

Abstract
BRCA1 maintains genome integrity and suppresses tumorigenesis by promoting homologous recombination (HR)-mediated repair of DNA double strand breaks (DSB) and DNA damage-induced cell cycle checkpoints. Phosphorylation of BRCA1 by ATM, ATR, CHK2, CDK, and PLK1 kinases has been reported to regulate its functions. Here we show that ATR and ATM-mediated phosphorylation of BRCA1 on T1394, a highly conserved but functionally uncharacterized site, is a key modification for its function in the DNA damage response. Following DNA damage, T1394 phosphorylation ensured faithful repair of DSBs by promoting HR and preventing single strand annealing, a deletion-generating repair process. BRCA1 T1394 phosphorylation further safeguarded chromosomal integrity by maintaining the G2/M checkpoint. Moreover, multiple patient-derived BRCA1 variants of unknown significance were shown to affect T1394 phosphorylation. These results establish an important regulatory mechanism of BRCA1 function in the DNA damage response and may have implications in the development or prognosis of BRCA1-associated cancers.
Introduction
BRCA1 is a major breast and ovarian cancer tumor suppressor. It plays key roles in the DNA damage response (DDR), particularly in homologous recombination (HR)-mediated repair of DNA double strand breaks (DSBs), DNA damage-induced cell cycle checkpoints, and stabilization of stalled replication forks (1,2). BRCA1 fulfills these functions by interacting with many other proteins involved in the DDR. One such protein is PALB2, which links BRCA1 and BRCA2, the other major breast cancer susceptibility protein, in HR and G2/M checkpoint control (3–6). Additionally, the direct interaction between BRCA1 and PALB2 is critical for preventing single strand annealing (SSA), a deletion-based DSB repair process (7). As such, the 3 proteins form a BRCA1-PALB2-BRCA2 axis that plays essential roles in maintaining genome integrity, thereby suppressing cancer development.
BRCA1 has an “SQ-cluster region” (SCR, Fig. 1A) that contains over a dozen “SQ” sites that can be phosphorylated by ATM and ATR, two apical kinases that coordinate the DDR and regulate numerous cellular process after DNA damage (8,9). Among the SQ sites, S1387, S1423, S1457 and S1524 have been subjected to considerable amount of study (10–12). Specifically, early studies showed that S1387 phosphorylation contributes to the intra-S phase checkpoint (10), whereas S1423 phosphorylation promotes the G2/M checkpoint activation (13). Together with S1423, S1524 phosphorylation was reported to regulate p53 acetylation and apoptosis (14,15). More recently, it was reported that a combined mutation of the above 4 sites almost completely abrogates BRCA1 function in HR (12).
Figure 1.
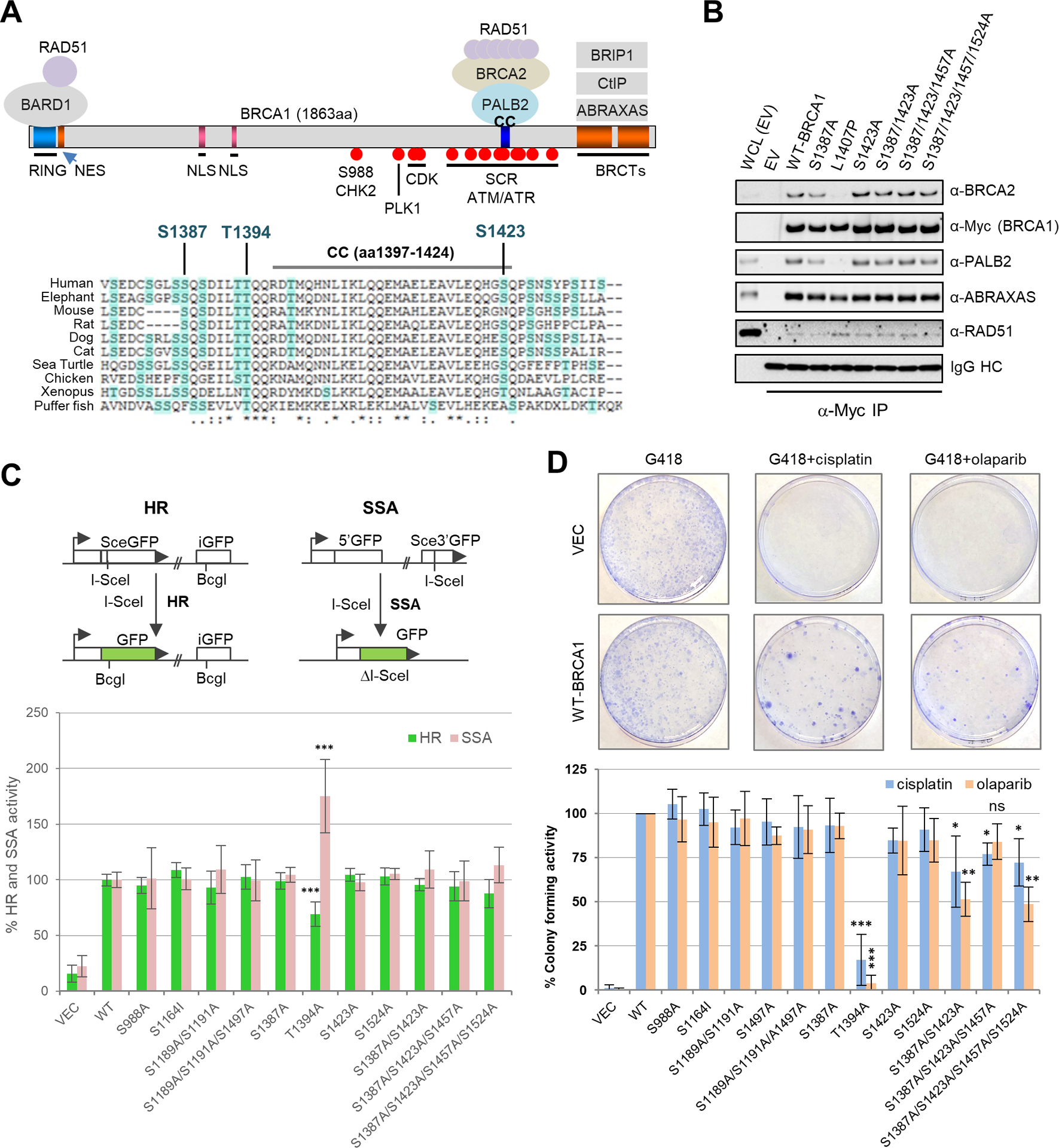
Functional characterization of 10 BRCA1 phosphorylation sites. (A) Domain structure of BRCA1 and the binding sites for its interacting partners. NES, nuclear export signal; NLS, nuclear localization signal. Relevant phosphorylation sites are indicated by filled red circles. Amino acid sequence alignment of the BRCA1 coiled-coil (CC) motif and its surrounding regions among different vertebrate species is shown below. (B) Stable association between BRCA1 SQ site mutants with PALB2, BRCA2 and RAD51. The Myc-tagged BRCA1 proteins were transiently expressed in 293T cells and IPed with anti-Myc paramagnetic beads. (C) HR and SSA activities of BRCA1 phosphorylation mutants. Error bars represent standard deviations (SDs) from at least 3 independent experiments. (D) Colony forming ability of MDA-MB-436 cells transfected with BRCA1 cDNA constructs in the presence of cisplatin and olaparib. Representative images of the plates/colonies are shown above the bar graph. Data presented are the means ± SDs from 2–4 independent experiments for each variant or mutant.
Besides the SCR, it has been reported that CDK phosphorylates BRCA1 at S1189, S1191 and S1497 and that these modifications promote BRCA1 focus formation and DNA repair activity (16). Similarly, PLK1-mediated phosphorylation of BRCA1 at S1164 has been reported to promote BRCA1 focus formation after DNA damage (17). Additionally, CHK2 phosphorylation of BRCA1 at S988 has been reported to regulate HR and mitotic spindle assembly (18,19). However, the different types of phosphorylation sites and events have not been studied together, and their relative contributions to BRCA1 DNA repair function remain unknown.
Here, we determined, in parallel, the functional significance of 10 BRCA1 phosphorylation sites, including the 9 sites mentioned above, in HR and drug resistance. Our results showed that the 9 sites have limited contribution to the HR function of BRCA1. Instead, we found that T1394, a highly conserved but heretofore functionally uncharacterized residue, is the most important BRCA1 phosphorylation site for DSB repair. Moreover, we identified several patient-derived BRCA1 variants of uncertain significance (VUS) that affect T1394 phosphorylation and generated a Brca1T1350I (mouse equivalent of the T1394I VUS) mouse strain. Characterization of the VUS in human cells and the mutant mice further substantiated the key role of T1394 phosphorylation for BRCA1 function in HR and G2/M checkpoint control.
Materials and Methods
Tissue culture
U2OS/DR-GFP and U2OS/SA-GFP cells were described before (20) and kindly provided by Dr. Jeremy Stark (City of Hope). MDA-MD-436 cells were originally obtained from Dr. Ceshi Chen (Albany Medical College) and later authenticated by Short Tandem Repeat (STR) analysis to be 91% match to American Type Culture Collection (ATCC) HTB-130. The U2OS reporter cells were cultured in Dulbecco’s Modified Eagle’s medium (DMEM) (D5796, Sigma-Aldrich), and MDA-MB-436 cells were cultured in DMEM/F12 (D8437, Sigma-Aldrich). Both media were supplemented with 10% fetal bovine serum and 1X Penicillin-Streptomycin (Pen-Strep), and cells were cultured at 37°C in a humidified incubator with 5% CO2. Mycoplasma was not tested, but the cells were closely monitored and periodically cultured in the presence of Plasmocin (ant-mpt, InvivoGen) to eliminate potential mycoplasma contamination. Primary mouse B lymphocytes were isolated from the spleens of 8 weeks old males and cultured as described (21).
Antibodies and reagents
Primary antibodies used are as follows: Myc (9E10, Covance), BRCA1 (D9, sc-6954, Santa Cruz), BRCA1-pS1423 (sc-101647, Santa Cruz), BRCA2 (OP95, EMD Millipore), BARD1 (H300, Santa Cruz), Abraxas (ab139191, AbCam), RAD51 (H-92, Santa Cruz), GAPDH (FL-335, Santa Cruz) and β-Actin (AC-15, Santa Cruz). Anti-human PALB2 was described before (22). Anti-mouse BRCA1 was a gift from Dr. Andre Nussenzweig (NCI). Key chemicals used are Olaparib (S1060, Selleckchem), cisplatin (S1166, Selleckchem), ATM inhibitors KU-55933 (S1092, Selleckchem), KU-60019 (S1570, Selleckchem), AZD1390 (S8680, Selleckchem); ATR inhibitors VE-821 (S8007, Selleckchem), AZ20 (S7050, Selleckchem), VE-822 (S7102, Selleckchem), and BAY-1895344 (S8666, Selleckchem).
Plasmids, transfection, immunoprecipitation (IP) and western blotting
Exogenous BRCA1 proteins were expressed using pcDNA-3xMyc-BRCA1 (23). Mutants and variants were generated by site-directed mutagenesis using the QuikChange protocol (Agilent). For testing protein-protein interactions, BRCA1 expression constructs were transfected into 293T cells plated at a density of 5×105 cells per well in six-well plates and transfected with 2 μg of plasmid per well using X-tremeGENE HP (Roche). Cells were collected ~32 h post-transfection and lysed with 350 μl of NETNG-250 (20 mM Tris-HCl [pH7.4], 250 mM NaCl, 1 mM EDTA, 10 mM NaF, 0.5% Nonidet P-40% and 10% Glycerol) containing Complete® protease inhibitor cocktail (Roche). The Myc-tagged BRCA1 proteins were IPed for 3 hr with Anti-c-Myc Magnetic Beads (88842, Thermo Scientific Pierce). For western blotting, proteins were resolved on 4–12% Tris-glycine SDS gels, transferred onto nitrocellulose membranes and probed with primary antibodies overnight at 4°C and then secondary antibodies for 1–1.5 h at room temperature (RT). Secondary antibodies used were horseradish peroxidase (HRP)-conjugated sheep anti-mouse IgG (NA931V, GE Healthcare) and donkey anti-rabbit IgG (NA9340V, GE Healthcare). Immobilon Western Chemiluminescent HRP Substrate (Millipore) was used to develop the blots.
Measurement of HR and SSA efficiency
HR and SSA activities of the BRCA1 variants were assessed as previously described (7). Briefly, U2OS/DR-GFP and U2OS/SA-GFP cells were plated in 10 cm dish at 1.2×106 cells per dish the day before siRNA transfection. Cells were transfected with an siRNA targeting BRCA1 3’-UTR using Lipofectamine RNAiMax (Life Technologies) (10 nM final concentration of siRNA and 12 μl of RNAiMax per dish). Cells were split into six-well plates (200,000 cells per well) 30 h after siRNA transfection. After another 18 h, cells were cotransfected with pcDNA3-Myc-BRCA1 expression constructs (1 μg) and the I-SceI expression plasmid pCBAsce (1.5 μg) using 6 μl of X-tremeGene 9 (Roche). Cells were harvested ~52 h post the second transfection. GFP-positive cells were scored by flow cytometry. For each sample, 1.5×104 events were counted.
Drug sensitivity assays
The ability of the BRCA1 artificial mutants and patient-derived variants to support cisplatin and olaparib resistance was determined using MDA-MB-236 cells and a colony formation assay as previously described (7). Two independent clones of each mutant or variant construct were used for all experiments, which were all conducted with duplicate plates. Sensitivity of mouse B cells to the drugs were determined using CellTiter-Glo® Luminescent Cell Viability Assay (Promega). Cells were seeded in 24-well plates at 10,000 cells per well, and different concentrations of drugs were added on the following day. Cells were incubated with drugs for 72 h, and aliquots of 35 μl were withdrawn and mixed with the reagent to measure ATP levels.
Generation of the Brca1T1350I knockin mice
Brca1T1350I mice were generated using CRISPR/Cas9 by the Genome Editing Shared Resource (GESR) of Rutgers University. Detailed procedures will be provided upon request. Genotypes were determined by PCR and verified by Sanger sequencing. Primers used for genotyping are GACTGCTCGCAGAGTGATATTTTAACCATC (forward) and GGTTCAGAGAATAAACATATC (reverse). Verified mutant mice were backcrossed to the C57BL/6 background for one generation and then intercrossed to generate wt and homozygous mutant mice for this study. All mouse work was approved by the Institutional Animal Care and Use Committee (IACUC) of the Rutgers Robert Wood Johnson Medical School under the protocol number ID999900493.
G2/M checkpoint assay
Mitotic indexes of activated B cells before and after IR were determined as previously described (3). In brief, cells were seeded in 6-well plates at 2×106 cells per well and treated with 3 Gy of IR the day after. Cells were collected at different time points and stained with anti-phospho-histone H3 (Ser10) (#9701, Cell Signaling), and positive cells were scored by flow cytometry. Prior to flow analysis, propidium iodide (PI) (Sigma, P4170) was added to a final concentration of 10 μg/mL to stain DNA. For each sample, 1.5 × 104 events were counted.
B cell metaphase spread
Chromosomal aberrations of activated B cells were determined as previously described (24). Briefly, cells were arrested with 100 ng/mL colcemid (Sigma) for 1 h prior to collection at each time point. This was followed by treatment with hypotonic solution (0.075 M KCl) and fixation with 3:1 methanol/acetic acid. Telomere-FISH analysis was performed with Cy3-labeled telomere peptide nucleic acid probe (Panagene). 50–55 images were analyzed per sample.
Immunohistochemistry
8–12 weeks old mice were treated with 3 Gy of whole-body gamma radiation and euthanized by CO2 asphyxiation. Tissues were collected immediately after euthanization, fixed in phosphate-buffered formalin, embedded in paraffin, and cross-sectioned at 5 μm thickness. Tissue sections were de-waxed in xylene and hydrated in a graded series of ethanol. Antigen retrieval was carried out by boiling the slides in 1x sodium citrate buffer (Millipore IHC Select® Citrate Buffer pH 6.0, #21545) with a commercial Panasonic microwave at medium power for 4 cycles (6 min each). Endogenous peroxidase activity was quenched by incubating the slides in 3% H2O2/methanol for 30 min. Slides were washed 3X with TBST, blocked with 2.5% horse serum for 1 h at RT, and then incubated with primary antibodies (diluted in SignalStain® Antibody Diluent, #8112, Cell Signaling) overnight at 4°C in a humidified container. Secondary antibody incubations were carried out using Dako LSAB2 System-HRP (Dako, K0675) or ImmPRESS® HRP Horse Anti-Rabbit IgG Polymer Detection Kit (Vector Laboratory, MP-7401) following the manufacturers’ instructions. Signals were developed with 3,3’-diaminobenzidine. Sections were counter-stained with hematoxylin, dehydrated in a graded series of ethanol followed by xylene, and mounted with Thermo Scientific™ Shandon™ Xylene Substitute Mountant (#9999122, Thermo Scientific). Primary antibodies used are anti-γH2A.X (Ser139) (clone JBW301, Millipore Sigma, 05-636, 1:200) and anti-phospho-Histone H3 (Ser10) (#9701L, Cell Signaling technology, 1:250).
Statistical analysis
Statistical significance was analyzed by either two-tailed student’s t-test or one-way ANOVA using GraphPad Prism 8.0 (GraphPad Software). *, p<0.05; **, p<0.01; ***, p<0.001.
Results
Identification of T1394 as the key phosphorylation site for BRCA1 function in DSB repair
To understand the precise roles and the relative contributions of the previously reported BRCA1 phosphorylation sites for HR and DSB repair, we mutated the 9 afore-mentioned sites, including the single CHK2 (S988) and PLK1 (S1161) sites, 3 CDK sites (S1189, S1191, S11497) and 4 ATM/ATR sites (S1387, S1423, S1457 and S1524), both individually and in combination (where applicable). Although the ATM/ATR sites are located within or in the vicinity of the PALB2 binding site (the CC motif), neither individual nor combined mutations of the 4 sites affected PALB2 (and BRCA2) binding to BRCA1 (Fig. 1B), consistent with previous reports (5,12). In addition, we also generated a mutation of T1394, a highly conserved residue immediately upstream of the CC motif (Fig. 1A). T1394 has been predicted to be an ATR site (25) but its phosphorylation has never been detected by mass spectrometry analyses, possibly due to a lack of two tryptic digestion sites (lysine or arginine) in its vicinity, nor has it been subjected to any functional studies.
We then tested the functional impact of the mutations on HR and SSA using U2OS cells harboring either DR-GFP or SA-GFP reporters (26) combined with a “protein replacement” approach, in which the endogenous BRCA1 is depleted with an siRNA targeting the 3’-UTR of its mRNA and the variants to be tested are transiently expressed from a cDNA vector (7). Among all the mutants tested, T1394A was the only one that significantly affected HR and SSA (Fig. 1C). Specifically, this mutation reduced HR and increased SSA, an outcome reminiscent of mutations that affect PALB2 binding (7). Remarkably, mutation of T1394 alone caused a larger effect than did the combined mutation of all 4 of the widely referenced SQ sites, which caused only a slight decrease in HR and a similarly minor increase in SSA.
Next, we assessed the ability of the mutants to support cellular resistance to cisplatin, a DNA crosslinker, and olaparib, a poly (ADP-ribose) polymerase (PARP) inhibitor, both of which cause DNA lesions that require HR to be resolved. Specifically, exogenous BRCA1 proteins were expressed in the BRCA1-deficient MDA-MB-436 breast cancer cells, and their abilities to support colony formation in the presence of the drugs were measured (7). In this setting, T1394 was again the most critical residue for this BRCA1 function, as the T1394A mutation severely reduced colony formation in the presence of either drug (Fig. 1D). Double mutation of S1387 and S1423 or quadruple mutation of all 4 SQ sites also moderately reduced colony formation in the presence of either agent. Finally, mutations of either the PLK1 site (S1161I) or the 3 CDK sites (S1189A/S1191A/S1497A) showed little impact on the ability of BRCA1 to confer resistance to either drug (Fig. 1D), consistent with their lack of effect on HR and SSA (Fig. 1C).
T1394 is phosphorylated by ATR and ATM following DNA damage
To prove that T1394 is phosphorylated in the cell, we immunoprecipitated (IPed) endogenous BRCA1 and subjected the protein to targeted mass spectrometry analysis. Indeed, pT1394 was detected (Fig. S1A–C). To study this modification in detail, we generated a phospho-specific antibody. The antibody detected a signal on immunoblots that matched the molecular weight of BRCA1, and the signal was more evident following ionizing radiation (IR) or upon overexpression of BRCA1, especially when the overexpressed BRCA1 was immunoprecipitated (IPed) (Fig. 2A). In the IPed materials, the antibody detected a double band, and the upper band was eliminated when cell lysate was treated with λ phosphatase prior to IP (Fig. 2B), indicating that this band represents T1394-phosphorylated BRCA1. Mutation of T1394 to an alanine largely abolished the signal detected by the antibody, further validating its specificity (Fig. 2C).
Figure 2.
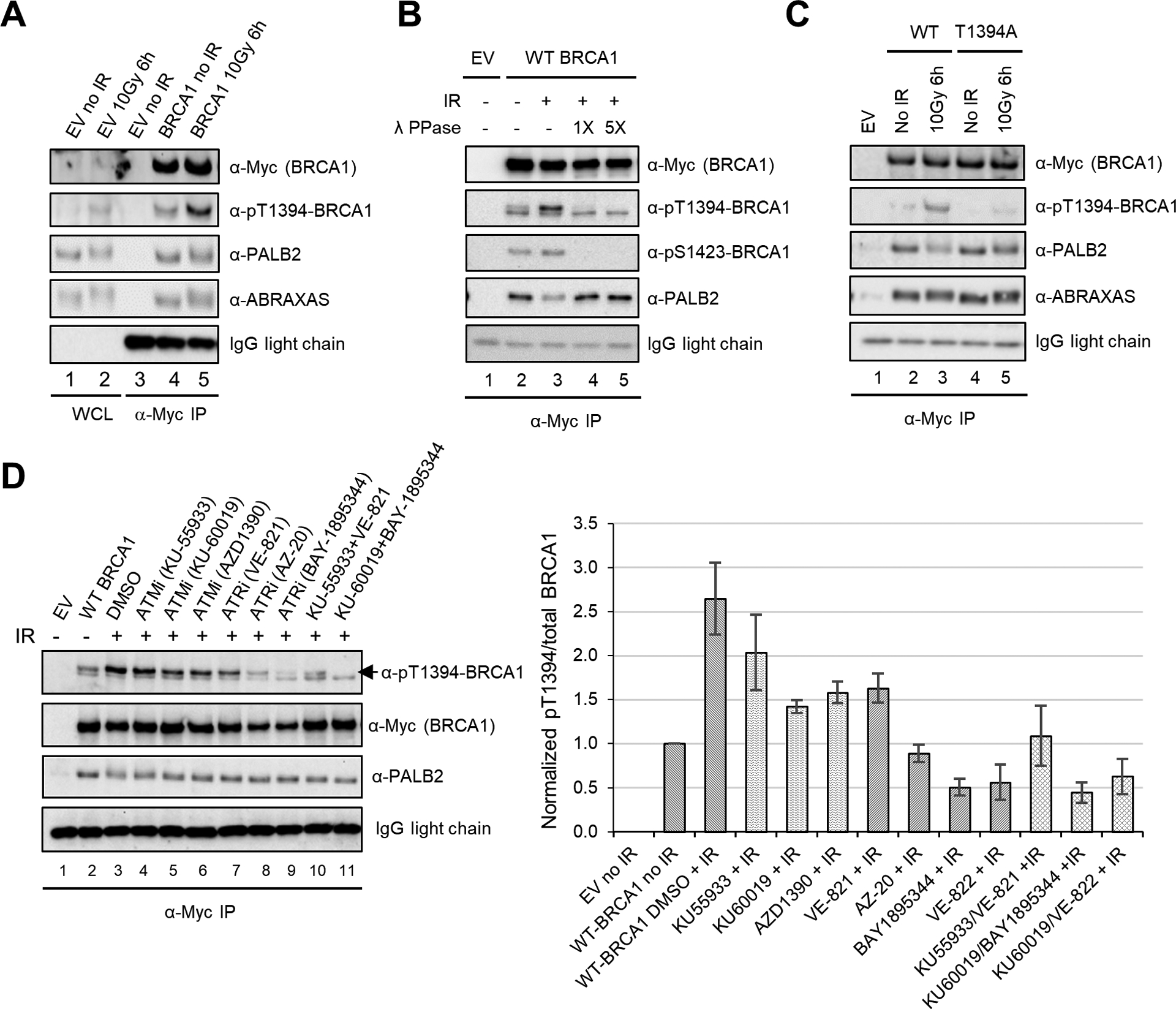
BRCA1 T1394 is phosphorylated by ATR and ATM. (A) DNA damage-induced phosphorylation of T1394. 293T cells were transfected with an empty vector (EV) or Myc-tagged BRCA1 construct. Lysates of cells collected before and after IR were subjected to direct western or IP-western as indicated. WCL, whole cell lysate. (B) Effect of lambda protein phosphatase (λ-PPase) treatment on IP-western signals detected by pT1394 phosphospecific antibody. Anti-pS1423 antibody was used as reference. (C) Specificity of the T1394 phosphospecific antibody. 293T cells were transfected with BRCA1 cDNAs and IP-western conducted as above. (D) Effect of ATM and ATR inhibition on T1394 phosphorylation and the stable association between BRCA1 with PALB2. 293T cells transfected with wt BRCA1 cDNA were treated with DMSO (vehicle) or ATM/ATR inhibitors (all at 1 μM) 30 min prior to IR. Cells were collected 3 h after IR and cell lysates subjected to IP-western. Quantification of BRCA1 pT1394 signals over total BRCA1 IP-ed is shown on the right. Error bars represent SDs from 3 independent experiments.
To confirm that T1394 is phosphorylated by ATM and ATR, we first tested the effects of several inhibitors of each. Inhibition of either enzyme reduced T1394 phosphorylation, but ATR inhibitors tended to be more effective, and simultaneous inhibition of both kinases with a combination of the most potent inhibitor for each largely eliminated the phosphorylation signal (Fig. 2D). Similarly, siRNA-mediated depletion of either kinase led to substantially reduced T1394 phosphorylation, with the impact of ATR knockdown being greater (Fig. S2A and B). Moreover, cell synchronization experiments showed that peak T1394 phosphorylation occurred in the S phase (Fig. S3A and B). These results demonstrate that while both ATM and ATR phosphorylate BRCA1 T1394, ATR is the primary kinase for this site.
Functional analyses of T1394 mutants and VUS affecting the conserved 1394TQ1395 motif
As T1394 is located immediately upstream of the CC motif, the PALB2 binding site, we performed co-IP to test whether its phosphorylation affects PALB2 binding. Neither the T1394A nor T1394E, a phosphomimetic mutant, affected the stable association of PALB2 with BRCA1 (Fig. 3A). In fact, even S1387/T1394/S1423 triple mutants failed to alter the stable association. However, these mutants all led to decreased HR and increased SSA (Fig. 3B). The extent to which they affected HR/SSA activities was similar to that elicited by a hypomorphic variant, M1400V, which partially reduces PALB2 binding to BRCA1, and less pronounced than that caused by L1407P, which almost completely abrogates PALB2 binding (Fig. 3A and B). Thus, T1394 phosphorylation may possibly affect the BRCA1-PALB2 interaction in ways that are beyond detection by co-IP.
Figure 3.
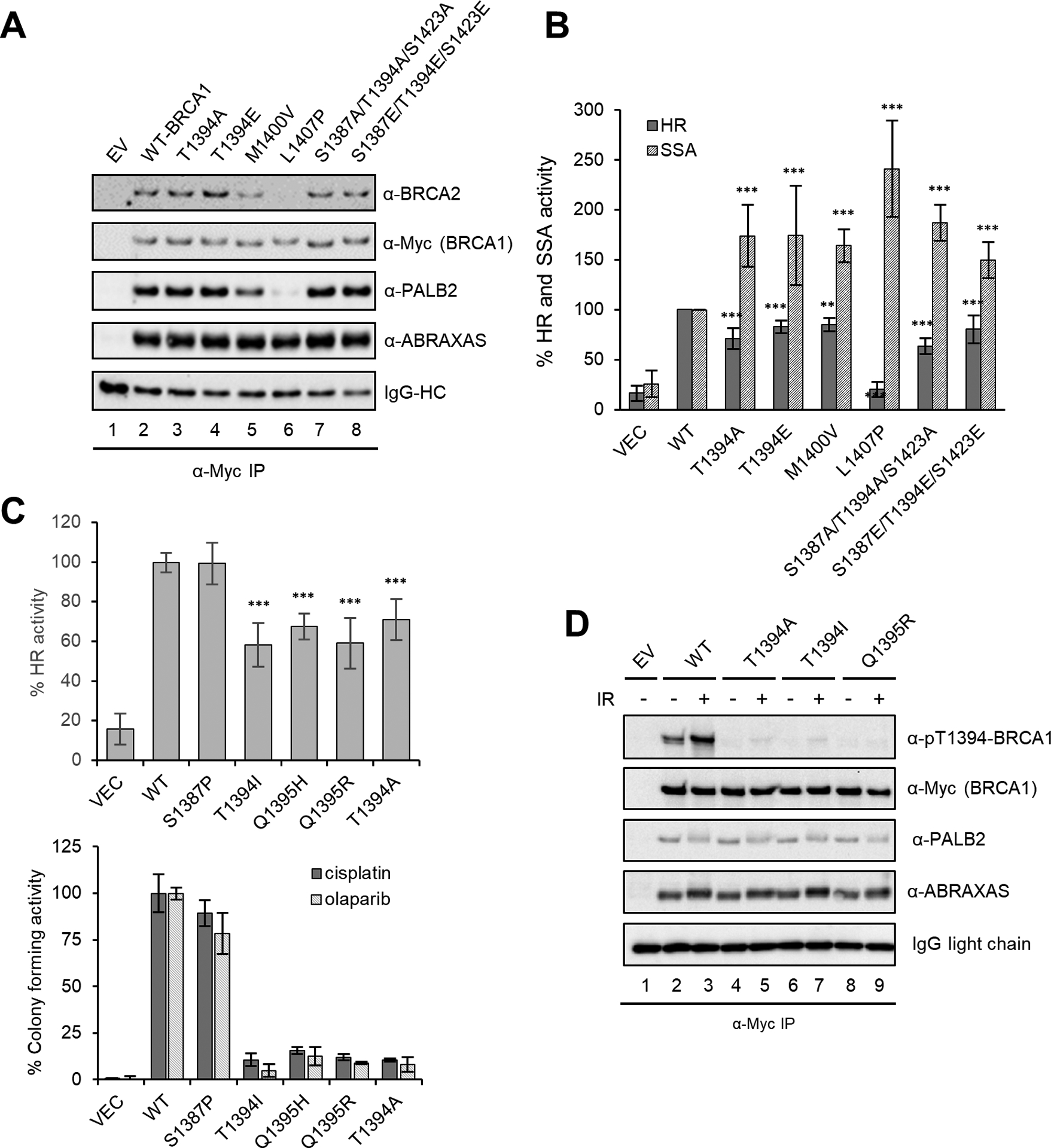
Characterization of BRCA1 patient-derived variants that affect the 1394TQ1395 motif. (A) Effect of T1394 mutations, individually and in combination with mutations of nearby SQ sites, on the stable association between BRCA1 and PALB2. Constructs were transfected into 293T cells and assay conducted as in Fig. 2. Previously reported BRCA1 M1400V and L1407P variants were included for comparison. (B) HR and SSA activities of the mutants and variants in A. Error bars represent SDs from at least 3 independent experiments. (C) HR activities of BRCA1 T1394I, T1395H and T1395R variants and their ability to confer resistance to olaparib and cisplatin. T1394A and S1387P, a neutral variant of S1387, were included for comparison. Error bars represent the SDs of 3 independent experiments for the HR assay and 2 for the colony formation assay. (D) Stale association between BRCA1 T1394A, T1394I and Q1395R variants and PALB2. 293T cells transfected with BRCA1 constructs and were either unirradiated or irradiated with 10 Gy of IR and collected 6 h after. IP-western was conducted as above.
To assess the possible role of T1394 phosphorylation in BRCA1 tumor suppression function, we searched the ClinVar and cBioPortal databases for patient-derived variants affecting T1394 and Q1395, which together form a “TQ” motif for ATM/ATR recognition. A total of 5 different variants of these 2 residues are listed in the databases, with at least 18 patient cases (Table S1). In particular, T1394I and Q1395R have each been found in at least 6 patients and are still classified as VUS. Like T1394A, these variants resulted in significant reduction of HR activity (Fig. 3C), failed to rescue the sensitivity of MDA-MB-436 cells to either cisplatin or olaparib (Fig. 3C), and showed no effect on the co-IP of PALB2 with BRCA1 (Fig. 3D).
DNA repair and G2/M checkpoint defects in Brca1T1350I knockin mouse cells and tissues
To understand the role of T1394 phosphorylation in vivo, we created a knockin mouse strain with a T1350I mutation (Figure S4A), the mouse equivalent of human T1394I, using CRISPR/Cas9. Although full inactivation of Brca1 leads to embryonic lethality (27), Brca1T1350I homozygous mutant mice were born in the expected Mendelian ratio, were viable and fertile, and did not appear to have any overt phenotypes. We first isolated splenic B cells from the mice. Following IR, an induction of pT1350 was observed in the wt but not mutant B cells (Fig. S4B), confirming the conserved nature of this modification and the effect of the mutation.
Next, we assessed genome instability and DNA damage response in the B cells. As shown in Fig 4A, the mutant cells showed a low level of chromosomal aberrations under normal conditions; after treatment with olaparib or cisplatin, while both wt and mutant cells showed a marked increase in chromatid breaks (CTBs) and radial chromosomes, the mutant cells had substantially higher levels of aberrations and were more sensitive to both agents. B cells were then treated with IR and stained with an antibody for phosphorylated histone H3 (Ser10), a marker of condensed chromosomes during mitosis, to assess the G2/M checkpoint. At 1 h after IR, both wt and mutant cells showed a dramatic reduction in mitotic index, indicative of checkpoint activation. At 3 h, while wt cells showed a further drop in mitotic index, mitosis had started to resume in the mutant cells. At 6 h, wt cells also resumed mitosis, but mitotic index of the mutant cells was still higher. Therefore, the mutant cells are capable of G2/M checkpoint activation but defective in checkpoint maintenance.
Figure 4.
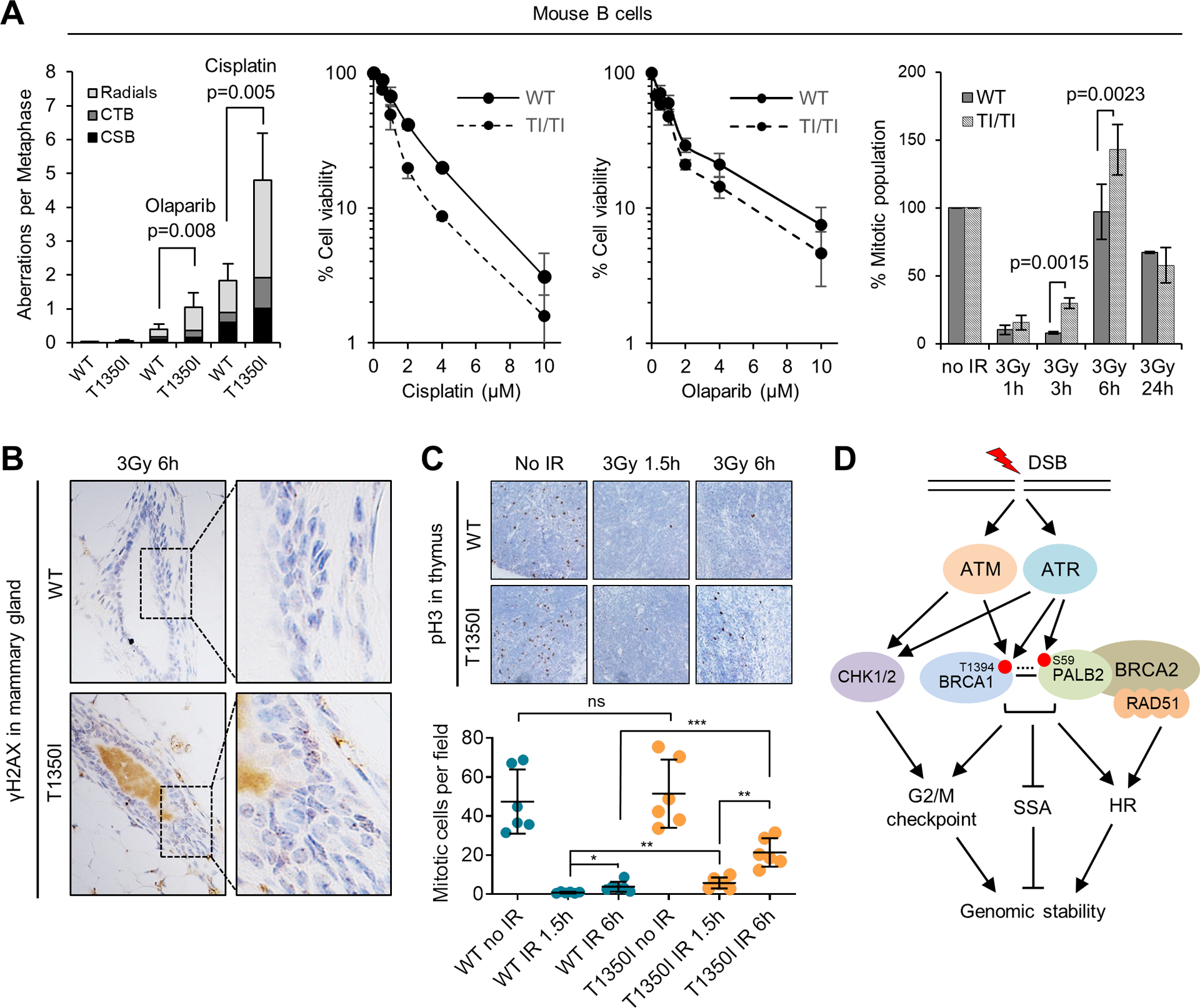
Characterization of Brca1T1350I/T1350I mice. (A) Genome instability, drug sensitivities and G2/M checkpoints of wt and mutant B cells. For genome instability measurement, cells were either untreated or treated with 5 μM cisplatin or 2 μM olaparib overnight. CSB, chromosome breaks; CTB, chromatid breaks. In all graphs, error bars represent SDs from 3 independent experiments each using 2 mice for each genotype. (B) Representative γH2AX IHC images of the mammary gland of wt and mutant mice at 6 h after 3 Gy of IR. Three pairs of mice were analyzed, and similar observations were made. (C) G2/M checkpoint in wt and mutant thymi. Upper panel: representative IHC images of pSer10-histone H3 in the thymi of wt and mutant mice before and after 3 Gy of IR; lower panel: quantification of mitotic cells. Each entry represents the mean of the numbers of mitotic cells per 10X field identified in individual mouse thymus. Error bars represent SDs from at least 5 mice for each genotype. (D) A model of the role of BRCA1 T1394 phosphorylation in genome maintenance. Solid line between BRCA1 and PALB2 denotes stable association and dotted line denotes proposed transient interaction. See text for details.
Finally, we subjected the mice to whole-body radiation and assessed their DNA repair capacity and G2/M checkpoint in vivo. Initially, levels of γH2AX, a marker of DSBs, and phospho-histone H3 in mammary glands were measured by immunohistochemistry (IHC). There was no discernible difference in γH2AX staining between the two genotypes either before or 1.5 h after IR (Fig. S5); however, at 6 h after IR, more γH2AX foci remained in the mutant tissue (Fig. 4B), suggestive of a reduced DSB repair capacity. Quantitative analysis of the G2/M checkpoint in the mammary gland was found to be difficult due to few M phase cells present. We therefore analyzed thymus, a highly proliferative tissue. Indeed, mutant thymi showed significantly more mitotic cells at both 1.5 and (especially) 6 h after IR (Fig. 4C), indicative of a G2/M checkpoint defect. These in vivo results further support a hypomorphic nature of the T1350I mutation.
Discussion
ATM/ATR, CHK2, CDK and PLK1 have been reported to phosphorylate BRCA1 and regulate its nuclear foci formation, DNA repair activity, or checkpoint function. Yet, the relative importance of the reported phosphorylation sites remains unclear. In this study, we analyzed 10 different sites in parallel for their importance for HR and SSA in human cells. Our results showed that mutations of the 9 previously reported sites, even in combination, have limited impact on the two homology-directed repair processes and on cellular resistance to HR-targeting drugs. Instead, we found that T1394, a highly conserved but heretofore functionally uncharacterized residue, is phosphorylated by ATR and ATM, and this modification is important for BRCA1 function in the DDR and genome maintenance after genotoxic insults.
We have shown that BRCA1 mutations in the RING and BRCT domains negatively affect both HR and SSA, while mutations in the CC motif that binds PALB2 lead to reduced HR but increased SSA (7). The findings indicate that the RING and BRCT domains are required for a common step in for both HR and SSA, such as resection, whereas BRCA1-mediated recruitment of PALB2 acts after resection and simultaneously promotes HR and prevents SSA. Interestingly, loss of T1394 phosphorylation resulted in a similar outcome, with T1394A causing nearly identical effect to that of M1400V, a patient derived BRCA1 variant that partially disrupts the binding of PALB2 (Fig. 3A and B). This would suggest that the phosphorylation status of T1394 may affect the interaction between BRCA1 and PALB2; however, no difference was found between T1394 mutant and wt proteins in co-IP experiments (Fig. 3A). This implies that T1394 phosphorylation may alter the conformation or activity of the BRCA1-PALB2 complex, rather than the amount of their stable complex. It is also possible that the BRCA1-PALB2 interplay may involve both stable and transient interactions and that T1394 phosphorylation affects the transient component. The fact that T1394E also caused reduced HR and increased SSA, although its activities were closer to wt levels, further suggests that timely dephosphorylation of the residue is also required to ensure optimal HR and to avoid or suppress SSA.
Approximately 30% of BRCA1 sequence alterations in humans are missense variants, most of which are considered as VUS (28). Due to its rarity and lack of functional characterization, clinical interpretation of a specific VUS can be challenging. Notably, there are 5 different VUS, reported for a total 18 times, that affect the conserved 1394TQ1395 site (Table S1). In silico predictions (Align-GVGD) also suggest worse clinical outcomes (Table S1). Our functional analyses showed that T1394I, Q1395R and Q1395H variants all caused reduced HR, increased SSA, and reduced cellular resistance to DNA damaging agents (Fig. 3C). Thus, these variants clearly have functional defects and may potentially increase cancer risk or affect prognosis.
In conclusion, our findings establish that ATR and ATM phosphorylate BRCA1 on T1394 after genotoxic insults, and this modification ensures faithful repair of DSBs by promoting HR, preventing SSA, and maintaining the G2/M checkpoint (Fig. 4D). At the same time, our results also provide increased clarity of the roles of other BRCA1 phosphorylation sites in homology-directed repair and cellular sensitivity to DNA damaging agents. Moreover, we identified several BRCA1 VUS that affect T1394 phosphorylation and demonstrated their functional defects, suggesting that these VUS could contribute to cancer development and/or influence therapy response. Notably, it has been reported that ATR phosphorylates PALB2 at S59 leading to increased PALB2-BRCA1 complex formation (29). Whether the two phosphorylation events are coordinated, the precise mechanisms that underlie the DNA repair and checkpoint defects of T1394 and Q1395 variants, and the pathogenicity of the variants await further investigation.
Supplementary Material
Significance.
This study identifies a BRCA1 phosphorylation event critical for its DNA repair function and reveals the functional defects of several BRCA1 variants of unknown significance.
Acknowledgements
We thank Dr. Andre Nussenzweig (NCI) for providing the anti-mouse BRCA1 antibody. This work was supported by the National Institutes of Health (R01CA138804 and P01CA250957 to BX, R01CA190858 to SFB, R01GM141091 to WZ, and P30NS046593 and S10OD025047 to HL) and New Jersey Health Foundation (PC 116-21 to BX and HL). This work was also supported by the Flow Cytometry and Cell Sorting, Genome Editing, and Biospecimen Repository and Histopathology shared resources of Rutgers Cancer Institute of New Jersey (P30CA072720). TKF was supported by a postdoctoral fellowship (DCHS19PPC010) from New Jersey Commission on Cancer Research (NJCCR).
Footnotes
The authors declare no conflicts of interest.
References
- 1.Chen CC, Feng W, Lim PX, Kass EM, Jasin M. Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer. Annu Rev Cancer Biol 2018;2:313–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhao W, Wiese C, Kwon Y, Hromas R, Sung P. The BRCA Tumor Suppressor Network in Chromosome Damage Repair by Homologous Recombination. Annual review of biochemistry 2019;88:221–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simhadri S, Vincelli G, Huo Y, Misenko S, Foo TK, Ahlskog J, et al. PALB2 connects BRCA1 and BRCA2 in the G2/M checkpoint response. Oncogene 2019;38:1585–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sy SM, Huen MS, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci U S A 2009;106:7155–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang F, Ma J, Wu J, Ye L, Cai H, Xia B, et al. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr Biol 2009;19:524–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang F, Fan Q, Ren K, Andreassen PR. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol Cancer Res 2009;7:1110–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anantha RW, Simhadri S, Foo TK, Miao S, Liu J, Shen Z, et al. Functional and mutational landscapes of BRCA1 for homology-directed repair and therapy resistance. Elife 2017;6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marechal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 2013;5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol Cell 2017;66:801–17 [DOI] [PubMed] [Google Scholar]
- 10.Xu B, O’Donnell AH, Kim ST, Kastan MB. Phosphorylation of serine 1387 in Brca1 is specifically required for the Atm-mediated S-phase checkpoint after ionizing irradiation. Cancer Res 2002;62:4588–91 [PubMed] [Google Scholar]
- 11.Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science (New York, NY: 1999;286:1162–6 [DOI] [PubMed] [Google Scholar]
- 12.Beckta JM, Dever SM, Gnawali N, Khalil A, Sule A, Golding SE, et al. Mutation of the BRCA1 SQ-cluster results in aberrant mitosis, reduced homologous recombination, and a compensatory increase in non-homologous end joining. Oncotarget 2015;6:27674–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu B, Kim S, Kastan MB. Involvement of Brca1 in S-phase and G(2)-phase checkpoints after ionizing irradiation. Molecular and cellular biology 2001;21:3445–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin SA, Ouchi T. BRCA1 phosphorylation regulates caspase-3 activation in UV-induced apoptosis. Cancer Res 2005;65:10657–62 [DOI] [PubMed] [Google Scholar]
- 15.Li Q, Hao Q, Cao W, Li J, Wu K, Elshimali Y, et al. PP2Cdelta inhibits p300-mediated p53 acetylation via ATM/BRCA1 pathway to impede DNA damage response in breast cancer. Sci Adv 2019;5:eaaw8417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson N, Cai D, Kennedy RD, Pathania S, Arora M, Li YC, et al. Cdk1 participates in BRCA1-dependent S phase checkpoint control in response to DNA damage. Mol Cell 2009;35:327–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chabalier-Taste C, Brichese L, Racca C, Canitrot Y, Calsou P, Larminat F. Polo-like kinase 1 mediates BRCA1 phosphorylation and recruitment at DNA double-strand breaks. Oncotarget 2016;7:2269–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang J, Willers H, Feng Z, Ghosh JC, Kim S, Weaver DT, et al. Chk2 Phosphorylation of BRCA1 Regulates DNA Double-Strand Break Repair. Molecular and Cellular Biology 2004;24:708–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stolz A, Ertych N, Kienitz A, Vogel C, Schneider V, Fritz B, et al. The CHK2-BRCA1 tumour suppressor pathway ensures chromosomal stability in human somatic cells. Nat Cell Biol 2010;12:492–9 [DOI] [PubMed] [Google Scholar]
- 20.Gunn A, Stark JM. I-SceI-based assays to examine distinct repair outcomes of mammalian chromosomal double strand breaks. Methods Mol Biol 2012;920:379–91 [DOI] [PubMed] [Google Scholar]
- 21.Simhadri S, Peterson S, Patel DS, Huo Y, Cai H, Bowman-Colin C, et al. Male fertility defect associated with disrupted BRCA1-PALB2 interaction in mice. The Journal of biological chemistry 2014;289:24617–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, et al. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell 2006;22:719–29 [DOI] [PubMed] [Google Scholar]
- 23.Chen J, Silver DP, Walpita D, Cantor SB, Gazdar AF, Tomlinson G, et al. Stable interaction between the products of the BRCA1 and BRCA2 tumor suppressor genes in mitotic and meiotic cells. Mol Cell 1998;2:317–28 [DOI] [PubMed] [Google Scholar]
- 24.Li M, Cole F, Patel DS, Misenko SM, Her J, Malhowski A, et al. 53BP1 ablation rescues genomic instability in mice expressing ‘RING-less’ BRCA1. EMBO Rep 2016;17:1532–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tibbetts RS, Cortez D, Brumbaugh KM, Scully R, Livingston D, Elledge SJ, et al. Functional interactions between BRCA1 and the checkpoint kinase ATR during genotoxic stress. Genes Dev 2000;14:2989–3002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stark JM, Pierce AJ, Oh J, Pastink A, Jasin M. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Molecular and cellular biology 2004;24:9305–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Evers B, Jonkers J. Mouse models of BRCA1 and BRCA2 deficiency: past lessons, current understanding and future prospects. Oncogene 2006;25:5885–97 [DOI] [PubMed] [Google Scholar]
- 28.Monteiro AN, Bouwman P, Kousholt AN, Eccles DM, Millot GA, Masson JY, et al. Variants of uncertain clinical significance in hereditary breast and ovarian cancer genes: best practices in functional analysis for clinical annotation. J Med Genet 2020;57:509–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buisson R, Niraj J, Rodrigue A, Ho CK, Kreuzer J, Foo TK, et al. Coupling of Homologous Recombination and the Checkpoint by ATR. Mol Cell 2017;65:336–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.