

Abstract
Rationale:
Preeclampsia (PE) is a potentially life-threatening, placenta-based hypertensive disorder during pregnancy, and the antiphospholipid syndrome (APS) frequently leads to PE. APS pregnancies are also complicated by fetal demise and intrauterine growth restriction (IUGR).
Objective:
Here we determined how the circulating antiphospholipid antibodies (aPL) characteristic of APS alter placental trophoblast function to cause PE and also endanger the fetus.
Methods and Results:
Experiments were performed in mice, in cultured human trophoblasts, and in human placenta samples. Effects of aPL and IgG from healthy subjects were compared. Based on prior findings in culture, in vivo studies were done in mice deficient in apolipoprotein E receptor 2 (ApoER2) in trophoblasts. Endpoints in tissues and cells were determined by enzymatic assay, Q-PCR, ELISA or immunoblotting. Whereas in wild-type mice aPL caused maternal hypertension and proteinuria, fetal demise and IUGR, mice lacking trophoblast ApoER2 were protected. In culture aPL attenuated trophoblast proliferation and migration via an ApoER2-related protein complex comprised of the protein phosphatase PP2A, Dab2, and JIP4. Via trophoblast ApoER2 in mice and in culture, aPL stimulated PP2A activity, leading to MMP14 and HIF1α upregulation and increased soluble endoglin (sEng) production. HIF1α and sEng upregulation was related to PP2A desphosphorylation of PHD2. In mice PP2A inhibition prevented aPL-induced maternal hypertension and proteinuria, and fetal demise and IUGR. Placentas from APS patients displayed PP2A hyperactivation, PHD2 dephosphorylation and HIF1α upregulation, and these findings were generalizable to placentas of women with PE from causes other from APS.
Conclusions:
In APS pregnancies trophoblasts are the critical cell target of aPL, and via ApoER2-dependent PP2A activation, aPL cause PE through MMP14 upregulation and PHD2 dephosphorylation leading to HIF1α and sEng upregulation. Moreover, parallel processes may be operative in PE in non-APS patients. Interventions targeting PP2A may provide novel means to combat APS-related PE and PE unrelated to APS.
Subject Terms: Animal Models of Human Disease, Basic Science Research, Cell Signaling/Signal Transduction
Graphical Abstract
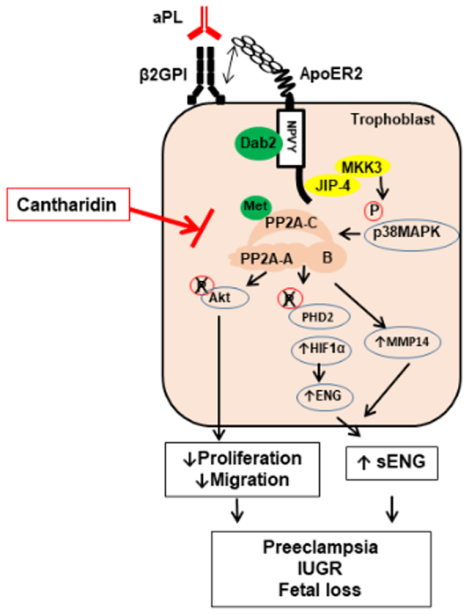
Preeclampsia (PE) is a leading cause of morbidity and mortality during pregnancy. The initiating processes in PE are poorly understood, and the only effective intervention is premature delivery, which threatens the well-being of the child. The Antiphospholipid Syndrome (APS), which is characterized by circulating antiphospholipid antibodies (aPL), is a common risk factor for PE, and also for fetal demise, premature birth and intrauterine growth restriction. This work presents an animal model of APS-related PE, revealing that aPL administration to pregnant mice induces PE and fetal compromise via ApoER2 in trophoblasts. Mechanistically, via ApoER2, aPL activate PP2A, which upregulates MMP14 and HIF1α, the latter via dephosphorylation of PHD2, leading to increased production of soluble endoglin, which is a known driver of PE. A role for PP2A in PE pathogenesis is revealed, and linkage of PP2A to PHD2 and HIF1α is demonstrated in placentas from both APS patients and women with PE from causes other than APS. As importantly, pharmacologic inhibition of PP2A affords full protection from both aPL-induced PE and the fetal complications. Future research should interrogate how PP2A is activated in PE not associated with APS, and how PP2A can be targeted to combat PE.
INTRODUCTION
Preeclampsia (PE), which is characterized by new-onset hypertension and proteinuria after 20 weeks gestation during pregnancy, is a leading cause of mortality and morbidity in mothers and fetuses that affects 5% to 7% of pregnant women worldwide1–3. PE is also associated with substantial increased risk of cardiovascular disease in the mother and child later in life4–6. One common risk factor for PE is the Antiphospholipid Syndrome (APS)7, 8, which is an autoimmune disorder characterized by the presence of circulating antiphospholipid antibodies (aPL) and elevated risk of thrombosis. In addition to PE, APS pregnancies are complicated by fetal loss, premature birth, and intrauterine growth restriction (IUGR)9–13. APS adversely impacts more than 10% of pregnancies, particularly in women with systemic lupus erythematosus (SLE)10, 14, and whereas 7% of normotensive pregnant women have detectable aPL, the antibodies are found in 29% of hypertensive pregnant women10. In addition, a recent prospective case-control study provided strong evidence for an association between the presence of aPL and early PE15, and a recent meta-analysis found that women with APS had the highest pooled event rate for PE among fourteen known risk factors including chronic hypertension, pregestational diabetes and obesity8. Thus, PE is a major health problem that places both the fetus and the mother in danger, and APS is a common underlying condition leading to PE and other pregnancy complications.
Although the adverse impacts of APS on pregnancy were initially believed to result from placental thrombosis, in most obstetric APS cases there is no evidence of placental thrombosis16–18. Other processes that have been considered are inflammation and complement activation in the placenta, particularly in response to aPL that recognize β2-glycoprotein I (β2GPI), which are the most pathogenic antibodies in APS9, 19. In vitro studies indicate that anti-β2GPI antibodies have direct detrimental impact on endothelial cells, monocytes and trophoblasts, which are all critical to placental development and function, and on platelets, which participate not only in thrombosis but also in inflammation and complement activation20–24. In culture the anti-β2GPI antibodies decrease trophoblast proliferation and migration, which are the hallmarks of the placental pathology observed in PE due to multiple primary conditions21. We previously found that the LDL receptor family member Apolipoprotein E receptor 2 (ApoER2, also known as LRP8) is abundantly expressed in human and mouse trophoblasts, and in cultured trophoblasts we determined that ApoER2 is required for aPL to impair cell growth and migration25. We also showed in the mouse model of maternal APS invoked by administration of aPL mid-pregnancy that fetal survival and bodyweight are protected by global deletion of ApoER225. However, whether aPL actions on trophoblasts underlie obstetric disorders in APS including PE, and the molecular mechanisms by which aPL disrupt normal trophoblast development and function in vivo leading to PE and other obstetric complications are entirely unknown.
Seeking to better understand the PE that complicates APS, experiments were designed in mice to identify the primary cellular target of aPL in the placenta participating in APS-related PE and also fetal demise and IUGR. Utilizing cell-specific gene silencing, we addressed the question whether the trophoblast is the critical cell target of aPL action, and whether ApoER2 in trophoblasts is the major driver of the maternal hypertension and proteinuria, and the fetal complications. Additional experiments were performed to 1) determine how ApoER2 mediates actions of aPL on trophoblasts leading to PE, 2) evaluate if the identified mechanisms are operative in human APS pregnancies, and 3) determine if the same processes may participate in PE in non-APS patients.
METHODS
Data Availability.
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Animal models of APS pregnancy.
In vivo studies were performed in wild-type C57BL/6 mice, or in littermate control mice (ApoER2fl/fl) or mice lacking ApoER2 specifically in trophoblasts (ApoER2ΔTR) on C57BL/6-FVB mixed background. The latter mouse strain was generated by crossing ApoER2fl/fl mice24 with mice expressing Cre recombinase under the regulation of Cyp19 promotor26. Virgin female mice (8–10 weeks old) were mated with males of the same genotypes, and vaginal plugs were checked (day 0). On day 8 and 12 of pregnancy, pregnant mice were randomly assigned to receive IP injection of NHIgG (10mg/mouse) or aPL (10mg/mouse), as previously described23, 25. On day 15, the mice were euthanized and the uteri were dissected, embryos were weighed and fetal resorption rate was calculated (number of resorptions/(number of live fetuses + number of resorptions) x100)(%)). The representative images were chosen as those that are most similar to all of the other images in the data set. Details are provided in Online Supplemental Materials and Methods.
Tail cuff systolic blood pressure (SBP) measurements were made using a Visitech System BP-2000 Series II (Apex, NC) BP monitor27, 28. Tail cuff SBP measurements were necessary to assess blood pressure because instrumentation for radiotelemetry was not feasible in pregnant mice at 8–10 weeks old in which the mouse model of aPL-induced pregnancy complications was established23, 25. Mice received tail cuff BP measurement training for 2 days (days 6 and 7 of pregnancy), undergoing 10 measurements over a 30-minute period each morning. Starting day 8 of pregnancy, daily the mean of the last 6 out of 16 readings was recorded as the SBP value for a given mouse. The experimenters were blinded by placing each mouse in a separate cage with no identifiers.
Human IgG and placenta preparation.
Normal human IgG (NHIgG) and aPL were isolated from healthy individuals and APS patients as previously described23, 25. Normal term placentas were obtained through the Ob-Gyn Tissue Procurement Facility at UT Southwestern Medical Center. Placentas from APS patients were obtained from UT Southwestern Medical Center. The relevant clinical and laboratory features of the APS patients and the individuals with PE unrelated to APS are provided in Online Supplemental Materials and Methods. Mouse monoclonal antibody directed against β2GPI (designated 3F8) and its isotype-matched control (designated BBG) were generated as previously described24, 25.
Cell culture, siRNA and adenoviral transfection.
Human trophoblast cell line HTR8/SVneo was maintained as described25. Additional human trophoblast cell line Sw.71 was kindly provided by Dr. Gil Mor (Yale University), and maintained as described previously29. In siRNA experiments, cells were transfected with the siRNAs (ThermoFisher) shown in Online Supplemental Materials and Methods using siPORT Amine transfection reagent as previously described24. Control siRNA was purchased from GE Dharmacon (ON-TARGETplus Non-targeting Control siRNA). For reconstitution experiments, cells were first transfected with siRNA targeting endogenous ApoER2, and subsequently the cells were transfected with adenoviral particles (1010 particles/ml) encoding ApoER2 constructs24. Twenty-four hours after adenoviral transfection, experiments were performed. Pharmacological inhibition studies were performed using HIF1α inhibitor (1μM, GN44028, TOCRIS), MMP14 inhibitor (10nM, NSC405020, Sigma-Aldrich), or p38 inhibitor SB 202190 (10 μM, Selleckchem) in the presence of NHIgG or aPL (100 μg/ml).
PP2A Phosphatase assay.
PP2A phosphatase activity was evaluated using PP2A immunoprecipitation and a phosphatase assay kit (EMD #17–313) as previously described24. In brief, trophoblasts were incubated with NHIgG or aPL (100μg/ml) for 2 h, or with C2-ceramide (30 μM) for 4 h to provide a positive control in select experiments. Mouse placentas were harvested on day 15 of pregnancy and tissues were stored at −80°C. Cell or tissue lysates were generated according to manufacturer’s instructions and immunoprecipitation was performed with anti-PP2A-C antibody immobilized on agarose beads for 2 h. Immunoprecipitated PP2A was incubated with phosphorylated peptide substrate for 20 min at 30°C. Malachite green reagent was then used to quantify sample free phosphate content.
Immunoprecipitation and immunoblot analysis.
HTR8/SVneo or Sw.71 cells were lysed and immunoprecipitation was performed as described in detail in Online Supplemental Materials and Methods. Immunoblot analyses were performed using appropriate primary antibodies. Detailed information regarding the antibodies used is provided in Online Supplemental Materials and Methods. All findings in cell culture were replicated in three independent experiments.
Mass spectrometry24.
Following protein separation by SDS PAGE, gel samples were digested overnight with trypsin (Pierce) followed by reduction and alkylation with dithiothreitol and iodoacetamide (Sigma–Aldrich). After solid-phase extraction cleanup with Oasis HLB plates (Waters), the samples were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) using an Orbitrap Fusion Lumos mass spectrometer (Thermo Electron) coupled to an Ultimate 3000 RSLC-Nano liquid chromatography system (Dionex). Raw MS data files were converted to a peak list format and analyzed using the central proteomics facilities pipeline (CPFP), Version 2.0.324, 30. Peptide identification was performed using the X! Tandem and Open MS Search Algorithm (OMSSA) search engines against a human protein database from Uniprot24, 31.
Statistical analysis.
With normally distributed data a two-way ANOVA with Tukey’s post hoc test was used for a dataset with two factors. In analyses of non-normally distributed data indicated by Shapiro-Wilk test a) a Mann-Whitney U test was used to compare two groups for a single factor, b) a Kruskal-Wallis test with Dunn’s post hoc test was used for datasets comparing three or more groups for a single factor, c) an aligned rank transform (ART) two-way ANOVA with Benjamini and Hochberg (BH) correction post hoc test was used for a dataset with two factors, d) an ART two-way mixed ANOVA with BH correction was used for a dataset with one between- subjects factor and one within-subjects factor, and an ART three-way mixed ANOVA with BH correction was used for a dataset with two between-subjects factors and one within-subjects factor. The statistical analysis was performed using the R Statistical Package, and the ART ANOVAs and BH post hoc tests were carried out with the R Statistical Package ARTool; the original non-normally distributed data first underwent aligned rank transformation, and then the ranked data was analyzed with the usual ANOVA procedure32–34. Values shown represent the mean ± SEM. In all graphs the specific comparisons made are shown by the horizontal lines below the P values provided, which are all corrected P values. A P value of less than 0.05 was considered statistically significant. No experiment-wide multiple test correction was applied.
Study Approval.
All animal experiments were approved by the Institutional Animal Care and Use Committees at the University of Texas Southwestern Medical Center. All human study protocols were approved by the Institutional Review Boards of the Hospital for Special Surgery and the University of Texas Southwestern Medical Center. Human subjects gave written informed consent before participating in these studies.
RESULTS
APL induce PE in a trophoblast ApoER2-dependent manner.
The placental pathology in APS is characterized by attenuated placentation, reduced decidual and vascular trophoblast invasion, and less spiral artery transformation12, 16, 17, 35, 36. To first determine if aPL impact trophoblasts in vivo, pregnant C57BL/6J mice received intraperitoneal (IP) injections of normal human IgG isolated from healthy subjects (NHIgG) or aPL (10mg/mouse) at day 8 of pregnancy, and the status of labyrinth trophoblasts and differentiating syncytiotrophoblasts was evaluated 48h later (Fig. 1A). We found that branching morphogenesis of the labyrinth was reduced by aPL, and that cell replication throughout the developing junctional zone and labyrinth layers was attenuated. Thus, mirroring findings in human placentas17, 35, trophoblast phenotype is disturbed in vivo in this murine model of APS during pregnancy.
Fig. 1.
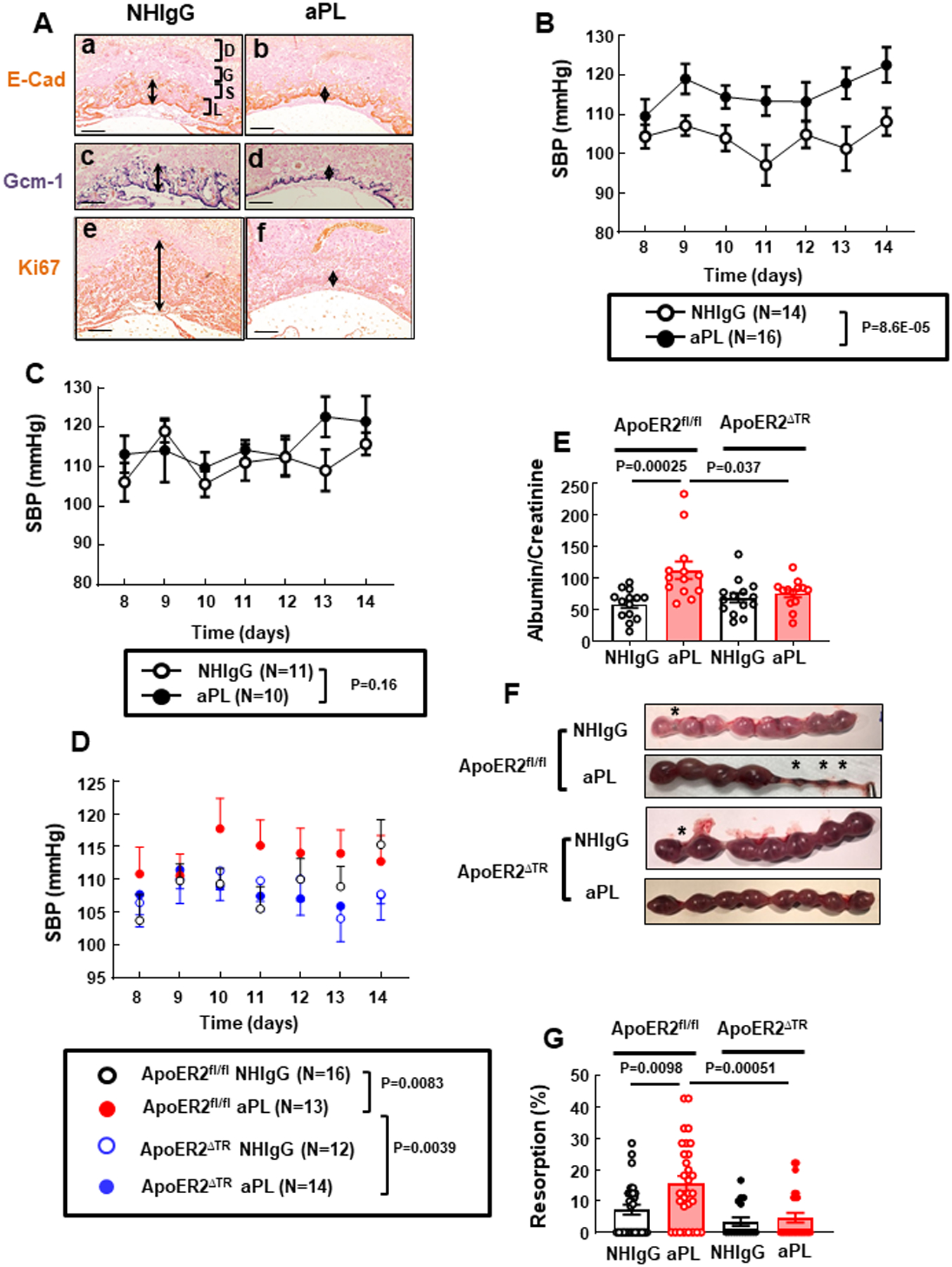
Mice lacking ApoER2 selectively in trophoblasts (ApoER2ΔTR) are protected from aPL-induced maternal hypertension and proteinuria, and fetal loss. (A) Pregnant C57BL/6 mice received NHIgG (a, c, e) or aPL (b, d, f) on day 8 of pregnancy, and 48h later uteri were harvested. Placental sections were immunostained with antibody against E-cadherin (a, b) and probed for Gcm-1 (c, d) using in situ hybridization, and proliferation was evaluated by Ki67 staining (e, f). D; Decidua, G; Giant cells, S; Spongiotrophoblasts, L; Labyrinth. Arrows indicate areas of positive staining. Representative images from N=3 mice/group in 2 independent experiments are shown. Scale bars = 100 μm. (B, C) Mated female C57BL/6 mice were injected with NHIgG or aPL on days 8 and 12 following evidence of a vaginal plug, and systolic blood pressure (SBP) was measured on days 8–14 in mice that were ultimately pregnant (B) or not pregnant (C). (D-G) Pregnant control ApoER2fl/fl mice or ApoER2ΔTR mice were injected with NHIgG or aPL as described in B and C, and SBP was monitored (D), or urine protein (E, N=14,13,14,13) and fetal resorption rates (F, G, N=28,30,17,26) were assessed at day 15. In F, asterisks indicate fetal resorption. Values are mean ± SEM. Statistical analyses were done by aligned rank transform two-way mixed ANOVA (B,C), by aligned rank transform three-way mixed ANOVA with Benjamini and Hochberg (BH) correction (D), and by aligned rank transform two-way ANOVA with BH correction (E, G).
We next tested whether aPL injections impact blood pressure (BP) in pregnant mice (Fig. 1B). Mice initially displaying vaginal plugs following mating (51 mice) were injected with NHIgG control (N=25) or aPL (N=26) 8 and 12 days later, and among them 30 were ultimately pregnant and 21 were not, and the latter mice served as non-pregnant controls. APL injections caused an increase in systolic blood pressure (SBP) in pregnant mice but not in non-pregnant mice (Fig. 1B, C), indicating that the hypertensive response to aPL requires pregnancy.
Having previously discovered that adverse actions of aPL in cultured trophoblasts require ApoER225, we next determined whether ApoER2 in trophoblasts is required for aPL-induced maternal hypertension. Compared to control ApoER2fl/fl mice, mice engineered to lack the receptor in trophoblasts, designated ApoER2ΔTR, displayed a 96% loss of ApoER2 mRNA in placenta (Online Fig. I A) and a parallel decline in trophoblast ApoER2 protein (Online Fig. I B). In contrast, in endothelial cells in which ApoER2 is also normally expressed, transcript level was unchanged (Online Fig. I C). In BP studies aPL induced an elevation in SBP and also relative proteinuria in ApoER2fl/fl mice but not in ApoER2ΔTR mice (Fig. 1D, E). In separate sets of mice, aPL caused a 2.2-fold increase in fetal resorption rate and a decline in the weight of surviving fetuses in ApoERfl/fl controls, but had no impact on these parameters in ApoER2ΔTR (Fig.1F, G and Online Table I). Thus, ApoER2 in trophoblasts mediates aPL-induced maternal hypertension and proteinuria, and fetal resorption and IUGR.
APL activation of PP2A underlies trophoblast dysfunction.
To delineate the molecular mechanisms whereby ApoER2 participates in aPL actions in trophoblasts, we interrogated the ApoER2 interactome in HTR8/SVneo human trophoblasts treated with NHIgG or aPL (OnlineTable II). Not surprisingly, β2GPI, the pathogenic antigen for aPL, was detected to interact with ApoER2 only in cells treated with aPL. In addition, the protein phosphatase 2A (PP2A) catalytic C subunit, and the PP2A scaffolding A subunit (designated Aα) displayed 5.6- and 4.1-fold greater interaction, respectively, with ApoER2 in aPL-treated cells. PP2A is a major protein phosphatase, and active PP2A is a heterotrimeric holoenzyme composed of catalytic (C), scaffold (A) and regulatory (B) subunits37, 38. Increased interaction between ApoER2 and the PP2A Aα subunit in response to aPL was confirmed by co-immunoprecipitation (Fig. 2A). We then showed that aPL from 4 different APS patients caused marked increases in PP2A activity in HTR8/SVneo cells (Fig. 2B), that the degree of activation with aPL was at least comparable to that observed with the known PP2A stimulant ceramide (Online Fig. II A), and that activation was also observed in a second human trophoblast cell line, Sw.71 (Online Fig. II B). In addition, comparable activation was induced by an aPL-mimicking monoclonal anti-β2GPI antibody, 3F8 (Fig. 2C)23, 25, providing evidence that aPL binding to β2GPI triggers the activation. Furthermore, injections of aPL but not NHIgG in pregnant mice resulted in PP2A activation in the placenta, indicating that the process occurs in vivo in mice (Fig. 2D). Moreover, in both HTR8/SVneo and Sw.71 cells, PP2A-Aα silencing prevented aPL attenuation of EGF-induced Akt activation (Fig. 2E, Online Fig. II C, D), which is a critical signaling event in normal trophoblast function25, 39–41. More importantly, whereas aPL blunted trophoblast proliferation and migration in response to serum in control cells, the knockdown of PP2A-Aα fully preserved normal proliferation and migration in aPL-treated cells (Fig. 2F, G).
Fig. 2.
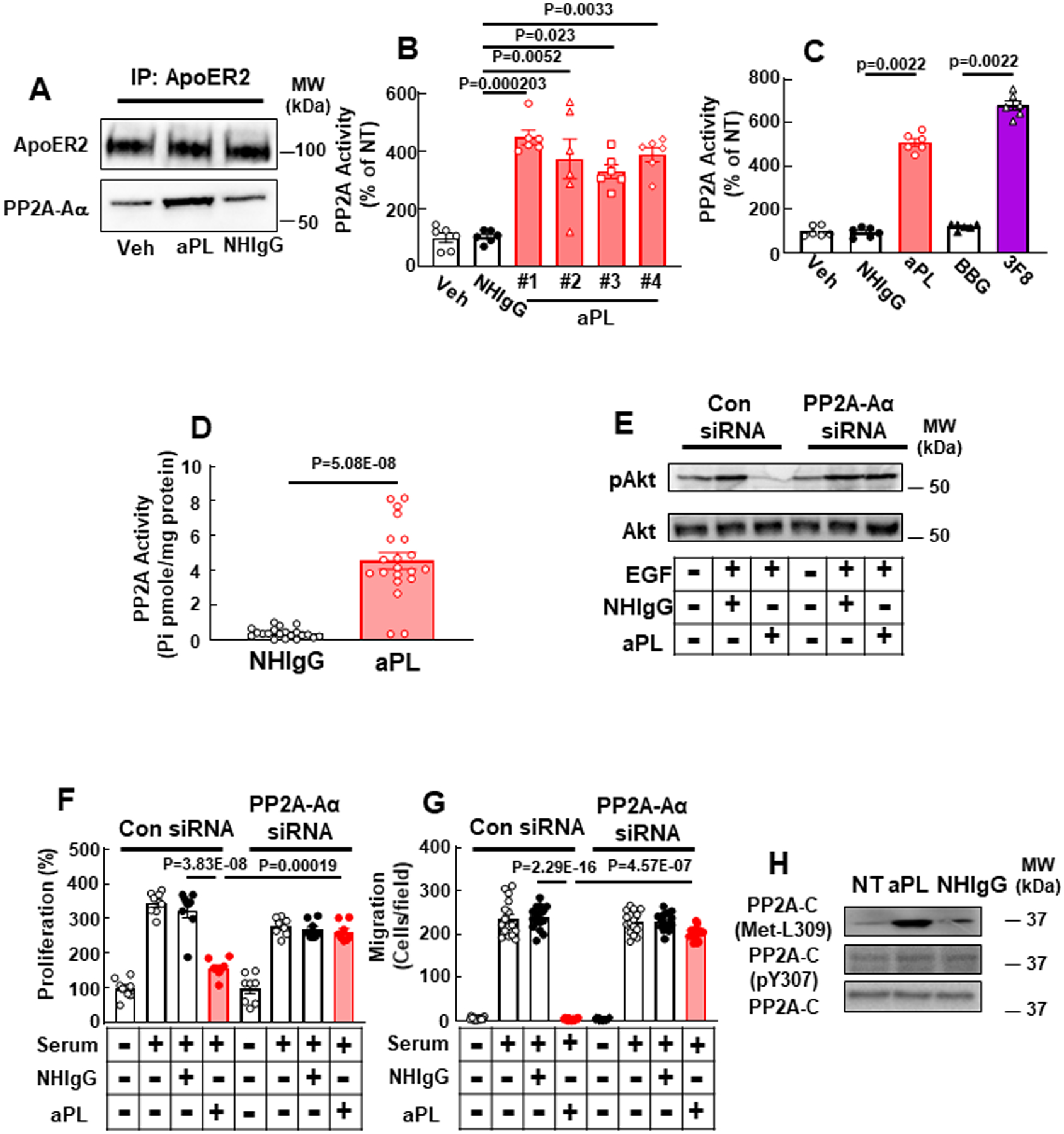
APL induces trophoblast dysfunction through the activation of PP2A. (A) HTR8/SVneo trophoblasts were incubated with PBS (Veh), aPL or NHIgG. ApoER2 was immunoprecipitated (IP), and ApoER2 and PP2A Aα subunit (PP2A-Aα) were detected. (B) Cells were incubated with PBS (Veh), NHIgG or aPL from 4 different APS patients (#1-#4), and PP2A activity was measured. N=6. (C) Cells were incubated with PBS (Veh), NHIgG, aPL, the anti-β2 GPI monoclonal antibody 3F8 or BBG (subclass-matched control IgG) and PP2A activity was measured. N=6. (D) Pregnant C57/BL6 mice were injected with NHIgG or aPL as in Fig. 1, and on day 15 PP2A activity in the placenta was measured. N=20. (E-G) Following transfection with control siRNA or siRNA targeting PP2A-Aα, cells were treated with vehicle (PBS) or EGF in the absence or presence of NHIgG or aPL, and Akt S473 phosphorylation was evaluated (E). Separate sets of cells were pretreated with NHIgG or aPL and then incubated in the presence or absence of 10% serum, and cell proliferation was quantified by BrdU incorporation (F, N=8) and cell migration was assessed by scratch assay (G, N=16). (H) PP2A-C Y307 phosphorylation and L309 methylation were assessed in cells treated with aPL or NHIgG. Values are Mean±SEM. For statistical analysis Kruskal-Wallis with Dunn’s post-hoc test (B), Mann-Whitney U test (C, D), or aligned rank transform two-way ANOVA with Benjamini and Hochberg correction (F, G) was used.
To delineate the processes by which PP2A enzymatic activity is stimulated by aPL, PP2A-B regulatory subunit participation was interrogated. PP2A-B subunits merge with PP2A-A and -C subunits to form functional heterotrimeric PP2A, and they afford specificity to the substrates dephosphorylated by PP2A. PP2A-B subunits are divided into four structurally distinct families designated B, B’, B” and B”’, and different cell types express unique sets of B subunits37. Characterizing the B subunits in trophoblasts for the first time, we found that B (α, β, γ and δ), B’ (α, β, γ, and δ) and B” (α, β, and γ) are expressed in HTR8/SVneo or Sw.71 cells (Online Fig. III A). To identify the B subunit(s) required for aPL activation of PP2A, the PP2A-C interactome was evaluated in HTR8/SVneo cells treated with NHIgG versus aPL (OnlineTable III). With aPL greater PP2A-Aα and PP2A-Aβ interaction with PP2A-C was observed, confirming aPL induction of PP2A A-C hetero-dimers, and PP2A-Bδ and PP2A-B’δ were recruited to PP2A-C. Then when either PP2A- Bδ or PP2A-B’δ was silenced in trophoblasts, EGF-induced Akt activation was preserved despite aPL treatment (Online Fig. III B, C). Thus, PP2A-Bδ and -B’δ subunits mediate the effects of aPL in human trophoblasts.
In addition to the B subunit-based direction of substrates to PP2A, substrate specificity and also enzyme activity are regulated by post-translational modifications of the PP2A-C subunit that govern the binding of the B subunits37, 38, 42, 43. The most common post-translational modifications of PP2A-C are Y307 phosphorylation, which is associated with attenuation of PP2A activity44, 45, and L309 methylation, which is mediated by leucine methyl-transferase 1 (LCMT-1) and enhances the affinity of PP2A-C for PP2A-B to provide substrate specificity and also promote enzyme activation43, 46, 47. We found that whereas PP2A-C Y307 phosphorylation was unaltered, aPL enhanced PP2A-C L309 methylation in both HTR8/SVneo cells (Fig. 2H) and Sw.71 cells (Online Fig. III D). Collectively these findings indicate that aPL cause trophoblast dysfunction by stimulating PP2A heterotrimer formation and enzyme activation.
APL activation of PP2A requires ApoER2, Dab2 and JIP4.
We next determined how aPL activate PP2A in trophoblasts. In HTR8/SVneo cells, ApoER2 knockdown prevented PP2A activation by aPL (Fig. 3A), thereby implicating the receptor. A requirement for ApoER2 in trophoblasts was also demonstrated in vivo in pregnant mice, with aPL activating PP2A in the placentas of control ApoER2fl/fl mice but not in the placentas of ApoER2ΔTR mice (Fig. 3B), mirroring the role of trophoblast ApoER2 in aPL-induced maternal hypertension, fetal loss and IUGR (Fig. 1, Online Table I). How ApoER2 is coupled to PP2A was then interrogated by first determining the role of Dab1 or Dab2, which are adaptor proteins for ApoER2 in neurons and in endothelial cells that interact with the receptor’s cytoplasmic NPXY domain and required for receptor-mediated intracellular signaling24, 48. We determined that Dab2, but not Dab1, is expressed in cultured human trophoblasts (HTR8/SVneo and Sw.71) and in human and mouse placentas (Online Fig. IV A). Using coimmunoprecipitation, it was then shown that aPL treatment increased Dab2-ApoER2 association both in cultured trophoblasts and in mouse placenta in vivo (Online Fig. IV B, C). Demonstrating a functional requirement for Dab2, its silencing in cultured trophoblasts suppressed not only aPL activation of PP2A, but also aPL antagonism of cell proliferation and migration (Online Fig. IV D–I).
Fig. 3.
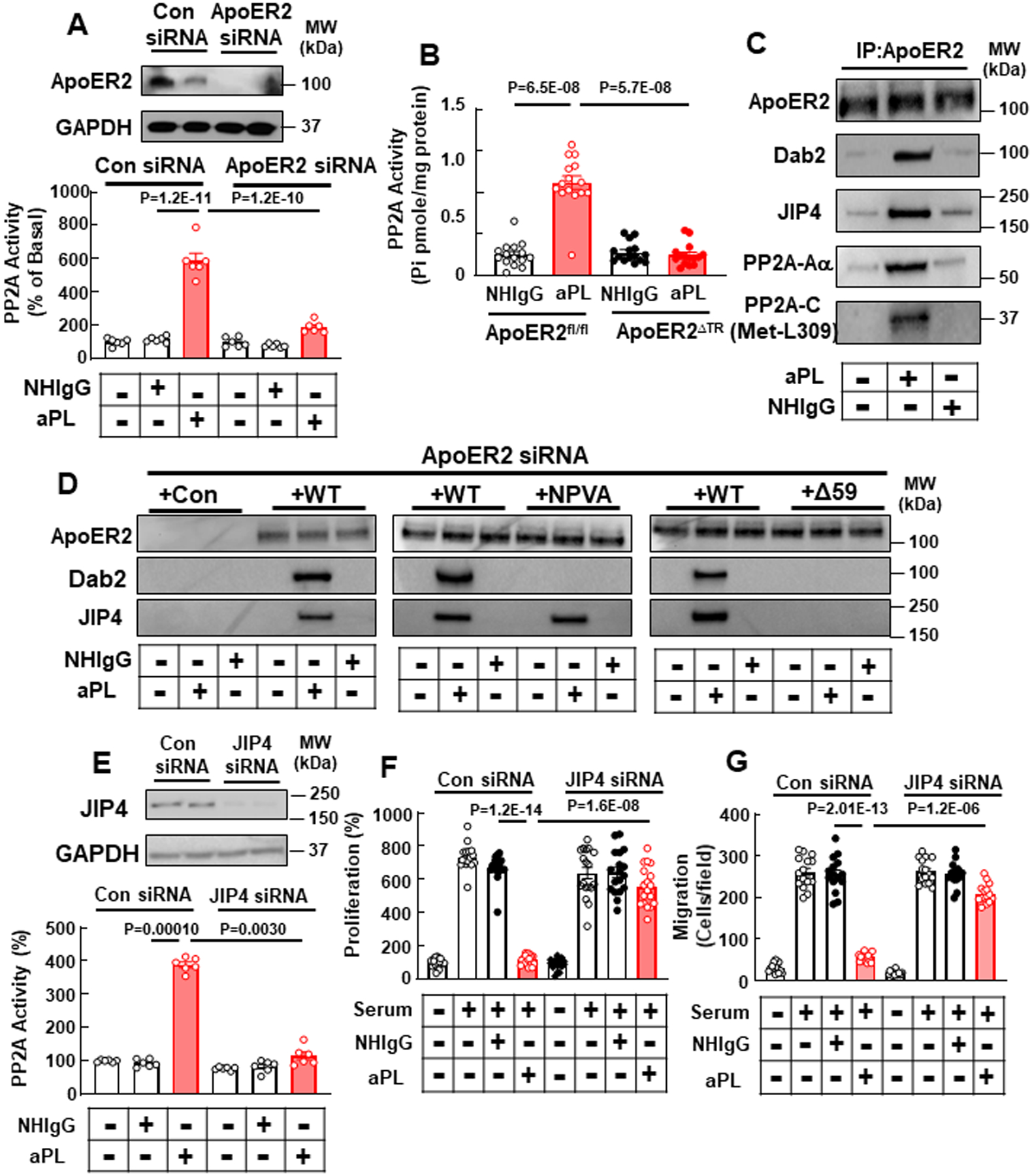
ApoER2 and JIP4 partnership is required for aPL activation of PP2A and inhibition of trophoblast cell proliferation and migration. (A) HTR8/SVneo trophoblasts were treated with control siRNA or siRNA targeting ApoER2, treated with NHIgG or aPL, and PP2A activity was measured. N=6. (B) ApoER2fl/fl or ApoERΔTR mice were injected with NHIgG or aPL as in Fig. 1, and PP2A activity in the placenta was measured on day 15. N=15. (C) Cells were treated with NHIgG or aPL, ApoER2 was immunoprecipitated, and ApoER2, Dab2, JIP4, PP2A-Aα and L309 methylation of PP2A-C were detected. (D) Cells previously transfected with siRNA targeting ApoER2 were then transfected with vector alone (+Con) or cDNA encoding wild-type apoER2 (+WT), ApoER2 with NPVA point mutation (+NPVA) or ApoER2 with C-terminal deletion (+Δ59). ApoER2 was immunoprecipitated, and ApoER2, Dab2 and JIP4 were detected. (E) Cells were transfected with control siRNA or siRNA targeting JIP4, treated with NHIgG or aPL, and PP2A activity was measured. N=6. (F, G) Similarly transfected cells were incubated with or without 10% serum and cell proliferation (F, N=18) and cell migration were assessed (G, N=15). Values are Mean±SEM. Two-way ANOVA with Tukey’s post-hoc test (A), or aligned rank transform two-way ANOVA with Benjamini and Hochberg correction (B, E-G) was used.
Revealed initially in the ApoER2 interactome in trophoblasts (OnlineTable II), we additionally identified aPL-induced recruitment to ApoER2 of C-Jun-N-terminal kinase-interacting protein 4 (JIP4), which is a member of the JIP protein family that regulates JNK and p38 MAPK activity49, 50. To investigate a possible role for JIP4 in aPL action, we first demonstrated JIP4 expression not only in cultured human trophoblasts (HTR8/SVneo and Sw.71) but also in mouse and human placenta (Online Fig. IV A). By coimmunoprecipitation we then confirmed JIP4 recruitment to ApoER2 in aPL-treated cultured trophoblasts, in a protein complex that included Dab2, PP2A-Aα, and L309 methylated PP2A-C (Fig. 3C, Online Fig. V A; HTR8/SVneo and Sw.71, respectively). Next we determined the ApoER2 C-terminal domain responsible for JIP4 interaction by silencing endogenous ApoER2 and replacing it with either wild-type ApoER2 (WT) or mutant forms of the receptor disrupting the NPXY motif (substitution with NPVA) or lacking the C-terminal 59 amino acid proline-rich domain (Δ59, Online Fig. V B, Fig. 3D). Whereas there was a predictable loss of ApoER2-Dab2 interaction with the NPVA mutant receptor, aPL-induced recruitment of JIP4 to the receptor was retained (Fig. 3D, middle panel). In contrast, there was a loss of aPL-induced JIP4 interaction with ApoER2-Δ59, and there was an accompanying loss of Dab2 interaction. Next we found that knockdown of JIP4 in HTR8/SVneo cells negated PP2A activation by aPL (Fig. 3E), and aPL antagonism of trophoblast proliferation and migration was also prevented (Fig. 3F, G).
Unlike other members of the JIP family, JIP4 does not participate in JNK signaling49. Alternatively, JIP4 binds to p38α MAPK and promotes its activation by a mechanism that requires the MAP kinase kinases MKK3 and MKK649. To delineate how JIP4 recruitment to ApoER2 causes PP2A activation, coimmunoprecipitation was employed to evaluate proteins associated with JIP4 (Fig. 4A). Whereas JNK was not coimmunoprecipitated with JIP4, MKK3 and p38 interaction with JIP4 was increased by aPL treatment. In a parallel manner, aPL had no impact on JNK-T183/Y185 phosphorylation (Online Fig. V C), and alternatively aPL increased p38 MAPK-T180/Y182 phosphorylation indicative of its activation (Fig. 4B). In addition, siRNA silencing of p38 or treatment with the p38 inhibitor SB202190 abolished aPL activation of PP2A (Fig. 4C, D), and SB202190 additionally suppressed aPL-stimulated methylation of PP2A-C-L309 (Fig. 4E). Furthermore, silencing of MKK3 resulted in a loss of aPL-induced p38 phosphorylation and PP2A activation (Fig. 4F, G). Collectively, these results indicate that aPL cause formation of an ApoER2-Dab2-JIP4-PP2A complex that results in PP2A activation via JIP4-related stimulation of MKK3 and p38 MAPK.
Fig. 4.
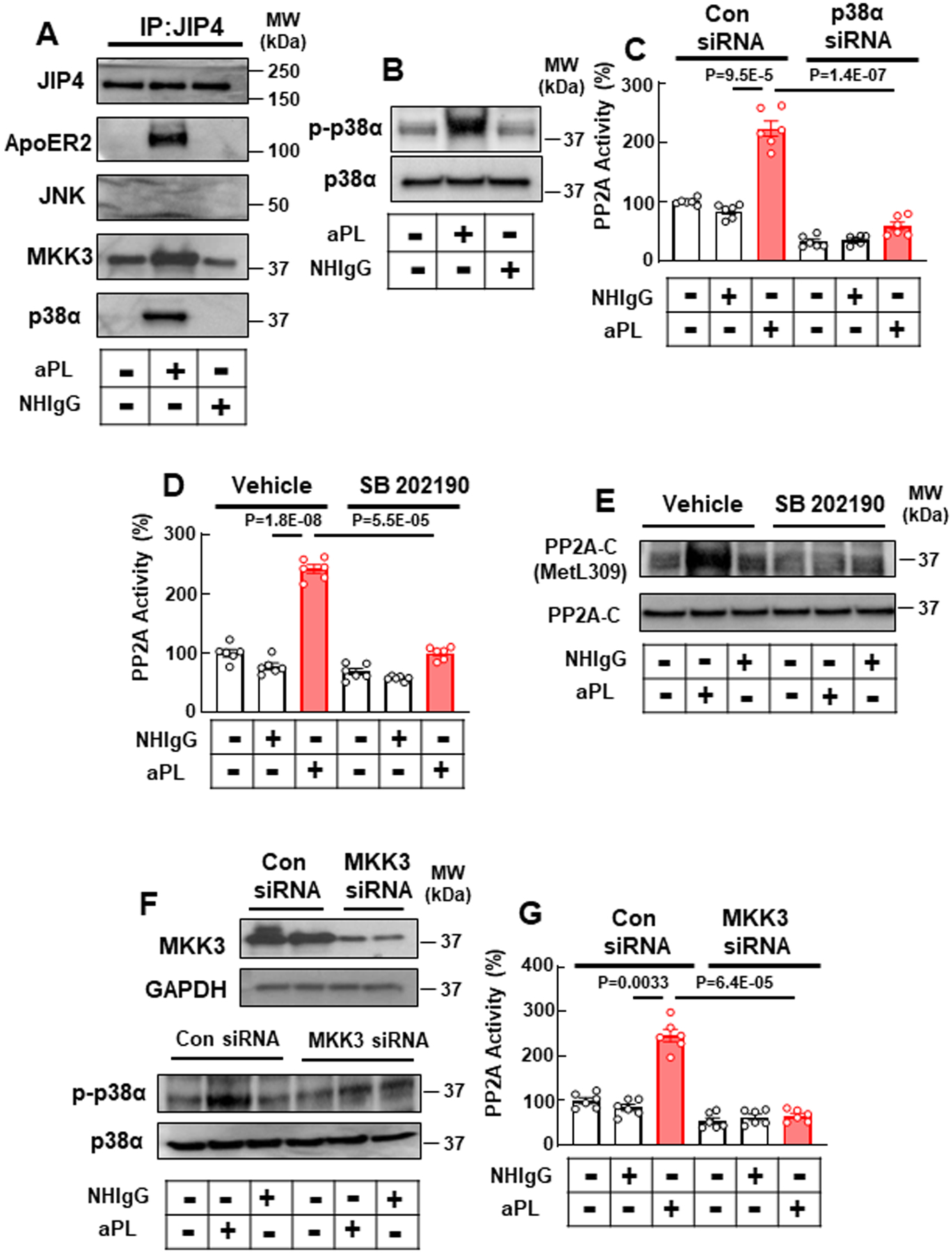
ApoER2, JIP4, MKK3 and p38α partner to mediate aPL activation of PP2A. (A) HTR8/SVneo trophoblasts were treated with NHIgG or aPL, JIP4 was immunoprecipitated and JIP4, ApoER2, JNK, MKK3 and p38α were detected. (B) Cells were treated with NHIgG or aPL, and phospho-p38α (p-p38α, Thr180/Tyr182) and total p38 abundance was determined. (C) Cells were transfected with control siRNA or siRNA targeting p38α, treated with NHIgG or aPL, and PP2A activity was evaluated. N=6. (D, E) Cells were pretreated with p38 inhibitor SB 202190 then treated with NHIgG or aPL, and either PP2A activity (D, N=6) or PP2A-C L309 methylation was determined (E). (F, G) Cells were transfected with control siRNA or siRNA targeting MKK3, treated with NHIgG or aPL, and p38 phosphorylation (F) or PP2A activation was assessed (G, N=6). Values are Mean±SEM. Aligned rank transform two-way ANOVA with Benjamini and Hochberg correction was performed (C, D, G).
APL induce MMP14 upregulation and soluble endoglin production in trophoblasts.
To next determine how aPL-ApoER2 actions in trophoblasts cause PE, we assessed whether aPL influence the production of soluble fms-like tyrosine kinase-1 (sFlt-1) or soluble endoglin (sEng), which are produced by dysfunctional placenta and implicated in the pathogenesis of PE in rodent models as well as in humans51–54. We found that aPL treatment increased sEng secretion in HTR8/SVneo cells in an ApoER2 dependent manner (Fig. 5A). In parallel, plasma levels of sEng were elevated by aPL in pregnant ApoER2fl/fl mice, but not in ApoER2ΔTR mice (Fig. 5B). APL administration did not affect plasma sFlt-1 level in either ApoER2fl/fl or ApoER2ΔTR mice (Online Fig. VI). Noting that the metalloproteinase MMP14 is responsible for the cleavage of full-length Eng to produce sEng55–58, we evaluated the expression of MMP14 in mouse placenta and found that it was upregulated by aPL in the placentas of ApoER2fl/fl mice, but not in the placentas of ApoER2ΔTR mice (Fig. 5C). We also showed that aPL induced the secretion of MMP14 by cultured trophoblasts in an ApoER2-dependent manner (Fig. 5D). Furthermore, siRNA knockdown of MMP14 or treatment with the MMP14 inhibitor NSC405020 abrogated the aPL-induced release of sEng (Fig. 5E, F), indicating that MMP14 is required for aPL-induced upregulation of sEng. Thus, via ApoER2, aPL upregulate MMP14 and sEng production by trophoblasts.
Fig. 5.
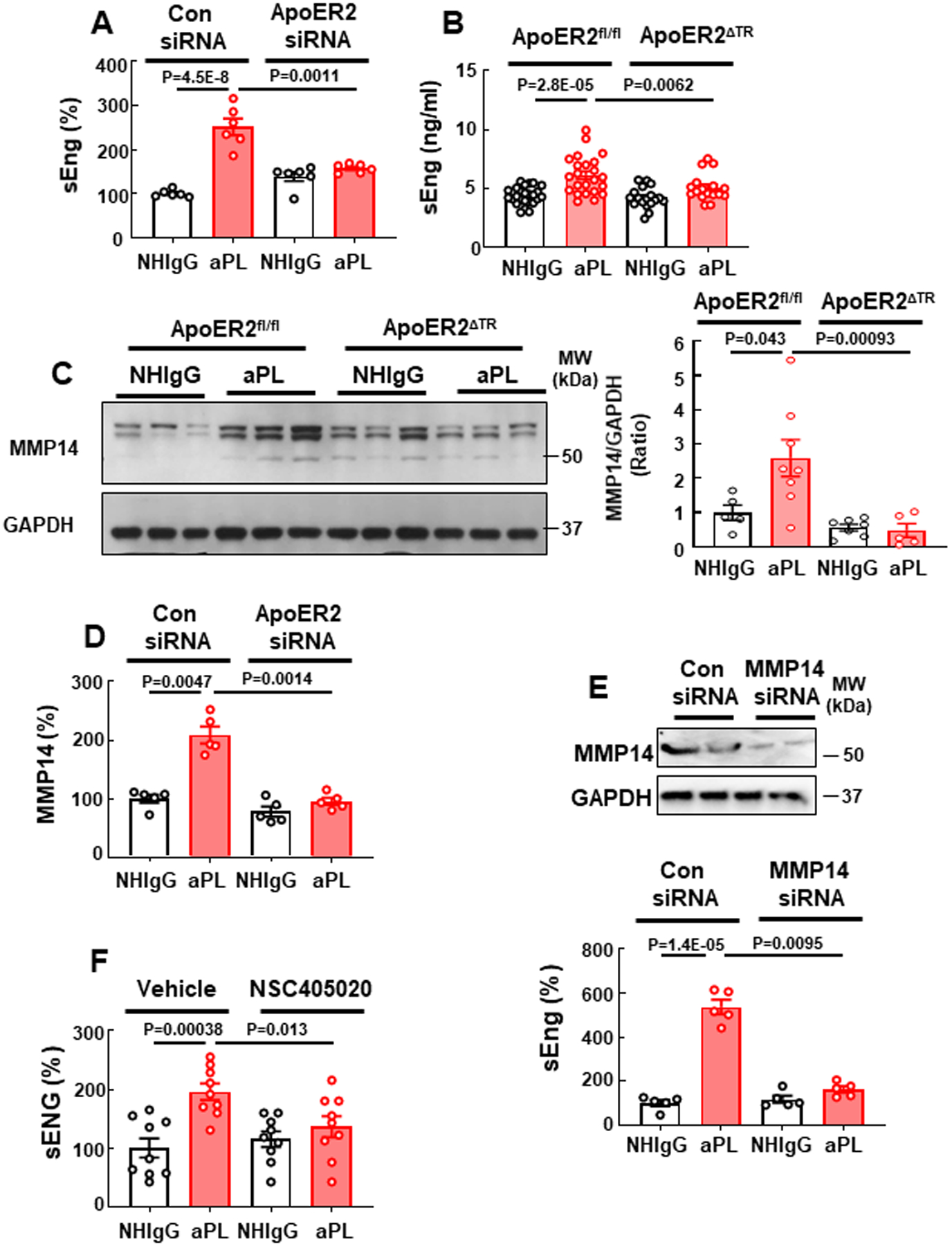
APL increase trophoblast soluble endoglin secretion through ApoER2-dependent upregulation of MMP14. (A) HTR8/SVneo trophoblasts were transfected with control siRNA or siRNA targeting ApoER2, treated with NHIgG or aPL, and soluble endoglin (sEng) release was measured. N=6. (B, C) ApoER2fl/fl or ApoER2ΔTR mice were injected with NHIgG or aPL as in Fig. 1, and on day 15 plasma sEng was measured (B, N=25,23,18,17) and placenta MMP14 content was determined (C, N=5,8,7,5). (D) Cells were transfected with control siRNA or siRNA targeting ApoER2, treated with NHIgG or aPL, and the release of soluble MMP14 was assessed. N=5. (E) Cells were transfected with control siRNA or siRNA targeting MMP14 (upper panel), treated with NHIgG or aPL and sEng release was measured. N=5. (F) Cells were incubated with aPL or NHIgG in the presence of vehicle or MMP14 inhibitor (NSC405020, 10 nM), and sEng release was quantified. N=9. Values are Mean±SEM. Aligned rank transform two-way ANOVA with Benjamini and Hochberg correction was used (A-F).
PP2A inhibition prevents APS-related PE in mice.
Next we sought to test whether pharmacological inhibition of PP2A by Cantharidin59–61 improves either the maternal or fetal outcomes in aPL-treated wild-type mice. Before attempting in vivo experiments, we determined that Cantharidin prevented aPL activation of PP2A, and aPL suppression of cell proliferation and migration in cultured trophoblasts (Fig. 6A–C). In vivo experiments were then performed in pregnant C57BL/6 mice, and Cantharidin decreased aPL-induced PP2A activation in the placenta, and it afforded protection from aPL-related systolic hypertension and proteinuria (Fig. 6D–F). Furthermore, Cantharidin prevented aPL-induced fetal demise and IUGR (Fig. 6G, H).
Fig. 6.
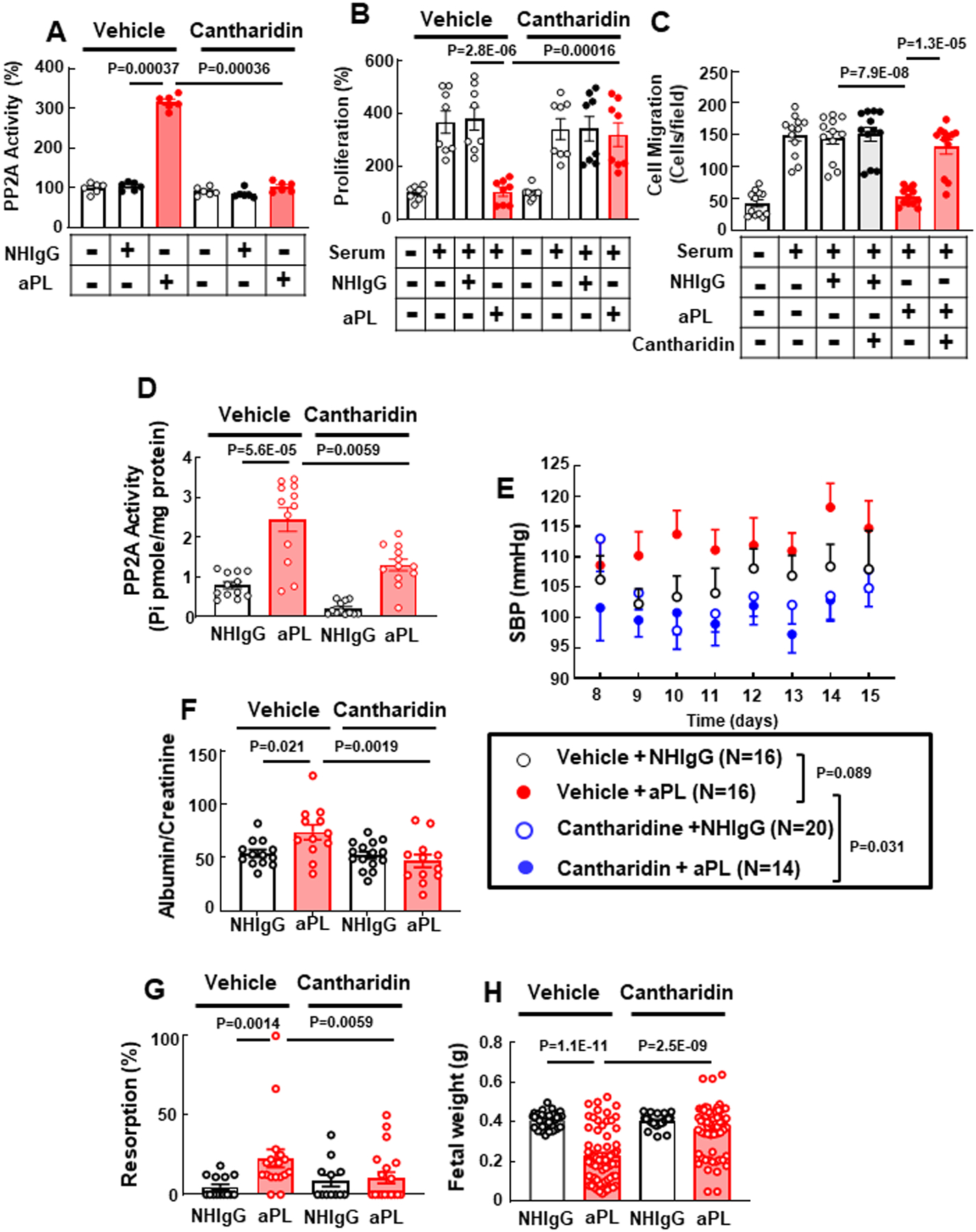
PP2A inhibitor Cantharidin prevents aPL inhibition of trophoblast cell proliferation and migration in culture, and aPL-induced maternal hypertension and proteinuria and fetal complications in vivo. (A-C) HTR8/SVneo trophoblasts were treated with vehicle or Cantharidin (5μM) and with NHIgG or aPL, and PP2A activity was measured (A, N=6). Similarly-treated cells were incubated in the presence or absence of 10% serum, and cell proliferation (B, N=8) and cell migration were assessed (C, N=12). (D) C57BL/6 mice treated with NHIgG or aPL on day 8 and 12 of pregnancy were injected with vehicle or Cantharidin on day 8, 10, 12, and 14, and placenta PP2A activity was determined on day 15. (E-H). C57BL/6 mice received vehicle or Cantharidin via minipump starting on day 7 of pregnancy and they were administered NHIgG or aPL on day 8 and 12. SBP was monitored from day 8 to 15 (E), and on day 15 urine protein (F, N=13,12,15,12), fetal resorption rate (G, N=14, 18, 12, 20) and body weights of surviving fetuses (H, N=40, 71, 23, 65) were evaluated. Values are Mean±SEM. Aligned rank transform two-way ANOVA with Benjamini and Hochberg correction was used (A-D, F-H), or aligned rank transform three-way mixed ANOVA with Benjamini and Hochberg correction was used (E).
To test whether PP2A inhibition blocks the mechanisms revealed to underlie aPL-mediated PE, the effect of Cantharidin on MMP14 and sEng was first evaluated in cultured trophoblasts. Whereas cells treated with vehicle showed increased release of MMP14 and sEng in response to aPL, Cantharidin prevented the increases in MMP14 and sEng (Fig. 7A, B). In addition, Cantharidin decreased the levels of MMP14 and Eng mRNA expression observed with aPL (Fig. 7C, D). Mirroring the findings in cell culture, in vivo levels of circulating sEng and MMP14 and transcript abundance in the placenta were increased by aPL in control-treated mice, but not in mice administered Cantharidin (Fig. 7E–H).
Fig. 7.
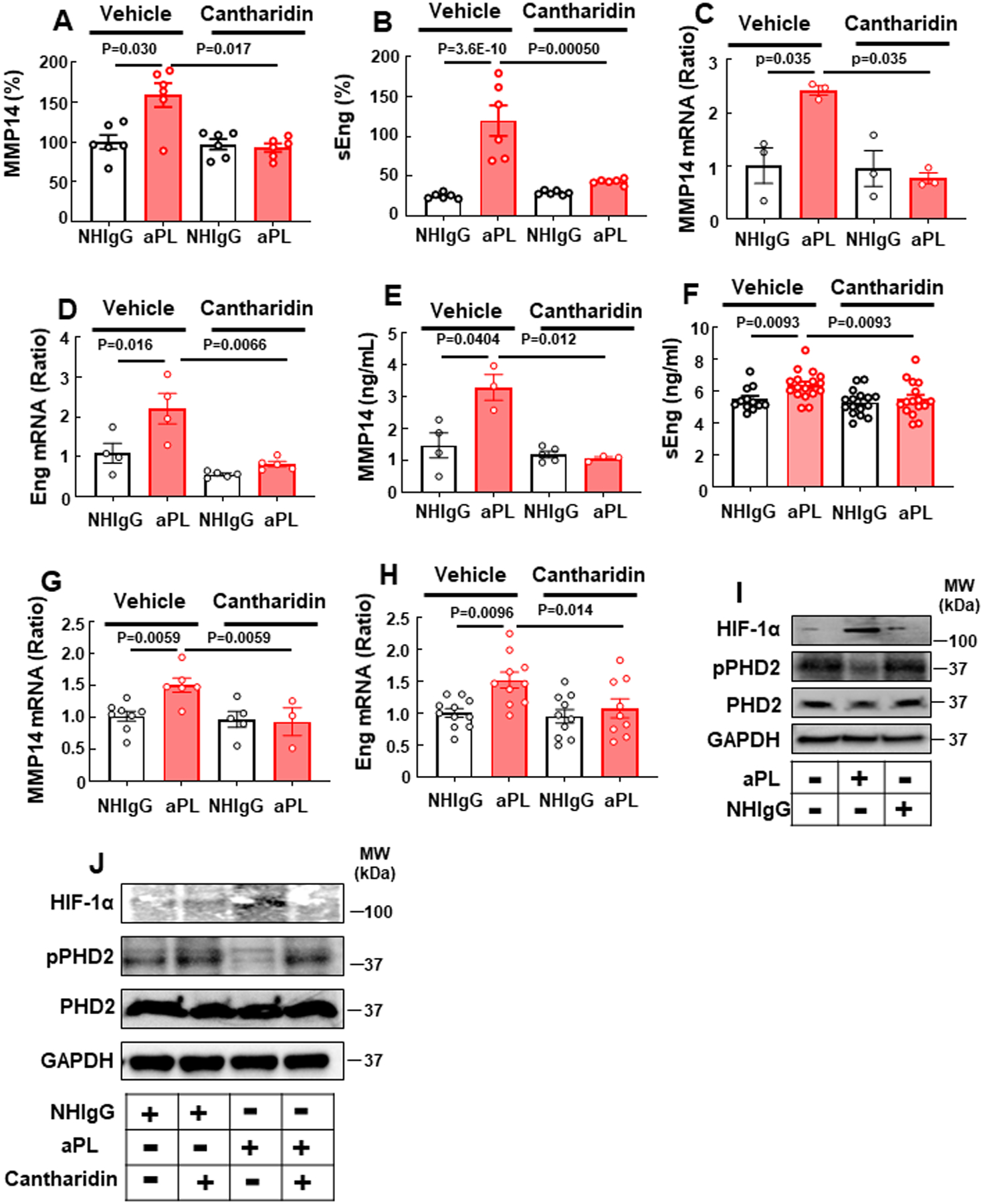
APL increase trophoblast sEng production via PP2A- and HIF-1α-dependent transcriptional upregulation of MMP14 and endoglin. (A-D) Following pretreatment with vehicle or Cantharidin, HTR8/SVneo trophoblasts were treated with NHIgG or aPL, and the release of MMP14 (A, N=6) and sEng (B, N=6) was assessed, and transcript levels for MMP14 (C, N=3) and Eng were quantified (D, N=4). (E-H) Pregnant C57BL/6 mice received vehicle or Cantharidin on day 8, 10, 12 and 14 of pregnancy, and they were injected with NHIgG or aPL on day 8 and 12. On day 15 plasma MMP14 (E, N=4,3,5,3) and sEng (F, N=12,17,16,16) were measured, and placenta MMP14 (G, N=8,6,5,3) and Eng (H, N=11,10,10,9) transcript abundance were determined. (I) Cells were treated with NHIgG or aPL, and HIF-1α, phospho-PHD2 (S125), total PHD2 and GAPDH abundance were evaluated. (J) Cells were pretreated with vehicle or Cantharidin, treated with NHIgG or aPL, and HIF-1α, phospho-PHD2, total PHD2 and GAPDH abundance was assessed. Values are Mean±SEM. Aligned rank transform two-way ANOVA with Benjamini and Hochberg correction was used (A-H).
Next, recognizing that the transcription factor hypoxia inducible factor-1 (HIF1α) plays a key role in PE pathogenesis62–66 and that HIF1α accumulation is regulated by HIF prolyl hydroxylase 2 (PHD2)67, we interrogated whether aPL impact HIFα and PHD2 in HTR8/SVneo cells. We found that aPL caused an upregulation of HIF1α, and there was a related decrease in PHD2 S125 phosphorylation, which promotes the accumulation of HIF1α67 (Fig. 7I). A link between PP2A and relative PHD2 phosphorylation and HIF1α was then sought in trophoblasts, and we found that Cantharidin prevented the effects of aPL on both PHD2 phosphorylation and HIF1α (Fig. 7J). Furthermore, whereas the HIF1α inhibitor GN44028 did not affect aPL upregulation of MMP14 mRNA or protein (Online Fig. VII A,B), HIF1α inhibition blocked aPL-induced upregulation of Eng mRNA and sEng release (Online Fig. VII C,D). Thus, aPL modulate HIF1α in trophoblasts, doing so via PP2A actions on PHD2 and resulting in increases in sEng, explaining why PP2A inhibition prevents aPL-induced PE in the mouse model.
Placental PP2A, PHD2 and HIF1α are dysregulated in human APS and in non-APS PE.
To determine if the induction of PP2A activity may contribute to the pathogenesis of APS in human pregnancies, PP2A activity was measured in human placental explants incubated with aPL or the anti-β2 monoclonal antibody 3F8. Both treatments activated PP2A (Fig. 8A). PP2A activity was then quantified in non-fixed placentas from an APS pregnancy and three normal pregnancies, and activity was markedly higher in the APS placenta (Fig. 8B). Since PP2A-C L309 methylation, an indicator of PP2A activation, can be evaluated in formalin-fixed tissue, PP2A-C L309 methylation was compared in formalin-fixed samples of placenta from four normal pregnancies and four pregnancies complicated by APS, and it was found that PP2A-C L309 methylation is increased in the APS placentas (Fig. 8C,D). In parallel, in the APS placentas there was a reduction in PHD2 S125 phosphorylation and upregulation of HIF1α (Fig. 8C,E, F), mirroring the findings for aPL actions on PP2A, PHD2 and HIF1α in cultured human trophoblasts (Fig. 7J).
Fig. 8.
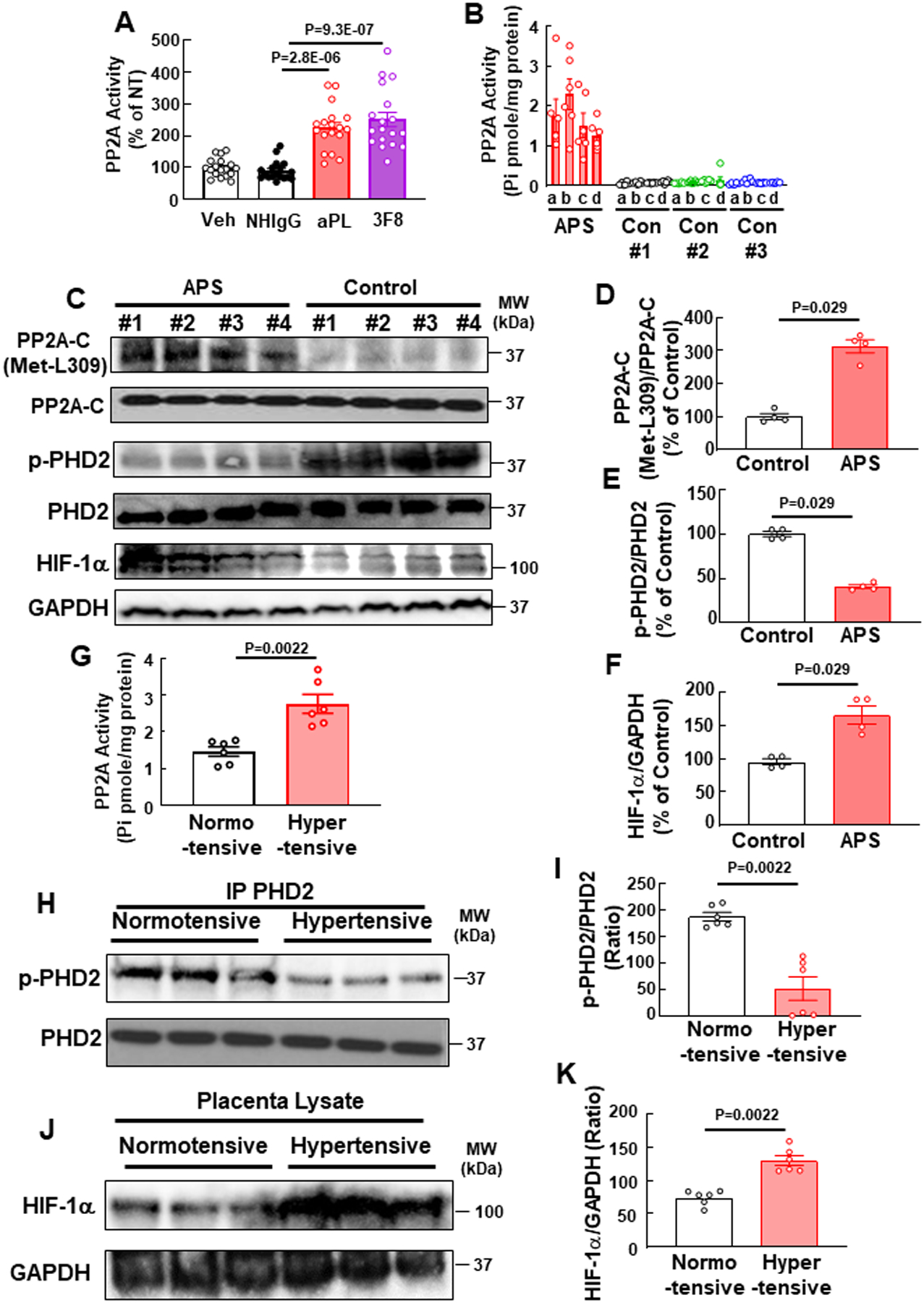
APL activate PP2A in human placental explants, and placentas from pregnancies affected by APS, and PE unrelated to APS display PP2A hyperactivation, phospho-PHD2 downregulation and HIF-1α upregulation. (A) Human placenta explants from normal term pregnancies were incubated ex vivo with PBS (Veh), NHIgG, aPL or 3F8, and PP2A activity was measured. N=18. (B) PP2A activity was measured in placentas from three normal term pregnancies (Con #1-#3) and one APS pregnancy. Samples were from 4 quadrants (a-d) in each placenta. (C-F) PP2A-C L309 methylation and total PP2A-C (C,D), PHD2 S125 phosphorylation and total PHD2 (C,E), and HIF-1α and GAPDH (C,F) were quantified in placentas from 4 normal term pregnancies and 4 APS pregnancies. N=4. (G-K) In placentas from normotensive and hypertensive mothers (N=6), PP2A activity was measured (G), or the abundance of phosphorylated PHD2 (S125) and total PHD2 (H, I) or HIF-1α and GAPDH (J,K) was assessed. Values are Mean±SEM. Kruskal-Wallis with Dunn’s post-hoc test (A), or Mann-Whitney U test (D-G, I, K) was performed.
Finally, having discovered a central role for PP2A in the pathogenesis of PE related to APS, we determined if the PP2A-related mechanism is generalizable to other forms of PE. To do so, studies were performed in placentas from six normal pregnancies and six pregnancies complicated by PE not related to APS. The systolic blood pressures in the normotensive pregnancies and the hypertensive pregnancies were 122.3±3.9 and 163.2±4.5, respectively (mean±S.E., P<0.0001), and the diastolic blood pressures were 79.0±2.1 and 98.8 (mean±S.E., P<0.0001). We found that PP2A activity was increased in the PE placentas (Fig. 8G), and there were parallel marked declines in PHD2 S125 phosphorylation (Fig 8H, I) and increases in HIF1α (Fig. 8J,K). Thus, the PP2A-driven processes discovered to be underlying PE in the mouse model of APS pregnancy may be operative in human APS pregnancies, and PP2A may additionally play a role in PE in non-APS patients.
DISCUSSION
Preeclampsia (PE) is one of the leading causes of maternal death and a major contributor to maternal and fetal and neonatal morbidity worldwide1–3. APS is a common risk factor for PE7, 8, and pregnancies in women with APS are also complicated by fetal demise, premature birth and IUGR9–13, 20. In the present work we established a model of APS-related PE in mice, and then employed the model to reveal that trophoblasts are the critical cellular target of aPL by which the antibodies induce maternal hypertension and proteinuria and fetal complications. We additionally demonstrated that ApoER2 in trophoblasts is a critical linchpin in the underlying pathogenetic processes.
The present studies additionally revealed a key role for trophoblast PP2A in APS-related PE and in APS-associated fetal loss and IUGR (Online Fig. IX). Polyclonal human aPL from multiple patients or the monoclonal anti-β2GPI antibody 3F8 stimulated PP2A in an ApoER2-dependent manner in cultured trophoblasts, and mice lacking ApoER2 selectively in trophoblasts were protected from aPL-induced activation of the phosphatase. In addition, PP2A activation was required for aPL antagonism of trophoblast cellular functions, and in the disease model pharmacologic inhibition of PP2A by Cantharidin prevented aPL-induced PE, fetal loss and IUGR. The latter findings link PP2A activation to the maternal and fetal complications of APS in vivo, and they provide proof-of-concept that PP2A inhibition may be an intervention to protect the health of both the mother and the fetus in an APS pregnancy.
The mechanistic underpinnings of PP2A activation by aPL via ApoER2 were also delineated (Online Fig. IX). The PP2A-B regulatory subunits affording PP2A substrate specificity37, 42 in aPL-exposed trophoblasts were identified to be βδ and β’δ. In addition, the ApoER2 adaptor molecule Dab2 was determined to be recruited to the NPVY sequence in the receptor cytoplasmic domain, and required for aPL activation of PP2A and inhibition of cell proliferation and migration in trophoblasts. Although the exact role of Dab2 in trophoblast is not yet clear, upon binding to NPXY sequence of ApoER2, Dab2 may mediate the activating L309 methylation of the PP2A catalytic subunit by leucine methyl transferase-1, as has been shown in endothelial cells24. We further identified JIP4 as a novel adaptor molecule for ApoER2 that upon aPL treatment binds to the C-terminal proline-rich region of the receptor and is necessary for PP2A activation and resulting adverse effects on trophoblast function. MKK3 and p38 MAPK were then found to be recruited to JIP4 in response to aPL and necessary for PP2A activation. PP2A modulation by p38 MAPK has been observed previously in cardiac stem cells; however, in contrast to cardiac stem cells in which p38 inhibits PP2A68, we found that in trophoblasts p38 promotes PP2A activation in response to aPL. Although how p38 MAPK stimulates PP2A activity in trophoblasts is yet to be determined, these findings reveal multiple previously unknown potential therapeutic targets worthy of further consideration in the management of PE during APS pregnancies.
In studies determining how PP2A activation promotes PE, the current work demonstrated that aPL induce sEng secretion from trophoblasts (Online Fig. IX). Maternal circulating levels of placenta-derived anti-angiogenic proteins such as sEng and sFLT1 are elevated in pregnancies complicated by PE53, 54, 69, 70. sEng is produced by proteolytic cleavage of transforming growth factor β (TGFβ) co-receptor endoglin (Eng), and elevated levels of circulating sEng interfere with TGFβ signaling in vascular endothelial cells, thereby inducing hypertension54. sEng is cleaved from full-length endoglin by matrix metalloprotease 14 (MMP14 or MT1)55–58. Our work revealed that aPL upregulates both MMP14 and endoglin/sEng in trophoblasts via ApoER2 and PP2A. We further obtained evidence that the Eng upregulation occurs via PP2A-mediated PHD2 S125 dephosphorylation, which attenuates the activity of PHD2 to degrade HIF1α, leading to HIF1α upregulation and resulting increases in endoglin gene transcription63, 65. The basis for the upregulation of MMP14 by aPL via ApoER2 and PP2A now deserves attention.
To determine the human relevance of the mechanisms discovered to underlie APS-related PE in the murine model, we leveraged the observations related to PP2A. We demonstrated that aPL potently stimulate PP2A activity in human placental explants, and more importantly, we discovered that activating PP2A-C subunit methylation, pPHD2 dephosphorylation and HIF1α upregulation are exaggerated in placentas from APS pregnancies in women. Furthermore, we demonstrated that PP2A hyperactivation and PHD2 dephosphorylation may also be proximal mechanisms driving HIF1α upregulation in cases of PE that are not related to APS. Along with confirmation of the current findings in larger datasets from pregnancies complicated by these conditions, the possible means by which trophoblast PP2A is activated in PE in non-APS patients should be pursued. If such efforts provide additional evidence linking PP2A dysregulation with HIF1α in the placental processes underlying PE in women both with and without APS, the insights gained in the current study of a specific cause of PE may have therapeutic implications for millions of pregnant women worldwide per year.
Supplementary Material
Novelty and Significance.
What Is Known?
Preeclampsia (PE) frequently complicates pregnancy in women with the antiphospholipid syndrome (APS).
APS, which is characterized by circulating antiphospholipid antibodies (aPL), also increases the risk of fetal demise, premature birth and intrauterine growth restriction (IUGR).
What New Information Does This Article Contribute?
aPL actions via Apolipoprotein E receptor 2 (ApoER2) in placental trophoblasts cause PE and fetal demise and IUGR in a mouse model of APS during pregnancy.
Via ApoER2, aPL activate trophoblast protein phosphatase 2A (PP2A), which upregulates matrix metallopeptidase 14 and hypoxia-inducible factor-1α (HIF1α), the latter via dephosphorylation of prolyl hydroxylase domain containing protein 2 (PHD2), leading to increased soluble endoglin production.
In placentas from both APS patients and women with PE from causes other than APS, PP2A is hyperactivated, PHD2 is dephosphorylated and HIF1α is upregulated, and PP2A inhibition protects pregnant mice from aPL-induced PE and fetal complications.
SOURCES OF FUNDING
This work was supported by National Institute of Health T32HL098040 (A.S.), R01HL109604 & R01094395 (C.M.), R01HL118001 (D.Y.H & P.W.S), R01HL147403 (D.Y.H.), R37HL63762, R01NS093382, NS108115 & RF1AG053391 (J.H.), 5R01HD100179-02 (D.R.N.), Canadian Institutes of Health Research 201203MOP-275374-CIA-CBBA-109624 (D.R.N.), the American Heart Association/the Harry S. Moss Heart Trust Postdoctoral fellowship 18POST33960170 (H.C.), the Brightfocus Foundation (J.H.), the Bluefield Project (J.H.), Harrington Scholar Innovator Award (J.H.), the Hartwell Foundation (P.W.S.), the Crystal Charity Ball Center for Pediatric Critical Care Research and the Children’s Medical Center Foundation (P.W.S).
Nonstandard Abbreviations and Acronyms:
- aPL
antiphospholipid antibodies
- ApoER2
Apolipoprotein E Receptor 2
- APS
Antiphospholipid Syndrome
- Dab2
Disabled-2
- Eng
endoglin
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- HIF-1α
hypoxia-inducible factor-1α
- MKK3
mitogen-activated protein kinase kinase 3
- MMP14
Matrix Metallopeptidase 14
- NHIgG
normal human IgG
- PHD2
prolyl hydroxylase domain containing protein 2
- PP2A
protein phosphatase 2A
- SBP
systolic blood pressure
Footnotes
REFERENCES
- 1.American College of O, Gynecologists and Task Force on Hypertension in P. Hypertension in pregnancy. Report of the American College of Obstetricians and Gynecologists’ Task Force on Hypertension in Pregnancy. Obstet Gynecol. 2013;122:1122–31. [DOI] [PubMed] [Google Scholar]
- 2.Hogan MC, Foreman KJ, Naghavi M, Ahn SY, Wang M, Makela SM, Lopez AD, Lozano R and Murray CJ. Maternal mortality for 181 countries, 1980–2008: a systematic analysis of progress towards Millennium Development Goal 5. Lancet. 2010;375:1609–23. [DOI] [PubMed] [Google Scholar]
- 3.Kuklina EV, Ayala C and Callaghan WM. Hypertensive disorders and severe obstetric morbidity in the United States. Obstet Gynecol. 2009;113:1299–306. [DOI] [PubMed] [Google Scholar]
- 4.Coutinho T, Lamai O and Nerenberg K. Hypertensive Disorders of Pregnancy and Cardiovascular Diseases: Current Knowledge and Future Directions. Curr Treat Options Cardiovasc Med. 2018;20:56. [DOI] [PubMed] [Google Scholar]
- 5.Tooher J, Thornton C, Makris A, Ogle R, Korda A and Hennessy A. All Hypertensive Disorders of Pregnancy Increase the Risk of Future Cardiovascular Disease. Hypertension. 2017;70:798–803. [DOI] [PubMed] [Google Scholar]
- 6.Sones JL and Davisson RL. Preeclampsia, of mice and women. Physiol Genomics. 2016;48:565–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rana S, Lemoine E, Granger JP and Karumanchi SA. Preeclampsia: Pathophysiology, Challenges, and Perspectives. Circ Res. 2019;124:1094–1112. [DOI] [PubMed] [Google Scholar]
- 8.Bartsch E, Medcalf KE, Park AL, Ray JG and High Risk of Pre-eclampsia Identification G. Clinical risk factors for pre-eclampsia determined in early pregnancy: systematic review and meta-analysis of large cohort studies. BMJ. 2016;353:i1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giannakopoulos B and Krilis SA. The pathogenesis of the antiphospholipid syndrome. N Engl J Med. 2013;368:1033–1044. [DOI] [PubMed] [Google Scholar]
- 10.Ruiz-Irastorza G, Crowther M, Branch W and Khamashta MA. Antiphospholipid syndrome. Lancet. 2010;376:1498–1509. [DOI] [PubMed] [Google Scholar]
- 11.Cervera R, Serrano R, Pons-Estel GJ, Ceberio-Hualde L, Shoenfeld Y, de RE, Buonaiuto V, Jacobsen S, Zeher MM, Tarr T, Tincani A, Taglietti M, Theodossiades G, Nomikou E, Galeazzi M, Bellisai F, Meroni PL, Derksen RH, de Groot PG, Baleva M, Mosca M, Bombardieri S, Houssiau F, Gris JC, Quere I, Hachulla E, Vasconcelos C, Fernandez-Nebro A, Haro M, Amoura Z, Miyara M, Tektonidou M, Espinosa G, Bertolaccini ML and Khamashta MA. Morbidity and mortality in the antiphospholipid syndrome during a 10-year period: a multicentre prospective study of 1000 patients. Ann Rheum Dis. 2015;74:1011–1018. [DOI] [PubMed] [Google Scholar]
- 12.de Jesus GR, Agmon-Levin N, Andrade CA, Andreoli L, Chighizola CB, Porter TF, Salmon J, Silver RM, Tincani A and Branch DW. 14th International Congress on Antiphospholipid Antibodies Task Force report on obstetric antiphospholipid syndrome. Autoimmun Rev. 2014;13:795–813. [DOI] [PubMed] [Google Scholar]
- 13.Levy RA, Dos Santos FC, de Jesus GR and de Jesus NR. Antiphospholipid Antibodies and Antiphospholipid Syndrome during Pregnancy: Diagnostic Concepts. Front Immunol. 2015;6:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levine JS, Branch DW and Rauch J. The antiphospholipid syndrome. N Engl J Med. 2002;346:752–763. [DOI] [PubMed] [Google Scholar]
- 15.Gibbins KJ, Tebo AE, Nielsen SK and Branch DW. Antiphospholipid antibodies in women with severe preeclampsia and placental insufficiency: a case-control study. Lupus. 2018;27:1903–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chighizola CB, Andreoli L, de Jesus GR, Banzato A, Pons-Estel GJ and Erkan D. The association between antiphospholipid antibodies and pregnancy morbidity, stroke, myocardial infarction, and deep vein thrombosis: a critical review of the literature. Lupus. 2015;24:980–984. [DOI] [PubMed] [Google Scholar]
- 17.Sebire NJ, Fox H, Backos M, Rai R, Paterson C and Regan L. Defective endovascular trophoblast invasion in primary antiphospholipid antibody syndrome-associated early pregnancy failure. Hum Reprod. 2002;17:1067–1071. [DOI] [PubMed] [Google Scholar]
- 18.Parke AL. Placental pathology in antiphospholipid syndrome. Hughes’ Syndrome (ed Khamashta, M A). 2006;Chapter28. [Google Scholar]
- 19.de Groot PG and Urbanus RT. The significance of autoantibodies against beta2-glycoprotein I. Blood. 2012;120:266–274. [DOI] [PubMed] [Google Scholar]
- 20.Schreiber K, Sciascia S, de Groot PG, Devreese K, Jacobsen S, Ruiz-Irastorza G, Salmon JE, Shoenfeld Y, Shovman O and Hunt BJ. Antiphospholipid syndrome. Nat Rev Dis Primers. 2018;4:17103. [DOI] [PubMed] [Google Scholar]
- 21.Meroni PL, Borghi MO, Raschi E and Tedesco F. Pathogenesis of antiphospholipid syndrome: understanding the antibodies. Nat Rev Rheumatol. 2011;7:330–339. [DOI] [PubMed] [Google Scholar]
- 22.Urbanus RT, Derksen RH and de Groot PG. Platelets and the antiphospholipid syndrome. Lupus. 2008;17:888–894. [DOI] [PubMed] [Google Scholar]
- 23.Mineo C, Lanier L, Jung E, Sengupta S, Ulrich V, Sacharidou A, Tarango C, Osunbunmi O, Shen YM, Salmon JE, Brekken RA, Huang X, Thorpe PE and Shaul PW. Identification of a Monoclonal Antibody That Attenuates Antiphospholipid Syndrome-Related Pregnancy Complications and Thrombosis. PLoS One. 2016;11:e0158757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sacharidou A, Chambliss KL, Ulrich V, Salmon JE, Shen YM, Herz J, Hui DY, Terada LS, Shaul PW and Mineo C. Antiphospholipid antibodies induce thrombosis by PP2A activation via apoER2-Dab2-SHC1 complex formation in endothelium. Blood. 2018;131:2097–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ulrich V, Gelber SE, Vukelic M, Sacharidou A, Herz J, Urbanus RT, de Groot PG, Natale DR, Harihara A, Redecha P, Abrahams VM, Shaul PW, Salmon JE and Mineo C. ApoE Receptor 2 Mediation of Trophoblast Dysfunction and Pregnancy Complications Induced by Antiphospholipid Antibodies in Mice. Arthritis Rheumatol. 2016;68:730–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wenzel PL and Leone G. Expression of Cre recombinase in early diploid trophoblast cells of the mouse placenta. Genesis. 2007;45:129–134. [DOI] [PubMed] [Google Scholar]
- 27.Peng J, Vongpatanasin W, Sacharidou A, Kifer D, Yuhanna IS, Banerjee S, Tanigaki K, Polasek O, Chu H, Sundgren NC, Rohatgi A, Chambliss KL, Lauc G, Mineo C and Shaul PW. Supplementation With the Sialic Acid Precursor N-Acetyl-D-Mannosamine Breaks the Link Between Obesity and Hypertension. Circulation. 2019;140:2005–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sundgren NC, Vongpatanasin W, Boggan BM, Tanigaki K, Yuhanna IS, Chambliss KL, Mineo C and Shaul PW. IgG receptor FcgammaRIIB plays a key role in obesity-induced hypertension. Hypertension. 2015;65:456–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Straszewski-Chavez SL, Abrahams VM, Alvero AB, Aldo PB, Ma Y, Guller S, Romero R and Mor G. The isolation and characterization of a novel telomerase immortalized first trimester trophoblast cell line, Swan 71. Placenta. 2009;30:939–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trudgian DC and Mirzaei H. Cloud CPFP: a shotgun proteomics data analysis pipeline using cloud and high performance computing. J Proteome Res. 2012;11:6282–6290. [DOI] [PubMed] [Google Scholar]
- 31.Elias JE and Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. [DOI] [PubMed] [Google Scholar]
- 32.Feys J New Nonparametric Rank Tests for Interactions in Factorial Designs with Repeated Measures. J Mod Appl Stat Meth. 2016;15:78–99. [Google Scholar]
- 33.Wobbrock JO, Findlater L, Gergle D and Higgins JJ. The Aligned Rank Transform for Nonparametric Factorial Analyses Using Only ANOVA Procedures. 29th Annual Chi Conference on Human Factors in Computing Systems. 2011:143–146. [Google Scholar]
- 34.Leys C and Schumann S. A nonparametric method to analyze interactions: The adjusted rank transform test. J Exp Soc Psychol. 2010;46:684–688. [Google Scholar]
- 35.Andreoli L, Chighizola CB, Banzato A, Pons-Estel GJ, Ramire de JG and Erkan D. Estimated frequency of antiphospholipid antibodies in patients with pregnancy morbidity, stroke, myocardial infarction, and deep vein thrombosis: a critical review of the literature. Arthritis Care Res (Hoboken). 2013;65:1869–1873. [DOI] [PubMed] [Google Scholar]
- 36.Bose P, Kadyrov M, Goldin R, Hahn S, Backos M, Regan L and Huppertz B. Aberrations of early trophoblast differentiation predispose to pregnancy failure: lessons from the anti-phospholipid syndrome. Placenta. 2006;27:869–875. [DOI] [PubMed] [Google Scholar]
- 37.Janssens V, Longin S and Goris J. PP2A holoenzyme assembly: in cauda venenum (the sting is in the tail). Trends Biochem Sci. 2008;33:113–121. [DOI] [PubMed] [Google Scholar]
- 38.Sontag E Protein phosphatase 2A: the Trojan Horse of cellular signaling. Cell Signal. 2001;13:7–16. [DOI] [PubMed] [Google Scholar]
- 39.Bass KE, Morrish D, Roth I, Bhardwaj D, Taylor R, Zhou Y and Fisher SJ. Human cytotrophoblast invasion is up-regulated by epidermal growth factor: evidence that paracrine factors modify this process. Dev Biol. 1994;164:550–561. [DOI] [PubMed] [Google Scholar]
- 40.LaMarca HL, Dash PR, Vishnuthevan K, Harvey E, Sullivan DE, Morris CA and Whitley GS. Epidermal growth factor-stimulated extravillous cytotrophoblast motility is mediated by the activation of PI3-K, Akt and both p38 and p42/44 mitogen-activated protein kinases. Hum Reprod. 2008;23:1733–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qiu Q, Yang M, Tsang BK and Gruslin A. EGF-induced trophoblast secretion of MMP-9 and TIMP-1 involves activation of both PI3K and MAPK signalling pathways. Reproduction. 2004;128:355–363. [DOI] [PubMed] [Google Scholar]
- 42.Sents W, Ivanova E, Lambrecht C, Haesen D and Janssens V. The biogenesis of active protein phosphatase 2A holoenzymes: a tightly regulated process creating phosphatase specificity. FEBS J. 2013;280:644–661. [DOI] [PubMed] [Google Scholar]
- 43.Xu Y, Xing Y, Chen Y, Chao Y, Lin Z, Fan E, Yu JW, Strack S, Jeffrey PD and Shi Y. Structure of the protein phosphatase 2A holoenzyme. Cell. 2006;127:1239–1251. [DOI] [PubMed] [Google Scholar]
- 44.Chen J, Martin BL and Brautigan DL. Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science. 1992;257:1261–1264. [DOI] [PubMed] [Google Scholar]
- 45.Chen J, Parsons S and Brautigan DL. Tyrosine phosphorylation of protein phosphatase 2A in response to growth stimulation and v-src transformation of fibroblasts. J Biol Chem. 1994;269:7957–7962. [PubMed] [Google Scholar]
- 46.Tolstykh T, Lee J, Vafai S and Stock JB. Carboxyl methylation regulates phosphoprotein phosphatase 2A by controlling the association of regulatory B subunits. EMBO J. 2000;19:5682–5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xing Y, Li Z, Chen Y, Stock JB, Jeffrey PD and Shi Y. Structural mechanism of demethylation and inactivation of protein phosphatase 2A. Cell. 2008;133:154–163. [DOI] [PubMed] [Google Scholar]
- 48.Beffert U, Durudas A, Weeber EJ, Stolt PC, Giehl KM, Sweatt JD, Hammer RE and Herz J. Functional dissection of Reelin signaling by site-directed disruption of Disabled-1 adaptor binding to apolipoprotein E receptor 2: distinct roles in development and synaptic plasticity. J Neurosci. 2006;26:2041–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kelkar N, Standen CL and Davis RJ. Role of the JIP4 scaffold protein in the regulation of mitogen-activated protein kinase signaling pathways. Mol Cell Biol. 2005;25:2733–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morrison DK and Davis RJ. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu Rev Cell Dev Biol. 2003;19:91–118. [DOI] [PubMed] [Google Scholar]
- 51.Karumanchi SA and Granger JP. Preeclampsia and Pregnancy-Related Hypertensive Disorders. Hypertension. 2016;67:238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burke SD, Zsengeller ZK, Khankin EV, Lo AS, Rajakumar A, DuPont JJ, McCurley A, Moss ME, Zhang D, Clark CD, Wang A, Seely EW, Kang PM, Stillman IE, Jaffe IZ and Karumanchi SA. Soluble fms-like tyrosine kinase 1 promotes angiotensin II sensitivity in preeclampsia. J Clin Invest. 2016;126:2561–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, Epstein FH, Sukhatme VP and Karumanchi SA. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111:649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Venkatesha S, Toporsian M, Lam C, Hanai J, Mammoto T, Kim YM, Bdolah Y, Lim KH, Yuan HT, Libermann TA, Stillman IE, Roberts D, D’Amore PA, Epstein FH, Sellke FW, Romero R, Sukhatme VP, Letarte M and Karumanchi SA. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med. 2006;12:642–649. [DOI] [PubMed] [Google Scholar]
- 55.Valbuena-Diez AC, Blanco FJ, Oujo B, Langa C, Gonzalez-Nunez M, Llano E, Pendas AM, Diaz M, Castrillo A, Lopez-Novoa JM and Bernabeu C. Oxysterol-induced soluble endoglin release and its involvement in hypertension. Circulation. 2012;126:2612–24. [DOI] [PubMed] [Google Scholar]
- 56.Hawinkels LJ, Kuiper P, Wiercinska E, Verspaget HW, Liu Z, Pardali E, Sier CF and ten Dijke P. Matrix metalloproteinase-14 (MT1-MMP)-mediated endoglin shedding inhibits tumor angiogenesis. Cancer Res. 2010;70:4141–50. [DOI] [PubMed] [Google Scholar]
- 57.Kaitu’u-Lino TJ, Palmer K, Tuohey L, Ye L and Tong S. MMP-15 is upregulated in preeclampsia, but does not cleave endoglin to produce soluble endoglin. PLoS One. 2012;7:e39864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kaitu’u-Lino TJ, Palmer KR, Whitehead CL, Williams E, Lappas M and Tong S. MMP-14 is expressed in preeclamptic placentas and mediates release of soluble endoglin. Am J Pathol. 2012;180:888–894. [DOI] [PubMed] [Google Scholar]
- 59.Li CC, Yu FS, Fan MJ, Chen YY, Lien JC, Chou YC, Lu HF, Tang NY, Peng SF, Huang WW and Chung JG. Anticancer effects of cantharidin in A431 human skin cancer (Epidermoid carcinoma) cells in vitro and in vivo. Environ Toxicol. 2016. [DOI] [PubMed] [Google Scholar]
- 60.Li W, Xie L, Chen Z, Zhu Y, Sun Y, Miao Y, Xu Z and Han X. Cantharidin, a potent and selective PP2A inhibitor, induces an oxidative stress-independent growth inhibition of pancreatic cancer cells through G2/M cell-cycle arrest and apoptosis. Cancer Sci. 2010;101:1226–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li YM, Mackintosh C and Casida JE. Protein phosphatase 2A and its [3H]cantharidin/[3H]endothall thioanhydride binding site. Inhibitor specificity of cantharidin and ATP analogues. Biochem Pharmacol. 1993;46:1435–1443. [DOI] [PubMed] [Google Scholar]
- 62.Rajakumar A, Brandon HM, Daftary A, Ness R and Conrad KP. Evidence for the functional activity of hypoxia-inducible transcription factors overexpressed in preeclamptic placentae. Placenta. 2004;25:763–9. [DOI] [PubMed] [Google Scholar]
- 63.Tal R, Shaish A, Barshack I, Polak-Charcon S, Afek A, Volkov A, Feldman B, Avivi C and Harats D. Effects of hypoxia-inducible factor-1alpha overexpression in pregnant mice: possible implications for preeclampsia and intrauterine growth restriction. Am J Pathol. 2010;177:2950–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Albers RE, Kaufman MR, Natale BV, Keoni C, Kulkarni-Datar K, Min S, Williams CR, Natale DRC and Brown TL. Trophoblast-Specific Expression of Hif-1alpha Results in Preeclampsia-Like Symptoms and Fetal Growth Restriction. Sci Rep. 2019;9:2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sanchez-Elsner T, Botella LM, Velasco B, Langa C and Bernabeu C. Endoglin expression is regulated by transcriptional cooperation between the hypoxia and transforming growth factor-beta pathways. J Biol Chem. 2002;277:43799–808. [DOI] [PubMed] [Google Scholar]
- 66.Yinon Y, Nevo O, Xu J, Many A, Rolfo A, Todros T, Post M and Caniggia I. Severe intrauterine growth restriction pregnancies have increased placental endoglin levels: hypoxic regulation via transforming growth factor-beta 3. Am J Pathol. 2008;172:77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Di Conza G, Trusso Cafarello S, Loroch S, Mennerich D, Deschoemaeker S, Di Matteo M, Ehling M, Gevaert K, Prenen H, Zahedi RP, Sickmann A, Kietzmann T, Moretti F and Mazzone M. The mTOR and PP2A Pathways Regulate PHD2 Phosphorylation to Fine-Tune HIF1alpha Levels and Colorectal Cancer Cell Survival under Hypoxia. Cell Rep. 2017;18:1699–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang Y, Xia Y, Kuang D, Duan Y and Wang G. PP2A regulates SCF-induced cardiac stem cell migration through interaction with p38 MAPK. Life Sci. 2017;191:59–67. [DOI] [PubMed] [Google Scholar]
- 69.Maynard S, Epstein FH and Karumanchi SA. Preeclampsia and angiogenic imbalance. Annu Rev Med. 2008;59:61–78. [DOI] [PubMed] [Google Scholar]
- 70.Levine RJ, Maynard SE, Qian C, Lim KH, England LJ, Yu KF, Schisterman EF, Thadhani R, Sachs BP, Epstein FH, Sibai BM, Sukhatme VP and Karumanchi SA. Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med. 2004;350:672–683. [DOI] [PubMed] [Google Scholar]
- 71.Murray SA, Morgan JL, Kane C, Sharma Y, Heffner CS, Lake J and Donahue LR. Mouse gestation length is genetically determined. PLoS One. 2010;5:e12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.He J, Luster TA and Thorpe PE. Radiation-enhanced vascular targeting of human lung cancers in mice with a monoclonal antibody that binds anionic phospholipids. Clin Cancer Res. 2007;13:5211–5218. [DOI] [PubMed] [Google Scholar]
- 73.Tanigaki K, Sacharidou A, Peng J, Chambliss KL, Yuhanna IS, Ghosh D, Ahmed M, Szalai AJ, Vongpatanasin W, Mattrey RF, Chen Q, Azadi P, Lingvay I, Botto M, Holland WL, Kohler JJ, Sirsi SR, Hoyt K, Shaul PW and Mineo C. Hyposialylated IgG activates endothelial IgG receptor FcgammaRIIB to promote obesity-induced insulin resistance. J Clin Invest. 2018;128:309–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Natale BV, Mehta P, Vu P, Schweitzer C, Gustin K, Kotadia R and Natale DRC. Reduced Uteroplacental Perfusion Pressure (RUPP) causes altered trophoblast differentiation and pericyte reduction in the mouse placenta labyrinth. Sci Rep. 2018;8:17162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Min S, Natale BV and Natale DRC. Temporal and spatial expression of glyceraldehyde 3-phosphate dehydrogenase (Gapdh) in the mouse placenta. Placenta. 2017;57:170–174. [DOI] [PubMed] [Google Scholar]
- 76.Natale BV, Schweitzer C, Hughes M, Globisch MA, Kotadia R, Tremblay E, Vu P, Cross JC and Natale DRC. Sca-1 identifies a trophoblast population with multipotent potential in the mid-gestation mouse placenta. Sci Rep. 2017;7:5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Anson-Cartwright L, Dawson K, Holmyard D, Fisher SJ, Lazzarini RA and Cross JC. The glial cells missing-1 protein is essential for branching morphogenesis in the chorioallantoic placenta. Nat Genet. 2000;25:311–4. [DOI] [PubMed] [Google Scholar]
- 78.Basyuk E, Cross JC, Corbin J, Nakayama H, Hunter P, Nait-Oumesmar B and Lazzarini RA. Murine Gcm1 gene is expressed in a subset of placental trophoblast cells. Dev Dyn. 1999;214:303–11. [DOI] [PubMed] [Google Scholar]
- 79.Simmons DG, Natale DR, Begay V, Hughes M, Leutz A and Cross JC. Early patterning of the chorion leads to the trilaminar trophoblast cell structure in the placental labyrinth. Development. 2008;135:2083–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kenchegowda D, Natale B, Lemus MA, Natale DR and Fisher SA. Inactivation of maternal Hif-1alpha at mid-pregnancy causes placental defects and deficits in oxygen delivery to the fetal organs under hypoxic stress. Dev Biol. 2017;422:171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.