

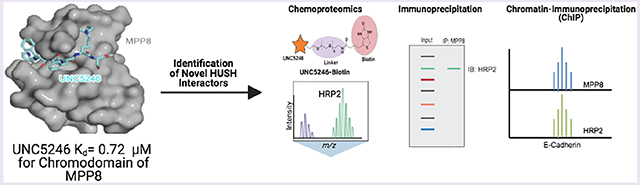
Abstract
The interpretation of histone post-translational modifications (PTMs), specifically lysine methylation, by specific classes of “reader” proteins marks an important aspect of epigenetic control of gene expression. Methyl-lysine (Kme) readers often regulate gene expression patterns through the recognition of a specific Kme PTM while participating in or recruiting large protein complexes that contain enzymatic or chromatin remodeling activity. Understanding the composition of these Kme-reader-containing protein complexes can serve to further our understanding of the biological roles of Kme readers, while small molecule chemical tools can be valuable reagents in interrogating novel protein–protein interactions. Here, we describe our efforts to target the chromodomain of M-phase phosphoprotein 8 (MPP8), a member of the human silencing hub (HUSH) complex and a histone 3 lysine 9 trimethyl (H3K9me3) reader that is vital for heterochromatin formation and has specific roles in cancer metastasis. Utilizing a one-bead, one-compound (OBOC) combinatorial screening approach, we identified UNC5246, a peptidomimetic ligand capable of interacting with the MPP8 chromodomain in the context of the HUSH complex. Additionally, a biotinylated derivative of UNC5246 facilitated chemoproteomics studies which revealed hepatoma-derived growth factor-related protein 2 (HRP2) as a novel protein associated with MPP8. HRP2 was further shown to colocalize with MPP8 at the E-cadherin gene locus, suggesting a possible role in cancer cell plasticity.
Graphical Abstract
INTRODUCTION
The maintenance of a specific chromatin landscape through the installation and interpretation of post-translational modifications (PTMs) on histone tails has been repeatedly shown to be important in mammalian gene expression.1,2 Lysine methylation is one of the most well-studied PTMs. Both the location and the degree of lysine methylation (mono- (Kme), di- (Kme2), and tri- (Kme3)) can influence gene expression by promoting either heterochromatin or euchromatin formation through the recruitment of a variety of protein complexes.3-5 For instance, trimethylation of histone 3 on lysine 9 (H3K9me3) is a signature of repressed genes and heterochromatin formation, while H3K36me3 is associated with active transcription.1,2,6 However, there is still much to be understood about the composition and detailed functions of the protein complexes that are recruited in response to such methylation signals and how they elicit and maintain changes in gene expression.
The human silencing hub (HUSH) complex is a multisubunit protein complex that participates in the maintenance of gene repression.7 The HUSH complex, composed of M-phase phosphoprotein 8 (MPP8), TASOR (FAM208A), and periphilin (PPHLN1), is thought to localize to chromatin rich in H3K9me3 through the recognition of this mark by the chromodomain of MPP8.7-9 Additionally, HUSH is essential for the targeted recruitment of a variety of auxiliary proteins such as SETDB1, which is a methyltransferase responsible for the maintenance of H3K9me3 and transcriptional silencing.10-12 HUSH-mediated gene silencing has more recently been shown to further rely on the recruitment of MORC2, an ATP-dependent chromatin remodeling enzyme, to sites of heterochromatin.13
The HUSH complex was first identified as a regulator of chromatin silencing through a screen for factors involved in position-effect variegation, and it was shown that the HUSH complex could maintain transcriptional silencing by facilitating the spread of H3K9me3 marks on integrated transgenes.11 In the same study, HUSH-mediated control of viral expression was demonstrated. Specifically, when Jurkat CD4+ lymphoid cells were infected with a standard HIV-1 reporter virus containing the HIV-1 LTR promoter, the proviruses were subject to HUSH-mediated repression as determined by knockdown of each HUSH subunit.11 This suggests that the HUSH complex may assist in regulating the persistence of a reservoir of latent cells in HIV infected individuals.14 Additionally, the HUSH complex and its associated machinery have been shown to be responsible for the silencing of endogenous retroviruses (ERVs) and transposable elements (TEs) which comprise up to 20% of the human genome, suggesting an important role in cellular development and genome maintenance.10,15,16
It has also been suggested that MPP8 can contribute to tumor progression, as MPP8 displays elevated expression levels in various human carcinoma cells and knock-down of MPP8 results in the reduction of tumor cell migration and invasiveness. Specifically, MPP8 has been shown to be important in promoting epithelial to mesenchymal transition (EMT), a hallmark of cancer metastasis, through the negative regulation of E-cadherin expression.7,17 This is achieved through the association of MPP8 with the DNA methyltransferase DNMT3A, allowing MPP8 to direct DNA methylation of the E-cadherin gene and subsequently repress this key tumor suppressor. Collectively, although both the HUSH complex and MPP8 have been implicated in disease, more clearly discerning the specific mechanisms behind HUSH-mediated gene repression and the auxiliary machinery associated with the HUSH complex could be critical in order to better evaluate opportunities for possible therapeutic intervention.18
The development of peptidomimetic ligands has emerged as a common strategy for targeting Kme readers. They can both facilitate extensive backbone hydrogen-bond networks and achieve unique geometries allowing them to span the large binding surfaces of many protein–protein interactions (PPIs).19-23 We have previously demonstrated the utility of peptidomimetic chemical tools that specifically target chromodomain containing Kme readers with the discovery of UNC3866 and UNC4976, potent cellular chemical probes of CBX4 and CBX7 which are members of the Polycomb repressive complex 1 (PRC1).20,24 Furthermore, a biotinylated derivative of UNC3866 was capable of pulling down all major PRC1 components through its interaction with the CBX chromodomain, demonstrating the utility of these chemical tools for studying larger protein complexes.25 In profiling the selectivity of UNC3866 against other related and unrelated protein targets, it was also revealed that UNC3866 has affinity for the CDYL chromodomains and the chromodomain of MPP8, likely due to the shared recognition motif across this protein family. Consequently, this provides a starting point for further exploration of potent and selective ligands targeting these related chromodomains. We previously implemented a combinatorial screening strategy to obtain novel chromodomain ligands with optimized potency and unique selectivity profiles. We reported submicromolar ligands for CDYL2 which were developed via a one-bead, one-compound (OBOC) combinatorial library based on UNC3866 and a target-class screening approach.23,26
Herein, we describe the development of a peptidic ligand of the MPP8 chromodomain (UNC5246), discovered through an affinity-based, target-class repurposing strategy.23 UNC5246 was shown to engage endogenous MPP8, as well as chemiprecipitate known MPP8 interacting proteins. To better understand the mechanisms of MPP8-mediated repression, we performed chemoproteomics with a cell line engineered to induce heterochromatin formation through MPP8 recruitment to a desired gene locus which revealed hepatoma-derived growth factor-related protein 2 (HRP2) as a novel protein associated with MPP8. Chromatin immunoprecipitation (ChIP) analysis further demonstrated that HRP2 is localized to the E-cadherin promoter and gene body at levels similar to that of MPP8. Overall, our efforts highlight the power of using chemical tools to better understand the mechanisms of chromatin regulation.
RESULTS AND DISCUSSION
Discovery and Characterization of UNC5246.
We previously reported a target-class screening strategy for the development of novel chromodomain ligands utilizing OBOC libraries23. This approach employs both negative selection and competitor exchange kinetics to achieve improved selectivity and potency, respectively. Briefly, based on our chemical probe of the PRC1 chromodomains (UNC3866), we previously generated a combinatorial library of ~14 000 UNC3866 derivatives with six unique points of diversity (Figure 1A).20,23 This library was rationally designed to balance conservative substitutions with riskier modifications because a combination of these features seemed most likely to yield differences in selectively while maintaining chromodomain binding. A preliminary selection was performed on the library with the chromodomain of MPP8 to isolate ligands with affinity for MPP8. To do so, the library was incubated with the His-tagged MPP8 chromodomain followed by magnetic Protein G precoated beads with an anti-His antibody, allowing only those peptide beads bound to MPP8 to be magnetically enriched.
Figure 1.

A combinatorial target class screening strategy affords potent and selective MPP8 ligands. (a) Overall structure of the OBOC UNC3866 derivative library including a linker and six unique points of variation. (b) Library screening strategy employing numerous negative selections against other chromodomain containing proteins and UNC3866 as a soluble competitor. (c) Selected MPP8 hits with >4-fold redundancy. (d) Structure of UNC5246 and the ITC isotherm showing potent binding to MPP8.
We next hypothesized that a cross-screening approach could be employed with these MPP8 hit beads to isolate those peptide ligands that are most potent and selective for MPP8 (Figure 1B). First, MPP8 hit beads were incubated with excess soluble UNC3866, which binds MPP8 with an affinity of ~3 μM, for 2 h.20,23 Upon addition of His-MPP8 followed by magnetic Protein G beads precoated with an anti-His antibody, the peptide ligands that were magnetized, presumably due to their ability to compete with UNC3866 for MPP8 binding, were isolated. The magnetized peptide beads were then stripped of MPP8 for subsequent chromodomain homologue negative selection screens, conducted to remove those ligands with pan activity across the chromodomain family.
We chose to cross-screen the set of UNC3866 competitive MPP8 hit beads against representative chromodomains from three different subfamilies (Figure 1B). We first cross-screened against CBX5 (HP1α) from the HP1 family of chromodomains, which similarly reads the H3K9me3 mark. This resulted in the removal of only a small number of magnetized peptides. Those peptide beads not bound by CBX5 were next cross-screened with CDYL2. On the basis of the fact that most of our prior UNC3866 analogs have some affinity for both MPP8 and CDYL2, it was not surprising that the majority of the peptide beads were magnetized by CDYL2, leading to very few unbound and putative MPP8 selective ligands. As a result, we added soluble UNC3866 and isolated only those peptide beads that became demagnetized after 15 min of incubation. We presumed that the CDYL2 bound ligands that were competed off more quickly by UNC3866 may have less affinity for CDYL2 because of their “fast-off” kinetics. Moving forward with this pool of peptide beads, a final negative selection was performed with the chromodomain of CBX8, a member of the polycomb family of chromodomains, which led to the removal of a small number of CBX8 bound peptides. These successive selection steps ultimately led to the isolation of a final pool of approximately 90 MPP8 hit beads that we hoped contained peptidic ligands with increased potency for MPP8 over UNC3866 and selectivity for MPP8 over CBX5, CDYL2, and CBX8.
Each bead from this final hit pool was then individually selected and treated with a CNBr peptide cleavage cocktail, and the cleaved peptides were subjected to MALDI-TOF/TOF mass spectrometry analysis (Figure S1). Following structural deconvolution, we prioritized those compounds with the highest level of redundancy. This left us with four hit compounds with a greater than 4-fold redundancy for further analysis, and encouragingly, there was significant structural similarity between the redundant hits (Figure 1C). The R1 and R2 positions showed conservation of a 4-piperdinyl capping group and phenylalanine, respectively. Further, all redundant hits contained an alanine at the R3 position. The R4 position demonstrated a preference for larger hydrophobic side chains as shown by the enrichment of phenylalanine and cyclohexylalanine. A preference for the closely related diethyl and ethyl-isopropyl lysine mimetics at the R5 position was also observed. Last, a serine was conserved at the R6 position.
Of the redundant hits, we prioritized the one with 2-fold higher redundancy relative to the others for further characterization (nine redundant hits versus five; Figure 1C, top left). A C-terminal methyl ester derivative was synthesized (UNC5246) via solution-phase peptide chemistry according to Scheme S1 and examined for binding to MPP8 via isothermal titration calorimetry (ITC), yielding a Kd of 0.72 ± 0.05 uM (Figure 1D). We were pleased to see this modest boost in affinity for MPP8 relative to UNC3866. To ensure that this was not related to the installation of the C-terminal methyl ester, we also prepared an analog with a C-terminal amide (UNC4848) which more closely resembles the compound on bead and found that UNC4848 is equipotent to UNC5246 (Figure S2A). To further evaluate the selectivity of UNC5246 against other chromodomains, we first examined the binding of UNC5246 to CDYL2. ITC revealed that UNC5246 binds CDYL2 about 4-fold more potently than MPP8 (Figure S2B), which was not surprising based on the fact that CDYL2 bound almost all MPP8 hit beads in the selection process likely due to the similarity of the two chromodomains. Additionally, a time-resolved fluorescence energy transfer (TR-FRET) assay revealed no significant binding (IC50 > 20 μM) of UNC5246 to CBX8 and CBX5, as well as CBX7, a primary target of UNC3866 (Figure S3). Overall, these binding data are closely aligned with our observations from the screening cascade, demonstrating the general effectiveness of this target-class combinatorial repurposing approach.
Evaluation of the UNC5246 Binding Mode by Crystallography.
To better understand the binding mode of UNC5246 and rationalize its improved affinity for MPP8 over other chromodomains, we solved the crystal structure of UNC5246 bound to MPP8 (Figure 2A and Table S1, PDB ID 7M5U) and compared it with our previously published structure of UNC3866 in complex with CBX7 (PDB ID 5EPJ). Overall, UNC3866 and UNC5246 are structurally very similar but contain unique N-terminal capping residues (Figure 2B). UNC3866 contains a hydrophobic tert-butyl benzyl cap, whereas UNC5246 contains a basic 4-piperidine (Pip) cap. The piperidine of UNC5246 lies in a strongly electronegative groove in MPP8 formed by Glu60, Glu62, Asp98, Glu101, and Glu105. Although the carboxylates of Glu60 and Glu101 are slightly outside of true salt bridge distances, each Glu orients one oxygen in range for a weak hydrogen bond with the amine cap. Interestingly, Glu60, Asp98, and Glu101 are Ala12, Leu49, and Arg52 residues in CBX7, respectively, which helps to explain CBX7’s preference for the hydrophobic N-terminal cap of UNC3866 over the basic endcap of UNC5246 (Figure 2B). Overall, we believe that the electronegative pocket of MPP8 is a key determinant in the selective binding of UNC5246 for the chromodomain of MPP8 over CBX7. UNC5246 also binds potently to CDYL2, which has a similar electronegative pocket to MPP8.
Figure 2.

Co-crystal structure of UNC5246 and the MPP8 chromodomain provides insight into potency and selectivity of binding. (a) UNC5246 (cyan) bound to the MPP8 chromodomain (gray) with a resolution of 2.02 Å. (b) Chemical structures of UNC5246 and UNC3866 highlighting differences in the N-terminal capping groups (top). The piperidine cap of UNC5246 lies in a strongly electronegative groove in MPP8 (bottom left), whereas the tert-butyl benzoyl cap of UNC3866 (green) binds in a more hydrophobic and electropositive groove in CBX7 (bottom right). (c) Key hydrogen bonds are formed between the peptide backbone of UNC5246 and residues Asp57, Phe59, Val61, His95, Asp98, and Glu91 of MPP8. (d) UNC5246 binds the MPP8 chromodomain via a surface-groove recognition mode. (e) The ethyl-isopropyl lysine mimetic of UNC5246 binds in the MPP8 aromatic cage with the isopropyl moiety pointing into the cage and the ethyl group pointing out toward solvent.
Further analysis of the UNC5246/MPP8 cocrystal structure revealed that key hydrogen bonds are formed between the peptide backbone of UNC5246 and residues Asp57, Phe59, Val61, His95, Asp98, and Glu91 of MPP8 (Figure 2C). Additionally, Glu91 of MPP8 forms two hydrogen bonds with Ser6 of UNC5246, an interaction that is conserved in CBX7 (Glu43) binding to UNC3866. The extensive hydrogen bond network between MPP8 and UNC5246 confirms the expected surface-groove binding interaction and highlights the benefits of a peptidic ligand strategy (Figure 2D). These hydrogen bonds are critical for binding and methylation of the amide backbone of UNC5246 (for example, UNC6084; Figure S4A) abolishes binding to MPP8 (Figure S4B). Additionally, the alanine side chain of UNC5246 exists in a very narrow groove, likely explaining why Ala was conserved at this location among all redundant hits that arose from the selection process. Our structural studies also revealed that the bulkier isopropyl moiety of the ethyl-isopropyl lysine (Lys(iPr,Et)) mimetic is projected into the MPP8 aromatic cage with the ethyl moiety pointing out toward solvent (Figure 2E); therefore, it is possible that this slightly bulkier lysine mimetic partially contributes to an increased affinity for MPP8.
Structure–Activity Relationship Studies of UNC5246.
With structural data on the binding mode of UNC5246 in hand, we chose to undergo a medical chemistry campaign to increase affinity and/or selectivity for MPP8. We largely focused on modifications to two key regions of UNC5246, the N-terminal cap due to the importance in this region for MPP8 selectivity and the Kme mimetic. On the basis of the preformed nature of the aromatic cage of MPP8, in contrast to CBX7 which utilizes an induced fit binding mechanism, we hypothesized that we may be able to improve MPP8 binding by optimizing the Kme mimetic. A common intermediate was synthesized to allow for facile modification of either the N-terminal capping group, the Kme mimetic, or both (Scheme S1). Final compounds were tested for binding to MPP8, CDYL2, and CBX7 by previously validated TR-FRET assays (Figure S3).27
As shown in Table 1, cap modifications led to only modest increases in potency and selectivity for MPP8 over CDYL2. In agreement with our crystal structure, the basic amine of the piperidine ring clearly contributes to binding as illustrated by compound UNC5247 which contains a cyclohexyl capping group and results in about a 6-fold loss in potency for MPP8 relative to UNC5246. Not surprisingly, replacing the piperidine nitrogen with a sulfur as in UNC5432 similarly decreases affinity for MPP8. Methylation of the piperidine amine did not result in a change in potency illustrated by UNC5430, indicating that the hydrogen-bond donating capabilities of the piperidine secondary amine are not required and that there is enough space in the binding pocket for alkylation of the amine. Interestingly, both UNC5432 and UNC5430 pick up weak affinity for CBX7. Increasing the flexibility of the piperidine cap and modifying the position of the basic amine, now slightly further away from the key acidic residues, as in UNC6215 results in a small decrease in potency. Further reducing the basicity of the amine in UNC6215 with a morpholine group in UNC6214 is also not beneficial. However, reducing the size of the piperidine ring in UNC6215 to a slightly smaller pyrrolidine as in UNC6113 results in a compound that is about equipotent to UNC5246 for both MPP8 and CDYL2. UNC6108 and UNC6109 demonstrate that extending the amine deeper into the electronegative pocket results in a 2-fold increase in potency for MPP8, suggesting potentially more favorable interactions with the acidic residues; however, relative to UNC6109, binding is reduced 10-fold with the addition of a single methylene in UNC6141, extending the amine further from the peptide backbone. We also found that a heterocycle is preferred over acyclic caps as highlighted by UNC6231, although only about a 2-fold loss in affinity for MPP8 is observed even with a significant increase in flexibility of the amine. Despite reduced basicity, the introduction of a range of aromatic capping groups resulted in a minimal change in binding for MPP8. Interestingly, many of these aromatic capping groups resulted in about equipotent binding for both MPP8 and CDYL2 (for example, UNC6220 and UNC6139). However, being that the cap of UNC6139 more closely resembles that of UNC3866, we were not surprised to observe a significant gain in potency for CBX7 relative to UNC5246, making this compound unattractive for further development. Additionally, UNC6142, UNC6263, and UNC6257 were plagued with poor aqueous solubility and were not pursued further.
Table 1.
SAR Studies of the N-Terminal Cap of UNC5246a

| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | R1 | IC50 MPP8 (μM) | IC50 CDYL2 | IC50 CBX7 | Entry | R1 | IC50 MPP8 (μM) | IC50 CDYL2 | IC50 CBX7 |
| UNC5246 |
|
0.5 ± 0.2 | 0.09 ± .04 | >30 | UNC6109 |

|
0.2 ± 0.1 | 0.1 ± 0.1 | >30 |
| UNC5247 |
|
3.2 ± 2 | 3.2 ± 0.6 | >30 | UNC6141 |

|
2.3 ± 0.4 | 0.4 ± 0.2 | >30 |
| UNC5432* |
|
3.3 ± 0.8 | 1.1 ± 0.5 | 16 ± 2 | UNC6231 |

|
0.9 ± 0.1 | 0.4 ± 0.1 | >30 |
| UNC5430* |
|
0.5 ± 0.1 | 0.1 ± 0.1 | 27 ± 3 | UNC6220 |
|
0.8 ± 0.3 | 0.4 ± 0.2 | >30 |
| UNC6215 |

|
1.3 ± 1 | 0.30 ± 0.4 | >30 | UNC6139 |

|
0.4 ± 0.1 | 0.3 ± 0.1 | 0.7 ± 0.1 |
| UNC6214 |

|
3.4 ± 1 | 0.5 ± 0.2 | >30 | UNC6142 |

|
0.2 ± 0.1 | 0.3 ± 0.2 | 5.0 ± 0.5 |
| UNC6113 |
|
0.3 ± 0.04 | 0.11 ± 0.1 | >30 | UNC6263 |

|
0.9 ± 0.3 | 0.2 ± 0.1 | 6 ± 1 |
| UNC6108 |
|
0.2 ± 0.05 | 0.1 ± 0.07 | >30 | UNC6257 |

|
0.4 ± 0.1 | 0.7 ± 0.2 | 7 ± 3 |
IC50 values are reported as the mean ± the standard deviation of three biological replicates. An * denotes that the compound has a C-terminal amide in place of ester.
Another region of UNC5246 that we wanted to explore further was the lysine mimetic at position R5. Highlighted in Table 2, a variety of UNC5246 analogs with unnatural lysine mimetics were synthesized and analyzed. Converting the ethyl-isopropyl amine of UNC5246 to a methyl-cyclopentyl amine as in UNC6216 resulted in no significant change in binding for MPP8 or CDYL2, suggesting that a slightly bulkier group can be tolerated but does not result in additional favorable interactions; however, an analogous pyrroline substituent was less well tolerated, resulting in about a 3-fold loss in binding for MPP8 (UNC6262). Larger cyclohexyl, tetrahydropyran, and piperidine substituents as in UNC6217, UNC6218, and UNC6127, respectively, were also well tolerated overall, but no increases in potency were observed. Similarly, increasing the steric bulk on the lysine amine as in UNC6254 was not effective at improving binding affinity for MPP8. UNC6251, which contains a 4-fluorobenzyl moiety, displayed equal potency to UNC5246 and may suggest that while the isopropyl moiety is engaging the aromatic cage of MPP8, the 4-fluorobenzene is solvent exposed and unable to make any productive or unproductive contacts. Interestingly, UNC6474, in which the two amine substituents have been replaced by a cyclic piperidine, is also equipotent to UNC5246 for both MPP8 and CDYL2. Collectively these analogs of UNC5246 further confirm the previously observed flexibility at this position to various modifications and that modification to the methyl-lysine mimetic has little effect on binding selectivity within the chromodomains tested. Overall, none of our structural modifications at either the N-terminus or lysine mimetic position resulted in significant improvements in potency for MPP8; however, this further supports the use of targeted OBOC libraries as an effective and efficient strategy in the discovery of potent peptide ligands. Additionally, through this focused SAR effort, we were unable to significantly decrease potency for CDYL2, suggesting high homology between the two proteins at the binding sites and that other regions of the molecule may warrant future exploration to enable selective binding to MPP8.
Table 2.
SAR Studies of the Methyl-Lysine Mimetic of UNC5246a

| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | R2 | IC50 MPP8 (μM) | IC50 CDYL2 | IC50 CBX7 | Entry | R2 | IC50 MPP8 (μM) | IC50 CDYL2 | IC50 CBX7 |
| UNC6216 |

|
0.45 ± 0.1 | 0.09 ± 0.03 | >30 | UNC6127 |

|
0.7 ± 0.03 | 0.2 ± 0.03 | >30 |
| UNC6262 |

|
1.4 ± 0.2 | 0.7 ± 0.2 | >30 | UNC6254 |
|
0.8 ± 0.2 | 0.2 ± 0.04 | >30 |
| UNC6217 |

|
0.4 ± 0.2 | 0.07 ± 0.05 | >30 | UNC6251 |

|
0.4 ± 0.1 | 0.09 ± 0.03 | >30 |
| UNC6218 |

|
0.5 ± 0.1 | 0.1 ± 0.04 | >30 | UNC6474 |
|
0.7 ± 0.1 | 0.07 ± 0.01 | >30 |
IC50 values are reported as the mean ± the standard deviation of three biological replicates.
Biotinylated UNC5246 Chemiprecipitates the HUSH Complex.
Having explored some SARs around UNC5246 and characterized the compound’s binding mode, we next sought to determine if our ligand could engage full length endogenous MPP8 in the context of the HUSH complex as this had not been done previously with UNC3866 despite its modest potency for MPP8. To do so, we synthesized a biotinylated analog of UNC5246 for use as a chemiprecipitation reagent. On the basis of the crystal structure, we chose to append a PEG-biotin tag to the C-terminus of UNC5246, which is relatively solvent exposed. UNC5246-Biotin (Figure 3A) was incubated with PC3 cell lysates and used to chemiprecipitate proteins bound to UNC5246 via conjugation to streptavidin magnetic beads. Subsequent immunoblotting was performed for MPP8 and the HUSH complex member TASOR.17 Periphilin was not evaluated in these studies due to a lack of availability of a suitable antibody. As illustrated in Figure 3B, UNC5246-Biotin was capable of effectively pulling down both MPP8 and TASOR. Importantly, the addition of 100 μM of UNC5246 as a soluble competitor greatly attenuated the ability of UNC5246-Biotin to pull down both MPP8 and TASOR, confirming the specificity of these interactions. Overall, this demonstrates that UNC5246 can engage full length endogenous MPP8 and that this binding event does not disrupt the interactions between MPP8 and TASOR.
Figure 3.

UNC5246-Biotin chemiprecipitates MPP8 and associated proteins from cell lysates. (a) Structure of UNC5246-Biotin. (b) UNC5246-Biotin chemiprecipitates MPP8 and TASOR from PC3 cell lysates, and both compounds are competed off with free UNC5246. (c) Identified hits from chemoproteomics analysis of UNC5246-Biotin pulldown experiment and the fold increase observed for each protein over enrichment with beads alone. (d) Chemiprecipitation using UNC5246-Biotin followed by Western blot analysis confirmed many of the protein hits identified by proteomics including the HP1 proteins, HRP2, and TRIM28. (e) MPP8 coimmunoprecipitates with all three HP1 isoforms.
Identification of HUSH-Associated Proteins by Proteomics.
To further expand these studies, we sought to explore MPP8 interacting proteins via an unbiased proteomics approach in order to better understand HUSH-mediated gene silencing and the role of MPP8 in biological functions. We utilized an engineered cell line in which MPP8 is recruited to a locus of interest to facilitate gene repression, with the hope that we would be more likely to identify direct and indirect protein–protein interactions that play a functional role in MPP8 mediated gene repression. Briefly, we developed a clonal HEK293T cell line (CiA EF1α HEK293T) expressing green fluorescent protein (GFP) with a Gal4 DNA binding site upstream of GFP enabling binding of a Gal4-MPP8 fusion protein (Figure S5A). HEK293T GFP+ cells were lentivirally infected to stably express the Gal4-MPP8 fusion. Upon recruitment of Gal4-MPP8, 48% of cells showed no GFP expression as determined by flow cytometry (Figure S5B). The GFP negative cell population was isolated and lysed, followed by incubation with UNC5246-Biotin conjugated to magnetic streptavidin beads. Those proteins isolated via direct or indirect interactions with UNC5246-Biotin were digested with trypsin and analyzed by quantitative LC-MS/MS using isobaric tandem mass tags (TMT). Proteins enriched in the UNC5246-Biotin pulldown samples as compared to a bead only control lacking UNC5246-Biotin were considered for further analysis (Supporting Information).
We were encouraged to see that MPP8 was the most highly enriched hit from the proteomics analysis with over 4-fold enrichment over the control (Figure 3C). Interestingly, a variety of proteins known to be associated with epigenetic gene silencing were also found to be enriched, including the previously described HUSH-interacting protein TRIM28 (KAP1), which helps to facilitate HUSH-mediated gene silencing via recruitment of SETDB1.10,12,16,28 All three isoforms of heterochromatin protein 1 (α, β, and γ or CBX5, CBX1, and CBX3, respectively), which are known binders of H3K9me3 and associated with transcriptional repression, were among the top enriched hits.8,28-31 Additionally, MPP8 has previously been implicated in HP1α mediated gene repression.32 Another protein found to be enriched by over 2-fold was hepatoma-derived growth factor-related protein 2 (HDGFRP2 or HRP2), which with closely related protein LEDGF has been shown to be an important driver of HIV integration and replication.33,34 Furthermore, HRP2 was previously found to be associated with H3K9me3 through a large scale proteomics effort conducted to define the chromatin interaction landscape in mouse tissues, making it an attractive target for further investigation.35 Prior studies have shown that HRP2 can be associated with CBX1 (HP1β).36 We were also intrigued to see the enrichment of high mobility group nucleosomal binding domain 2 (HMGN2) due to its known association with active transcription and euchromatin maintenance.
We next sought to further validate our proteomics results by Western blot analysis. We performed chemiprecipitation experiments with UNC5246-Biotin in normal HEK293T cells and immunoblotted for those proteins identified via proteomics (Figure 3D).17 Encouragingly, we confirmed that those proteins associated with heterochromatin formation including the three HP1 isoforms, HRP2, and TRIM28 could be successfully pulled down by UNC5246-Biotin; however, we were unable to chemiprecipitate HMGN2. This highlights the importance of validating our proteomics results by an orthogonal method and was not overly unexpected due to the association of HMGN2 with transcriptionally active chromatin. We hypothesized that successful chemiprecipitation of these proteins could be occurring for numerous reasons: (1) through direct binding to UNC5246-Biotin, (2) through direct or indirect interactions with MPP8, or (3) through direct or indirect interactions with CDYL2, which also binds UNC5246. Consequently, we next investigated if UNC5246 binds the HP1 proteins or HRP2 directly, as the HP1 proteins contain a chromodomain that, like MPP8, recognizes H3K9me3 and HRP2 has an N-terminal PWWP Kme reader domain. We performed ITC with UNC5246 and the chromodomains of HP1α/β/γ and the PWWP domain of HRP2. Weak binding was observed for the HP1 proteins with Kd values ranging from 8 to 20 μM (Figure S6A), while no binding was observed for the PWWP domain of HRP2 (Figure S6B). This indicates that chemiprecipitation of the HP1 proteins could possibly be due to weak binding interactions with UNC5246. On the basis of this result and literature precedence, we also wanted to examine if HP1 interacts directly with MPP8 by coimmunoprecipitation experiments. All three HP1 isoforms were found to coimmunoprecipitate with both MPP8 and TASOR, further suggesting a bona fide association of HP1 with the HUSH complex (Figure 3E, Figure S7). This is further supported by the known ability of HP1 to interact with TRIM28, a reported HUSH interactor, and helps to validate our chemoproteomics approach as a powerful method for identifying complex protein–protein interactions.28,30,31
HRP2 Associates with the HUSH Complex.
In contrast to HP1, the association of HRP2 with the HUSH complex has not been suggested previously; therefore, we took a similar approach to investigate the potential interaction between HRP2 and MPP8. In HEK293T cells expressing Myc-HRP2, we found that both MPP8 and TASOR coimmunoprecipitate with HRP2 by Western blot (Figure 4A). This result was consistent with varying Myc-HRP2 transfection levels (Figure S8A), while flag-HRP2 similarly coimmunoprecipitated with MPP8, further validating this interaction (Figure S8B). In contrast, CDYL2 did not coimmunoprecipitate with Myc-HRP2 (Figure 4A). Last, to further control for potential overexpression artifacts, we overexpressed an unrelated protein, Myc(His-LRRC33), in HEK293T cells and found that it did not coimmunoprecipitate with MPP8 (Figure S8C). Together, our chemoproteomics, ITC, and coimmunoprecipitation data support a newly identified association between HRP2 and the HUSH complex.
Figure 4.

HUSH complex associates with HRP2. (a) Myc-HRP2 expressed in HEK293T cells coimmunoprecipitates with MPP8 and TASOR but not CDYL2. (b) Myc-HRP2 truncation constructs expressed in HEK293T cells (left) followed by IP-Western blot analysis demonstrating association between HPR2 truncations and MPP8 (right). (c) Flag-MPP8 truncation constructs expressed in HEK293T cells (left) followed by IP-Western blot analysis demonstrating association between MPP8 truncations and HRP2 (right).
To further investigate this interaction, we generated a series of Myc-HRP2 truncation constructs, expressed them in HEK293T cells, and performed IP-western analysis. Interestingly, all HRP2 deletions which include a loss of the N-terminal PWWP domain or the C-terminal integrase binding domain (IBD) were coimmunoprecipitated with MPP8 as well as TASOR with similar efficiency to that of wildtype Myc-HRP2 (Figure 4B, Figure S9A). This suggests that HRP2’s interaction with the HUSH complex could span more than one protein within the complex and is likely facilitated by multiple protein–protein interactions. This observation was further validated through the use of a variety of flag-MPP8 truncations which included deletion of the N-terminal chromodomain or C-terminal ankyrin repeat domain.7,17 Upon coexpression with full length Myc-HRP2 in HEK293T cells, we found that all MPP8 truncations evaluated coimmunoprecipitated with full length Myc-HRP2 (Figure 4C, Figure S9B).
HRP2 and MPP8 Colocalize at the E-cadherin Locus.
Last, we sought to determine if MPP8 and HRP2 colocalize at a known gene locus. MPP8 has previously been shown to target the E-cadherin gene promoter to promote tumor cell motility, invasiveness, and epithelial to mesenchymal transition (EMT). Thus, we chose to examine HPR2 localization on the E-cadherin locus by chromatin immunoprecipitation (ChIP) in MDA-MB-231 cells.7,17 Before doing so, we confirmed that the association between HRP2 and MPP8 is conserved in MDA-MB-231 cells by coimmunoprecipitation (Figure 5A). Next, Myc-HRP2 was stably expressed in MDA-MB-231 cells and ChIP was performed at the E-cadherin locus, probing for Myc-HRP2 and MPP8 occupancy as well as H3K9me3. Enrichment was observed for both Myc-HRP2 and MPP8 at E-cadherin and to a similar extent as what has been previously reported for MPP8 (Figure 5B).7,17 Additionally, we observed significant decreases in MPP8 and HRP2 enrichment at SPINK1, a locus at which H3K9me3 deposition is directed by polycomb repressive complex 2 (PRC2) in MDA-MB-231 cells, relative to E-cadherin (Figure S10).37,38 This suggests that localization of HRP2 to heterochromatin is specific to its association with MPP8. These results further support the association of HRP2 with the HUSH complex and suggest that, in coordination with MPP8, HRP2 may play a role in gene silencing at the E-cadherin locus.
Figure 5.

HRP2 and MPP8 colocalize at the E-cadherin locus. (a) The HUSH complex coimmunoprecipitates with HRP2 in MDA-MB-231 cells. (b) ChIP-qPCR analysis of MPP8, HRP2, and H3K9me3 at E-cadherin in MDA-MB-231 cells demonstrating enrichment of both MPP8 and HRP2 at similar levels. TSS indicates the position (in bp) from the transcriptional start site. Statistical significance was calculated using a paired t test (n = 6; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001).
CONCLUSION
We have demonstrated the effectiveness of using a one-bead, one-compound target-class screening approach toward the discovery of novel peptidomimetic ligands for the MPP8 chromodomain.23,39 UNC5246 binds MPP8 with submicromolar potency and is greater than 70-fold selective for MPP8 over CBX7, the primary target of UNC3866, which was the starting point for the OBOC library.20,23 Crystallographic studies revealed key acidic residues in MPP8 that create unique interactions with the N-terminal piperidine cap of UNC5246 and drive selectivity, further demonstrating the importance of the N-terminal capping residue in achieving chromodomain selectivity for this class of peptidomimetics. UNC5246 also has affinity for the closely related protein CDYL2, and additional structural insight into UNC5246 binding to CDYL2 may be required for the development of high-quality chemical probes with improved selectivity within the chromodomain family.
On the basis of our discovery of UNC5246, we developed a unique chemiprecipitation tool, UNC5246-Biotin, which we were able to successfully use to demonstrate engagement of endogenous MPP8 in the context of the HUSH complex, as well as further interrogate MPP8 and HUSH-interacting proteins through a chemoproteomics approach. Using a cell line specifically engineered to induce heterochromatin formation through MPP8 recruitment, we revealed that MPP8 is associated with a wide array of gene silencing machinery. This includes proteins previously implicated as being associated with MPP8 such as the HP1 family of proteins, as well as newly discovered protein interactors such as HRP2. HRP2 was found to coimmunoprecipitate with both MPP8 and TASOR, and these interactions were not disrupted by the truncation of either HRP2 or MPP8, suggesting that multiple protein–protein interactions facilitate HRP2’s association with HUSH. This is supported by recent reports describing the structural and biochemical basis of the HUSH assembly and epigenetic regulation.40-42 ChIP analysis further demonstrated that HRP2 is localized to the E-cadherin locus at levels similar to that of MPP8, suggesting a potential role for HRP2 in E-cadherin gene silencing.
Although our chemical biology approach revealed a previously unknown association between MPP8 and HRP2, there is still much to be understood regarding the biological role of HRP2 and the specific mechanism by which HRP2 interacts with the HUSH complex. The PWWP domain of HRP2 has been shown to weakly recognize post-translational modifications on chromatin and therefore may assist with localization to specific genomic loci.35 MPP8 has also been previously shown to recruit other PWWP-containing proteins such as DNMT3a to regulate DNA methylation and gene silencing, while also mediating interactions between DNMT3a and the H3K9 methyltransferases G9a and GLP.17,18 Therefore, further investigation into HRP2’s direct protein contacts and association with MPP8 will likely provide additional insight into how the HUSH complex induces and maintains epigenetic silencing.
METHODS
Protein Expression and Purification for Screening and Biochemical Assays.
The chromodomains of CBX1 (residues 20–73 of NP_006798), CBX3 (residues 29–81 of NP_009207), CBX5 (residues 18–75 of NP_036429), CBX8 (residues 8–61 of NP_065700), and MPP8 (residues 55–116 of NP_059990) were expressed with N-terminal His-tags in pET28 expression vectors. The chromodomains of CBX2 (residues 9–66 of NP_001580), CBX7 (residues 8–62 of NP_783640), and CDYL2 (residues 1–75 of NP_689555) were expressed with C-terminal His-tags in pET30 expression vectors. The PWWP domain of HRP2 (residues 1–93 of NP_001001520) was expressed with an N-terminal His-tag in a pET28 expression vector. All expression constructs were transformed into Rosetta2 BL21(DE3)pLysS competent cells (Novagen, EMD Chemicals, San Diego, CA). Protein expression was induced by growing cells at 37 °C with shaking until the OD600 reached ~0.6–0.8, at which time the temperature was lowered to 18 °C and expression was induced by adding 0.5 mM IPTG and continuing shaking overnight. Cells were harvested by centrifugation, and pellets were stored at −80 °C.
His-tagged proteins were purified by resuspending thawed cell pellets in 30 mL of lysis buffer (50 mM sodium phosphate pH 7.2, 50 mM NaCl, 30 mM imidazole, 1X EDTA free protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN)) per liter of culture. Cells were lysed on ice by sonication with a Branson Digital 450 Sonifier (Branson Ultrasonics, Danbury, CT) at 40% amplitude for 12 cycles with each cycle consisting of a 20 s pulse followed by a 40 s rest. The cell lysate was clarified by centrifugation and loaded onto a HisTrap FF column (GE Healthcare, Piscataway, NJ) that had been pre-equilibrated with 10 column volumes of binding buffer (50 mM sodium phosphate pH 7.2, 500 mM NaCl, 30 mM imidazole) using an AKTA FPLC (GE Healthcare, Piscataway, NJ). The column was washed with 15 column volumes of binding buffer, and protein was eluted in a linear gradient to 100% elution buffer (50 mM sodium phosphate pH 7.2, 500 mM NaCl, 500 mM imidazole) over 20 column volumes. Peak fractions containing the desired protein were pooled and concentrated to 2 mL in Amicon Ultra-15 concentrators with a 3000 molecular weight cutoff (Merck Millipore, Carrigtwohill, Co. Cork, IRL). Concentrated protein was loaded onto a HiLoad 26/60 Superdex 75 prep grade column (GE Healthcare, Piscataway, NJ) that had been pre-equilibrated with 1.2 column volumes of sizing buffer (25 mM Tris pH 7.5, 250 mM NaCl, 2 mM DTT, 5% glycerol) using an ATKA Purifier (GE Healthcare, Piscataway, NJ). Protein was eluted isocratically in sizing buffer over 1.3 column volumes at a flow rate of 2 mL/min collecting 3 mL fractions. Peak fractions were analyzed for purity by SDS-PAGE, and those containing pure protein were pooled and concentrated using Amicon Ultra-15 concentrators with a 3000 molecular weight cutoff (Merck Millipore, Carrigtwohill, Co. Cork, IRL).
Library Synthesis.
Fmoc-protected amino acids were purchased from Chem-Impex and Sigma-Aldrich with the exception of Fmoc lysine derivatives (synthesized as previously described23). All other chemicals and solvents were purchased from TCI America and Sigma-Aldrich, unless otherwise stated. Syringe reaction vessels were made in-house with 2 and 10 mL syringes (Norm-Ject) and frits (Teledyne). Approximately 200 mg of PEGA resin (140–300 μm; EMD Millipore) swollen in ethanol was removed by pipet to a 10 mL syringe reaction vessel. The resin was rinsed once with N,N-dimethylformamide (DMF), drained, and left to equilibrate in DMF for 2 h. Standard Fmoc solid phase peptide synthesis was combined with the split-and-pool method to generate the library. Briefly, Fmoc-protected amino acids (15 equiv) were mixed for 5 min with HBTU (15 equiv), HOAt (15 equiv), and DIPEA (30 equiv) in 5 mL of DMF and 3 mL of dichloromethane (DCM). The solution was then added to the resin and left on a shaker at RT for 1 h. The resin was filtered and washed twice with DCM, DMF, methanol, and DMF again. Fmoc protecting groups were removed in a solution of 2.5% 1,8-diazabicycloundec-7-ene and 2.5% pyrrolidine in DMF for 10 min. The resin was filtered and washed twice with DMF, methanol, DMF, and DCM before adding the next amino acid for coupling. After completion of the linker synthesis, splitting of the library was conducted following Fmoc deprotection. Resin in DMF was split equally by pipet into 2 mL syringe reaction vessels. Following the coupling reactions, the resin in DMF was pooled back together into a 10 mL syringe reaction vessel, and Fmoc was deprotected before splitting again. The carboxylic acid caps were coupled under the same conditions as the amino acids described above, but the couplings were left overnight. The resin was then pooled back into a new 10 mL syringe reaction vessel. Following several DCM rinses, the on-bead peptides were deprotected in 95% trifluoroacetic acid, 2.5% triisopropylsilane, and 2.5% water for 3 h. The resin was rinsed again with DCM 10 times followed by several ethanol washes. The on-bead library was stored in ethanol until use.
Library Screening.
Library screening was completed as described previously with the following modifications.23 Half of the synthesized library was removed to a 15 mL conical tube and equilibrated in 25 mM Tris, at pH 7.8, 150 mM NaCl, and 0.1% Tween-20 (TBST) overnight. For 1 h, the beads were blocked in 5% BSA in TBST (10 mL) at RT. The resin was washed once (10 mL TBST) and then incubated with 1 μM His-tagged MPP8 in 2.5% BSA in TBST (10 mL) for 1 h. Simultaneously, mouse anti-His antibody (10 μL of a 1 μg/μL solution; Pierce MA1- 21315) was incubated with Protein G Dynabeads (50 μL; Life Technologies) in 2.5% BSA in TBST (200 μL). After incubation, the Dynabeads were washed three times with TBST (10 mL), and the library was washed three times with TBST (10 mL). 2.5% BSA in TBST (10 mL) was added to the library followed by Dynabead addition. The library beads and magnetic beads were left to mix at RT for 1 h. A magnet was used to remove initial MPP8 hit beads which were then stripped with 1% SDS (10 mL) at 95 °C for 3 min and rinsed repeatedly with TBST. The beads were then left in TBST overnight at 4 °C on the rotator. The next day the library was screened under the same conditions as above with the addition of soluble competitor UNC3866 at a final concentration of 1 μM. Magnetized hits from this screen were isolated and stripped as described above. A negative selection screen using His-CBX5 was performed, and any magnetized hits were discarded. Nonmagnetized beads were collected and stripped of protein. These beads were then incubated with HIS-CDYL2 as a negative selection screen followed by incubation of 1 μM UNC3866 for 15 min. Again, the nonmagnetized beads were collected and stripped of protein. A final negative selection was performed against His-CBX8 beads and then with HIS-CBX8 to yield the final pool of nonmagnetized beads as MPP8 hits which were rinsed repeatedly in ethanol and allowed to equilibrate overnight.
Hit Cleavage and MALDI-TOF/TOF Sequencing.
Hits were cleaved and sequenced as previously described.23 Briefly, 1–2 hit beads per well were loaded into a 96-well plate (Thermo Scientific 0.2 mL 96-well PCR plate). The ethanol solutions were left to evaporate. Twenty microliters of a mixture of 0.25 M cyanogen bromide in 10% water, 40% acetic acid, and 50% acetonitrile was added to each well. The plates were loosely covered and left to mix overnight on a shaker platform at RT in a fume hood. Cleaved compounds were redissolved in 10 μL of 1:1 water/acetonitrile. From this solution, 0.5 μL was spotted on an Opti-TOF 384-well MALDI plate, and 0.5 μL of a saturated solution of α-cyano-4-hydroxycinnamic acid in 50% acetonitrile and 50% water with 0.1% TFA was spotted on top. The solutions were left to evaporate. Spectra were recorded on an ABSciex 5800 MALDITOF/TOF (housed in the UNC Proteomics Core Facility) in positive, linear mode with a laser power between 2200 and 2600.
Isothermal Titration Calorimetry.
ITC measurements were recorded at 25 °C using an AutoITC200 microcalorimeter (MicroCal Inc., MA). Protein was dialyzed into ITC buffer (25 mM Tris-HCl, at pH 7.5, 150 mM NaCl, and 2 mM β-mercaptoethanol) and then diluted into ITC buffer to achieve a final concentration of 100 or 150 μM (325 μL). Peptides were dissolved in ITC buffer at a concentration of 10 mM and then diluted to the final concentration of 1 or 1.5 mM. Protein concentrations were 10-fold lower than the concentration of peptide. A typical experiment included a single 0.2 μL compound injection into a 200 μL cell filled with protein, followed by 26 subsequent 1.5 μL injections of compound. Injections were performed with a spacing of 180 s and a reference power of 8 μcal/s. The titration data were analyzed using Origin Software (MicroCal Inc., USA) by nonlinear least-squares, fitting the heats of binding as a function of the compound/protein ratio to a one site binding model. The first data point was deleted from all analyses. All assays were run in duplicate or triplicate. The data were fit separately for each experiment, and the reported Kd is the average of all of the runs. Error was calculated as the standard deviation of the various Kd values.
Time-Resolved Fluorescence Energy Transfer Assays.
The TR-FRET assays were performed as previously reported.27 Briefly, assays were completed using Kme reader buffer containing 20 mM Tris at pH 7.5, 150 mM NaCl, 0.05% Tween 20, and 2 mM dithiothreitol (DTT). White, low-volume, flat-bottom, nonbinding, 384-well microplates (Greiner, #784904) were used for screening with a total assay volume of 10 μL. 384-well, V-bottom polypropylene plates (Greiner, #781280) were used for compound serial dilutions and for transfer of assay mixtures. Compound serial dilutions were made using DMSO. Following addition of all assay components, plates were sealed with clear covers, gently mixed on a tabletop shaker for 1 min, centrifuged at 1000g for 2 min, and allowed to equilibrate in a dark space for 1 h before reading. Measurements were taken on an EnVision 2103 Multilabel Plate Reader (PerkinElmer) using an excitation filter at 320 nm and emission filters at 615 and 665 nm. 615 and 650 nm emission signals were measured simultaneously using a dual mirror at D400/D630. The TR-FRET output signal was expressed as emission ratios of acceptor/donor (665/615 nm) counts. Percent inhibition was calculated on a scale of 0% (i.e., activity with DMSO vehicle only) to 100% (100 μM UNC5246) using full column controls on each plate. The interquartile mean of control wells was used to calculate Z′ values. For dose–response curves, data were fit with a four-parameter nonlinear regression analysis using GraphPad Prism 7.0 or ScreenAble software to obtain IC50 values.
Protein Expression and Purification for Crystallization.
For crystallization, the MPP8 chromodomain was expressed and purified as described above with the following modifications. The expression plasmid was transformed into BL21(DE3)pLysS and grown in 2XTY media at 37 °C to an OD600 of ~0.6. One hour prior to induction, the incubator temperature was lowered to 18 °C. Cells were induced with 0.2 mM IPTG for 16 h. Pelleted cells were resuspended in P300 (50 mM NaHPO4, at pH 7.0, 300 mM NaCl, 10 mM 2-mercaptoethanol) supplemented with 1 mM benzamidine, flash frozen in liquid nitrogen and stored at −20 °C. Thawed cells were lysed by sonication on ice as above using four rounds of 14 × 0.5 s pulses at 50% amplitude. Cleared lysates were diluted to 100 mL with P300 and purified using Talon resin (Takara, Mountain View, CA) on an AKTA Start (GE Healthcare, Piscataway, NJ), eluting with P300 supplemented with 200 mM imidazole. The HIS tag was cleaved with a 1:100 molar ratio of TEV protease overnight at RT, and the protein was subsequently purified by anion exchange chromatography (Source Q, GE Healthcare) using an AKTA Pure 25 M HPLC (GE Healthcare). Immediately prior to setting up crystal trays, MPP8 was purified by gel filtration using a Superdex 75 increase 10/300 column (GE Healthcare) pre-equilibrated in T400 (20 mM Tris-Cl, at pH 8.0, 400 mM NaCl, 1 mM DTT) and concentrated to 25 mg mL−1 using a centrifuge concentrator (Vivaspin 20, 5 kDa molecular weight cutoff, Vivascience).
Crystallization and Data Collection.
The concentrated MPP8 chromodomain was mixed with UNC5246 resuspended in DMSO (Fisher) at a 1:1.7 molar ratio and crystallized in modified microbatch under oil at RT.43 Briefly, 1 μL of MPP8-UNC5246 was added to 1 μL of 100 mM sodium citrate tribasic dihydrate, at pH 5.6 (Hampton), 100 mM potassium sodium tartrate tetrahydrate (Hampton), and 2.0 M AmSO4 (Hampton) and overlaid with 200 μL of Al’s oil (1:1 silicon oil (Clearco) and mineral oil (Fisher)) in a 96-well plate (Corning). After ~4 weeks, crystals were harvested into fresh crystallization solution prior to soaking in the same solution supplemented with 20% glycerol (Fisher) for cryoprotection. After 5 min, crystals were flash frozen in liquid nitrogen.
Diffraction data were collected at Advanced Photon Source’s SER-CAT beamline 22-ID-D (1 Å, 100 K) equipped with an Eiger 16 M detector. Diffraction data were collected with 360° rotation in 0.3° wedges with an exposure time of 0.25 s. Beam aperture was set to 50 μm, and a beam intensity of 9% was used for data collection. Reflections were indexed, integrated, and scaled using XDS and Aimless.44,45 The structure was phased and solved by molecular replacement using PhaserMR and a polyalanine version of the MPP8 chromodomain generated using chainsaw in the CCP4 software package (PDB 3LWE) as a search model.8,46-48 MPP8 side chains were manually built into the model in Coot and refined in Refmac5.49,50 Ligand restraints were generated in jLigand and fit into the model using the Ligand Fit module in Coot. The ligand-bound model was iteratively refined by manual model building in Coot and automatic refinement in Phenix using TLS parameters.51,52 The structure of MPP8 bound to UNC5246 was solved at a resolution of 2.02 Å with Rwork/Rfree = 21.6%/22.8%. The final model includes MPP8 (D57-K114) in chain A and UNC5246 in chain B.
Cell Culture and Lysis.
HEK293T cells were obtained from ATCC (CRL-11268). Cells were cultured in DMEM (Gibco, 11995–065), 1% pen/strep, and 10% FBS. PC3 cells were obtained from ATCC (CRL-1435). Cells were cultured using DMEM/F12 (Ham; Gibco 11765–054), [+] l-glutamine, [+] 15 mM HEPES, and 10% FBS media. All cells were maintained in a humidified incubator at 37 °C, at 5% CO2.
HEK293T or PC3 cells were harvested with 0.25% trypsin, and the cell pellets were washed 2 × with PBS. HEK293T or PC3Cell Pellets (~30 million) were suspended in 500 μL of CytoBuster Protein Extraction Reagent (Millipore Sigma) containing 5 μL of 100× protease inhibitor cocktail and 1 μL of Benzonase (≥250 units/μL, Millipore Sigma) and incubated in a water bath at 37 °C for 10 min. The samples were then rotated at RT for 20 min followed by centrifugation for 1 min at 14 000 rpm, and the supernatant was removed. Total protein was quantified using a Bio-Rad protein assay by generating a standard curve from stock BSA solutions.
Chemiprecipitation Experiments with UNC5246-Biotin Followed by Western Blot Analysis.
Magnetic streptavidin M-280 Dynabeads (30 μL beads per pulldown) were incubated with UNC5246-Biotin (1 μL of 10 mM stock) in TBST. The beads were left to rotate for 45 min at RT. Concurrently, 1 mg of cell lysate generated from HEK293T or PC3 cells as described above was diluted to 500 μL in TBST. Next, the Dynabeads were washed with TBST to remove excess biotin ligand and then added to the lysate solution. The mixture was rotated at 4 °C overnight, and then the beads were isolated by magnetization and washed with TBST. For experiments using transfected cell lysates, beads were washed with TBST containing 500 mM NaCl. Beads were then resuspended with 15 μL of TBST and 15 μL of 2 × Laemmli sample buffer and heated at 95 °C for 5 min. In parallel, an input of 10–15 μg samples of cell lysate was diluted to 7.5 μL, and 2 × Laemmi sample buffer was added and heated at 95 °C for 5 min. Half of each pull down sample was used for analysis by SDS-PAGE (BioRad any kD) and Western blotting. When blotting for HRP2, the full 1 mg sample was loaded onto the gel. Following membrane transfer, membranes were incubated with the indicated primary antibody at 4 °C overnight, washed with TBST, and then treated with the complementary secondary antibody-IRDye conjugate (Li-COR; 1:10 000 TBST) for 1 h at RT and visualized on a Li-COR Odyssey instrument.
Development of CiA EF1alpha HEK293T.
To develop clonal cells expressing GFP, we used a plasmid, “P024” (targeting the Ey locus with the following features loxp, DSCP spacer, ZF and Gal4, EF1alpha promoter, CpG free GFP:Sh fusion gene, Balb/C arm1, and Balb/C arm2), previously described.53 Instead of making a targeted knock-in line with this construct, we transduced cells with lentivirus infected HEK293T cells with P024, selected the cells that were infected, and plated the cells into 96-well plates with 1 cell/well. Clonal lines were expanded and validated for GPF-positivity with flow. Using an expanded clonal line, we transduced cells with a lentivirus construct expressing Gal4-MPP8 (“P037”), selected the cells, and expanded.
LC-MS/MS Analysis of Chemiprecipitation Experiments.
Each pull-down eluate was reduced with 5 mM DTT for 45 min at 37 °C, alkylated with 15 mM iodoacetamide for 1 h in the dark at RT, then digested with trypsin (Promega Gold) overnight at 37 °C. The peptide samples were desalted using C18 spin columns (Pierce), then labeled with 6plex TMT reagents according to the manufacturer’s protocol (Thermo). After labeling, the samples were combined then dried down via vacuum centrifugation.
The combined TMT sample was reconstituted in 0.1% formic acid, then analyzed (n = 2) by LC/MS/MS using an Easy nLC 1000 coupled to a QExactive HF (Thermo Scientific). Samples were injected onto an Easy Spray PepMap C18 column (75 μm id × 25 cm, 2 μm particle size; Thermo Scientific) and separated over a 120 min method. The gradient for separation consisted of 5–35% mobile phase B at a 250 nL/min flow rate, where mobile phase A was 0.1% formic acid in water and mobile phase B consisted of 0.1% formic acid in ACN. The QExactive HF was operated in data-dependent mode where the 20 most intense precursors were selected for subsequent HCD fragmentation. Resolution for the precursor scan (m/z 350–1600) was set to 120 000 (max IT, 50 ms; target AGC, 3e6), while MS/MS scan resolution was set to 15 000 (max IT, 100 ms; target AGC, 1e5). For MS/MS, the normalized collision energy for HCD was set to 30, with a fixed first mass of 110 m/z and an isolation window of 1.2 m/z. Precursors with unknown charge or a charge state of 1 and ≥8 were excluded.
Raw data were searched against a Uniprot human database using Sequest HT within Proteome Discoverer 2.1 (Thermo Scientific). The following parameters were used to identify tryptic peptides for protein identification: a precursor mass tolerance of 10 ppm; product mass tolerance of 0.02 Da; up to two missed cleavages; carbamidomethylation (C) set as a fixed modification; and oxidation (M) and TMT 6plex (N-term, K) set as variable modifications. The percolator node was used to calculate peptide false discovery rates (FDR), and a < 5% FDR was used to filter all results. Proteins were reported only if ≥2 peptides were identified with a ≤50% coisolation interference. The TMT abundance ratio for each experimental comparison (sample versus controls) was calculated, and a ±1.5-fold change threshold was applied.
Antibodies.
The following antibodies were purchased from the indicated vendors and used for both IB and IP unless otherwise stated: MPP8 (Proteintech, 16796-1-AP), TASOR (Atlas, HPA006735), HRP2 (Abcam, ab156406, IB), HP1α (Abcam, ab109028, IB), HP1β (Santa Cruz, SC-517288, IB), HP1γ (Santa Cruz, SC398562, IB), CDYL2 (Abcam, ab183854), HMGN2 (Abcam, ab199679), TRM28 (Abcam, ab10484), SETDB1 (Abcam, ab12317), MYC (Millipore Sigma, 05–724), FLAG (Abcam, 205606), HP1α (Abcam, ab77256, IP), HP1β (Abcam, ab10811, IP), HP1γ (Milliore Sigma, EMD-05-690, IP), HRP2 (ThermoFisher, 15134-1-AP, IP), and GAPDH (Abcam, ab181602, IB).
Co-Immunoprecipitation Experiments.
Co-IP experiments were performed similarly to chemiprecipitation experiments described above using magnetic Protein G coated Dynabeads that were incubated with 3 μg of desired antibody at RT for 1 h, washed, and then added to HEK293T lysates and analyzed by SDS-PAGE and Western blot as described above.
Generation of Expression Constructs.
The HRP2-Myc construct was cloned as previously described.32 HRP2 truncation plasmids were subcloned from the HRP2-Myc parent vector and inserted into the NotI restriction site of lentiviral transfer plasmid nLV_N103 through In-Fusion cloning. MPP8-FLAG full-length and truncation plasmids were generously provided by the Fang laboratory at Roswell Park Comprehensive Cancer Center. LRRCC3 plasmid was purchased from Addgene (pcDNA3.1(+)-human LRRC33, plasmid #111602).
Cell Transfection.
The day before transfection, 18 × 106 HEK 293T cells were seeded in a 15 cm tissue culture plate. The HRP2-Myc plasmid, MPP8/HRP2 truncation plasmid, or LRRCC3 plasmid was transfected into cells using polyethylenimine (PEI; Polysciences) in Opti-MEM (Gibco) at a 1:3:50 (μg DNA/μL PEI/μL Opti-MEM) ratio. After 48 h, cells were harvested for co-IP experiments.
Lentiviral Infection.
The day before transfection, 18 × 106 Lenti-X 293T cells were seeded in a 15 cm tissue culture plate. The HRP2-Myc plasmid (18 μg), a plasmid expressing Gag–Pol (13.5 μg; Addgene, 12260), and a plasmid expressing VSV-G envelope protein (4.5 μg; Addgene, 12259) were PEI-transfected into cells (as described in “Cell Transfection” method above). After 48 h, the viral medium was carefully removed from Lenti-X 293T cells and centrifuged at 1000 rpm. The supernatant was transferred to an Ultra-Clear Centrifuge tube (Beckman Coulter) using a 0.45 μm syringe filter (Corning) and spun at 20 000 rpm for 2.5 h at 4 °C. The resulting viral pellet was resuspended in 100 μL of PBS and added to MDA-MB-231 cells with 0.5 μg/mL Polybrene (Santa Cruz Biotechnology) added to growth media. HRP2-Myc selection was performed using 6 μg/mL puromycin.
ChIP-qPCR.
Chromatin isolation was performed on MDA-MB-231 cells stably infected with HRP2-Myc. Cells were trypsinized with 0.05% trypsin-EDTA (Gibco), and 16 × 106 cells per biological triplicate were pelleted at 300g and washed with 10 mL of PBS. Cells were formaldehyde cross-linked with 1 mL of 11× Fix Buffer (11% PFA, 50 mM HEPES at pH 8.0, 1 mM EDTA, 0.5 mM EGTA, 100 mM NaCl) for 10 min, and the reaction was quenched on ice with 2.5 M glycine (0.125 M final) for 5 min. Cells were then pelleted at 1200g for 5 min at 4 °C. To isolate nuclei, pellets were resuspended in Rinse Buffer 1 (50 mM HEPES at pH 8.0, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100), incubated on ice for 10 min, spun down at 1200g for 5 min, resuspended in Rinse Buffer 2 (10 mM Tris at pH 8.0, 1 mM EDTA, 0.5 mM EGTA, 200 mM NaCl), and spun down at 1200g for 5 min. Pellets were gently rinsed twice with Shearing Buffer (0.1% SDS, 1 mM EDTA at pH 8.0, 10 mM Tris HCl pH 8.0) then resuspended in 90 μL of Shearing Buffer with protease inhibitors (1:1000; Active Motif) and 10 μL nanodroplets (generously provided by Samantha Pattenden).54 Covaris E110 was used to sonicate chromatin for 9.5 min per sample at 200 cycles/burst or until chromatin was sheared between 200 and 500 bp. A total of 12.5 μL of sheared chromatin was diluted in Shearing Buffer and incubated with 1 μL RNAase A (10 mg mL−1) and 1 μL Proteinase K (10 mg mL−1) overnight. Inputs were purified with a Qiagen MinElute PCR Purification Kit. Remaining chromatin (IP) was suspended in 5 × IP Buffer (250 mM HEPES/KOH at pH 7.5, 1.5 M NaCl, 5 mM EDTA, 5% Triton X-100, 0.5% DOC, 0.5% SDS) to a 1 × final adjusted salt concentration. Five micrograms of antibody (MYC, Millipore Sigma, 05–724; MPP8, Proteintech, 16796-1-AP; H3K9me3, Abcam, ab8898; IgG, Jackson ImmunoResearch, 011-000-003) and 40 μL of prewashed Protein G magnetic Dynabeads (Thermo Fisher Scientific) were added to chromatin and incubated at 4 °C with overhead rotation overnight. Beads were washed with 1 mL of 1 × IP Buffer and incubated at RT with overhead rotation for 3 min, then the wash step was repeated. Beads were washed with 1 mL of DOC Buffer (10 mM Tris at pH 8.0, 0.25 M LiCl, 0.5% NP-40, 0.5% DOC, 1 mM EDTA) and 1 mL of TE at pH 7.4, then resuspended in 100 μL of TE with 2.5 μL of 10% SDS and 5 μL of Proteinase K (10 mg mL−1) and incubated overnight. IP samples were purified with a Qiagen MinElute PCR Purification Kit. QuantStudio ViiA 7 System was used for qPCR. Samples were analyzed using the percent input method.
ChIP Primers.
E-cadherin Promoter Region (TSS – 127). Forward: 5′ CTCAGCCAATCAGCGGTACG 3′. Reverse: 5′ GCGGGCTGGAGTCTGAAC 3′
E-cadherin Exon 2 Region (TSS + 988). Forward: 5′ AGCTACACGTTCACGGTGC 3′. Reverse: 5′ CCCTCCCTACTCCGCCCA 3′
SPINK1. Forward: 5′ TTGCCTAGTGTGTGATGCAA 3′. Reverse: 5′ GCGAAATCCATGCCTTCTAA 3′
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institute on Drug Abuse, NIH (grant R61DA047023) and the CNIHR Program, NIH, to L.I.J. This work was also supported by the National Institute of General Medical Sciences, NIH (grant R01GM118653) to N.A.H. and (grant R35GM133498) to R.K.M. J.M.W. is supported by the National Cancer Institute’s Cancer Epigenetic Training Program (T32CA217824) awarded through the UNC Lineberger Comprehensive Cancer Center. G.R.B. was supported by the MiBio Graduate Training Program (T32GM119999). This research is based in part upon work conducted using the UNC Proteomics Core Facility, which is supported in part by a P30CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center. The authors would like to thank N. Dicheva at the UNC Proteomics Core for preparing samples for proteomics analysis. The authors thank J. Fang for his generous donation of the MPP8 truncation plasmids. The authors thank S. Frye for helpful discussions, R. Hanley for review of all primary chemistry data, and C. Foley for review of all primary biological data. The authors thank B. Hardy for assembly of all screening plates for TR-FRET assays. The TOC graphic was created with BioRender.com.
ABBREVIATIONS
- Kme
Methyl lysine
- Kme2
Dimethyl lysine
- Kme3
Trimethyl lysine
- PTM
Post-translational modification
- HRP2
Hepatoma-derived growth factor-related protein 2
- H3K9me3
Histone 3 lysine 9 trimethyl
- OBOC
One-bead, one-compound
- MPP8
M-phase phosphoprotein 8
- PPHLN1
Periphilin
- TASOR
FAM208A
- HUSH
Human silencing hub
- SETDB1
SET domain bifurcated histone lysine methyltransferase 1
- MORC2
MORC family CW-type zinc finger 2
- LTR
Long terminal repeat
- HIV
Human immunodeficiency virus
- ERV
Endogenous retrovirus
- TE
Transposable elements
- PRC1
Polycomb repressive complex 1
- PPI
Protein protein interaction
- CBX4
Chromobox protein homologue 4
- CBX7
Chromobox protein homologue 7
- CDYL
Chromodomain Y-like protein
- CDYL2
Chromodomain Y-like protein 2
- ChIP
Chromatin immunoprecipitation
- CBX1/HP1β
Chromobox protein homologue 1
- CBX3/HP1γ
Chromobox protein homologue 3
- CBX5/HP1α
Chromobox protein homologue 5
- CBX8
Chromobox protein homologue 8
- CNBr
Cyanogen bromide
- MALDI-TOF
Matrix-assisted laser desorption/ionization time-of-flight
- Lys(iPr,Et)
Ethyl-isopropyl lysine
- TR-FRET
Time-resolved fluorescence energy transfer
- SAR
Structure–activity relationship
- PEG
Polyethylene glycol
- GFP
Green fluorescent protein
- LC-MS
Liquid chromatography mass spectrometry
- TRIM28 (KAP1)
Tripartite motif containing 28
- LEDGF
SFRS1-interacting protein 1
- HMGN2
High mobility group nucleosomal binding domain 2
- PWWP
Pro–Trp–Trp–Pro
- ITC
Isothermal titration calorimetry
- Kd
Dissociation constant
- CO-IP
Co-immunoprecipitation
- IB
Immunoblot
- IP
Immunoprecipitation
- IBD
Integrase binding domain
- DNM3TA
DNA (cytosine-5)-methyltransferase 3A
- G9a
Euchromatic histone-lysine N-methyltransferase 2
- NMR
Nuclear magnetic resonance
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.1c00429.
supplementary figures, synthesis and characterization of all compounds (PDF)
Chemoproteomic analysis of UNC5246 targets (XLSX)
The authors declare no competing financial interest.
Published as part of the ACS Chemical Biology special issue “Epigenetics 2022”.
Contributor Information
Jarod M. Waybright, Center for Integrative Chemical Biology and Drug Discovery, Division of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Sarah E. Clinkscales, Center for Integrative Chemical Biology and Drug Discovery, Division of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Kimberly D. Barnash, Center for Integrative Chemical Biology and Drug Discovery, Division of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy, Department of Biochemistry and Biophysics, School of Medicine, UNC Proteomics Core Facility, Department of Pharmacology, and Lineberger Comprehensive Cancer Center, School of Medicine, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States; Present Address: Foghorn Therapeutics, Cambridge, MA 02139, USA.
Gabrielle R. Budziszewski, Department of Biochemistry and Biophysics, School of Medicine, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States
Justin M. Rectenwald, Department of Biochemistry and Biophysics, School of Medicine, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States
Anna M. Chiarella, Center for Integrative Chemical Biology and Drug Discovery, Division of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States
Jacqueline L. Norris-Drouin, Center for Integrative Chemical Biology and Drug Discovery, Division of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States
Stephanie H. Cholensky, Center for Integrative Chemical Biology and Drug Discovery, Division of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States
Kenneth H. Pearce, Center for Integrative Chemical Biology and Drug Discovery, Division of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States
Laura E. Herring, UNC Proteomics Core Facility, Department of Pharmacology, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Robert K. McGinty, Center for Integrative Chemical Biology and Drug Discovery, Division of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy, UNC Proteomics Core Facility, Department of Pharmacology, and Lineberger Comprehensive Cancer Center, School of Medicine, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States
Nathaniel A. Hathaway, Center for Integrative Chemical Biology and Drug Discovery, Division of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy and Lineberger Comprehensive Cancer Center, School of Medicine, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States
Lindsey I. James, Center for Integrative Chemical Biology and Drug Discovery, Division of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy and Lineberger Comprehensive Cancer Center, School of Medicine, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
REFERENCES
- (1).Strahl BD; Allis CD The language of covalent histone modifications. Nature 2000, 403, 41–45. [DOI] [PubMed] [Google Scholar]
- (2).Hake SB; Xiao A; Allis CD Linking the epigenetic ‘language’ of covalent histone modifications to cancer. Br. J. Cancer 2007, 761–796. [PubMed] [Google Scholar]
- (3).Tolsma TO; Hansen JC Post-translational modifications and chromatin dynamics. Essays Biochem. 2019, 63, 89–96. [DOI] [PubMed] [Google Scholar]
- (4).Shilatifard A Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu. Rev. Biochem 2006, 75, 243–269. [DOI] [PubMed] [Google Scholar]
- (5).Hyun K; Jeon J; Park K; Kim J Writing, erasing and reading histone lysine methylations. Exp. Mol. Med 2017, 49, No. e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Martin C; Zhang Y The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol 2005, 6, 838–849. [DOI] [PubMed] [Google Scholar]
- (7).Kokura K; Sun L; Bedford MT; Fang J Methyl-H3K9-binding protein MPP8 mediates E-cadherin gene silencing and promotes tumour cell motility and invasion. EMBO J. 2010, 29, 3673–3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Li J; Li Z; Ruan J; Xu C; Tong Y; Pan PW; Tempel W; Crombet L; Min J; Zang J Structural basis for specific binding of human MPP8 chromodomain to histone H3 methylated at lysine 9. PLoS One 2011, 6, No. e25104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Chang Y; Horton JR; Bedford MT; Zhang X; Cheng X Structural insights for MPP8 chromodomain interaction with histone H3 lysine 9: potential effect of phosphorylation on methyl-lysine binding. J. Mol. Biol 2011, 408, 807–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Robbez-Masson L; Tie CHC; Conde L; Tunbak H; Husovsky C; Tchasovnikarova IA; Timms RT; Herrero J; Lehner PJ; Rowe HM The HUSH complex cooperates with TRIM28 to repress young retrotransposons and new genes. Genome Res. 2018, 28, 836–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Tchasovnikarova IA; Timms RT; Matheson NJ; Wals K; Antrobus R; Gottgens B; Dougan G; Dawson MA; Lehner PJ Epigenetic silencing by the HUSH complex mediates position-effect variegation in human cells. Science 2015, 348, 1481–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Timms RT; Tchasovnikarova IA; Antrobus R; Dougan G; Lehner PJ ATF7IP-Mediated Stabilization of the Histone Methyltransferase SETDB1 Is Essential for Heterochromatin Formation by the HUSH Complex. Cell Rep. 2016, 17, 653–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Tchasovnikarova IA; Timms RT; Douse CH; Roberts RC; Dougan G; Kingston RE; Modis Y; Lehner PJ Hyperactivation of HUSH complex function by Charcot-Marie-Tooth disease mutation in MORC2. Nat. Genet 2017, 49, 1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Chougui G; Margottin-Goguet F HUSH, a Link Between Intrinsic Immunity and HIV Latency. Front. Microbiol 2019, 10, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Liu N; Lee CH; Swigut T; Grow E; Gu B; Bassik MC; Wysocka J Selective silencing of euchromatic L1s revealed by genome-wide screens for L1 regulators. Nature 2018, 553, 228–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Kato M; Takemoto K; Shinkai Y A somatic role for the histone methyltransferase Setdb1 in endogenous retrovirus silencing. Nat. Commun 2018, 9, 1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sun L; Kokura K; Izumi V; Koomen JM; Seto E; Chen J; Fang J MPP8 and SIRT1 crosstalk in E-cadherin gene silencing and epithelial-mesenchymal transition. EMBO Rep. 2015, 16, 689–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Chang Y; Sun L; Kokura K; Horton JR; Fukuda M; Espejo A; Izumi V; Koomen JM; Bedford MT; Zhang X; Shinkai Y; Fang J; Cheng X MPP8 mediates the interactions between DNA methyltransferase Dnmt3a and H3K9 methyltransferase GLP/G9a. Nat. Commun 2011, 2, 533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).James LI; Frye SV Chemical probes for methyl lysine reader domains. Curr. Opin. Chem. Biol 2016, 33, 135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Stuckey JI; Dickson BM; Cheng N; Liu Y; Norris JL; Cholensky SH; Tempel W; Qin S; Huber KG; Sagum C; Black K; Li F; Huang XP; Roth BL; Baughman BM; Senisterra G; Pattenden SG; Vedadi M; Brown PJ; Bedford MT; Min J; Arrowsmith CH; James LI; Frye SV A cellular chemical probe targeting the chromodomains of Polycomb repressive complex 1. Nat. Chem. Biol 2016, 12, 180–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Barnash KD; The J; Norris-Drouin JL; Cholensky SH; Worley BM; Li F; Stuckey JI; Brown PJ; Vedadi M; Arrowsmith CH; Frye SV; James LI Discovery of Peptidomimetic Ligands of EED as Allosteric Inhibitors of PRC2. ACS Comb. Sci 2017, 19, 161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Arrowsmith CH; Bountra C; Fish PV; Lee K; Schapira M Epigenetic protein families: a new frontier for drug discovery. Nat. Rev. Drug Discovery 2012, 11, 384–400. [DOI] [PubMed] [Google Scholar]
- (23).Barnash KD; Lamb KN; Stuckey JI; Norris JL; Cholensky SH; Kireev DB; Frye SV; James LI Chromodomain Ligand Optimization via Target-Class Directed Combinatorial Repurposing. ACS Chem. Biol 2016, 11, 2475–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Lamb KN; Bsteh D; Dishman SN; Moussa HF; Fan H; Stuckey JI; Norris JL; Cholensky SH; Li D; Wang J; Sagum C; Stanton BZ; Bedford MT; Pearce KH; Kenakin TP; Kireev DB; Wang GG; James LI; Bell O; Frye SV Discovery and Characterization of a Cellular Potent Positive Allosteric Modulator of the Polycomb Repressive Complex 1 Chromodomain, CBX7. Cell Chemical Biology 2019, 26, 1365–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Waybright JM; James LI Getting a handle on chemical probes of chomatin readers. Future Med. Chem 2021, 13 (8), 749–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Barnash KD; James LI; Frye SV Target class drug discovery. Nat. Chem. Biol 2017, 13, 1053–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Rectenwald JM; Hardy PB; Norris-Drouin JL; Cholensky SH; James LI; Frye SV; Pearce KH A General TR-FRET Assay Platform for High-Throughput Screening and Characterizing Inhibitors of Methyl-Lysine Reader Proteins. SLAS Discovery 2019, 24 (6), 693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Schultz DC; Ayyanathan K; Negorev D; Maul GG; Rauscher FJ 3rd SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002, 16, 919–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Hathaway NA; Bell O; Hodges C; Miller EL; Neel DS; Crabtree GR Dynamics and memory of heterochromatin in living cells. Cell 2012, 149, 1447–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Smothers JF; Henikoff S The HP1 chromo shadow domain binds a consensus peptide pentamer. Curr. Biol 2000, 10, 27–30. [DOI] [PubMed] [Google Scholar]
- (31).Lechner MS; Begg GE; Speicher DW; Rauscher FJ 3rd Molecular determinants for targeting heterochromatin protein 1-mediated gene silencing: direct chromoshadow domain-KAP-1 corepressor interaction is essential. Mol. Cell. Biol 2000, 20, 6449–6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).MacDonald IA; Butler KV; Herring LE; Clinkscales SE; Yelagandula R; Stecher K; Bell O; Graves LM; Jin J; Hathaway NA Pathway-Based High-Throughput Chemical Screen Identifies Compounds That Decouple Heterochromatin Transformations. SLAS Discovery 2019, 24, 802–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Vandegraaff N; Devroe E; Turlure F; Silver PA; Engelman A Biochemical and genetic analyses of integrase-interacting proteins lens epithelium-derived growth factor (LEDGF)/p75 and hepatoma-derived growth factor related protein 2 (HRP2) in preintegration complex function and HIV-1 replication. Virology 2006, 346, 415–426. [DOI] [PubMed] [Google Scholar]
- (34).Wang H; Jurado KA; Wu X; Shun MC; Li X; Ferris AL; Smith SJ; Patel PA; Fuchs JR; Cherepanov P; Kvaratskhelia M; Hughes SH; Engelman A HRP2 determines the efficiency and specificity of HIV-1 integration in LEDGF/p75 knockout cells but does not contribute to the antiviral activity of a potent LEDGF/p75-binding site integrase inhibitor. Nucleic Acids Res. 2012, 40, 11518–11530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Eberl HC; Spruijt CG; Kelstrup CD; Vermeulen M; Mann M A map of general and specialized chromatin readers in mouse tissues generated by label-free interaction proteomics. Mol. Cell 2013, 49, 368–378. [DOI] [PubMed] [Google Scholar]
- (36).Baude A; Aaes TL; Zhai B; Al-Nakouzi N; Oo HZ; Daugaard M; Rohde M; Jaattela M Hepatoma-derived growth factor-related protein 2 promotes DNA repair by homologous recombination. Nucleic Acids Res. 2016, 44, 2214–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Mozzetta C; Pontis J; Fritsch L; Robin P; Portoso M; Proux C; Margueron R; Ait-Si-Ali S The histone H3 lysine 9 methyltransferases G9a and GLP regulate polycomb repressive complex 2-mediated gene silencing. Mol. Cell 2014, 53, 277–289. [DOI] [PubMed] [Google Scholar]
- (38).Curry E; Green I; Chapman-Rothe N; Shamsaei E; Kandil S; Cherblanc FL; Payne L; Bell E; Ganesh T; Srimongkolpithak N; Caron J; Li F; Uren AG; Snyder JP; Vedadi M; Fuchter MJ; Brown R Dual EZH2 and EHMT2 histone methyltransferase inhibition increases biological efficacy in breast cancer cells. Clin. Epigenet 2015, 7, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Barnash KD; James LI; Frye SV Target class drug discovery. Nat. Chem. Biol 2017, 13, 1053–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Douse CH; Tchasovnikarova IA; Timms RT; Protasio AV; Seczynska M; Prigozhin DM; Albecka A; Wagstaff J; Williamson JC; Freund SMV; Lehner PJ; Modis Y TASOR is a pseudo-PARP that directs HUSH complex assembly and epigenetic transposon control. Nat. Commun 2020, 11, 4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Prigozhin DM; Douse CH; Farleigh LE; Albecka A; Tchasovnikarova IA; Timms RT; Oda SI; Adolf F; Freund SMV; Maslen S; Lehner PJ; Modis Y Periphilin self-association underpins epigenetic silencing by the HUSH complex. Nucleic Acids Res. 2020, 48, 10313–10328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Villasenor R; Pfaendler R; Ambrosi C; Butz S; Giuliani S; Bryan E; Sheahan TW; Gable AL; Schmolka N; Manzo M; Wirz J; Feller C; von Mering C; Aebersold R; Voigt P; Baubec T ChromID identifies the protein interactome at chromatin marks. Nat. Biotechnol 2020, 38, 728–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).D’Arcy A; Sweeney AM; Haber A Practical aspects of using the microbatch method in screening conditions for protein crystallization. Methods 2004, 34, 323–328. [DOI] [PubMed] [Google Scholar]
- (44).Kabsch W Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Evans P Scaling and assessment of data quality. Acta Crystallogr., Sect. D: Biol. Crystallogr 2006, 62, 72–82. [DOI] [PubMed] [Google Scholar]
- (46).McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser crystallographic software. J. Appl. Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Stein N CHAINSAW: a program for mutating pdb files used as templates in molecular replacement. J. Appl. Crystallogr 2008, 41, 641–643. [Google Scholar]
- (48).Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AG; McCoy A; McNicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr 2011, 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Emsley P; Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- (50).Murshudov GN; Vagin AA; Dodson EJ Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr 1997, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- (51).Lebedev AA; Young P; Isupov MN; Moroz OV; Vagin AA; Murshudov GN JLigand: a graphical tool for the CCP4 template-restraint library. Acta Crystallogr., Sect. D: Biol. Crystallogr 2012, 68, 431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Liebschner D; Afonine PV; Baker ML; Bunkoczi G; Chen VB; Croll TI; Hintze B; Hung LW; Jain S; McCoy AJ; Moriarty NW; Oeffner RD; Poon BK; Prisant MG; Read RJ; Richardson JS; Richardson DC; Sammito MD; Sobolev OV; Stockwell DH; Terwilliger TC; Urzhumtsev AG; Videau LL; Williams CJ; Adams PD Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct Biol 2019, 75, 861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Vignaux PA; Bregio C; Hathaway NA Contribution of promoter DNA sequence to heterochromatin formation velocity and memory of gene repression in mouse embryo fibroblasts. PLoS One 2019, 14, No. e0217699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Kasoji SK; Pattenden SG; Malc EP; Jayakody CN; Tsuruta JK; Mieczkowski PA; Janzen WP; Dayton PA Cavitation Enhancing Nanodroplets Mediate Efficient DNA Fragmentation in a Bench Top Ultrasonic Water Bath. PLoS One 2015, 10, No. e0133014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.