,4-dihydroxy-α-cyanocinnamate (CDC; 10−6 mol/L), a potent 5/12-lipoxygenase inhibitor. MRA were incubated with the inhibitors for 60 minutes in the wire myograph chamber prior to concentration response curves to PE or ACh.
2.4. Measurements of Thromboxane A2 and Prostaglandin E2 in Resistance Arteries
To evaluate thromboxane A2 (TXA2) and prostaglandin E2 (PGE2) in the MRA supernatant, the mesenteric bed from each strain was perforated and divided into separate eppendorf tubes, as shown in Fig. 2. One mL of fresh oxygenated Krebs solution was added to two vials. The experimental vial was incubated with 10 μmol/L ACh and the control trial received an equivalent volume of deionized water (vehicle) for 5 minutes. The supernatant was then collected from the vials and flash frozen in liquid nitrogen. Serum was also collected from the strains and flash frozen in liquid nitrogen, as previously described in 2.2 Harvested Tissues. The supernatant and serum were then stored in multiple aliquots at −80°C until analysis. To detect TXA2 or PGE2 in the supernatant, we used the monoclonal ELISA Kit for thromboxane B2 (TXB2, Item No. 19030, Cayman Chemical, USA) or for PGE2 (Item No. 514010, Cayman Chemical, USA) according to the instructions provided by the manufacturer. TXB2 is a stable, biologically inert metabolite formed from the non-enzymatic hydrolysis of TXA2, which has a half-life of about 30 seconds.
Figure 2.
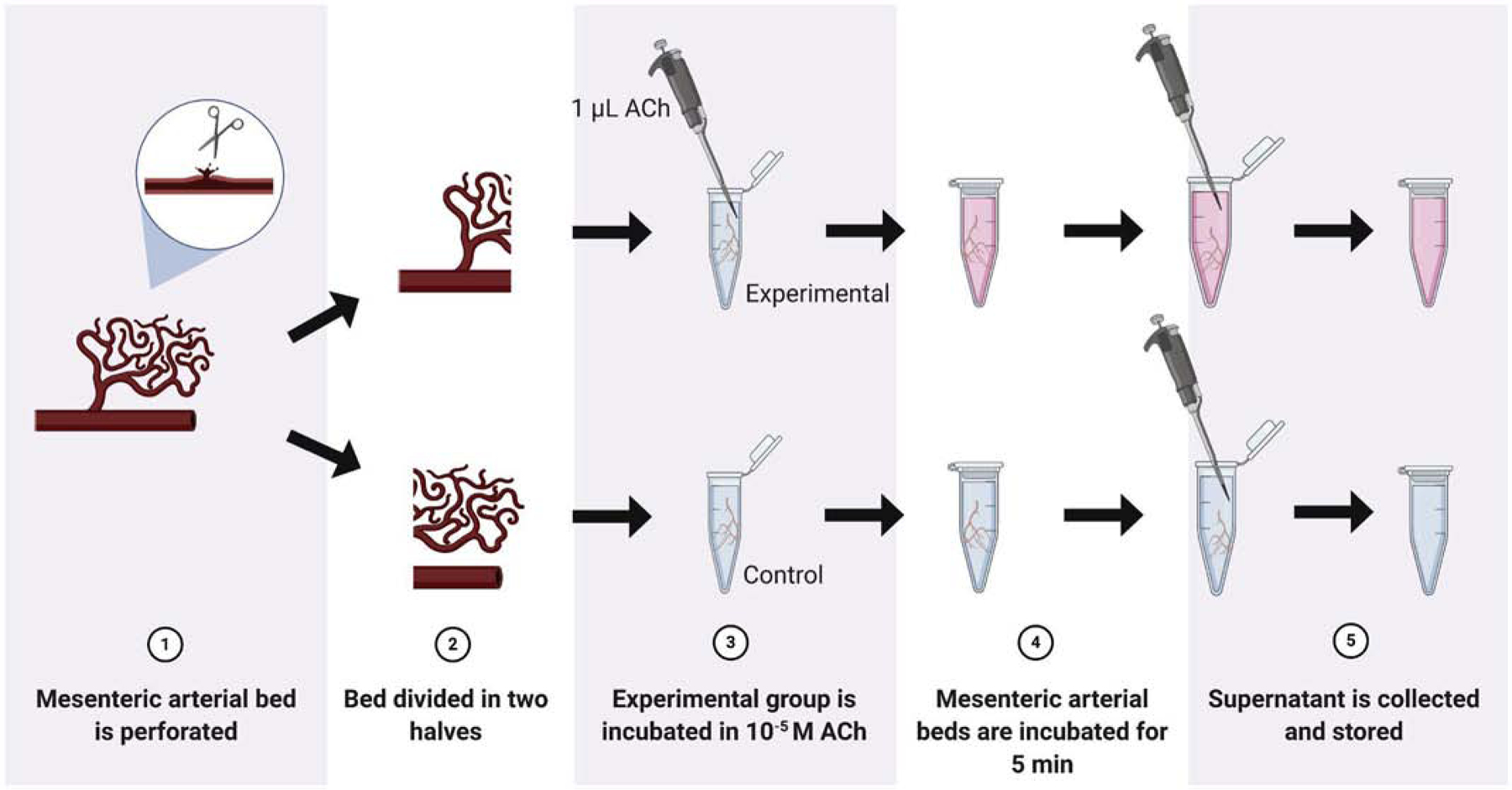
Methodological procedure for the measurement of thromboxane A2 and prostaglandin E2 in the supernatant of mesenteric resistance arteries (MRA). For this, MRA were cleaned from the surrounding tissue and perforated using ophthalmological scissors. This allows the AA-metabolites to be released into the supernatant. MRA were then stimulated with acetylcholine (10−6 mol/L) or vehicle (deionized water).
2.5. Statistical Analysis
All statistical analyses were performed using GraphPad Prism 8.2 (La Jolla, CA, USA). Data are presented as mean ± S.E.M. and statistical significance was set at p < 0.05. Specific procedures used include: Student’s unpaired t-test to compare the means between 2 samples; one-way or two-way analysis of variance (ANOVA) to compare more than 2 conditions and concentration response curves, respectively. Tukey’s post-hoc test and Bonferroni post-hoc test were used in one-way ANOVA and two-way ANOVA, respectively. To test data for normal distribution, we used a normality test (D’ Agostino & Person and/or Shapiro-Wilk test). The sample size indicated per experiment is the number of independent rats used.
3. Results
3.1. Body, Fat Pad, and Heart Weights
We observed that LCR presented greater body and ovarian fat pad weight when compared to HCR (Fig. 3a and b). HCR displayed increased whole heart, right ventricle (RV), and increased left ventricle weight (LV) (Fig. 3c–e).
Figure 3.

Female rats with low intrinsic aerobic capacity (LCR) display higher body weight and fat pad. Female rats with high intrinsic aerobic capacity (HCR) display increased cardiac mass. (a) Body weight; (b) ovarian fat pad weight; (c) total heart weight (HW); (d) right ventricle weight (RV); and (e) left ventricle weight (LV). Number of animals and p values are indicated in the graphs. Data are presented as mean ± SEM. T-test was used.
3.2. Resistance arteries from LCR present with hyporeactivity independent of endothelium dysfunction
No changes were observed in KCl-induced contraction (Fig. 4a), endothelium-dependent and -independent relaxation (Fig. 4b and c) and lumen diameter [HCR: 271 ± 13 μm (n=9) vs. LCR: 266 ± 13 μm (n=9) p>0.05] between groups. However, arteries from LCR presented decreased PE-induced contraction when compared to HCR (Fig. 4d). Similarly, the concentration-response curve to U46619, a potent and stable thromboxane A2 receptor agonist, was decreased in arteries from LCR compared to HCR (Fig. 4e).
Figure 4.

Arteries from female rats with low intrinsic aerobic capacity (LCR) display decreased phenylephrine-induced contraction when compared to female rats with high intrinsic aerobic capacity (HCR). Contractile response to KCl (120 mM) (a); Concentration-response curves to acetylcholine (b); sodium nitroprusside (SNP) (c); phenylephrine (d); and U46619 (e). Number of animals and p values are indicated in the graphs. Data are presented as mean ± SEM. Two-way ANOVA and Bonferroni post-hoc tests were used.
3.3. LCR present dual enzyme activation via COX and 5/12 LOX to induce pro-inflammatory molecules and vasoconstriction in resistance arteries
In the present study, the use of a non-specific COX inhibitor (Indo, 10−5 mol/L) presented a minor effect in PE-induced contraction in arteries from HCR (Fig. 5a and c). However, Indo abolished contraction in arteries from LCR (Fig. 5b and c), suggesting that COX-activity is increased in arteries from LCR. Interestingly, a similar pattern was observed in arteries from HCR and LCR treated with 5/12-LOX inhibitor (CDC, 10−6 mol/L), which blocks the pro-inflammatory mediator LTB4 formation (Fig. 5 d–f). On the other hand, arteries from LCR and HCR treated with cytochrome P450 (CYP450) inhibitor (17-ODYA, 10−6 mol/L) did not change PE-induced contraction (Fig. 5g–i). Image (Fig. 5j) shows the arachidonic acid metabolism pathway with COX, 5/12-LOX, and CYP450 inhibitors.
Figure 5.
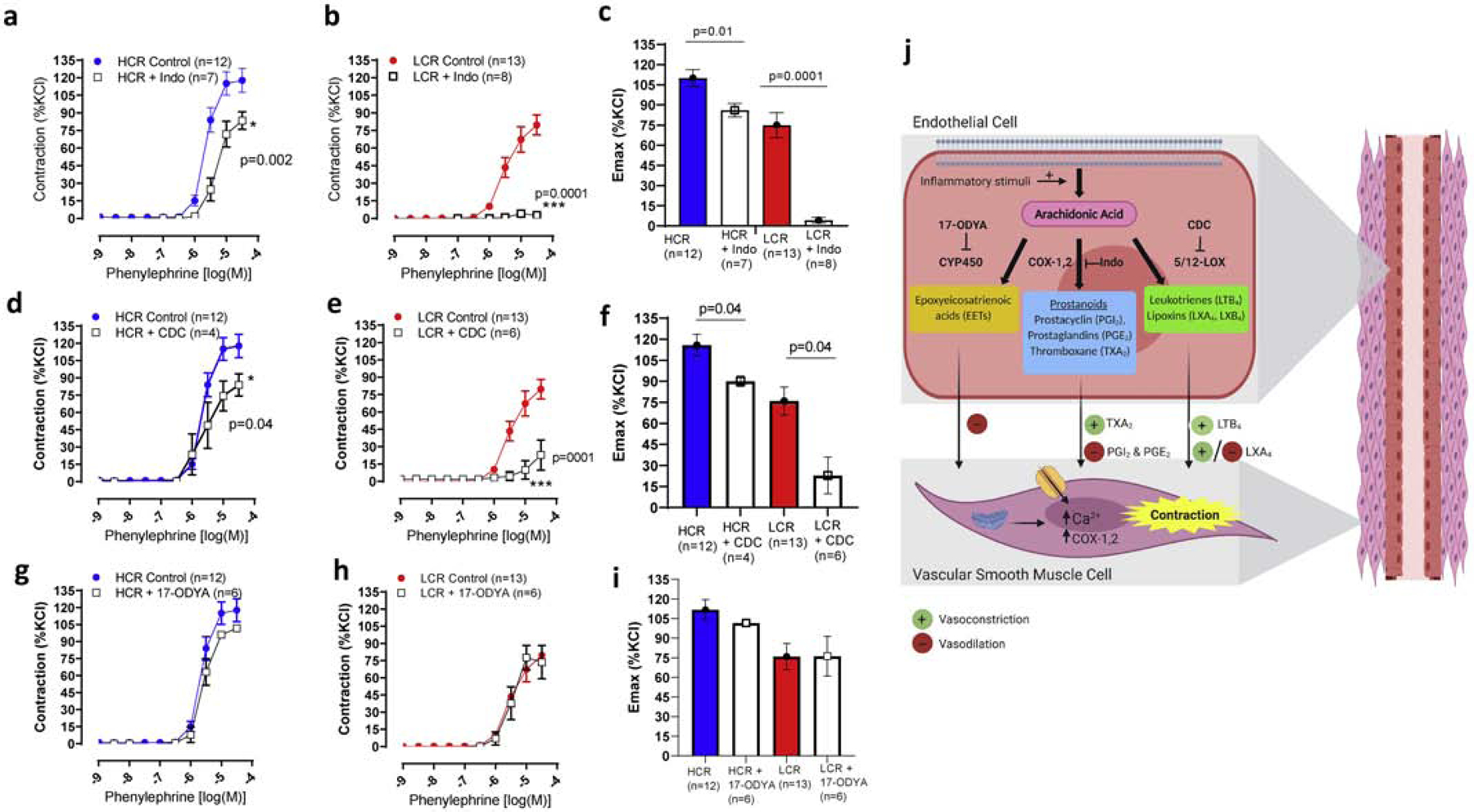
Cyclooxygenase (COX) and lipoxygenase (LOX)-derived vasoconstrictors are increased in arteries from female rats with low intrinsic aerobic capacity (LCR). Concentration-response curves to phenylephrine and maximum response (Emax) in the presence and absence of cyclooxygenase inhibitor, indomethacin (Indo) (a-c), 5/12-lipoxygenase inhibitor (d-f) or cytochrome P450 (CYP450, 17-ODYA) (g-i) in arteries from female LCR and high intrinsic aerobic capacity (HCR) rats. Image shows COX, LOX, CYP450 inhibitor, and endothelium-derived arachidonic acid metabolites (j). Number of animals and p values are indicated in the graphs. Data are presented as mean ± SEM. Two-way ANOVA and Bonferroni post-hoc tests were used.
3.4. Inhibition of CYP450-derived metabolites decreased endothelium-relaxation in arteries from LCR
The presence of the COX or 5/12-LOX inhibitor did not change concentration-response curves to ACh in arteries from LCR and HCR (Fig. 6a–d). Interestingly, arteries from LCR treated with CYP450 inhibitor (17-ODYA, 10−5 mol/L) significantly reduced endothelium-induced relaxation when compared to HCR (Fig. 6e and f).
Figure 6.

A potent cytochrome P450 inhibitor (17-ODYA) impaired acetylcholine-induced relaxation in arteries from female rats with low intrinsic aerobic capacity (LCR). Concentration-response curves to acetylcholine in the presence and absence of cyclooxygenase inhibitor, indomethacin (Indo) (a and b), 5/12-lipoxygenase inhibitor, (CDC) (c and d) or cytochrome P450 inhibitor (17-ODYA) (e and f) in arteries from female LCR and high intrinsic aerobic capacity (HCR) rats. Number of animals and p values are indicated in the graphs. Data are presented as mean ± SEM. Two-way ANOVA and Bonferroni post-hoc tests were used.
3.5. Lipoxin (LXA4), a LOX-derived mediator of resolution of inflammation, is important to maintain vascular tone in arteries from HCR
Given that inhibition of COX-derived prostaglandins and LTB4 had minor or no effect in PE-induced contraction in arteries from HCR, but significantly reduced contraction in arteries from LCR, we questioned whether another factor would be important to maintain vascular tone in HCR arteries. We observed that LXA4 induced contraction in arteries from HCR, but induced small relaxation in arteries from LCR (Fig. 7a and b).
Figure 7.

Lipoxin A4 (LXA4), an important arachidonic acid metabolite, maintains vascular tone in arteries from female rats with high intrinsic aerobic capacity (HCR). Concentration-response curves to LXA4 in arteries from female HCR and low intrinsic aerobic capacity (LCR) rats (a); Maximum response (Emax) to LXA4 (b). Number of animals and p values are indicated in the graphs. Data are presented as mean ± SEM. Two-way ANOVA and Bonferroni post-hoc tests and t-test were used.
3.6. Release of prostaglandin E2 is increased in arteries from HCR
Although we observed that COX inhibition did not change ACh-induced relaxation in arteries from HCR and LCR, we have found that prostaglandin E2 (PGE2), a primary product of arachidonic acid metabolism, is elevated in arteries from HCR after stimulation with ACh compared to LCR (Fig. 8a). No changes were observed in the release of TXB2 (Fig. 8b).
Figure 8.

Measurements of (a) thromboxane B2 (TXB2) and (b) prostaglandin E2 (PGE2) in the supernatant from non-stimulated (basal conditions) and stimulated with acetylcholine (ACh, 10−6 mol/L) in arteries from female rats with low intrinsic aerobic capacity (LCR) and high intrinsic aerobic capacity (HCR) rats. Number of animals and p values are indicated in the graphs. Data are presented as mean ± SEM. T-test was used for basal condition vs. stimulated condition.
4. Discussion
A study that analyzed data from the National Health and Nutrition Examination Survey (NHANES) from 1988 through 201221 observed that metabolic syndrome prevalence increased since 1988 among US adults, particularly among non-hispanic white women and non-hispanic black women. Without immediate intervention, this scenario will get worse, especially because 1) physical activity declines dramatically as we age25 and 2) and intrinsic exercise capacity is inheritable18–19. In the present study, we observed that female LCR rats displayed increased body weight and ovarian fat pad weight. This is consistent with previous research associating visceral adiposity with metabolic syndrome7. On the other hand, female HCR display a lower fad pad and body weight. The increase and decrease in body weight of LCR and HCR is not associated with food intake. For instance, it has been shown that male HCR rats consumed more calories after mass correction compared to LCR rats26. In male rats, the lean phenotype is not due to low caloric intake. Overall, leanness and high physical activity levels may be a byproduct of natural selection of high capacity for running endurance26,27.
“Athlete’s heart” is a concept that has been used to describe the changes that occur to cardiac structure and function induced by long-term physical exercise28. Accordingly, it has been shown that chronic physical exercise leads to physiological cardiac remodeling and hypertrophy as an adaptive response28. Aerobic exercise training, such as long-distance running, may increase cardiac mass with left ventricle chamber dilation, known as eccentric hypertrophy28. Conversely, resistance training (i.e. weightlifting) may increase cardiac mass without left ventricle chamber dilation, referred to as concentric hypertrophy28. In our study, we observed that female HCR presented an increase in left and right ventricle weights compared to LCR. These data are consistent with our previous study showing that male HCR presented an increase in cardiac mass27, suggesting that physical activity-induced cardiac remodeling can also be inherited regardless of sex. As a mechanism for this phenomenon, it was observed that mitochondrial DNA was a heritable and important factor in determining this cardiac phenotype in HCR27. Overall, these findings open a new frontier for studying inherited physiological vs. pathological cardiac hypertrophy present in exercise and cardiovascular diseases in women, respectively.
It has been shown that obesity and metabolic syndrome lead to a decrease in vascular contractility29–31. Specifically, it was observed that male Wistar rats, fed a high-fat and high-sucrose diet for 15 weeks, present α1-adrenergic hyporesponsiveness without changes in blood pressure29. Further, contractile responses to PE were decreased in aortas from obese mice30, and exercise training attenuated the inducible nitric oxide synthase (iNOS)-dependent reduction in contraction30. Corroborating these data, in the present study we observed that resistance arteries from female LCR present with vascular hyporeactivity to both α1-adrenergic and thromboxane receptor agonists, suggesting that an intrinsic and common alteration in the vascular smooth muscle cell (VSMC) signaling components and/or structure is responsible for this response. Supporting this, it is known that physical inactivity and obesity are characterized by muscle atrophy27,32 and vascular remodeling9. Previously, we observed that male LCR presented with inward hypotrophic remodeling or vascular atrophy27. Accordingly, vascular remodeling can be defined as an increase (hypertrophic), no change (eutrophic), or a decrease (hypotrophic) of the cross-sectional area. These forms of remodeling can result in a reduction (inward) or increase (outward) of the lumen diameter33. Inward hypotrophic remodeling or vascular atrophy occurs due to low blood flow which leads to vascular hyporeactivity34. The accompanying hyporeactivity results from prolonged excessive inflammation and vasoconstriction34. Inflammation is considered one of the hallmarks of muscle atrophy and cachexia35,36. In vascular atrophy, pro-inflammatory molecules, such as TNF-α, lead to apoptosis by direct and autocrine mechanisms36. Furthermore, long-term exposure to vasoconstrictors, through adrenergic stimulation and/or proinflammatory molecules, results in reduced vascular contractility37,38. Given that COX- and LOX-derived products are important for vascular contraction and inflammation, we suggested that these enzymes lead to low grade chronic inflammation and subsequent vascular hyporeactivity in LCR.
Arachidonic acid (AA) is the most common precursor of biologically active lipid metabolites, and it plays a vital role in the initiation of inflammation and the resolution of inflammation in physiological states. As shown previously in Fig. 5j, AA is the preferred substrate for the COX, LOX, and CYP450 pathways to produce prostaglandins, leukotrienes, and epoxyeicosatrienoic acids (EETs), respectively13,39–40. In physiological conditions, flow, tension, and vasodilator agonists promote the synthesis of eicosanoids (e.g. prostacyclin) or pro-resolving molecules (e.g. lipoxin A4)13. However, in diseases such as obesity, there is aberrant AA metabolism13. Accordingly, vasoconstrictors, such as norepinephrine, promote an exacerbated synthesis of inflammatory metabolites, predominantly from COX-2 [e.g., thromboxane A2, (TXA2)] and 5-LOX [e.g., leukotriene B4 (LTB4)] enzymes that act on VSMC. In the present study, the use of a non-specific COX inhibitor, indomethacin, abolished PE-induced contraction in arteries from female LCR, suggesting that COX-activity is increased in arteries from LCR. Further, 5/12-LOX inhibitor, CDC, which blocks the formation of pro-inflammatory mediator LTB4, also reduced contraction in arteries from LCR, but in a higher magnitude compared to HCR. These data suggest that LCR presents dual enzyme activation through COX and 5-LOX, to synthetize pro-inflammatory and vasoconstricting molecules, such as prostaglandins and LTB4. When compared to arteries from LCR, COX and 5/12-LOX inhibitors only had a minor effect in PE-induced contraction in arteries from female HCR. Interestingly, we observed that prostaglandin E2 (PGE2) is increased in arteries from HCR when compared to LCR. Traditionally, PGE2 is well known because this molecule is a potent inflammatory mediator that is generated by COX-2 conversion of AA13. However, several studies support the new paradox about the importance of PGE2 in physiological processes without eliciting inflammation41. For instance, a physiological process where prostaglandins play an essential role is human parturition. Intra-amniotic concentrations of PGE2 are rapidly increased, within hours, leading to the initiation of labor at term42. Given that we did not observed vascular dysfunction in arteries from female HCR, we suggest that increased PGE2 levels in arteries from HCR may be associated with maintenance of vascular tone in physiological conditions. Similarly, we observed that LXA4 induced contraction in arteries from HCR, but slight relaxation in arteries from LCR. These data suggest that a non-prostaglandin and non-leukotriene factor protects the arteries and plays a major role in vascular contractility in HCR. These data reveal a new, dichotomous mechanism for vascular contractility and inflammation in LCR and HCR rats.
LXA4 is a biologically active product generated from AA by 15-LOX and has opposite effects to LTB412,13. The generation of LXA4 is a rapid process and aspirin does not inhibit its formation. In fact, aspirin triggers the production of LXA4 through acetylation of COX-2, also known as aspirin-triggered LXA4. We previously observed that LXA4 induces contraction in the aorta43 and it is not inhibited by TXA2/PGH2 receptor antagonist and COX-1 specific inhibitor. However, COX-2 inhibitor decreases this contraction, suggesting that LXA4 may have a positive feedback on COX-243. We speculate that this is to induce the release of PGE2 in physiological conditions in HCR arteries. Therefore, we suggest that HCR present beneficial vascular plasticity via a “shutdown” of COX-induced vascular contraction and increased resolution of inflammation via LXA4, to maintain vascular tone.
Furthermore, we did not observe changes in ACh-induced relaxation in arteries from female LCR and HCR. However, we found that arteries from female rats that were artificially selected for low and high intrinsic aerobic capacity present a swap mechanism to regulate endothelium-dependent vasodilation. Accordingly, we observed that 17-ODYA, a potent inhibitor of cytochrome P450, significantly reduced endothelium-induced relaxation in arteries from LCR, but not HCR. These data suggest that CYP450-derived eicosanoids play a major role in maintaining vascular homeostasis in resistance arteries from female LCR. In healthy conditions, nitric oxide plays the predominant role in vascular tone regulation in larger arteries, such as the aorta, when compared to smaller arteries. In another words, nitric oxide-mediated relaxation is reduced with decreasing artery size44. This means that other factors, such as endothelium-dependent hyperpolarization (EDH) and AA metabolites, play a major role in controlling resistance artery tone. It is well known that CYP450 epoxygenases generate epoxyeicosatrienoic acids (EETs) which affect EDH-mediated responses45. Interestingly, it has been shown that depending on the type of cardiovascular disorder, increased EDH responses may contribute to, or compensate for, endothelial abnormalities associated with the pathogenesis of certain diseases46,47. Further, in eNOS/COX-1 double knockout mice, EDH-mediated responses appeared to compensate for the absence of endothelial NO in females but not in males47. Therefore, we suggest that arteries from female LCR present with an increase in EDH-mediated relaxation via CYP450-derived metabolites.
A limitation in our present study is a phenotype that is comparable to a “normal” control rat, as the LCR and HCR both display aerobic capacity selected for generations. Due to limitations in the selection process, we do not have a strain that has been selected for “normal” aerobic capacity. However, we expect that the inherent, genotypic differences in the LCR and HCR are more translational to human studies as they could have a stronger resemblance with genetic changes seen in underserved communities. Another important point is that the level of activity in the cage could change vascular responses. However, Koch et al.48 demonstrated that activity levels, outside aerobic capacity testing, were not different between LCR and HCR while pair-housed in separate cages.
Overall, intrinsic exercise capacity induces divergent vascular plasticity. LCR presents deleterious vascular hypocontractility via increased endothelial COX- and LOX-derived pro-inflammatory factors. On the other hand, HCR presents beneficial vascular plasticity via a “shutdown” of COX-induced vasoconstriction, and enhanced resolution of inflammation to maintain vascular tone and homeostasis.
Acknowledgements
We wish to acknowledge the excellent veterinary and husbandry services provided at The University of Toledo College of Medicine and Life Sciences Department of Laboratory Animal Resources. Especially, we thank Mariah DuPree and Samantha McKee from the Koch lab for the phenotyping and colony maintenance of the LCR/HCR rat strains.
Funding
This work was supported by National Institutes of Health (R01HL149762 and R00GM118885 to C.F.W., K99HL151889 to C.G.M., R01HL1430820 to B.J.,), American Heart Association (18POST34060003 to C.G.M.) and NSF (AGEP1432878 to J.M.E.). The LCR and HCR rat model system was funded by the Office of Research Infrastructure Programs grant (P40OD021331 to L.G.K.). The rat models for low and high exercise capacity are maintained as an international resource with support from the Department of Physiology & Pharmacology, University of Toledo College of Medicine, Toledo, OH. Contact LGK Lauren.Koch2@UToledo.Edu for information on the rat models.
COI
This study was supported by NIH, American Heart Association and NSF. The LCR and HCR rat model system was funded by the Office of Research Infrastructure Programs grant (P40OD021331 - L.G.K.). The rat models for low and high exercise capacity are maintained as an international resource with support from the Department of Physiology & Pharmacology, University of Toledo College of Medicine, Toledo, OH.
None of the authors have any further (real or perceived) conflicts of interest to declare.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.The World Health Organization report - Global action plan on physical activity 2018–2030: more active people for a healthier world. 2018. Licence: CC BY-NC-SA 3.0 IGO.
- 2.Division of Nutrition, Physical Activity, and Obesity, National Center for Chronic Disease Prevention and Health Promotion. Center for Diseases Control and Prevention. 2019, https://www.cdc.gov/physicalactivity/about-physical-activity/why-it-matters.html [Google Scholar]
- 3.Young DR, Hivert MF, Alhassan S, Camhi SM, Ferguson JF, Katzmarzyk PT, Lewis CE, Owen N, Perry CK, Siddique J, Yong CM; Physical Activity Committee of the Council on Lifestyle and Cardiometabolic Health; Council on Clinical Cardiology; Council on Epidemiology and Prevention; Council on Functional Genomics and Translational Biology; and Stroke Council. Sedentary Behavior and Cardiovascular Morbidity and Mortality: A Science Advisory from the American Heart Association. Circulation. 2016;134(13):e262–79. [DOI] [PubMed] [Google Scholar]
- 4.Pober JS, Sessa WC. Inflammation and the blood microvascular system. Cold Spring Harb Perspect Biol. 2014; 7(1):a016345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wenceslau CF, Rossoni LV. Rostafuroxin ameliorates endothelial dysfunction and oxidative stress in resistance arteries from deoxycorticosterone acetate-salt hypertensive rats: the role of Na+K+-ATPase/ cSRC pathway. J Hypertens. 2014;32(3):542–54. [DOI] [PubMed] [Google Scholar]
- 6.Stapleton PA, James ME, Goodwill AG, Frisbee JC. Obesity and vascular dysfunction. Pathophysiology. 2008;15(2):79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belin de Chantemele EJ, Stepp DW. Influence of obesity and metabolic dysfunction on the endothelial control in the coronary circulation. J Mol Cell Cardiol. 2012;52(4):840–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davel AP, Lu Q, Moss ME, Rao S, Anwar IJ, DuPont JJ, Jaffe IZ. Sex‐Specific Mechanisms of Resistance Vessel Endothelial Dysfunction Induced by Cardiometabolic Risk Factors. J Am Heart Assoc. 2018;7(4): e007675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vessières E, Belin de Chantemèle EJ, Guihot AL, Jardel A, Toutain B, Loufrani L, Henrion D. Cyclooxygenase-2-derived prostanoids reduce inward arterial remodeling induced by blood flow reduction in old obese Zucker rat mesenteric arteries. Vascul Pharmacol. 2013;58(5–6):356–62. [DOI] [PubMed] [Google Scholar]
- 10.Nosova EV, Yen P, Chong KC, Alley HF, Stock EO, Quinn A, Hellmann J, Conte MS, Owens CD, Spite M, Grenon SM. Short-term physical inactivity impairs vascular function. J Surg Res. 2014;190(2):672–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laufs U, Wassmann S, Czech T, Münzel T, Eisenhauer M, Böhm M, Nickenig G. Physical Inactivity Increases Oxidative Stress, Endothelial Dysfunction, and Atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:809–814. [DOI] [PubMed] [Google Scholar]
- 12.Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510(7503):92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edwards JM, McCarthy CG, Wenceslau CF. The Obligatory Role of the Acetylcholine-Induced Endothelium-Dependent Contraction in Hypertension: Can Arachidonic Acid Resolve this Inflammation? Curr Pharm Des. 2020;26(30):3723–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh NK, Rao GN. Emerging role of 12/15-Lipoxygenase (ALOX15) in human pathologies. Prog Lipid Res. 2019;73:28–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bishop-Bailey D, Mitchell JA, Warner TD. COX-2 in cardiovascular disease. Arterioscler Thromb Vasc Biol. 2006;26(5):956–8. [DOI] [PubMed] [Google Scholar]
- 16.Bäck M, Avignon A, Stanke-Labesque F, Boegner C, Attalin V, Leprieur E, Sultan A. Leukotriene Production Is Increased in Abdominal Obesity. PLoS ONE. 2014;(12): e104593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chiang N, Arita M, Serhan CN. Anti-inflammatory circuitry: lipoxin, aspirin-triggered lipoxins and their receptor ALX. Prostaglandins Leukot. Essent. Fatty Acids 2005; 73:163–177. [DOI] [PubMed] [Google Scholar]
- 18.Koch LG, Britton SL. Divergent selection for aerobic capacity in rats as a model for complex disease. Integr Comp Biol. 2005;45(3):405–15. [DOI] [PubMed] [Google Scholar]
- 19.Bouchard C, Daw EW, Rice T, Pérusse L, Gagnon J, Province MA, Leon AS, Rao DC, Skinner JS, Wilmore JH. Familial resemblance for VO2max in the sedentary state: the HERITAGE family study. Med. Sci. Sports Exerc 1998;252–8. [DOI] [PubMed] [Google Scholar]
- 20.Reusch JEB, Kumar TR, Regensteiner JG, Zeitler PS; Conference Participants. Identifying the Critical Gaps in Research on Sex Differences in Metabolism Across the Life Span. Endocrinology. 2018;159(1):9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moore JX, Chaudhary N, Akinyemiju T. Metabolic Syndrome Prevalence by Race/Ethnicity and Sex in the United States, National Health and Nutrition Examination Survey, 1988–2012. Prev Chronic Dis. 2017;14:E24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Craft BB, Carroll HA, Lustyk MK. Gender Differences in Exercise Habits and Quality of Life Reports: Assessing the Moderating Effects of Reasons for Exercise. Int J Lib Arts Soc Sci. 2014;2(5):65–76. [PMC free article] [PubMed] [Google Scholar]
- 23.Hagdorn QAJ, Bossers GPL, Koop AMC. A novel method optimizing the normalization of cardiac parameters in small animal models: the importance of dimensional indexing. Am J Physiol Heart Circ Physiol. 2019;316:H1552–H1557. [DOI] [PubMed] [Google Scholar]
- 24.Wenceslau CF, McCarthy CG, Szasz T, Calmasini FB, Mamenko M, Webb RC. Formyl peptide receptor-1 activation exerts a critical role for the dynamic plasticity of arteries via actin polymerization. Pharmacol Res. 2019;141:276–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Troiano RP, Berrigan D, Dodd KW, Mâsse LC, Tilert T, McDowell M. Physical activity in the United States measured by accelerometer. Med Sci Sports Exerc. 2008;40(1):181–8. [DOI] [PubMed] [Google Scholar]
- 26.Novak CM, Escande C, Gerber SM, Chini EN, Zhang M, Britton SL, Koch LG, Levine JA. Endurance capacity, not body size, determines physical activity levels: role of skeletal muscle PEPCK. PLoS One 2009;12;4(6):e5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roy S, Edwards JM, Tomcho JC, Schreckenberger Z, Bearss NR, Zhang Y, Morgan EE, Cheng X, Spegele AC, Vijay-Kumar M, McCarthy CG, Koch LG, Joe B, Wenceslau CF. Intrinsic exercise capacity and mitochondrial DNA lead to opposing vascular-associated risks. Function, 2021;2(1):zqaa029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fernandes T, Soci UP, Oliveira EM. Eccentric and concentric cardiac hypertrophy induced by exercise training: microRNAs and molecular determinants. Braz J Med Biol Res. 2011;44(9):836–47. [DOI] [PubMed] [Google Scholar]
- 29.Battault S, Meziat C, Nascimento A, Braud L, Gayrard S, Legros C, De Nardi F, Drai J, Cazorla O, Thireau J, Meyer G, Reboul C. Vascular endothelial function masks increased sympathetic vasopressor activity in rats with metabolic syndrome. Am J Physiol Heart Circ Physiol. 2018;314(3):H497–H507. [DOI] [PubMed] [Google Scholar]
- 30.Silva JF, Correa IC, Diniz TF, Lima PM, Santos RL, Cortes SF, Coimbra CC, Lemos VS. Obesity, Inflammation, and Exercise Training: Relative Contribution of iNOS and eNOS in the Modulation of Vascular Function in the Mouse Aorta. Front Physiol. 2016;7:386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Romanko OP, Stepp DW. Reduced constrictor reactivity balances impaired vasodilation in the mesenteric circulation of the obese Zucker rat. Am J Physiol Heart Circ Physiol, 2005;289:H2097–H2102. [DOI] [PubMed] [Google Scholar]
- 32.Zhu S, Tian Z, Torigoe D, Zhao J, Xie P, Sugizaki T, Sato M, Horiguchi H, Terada K, Kadomatsu T, Miyata K, Oike Y. Aging- and obesity-related peri-muscular adipose tissue accelerates muscle atrophy. PLoS One, 2019;14(8): e0221366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mulvany MJ. Small artery remodelling in hypertension: causes, consequences and therapeutic implications. Med. Biol. Eng. Comput, 2008;46:461–467. [DOI] [PubMed] [Google Scholar]
- 34.Pourageaud F, DeMey JGR. Structural properties of rat mesenteric small arteries after 4-week exposure to elevated or reduced blood flow. Am J Physiol. 1997;273:H1699–H1706.. [DOI] [PubMed] [Google Scholar]
- 35.Stenholm S, Harris TB, Rantanen T, Visser M, Kritchevsky SB, Ferrucci L. Sarcopenic obesity: definition, cause and consequences. Curr Opin Clin Nutr Metab Care 2008;11(6):693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boyle JJ, Weissberg PL, Bennett MR. Tumor necrosis factor-alpha promotes macrophage-induced vascular smooth muscle cell apoptosis by direct and autocrine mechanisms. Arterioscler. Thromb. Vasc. Biol 2003;23:1553–1558. [DOI] [PubMed] [Google Scholar]
- 37.Boonen HC, Daemen MJ, Eerdmans PH, Fazzi GE, van Kleef EM, Schiffers PM, De Mey JG. Mesenteric small artery changes after vasoconstrictor infusion in young rats. J Cardiovasc Pharmacol. 1993;22(3):388–95. [DOI] [PubMed] [Google Scholar]
- 38.Hilgers RH, Xing D, Gong K, Chen YF, Chatham JC, Oparil S. Acute O-GlcNAcylation prevents inflammation-induced vascular dysfunction. Am J Physiol Heart Circ Physiol. 2012;303(5):H513–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang EH, Vanhoutte PM. Prostanoids and reactive oxygen species: team players in endothelium-dependent contractions. Pharmacol Ther. 2009;122(2):140–149. [DOI] [PubMed] [Google Scholar]
- 40.Campbell WB, Fleming I. Epoxyeicosatrienoic acids and endothelium-dependent responses. Pflugers Arch. 2010;459: 881–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maddipati KR. Non-inflammatory Physiology of “Inflammatory” Mediators - Unalamation, a New Paradigm. Front Immunol. 2020;11:580117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Romero R, Munoz H, Gomez R, Parra M, Polanco M, Valverde V, Hasbun J, Garrido J, Ghezzi F, Mazor M, Tolosa JE, Mitchell MD. Increase in prostaglandin bioavailability precedes the onset of human parturition. Prostaglandins Leukotrienes Essent Fatty Acids. 1996;54:187–91. [DOI] [PubMed] [Google Scholar]
- 43.Wenceslau CF, McCarthy CG, Szasz T, Webb RC. Lipoxin A4 mediates aortic contraction via RHOA/RHO kinase, endothelial dysfunction and reactive oxygen species. J Vasc Res. 2014;51(6):407–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hwa JJ, Ghibaudi L, Williams P, Chatterjee M. Comparison of acetylcholine-dependent relaxation in large and small arteries of rat mesenteric vascular bed. Am J Physiol. 1994;266(3 Pt 2):H952–8. [DOI] [PubMed] [Google Scholar]
- 45.Fleming I Cytochrome P450 epoxygenases as EDHF synthase(s). Pharmacol Res. 2004;49(6):525–33. [DOI] [PubMed] [Google Scholar]
- 46.Luksha L, Agewall S, Kublickiene K. Endothelium-derived hyperpolarizing factor in vascular physiology and cardiovascular disease. Atherosclerosis. 2009;202(2):330–44. [DOI] [PubMed] [Google Scholar]
- 47.Scotland RS, Madhani M, Chauhan S, Moncada S, Andresen J, Nilsson H, Hobbs AJ, Ahluwalia A. Investigation of vascular responses in endothelial nitric oxide synthase/cyclooxygenase-1 double-knockout mice: key role for endothelium-derived hyperpolarizing factor in the regulation of blood pressure in vivo. Circulation. 2005;15;111(6):796–803. [DOI] [PubMed] [Google Scholar]
- 48.Koch LG, Kemi OJ, Qi N, Leng SX, Bijma P, Gilligan LJ, Wilkinson JE, Wisløff H, Høydal MA, Rolim N, PM Abadir, van Grevenhof EM, Smith GL, Burant CF, Ellingsen O, Britton SL, Wisløff U. Intrinsic aerobic capacity sets a divide for aging and longevity. Circulation research. 2011;109(10), 1162–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]