

Abstract
Osimertinib (AZD9291 or TAGRISSO™) is a promising and approved third-generation epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) for treating patients with advanced non-small cell lung cancer (NSCLC) harboring EGFR-activating mutations or the resistant T790M mutation. However, the inevitable emergence of acquired resistance limits its long-term efficacy. A fuller understanding of the mechanism of action of osimertinib and its linkage to acquired resistance will enable the development of more efficacious therapeutic strategies. Consequently, we have identified a novel connection between osimertinib or other EGFR-TKI and c-Myc. Osimertinib rapidly and sustainably decreased c-Myc levels primarily via enhancing protein degradation in EGFR-mutant (EGFRm) NSCLC cell lines and xenograft tumors. c-Myc levels were substantially elevated in different EGFRm NSCLC cell lines with acquired resistance to osimertinib in comparison with their corresponding parental cell lines and could not be reduced any further by osimertinib. Consistently, c-Myc levels were elevated in the majority of EGFRm NSCLC tissues relapsed from EGFR-TKI treatment compared to their corresponding untreated baseline c-Myc levels. Suppression of c-Myc through knockdown or pharmacological targeting with BET inhibitors restored the response of resistant cell lines to osimertinib. These findings indicate that c-Myc modulation mediates the therapeutic efficacy of osimertinib and the development of osimertinib-acquired resistance. Furthermore, they establish c-Myc as a potential therapeutic target and warrant clinical testing of BET inhibition as a potential strategy to overcome acquired resistance to osimertinib or other EGFR inhibitors.
Keywords: EGFR, c-Myc, osimertinib/osimertinib, resistance, BET inhibitors, lung cancer
Introduction
Development of epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (EGFR-TKIs) based on the discovery of EGFR activating mutations is an important milestone in the targeted therapy of non-small cell lung cancer (NSCLC), which constitutes over 80% of lung cancer cases and has a low 5-year survival rate of about 19% (1,2). Over the past decades, the development of EGFR-TKIs has rapidly progressed from the first generation to the current third generation. Osimertinib (AZD9291 or TAGRISSO™) is a successful example of 3rd generation EGFR-TKIs, which selectively and irreversibly inhibit activating EGFR mutations and the resistant T790M mutation while sparing wild-type (WT) EGFR. Osimertinib is now an FDA-approved drug for treating patients with advanced NSCLC that has become resistant to 1st generation EGFR-TKIs through the T790M mutation and for EGFR mutation-positive advanced NSCLC as a first line treatment. Very recently, osimertinib was approved by the FDA for adjuvant therapy after tumor resection in patients with NSCLC whose tumors have activating EGFR mutations. When used as a first-line treatment, osimertinib significantly improves overall survival (38.6 months) in EGFR-mutant (EGFRm) NSCLC as reported recently (3). Despite this, almost all patients eventually relapse from osimertinib treatment due to the emergence of acquired resistance, resulting in disease progression (4). Hence, understanding the underlying resistance mechanisms and developing effective strategies to overcome acquired resistance to osimertinib is highly desirable and urgently needed in the clinic.
c-MYC (or MYC) was the first Myc family member found in the human genome and was originally identified as a cellular homologue of the avian myelocytomatosis retroviral oncogene (v-Myc). Additional Myc family members include MYCN and MYCL, which encode N-Myc and L-Myc, respectively. The MYC product, c-Myc, functions as a basic-helix–loop–helix leucine-zipper (bHLHZip) transcription factor, which primarily forms a heterodimer with Myc associated factor X (MAX), binds the E-box element CACGTG, and regulates the expression of many genes whose products are involved in regulation of various physiological processes such as cell growth, differentiation, survival, metabolic reprogramming, cell adhesion, angiogenesis and immune surveillance (5–7). MYC is genetically activated and/or overexpressed through gene amplification, chromosomal translocation, or mutation in most types of human cancer including lung cancer, thereby driving autonomous proliferation, growth, and self-renewal, blocking differentiation, promoting metabolic reprogramming and inducing genomic destabilization. Thus, MYC is a central driver of malignant cellular growth and proliferation (8).
There are some previous studies that identified MYC amplification at the time of resistance to EGFR-TKIs in some EGFRm patients, which is associated with shorter progression-free survival (PFS) when present as a co-mutation at time of diagnosis (9–13). However, a connection between c-Myc modulation and osimertinib-mediated targeted cancer therapy has not been established. In an effort to understand the biology or mechanism of osimertinib in EGFRm NSCLC cells, we found that osimertinib rapidly and sustainably decreased c-Myc levels primarily in NSCLC lines with activating EGFR mutations. Moreover, c-Myc elevation was tightly associated with osimertinib acquired resistance and suppression of c-Myc through gene knockdown or BET inhibition, which is known to inhibit MYC transcription (14), restored the response of osimertinib-resistant cell lines to osimertinib. Hence the current study focuses on osimertinib modulation of c-Myc in EGFRm NSCLC cell lines, understanding the underlying mechanisms, involvement of c-Myc elevation in osimertinib acquired resistance and the development of clinically testable strategies for overcoming acquired resistance via targeting c-Myc.
Material and Methods
Reagents.
Osimertinib, CO1686, erlotinib, MG132, actinomycin D (Act D), trametinib, selumetinib (AZD6244), cycloheximide (CHX), GDC0994 (ravoxertinib) and VRT752271 (ulixertinib or BVD-523) were the same as described previously (15–17). EGF816 and afatinib were purchased from MedChem Express (MCE; Monmouth Junction, NJ). JQ1, OTX015, ZBC260 and dBET were described in our previous studies (18,19). SB216763 and CHIR99021 were purchased from Sigma Chemical Co. (St. Louis, MO) and LC Laboratories (Woburn, MA), respectively. c-Myc (#5605), p-c-Myc (S62; #13748) and p-c-Myc (T58/S62; #9401) antibodies were purchased from Cell Signaling Technology, Inc. (Beverly, MA). FBXW7 (A301-721A) antibody was purchased from Bethyl Laboratories, Inc. (Montgomery, TX). Other antibodies were the same as described in our previous studies (15,16,20,21).
Cell lines and cell culture.
SH416, SH416/AR (AZD9291-resistant), SH450 and SH450/AR cell lines were kindly provided by Dr. Christine M. Lovly (Vanderbilt University School of Medicine, Nashville, TN). Bim knockout (KO) cell lines established from PC-9/AR (BimKO#2 and BimKO#3) were the same as described in our previous study (22). PC-9/AR (resistant to osimertinib with unknown resistance mechanism), PC-9/GR/AR (resistant to gefitinib and then osimertinib with T790M mutation), PC-9/3M (resistant to osimertinib with 19del, T790M and C797S triple mutations), HCC827/AR (resistant to osimertinib with MET amplification) and other cell lines used in this study and cell culture conditions were described previously (15,16). These cell lines were not genetically authenticated. PC-9/V, PC-9/Myc and PC-9/MycS62A were established with retroviral infection of PC-9 cells using retroviruses carrying pBabe, pBabe-Myc and pBabe-Myc/S62A, respectively, which were generously provided by Dr. Junran Zhang (The Ohio State University James Comprehensive Cancer Center and College of Medicine, Columbus, Ohio), as described previously (23).
Cell survival assay.
Cells were seeded in 96-well cell culture plates and on the second day were exposed to various treatments for 3 days. Cell numbers were then estimated by sulforhodamine B (SRB) assay as previously described (24). Combination index (CI) for drug interaction (e.g., synergy) was calculated using CompuSyn software (ComboSyn, Inc.).
Detection of apoptosis.
Apoptosis was detected with an Annexin V/7-AAD apoptosis detection kit (BD Biosciences; San Jose, CA) following the manufacturer’s instructions. Protein cleavage were detected by Western blot analysis as additional indicators of apoptosis.
Western blot analysis.
Preparation of whole-cell protein lysates and Western blot analysis were described previously (15,16). Band intensities were scanned and quantified by NIH image J software.
Gene knockdown using small interfering RNA (siRNA) and short hairpin RNA (shRNA).
Non-silencing control, GSK3α/β and FBXW7 siRNAs and their transfection were the same as described previously (25,26). Lentiviral c-Myc shRNAs (#1 to #4) in pLKO.1 were purchased from Sigma (St. Louis, MO) and used according to the manufacturer’s instructions.
Quantitative reverse transcription PCR (qRT-PCR) and RNA sequencing (RNA-seq).
Cellular total RNA was extracted with TRIzol (ThermoFisher Scientific/Invitrogen; Grand Island, NY) and cDNAs were synthesized using a Reverse Transcriptase M-MLV (RNase H-) kit (Applied Biological Materials Inc.; Richmond, BC, Canada). Real-time PCRs were performed using IQTM SYBR Green Supermix (Bio-Rad Laboratories; Hercules, CA) for 40 cycles of 95°C for 15 s and 60°C for 40 s with the QuantStudio 3 and 5 systems (ThermoFisher Scientific). The primer pairs for c-Myc were 5’-GGCTCCTGGCAAAAGGTCA-3’ (forward) and 5’-CTGCGTAGTTGTGCTGATGT-3’ (reverse). GAPDH was used as an internal control and detected with the primers of 5’-GACATCAAGAAGGGGTGAA-3’ (forward) and 5’-TGTCAT ACCAGGAAATGAGC-3’ (reverse). All primers were synthesized from Integrated DNA Technologies (Coralville, Iowa). mRNA expression was also detected with RNA-seq analysis by MedGenome Inc. (Foster City, CA). Differential expression analysis was performed using DESeq2. The expression values for each gene were presented in FPKM (fragments per kilobase per million) units.
Animal xenograft and treatments.
Animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Emory University. For dynamic c-Myc detection in xenografts, treatments including vehicle control and osimertinib (10 mg/kg/day, og) were started when xenografts reached around 300 mm3. On days 1, 3, 6 and 9 post treatment, 3 mice in each group were euthanized with CO2 asphyxia for collecting tissues, which were frozen in liquid nitrogen. Tumor tissue aliquots were homogenized in protein lysis buffer for preparation of whole-cell protein lysates for Western blotting to detect the given proteins. Other therapeutic experiments were conducted as described previously (15,16). Treatments in the first experiment included vehicle control and osimertinib (5 mg/kg/day, og). Treatments in the second experiment included vehicle control, osimertinib (5 mg/kg, day, og), JQ1 (50 mg/kg, day, ip), ZBC260 (2 mg/kg, three times or twice a week, iv and ip), osimertinib plus JQ1 and osimertinib plus ZBC260.
Human NSCLC tissues.
Paired tissue samples from EGFRm NSCLC patients before treatment (i.e., baseline) and after relapse from treatment with first generation EGFR-TKIs including gefitinib, erlotinib or icotinib were primarily collected at the Second Xiangya Hospital (Changsha, Hunan, China), Henan Cancer Hospital (Zhengzhou, Henan, China) and Daping Hospital (Chongqing, China) under Ethics Review Committee (IRB)-approved protocols (2019-009, 2019-067 and 2019-274, respectively). The majority of tissues were collected from the lung. A few samples were collected from neck or clavicular lymph nodes, pleura or pleural fluids. All tissues were sent to and stained at the Second Xiangya Hospital.
Immunohistochemistry (IHC).
Human NSCLC tissues were stained with IHC using the EnVision™ + Dual Link System-HRP Kit (Dako; Carpinteria, CA) with rabbit monoclonal c-Myc antibody (EP121) purchased from MXB Biotechnologies (cat # RMA-0803; Fuzhou, China) at 1:100 dilutions overnight at 4°C. Both the percentage of positive staining in tumor cells and intensity of staining were scored. The intensity of IHC staining was measured by using a numerical scale (0 = no expression, 1 = weak expression, 2 = moderate expression and 3 = strong expression). The staining data were finally quantified as the weighted index (WI) (WI = % positive staining in tumor × intensity score) as previously described (27). The WI was determined by 2 individuals, and the final values were the average of the two readings.
Statistical analysis.
The statistical significance of differences between two experimental groups was analyzed with two-sided unpaired Student’s t tests (for equal variances) or with Welch’s corrected t test (unequal variances) by use of Graphpad InStat 8 software. Results were considered to be statistically significant at P < 0.05.
Results
Osimertinib and other EGFR-TKIs decrease c-Myc levels in EGFRm NSCLC cells and tumors.
In two NSCLC cell lines with mutant EGFR, PC-9 and HCC827, osimertinib at 100 nM effectively exerted time-dependent effects on suppressing the phosphorylation of EGFR and ERK1/2 and on decreasing c-Myc levels starting from 2 h post treatment and being sustained up to 24 h at the longest time tested (Fig. 1A). Osimertinib could effectively and concentration-dependently decrease c-Myc levels starting at 50 nM, which was in parallel with its effects on inhibiting the phosphorylation of EGFR and ERK1/2 (Fig. 1B). Osimertinib decreased c-Myc levels in another 3 EGFRm NSCLC cell lines: i.e., H1650, H1975 and SH450, but not in other NSCLC cell lines (A549, H1299 and H460) with WT EGFR even at 500 nM (Fig. 1C), suggesting an EGFR mutation-dependent modulatory effect on c-Myc. Beyond osimertinib, other EGFR-TKIs, including the 3rd generation EGFR-TKIs, EGF816 and CO1686, the 2nd generation EGFR TKI, afatinib, and the 1st generation EGFR-TKI, erlotinib, all effectively reduced c-Myc levels in both PC-9 and HCC827 cells (Fig. 1D), indicating that c-Myc reduction is a common event modulated by EGFR-TKIs. In PC-9 xenograft tumors receiving osimertinib treatment, substantial c-Myc reduction was detected after 1 day treatment and sustained for 9-days treatment (Figs. 1E and 1F), clearly indicating that osimertinib actively reduces c-Myc in vivo. In RNA-seq analysis with HCC827 cells exposed to osimertinib, we found that the expression of several c-Myc target genes such as CDC25A, ODC and CCNE (28,29) was significantly inhibited (Figs. 1G and H), consistent with osimertinib’s effect on downregulation of c-Myc in EGFRm NSCLC cells.
Figure 1. Osimertinib and other EGFR-TKIs decrease c-Myc levels in EGFRm NSCLC cell lines (A–D) and tumors (E and F), and suppress downstream target genes (G and H).
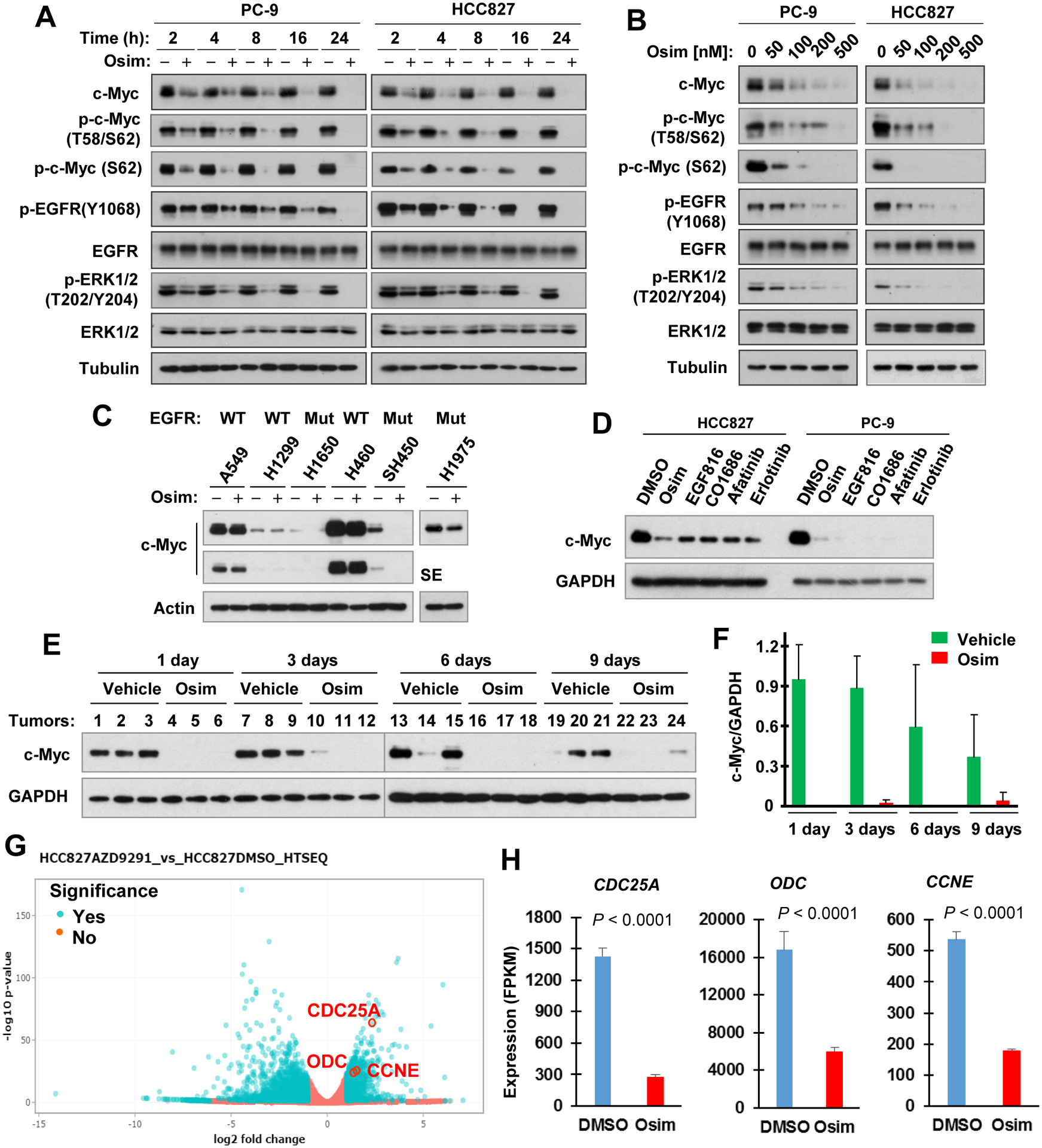
A and B, Both PC-9 and HCC827 cell lines were exposed to 100 nM osimertinib (Osim) for different times as indicated (A) or to the indicated concentrations of osimertinib for 6 h (B). C and D, The indicated cell lines were treated with 500 nM for 6 h (C) or with 400 nM different EGFR-TKIs for 12 h (D). After the aforementioned treatments, whole-cell protein lysates were prepared from these cells and used for detection of different proteins with Western blotting. E and F, Whole-cell protein lysates were prepared from tumors in nude mice exposed to osimertinib for different times as labeled and then subjected to Western blot analysis for detection of the given proteins (E), which were further quantified (F). SE, shorter exposure. G and H, HCC827 cells were exposed to 100 nM osimertinib for 14 h and then harvested for preparation of total cellular RNA and subsequent RNA-seq analyses. Data are means ± SDs of triplicate samples.
Osimertinib facilitates c-Myc proteasomal degradation and suppresses c-Myc transcription to some extent.
RNA-seq analysis showed MYC mRNA expression was weakly, but significantly decreased (approximately 27%; Fig. 2A) in HCC827 cells treated with osimertinib for a prolonged time (14 h). This weak decrease (20–35%) was further confirmed in both PC-9 and HCC827 cells treated with osimertinib for a short time (4 h) using qRT-PCR (Fig. 2B), suggesting a degree of transcriptional regulation. Considering that c-Myc is an unstable protein subject to proteasomal degradation and the rapid effect of osimertinib on reduction of c-Myc, we further determined whether osimertinib enhances c-Myc proteasomal degradation. The presence of the proteasome inhibitor, MG132, not only elevated basal levels of c-Myc, but also rescued c-Myc reduction induced by osimertinib in the four tested EGFRm NSCLC cell lines (Fig. 2C). In the complementary CHX chase assay, c-Myc clearly decayed faster in osimertinib-treated HCC827 cells than in the control DMSO-treated cells (Fig. 2D). Similar results were also generated in PC-9 cells (Fig. 2D). These results clearly demonstrate that osimertinib prompts c-Myc degradation.
Figure 2. Osimertinib suppresses c-Myc transcription to some degree (A and B) and effectively induces c-Myc proteasomal degradation in EGFRm NSCLC cells (C and D) independent of GSK3 and FBXW7 (E–H).
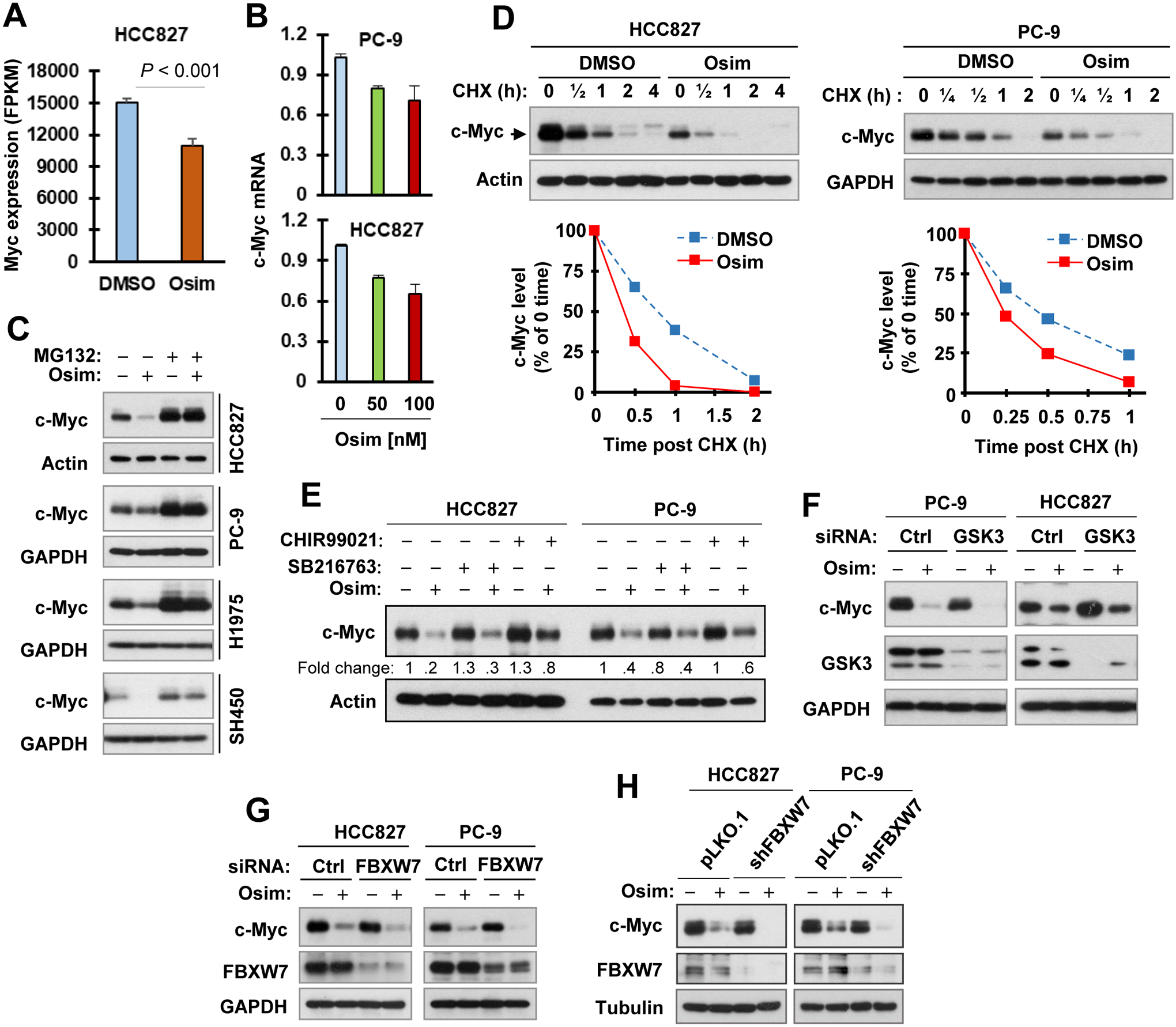
A, RNA-seq analysis of c-Myc mRNA regulation in HCC827 cells treated with 100 nM osimertinib (Osim) for 14 h. Data are means ± SDs of triplicate samples. B, Both PC-9 and HCC827 cells were exposed to the indicated concentrations of osimertinib for 4 h and then harvested for preparation of cellular total RNA and subsequent qRT-PCR. The data are means ± SE of triplicate experiments. C, The indicated EGFRm cell lines were pre-treated with 10 μM MG132 for 30 min and then co-treated with 100 nM osimertinib for 4 h. D, Both PC-9 and HCC827 cells were exposed to 100 nM (PC-9) or 200 nM (HCC827) osimertinib for 4 h followed by adding 10 μg/ml CHX to all dishes and then harvested at different times post CHX addition. Whole-cell protein lysates were then prepared from the aforementioned treated cells and used for detection of different proteins with Western blotting. c-Myc levels were plotted relative to those at time 0 of CHX treatment after being quantified by NIH Image J software and normalized to Actin or GAPDH. E, PC-9 and HCC827 cells were pretreated with 10 μM CHIR99021 or SB216763 for 30 min and then co-treated with 100 nM (PC-9) or 200 nM (HCC827) osimertinib for an additional 6 h. F and G, PC-9 and HCC827 cells were transfected with the given siRNAs for 48 h followed with treatment with 100 nM (PC-9) or 200 nM osimertinib (HCC827) for another 4 h. H, The indicated cell lines were treated with DMSO or 200 nM osimertinib for 4 h. The indicated proteins were detected with Western blotting.
Osimertinib induces GSK3- and FBXW7-independent c-Myc degradation, albeit with suppression of phosphorylation of Akt, GSK3 and c-Myc, in EGFRm NSCLC cells.
c-Myc is known to undergo GSK3-dependent and FBXW7-mediated protein degradation (30,31). Given the Akt/GSK3 axis is downstream of EGFR, it is plausible to speculate that osimertinib inhibits Akt, resulting in activation of GSK3 and subsequent GSK3-dependent, FBXW7-mediated c-Myc degradation. Indeed, osimertinib decreased the levels of p-Akt (S473) and p-GSK3 in both PC-9 and HCC827 cells starting early at 1 h or 2 h post treatment (Fig. S1), indicating suppression of Akt and activation of GSK3. Moreover, osimertinib also effectively decreased the levels of p-c-Myc (T58/S62) and p-c-Myc (S62) in these cell lines (Figs. 1A, 1B and S1). However, suppression of GSK3 with both small molecule inhibitors (e.g., SB216763 and CHIR99021) and siRNA-mediated GSK3 knockdown minimally protected c-Myc from reduction induced by osimertinib in both PC-9 and HCC827 cells (Figs. 2E and 2F). Moreover, knockdown of FBXW7 in these cell lines with both siRNA and shRNA also failed to rescue osimertinib-induced c-Myc reduction (Figs. 2G and 2H). Collectively, these findings suggest that osimertinib likely induces GSK3- and FBXW7-independent c-Myc reduction or degradation in EGFRm NSCLC cells.
c-Myc levels are elevated in EGFRm NSCLC cell lines with acquired osimertinib-resistance and in EGFRm NSCLC tissues relapsed from EGFR-TKI treatment.
In contrast to the sensitive EGFRm NSCLC cells, osimertinib even at 500 nM weakly reduced the levels of c-Myc in a few osimertinib-resistant cell lines (Fig. 3A). Interestingly, we noticed that osimertinib-resistant cell lines derived from different parental EGFRm NSCLC cell lines possessed elevated levels of c-Myc in comparison with their corresponding parental cell lines (Fig. 3B). IHC detection of c-Myc in 50 paired EGFRm NSCLC tissues pre and post relapse from treatment with EGFR-TKIs showed that c-Myc levels were elevated in more than half of the resistant cases (29/50, 58%; Fig. 3C), decreased in a small portion of the resistant cases (7/50, 14%; Fig. 3D) and remained unchanged in the rest of the resistant cases (14/50, 28%; Fig. 3E) in comparison with those in their corresponding baseline counterpart tissues (Figs. 3C–F). This analysis suggests that c-Myc levels have a high likelihood to be elevated in most EGFRm NSCLCs with acquired resistance to osimertinib and other EGFR-TKIs. By analyzing 40 cases with available PFS information, we found that patients with elevated c-Myc tended to have shorter times of PFS (2.1 months) than those with decreased c-Myc (Fig. S2), albeit not being statistically significant given the limited sample size.
Figure 3. c-Myc levels are minimally reduced by osimertinib in osimertinib-resistant EGFRm NSCLC cell lines (A) and are elevated in EGFRm NSCLC cell lines with acquired resistance to osimertinib (B) and in EGFRm NSCLC tissues from patients relapsed from EGFR-TKI treatment (C–F).
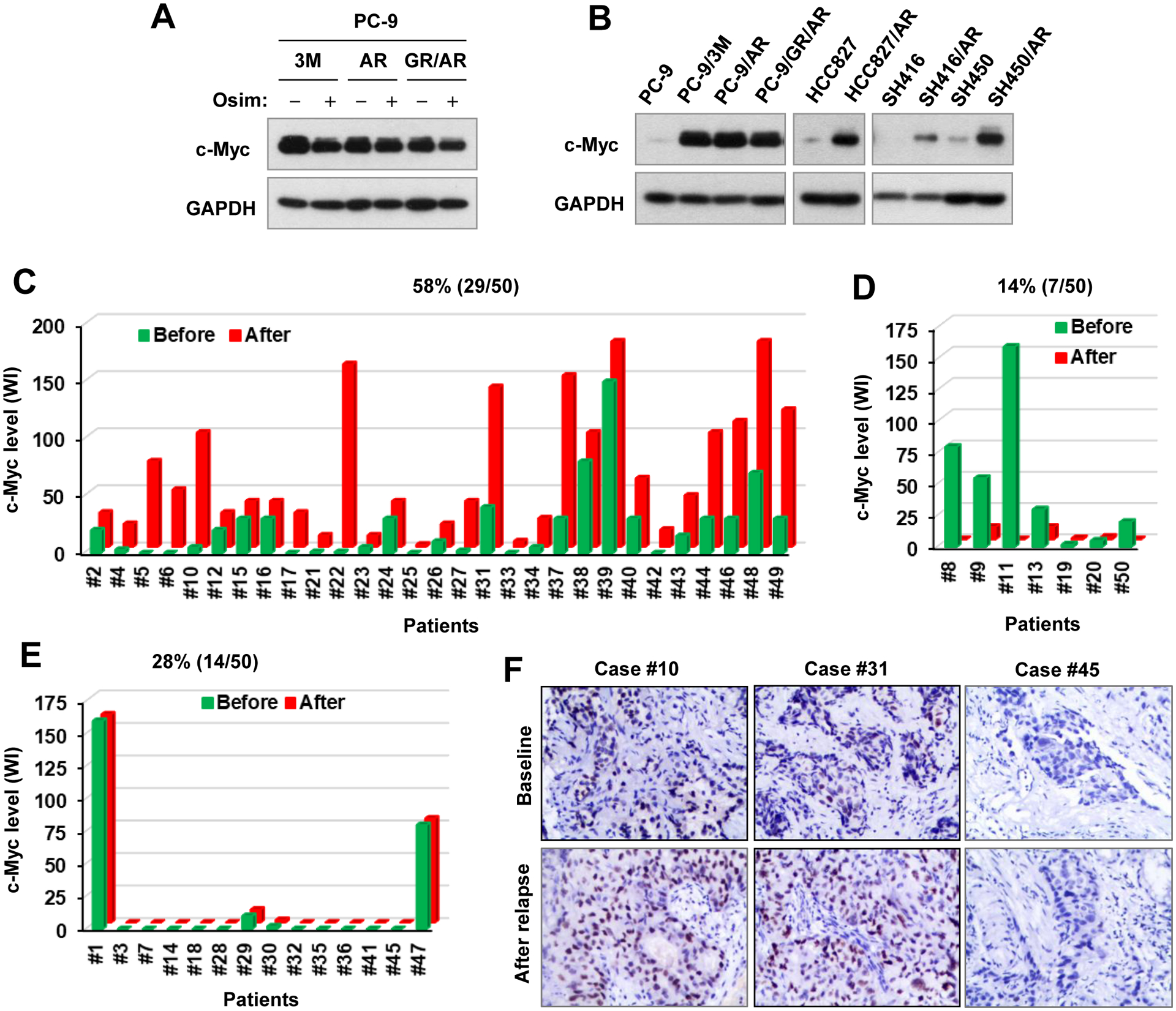
A and B, Whole-cell protein lysates were prepared from the given osimertinib-resistant cell lines exposed to 1000 nM osimertinib (Osim) for 6 h (A) or from untreated given cell lines with similar densities (B). The indicated proteins were detected with Western blotting. C–F, c-Myc in human NSCLC tissues before and after relapse from treatment using first generation EGFR-TKIs was stained with IHC.
MEK or ERK inhibition combined with osimertinib enhances c-Myc degradation with augmented induction of apoptosis.
We previously demonstrated that MEK inhibition with different MEK or inhibitors effectively restores the sensitivities of different osimertinib-resistant cell lines and tumors to osimertinib with enhanced induction of apoptosis (15,17). Although osimertinib exerted limited effects on decreasing c-Myc levels in different osimertinib-resistant cell lines as demonstrated above (also see Figs. 3A), the presence of the MEK inhibitor, trametinib, restored the ability of osimertinib to reduce c-Myc levels with enhanced PARP cleavage in osimertinib-resistant cell lines (Figs. 4A and B). c-Myc reduction induced by the combination of osimertinib and trametinib could be rescued by the presence of MG132 (Fig. 4C). Moreover, the combination of osimertinib and trametinib was more potent than either agent alone in facilitating c-Myc degradation as evaluated in the CHX assay (Fig. 4D). These data collectively suggest that the combination of osimertinib and trametinib enhances proteasomal degradation of c-Myc in osimertinib-resistant cells; this effect is correlated with augmented induction of apoptosis. In agreement, the combination of osimertinib with another MEK inhibitor named selumetinib or the ERK inhibitor, GDC0994 or VRT752271, exerted an enhanced effect on suppressing the phosphorylation of p90RSK, a known substrate of ERK1/2 and augmented the reduction of c-Myc and cleavage of PARP in both osimertinib-resistant cell lines (Fig. 4E), furthering the notion that suppression of the MEK/ERK signaling restores the response of osimertinib-resistant cells to undergo osimertinib-induced c-Myc reduction and apoptosis.
Figure 4. Osimertinib combined with trametinib or other MEK (A–D) or ERK inhibitors (E) enhances c-Myc reduction with enhanced PARP cleavage (A, B and E) via promoting c-Myc degradation (C and D) in osimertinib-resistant EGFRm NSCLC cells.
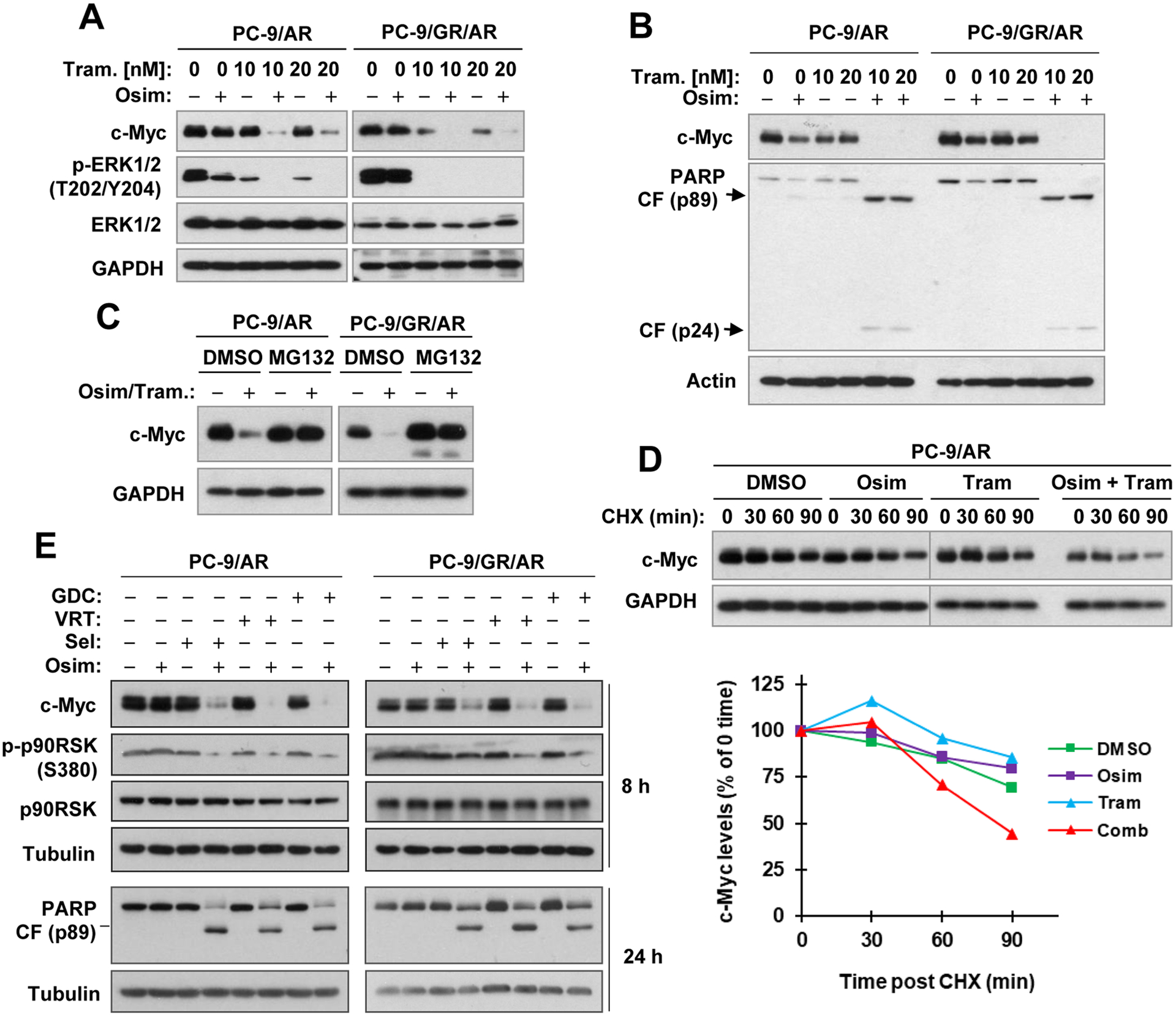
A and B, The tested cell lines were exposed to DMSO, 100 nM osimertinib (Osim), 10 nM or 20 nM trametinib (Tram) and the combinations of osimertinib and trametinib for 8 h (A) or 24 h (B). C, The indicated cell lines were pretreated with 10 μM MG132 and co-treated with DMSO or osimertinib (100 nM) plus trametinib (10 nM) for an additional 8 h. D and E, PC-9/AR cells were exposed to DMSO, 100 nM osimertinib, 10 nM trametinib or osimertinib plus trametinib for 2 h followed by adding 10 μg/ml CHX to all dishes and then harvested at different times post CHX addition. E. The indicated cell lines were treated with DMSO, 100 nM osimertinib, 100 nM selumetinib (Sel), 500 nM VRT752271 (VRT), 2 μM GDC0994 (GDC) and different combinations as indicated for 8 and 24 h respectively. After the aforementioned treatments, whole-cell protein lysates were prepared and used for detection of different proteins with Western blotting. c-Myc levels were plotted relative to those at time 0 of CHX treatment after being quantified by NIH Image J software and normalized to GAPDH (D).
Genetic manipulation of c-Myc levels alters the responses of EGFRm NSCLC cells to osimertinib.
The above findings of elevated c-Myc levels in osimertinib-resistant cell lines and enhanced effects of osimertinib combined with trametinib or other MEK or ERK inhibitor on reducing c-Myc levels and inducing apoptosis strongly suggest that c-Myc elevation may contribute to the development of acquired resistance to osimertinib. If so, enforced manipulation of c-Myc levels in EGFRm NSCLC cells should alter cell responses to osimertinib. To this end, we first enforced expression of ectopic c-Myc and c-Myc mutant (S62A) in the sensitive PC-9 cells and then tested their impact on cell responses to osimertinib. The expression of both c-Myc and particularly S62A mutant in part, but significantly, protected PC-9 cells from undergoing apoptosis upon osimertinib treatment in comparison with PC-9 vector control cells as evidenced by measuring PARP cleavage (Fig. 5A) and annexin V-positive cells (Fig. 5B). In agreement, the protective effects of both c-Myc and S62A mutant on osimertinib-induced cell killing were confirmed by measuring cell number reduction (Fig. 5C). Under the tested condition of prolonged treatment (48 h), ectopic c-Myc and S62A protein were still reduced in cells exposed to osimertinib, but the reduced levels were still higher than the endogenous c-Myc levels of the tested cell line (Fig. 1A). However, osimertinib, under the condition with a short time of treatment (e.g., 6 h), had minimal effect on decreasing the levels of c-Myc S62A mutant when compared with endogenous c-Myc or ectopic WT c-Myc (Fig. 5A). Next, we asked whether enforced c-Myc reduction in osimertinib-resistant cells may restore cell response to osimertinib. To test this speculation, we utilized c-Myc shRNAs to genetically knock down c-Myc expression in osimertinib-resistant cells and then examined their impact on cell response to osimertinib. Among four different c-Myc shRNAs tested, #2 and #3 c-Myc shRNAs effectively decreased c-Myc levels in different osimertinib-resistant cell lines (Fig. 5D). The cell lines expressing these c-Myc shRNAs in PC-9/AR and PC-9/GR/AR cells were significantly more sensitive than their corresponding control cell lines to osimertinib in terms of apoptosis induction as determined by detection of both annexin V-positive cells (Fig. 5E) and cleavage of caspase-3 and PARP (Fig. 5F) and cell number reduction (Fig. 5G). Similar results were also generated in HCC827/AR cells (Fig. S3A and B). We noted that osimertinib treatment in c-Myc shRNA-transfected cells further enhanced reduction of c-Myc (Fig. 5F).
Figure 5. Enforced expression of ectopic c-Myc partially protects sensitive EGFRm NCLC cells from undergoing osimertinib-induced apoptosis (A and B) and decrease in cell survival (C), whereas genetic knockdown of c-Myc in osimertinib-resistant cells (D) restores their sensitivities to osimertinib both in vitro (E–G) and in vivo (H and I).
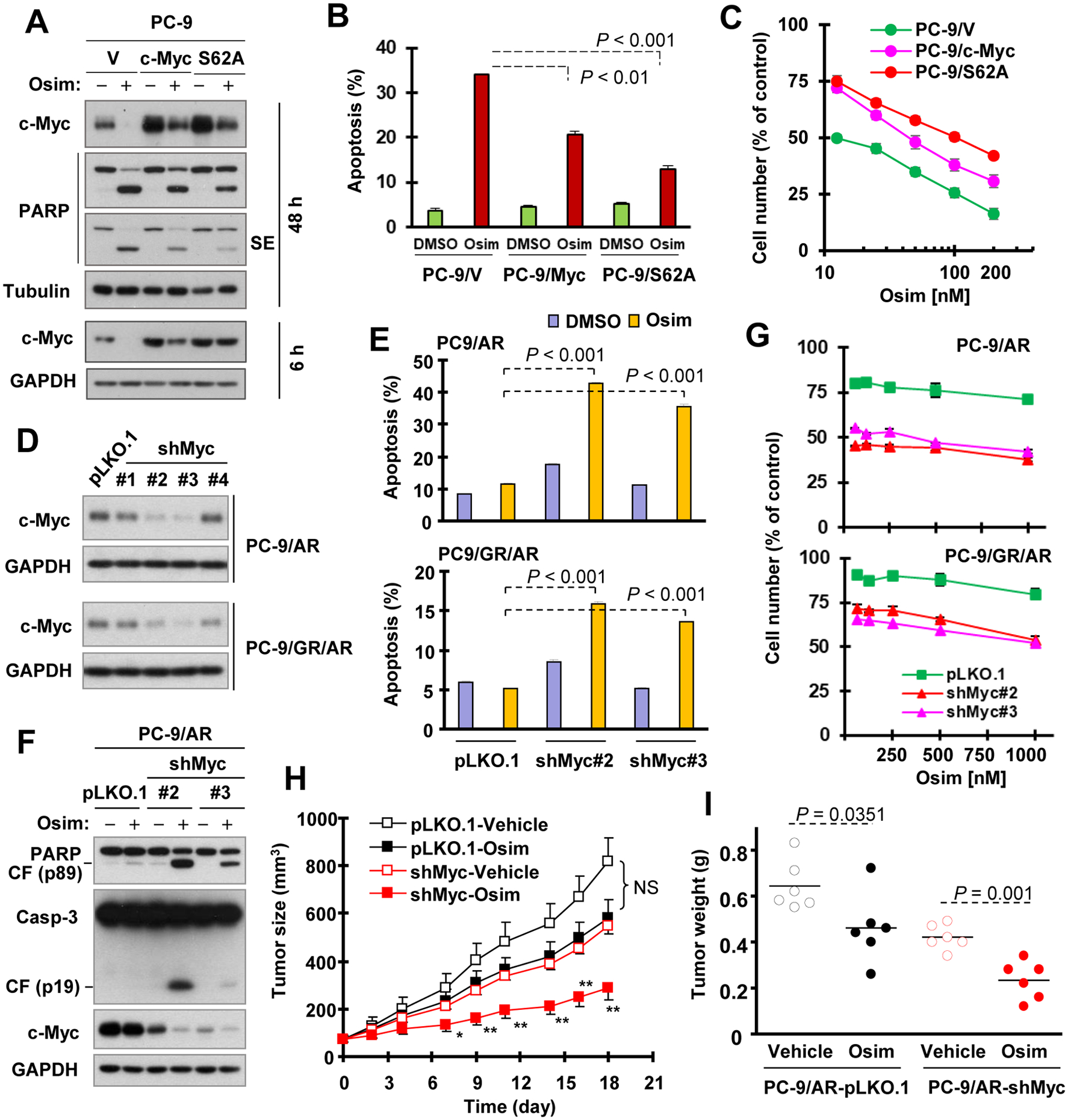
A and B. The indicated cell lines were exposed to DMSO or 100 nM osimertinib (Osim) for 48 h. C and G, The indicated cell lines in 96-well plates were exposed to the varied concentrations of osimertinib for 3 days. D, c-Myc knockdown in the given cell lines was confirmed with Western blotting. E and F, The indicated cell lines were exposed to 400 nM osimertinib for 72 h (E) or 48 h (F). After the aforementioned treatments, the cells were harvested for detection of the given proteins with Western blotting (A, D and F), of apoptosis using annexin V/Flow cytometry (B and E) or of cell number using the SRB assay (C and G). Each column is the mean ± SD of duplicate determinations (B and E). Data in C and G are means ± SDs of four replicate determinations. H and I. PC-9/AR-pLKO.1 and PC-9/AR-shMyc #2 xenografts in nude mice were treated with vehicle or 5 mg/kg osimertinib (daily, og) for the indicated days. The data in each group are means ± SEs of tumors from 6 mice. NS, not significant at any days measured; *, P < 0.05; **, P < 0.01 compared with shMyc-Vehicle.
Following the in vitro studies, we further determined whether c-Myc knockdown exerts similar effects on tumor response to osimertinib in vivo. We noted that PC-9/AR-shMyc#2 xenografts in nude mice grew slower than PC-9/AR-pLKO.1 tumors. Treatment of these xenografts with osimertinib for a period of 18 days showed that osimertinib significantly inhibited the growth of PC-9/AR-shMyc#2 tumors by measuring both tumor sizes and weights, whereas it had limited suppressive effect on the growth of PC-9/AR-pLKO.1 tumors (Figs. 5H and I). Under the tested conditions, there was no difference in mouse body weights between control and osimertinib-treated mice (Fig. S4). Hence, it is clear that enforced manipulation of c-Myc levels in EGFRm NSCLC cells through different genetic approaches alters cell responses to osimertinib.
Targeting BET synergizes with osimertinib in decreasing the survival and enhancing apoptosis of osimertinib-resistant cells and in suppressing the growth of osimertinib-resistant tumors in vivo.
Considering the translation of our findings into clinical practice, we sought to target c-Myc with small molecule inhibitors to overcome osimertinib acquired resistance. Among the potential compounds with c-Myc inhibitory activity we screened, including BET inhibitors, we found that the BET inhibitor JQ1, when combined with osimertinib, synergistically decreased the survival of osimertinib-resistant cells (e.g., PC-9/AR and PC-9/GR/AR; Fig. 6A). Similarly, the BET inhibitor OTX015, when combined with osimertinib, also synergistically decreased the survival of these osimertinib-resistant cell lines (Fig. S5A). The combination of JQ1 and osimertinib clearly enhanced the induction of apoptosis in both PC-9/AR and PC-9/GR/AR cell lines since each agent alone induced minimal or no apoptosis, as evidenced by enhancing annexin V-positive cells (Fig. 6B) and cleavage of PARP, caspase-3 and caspase-8 (Fig. 6C). In addition to the BET inhibitors, the BET degraders, ZBC260 (Fig. 6D) and dBET (Fig. S5B), also exerted similar effects in synergistically decreasing the survival of osimertinib resistant cells when combined with osimertinib. Similar results were also generated in HCC827/AR with these BET inhibitors or degraders (Fig. S6). Beyond these in vitro effects, the combination of osimertinib with JQ1 or ZBC260 significantly inhibited the growth of PC-9/AR xenograft tumors, whereas each agent alone had limited or no inhibitory effect under the tested conditions (Figs. 6E and 6F). The combination of osimertinib and JQ1 had comparable effects to vehicle on mouse body weights (Fig. 6G). The combination of osimertinib and ZBC260 slightly decreased mouse body weights in the initial treatment times when ZBC260 was given via iv injection for the first week; however, this suppression was eventually recovered at the end of treatment after ZBC260 was changed to ip administration (Fig. 6G). Therefore, BET inhibition combined with osimertinib is well tolerated in mice with significant activity against the growth of osimertinib-resistant tumors.
Figure 6. BET inhibition combined with osimertinib synergistically decreases survival (A and D) and enhances apoptosis in osimertinib-resistance cells (B and C) and augments suppressive effects against osimertinib-resistant tumors in nude mice (E–G).

A and D, The given cell lines were exposed to varied concentrations of JQ1 or ZBC260 alone, osimertinib (Osim) alone or the combinations of osimertinib with JQ1 or ZBC260 as indicated for 3 days. Cell numbers were then estimated with the SRB assay. The data are means ± SDs of four replicates. Numbers inside the graphs are CIs. B and C, The indicated cell lines were exposed to 200 nM osimertinib alone, 2 μM (PC-9/AR) or 5 μM (PC-9/GR/AR) JQ1 alone or their combination for 72 h (B) and then harvested for detection of the given proteins (C) or apoptosis using annexin V/Flow cytometry (B). Each column is the mean ± SD of duplicate determinations (B). E–G, PC-9/AR tumors in nude mice (6 mice/group) were treated with vehicle, osimertinib alone, JQ1 alone, ZBC260 alone or the combination of osimertinib with JQ1 or ZBC260. ZBC260 was given iv for the first week and then switched to ip administration. The data are presented as means ± SEs of 6 tumors. CF, Cleaved fragment; ZBC, ZBC260.
BET inhibition enhances c-Myc reduction and Bim-dependent induction of apoptosis in osimertinib-resistant NSCLC cells.
In the tested osimertinib-resistant cell lines, JQ1 effectively decreased c-Myc levels and further reduced c-Myc levels when combined with osimertinib (Fig. 7A), indicating that JQ1 indeed inhibits c-Myc expression in osimertinib-resistant cell lines. We then determined the mechanism by which c-Myc suppression combined with osimertinib enhances apoptosis in osimertinib-resistant cell lines. We examined the effects of JQ1 and osimertinib combination on modulation of Bim and Mcl-1 in osimertinib-resistant cell lines, a key mechanism for osimertinib to induce apoptosis in sensitive EGFRm NSCLC cells as we recently demonstrated (15). Interestingly, the combination enhanced elevation of not only Bim, but also Mcl-1, in both PC-9/AR and PC-9/GR/AR cells in comparison with each agent alone (Fig. 7A). Consistently, osimertinib elevated the levels of Bim and Mcl-1 in both PC-9/AR and PC-9/GR/AR cells expressing c-Myc shRNAs, but not in their corresponding control cell lines (Fig. 7B). In xenograft tissues, the highest levels of Mcl-1 and particularly Bim were detected both in tumors receiving the combined treatment of osimertinib and JQ1 and in xenografts expressing c-Myc shRNA with osimertinib treatment in comparison with other groups (Fig. 7C). These results together suggest that elevation of Bim and Mcl-1 by JQ1 and osimertinib combination is very likely to be associated with c-Myc inhibition.
Figure 7. Osimertinib, when combined with BET inhibition or c-Myc knockdown, enhances elevation of Bim and Mcl-1 levels in osimertinib-resistant cells (A and B) and in tumors (C) and Bim knockout attenuates enhanced induction of apoptosis (D and E) and decrease in cell survival (F) caused by osimertinib and JQ1 combination.
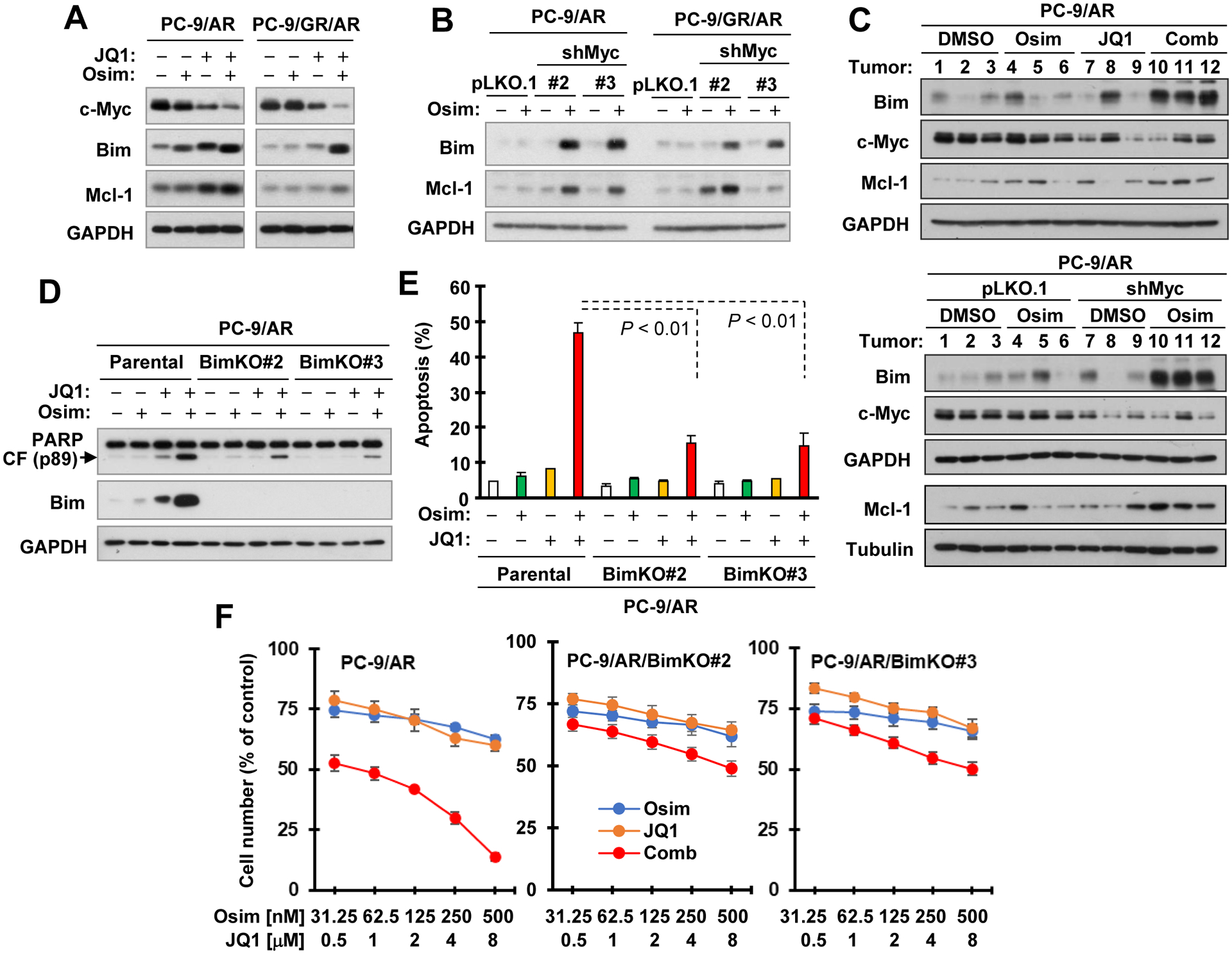
A and B, Whole-cell protein lysates were prepared from the indicated cell lines exposed to DMSO, 200 nM osimertinib (Osim), 2 μM JQ1 or osimertinib plus JQ1 for 16 h (A) or treated with 400 nM osimertinib for 72 h (B) and then used for detection of the given proteins with Western blotting. C, Tissue lysates were prepared from 3 tumors of each group as depicted in Figs. 5 and 6 and applied to detection of the indicated proteins with Western blotting. D and E, The indicated cell lines were exposed to DMSO, 200 nM osimertinib, 2 μM JQ1 or osimertinib plus JQ1 for 72 h. Apoptosis was evaluated with annexin V/flow cytometry and expressed as means ± SDs of duplicate determinations (E). The given proteins were detected with Western blotting (D). CF, cleaved fragment. F, The given cell lines in 96-well plates were treated with varied concentrations of osimertinib or JQ1 alone as indicated and the respective combinations of osimertinib and JQ1 for 3 days. Cell numbers were estimated with the SRB assay and data were presented as means ± SDs of four replicate determinations.
We further determined whether Bim elevation plays a critical role in mediating enhanced induction of apoptosis by the combination of JQ1 and osimertinib. We found that Bim knockout in PC-9/AR cells (Fig. 7D) significantly attenuated the ability of the combination to enhance PARP cleavage (Fig. 7D) and annexin V-positive cells (Fig. 7E). Consistently, the combinational effects of osimertinib and JQ1 on decreasing survival of PC-9/AR cell were substantially compromised in PC-9/AR Bim KO cells (Fig. 7F). Hence, the findings suggest that the combination of JQ1 and osimertinib enhances Bim-dependent induction of apoptosis and cell-killing in osimertinib-resistant cells.
Discussion
The current study reports an interesting and novel finding that osimertinib and other EGFR-TKIs rapidly and potently decreased the levels of c-Myc primarily in EGFRm NSCLC cell lines and in EGFRm NSCLC xenograft tumors. The c-Myc reduction occurred early at 2 h in cell lines and after 1 day in xenografts post osimertinib treatment, suggesting a very early event ahead of the induction of apoptosis and tumor suppression. Therefore, c-Myc reduction can be an early sign or marker predicting tumor response to osimertinib or other EGFR-TKI treatment.
Our results have clearly shown that osimertinib decreases c-Myc levels primarily via enhancing protein degradation and suppressing gene transcription to some degree. However, we have not been able to demonstrate the mechanism accounting for c-Myc degradation induced by osimertinib. GSK3-depenent phosphorylation and FBXW3-mediated ubiquitination is a well-known key mechanism accounting for c-Myc degradation (30,31). c-Myc phosphorylation at T58 and S62 affects c-Myc degradation and stability. In general, MAPK/ERK phosphorylates c-Myc at S62, resulting in protein stabilization, whereas GSK3β phosphorylates c-Myc at T58, promoting ubiquitin-dependent c-MYC degradation once S62 is dephosphorylated (32,33). In this study, we did find that osimertinib suppressed phosphorylation of Akt and GSK3, resulting in GSK3 activation. We also observed suppression of c-Myc phosphorylation at S62 and S62/T58. However, inhibition of GSK3 with both small molecule inhibitors and siRNA and knockdown of FBXW7 failed to rescue c-Myc reduction induced by osimertinib, suggesting a GSK3- and FBXW7-independent degradation mechanism. ERK-dependent phosphorylation of c-Myc at S62 has been linked to stabilization of c-Myc (34). Since osimertinib potently inhibits MEK/ERK signaling (15,35), it is likely that c-Myc S62 suppression plays a dominant role in triggering c-Myc degradation. Our finding of enhanced c-Myc degradation accompanied with augmented reduction of c-Myc levels in osimertinib-resistant cell lines treated with the combination of osimertinib and trametinib strongly supports this likelihood. The result on partial resistance of c-Myc S62A mutant to osimertinib also supports this mechanism.
Beyond FBXW7, many other E3 ubiquitin ligases are reported to mediate c-Myc ubiquitination and proteasomal degradation, including HUWE1 (36), TRIM32 (37), SPOP (38,39), FBXL14 (40,41), FBXL3 (41), ELL (42), SKP2 (43–45), FBXO32 (46) and CHIP (47). Moreover, some deubiquitinases also regulate c-Myc degradation (37,40,48). Therefore, future work will focus on identifying the E3 ubiquitin ligase or deubiquitinase that mediates osimertinib-induced c-Myc degradation.
There is a rebound upregulation of c-Myc in osimertinib-resistant EGFRm NSCLC cell lines and most EGFR-TKI-resistant EGFRm NSCLC tissues, which was also resistant to osimertinib modulation. Pilot analysis of tumor tissues from a small size of patients revealed a trend for patients with elevated c-Myc post relapse from EGFR-TKI treatment to have shorter PFS (over 2 months) than those whose tumors had reduced c-Myc, although the difference was not statistically significant. Further validation of this finding with a larger size of patient samples is warranted. MEK inhibition combined with osimertinib enhanced apoptosis (e.g., PARP cleavage) accompanied with augmented reduction of c-Myc in osimertinib-resistant cell lines. Moreover, enforced suppression of c-Myc in osimertinib-resistant NSCLC cells and tumors through gene knockdown restored cell and tumor response to osimertinib. These findings suggest that rebound upregulation of c-Myc likely plays a critical role in mediating the development of acquired resistance to osimertinib. This is supported by our data that enforced expression of ectopic c-Myc in sensitive EGFRm NSCLC cells conferred partial resistance to osimertinib-induced apoptosis. Therefore, c-Myc elevation may be a potential biomarker predicting treatment relapse. Additionally, c-Myc may also serve as a potential therapeutic target for overcoming osimertinib acquired resistance. We noted that the majority of cases (79%; 11/14) among those in which c-Myc expression was unchanged after relapse from EGFR-TKI treatment had undetectable baseline expression of c-Myc (Fig. 3E). It is very likely that c-Myc may play a minimal role in regulation of cell response and resistance to EGFR-TKi treatment in this type of tumor.
Although c-Myc inhibition would be a powerful cancer therapeutic approach, direct targeting of c-Myc has been a challenge for decades owing to its “undruggable” protein structure and nuclear location. Accordingly, alternatives to indirectly target Myc have been widely explored to achieve desirable anti-tumor effects, including Myc/Max complex disruption, MYC transcription and/or translation inhibition, and Myc destabilization as well as the synthetic lethality associated with c-Myc overexpression (5,29). The current study has identified BET inhibition with either a BET inhibitor (e.g., JQ1) or degrader (e.g., ZBC260) as an effectively strategy to overcome osimertinib acquired resistance evidenced by enhanced induction of apoptosis in vitro and augmented tumor suppressive effects in vivo. We have realized that BET inhibition does not exclusively inhibit c-Myc. Considering the fact that JQ1 effectively decreased c-Myc levels in osimertinib-resistant cells (Fig.7A) and c-Myc knockdown restored the responses of osimertinib-resistant cells and tumors to osimertinib (Fig. 5) as demonstrated in this study, it is fair to assume that c-Myc suppression at least contributes to the effects of BET inhibition on overcoming osimertinib acquired resistance. The involvement of other mechanisms beyond c-Myc inhibition in the enhanced therapeutic efficacies against osimertinib-resistant cells and tumors by the combination of osimertinib with BET inhibition cannot be excluded. Currently, there are several BET inhibitors or degraders tested in clinical trials (49,50); our findings warrant the clinical validation of BET inhibition as a novel strategy for overcoming acquired resistance to osimertinib and possibly other EGFR-TKIs, particularly those with elevated c-Myc.
In summary, the current study has demonstrated a previously unrevealed finding that osimertinib and other EGFR-TKIs effectively downregulate the levels of c-Myc, a well-known oncogenic protein, primarily though enhancing its degradation in EGFRm NSCLC cells and tumors. This modulation critically contributes to the therapeutic efficacy of osimertinib and likely other EGFR-TKIs against EGFRm NSCLCs. Moreover, loss of this modulation and late stage rebound upregulation of c-Myc is tightly associated with the development of osimertinib acquired resistance. Accordingly, targeting c-Myc either directly or indirectly is likely to be an effective strategy for overcoming acquired resistance to osimertinib. Toward this direction, our findings warrant the clinical testing of BET inhibition as a potential strategy to overcome acquired resistance to osimertinib or other EGFR inhibitors.
Supplementary Material
Significance.
This study demonstrates a critical role of c-Myc modulation in mediating therapeutic efficacy of osimertinib including osimertinib acquired resistance and suggests targeting c-Myc as a potential strategy to overcome osimertinib acquired resistance.
Acknowledgement
We are grateful to Dr. Shaomeng Wang at University of Michigan (Michigan, MI, USA) for providing ZBC260, Dr. Christine M. Lovly at Vanderbilt University School of Medicine (Nashville, TN, USA) for providing some cell lines, Dr. Junran Zhang at The Ohio State University James Comprehensive Cancer Center and College of Medicine (Columbus, OH, USA) and Dr. Anthea Hammond in our department for editing the manuscript.
Grant Support:
NIH/NCI R01 CA223220 (to SYS), R01 CA245386 (to SYS) and UG1 CA233259 (to SSR) and Emory Winship Cancer Institute lung cancer research pilot funds (to SYS).
Footnotes
Disclosure of potential conflicts of interest:
SSR is on consulting/advisory board for AstraZeneca, BMS, Merck, Roche, Tesaro and Amgen. TKO is on consulting/advisory board for Novartis, Celgene, Lilly, Sandoz, Abbvie, Eisai, Takeda, Bristol-Myers Squibb, MedImmune, Amgen, AstraZeneca and Boehringer Ingelheim. No potential conflicts of interest were disclosed for other people.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians 2018;68:394–424 [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA: a cancer journal for clinicians 2020;70:7–30 [DOI] [PubMed] [Google Scholar]
- 3.Ramalingam SS, Vansteenkiste J, Planchard D, Cho BC, Gray JE, Ohe Y, et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N Engl J Med 2020;382:41–50 [DOI] [PubMed] [Google Scholar]
- 4.Ramalingam SS, Yang JC, Lee CK, Kurata T, Kim DW, John T, et al. Osimertinib As First-Line Treatment of EGFR Mutation-Positive Advanced Non-Small-Cell Lung Cancer. J Clin Oncol 2017:JCO2017747576 [DOI] [PubMed] [Google Scholar]
- 5.Chen H, Liu H, Qing G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal transduction and targeted therapy 2018;3:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trop-Steinberg S, Azar Y. Is Myc an Important Biomarker? Myc Expression in Immune Disorders and Cancer. The American journal of the medical sciences 2018;355:67–75 [DOI] [PubMed] [Google Scholar]
- 7.Gnanaprakasam JN, Wang R. MYC in Regulating Immunity: Metabolism and Beyond. Genes 2017;8:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Casey SC, Baylot V, Felsher DW. MYC: Master Regulator of Immune Privilege. Trends in immunology 2017;38:298–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blakely CM, Watkins TBK, Wu W, Gini B, Chabon JJ, McCoach CE, et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nature genetics 2017;49:1693–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhong J, Li L, Wang Z, Bai H, Gai F, Duan J, et al. Potential Resistance Mechanisms Revealed by Targeted Sequencing from Lung Adenocarcinoma Patients with Primary Resistance to Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitors (TKIs). Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 2017;12:1766–78 [DOI] [PubMed] [Google Scholar]
- 11.Jin Y, Shi X, Zhao J, He Q, Chen M, Yan J, et al. Mechanisms of primary resistance to EGFR targeted therapy in advanced lung adenocarcinomas. Lung Cancer 2018;124:110–6 [DOI] [PubMed] [Google Scholar]
- 12.Chang N, Duan J, Wang L, Dong Z, Liu Z. Patients with advanced non-small cell lung cancer with EGFR mutations in addition to complex mutations treated with osimertinib have a poor clinical outcome: A real-world data analysis. Oncol Lett 2020;20:2266–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bordi P, Del Re M, Minari R, Rofi E, Buti S, Restante G, et al. From the beginning to resistance: Study of plasma monitoring and resistance mechanisms in a cohort of patients treated with osimertinib for advanced T790M-positive NSCLC. Lung Cancer 2019;131:78–85 [DOI] [PubMed] [Google Scholar]
- 14.Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell 2014;54:728–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi P, Oh YT, Deng L, Zhang G, Qian G, Zhang S, et al. Overcoming Acquired Resistance to AZD9291, A Third-Generation EGFR Inhibitor, through Modulation of MEK/ERK-Dependent Bim and Mcl-1 Degradation. Clin Cancer Res 2017;23:6567–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi P, Oh YT, Zhang G, Yao W, Yue P, Li Y, et al. Met gene amplification and protein hyperactivation is a mechanism of resistance to both first and third generation EGFR inhibitors in lung cancer treatment. Cancer Lett 2016;380:494–504 [DOI] [PubMed] [Google Scholar]
- 17.Li Y, Zang H, Qian G, Owonikoko TK, Ramalingam SR, Sun SY. ERK inhibition effectively overcomes acquired resistance of epidermal growth factor receptor-mutant non-small cell lung cancer cells to osimertinib. Cancer 2020;126:1339–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qian G, Yao W, Zhang S, Bajpai R, Hall WD, Shanmugam M, et al. Co-inhibition of BET and proteasome enhances ER stress and Bim-dependent apoptosis with augmented cancer therapeutic efficacy. Cancer Lett 2018;435:44–54 [DOI] [PubMed] [Google Scholar]
- 19.Zong D, Gu J, Cavalcante GC, Yao W, Zhang G, Wang S, et al. BRD4 levels determine the response of human lung cancer cells to BET degraders that potently induce apoptosis through suppression of Mcl-1. Cancer Res 2020;80:2380–93 [DOI] [PubMed] [Google Scholar]
- 20.Yao W, Oh YT, Deng J, Yue P, Deng L, Huang H, et al. Expression of Death Receptor 4 Is Positively Regulated by MEK/ERK/AP-1 Signaling and Suppressed upon MEK Inhibition. J Biol Chem 2016;291:21694–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oh YT, Liu X, Yue P, Kang S, Chen J, Taunton J, et al. ERK/ribosomal S6 kinase (RSK) signaling positively regulates death receptor 5 expression through co-activation of CHOP and Elk1. J Biol Chem 2010;285:41310–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zang H, Qian G, Zong D, Fan S, Owonikoko TK, Ramalingam SS, et al. Overcoming acquired resistance of epidermal growth factor receptor-mutant non-small cell lung cancer cells to osimertinib by combining osimertinib with the histone deacetylase inhibitor panobinostat (LBH589). Cancer 2020;126:2024–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qiu Z, Fa P, Liu T, Prasad CB, Ma S, Hong Z, et al. A Genome-Wide Pooled shRNA Screen Identifies PPP2R2A as a Predictive Biomarker for the Response to ATR and CHK1 Inhibitors. Cancer Res 2020;80:3305–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun SY, Yue P, Dawson MI, Shroot B, Michel S, Lamph WW, et al. Differential effects of synthetic nuclear retinoid receptor-selective retinoids on the growth of human non-small cell lung carcinoma cells. Cancer Res 1997;57:4931–9 [PubMed] [Google Scholar]
- 25.Liu X, Yue P, Zhou Z, Khuri FR, Sun SY. Death receptor regulation and celecoxib-induced apoptosis in human lung cancer cells. J Natl Cancer Inst 2004;96:1769–80 [DOI] [PubMed] [Google Scholar]
- 26.Ren H, Koo J, Guan B, Yue P, Deng X, Chen M, et al. The E3 ubiquitin ligases beta-TrCP and FBXW7 cooperatively mediates GSK3-dependent Mcl-1 degradation induced by the Akt inhibitor API-1, resulting in apoptosis. Mol Cancer 2013;12:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fan S, Ramalingam SS, Kauh J, Xu Z, Khuri FR, Sun SY. Phosphorylated eukaryotic translation initiation factor 4 (eIF4E) is elevated in human cancer tissues. Cancer Biol Ther 2009;8:1463–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol 1999;19:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dang CV. MYC on the path to cancer. Cell 2012;149:22–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, et al. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci U S A 2004;101:9085–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, et al. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J 2004;23:2116–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gregory MA, Qi Y, Hann SR. Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. J Biol Chem 2003;278:51606–12 [DOI] [PubMed] [Google Scholar]
- 33.Amati B Myc degradation: dancing with ubiquitin ligases. Proc Natl Acad Sci U S A 2004;101:8843–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev 2000;14:2501–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gu J, Yang W, Shi P, Zhang G, Owonikoko TK, Ramalingam SR, et al. MEK or ERK inhibition effectively abrogates emergence of acquired osimertinib resistance in the treatment of EGFR-mutant lung cancers Cancer 2020;126:3788–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin TP, Li J, Li Q, Li X, Liu C, Zeng N, et al. R1 Regulates Prostate Tumor Growth and Progression By Transcriptional Suppression of the E3 Ligase HUWE1 to Stabilize c-Myc. Molecular cancer research : MCR 2018;16:1940–51 [DOI] [PubMed] [Google Scholar]
- 37.Nicklas S, Hillje AL, Okawa S, Rudolph IM, Collmann FM, van Wuellen T, et al. A complex of the ubiquitin ligase TRIM32 and the deubiquitinase USP7 balances the level of c-Myc ubiquitination and thereby determines neural stem cell fate specification. Cell Death Differ 2019;26:728–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geng C, Kaochar S, Li M, Rajapakshe K, Fiskus W, Dong J, et al. SPOP regulates prostate epithelial cell proliferation and promotes ubiquitination and turnover of c-MYC oncoprotein. Oncogene 2017;36:4767–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luo L, Tang H, Ling L, Li N, Jia X, Zhang Z, et al. LINC01638 lncRNA activates MTDH-Twist1 signaling by preventing SPOP-mediated c-Myc degradation in triple-negative breast cancer. Oncogene 2018 [DOI] [PubMed] [Google Scholar]
- 40.Fang X, Zhou W, Wu Q, Huang Z, Shi Y, Yang K, et al. Deubiquitinase USP13 maintains glioblastoma stem cells by antagonizing FBXL14-mediated Myc ubiquitination. J Exp Med 2017;214:245–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huber AL, Papp SJ, Chan AB, Henriksson E, Jordan SD, Kriebs A, et al. CRY2 and FBXL3 Cooperatively Degrade c-MYC. Mol Cell 2016;64:774–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen Y, Zhou C, Ji W, Mei Z, Hu B, Zhang W, et al. ELL targets c-Myc for proteasomal degradation and suppresses tumour growth. Nature communications 2016;7:11057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee S, Kim W, Ko C, Ryu WS. Hepatitis B virus X protein enhances Myc stability by inhibiting SCF(Skp2) ubiquitin E3 ligase-mediated Myc ubiquitination and contributes to oncogenesis. Oncogene 2016;35:1857–67 [DOI] [PubMed] [Google Scholar]
- 44.von der Lehr N, Johansson S, Wu S, Bahram F, Castell A, Cetinkaya C, et al. The F-box protein Skp2 participates in c-Myc proteosomal degradation and acts as a cofactor for c-Myc-regulated transcription. Mol Cell 2003;11:1189–200 [DOI] [PubMed] [Google Scholar]
- 45.Kim SY, Herbst A, Tworkowski KA, Salghetti SE, Tansey WP. Skp2 regulates Myc protein stability and activity. Mol Cell 2003;11:1177–88 [DOI] [PubMed] [Google Scholar]
- 46.Mei Z, Zhang D, Hu B, Wang J, Shen X, Xiao W. FBXO32 Targets c-Myc for Proteasomal Degradation and Inhibits c-Myc Activity. J Biol Chem 2015;290:16202–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paul I, Ahmed SF, Bhowmik A, Deb S, Ghosh MK. The ubiquitin ligase CHIP regulates c-Myc stability and transcriptional activity. Oncogene 2013;32:1284–95 [DOI] [PubMed] [Google Scholar]
- 48.Sun XX, He X, Yin L, Komada M, Sears RC, Dai MS. The nucleolar ubiquitin-specific protease USP36 deubiquitinates and stabilizes c-Myc. Proc Natl Acad Sci U S A 2015;112:3734–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stathis A, Bertoni F. BET Proteins as Targets for Anticancer Treatment. Cancer Discov 2018;8:24–36 [DOI] [PubMed] [Google Scholar]
- 50.Yang CY, Qin C, Bai L, Wang S. Small-molecule PROTAC degraders of the Bromodomain and Extra Terminal (BET) proteins - A review. Drug discovery today Technologies 2019;31:43–51 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.