

Abstract
Purinergic signaling is a fundamental mechanism used by all cells to control their internal activities and interact with the environment. As a key component of the purinergic system, the enzyme ecto-5′-nucleotidase (CD73) catalyzes the last step in the extracellular metabolism of adenosine triphosphate (ATP) to form adenosine. Efforts to harness the therapeutic potential of endogenous adenosine in cancer have culminated in the ongoing clinical development of multiple CD73-targeting antibodies and small-molecule inhibitors. However, recent studies are painting an increasingly complex picture of CD73 mRNA and protein regulation and function in cellular homeostasis, physiological adaptation, and disease development. This review discusses the latest conceptual and methodological advances helping to unravel the complexity of this important enzyme, identified nearly 90 years ago.
Keywords: purinergic system, adenosine, inflammation, cancer, digestive system, zonation
CD73 is an integral component of the purinergic system
Cells produce and consume adenosine triphosphate (ATP) in a tightly regulated manner to ensure optimal organismal function. In addition to being used as fuel for essential activities within the cell, ATP is also released outside of the cell, where the sequential removal of its phosphate groups results in the formation of the nucleoside adenosine. Extracellular ATP and adenosine, together with the associated synthetic and catabolic enzymes, receptors, and transporters are part of the evolutionarily conserved purinergic system, which links cellular metabolism to a myriad of other processes, including proliferation, differentiation, and cell death [1].
Extracellular ATP is rapidly metabolized by a number of enzymes collectively known as ectonucleotidases, which include members of the ecto-nucleoside triphosphate diphosphohydrolase (ENTPD) and ecto-nucleotide pyrophosphatase/phosphodiesterase (ENPP) protein families [2]. ENTPD1 (CD39) and ENPP1 generate adenosine monophosphate (AMP) from ATP (Figure 1). Subsequent hydrolysis of AMP to adenosine is primarily, but not exclusively, carried out by the enzyme 5’-nucleotidase (CD73) [3] (Figure 1). Extracellular adenosine activates four G-protein coupled adenosine receptors (ARs), which have different affinities for adenosine (A1R>A2AR>A3R>>A2BR) and activate numerous signaling pathways to control oxygen supply/demand, inflammation, and other activities, dependent on the cell type and receptor expression pattern [4] (Figure 1). Adenosine can also be taken up via equilibrative nucleoside transporters (ENTs) and re-phosphorylated to AMP inside the cell [5]. As the major enzymatic source of extracellular adenosine, CD73 is a key regulator of cellular homeostasis, stress responses, injury, and disease mechanisms across many tissues [6].
Figure 1. CD73 is an Essential Component of Purinergic Signaling and a Disease Target.
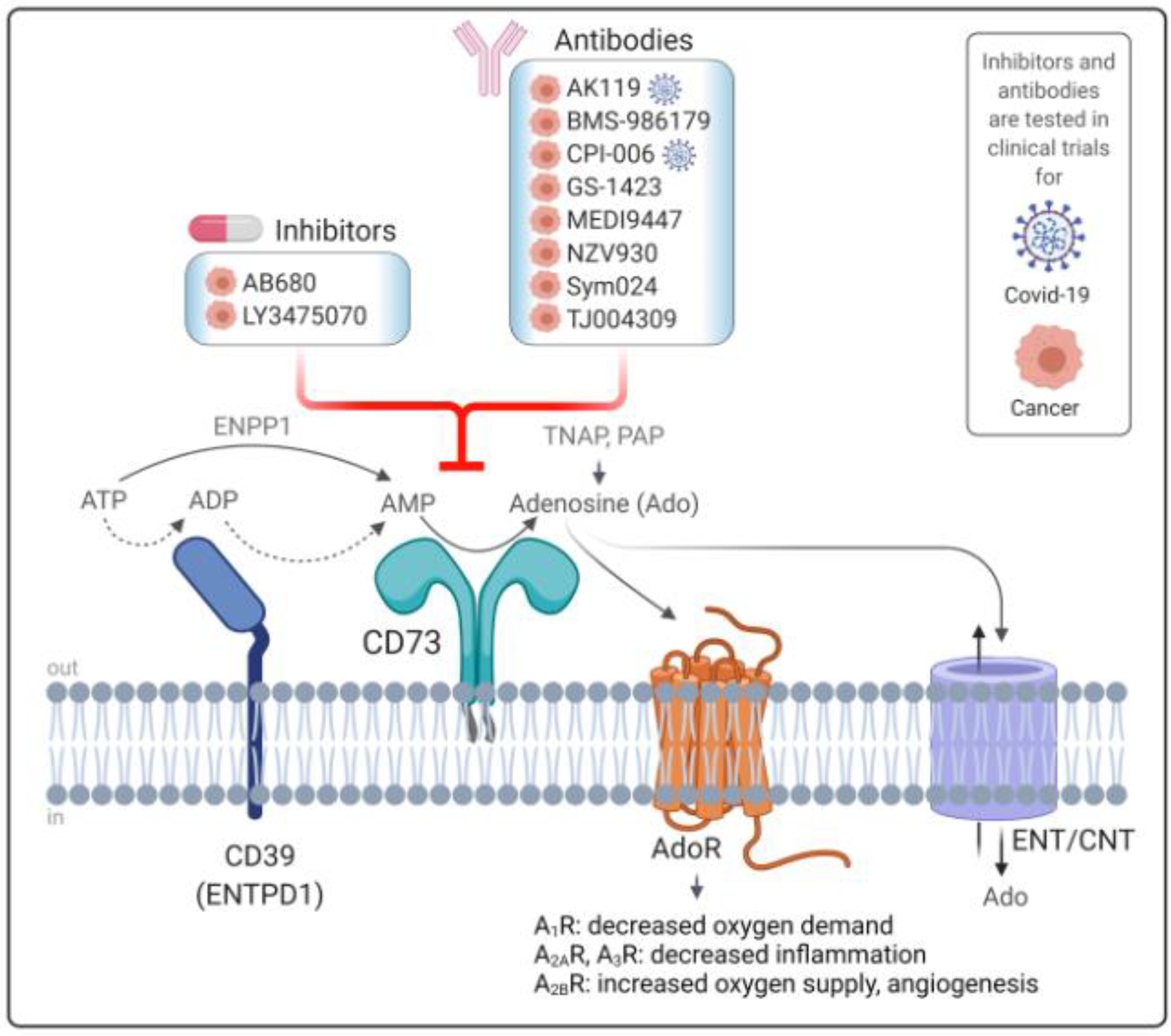
CD73 is a ubiquitously expressed ecto-nucleotidase of the purine metabolism pathway. As a glycosylphosphatidylinositol (GPI) -anchored glycoprotein on the plasma membrane, CD73 works in tandem with ectonucleoside triphosphate diphosphohydrolase-1 (CD39), which breaks down adenosine triphosphate (ATP) to form adenosine 5’-monophosphate (AMP) in a two-step process. Alternatively, AMP can be generated via direct conversion from ATP by the enzyme ecto-nucleotide pyrophosphatase/phosphodiesterase-1 (ENPP1). CD73 is the major enzyme that dephosphorylates AMP to generate extracellular adenosine (Ado), but this reaction can also be carried out by tissue non-specific alkaline phosphatase (TNAP) or prostatic acid phosphatase (PAP). CD73-generated adenosine directly exerts tissue-specific functions by binding to four different types of G-coupled adenosine receptors (AdoR), which regulate oxygen supply/demand ratios, inflammation and angiogenesis in a receptor-dependent manner. Additionally, adenosine is transported into the cytoplasm through equilibrative and concentrative nucleoside transporters (ENTs and CNTs). Due to its role in inflammatory responses and tumor growth and metastasis, small molecule inhibitors and monoclonal antibodies against CD73 are currently being tested in clinical trials for cancer immunotherapy and COVID-19 therapy.
Currently, blocking the enzymatic activity of CD73 is regarded as an important avenue for cancer therapy because adenosine suppresses anti-tumor immunity [7]. Multiple CD73-targeting antibodies and small molecule inhibitors are undergoing clinical testing (Figure 1). However, the long-term safety of systemic interventions blocking CD73 is an important consideration, because adenosine is critical for normal physiology, and loss-of-function mutations in the CD73-encoding gene (NT5E) cause a rare vascular disease in humans [8]. Moreover, studies from the last 2–4 years have illuminated significant biological complexity in human CD73 regulation and function, which need to be taken into account as the field moves forward. The purpose of this review is to render the latest discoveries on CD73 biology in a historical context and highlight CD73 functions that are important for normal cell biology and physiological homeostasis.
From 5’-NT to CD73: the evolution of CD73 biology and its links to human disease
CD73 is a glycosylphosphatidylinositol (GPI)-anchored glycoprotein that functions as a dimer on the plasma membrane [9]. Discovered more than 80 years ago as 5′-nucleotidase (5′-NT), it received the designation ‘cluster of differentiation 73 (CD73)’ just prior to the cloning of its cDNA in 1990 [3, 6]. To distinguish it from functionally similar cytoplasmic enzymes [10], it is also called ecto-5′-nucleotidase, abbreviated eN or eNT. Currently, 5′-NT, eN, eNT and CD73 are all used to refer to the protein product (P21589; NP_002517) of the same gene, which is NT5E (Gene ID: 4907). The name CD73 is most commonly used in the recent literature (last 10–15 years) and coincides with a shift in focus on immune functions, especially in the context of cancer. However, CD73 is ubiquitously expressed and involved in virtually every aspect of normal physiology and many disease-associated mechanisms [6]. Moreover, non-immune cells that normally express high levels of CD73, including fibroblasts, epithelial, and endothelial cells, are epigenetically primed to elicit tissue-specific immune responses [11]. This is an important consideration, but it is currently underappreciated.
CD73-generated adenosine controls cellular stress adaptation
Tissues with the highest level of NT5E expression include smooth muscle, the female reproductive system, liver, and gastrointestinal tract [6, 12]. In addition to its abundance on epithelial and endothelial cells, CD73 is also active on neurons, glia, myocytes, and fibroblasts [6]. Based on many studies in whole-body Nt5e−/− mice, CD73 activity is important for maintaining tissue integrity, especially endothelial and epithelial barrier functions, and for facilitating recovery following hypoxia, ischemia/reperfusion, and inflammatory injury in the brain, heart, lung, kidney, liver, and digestive tissues [13, 14]. A comprehensive description of the cell type- and tissue-specific functions of CD73 is beyond the scope of the current article, and we refer readers to previous reviews for more details [6, 15]. Of note, there is existing evidence that CD73 also works non-enzymatically to regulate T-cell receptor activation, immune-endothelial interactions, apoptosis, intracellular kinase signaling and other cellular functions [6, 15]. Still, most studies to date have attributed the physiological functions of CD73 to its ability to control the extracellular ATP-adenosine balance.
The basal release of ATP triggers activation of purinergic signaling to control diverse cellular processes, including ion transport, cell volume regulation, intercellular communication, blood flow, and neuronal activity [16]. Adaptability in this pathway is achieved through altered expression and activity of enzymes and receptors and it is crucial for regaining homeostasis following exposure to stress, including mechanical, inflammatory, hypoxic, metabolic, and other types. For example, the mechanical stress-dependent release of ATP is particularly well documented across many cell types, such as erythrocytes [17], airway epithelial cells [18], vascular endothelial cells [19], astrocytes [20], and neurons [21]. Increased ATP release from stressed or dying cells represents a ‘danger’ pro-inflammatory signal, and is terminated upon ATP metabolism to the anti-inflammatory mediator adenosine in order to avoid excessive activation of tissue defense mechanisms [22, 23].
Adenosine performs life-preserving functions inside and outside of the cell
Adenosine has fascinated biologists for decades because it controls virtually every system in the body. It has been named a “retaliatory metabolite” because it enables target cells to respond to stress and adjust their energy supply, thereby retaliating against external stimuli that would otherwise promote the excessive breakdown of ATP [24]. External stimuli can be physiological or stress-related, and vary by cell and tissue type (e.g. altered blood flow or tissue oxygenation, exposure to hormones, neurotransmitters, and inflammatory mediators) [25]. More recently, adenosine has been called a “multi-signaling guardian angel” because it restores the oxygen supply-and-demand balance and exerts potent anti-inflammatory effects to guard against cellular damage [26]. Adenosine is taken up into the cell and re-phosphorylated to replenish intracellular ATP stores, which is an important mechanism for purine salvage [27]. In addition, the transport-dependent adenosine uptake and phosphorylation by adenosine kinase promote increased levels of AMP and activation of the master metabolic regulator AMP-activated protein kinase (AMPK) [28], which is important for cellular and tissue homeostasis. For example, mice with a targeted deletion of Nt5e in hepatocytes exhibit significant hypoactivation of AMPK and develop spontaneous hepatocyte degeneration and liver injury [29].
The four ubiquitously-expressed metabotropic adenosine receptors and their G protein-coupled activities regulate cardiovascular, respiratory and immune functions, metabolism, neurological activity, gastrointestinal and liver biology [4]. Direct activation of adenosine receptors by small molecules with selective affinity for each receptor type represents an important avenue for drug development for cardiovascular diseases, pain, cancer, diabetes, liver disease and other disorders [30]. Recently, mice that lack all four adenosine receptors were generated and reported to have significantly shorter lifespan [31]. The decline in survival began at 15 weeks of age, reaching 50% by the time the mice were 30 weeks old [31]. While this mouse model reveals that baseline adenosine signaling via adenosine receptors collectively is critical for long-term organismal viability, the mechanisms leading to shortened life span are unknown. Going forward, this mouse model will be a useful tool to address the role of adenosine receptor signaling not only in homeostasis, but also in allostasis - the process by which regulatory systems adapt under stress in order to regain homeostasis [32]. Shifting away from whole-body to tissue-specific Nt5e−/− mice will provide additional clarity on where CD73 function fits with respect to homeostatic and allostatic adenosine receptor signaling across different tissues.
CD73-generated adenosine controls cancer progression
Persistent changes in the activation of purinergic signaling pathways can promote the development of diseases that are driven by metabolic perturbations and chronic inflammation, such as cancer [33]. Mechanical forces and other stress-related mechanisms are particularly relevant to growing tumors because they promote the release of ATP from cancer cells [34]. Pharmacologic or genetic ablation of CD73 in mice leads to decreased conversion of extracellular ATP to adenosine and promotes anti-tumor immunity [7]. The persistence of an immunosuppressive environment in the presence of active CD73 is largely due to increased A2AR and, to a lesser extent, A2BR activation on multiple immune cell types, including natural killer (NK) cells, and effector and regulatory T (Treg) cells. Specifically, A2AR signaling inhibits effector T cell proliferation and cytotoxic function [35–37], enhances Treg expansion and immunosuppressive activity [38], and inhibits NK cell maturation and target cell killing [39, 40]. Signaling via A2BR facilitates expansion of myeloid derived suppressor cells (MDSC) [41], and CD73 activity on MDSCs inhibits T cells and NK cells [42]. Persistent hypoxia and inflammation within the tumor microenvironment boost immunosuppressive responses by elevating extracellular adenosine through modulation of adenosine-related genes [43, 44]. Currently there is a strong emphasis on understanding the functional role of CD73 as an immune checkpoint modulator [45–48], while simultaneously testing if CD73 inhibitors can provide benefit to cancer patients when used in combinatorial immunotherapy regimens [7].
Despite these efforts, we do not yet have a thorough understanding of all of the mechanisms by which CD73 controls tumor biology, especially in settings where its functions are tumor-suppressing. It is known that sustained expression of CD73 on epithelial cells is associated with favorable outcomes in endometrial and bladder cancer patients [49–51]. In endometrial cancer, epithelial CD73-generated adenosine is necessary for A1R-dependent cortical actin polymerization and cell–cell adhesion, while CD73 loss is pro-tumorigenic because it compromises epithelial barrier function [51]. Therefore, future studies need to reconcile these opposing, cell type-specific roles of CD73 in cancer. Particular care should be exercised to avoid over-emphasis of CD73 function on immune cells over other cell types, given its ubiquitous expression.
Genetic deficiency of CD73 alters kinase signaling and gene regulation in a rare human disease
The critical role of CD73 in human biology is illustrated by the fact that loss-of-function mutations in NT5E cause the rare adult-onset vascular disease ‘calcification of joint and arteries’ (CALJA; OMIM: 211800) [52]. Also known as ‘arterial calcification due to deficiency of CD73’ (ACDC), the disease is characterized by painful calcifications that affect the lower and upper extremities [8, 53, 54] due to diminished CD73 activity on smooth muscle cells. The tissue-specific ACDC presentation is in line with recent quantitative profiling studies that reveal the highest NT5E/CD73 expression in human arteries, in a comparison of 32 normal tissues [55].
Mechanisms linking NT5E mutations to clinical presentations are not fully understood, partly because genetic mouse models of CD73 deficiency do not reflect the human phenotype [52]. To overcome this limitation, ACDC patient fibroblasts and induced pluripotent stem cell (iPSC)-derived mesenchymal stromal cells (MSCs) have been used to study signaling mechanisms altered in the absence of functional CD73 [56, 57]. These studies reveal that ACDC patient fibroblasts have dysregulated transcription factor Forkhead Box O1 Protein (FOXO1) activity [57]. Furthermore, ACDC patient MSCs display increased activation of AKT kinase, mechanistic target of rapamycin (mTOR), and the 70-kDa ribosomal protein S6 kinase (p70S6K) in the presence of osteogenic stimuli [56]. In the absence of functional CD73, there is increased phosphorylation of AKT, leading to FOXO1 activation, which in turn promotes expression of TNAP. Increased TNAP activity is a known factor in promoting ectopic calcification in ACDC [56]. Moreover, decreased levels of intracellular adenosine due to elevated activity of adenosine kinase, which phosphorylates adenosine to AMP, exacerbate vascular inflammation in mice via epigenetic reprogramming of histone methylation [58]. This mechanism was shown to be dependent on the uptake of extracellular adenosine, further supporting the hypothesis that CD73 provides a key link between extracellular purinergic signaling and gene regulation.
It will be important to assess additional stress response mechanisms in ACDC model systems, as CD73 regulates stress recovery of bone marrow stromal cells [59]. Specifically, it was shown that depletion of CD73 from stromal cells impairs early hematopoietic cell regeneration following irradiation in mice. Understanding the detailed mechanisms behind these observations will be important in oncology settings, as systemic use of CD73 inhibitors could affect the ability of cancer patients to recover from cytotoxic stress due to chemotherapy or radiation. As the natural history of ACDC becomes defined, patient-derived cellular tools to model aspects of the disease will enable further refinement of the cell biological consequences of CD73 genetic inactivation or inhibition.
CD73 is zonally expressed on subsets of cells within epithelial tissues
Despite the noted species differences, animal models will continue to play an important role in advancing CD73 biology because we are also beginning to recognize the importance of zonal distribution of CD73 on subsets of cells within a given tissue, in particular digestive epithelia (Figure 2). Advances in single cell sequencing technologies are revealing how spatial dynamics may play a role in the physiological homeostatic functions of CD73, particularly in response to tissue oxygenation. Under homeostasis, CD73 defines populations of cells residing in low-oxygen areas, such as the villus tip enterocytes of the small intestine [60], the pericentral hepatocytes in the liver [61], and erythropoietin-producing interstitial cells of the kidney [62]. The zonal distribution of CD73 is consistent with earlier findings that its expression is strongly responsive to hypoxia because of the presence of hypoxia response element - 1 (HIF-1) binding sites in the NT5E promoter [63]. This regulatory mechanism is similar between human and mouse. Nt5e−/− mice display vascular leakage in response to normobaric hypoxia in multiple tissues (lung, liver, gut, muscle, heart, kidney, brain) [13]. Furthermore, Nt5e−/− mice have increased susceptibility to cardiovascular, respiratory, gastrointestinal (GI) and liver injury, in large part because they lack the adaptive mechanisms afforded by extracellular adenosine in response to hypoxia stress [13, 64–67]. Therefore, expression of CD73 in low oxygen environments provides a physiological benefit that is suited to the architecture of the particular tissue type.
Figure 2. Zonal Expression of CD73 Supports Tissue-Specific Homeostasis.
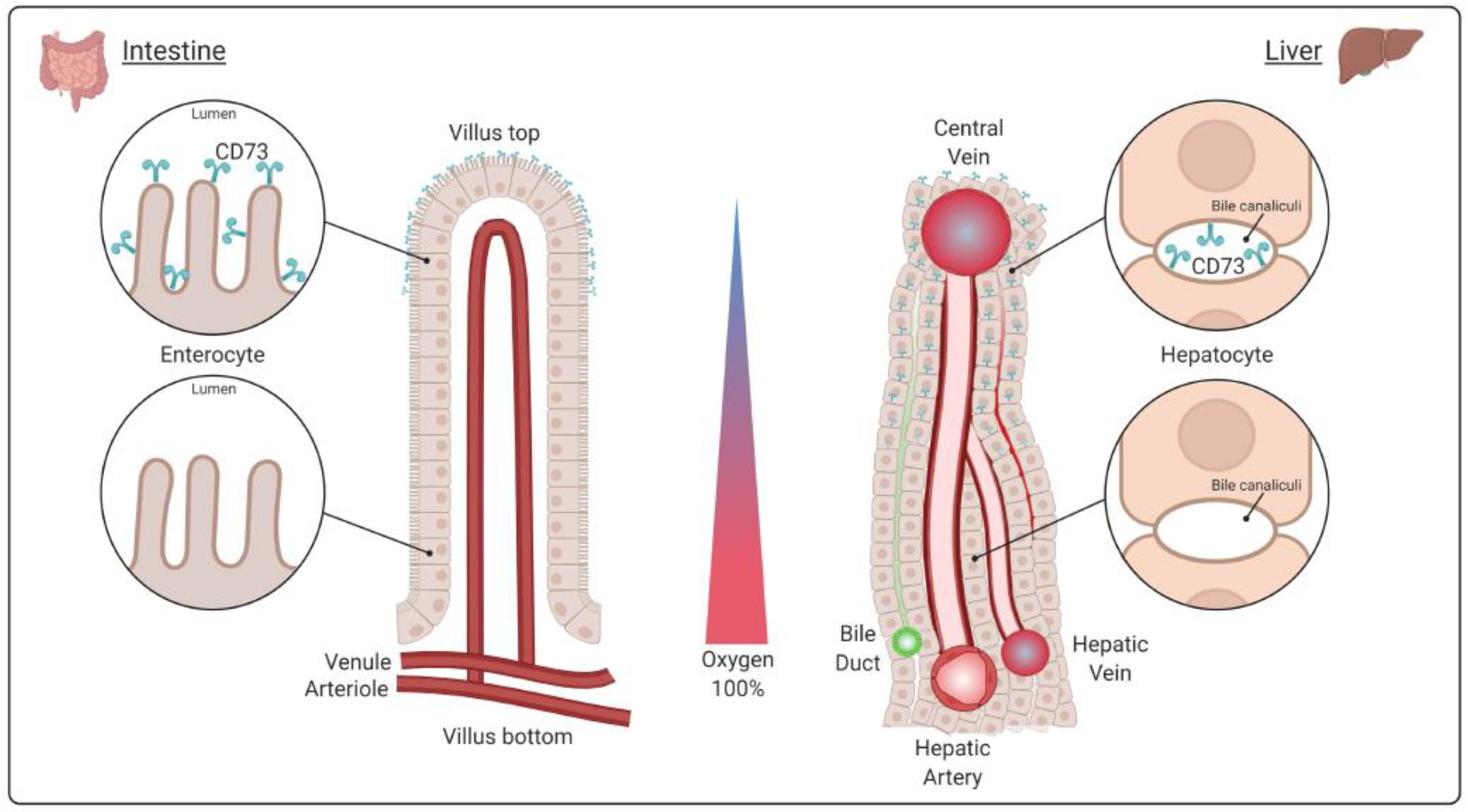
A hypoxic environment induces CD73 expression in the apical membrane of different tissues. In the intestinal epithelia, for example, CD73 is present on villus tip enterocytes, which are subjected to low oxygen conditions. The villus tips face the intestinal lumen where anaerobic bacteria normally reside. Similarly, CD73 is zonally expressed in hepatocytes in the liver. In the hepatic lobule, periportal hepatocytes are located next to the portal triad on one side (encompassing the hepatic artery, vein, and bile duct), while pericentral hepatocytes reside adjacent to the central vein. This arrangement follows the oxygen gradient: highly oxygenated blood enters the liver via the hepatic artery from the portal triad, mixes with deoxygenated blood through the sinusoids between hepatocytes and is returned to the circulation via the central vein.
One reason for the preferential localization of CD73 at the villus tip enterocytes is to counteract the ATP released by bacteria in the intestinal lumen and thereby control inappropriate immune activation by the host microbiome. Other potential functions may include the metabolism of cyclic dinucleotides to regulate host defense mechanisms at mucosal surfaces and serve as a source of antimicrobial adenosine to prevent bacterial colonization and infection [68]. CD73 distribution on pericentral hepatocytes in the mammalian liver likely enables the cells to calibrate their metabolic activities under physiologically low oxygen conditions, since genetic deletion of hepatocyte CD73 results in metabolic and inflammatory liver injury [29]. Since pericentral hepatocytes are involved in the homeostatic renewal of the liver in response to Wnt ligands [69], it will be of interest to examine in the future whether CD73 modulates these pathways as part of liver mass maintenance.
Given these observations, it will be important to examine how purinergic intermediates in the gut, liver, and systemic circulation are affected by anti-CD73 interventions, as this may affect patients’ response to, and effects from, immunotherapy. It was shown recently that the nucleoside inosine significantly potentiates the anti-cancer effects of checkpoint inhibitors [70]. Inosine is derived metabolically from the gut microbiome [70] and it can also be produced extracellularly from adenosine by cell surface-localized adenosine deaminase (ADA) [71]. It was observed that inosine translocates from the gut lumen to the systemic circulation and activates T cell–specific A2AR signaling to promote anti-tumor TH1 cell activation [70]. This raises a potential concern that co-administration of CD73 inhibitors (e.g. oleclumab) and checkpoint inhibitors (e.g. the PD-1 inhibitor durvalumab), as it is being done in clinical trials, may reduce treatment efficacy by lowering inosine levels. Moreover, combining CD73 inhibitors with checkpoint inhibitors, which are known to cause acute hepatitis [72], may lower the threshold for liver injury and result in poor outcomes in the long-term [73], especially in patients with other predisposing factors for liver disease, such as genetic makeup, age, and biological sex.
Sex and age are important variables in CD73 biology
An important question moving forward is how biological sex will impact the safety and effectiveness of CD73-targeted cancer immunotherapies. Although there is some evidence based on meta-analyses [74] and multi-omics datasets [75] to support sex differences, this is still a highly debated issue and it remains an open question. At the basic science level, most in vivo studies on CD73 have been done only in male mice, or biological sex was not explicitly considered as a potential variable. However, an emerging concept in the field is that there are critical hormonal influences, particularly estrogen-derived, in how males and females metabolize extracellular adenosine and cope with deficiency of CD73. Sex differences in adenosine signaling play a role in neuromodulation within the hippocampus, which has a high frequency of spontaneous transient adenosine events that regulate synaptic transmission, glia-neuron interactions, and other important functions [76]. Female Nt5e−/− mice have dramatically lower numbers of spontaneous transient adenosine events compared to WT mice, whereas male WT and Nt5e−/− mice have similar frequencies because of compensatory upregulation in TNAP in the latter [76]. Thus, female mice appear to be more reliant on CD73 for spontaneous adenosine transients, which is in line with earlier observations that CD73 expression and activity in the hippocampus are positively regulated by estrogen receptors (ER) α and β [77, 78].
ERβ-mediated signaling impacts CD73 biology in the GI tract via Tregs, which rely on CD73-generated adenosine for their immunosuppressive functions [79]. Specifically, estrogen mediates the differentiation of peripheral Tregs in an ERβ-dependent mechanism, and deletion of ERβ reduces the numbers of CD39/CD73-positive Tregs in female, but not in male mice affected by chronic intestinal inflammation [80]. This finding has potential implications for the management of female versus male patients with inflammatory GI conditions because ERβ expression is selectively downregulated in the intestinal mucosa and circulating T cells of female Crohn’s disease patients [80]. In the liver, hepatocyte-specific genetic deletion of CD73 leads to spontaneous injury characterized by metabolic dysfunction, hepatocyte swelling and ballooning, steatosis, and inflammation in middle-aged male mice [29]. Female mice lacking hepatocyte CD73 are relatively protected, potentially via compensatory upregulation of Entpd1 and all four adenosine receptors [29]. Thus sex-dependent differences in vulnerability versus resilience in coping with CD73 deficiency appear to be highly tissue-specific, and this will need to be resolved in future work.
Age and aging-related stress responses will be other important considerations for future studies, given recent findings that CD73 activity can be beneficial or harmful in atherosclerosis settings, depending on the age of mice [81] and that CD73 expression levels change throughout the human lifespan [82].
CD73 expression and activity are regulated at multiple levels
While many studies report that CD73 is altered in stress, chronic diseases and cancer, a few address the full spectrum of changes at the mRNA level, protein expression and localization, or enzymatic activity (Figure 3). The specific mechanism by which CD73 is affected in stress and disease is an important consideration because upregulation at the mRNA level does not necessarily result in increased protein expression, while increased protein expression does not always correlate with increased enzymatic activity [83]. Moreover, the possibility that NT5E mRNA upregulation in cancer, as it is frequently reported, could act independently of CD73 activity has not been fully explored. Aside from HIF-1, several other transcription factors are known to induce CD73 expression, including SP1, SMAD and c-Jun/AP-1 [84, 85].
Figure 3. Molecular Regulation of CD73.
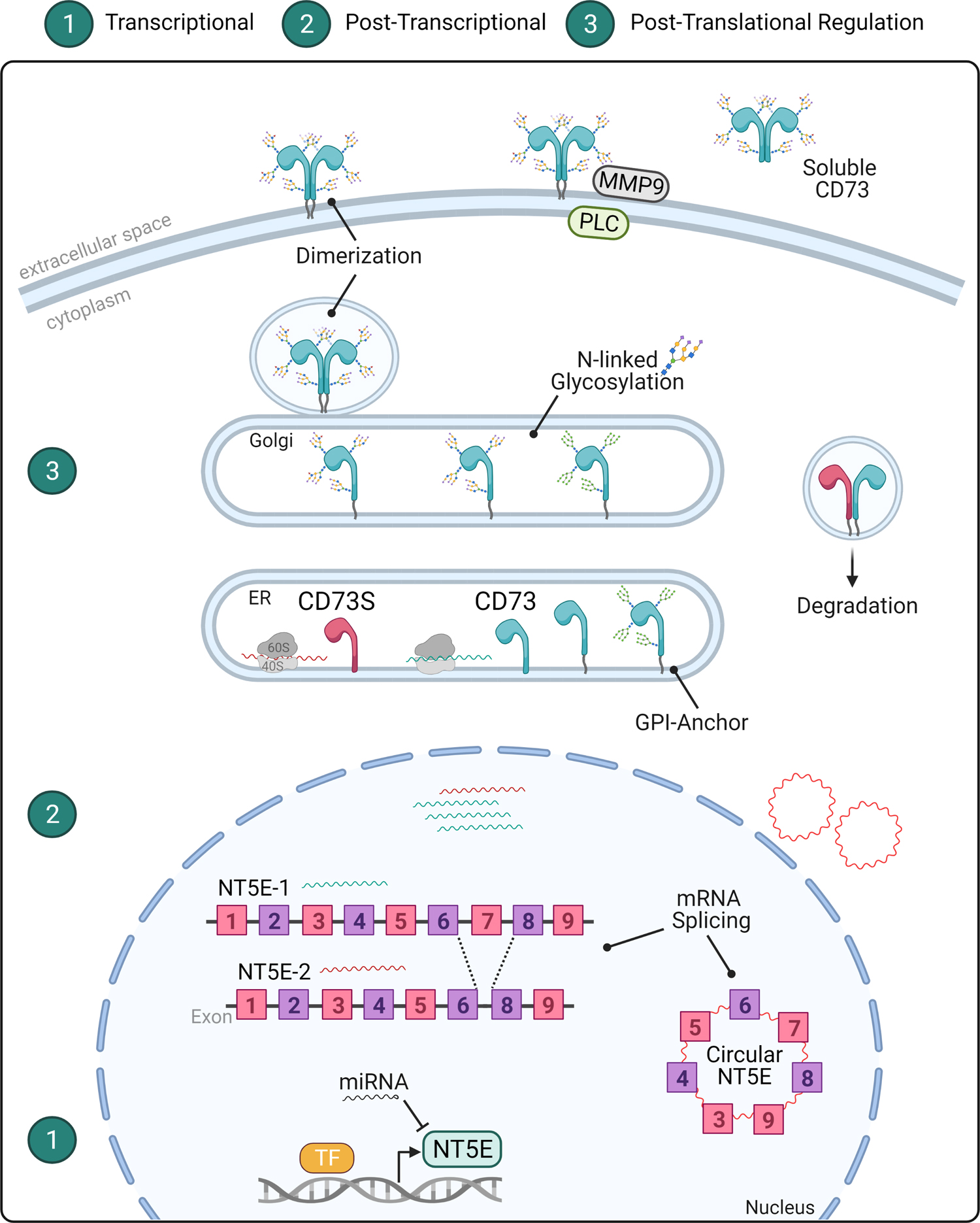
The important functions of CD73 across cell- and tissue-types warrant different levels of molecular regulation. 1) At the transcriptional level, the expression of the CD73-encoding gene NT5E is upregulated by transcription factors (TF) such as HIF1α, SP1, SMAD, and AP1 and several micro RNAs (miRNAs), as noted in the text. In the context of cancer, NT5E mRNA undergoes 2) post-transcriptional splicing to generate an alternative NT5E-2 transcript or the circular RNA circNT5E. Subsequently, NT5E-1 mRNA is translated into CD73 and NT5E-2 mRNA into CD73S, which is an intracellular enzymatically-inactive isoform that targets canonical CD73 for proteosomal degradation. The circNT5E transcript is oncogenic and expressed in glioma and lung cancer. 3) At the post-translational level, CD73 protein is modified by the addition of a glycosylphosphatidylinositol (GPI)-anchor and by asparagine (N)-glycosylation in the endoplasmic reticulum (ER) and Golgi apparatus. The mature CD73 protein dimerizes and is transported to the plasma membrane facing the extracellular space. CD73 can be cleaved by phospholipase C (PLC) or matrix metallopeptidase 9 (MMP9) to generate a soluble form of the protein.
Recently, a novel tumor-promoting non-coding circular RNA with oncogenic activity called circNT5E was discovered in glioblastoma [86] and non-small cell lung cancer [87] (Figure 3). The circNT5E mRNA arose from exon 3–9 region of NT5E through the activity of the double-stranded RNA-specific editase B2 (ADARB2). The pro-tumorigenic activity of circNT5E due to its ability to act as a sponge, or sink, for tumor suppressor micro RNAs (miRNAs), including miR-422a [86]. Adding to that complexity, miR-442a, miR-30a, miR-30b, miR-30a-5p, and miR-340 directly target and inhibit NT5E expression in head and neck, colorectal, gallbladder, glioma, lung, and pancreas cancer [88–90]. Other ways in which NT5E is dysregulated in cancer is via alternative splicing of exon 7 to produce a shorter enzymatically-inactive intracellular protein isoform (CD73S), which acts as a dominant negative to the canonical form [91] (Figure 3). CD73S is a human-specific isoform and its exact functions remain to be determined. Unravelling these transcriptional mechanisms may open possibilities for selectivity in targeting the pro-tumorigenic effects of NT5E without interfering with the normal enzymatic functions of CD73.
At the protein level, canonical CD73 undergoes a number of post-translational modifications (PTMs) that can significantly impact its localization and activity, including cleavage from the membrane to form a soluble enzyme [92–94] (Figure 3). This is a key consideration in studies that involve tissue digestion, since that removes the membrane-bound form of CD73, as previously shown to occur immediately following hepatocyte isolation [83]. Another important consideration is that CD73 is N-glycosylated at four different residues (N53, N311, N333 and N403) and site-specific changes in the abundance and composition of glycans alter its sub-cellular localization and enzymatic activity [94]. Aside from human cirrhosis and liver cancer, presently little is known about how alternative splicing and glycosylation impact CD73 expression, localization, and activity in other chronic diseases and in different cancer types. Importantly, the transcriptional and post-translational regulation mechanisms of CD73 under homeostatic conditions are not well defined, but it is likely that they exert important effects on its function.
Development of CD73 inhibitors and other tools to support further research
Active efforts to block the adenosine-producing CD73 activity for therapeutic purposes of limiting cancer growth and metastasis include monoclonal antibodies and small molecule inhibitors [95]. The initial proof-of-concept pre-clinical study using an inhibitory antibody against CD73 was done 11 years ago [96] and there are now at least five different anti-CD73 antibodies (BMS-986179, CPI-006, MEDI9447, NZV930 and TJ004309) and two small molecule inhibitors (AB122 and LY3475070) undergoing Phase I/II clinical trials [95]. Many similar agents are in early stage discovery and pre-clinical development [95, 97–102]. Somewhat surprisingly, some anti-CD73 antibodies are already being tested for COVID-19 therapy [103, 104], despite evidence of clinical benefits of CD73 and adenosine in lung injury [105], and benefit of adenosine in pneumonia associated with COVID-19 [106]. CD73 activity on immune cells versus other types of cells (endothelial, epithelial) need to be carefully considered in order to advance safe and effective treatments for COVID-19.
Elevating, rather than suppressing, the function of CD73 is going to be beneficial in many situations where tissue inflammation needs to be reduced. To that end, bi-functional proteins were engineered by fusing the extracellular domains of CD39 and CD73 [107]. The fusion proteins exhibited high phosphohydrolysis activity towards extracellular ATP and anti-platelet activity in vitro, suggesting they could potentially be developed to treat inflammatory diseases [107]. In addition, extracellular ATP release can be directly visualized in live animals using a newly developed optical sensor (ATPOS) [108], which represents another important methodological advance toward understanding the dynamics of the purinergic signaling components in vivo.
Radiolabeled antibodies [109] and fluorescent probes [110] are among the latest tools that were developed to monitor CD73 distribution and regulation in various settings. An Nt5e reporter mouse was also generated, and it appears to be a useful tool for studying CD73 on multipotent stromal cells and sinusoidal endothelial cells [111]. The availability of multiple approaches to target, manipulate, and track CD73 will undoubtedly open new opportunities to understand its biology and regulation during physiological adaptation.
Conclusions and future perspectives
Decades of research breakthroughs on the release and metabolism of ATP to adenosine outside of the cell have revealed critical functions that are independent of the essential metabolic activities occurring within the cell. Adenosine controls numerous homeostatic processes and stress adaptation mechanisms, which would be rendered ineffective in the setting of chronic CD73 inhibition, an effort currently being undertaken in clinical research [7]. In order to successfully advance therapies around CD73, now is the time to take a step back and understand the fundamental biology behind this fascinating molecule (see Outstanding Questions). Priorities for future work include the generation of additional human-specific tools to study CD73 regulation – such as iPSC-derived cells and tissue organoids [52]. These tools can help resolve species-specific mechanisms, such as alternative splicing [91], and help streamline the process of translating pre-clinical discoveries to the clinic. It will be important for future studies to carefully consider the mechanism by which CD73 expression and activity are altered in disease, such as mRNA expression and processing, protein expression and localization, and enzymatic activity, as these are often discordant under pathological conditions. The current anti-CD73 targeting strategies rely on the presence of cell surface - expressed, enzymatically active form of CD73 but do not address alternative splice isoforms and PTM variants that can affect localization and activity. Ideally, CD73 targeting in disease and cancer should be tailored to specific cell types to avoid untoward effects. The whole-body knockout mouse model has been instrumental in understanding CD73 function and for disease modeling [13], but given the ubiquitous expression and complex interplay between CD73 on different cell types [6], it is critical to move forward using tissue-specific knockout models, as has already been done in intestinal [112], kidney [113] and liver models [29]. All of these questions are addressable with the availability of new iPSC technologies, genetic mouse models, highly selective and potent inhibitors, and imaging probes, which are creating new opportunities to monitor, target and manipulate CD73 (Figure 4). Future studies aimed at unravelling the biological complexity of CD73 regulation and functions will help guide translational and clinical efforts for cancer and other human diseases.
Outstanding Questions.
Which aspects of CD73 regulation and function are conserved across species and which ones are unique to humans?
What are the influences of age and biological sex, including hormones, on CD73 function, regulation and roles in disease?
What is the significance of CD73 metabolic zonation, and how does CD73 mediate physiological adaptation in epithelial tissues?
How are the transcriptional, post-transcriptional and post-translational mechanisms integrated to control CD73 expression and activity in normal cells? How are these mechanisms altered during stress and in disease conditions?
What are the functions of the circNT5E mRNA in malignant neoplastic cells?
Which RNA-binding proteins control NT5E expression and splicing in homeostasis and stress?
How are kinase signaling pathways (e.g. AKT, mTOR, AMPK) altered in the absence of functional CD73?
Does CD73 have adenosine-independent functions and, if so, how are they altered by CD73-targeting antibodies and small molecule inhibitors?
Figure 4. New Tools to Study CD73 Regulation and Function.
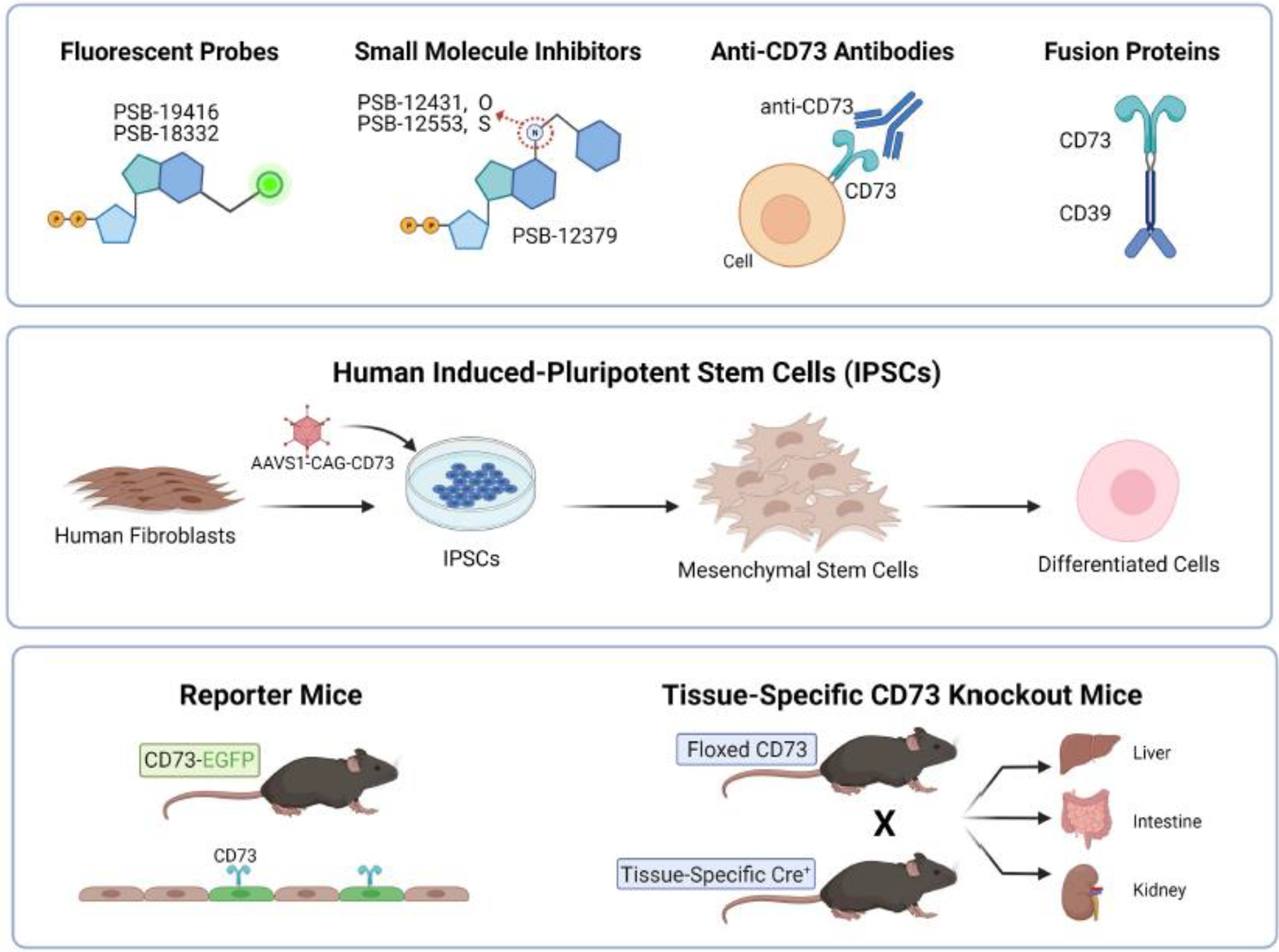
The ubiquitous nature of CD73 and the purinergic signaling complexity conceal important tissue-specific mechanisms, which warrants development of new tools to study this ecto-enzyme. (Top Row) Recently synthesized fluorescent probes and small molecule inhibitors were designed based on the lead structure of the most common CD73 inhibitor, adenosine 5’-(α,β-methylene)diphosphate or APCP. These newer probes exhibit higher potency while also enabling visualization and monitoring of CD73. Additionally, studies demonstrating pro-tumorigenic functions of CD73 led to the advent of new monoclonal antibodies tested in clinical trials. In contrast, promoting CD73 activity may alleviate inflammation and platelet aggregation. To that end, a CD39-CD73 fusion protein was shown to sequentially hydrolyze pro-inflammatory ATP to anti-inflammatory adenosine. (Middle Row) To interrogate relevant disease mechanisms, patient-derived induced pluripotent stem cells (iPSCs) have become a robust model system. For example, fibroblasts derived from patients with a rare genetic mutation of CD73 can be reprogrammed to generate iPSCs. These, in turn, can be differentiated into affected cell types to study pathological mechanisms the rare disease Arterial Calcification Due to Deficiency of CD73 (ACDC). (Bottom Row) To elucidate tissue-specific functions of proteins, reporter mouse lines and targeted gene deletion have been instrumental. A new reporter mouse line called CD73-EGFP enables tracking cell lineage and identification of CD73+ cells. Another useful model is the floxed CD73 mouse line, which enables targeted deletion of CD73 in specific tissues when mated with Cre recombinase mice. Specifically, deletion of CD73 in the liver, intestines, and kidney demonstrated tissue-specific protection under physiological and pathological conditions.
Highlights.
The ecto-enzyme CD73, and its enzymatic product adenosine, control cellular homeostasis and allostasis by integrating extracellular purinergic signaling with intracellular kinase activities and gene transcription.
CD73 is complex and coordinated at multiple levels, including transcriptional (by hypoxia and Hippo signaling), post-transcriptional (by production of an alternative splice isoform and circular mRNA), and post-translational (by N-glycosylation and shedding from the membrane to produce a soluble form).
An oxygen gradient patterns zonal expression of CD73 to regulate the long-term metabolic and immune stability of epithelial cells and tissues.
Multiple CD73 inhibitors are undergoing clinical development for cancer. Given the complex regulation and homeostatic functions of CD73 and adenosine, caution around the long-term safety of systemic inhibition of CD73 is warranted.
Acknowledgments:
The authors’ work is supported by NIH grants DK110355, DK034987, and T32CA071341. The authors thank Kenneth Negy for editing and creative input on the review manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Burnstock G (2020) Introduction to purinergic signaling. In Purinergic Signaling, pp. 1–15, Springer. [DOI] [PubMed] [Google Scholar]
- 2.Zimmermann H (2020) History of ectonucleotidases and their role in purinergic signaling. Biochem Pharmacol, 114322. [DOI] [PubMed] [Google Scholar]
- 3.Zimmermann H (2020) Ectonucleoside triphosphate diphosphohydrolases and ecto-5′-nucleotidase in purinergic signaling: how the field developed and where we are now. Purinergic Signalling, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borea PA et al. (2018) Pharmacology of Adenosine Receptors: The State of the Art. Physiol Rev 98 (3), 1591–1625. [DOI] [PubMed] [Google Scholar]
- 5.Pastor-Anglada M and Perez-Torras S (2018) Who Is Who in Adenosine Transport. Front Pharmacol 9, 627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Minor M et al. (2019) Cell type- and tissue-specific functions of ecto-5’-nucleotidase (CD73). Am J Physiol Cell Physiol 317 (6), C1079–C1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allard B et al. (2020) The adenosine pathway in immuno-oncology. Nature Reviews Clinical Oncology 17 (10), 611–629. [DOI] [PubMed] [Google Scholar]
- 8.St. Hilaire C et al. (2011) NT5E mutations and arterial calcifications. New England Journal of Medicine 364 (5), 432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knapp K et al. (2012) Crystal structure of the human ecto-5’-nucleotidase (CD73): insights into the regulation of purinergic signaling. Structure 20 (12), 2161–73. [DOI] [PubMed] [Google Scholar]
- 10.Bianchi V and Spychala J (2003) Mammalian 5′-nucleotidases. Journal of Biological Chemistry 278 (47), 46195–46198. [DOI] [PubMed] [Google Scholar]
- 11.Krausgruber T et al. (2020) Structural cells are key regulators of organ-specific immune responses. Nature 583 (7815), 296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han X et al. (2020) Construction of a human cell landscape at single-cell level. Nature 581 (7808), 303–309. [DOI] [PubMed] [Google Scholar]
- 13.Thompson LF et al. (2004) Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. The Journal of experimental medicine 200 (11), 1395–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bowser JL and Broaddus RR (2016) CD73s protection of epithelial integrity: thinking beyond the barrier. Tissue Barriers 4 (4), 220–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colgan SP et al. (2006) Physiological roles for ecto-5’-nucleotidase (CD73). Purinergic signalling 2 (2), 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corriden R and Insel PA (2010) Basal release of ATP: an autocrine-paracrine mechanism for cell regulation. Science signaling 3 (104), re1–re1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wan J et al. (2008) Dynamics of shear-induced ATP release from red blood cells. Proceedings of the National Academy of Sciences 105 (43), 16432–16437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Homolya L et al. (2000) Cell to cell communication in response to mechanical stress via bilateral release of ATP and UTP in polarized epithelia. The Journal of cell biology 150 (6), 1349–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bodin P and Burnstock G (2001) Evidence that release of adenosine triphosphate from endothelial cells during increased shear stress is vesicular. Journal of cardiovascular pharmacology 38 (6), 900–908. [DOI] [PubMed] [Google Scholar]
- 20.Joseph SM et al. (2003) Colocalization of ATP release sites and ecto-ATPase activity at the extracellular surface of human astrocytes. Journal of Biological Chemistry 278 (26), 23331–23342. [DOI] [PubMed] [Google Scholar]
- 21.Xia J et al. (2012) Neurons respond directly to mechanical deformation with pannexin‐mediated ATP release and autostimulation of P2X7 receptors. The Journal of physiology 590 (10), 2285–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stagg J and Smyth M (2010) Extracellular adenosine triphosphate and adenosine in cancer. Oncogene 29 (39), 5346–5358. [DOI] [PubMed] [Google Scholar]
- 23.Fredholm B (2007) Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death & Differentiation 14 (7), 1315–1323. [DOI] [PubMed] [Google Scholar]
- 24.Newby AC (1984) Adenosine and the concept of ‘retaliatory metabolites’. Trends in Biochemical Sciences 9 (2), 42–44. [Google Scholar]
- 25.Haskó G and Cronstein BN (2004) Adenosine: an endogenous regulator of innate immunity. Trends in immunology 25 (1), 33–39. [DOI] [PubMed] [Google Scholar]
- 26.Borea PA et al. (2016) Adenosine as a multi-signalling guardian angel in human diseases: when, where and how does it exert its protective effects? Trends in pharmacological sciences 37 (6), 419–434. [DOI] [PubMed] [Google Scholar]
- 27.Burnstock G (1972) Purinergic nerves. Pharmacol Rev 24 (3), 509–81. [PubMed] [Google Scholar]
- 28.Aymerich I et al. (2006) Extracellular adenosine activates AMP-dependent protein kinase (AMPK). Journal of cell science 119 (8), 1612–1621. [DOI] [PubMed] [Google Scholar]
- 29.Alcedo KP et al. (2021) CD73 maintains hepatocyte metabolic integrity and mouse liver homeostasis in a sex-dependent manner. Cellular and Molecular Gastroenterology and Hepatology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jacobson KA et al. (2019) Historical and current adenosine receptor agonists in preclinical and clinical development. Frontiers in cellular neuroscience 13, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiao C et al. (2019) Physiology and effects of nucleosides in mice lacking all four adenosine receptors. PLoS biology 17 (3), e3000161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McEwen BS and Wingfield JC (2003) The concept of allostasis in biology and biomedicine. Hormones and behavior 43 (1), 2–15. [DOI] [PubMed] [Google Scholar]
- 33.Kotas ME and Medzhitov R (2015) Homeostasis, inflammation, and disease susceptibility. Cell 160 (5), 816–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burnstock G and Di Virgilio F (2013) Purinergic signalling and cancer. Purinergic signalling 9 (4), 491–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang S et al. (1997) Role of A2a extracellular adenosine receptor-mediated signaling in adenosine-mediated inhibition of T-cell activation and expansion. Blood 90 (4), 1600–10. [PubMed] [Google Scholar]
- 36.Ohta A et al. (2009) A2A adenosine receptor may allow expansion of T cells lacking effector functions in extracellular adenosine-rich microenvironments. J Immunol 183 (9), 5487–93. [DOI] [PubMed] [Google Scholar]
- 37.Lappas CM et al. (2005) A2A adenosine receptor induction inhibits IFN-gamma production in murine CD4+ T cells. J Immunol 174 (2), 1073–80. [DOI] [PubMed] [Google Scholar]
- 38.Ohta A et al. (2012) The development and immunosuppressive functions of CD4(+) CD25(+) FoxP3(+) regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front Immunol 3, 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beavis PA et al. (2013) Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. Proc Natl Acad Sci U S A 110 (36), 14711–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Young A et al. (2018) A2AR Adenosine Signaling Suppresses Natural Killer Cell Maturation in the Tumor Microenvironment. Cancer Res 78 (4), 1003–1016. [DOI] [PubMed] [Google Scholar]
- 41.Ryzhov S et al. (2011) Adenosinergic regulation of the expansion and immunosuppressive activity of CD11b+Gr1+ cells. J Immunol 187 (11), 6120–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li J et al. (2017) CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-beta-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology 6 (6), e1320011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lan J et al. (2018) Hypoxia-inducible factor 1-dependent expression of adenosine receptor 2B promotes breast cancer stem cell enrichment. Proc Natl Acad Sci U S A 115 (41), E9640–E9648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hatfield SM et al. (2014) Systemic oxygenation weakens the hypoxia and hypoxia inducible factor 1alpha-dependent and extracellular adenosine-mediated tumor protection. J Mol Med (Berl) 92 (12), 1283–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neo SY et al. (2020) CD73 immune checkpoint defines regulatory NK cells within the tumor microenvironment. The Journal of clinical investigation 130 (3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu M et al. (2020) CD73 on cancer-associated fibroblasts enhanced by the A 2B-mediated feedforward circuit enforces an immune checkpoint. Nature communications 11 (1), 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goswami S et al. (2020) Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nature medicine 26 (1), 39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roh M et al. (2020) Targeting CD73 to augment cancer immunotherapy. Current Opinion in Pharmacology 53, 66–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wettstein MS et al. (2015) CD73 Predicts Favorable Prognosis in Patients with Nonmuscle-Invasive Urothelial Bladder Cancer. Dis Markers 2015, 785461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koivisto MK et al. (2019) Cell-type-specific CD73 expression is an independent prognostic factor in bladder cancer. Carcinogenesis 40 (1), 84–92. [DOI] [PubMed] [Google Scholar]
- 51.Bowser JL et al. (2016) Loss of CD73-mediated actin polymerization promotes endometrial tumor progression. The Journal of clinical investigation 126 (1), 220–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Joolharzadeh P and St. Hilaire C (2019) CD73 (cluster of differentiation 73) and the differences between mice and humans. Arteriosclerosis, thrombosis, and vascular biology 39 (3), 339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yoshioka K et al. (2017) Calcification of joints and arteries with novel NT5E mutations with involvement of upper extremity arteries. Vascular Medicine 22 (6), 541–543. [DOI] [PubMed] [Google Scholar]
- 54.Zhang Z et al. (2015) Calcification of joints and arteries: second report with novel NT5E mutations and expansion of the phenotype. Journal of human genetics 60 (10), 561–564. [DOI] [PubMed] [Google Scholar]
- 55.Jiang L et al. (2020) A Quantitative Proteome Map of the Human Body. Cell 183 (1), 269–283 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jin H et al. (2016) Increased activity of TNAP compensates for reduced adenosine production and promotes ectopic calcification in the genetic disease ACDC. Sci Signal 9 (458), ra121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moorhead III WJ et al. (2020) Dysregulation of FOXO1 (Forkhead Box O1 Protein) drives calcification in arterial calcification due to deficiency of CD73 and is present in peripheral artery disease. Arteriosclerosis, thrombosis, and vascular biology 40 (7), 1680–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu Y et al. (2017) Regulation of endothelial intracellular adenosine via adenosine kinase epigenetically modulates vascular inflammation. Nature communications 8 (1), 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Severe N et al. (2019) Stress-induced changes in bone marrow stromal cell populations revealed through single-cell protein expression mapping. Cell Stem Cell 25 (4), 570–583. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moor AE et al. (2018) Spatial reconstruction of single enterocytes uncovers broad zonation along the intestinal villus axis. Cell 175 (4), 1156–1167. e15. [DOI] [PubMed] [Google Scholar]
- 61.Ben-Moshe S et al. (2019) Spatial sorting enables comprehensive characterization of liver zonation. Nature metabolism 1 (9), 899–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Broeker KA et al. (2020) Different subpopulations of kidney interstitial cells produce erythropoietin and factors supporting tissue oxygenation in response to hypoxia in vivo. Kidney International 98 (4), 918–931. [DOI] [PubMed] [Google Scholar]
- 63.Synnestvedt K et al. (2002) Ecto-5’-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest 110 (7), 993–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eckle T et al. (2007) Cardioprotection by ecto-5’-nucleotidase (CD73) and A2B adenosine receptors. Circulation 115 (12), 1581. [DOI] [PubMed] [Google Scholar]
- 65.Khoury J et al. (2007) Antiinflammatory adaptation to hypoxia through adenosine-mediated cullin-1 deneddylation. The Journal of clinical investigation 117 (3), 703–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hart ML et al. (2008) Extracellular adenosine production by ecto-5′-nucleotidase protects during murine hepatic ischemic preconditioning. Gastroenterology 135 (5), 1739–1750. e3. [DOI] [PubMed] [Google Scholar]
- 67.Bowser JL et al. (2018) The Hypoxia-Adenosine Link during Intestinal Inflammation. J Immunol 200 (3), 897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chang D et al. (2020) Extracellular cyclic dinucleotides induce polarized responses in barrier epithelial cells by adenosine signaling. Proceedings of the National Academy of Sciences 117 (44), 27502–27508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang B et al. (2015) Self-renewing diploid Axin2+ cells fuel homeostatic renewal of the liver. Nature 524 (7564), 180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mager LF et al. (2020) Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 369 (6510), 1481–1489. [DOI] [PubMed] [Google Scholar]
- 71.Kameoka J et al. (1993) Direct association of adenosine deaminase with a T cell activation antigen, CD26. Science 261 (5120), 466–469. [DOI] [PubMed] [Google Scholar]
- 72.De Martin E et al. (2018) Characterization of liver injury induced by cancer immunotherapy using immune checkpoint inhibitors. Journal of hepatology 68 (6), 1181–1190. [DOI] [PubMed] [Google Scholar]
- 73.Yokohama K et al. (2020) Liver dysfunction is associated with poor prognosis in patients after immune checkpoint inhibitor therapy. Scientific Reports 10 (1), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Conforti F et al. (2018) Cancer immunotherapy efficacy and patients’ sex: a systematic review and meta-analysis. The Lancet Oncology 19 (6), 737–746. [DOI] [PubMed] [Google Scholar]
- 75.Ye Y et al. (2020) Sex-associated molecular differences for cancer immunotherapy. Nature communications 11 (1), 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang Y et al. (2020) CD73 or CD39 Deletion Reveals Different Mechanisms of Formation for Spontaneous and Mechanically Stimulated Adenosine and Sex Specific Compensations in ATP Degradation. ACS chemical neuroscience 11 (6), 919–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mitrovid N et al. (2019) Estrogen receptors modulate ectonucleotidases activity in hippocampal synaptosomes of male rats. Neurosci Lett 712, 134474. [DOI] [PubMed] [Google Scholar]
- 78.Mitrovic N et al. (2016) 17beta-Estradiol upregulates ecto-5’-nucleotidase (CD73) in hippocampal synaptosomes of female rats through action mediated by estrogen receptor-alpha and -beta. Neuroscience 324, 286–96. [DOI] [PubMed] [Google Scholar]
- 79.Deaglio S et al. (2007) Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med 204 (6), 1257–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Goodman WA et al. (2020) Impaired estrogen signaling underlies regulatory T cell loss-of-function in the chronically inflamed intestine. Proceedings of the National Academy of Sciences 117 (29), 17166–17176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sutton NR et al. (2020) CD73 promotes age-dependent accretion of atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology 40 (1), 61–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Crooke A et al. (2017) Low expression of CD39 and CD73 genes in centenarians compared with octogenarians. Immunity & Ageing 14 (1), 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Snider NT et al. (2013) CD73 (ecto-5’-nucleotidase) hepatocyte levels differ across mouse strains and contribute to mallory-denk body formation. Hepatology 58 (5), 1790–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fausther M et al. (2012) Activated hepatic stellate cells upregulate transcription of ecto-5′-nucleotidase/CD73 via specific SP1 and SMAD promoter elements. American Journal of Physiology-Gastrointestinal and Liver Physiology 303 (8), G904–G914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Reinhardt J et al. (2017) MAPK signaling and inflammation link melanoma phenotype switching to induction of CD73 during immunotherapy. Cancer research 77 (17), 4697–4709. [DOI] [PubMed] [Google Scholar]
- 86.Wang R et al. (2018) CircNT5E acts as a sponge of miR-422a to promote glioblastoma tumorigenesis. Cancer research 78 (17), 4812–4825. [DOI] [PubMed] [Google Scholar]
- 87.Dong L et al. (2020) The circular RNA NT5E promotes non-small cell lung cancer cell growth via sponging microRNA-134. Aging (Albany NY) 12 (4), 3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kordaß T et al. (2018) Controlling the immune suppressor: transcription factors and microRNAs regulating CD73/NT5E. Frontiers in immunology 9, 813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Peng L et al. (2020) MicroRNA‐30a suppresses self‐renewal and tumorigenicity of glioma stem cells by blocking the NT5E‐dependent Akt signaling pathway. The FASEB Journal 34 (4), 5128–5143. [DOI] [PubMed] [Google Scholar]
- 90.Zhou L et al. (2019) The distinct role of CD73 in the progression of pancreatic cancer. J Mol Med (Berl) 97 (6), 803–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Snider NT et al. (2014) Alternative splicing of human NT5E in cirrhosis and hepatocellular carcinoma produces a negative regulator of ecto-5’-nucleotidase (CD73). Mol Biol Cell 25 (25), 4024–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schneider E et al. (2019) Generation and Function of Non-cell-bound CD73 in Inflammation. Frontiers in immunology 10, 1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang W et al. (2018) Multiple steps determine CD73 shedding from RPE: lipid raft localization, ARA1 interaction, and MMP-9 up-regulation. Purinergic signalling 14 (4), 443–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Alcedo KP et al. (2019) Tumor‐Selective Altered Glycosylation and Functional Attenuation of CD73 in Human Hepatocellular Carcinoma. Hepatology communications 3 (10), 1400–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Boison D and Yegutkin GG (2019) Adenosine metabolism: emerging concepts for cancer therapy. Cancer Cell 36 (6), 582–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stagg J et al. (2011) CD73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res 71 (8), 2892–900. [DOI] [PubMed] [Google Scholar]
- 97.Du X et al. (2020) Orally Bioavailable Small-Molecule CD73 Inhibitor (OP-5244) Reverses Immunosuppression through Blockade of Adenosine Production. Journal of Medicinal Chemistry 63 (18), 10433–10459. [DOI] [PubMed] [Google Scholar]
- 98.Jin R et al. (2020) Dual Mechanisms of Novel CD73-Targeted Antibody and Antibody–Drug Conjugate in Inhibiting Lung Tumor Growth and Promoting Antitumor Immune-Effector Function. Molecular Cancer Therapeutics 19 (11), 2340–2352. [DOI] [PubMed] [Google Scholar]
- 99.Stefano JE et al. (2020) A highly potent CD73 biparatopic antibody blocks organization of the enzyme active site through dual mechanisms. Journal of Biological Chemistry 295 (52), 18379–18389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lawson KV et al. (2020) Discovery of AB680: A potent and selective inhibitor of CD73. Journal of Medicinal Chemistry 63 (20), 11448–11468. [DOI] [PubMed] [Google Scholar]
- 101.Beatty JW et al. (2020) Discovery of potent and selective non-nucleotide small molecule inhibitors of CD73. Journal of medicinal chemistry 63 (8), 3935–3955. [DOI] [PubMed] [Google Scholar]
- 102.Bhattarai S et al. (2020) 2-Substituted α, β-methylene-ADP derivatives: Potent competitive ecto-5′-nucleotidase (CD73) inhibitors with variable binding modes. Journal of medicinal chemistry 63 (6), 2941–2957. [DOI] [PubMed] [Google Scholar]
- 103.Willingham SB et al. (2020) Characterization and Phase 1 Trial of a B Cell Activating Anti-CD73 Antibody for the Immunotherapy of COVID-19. medRxiv. [Google Scholar]
- 104.Zhong T et al. , 476 AK119, a humanized anti-CD73 monoclonal antibody, as Immunotherapy for COVID-19, BMJ Specialist Journals, 2020. [Google Scholar]
- 105.Eckle T et al. (2007) Identification of ectonucleotidases CD39 and CD73 in innate protection during acute lung injury. The Journal of Immunology 178 (12), 8127–8137. [DOI] [PubMed] [Google Scholar]
- 106.Correale P et al. (2020) Therapeutic effects of adenosine in high flow 21% oxygen aereosol in patients with Covid19-pneumonia. Plos one 15 (10), e0239692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhong EH et al. (2021) Structural and functional characterization of engineered bifunctional fusion proteins of CD39 and CD73 ectonucleotidases. American Journal of Physiology-Cell Physiology 320 (1), C15–C29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kitajima N et al. (2020) Real-time in vivo imaging of extracellular ATP in the brain with a hybrid-type fluorescent sensor. Elife 9, e57544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sudo H et al. (2020) Radiolabeled human monoclonal antibody 067–213 has the potential for noninvasive quantification of cd73 expression. International journal of molecular sciences 21 (7), 2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schmies CC et al. (2020) Fluorescent Probes for Ecto-5′-nucleotidase (CD73). ACS Medicinal Chemistry Letters 11 (11), 2253–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Breitbach M et al. (2018) In vivo labeling by CD73 marks multipotent stromal cells and highlights endothelial heterogeneity in the bone marrow niche. Cell Stem Cell 22 (2), 262–276. e7. [DOI] [PubMed] [Google Scholar]
- 112.Kao DJ et al. (2017) Intestinal epithelial ecto-5′-nucleotidase (CD73) regulates intestinal colonization and infection by nontyphoidal salmonella. Infection and immunity 85 (10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sun-sang JS et al. (2017) Proximal tubule CD73 is critical in renal ischemia-reperfusion injury protection. Journal of the American Society of Nephrology 28 (3), 888–902. [DOI] [PMC free article] [PubMed] [Google Scholar]