

Abstract
Purpose:
Systems biology approaches can identify critical targets in complex cancer signaling networks to inform new therapy combinations that may overcome conventional treatment resistance.
Experimental Design:
We performed integrated analysis of 1,046 childhood B-ALL cases and developed a data-driven network controllability-based approach to identify synergistic key regulator targets in Philadelphia chromosome–like B-acute lymphoblastic leukemia (Ph-like B-ALL), a common high-risk leukemia subtype associated with hyperactive signal transduction and chemoresistance.
Results:
We identified 14 dysregulated network nodes in Ph-like ALL involved in aberrant JAK/STAT, Ras/MAPK, and apoptosis pathways and other critical processes. Genetic cotargeting of the synergistic key regulator pair STAT5B and BCL2-associated athanogene 1 (BAG1) significantly reduced leukemia cell viability in vitro. Pharmacologic inhibition with dual small molecule inhibitor therapy targeting this pair of key nodes further demonstrated enhanced antileukemia efficacy of combining the BCL-2 inhibitor venetoclax with the tyrosine kinase inhibitors ruxolitinib or dasatinib in vitro in human Ph-like ALL cell lines and in vivo in multiple childhood Ph-like ALL patient-derived xenograft models. Consistent with network controllability theory, co-inhibitor treatment also shifted the transcriptomic state of Ph-like ALL cells to become less like kinase-activated BCR-ABL1–rearranged (Ph+) B-ALL and more similar to prognostically favorable childhood B-ALL subtypes.
Conclusions:
Our study represents a powerful conceptual framework for combinatorial drug discovery based on systematic interrogation of synergistic vulnerability pathways with pharmacologic inhibitor validation in preclinical human leukemia models.
Translational Relevance.
We performed unbiased integrated network analysis of large-scale patient genomic and transcriptomic datasets to identify previously unrecognized targetable synergistic regulators in human Ph-like ALL. We then queried drug databases for clinically available drugs and discovered synergistic efficacy of cotargeting the pair of top-ranked regulators BCL-2 and STAT5 with venetoclax and ruxolitinib or dasatinib, respectively, in vitro in human Ph-like ALL cell lines and in vivo in Ph-like ALL patient-derived xenograft models. This dual inhibitor precision medicine strategy is imminently translatable to the clinic given the established therapeutic dosing of these drugs and the high rates of chemoresistance and relapse in patients with Ph-like ALL. Our combinatorial genetic target discovery and pharmacologic validation approach may be broadly applicable to interrogation of other human cancers.
Introduction
Cancer cells exploit multiple deregulated pathways to evade the selective pressure of single-agent drugs, promoting therapeutic resistance and clinical relapse. However, combination therapy regimens for cancer have traditionally been nonspecific with broad toxicity profiles and developed in an ad hoc manner. More rational identification of new targets in human cancers for combination drug regimens is an essential next step. There is growing interest in identifying synergistic genetic interactions as targets for combination therapy (1), but large-scale experimental screening for genetic interactions has been technically challenging and expensive given the large number of candidate gene pairs one has to screen. As a result, existing RNA-interference and CRISPR-based screenings have been limited to only a few hundred genes (2, 3), far from saturating the search space of all possible (∼4×108) pairwise interactions in the human genome. Given the above challenges, we developed a systems biology approach that enables efficient in silico genetic screening and prioritization of co-targetable pathways for combinatorial therapeutics followed by rigorous in vitro and in vivo pharmacologic validation in a difficult-to-cure subtype of leukemia.
Philadelphia chromosome–like acute lymphoblastic leukemia (Ph-like ALL) comprises 15% to 30% of high-risk B-ALL cases in children and adolescents/young adults (AYA) and 20% to 40% in older adults (4–6), and is associated with high rates of conventional chemotherapy resistance and poor clinical outcomes (6, 7). Ph-like ALL is defined by a kinase-activated transcriptomic signature resembling that of Philadelphia chromosome-positive (Ph+) ALL, but lacks the BCR-ABL1 rearrangement (8). Ph-like ALL is instead driven by alternative genetic alterations in two major subclasses: (i) JAK/STAT pathway alterations involving CRLF2, JAK2, EPOR, IL7R, or SH2B3 rearrangements or indels and (ii) ABL-class kinase fusions involving ABL1, ABL2, CSF1R, or PDGFRB rearrangements (7). Preclinical studies of tyrosine kinase inhibitor (TKI) monotherapy in Ph-like ALL models have expectedly demonstrated incomplete anti-leukemia activity (9–12) likely via compensatory signaling mechanisms, emphasizing the need for more rationally-designed combination therapy approaches to achieve cure. In the present studies, we hypothesized that an unbiased systems biology approach could effectively elucidate optimal target pairings. Our network-based analysis is optimal to address the unique challenges of Ph-like ALL given its known dysregulation of multiple intracellular pathways that maintain a high degree of crosstalk.
A main goal of effective multiagent therapy is identifying drug combinations with synergistic efficacies, but not synergistic toxicities. We recently reported our Optimal Control algorithm (OptiCon) (13) that is capable of discovering novel disease-specific synergistic regulators by integrating a molecular interaction network with large-scale patient genomic and transcriptomic data. OptiCon is based on the theory of network controllability, a mathematically validated framework for identifying a set of driver nodes in a complex network that can guide the system from an initial state to any desired final state (14). OptiCon integrates clinical, genomic and expression data, as well as a gene regulatory network to identify critical network nodes termed optimal control nodes (OCN) that control a maximal number of deregulated genes (for optimal therapeutic efficacy) and a minimal number of unperturbed genes (for toxicity minimization). Synergistic regulators (OCN pairs) are then nominated on the basis of the synergy score, which quantifies the degree of crosstalk between pathways downstream of the two OCNs and the amount of enrichment for deregulated and mutated genes in the optimal control regions (OCR) of the two OCNs (13). In the current study, we leveraged this powerful computational tool to identify key oncogenic dependencies in Ph-like ALL and to prioritize pathways for pharmacologic targeting in vitro and in vivo using human cell lines and various preclinical patient-derived xenograft (PDX) models of CRLF2-R or ABL-class Ph-like ALL.
Materials and Methods
Data and code availability
The results published here are in part based upon data generated by the Therapeutically Applicable Research to Generate Effective Treatments (TARGET) initiative (https://ocg.cancer.gov/programs/target). Whole genome sequencing (WGS), whole exome sequencing (WES), and microarray expression data were downloaded from dbGaP with study identifier phs000218 (including phs000463, phs000464). The data used for these analyses are available at https://portal.gdc.cancer.gov/projects. Transcriptomic datasets of childhood B-ALL samples were downloaded from publicly available databases as specified in “Source Details” of Supplementary Table S1. The drug/gene databases were downloaded from the Therapeutic Target Database (TTD; ref. 15), DrugBank (16), and DGIdb (17). The RNA sequencing data of untreated human B-ALL cell lines (except TVA-1) were downloaded from the Broad Institute Cancer Cell Line Encyclopedia (CCLE) available at https://portals.broadinstitute.org/ccle. The RNA sequencing data generated in this study is deposited at Gene Expression Ominbus (GEO) under the accession number GSE161939. All software supporting the analysis in this study can be found in public repositories. Software package implementing the OptiCon algorithm has been deposited at GitHub (https://github.com/tanlabcode/OptiCon).
Prediction of candidate combination therapeutic targets using OptiCon
We used our recently developed computational algorithm, OptiCon (13), to nominate candidate combination therapeutic targets. Inputs to the algorithm consists of a gene regulatory network, genetic mutation data, and gene expression data. A high-quality gene regulatory network was generated by combining known expert-curated pathway annotations from three public pathway databases: KEGG (1,597 pathways; ref. 18), Reactome (195 pathways; ref. 19), and NCI Pathway Interaction Database (PID; 745 pathways; Supplementary Table S5; ref. 20). All pathways were downloaded in the Simple Interaction Format from Pathway Commons 252. We also removed undirected, redundant, and small-molecule-associated interactions to generate a regulatory network comprising 5959 nodes (genes) and 108,281 directed edges (regulatory links). Mutation information used included gene fusions (Supplementary Table S2), copy number variations, and missense or nonsense mutations, called using the pipeline detailed in He and colleagues (21) to analyze TARGET and Pediatric Cancer Genome Project (PCGP) WGS, WES, and microarray data, to generate a list of recurrently mutated genes in B-ALL patients (Supplementary Table S3). The third input includes differential gene expression data (Supplementary Table S4). Patient gene expression microarray datasets were downloaded from the TARGET project and PCGP (Supplementary Table S1). Differential gene expression between Ph-like ALL and favorable-risk genetic subtypes of B-ALL samples [ETV6-RUNX1 (22), ERG-deleted (23), and hyperdiploid (24)] was performed using linear models for microarray data (LIMMA). Default parameter setting of OptiCon was used and a deregulation score (DScore)-weighted network was obtained. Synergy score between two OCNs in the network is calculated and consists of two parts: the enrichment of recurrently mutated cancer genes in the OCR of each OCN and the interaction density between genes in the OCRs of the two OCNs. Identification of OCR genes of each OCN and calculation of DScore and synergy scores are defined as in the OptiCon manuscript (13). To identify significantly synergistic OCN pairs, we generated a null distribution of synergy scores based on 10 million randomly selected gene pairs from the input regulatory network. The OCN pairs with an empirical P value < 0.05 were predicted as significantly synergistic. P values were adjusted for multiple testing using the method of Benjamini–Hochberg.
Chemicals and reagents
Venetoclax, dasatinib, and ruxolitinib were purchased from LC Laboratories (catalog nos. V-3579, D-3307, and R-6688, respectively) and solubilized at 50 mmol/L stock in DMSO. For in vivo PDX model treatment, dasatinib was dissolved in 10% citric acid in 80 mmol/L sodium citrate (dasatinib vehicle), and venetoclax was dissolved in 60% phospho-polyethylene glycol 50, 30% polyethylene glycol 400, 10% ethanol (venetoclax vehicle). Ruxolitinib 2 g/kg chow was kindly provided by the Incyte Corporation. Annexin V assays were performed using Annexin V-FITC and PI co-staining (BioSciences, #640914) on a BD FACSVerse flow cytometer (Supplementary Fig. S5).
Cell culture of leukemia cell lines
The Ph-like B-ALL cell lines MUTZ5 (IGH-CRLF2 translocation and JAK2 R683G) and MHH-cALL4 (IGH-CRLF2 translocation and JAK2 I682F), and the non-Ph-like B-ALL cell lines REH (ETV6-RUNX1 translocation) and NALM6 (ETV6-PDGFRB translocation) were obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ; https://www.dsmz.de/) cell biorepository. TVA-1 cells with ETV6-ABL1 fusion were immortalized in vitro as a cell line from a PDX model established by the laboratory of Dr. David Fruman at the University of California, Irvine (25). Cells were cultured in RPMI1640 (31870–025; Invitrogen) supplemented with 20% FBS, 1% Glutamax, 1% HEPES, 1% NEAA, and 100 unit/ml penicillin/streptomycin (MUTZ5 and MHH-cALL4) or RPMI supplemented with 10% FBS and 100 unit/mL penicillin/streptomycin (TVA-1, REH, and NALM6), and fresh cells were thawed for experiments every 3 months. MUTZ5, MHH-cALL-4, REH, and NALM6 cell lines were validated by ATCC STR profiling. All cell lines were confirmed to be Mycoplasma-free every 6 months.
CRISPR/cas9-mediated double knockout of predicted gene pairs
Electroporation of guide RNA/Cas9 ribonucleoprotein complex was performed according to IDT's Alt-R CRISPR-Cas9 protocol. sgRNA was mixed with recombinant Cas9 (TrueCut Cas9 Protein v2; Thermo Fisher Scientific) in a 1:2 ratio to produce RNA ribonucleoprotein (RNP) complexes. Individually complexed RNPs targeting each gene of a pair were then combined for simultaneous targeting of a gene pair (for a final ratio of 2.5 μg total gRNA and 5 μg Cas9 protein per 1 million cells). For single gene targeting, RNP complexed to sgRNA against a single gene was combined with RNP complexed to nontargeting control (NTC) sgRNA. Leukemia cells underwent nucleofection with RNP complexes using the Amaxa 4D-Nucleofector system and pulse code EO-117. Cells electroporated with RNP complexed to nontargeting control (NTC) gRNA was used as the negative control. After recovery overnight, cells were replated in triplicate for viability assays using the Cell-TiterGlo assay, at 4 days and 9 days post-nucleofection. Gene editing efficiency for each reaction was calculated using the Tracking of Indels by Decomposition (TIDE) assay (26); Q5 DNA polymerase (NEB #M0492S) was used and TIDE software is available at https://tide.nki.nl/. sgRNA guide sequences were synthesized by IDT and are as follows: TAAGAGGTCAGACCGTCGTG (STAT5B), TGAACCAGTTGTCCAAGACC (BAG1), CGTTAATCGCGTATAATACG (nontargeting control, IDT no. 1072544/Alt-R CRISPR Negative Control No. 1). Primer sequences for Sanger sequencing-based TIDE assay are as follows: AAGTGAAACAGTTCTCAGGG (STAT5B Forward), TTGAACAACTGCTGCGT (STAT5B Reverse), GGCCATAAGGAAAAGCCGG (BAG1 Forward), GAGTGACCTTGGGATGGACG (BAG1 Reverse).
Drug synergy testing in cell lines
Human ALL cell lines were incubated at drug concentrations ranging between 1 nmol/L and 50 μmol/L for 72 hours, and cell viability was assessed by Cell-Titer Glo viability assays (Promega). Cells were plated in triplicate in 96-well plates at 50,000 cells/well and treated with drug at the indicated concentrations individually or in combination. Percent cell growth was calculated relative to 0.1% DMSO vehicle treated cells and normalized (using values of vehicle-treated cells set to 100% and medium alone without viable cells set to 0%) and displayed graphically in Prism. IC50 values were determined using GraphPad Prism and Compusyn software. Combination index (CI) values were calculated using Compusyn software (27). Each monotherapy and combination drug assay was repeated at least three times for each studied cell line.
RNA sequencing of drug-treated cells
MUTZ5, MHH-cALL-4, and TVA-1 cells were cultured in medium containing vehicle, TKI alone, venetoclax alone, or in combination at indicated concentrations for 72 hours. Total RNA was extracted using RNeasy Micro Kit (Qiagen) and treated with DNAse, and SMART-seq v4 Ultra Low Input RNA Kit (Takara) and Nextera XT DNA Library Prep Kit (Illumina) were used for library preparation using 10 ng of total RNA as input. Paired-end sequencing was performed using the Illumina NovaSeq 6000 platform with a 150-bp read length at the Children's Hospital of Philadelphia. Sequencing reads were aligned to the human genome hg38 reference sequence using STAR v3.5.3a. Samples all had >70% reads mapped to exonic regions and no 3′-bias. Gene-based FPKM and read count matrices were performed with Cufflinks v2.2.1 and featureCounts v1.3.6.
Transcriptome analysis methods
Differential gene expression analysis
In the Ph-like ALL cell line studies, differentially expressed genes (DEG) between drug treatment conditions were identified using LIMMA-voom after filtering to include protein-coding genes with at least five reads in greater than or equal to 50% of samples. DEGs were identified using a cutoff of FDR <0.1 and absolute log2 fold change > 1. Adjusted P values were computed using the Benjamini–Hochberg method.
Gene ontology analysis
Genes from OCR of each OCN were input into DAVID (28) for enrichment analysis. The top three most significant biological process (ranked by FDR) terms for each OCR set were combined and redundant terms were merged.
Dimensionality reduction
To generate a co-embedding between microarray expression profiles of publicly-available primary childhood B-ALL specimens (TARGET and PCGP datasets) and our Ph-like ALL cell line RNA-Seq data, we first performed quantile normalization on combined gene expression matrices consisting of the microarray-based primary patient specimen expression profiles and log-transformed FPKM gene expression values from the RNA-seq. ComBat was used to account for technical variation between technologies, treating the microarray and RNA-seq technologies as a batch covariate. Principal component analysis (PCA) was performed on the microarray component of the jointly normalized expression matrix. Uniform Manifold Approximation and Projection (UMAP) was performed with 20 neighbors on principal components 2 to 20, which we found to clearly separate expression profiles by B-ALL subtype. Cell line RNA-seq data were projected onto principal component space using the loadings matrix and subsequently projected onto the UMAP embedding.
Gene signature scores for B-ALL subtypes
Gene signature scores were developed for each B-ALL subtype in comparison to Ph+/Ph-like B-ALL. To define the gene signatures for each subtype, we performed LIMMA-voom on TARGET and PCGP patient specimen microarray expression profiles using B-ALL subtype as a covariate with the combined Ph+/Ph-like subtypes as baseline. For the ETV6-RUNX1, hyperdiploidy, TCF3-PBX1, and KMT2A-rearranged subtypes, gene signatures were defined as the top 500 significantly DEGs compared with the Ph+/Ph-like subtypes, with FDR < 0.05 and log-fold change greater than 0. Using these gene signatures, we performed single-sample gene set enrichment analysis (29) on the cell-line RNA-seq data, which is a rank-based procedure that produces a single-sample score for each B-ALL subtype. Statistical significance between gene signature scores for pairs of drug treatment groups was assessed with Welch t test.
Immunoblotting
Protein and phosphoprotein levels were measured by Western blot analysis. Whole cell protein lysates were extracted using RIPA buffer supplemented with protease and phosphatase inhibitor cocktail (Santa Cruz Biotechnology), and protein concentrations were determined using BCA protein assay reagent (Bio-Rad). Primary unconjugated antibodies for immunoblotting were obtained from Cell Signaling Technology (CST), including anti-pSTAT5 Y694 (CST-9351), total STAT5 (CST-94205), pERK T202/Y204 (CST-4370), total ERK (CST-9102), pJNK T183/Y185 (CST-4668), total JNK (CST-9252), BCL-2 (CST-4223), MCL-1 (CST-94296), BCL-xL (CST-2762), BIM (CST-2933), β-actin (rabbit; CST-8457), β-tubulin (CST-5346), and GAPDH (CST-5174). Antibodies against BAG1 and β-actin (mouse) were obtained from Santa Cruz Biotechnology (sc-56003) and from Millipore (MAB1501), respectively. Primary antibodies were used in combination with anti-rabbit or anti-mouse HRP-linked secondary antibodies (Cell Signaling Technology) and SuperSignal West Pico PLUS ECL detection reagent (Thermo Fisher Scientific). Immunoblot signals were quantified by densitometry using ImageStudio software. Target protein densitometry signals were normalized to its individual lane β-actin or β-tubulin or GAPDH loading controls, and displayed graphically relative to 0.1% DMSO control. Apoptosis protein arrays were also performed on treated cell lysates using the Human Apoptosis Array Kit according to the manufacturer's protocol (R&D Systems, #ARY009).
PDX modeling and in vivo drug combination trials
PDX models of childhood Ph-like ALL were established as described previously (9, 10, 30). Briefly, viably cryopreserved diagnostic bone marrow ALL cells (informed written consent obtained from patients or guardians on IRB-approved Children's Oncology Group or Children's Hospital of Philadelphia biobanking research protocols in accordance with the Declaration of Helsinki) were engrafted into NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice. Murine peripheral blood was monitored for engraftment of human ALL cells by flow cytometry using antibodies (CD10-PE Cy7 Invitrogen #25-0106-2, CD19-PE Invitrogen #12-0199-42, CD45-APC Invitrogen #17-9459-42). Once ≥1% human ALL was detected in murine peripheral blood, PDX mice were randomized to receive either vehicle (see “Chemicals and Reagents” above), single agent treatment with TKI alone, venetoclax (50 or 100 mg/kg once daily oral gavage 5 days a week) alone, or both drugs at indicated doses as specified in figure legends. Dosing for dasatinib, ruxolitinib, and venetoclax was selected based upon prior studies by our group or others in leukemia PDX models (10, 31, 32). PDX models with CRLF2/JAK2 alterations (UP_ALL4988, ALL4364, JH331, ALL2128) received ruxolitinib, whereas ABL1-rearranged PDX models (TVA-1, NH011) were treated with dasatinib. Ruxolitinib dosing was administered as a 2 g/kg rodent chow orally ad libitum. The other inhibitors were administered at either full dose (100 mg/kg/day venetoclax, 10 mg/kg/day dasatinib) or half dose (50 mg/kg/day venetoclax, 5 mg/kg/dose dasatinib) once daily via oral gavage for 5 days per week to assess for potential dose-dependent combinatorial effects. For non–luciferase-expressing PDX models, human ALL cells were enumerated weekly by quantitative flow cytometric analysis of retro-orbital murine venous blood samples as above. Total human ALL burden was quantified in spleens of sacrificed animals after 21 to 28 days of treatment. For the luciferase-expressing TVA-1 PDX model, mice were followed by weekly bioluminescent imaging to quantify total human ALL burden as described previously (33) and shown in Fig. 5F. All PDX studies were performed on protocols approved by the Institutional Animal Care and Use Committee (IACUC) of The Children's Hospital of Philadelphia in accordance with NIH standards. Flow cytometry data were analyzed using Cytobank. Statistical analysis via ANOVA (two-way for blood analyses, one-way for spleen analyses) with Dunnett's post-test for multiple comparisons were performed with GraphPad Prism.
Figure 5.
![Figure 5. Combined TKI and venetoclax treatment inhibits Ph-like ALL proliferation in vivo. A–D, CRLF2-rearranged (n = 4) and (E) ABL1-rearranged (n = 1) Ph-like ALL PDX models were treated with vehicle or inhibitors (5 mice/cohort) as delineated above and followed by flow cytometric quantification of human CD10+/CD19+ ALL in murine peripheral blood (left) and in end-study spleens (middle and right). F, The luciferase-expressing ABL1-rearranged TVA-1 PDX model was followed by bioluminescent imaging [flux measured in photons per second (p/s) with scales as designated] with terminal splenic leukemia burden quantified by flow cytometry as in the other five models. TKI and venetoclax cotreatment significantly inhibited leukemia proliferation in vivo in most PDX models versus inhibitor monotherapy. Error bars represent ± SD. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001 using 2-way ANOVA for peripheral blood analyses over time and one-way ANOVA for end-study spleen analyses. ns, not significant.](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=9401533_5109fig5.jpg)
Combined TKI and venetoclax treatment inhibits Ph-like ALL proliferation in vivo. A–D, CRLF2-rearranged (n = 4) and (E) ABL1-rearranged (n = 1) Ph-like ALL PDX models were treated with vehicle or inhibitors (5 mice/cohort) as delineated above and followed by flow cytometric quantification of human CD10+/CD19+ ALL in murine peripheral blood (left) and in end-study spleens (middle and right). F, The luciferase-expressing ABL1-rearranged TVA-1 PDX model was followed by bioluminescent imaging [flux measured in photons per second (p/s) with scales as designated] with terminal splenic leukemia burden quantified by flow cytometry as in the other five models. TKI and venetoclax cotreatment significantly inhibited leukemia proliferation in vivo in most PDX models versus inhibitor monotherapy. Error bars represent ± SD. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001 using 2-way ANOVA for peripheral blood analyses over time and one-way ANOVA for end-study spleen analyses. ns, not significant.
Results
Network-controllability analysis of patient omics datasets identifies targetable synergistic regulators in the Ph-like ALL gene network
We applied our unbiased OptiCon algorithm (13) to the study of Ph-like ALL with an overarching goal of identifying synergistic target pairings for biologically-rational combination therapy (Fig. 1A). To this end, we analyzed whole genome sequencing, whole exome sequencing, and gene expression microarray data from 1,046 primary childhood/AYA B-ALL specimens (of which 289 were Ph-like) generated by the NIH-sponsored Therapeutically Applicable Research to Generate Effective Treatments (TARGET) project (34) and the Pediatric Cancer Genome Project (PCGP; Materials and Methods; Supplementary Table S1; ref. 4). We identified structural variants, small indels, and point mutations (Supplementary Tables S2 and S3) and DEGs (Supplementary Table S4) in Ph-like B-ALL as compared with prognostically-favorable B-ALL subtypes that are highly curable with conventional chemotherapy, including the ETV6-RUNX1 (22), high hyperdiploidy (24), and DUX4-rearranged/ERG-dysregulated subtypes (23), to elucidate the key genetic dependencies that may render Ph-like ALL less sensitive to conventional chemotherapy. Using these data and a high-quality curated gene regulatory network (Supplementary Table S5) as inputs, OptiCon analysis predicted 81 key regulator gene pairs to be significantly synergistic in Ph-like ALL (synergy score P-values <0.05; Supplementary Table S6), which represent specific pairings of 14 OCNs (Table 1; Supplementary Fig. S1B).
Figure 1.
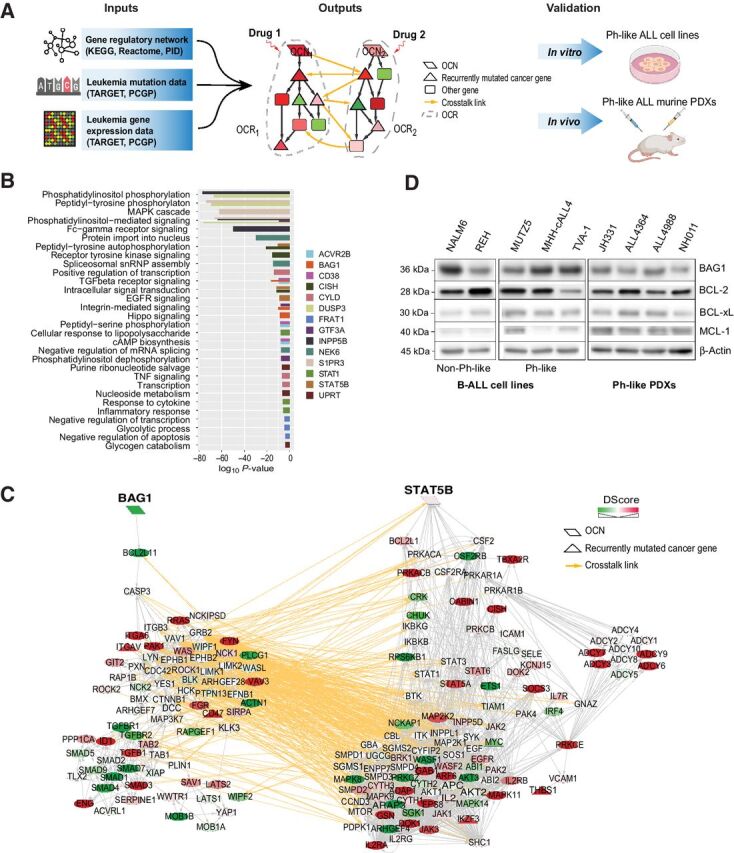
A systems biology approach to discovery and testing of synergistic therapeutic targets. A, Overview of the Optimal Control (OptiCon) network-based approach toward identifying and validating synergistic drug targets in Ph-like B-ALL. OptiCon input data include a high-quality human gene regulatory network integrating expert-curated pathway annotations from three public pathway databases (KEGG, Reactome, and NCI-Nature Pathway Interaction Database), patient genetic mutation data, and gene expression data from TARGET and PCGP consortia (details in Materials and Methods). Output of OptiCon identifies synergistic OCN pairs. Druggable pathways defined by OCNs and their respective OCRs are then validated in vitro in Ph-like ALL cell lines and in vivo in murine PDX models. B, Enriched gene ontology (GO) terms among predicted OCRs. Each color represents the OCR of a predicted OCN. Thickness of bars varies since certain terms were enriched among multiple OCRs. C, Synergistic OCN pair STAT5B and BAG1 predicted for Ph-like ALL. Gene crosstalk links between their specific OCRs are shown in yellow. Shade of a node represents the deregulation score (DScore) of the corresponding gene. Red, up-regulated in Ph-like ALL; green, down-regulated in Ph-like ALL. D, Baseline protein expression of BAG1 and the anti-apoptotic BCL2-family proteins BCL2, BCL-xL, and MCL1 in several B-ALL and Ph-like ALL cell lines and Ph-like PDXs.
Table 1.
OCNs predicted to be synergistic key regulators in Ph-like ALL and their known drugs or small molecule inhibitors.
| OCN | Name | Drug/compound | Source |
|---|---|---|---|
| ACVR2B | Activin A receptor type 2B | Bimagrumab | DGIdb |
| BAG1 | BCL2 associated athanogene 1 | (2R,3R,4S,5R)-2-[6-amino-8-[(3,4-dichlorophenyl)methylamino]purin-9-yl]-5-(hydroxymethyl)oxolane-3,4-diol | DrugBank |
| CD38 | CD38 molecule | Daratumumab, isatuximab, MOR-202, SAR-650984 | DGIdb, TTD, DrugBank |
| CISH | Cytokine inducible SH2 containing protein | Epoetin alfa | DGIdb |
| CYLD | CYLD lysine 63 deubiquitinase | NA | NA |
| DUSP3 | Dual specificity phosphatase 3 | NA | NA |
| FRAT1 | FRAT1, WNT signaling pathway regulator | NA | NA |
| GTF3A | General transcription factor IIIA | NA | NA |
| INPP5B | Inositol polyphosphate-5-phosphatase B | D-myo-inositol-1,4-bisphosphate | DrugBank |
| NEK6 | NIMA-related kinase 6 | 8205, (5Z)-2-hydroxy-4-methyl-6-oxo-5-[(5-phenylfuran-2-yl)methylidene]-5,6-dihydropyridine-3-carbonitrile | DGIdb, PubChem |
| S1PR3 | Sphingosine-1-phosphate receptor 3 | AFD(R), AUY954, EDD7H9, FTY720-phosphate, VPC03090-P, VPC12249, VPC23019, VPC44116, compound 26 | DGIdb, TTD |
| STAT1 | Signal transducer and activator of transcription 1 | AVT-02 UE | DGIdb,TTD |
| STAT5B | Signal transducer and activator of transcription 5B | Dasatinib, ruxolitinib | DGIdb, DrugBank |
| UPRT | Uracil phosphoribosyltransferase homolog | NA | NA |
Pathway enrichment analysis of these Ph-like ALL-specific OCNs and their respective OCR genes showed enrichment in multiple kinase signaling pathways, in regulation of transcription and apoptosis, and in metabolic processes such as glycolysis and nucleoside metabolism (Fig. 1B). Importantly, OptiCon identified STAT5B and CISH (cytokine-inducible SH2-containing protein, a known negative regulator of STAT5B) as OCNs, supporting the robustness of our methodology because STAT5B is a major known effector in Ph-like ALL (4, 35). Relatedly, PI3K and SRC family kinases (LYN, LCK, SRC) were also identified in the downstream OCRs of several predicted OCNs in Ph-like ALL (Fig. 1C; Supplementary Table S7). These findings collectively serve as unbiased support for our and others' prior pharmacologic studies that demonstrated effective targeting of JAK/STAT, PI3K, and SRC kinase pathway signaling in primary Ph-like ALL cells and preclinical models (12, 25, 30). Importantly, OptiCon identified novel OCNs not previously known to be targets in Ph-like ALL, including BAG1 (BCL2-associated athanogene 1), DUSP3 (dual specificity phosphatase 3), CD38, and NEK6 (never in mitosis gene A-related kinase 6); these genes have all been implicated in other leukemias or in tumorigenesis (36–39). We further identified several anti- and pro-apoptotic genes, including BCL2, BCL2L11, BAD, BAX, and BCL2L1, in the OCRs of several OCNs in Ph-like ALL (Supplementary Table S7).
Next, we queried the drug databases therapeutic target database (TTD) (15), DrugBank (16), DGIdb (17), and NIH PubChem to identify known drugs or new chemical compounds that could be used for potential pharmacologic targeting of predicted nodes. Nine of our identified 14 OCNs matched with drug compounds (Table 1). Some agents have only preclinical testing data available in human cancer, such as the recently-described NEK6 inhibitor (40). Others are more advanced, including the anti-CD38 mAb daratumumab (37) that is FDA-approved for adults with multiple myeloma and under current phase 1/2 clinical study in children with relapsed/refractory leukemias. Amongst the 81 OCN gene pairs predicted to be significantly synergistic, 32 (40%) were found to have known drugs for both members of the pair (hypergeometric test P-value < 0.0001, Supplementary Table S6). In addition, 559 of the 973 predicted OCR genes (57%) were found to be targets of known drugs or experimental compounds (hypergeometric test P-value < 0.0001). Taken together, these results suggest that computationally predicted synergistic regulators and their target pathways in a disease-specific network are valuable sources for identifying novel drug targets.
Because STAT5 signaling is known to be hyperactivated via both JAK and ABL class kinases in Ph-like ALL (4, 12), OptiCon's nomination of STAT5B provided an attractive pathway for subsequent validation efforts. Several clinical trials are currently investigating the addition of the Janus kinase 1/2 inhibitor (JAKi) ruxolitinib to treatment of Ph-like ALL patients with CRLF2 rearrangements and other JAK/STAT pathway alterations (NCT02723994) or the addition of the SRC/ABL kinase inhibitor (ABLi) dasatinib to therapy for patients with ABL-class alterations (NCT02883049). Given these ongoing trials incorporating single-agent TKIs, we focused on other key regulons predicted to be synergistic with STAT5B that may further optimize combination therapy. One OCN pair that ranked as highly synergistic was STAT5B and BAG1 (synergy score 0.023, adjusted P value = 0.016). The BAG1 protein is known to bind to and enhance the anti-apoptotic effect of BCL-2, likely by preventing its degradation (39, 41).
Genetic and pharmacologic cotargeting of STAT5 and BAG1/BCL-2 has synergistic antileukemia efficacy in Ph-like ALL
We next validated our computational predictions in vitro in the two known human CRLF2-rearranged Ph-like ALL cell lines MUTZ5 (IGH-CRLF2, JAK2 R683G) and MHH-cALL-4 (IGH-CRLF2, JAK2 I682F) and in a third cell line that we immortalized for in vitro studies from a recently-established ABL-class Ph-like ALL PDX model TVA-1 harboring an ETV6-ABL1 kinase fusion (25). PCA of gene expression data from these cell lines and primary pediatric B-ALL patient specimens included in the TARGET and PCGP datasets showed that these Ph-like ALL cell lines clustered together with Ph-like and Ph+ ALL primary patient samples (Supplementary Fig. S1A) and separate from the other B-ALL subtypes, recapitulating expression signatures of primary Ph-like ALL samples used in the OptiCon analysis. We investigated the baseline protein expression of BAG1 and anti-apoptotic BCL-2 family members in Ph-like ALL cell lines and PDX models (Supplementary Table S8) and detected high levels of BAG1, BCL-2, BCL-xL, and/or MCL-1 in all tested Ph-like ALL cell lines and PDX cells (Fig. 1D).
Genetic validation of our computational prediction via Cas9-mediated double-knockout of STAT5B and BAG1 pair demonstrated significant reduction in leukemia cell proliferation in both the CRLF2-R MUTZ5 and the ABL1-R TVA-1 Ph-like cell lines compared with single-gene knockouts or non-targeting control (Fig. 2A; Supplementary Fig. S2). These results suggest that cotargeting of these essential pathways could indeed be superior. BAG1 is known to prevent apoptosis via preserving high BCL-2 levels, and our observed enrichment of several apoptosis-related genes in the predicted OCRs highlights the potential importance of cell death pathways in Ph-like ALL. However, the BAG1 protein does not have a readily-available pharmacologic inhibitor. We alternatively focused upon inhibition of BCL-2 as a more clinically-relevant pharmacologic surrogate for BAG1 and investigated the therapeutic efficacy of venetoclax, a potent and highly-selective BCL-2 inhibitor approved by the FDA for treatment of adults with relapsed/refractory chronic lymphocytic leukemia or acute myeloid leukemia (AML).
Figure 2.
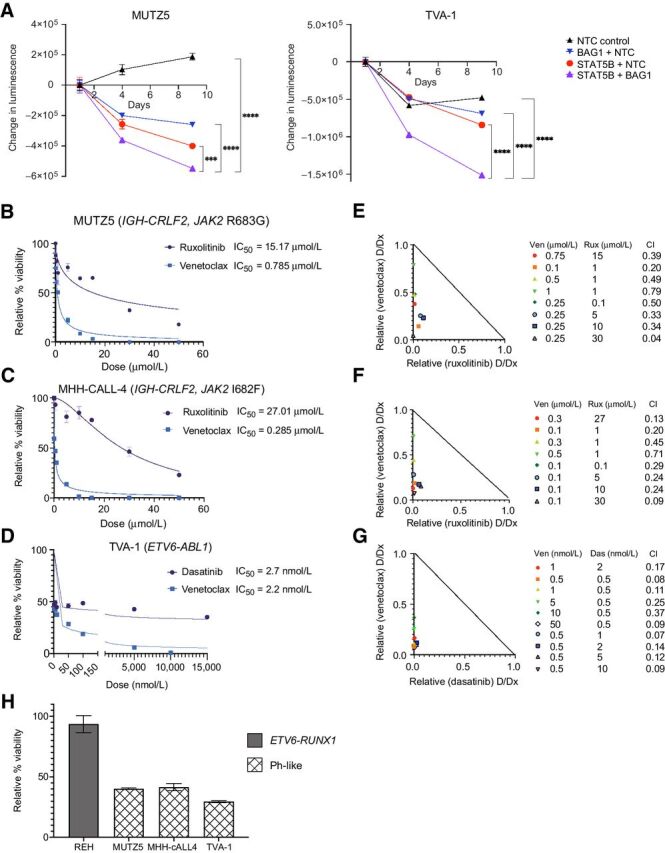
Genetic and pharmacologic cotargeting of BAG1 or BCL-2 and STAT5 signaling is synergistic in vitro in Ph-like ALL. A, Double knockout of STAT5B and BAG1 by CRISPR/Cas9- enhances inhibition of cell viability as measured by CellTiter-Glo assay in MUTZ5 (left) and TVA-1 (right) ALL cells; x-axis represents days after sgRNA/Cas9 RNP nucleofection; y-axis represents change in absolute luminescence; NTC, nontargeting control sgRNA. Experiments were repeated with two different sgRNAs per target; representative data from a single experiment are shown for each cell line. Each data point represents the mean of three technical replicates ± SD. Error bars not displayed when shorter than the size of the symbol. ***P ≤ 0.001 and ****P ≤ 0.0001 using one-way ANOVA and Dunnett posttest correction. B–D, Individual dose response curves and IC50 values for ruxolitinib (rux) and venetoclax (ven) treatment of (B) MUTZ5 and (C) MHH-cALL4 cell lines and dasatinib (das) and venetoclax treatment of (D) TVA-1 cells. Viability data are shown relative to 0.1% DMSO vehicle assayed at 72 hours using CellTiter-Glo absorbance assays. Each data point represents the mean of six replicate measures ± SD. E–G, Isobolograms for combination of TKIs with venetoclax at various dose combinations in MUTZ5, MHH-cALL-4, and TVA-1 cell lines, respectively, along with CI values for each dose combination that were generated using Compusyn. CI values were less than 1.0 (synergistic effect) for all dose combinations tested. H, Effect sizes (percentage cell viability relative to DMSO control) of combination treatment using 1 μmol/L ruxolitinib with 0.1 μmol/L venetoclax in REH, MUTZ5, and MHH-cALL-4 cells and of 0.5 nmol/L dasatinib combined with 50 nmol/L venetoclax in TVA-1 cells.
To evaluate potential synergy in vitro, we treated Ph-like ALL cell lines with venetoclax and ruxolitinib (MUTZ5, MHH-cALL-4) or dasatinib (TVA-1) and measured cell viability. Figure 2B–D show individual drug response curves for each cell line with corresponding IC50 values. We confirmed high ruxolitinib IC50 values for MUTZ5 and MHH-cALL-4 (15 and 27 μmol/L, respectively) and low dasatinib IC50 (2.7 nmol/L) for TVA-1 (25, 42). However, limited cell killing was observed at up to 72 hours of drug incubation despite supratherapeutic TKI dosing (Fig. 2B–D). In contrast, all three Ph-like ALL cell lines showed sensitivity to venetoclax with IC50 values <800 nmol/L and near-complete cell killing at 72 hours at higher drug doses, suggesting a strong BCL-2 dependency.
On the basis of these initial monotherapy IC50 data, we then chose a range of drug doses to test in combination and assessed potential drug synergy. Figure 2E–G show the isobolograms and CI values of the indicated venetoclax and ruxolitinib or dasatinib combinations; all dosage combinations tested were determined to be significantly synergistic using the Chau–Talalay method (CI values < 1.0) in the three Ph-like ALL cell lines. The percent reduction in cell viability (effect size) for each drug combination exposure are shown in Supplementary Fig. S3A. As a negative control, we tested ruxolitinib and venetoclax alone and in combination in the non-Ph-like B-ALL cell line REH (which harbors the clinically favorable ETV6-RUNX1 fusion) and interestingly observed that most dose combinations were significantly antagonistic (CI values >1.0; Supplementary Fig. S3D). Combined treatment with subtherapeutic doses of 1 μmol/L ruxolitinib and 0.1 μmol/L venetoclax further significantly reduced cell viability in MUTZ5 and MHH-cALL-4 cells with expectedly nominal effects on REH cells (Fig. 2H). These findings support specificity of our prediction and likely preferential activity of dual STAT5 and BCL-2 targeting in Ph-like versus other B-ALL subtypes. In subsequent investigations into mechanisms of drug synergy detailed below, we used 1 μmol/L ruxolitinib with 0.1 μmol/L venetoclax in MUTZ5 and MHH-cALL-4 cell lines and 0.5 nmol/L dasatinib with 50 nmol/L venetoclax in the TVA-1 cell line. These particular dose combinations were chosen because they display high synergy (low CI values) in these Ph-like ALL cell lines (Fig. 2E–G), are sublethal (effect sizes shown in Fig. 2H), and are not expected to induce appreciable off-target kinase inhibition (43).
Combination inhibitor treatment shifts the transcriptome of Ph-like cells away from kinase-activated chemoresistant subtypes and toward more chemosensitive B-ALL subtypes
To elucidate the transcriptomic effects of targeted therapy on Ph-like ALL cells, we performed RNA sequencing of the three Ph-like cell lines treated with venetoclax, TKI, or both drugs at our optimized dosing. Analysis of DEGs and PCA revealed that venetoclax monotherapy effect on the Ph-like ALL transcriptome was minimal (DEGs = 20 using FDR < 0.1, absolute log2FC > 1; Supplementary Fig. S4A). In contrast, TKI monotherapy and combined TKI and venetoclax exposure had a large, but similar, effect on the transcriptome compared with DMSO control treatment (DEGs = 9651 and 10378, respectively; Supplementary Fig. S4A).
We next compared transcriptomes of inhibitor-treated Ph-like ALL cells to those of different B-ALL patient samples using UMAP to project gene expression data to lower dimensions (Fig. 3A). We observed that the transcriptomes of vehicle control-treated and venetoclax monotherapy-treated Ph-like ALL samples, as well as some TKI monotherapy-treated ones, remained similar to those of untreated Ph+ and Ph-like ALL patient samples. Conversely, we found that combined venetoclax and TKI treatment altered the transcriptomic state of Ph-like ALL cells, causing them to cluster closer to B-ALL leukemia subtypes with more favorable cytogenetic alterations. This shift effectively made cells “less Ph-like” and more similar to subtypes that are sensitive to conventional chemotherapy, thereby supporting our network controllability theory (Fig. 3B). To quantify further this transcriptome shift in treated Ph-like ALL cells, we developed comparator gene signature scores (Materials and Methods) for several common B-ALL genetic subtypes. We observed that treating Ph-like ALL cell lines with TKI monotherapy or simultaneous TKI and venetoclax altered their transcriptomes to have higher ETV6-RUNX1, hyperdiploidy, and TCF3-PBX1 gene signature scores (representing favorable risk subtypes) and lower KMT2A-rearranged scores (a prognostically-unfavorable subtype) in comparison to control-treated cells (Fig. 3C). Therefore, both UMAP and gene signature score analyses support the specific shift of transcriptomic state in Ph-like ALL cells posttreatment.
Figure 3.
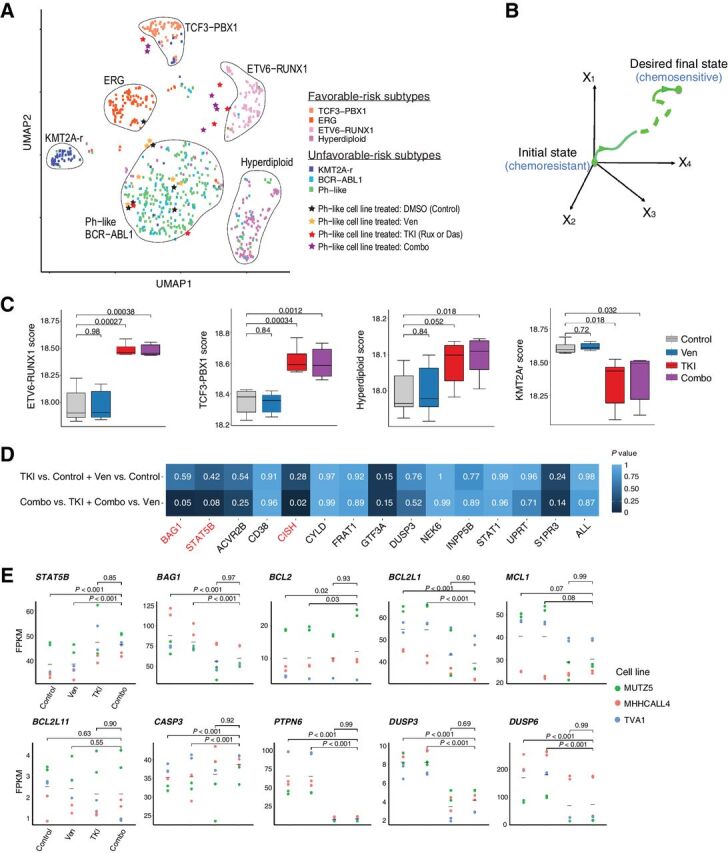
Combination treatment elicits specific and unique changes in the transcriptome of Ph-like cell lines. A, UMAP based on RNA-seq data from drug-treated Ph-like cell lines and B-ALL patient microarray expression data show that combination treatment shifts transcriptome of Ph-like cells to resemble that of favorable-risk B-ALL subtypes. Treatment with 1 μmol/L ruxolitinib and 0.1 μmol/L venetoclax was used for MUTZ5 and MHH-cALL-4, and 0.5 nmol/L dasatinib and 50 nmol/L venetoclax was used for TVA-1. B, Network controllability theory posits that one can use control nodes in a gene network to guide a system from an initial state (in this case relatively chemoresistant Ph-like ALL) to a final state (chemosensitive favorable-risk subtype B-ALL). C, Signature genes of non–Ph-like B-ALL subtypes (ETV6-RUNX1, TCF3-PBX1, and hyperdiploidy subtypes) are enriched in Ph-like ALL cells treated with venetoclax and ruxolitinib or dasatinib. Enrichment scores were computed using single-sample gene set enrichment analysis (ssGSEA). D, Predicted synergistic OCRs STAT5B and BAG1 were significantly perturbed only by combination drug treatment, but not by monotherapy. The degree of perturbation was measured by the overlap between genes in the OCR of each specified OCN and DEGs in the specified comparison (TKI or venetoclax monotherapy versus control, or combination therapy versus either TKI or venetoclax monotherapy). Significance of overlap was determined using hypergeometric test P values. Significant P values were observed for the STAT5B, BAG1, and CISH OCRs in dual inhibitor-treated cells, but not with venetoclax or ruxolitinib monotherapy or in other OCRs. E, Gene expression changes during monotherapy or combination inhibitor therapy in OptiCon-nominated OCNs and some OCR genes. P values were calculated using one-way ANOVA implemented in LIMMA software and adjusted for multiple testing using the Benjamini–Hochberg method.
The network controllability theory underlying OptiCon suggests that cotargeting specific OCNs will lead to perturbation of their corresponding OCRs. We thus hypothesized that expression of genes within the identified OCRs of STAT5B and BAG1 would change after combination treatment, but not after monotherapy. Indeed, we observed that STAT5B and BAG1 OCRs were significantly enriched for DEGs only with dual BCL-2 and STAT5 inhibition, but not with single-drug treatment (Fig. 3D). On the contrary, none of the OCRs were enriched for DEGs when comparing monotherapy versus control. Interestingly, the OCR of the OCN CISH (cytokine inducible SH2 containing protein, a known negative regulator of JAK/STAT signaling) was also enriched for DEGs when comparing dual-inhibitor versus single-agent treatment, although the other 11 OCN-associated OCRs were not appreciably perturbed. Taken together, these findings suggest that Ph-like ALL cells may be shifted towards a transcriptomic state with potential prognostic significance by direct pharmacologic perturbation of STAT5B and BAG1 and their downstream control regions (Fig. 3B).
Effective in vitro cotargeting of apoptosis and cytokine signaling pathways in Ph-like ALL cells
We next interrogated the transcriptional, translational, and functional effects of inhibitor therapy on specific pathways nominated by OptiCon. Interestingly, both BCL2 and STAT5B gene expression were upregulated in combined venetoclax/TKI conditions as compared with control (Fig. 3E), which we hypothesize could be due to negative feedback mechanisms such as downregulation of PTPN6 following TKI treatment. Conversely, other anti-apoptotic genes BAG1, MCL1, and BCL2L1 (encoding BCL-xL) were significantly downregulated in combination drug-treated cells versus vehicle control. We also detected significant expression changes in several other genes involved in the PI3K/Akt/mTOR, Ras/MAPK pathways, and intrinsic and effector apoptosis mechanisms following in vitro inhibitor exposure (Supplementary Fig. S4B).
On a posttranslational level, ruxolitinib or dasatinib treatment was sufficient to completely abrogate activated pSTAT5 and MAPK targets pERK and/or pJNK (Fig. 4A and B). Unexpectedly, BCL-2 expression was observed to be highest in venetoclax-treated and combination drug-treated conditions, which could potentially be interpreted as a survival advantage of BCL-2 overexpressing leukemia cells and elimination of low-BCL-2 cells, as has been reported in AML (44), although other studies have shown that elevated BCL-2 expression is not a reliable biomarker of venetoclax sensitivity or resistance (45). Conversely, MCL-1 is an anti-apoptotic BCL-2 family protein that is tightly transcriptionally regulated and whose high expression is known to mediate venetoclax resistance by binding to BIM (46). We thus noted with interest that levels of the anti-apoptotic proteins MCL-1 and BAG1 noticeably decreased after combination treatment, whereas levels of the pro-apoptotic BH3-only protein BIM increased (Fig. 4A and B).
Figure 4.
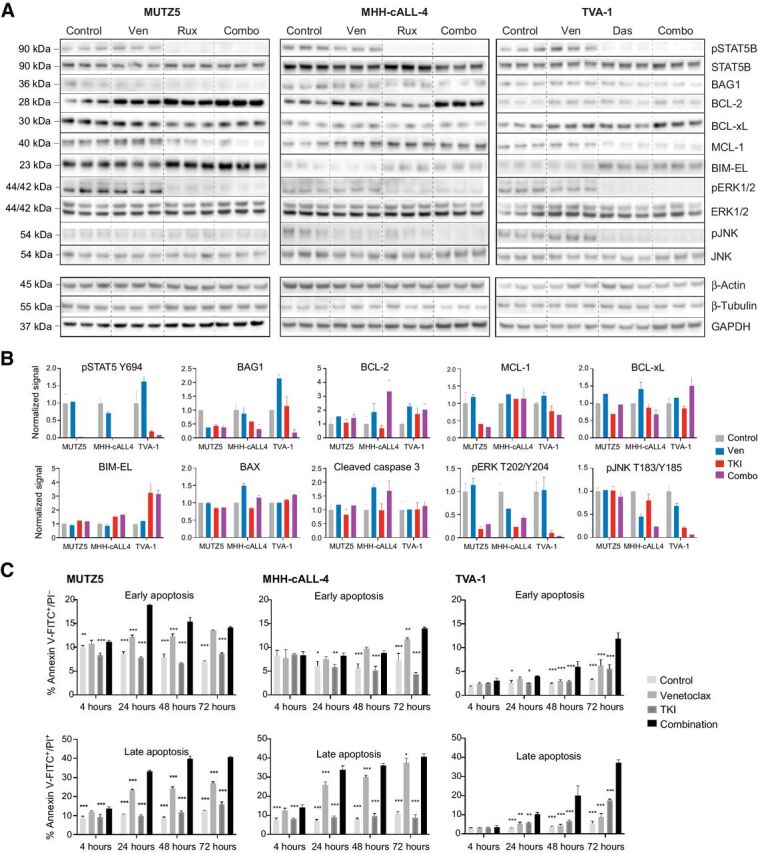
Effects of combined kinase and BCL-2 inhibition on intracellular phosphosignaling, apoptosis proteins, and functional apoptosis. A, Immunoblot images and (B) normalized immunoblotting signal intensities of phosphorylated (p) STAT5, pERK, and pJNK (along with total protein levels below their respective phosphoprotein levels), and BAG1 and BCL-2 family proteins in Ph-like ALL cell lines treated in vitro with single-agent venetoclax, single-agent TKI (ruxolitinib for MUTZ5 and MHH-cALL-4, dasatinib for TVA-1), or both drugs. Densitometry signals were first normalized to either β-actin, β-tubulin, or GAPDH loading controls (representative strips shown) and displayed graphically relative to 0.1% DMSO control treatment. Each bar represents mean ± SD of three technical replicates. BAX and cleaved caspase-3 targets were assessed by apoptosis protein arrays. C, Time course of apoptosis under single or combination drug conditions was assessed by annexin V/propidium iodide (PI) co-staining and flow cytometric analysis. MUTZ5 and MHH-cALL-4 cells were treated with 0.1 μmol/L venetoclax, 1 μmol/L ruxolitinib, both drugs, or 0.1% DMSO control. TVA-1 cells were treated with 50 nmol/L venetoclax, 0.5 nmol/L dasatinib, both drugs, or 0.1% DMSO control. Early apoptosis (assessed by percent annexin V+/PI−, left) and late apoptosis/necrosis (assessed by percent annexin V+/PI+, right) are shown for each time point. Each bar represents mean ± SD of three replicates. Significance of control or single drug as compared with combination indicated by asterisks above bars: *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 using two-way ANOVA and Dunnett post-test correction.
We next hypothesized that the synergistic decrease in cell viability seen with combined TKI and venetoclax treatment could be due to augmentation of apoptosis. We thus determined the proportion of cells undergoing early apoptosis (Annexin V+/PI−) and late apoptosis/necrosis (Annexin V+/PI+) and found that combining ruxolitinib or dasatinib with venetoclax led to significantly greater apoptosis than either monotherapy with effects detected within 4 to 24 hours of drug exposure (Fig. 4C). Increased cleaved caspase-3 was detected in both venetoclax and combination drug-treated cells, consistent with the observed increase in apoptosis at 72 hours in these conditions (Fig. 4C; Supplementary Fig. S5). Importantly, despite its strong effects on the expression of apoptosis pathway targets, TKI monotherapy did not appreciably increase apoptosis. Together, these data suggest that the antileukemia effects observed from combination drug treatment may be due to TKI-mediated effects of decreasing anti-apoptotic proteins while increasing pro-apoptosis proteins in leukemia cells, thereby resulting in enhanced apoptosis when combined with venetoclax.
Combined venetoclax and TKI treatment has potent in vivo antileukemia activity in preclinical Ph-like ALL models
To validate pharmacologically our OptiCon-predicted pairing in vivo, we next investigated the potential efficacy of combined venetoclax and ruxolitinib or dasatinib treatment in six different PDX Ph-like ALL models comprised of CRLF2-rearranged or ABL1-rearranged genetic backgrounds (Supplementary Table S8). Given that optimal inhibitor dosing for combination therapy may differ from optimal monotherapy dosing, we tested two dose levels (50% and 100%) of inhibitors in several PDX models to model potential synergy and to assess if antileukemia benefit could be achieved with lower dosing.
As hypothesized, we observed significant inhibition of leukemia proliferation in peripheral blood and end-study spleens in most CRLF2-rearranged and ABL1-rearranged Ph-like ALL PDX models treated with combined venetoclax and ruxolitinib or dasatinib (Fig. 5). Detected combination treatment effects were superior to TKI and/or venetoclax monotherapy in several models with near-curative effects of ruxolitinib and venetoclax or dasatinib and venetoclax detected in end-study spleens of three PDX models (CRLF2-rearranged/JAK2-mutant JH331, ABL1-rearranged NH011 and TVA1). Interestingly, combined TKI and venetoclax at 50% “half-dosing” also had more potent anti-leukemia effects than full monotherapy dosing of either agent, suggesting enhanced STAT5 and BCL-2 cotargeting in these leukemias. Although most models did not demonstrate appreciable single-agent venetoclax activity, our surprisingly ruxolitinib-resistant JAK-mutant JH331 PDX model was exquisitely sensitive to BCL-2 inhibition and further showed superior inhibition of leukemia proliferation with dual ruxolitinib/venetoclax treatment, highlighting potential for JAKi resensitization or resistance reversal. Other CRLF2-rearranged models (ALL4364, ALL2128) demonstrated marked sensitivity to ruxolitinib with >50% reduction of ALL burden in end-study spleens, although combined treatment with venetoclax did not further augment antileukemia response. Importantly, we also observed excellent in vivo tolerability of dual inhibitor treatment for up to 4 weeks in our PDX models with stability of murine physical health parameters. In addition, the dual-inhibitor treated mice did not experience more significant weight loss (>20% of entry weight) compared with monotherapy arms (Supplementary Fig. S6).
Discussion
Omics analyses in the study of human cancers often generate large candidate gene lists that can be difficult to prioritize for pharmacologic targeting. We employed an innovative systems biology strategy for unbiased identification of synergistic pathways as candidates for combination therapy. This approach efficiently narrowed the search space of relevant oncogenic pathways in these Ph-like ALL studies. In our development of the OptiCon algorithm, we found that a high proportion of the nominated gene pairs in three separate adult cancer types has known synthetic lethal genetic interactions (13). Here we applied our network-controllability approach to a clinically high-risk leukemia subtype and identified 14 novel OCNs in Ph-like ALL. The largest fraction of the OCNs is involved in kinase signaling, followed by metabolism, transcriptional regulation, splicing, and apoptosis (Fig. 1B), suggesting relative dependencies of Ph-like ALL on these biological processes. We validated our prediction of the highly ranked OCN pair STAT5B and BAG1 via CRISPR/Cas9-mediated double-knockout and found enhanced antileukemia efficacy. To directly translate our findings to the clinic, we focused further pharmacologic validation studies upon STAT5B and BCL2 given the strong biologic rationale for this Ph-like ALL-specific OCN pairing. Furthermore, there is pragmatic translational potential for investigating combination therapy using the TKIs ruxolitinib or dasatinib and the BCL-2 inhibitor venetoclax given their clinical availability and established adult and pediatric dosing. Venetoclax has not been extensively investigated in Ph-like ALL, although several preclinical studies have reported preliminary efficacy of combining BCL-2 inhibitors with MCL1-inhibitors (47) or with TKIs in other high-risk ALL types, such as BCR-ABL1–rearranged (Ph+) models (31) and IL7R-mutant T-ALL models (48). OptiCon's nomination of STAT5B and BCL2-related pathways from integrative-omics analysis of primary Ph-like ALL samples provides independent rationale for cotargeting these specifically in the Ph-like ALL subtype.
Intriguingly, we demonstrated that “precision drugging” of the STAT5B and BAG1 pair of synergistic regulators altered the transcriptomic state of Ph-like ALL to become more similar to other chemosensitive B-ALL subtypes. These results support the basis of network controllability theory that successful identification of key control nodes can shift cells from one transcriptomic state to another desired one via specific perturbation. Although the transcriptomic effects of joint BCL-2 and kinase inhibition appear to be driven in large part by the TKI, the phenotypic effects of combined therapy in enhancing cell death seem driven more by venetoclax. Our functional assays of apoptosis and cell viability combined with transcriptional and protein analyses suggest that TKI monotherapy may promote apoptosis by altering the fine balance between pro-apoptotic and anti-apoptotic protein expression, but is insufficient to achieve apoptosis on its own.
OptiCon analysis also elucidated potential crosstalk pathways that mediate synergy between the STAT5 signaling and BCL-2 pathways in Ph-like ALL. We found that several MAPK pathway genes [e.g., MAPK8 (JNK1) and MAPK11 (p38)] participate in crosstalk between the OptiCon-predicted OCNs STAT5B and BAG1. Although mutations in MAPK pathway genes have been reported in Ph-like ALL (4), the mechanistic relevance of deregulated MAPK signaling in Ph-like ALL is not well-understood. JNKs are known to phosphorylate and regulate BCL-2, BIM, and BAD with effects on apoptosis in a context and cell-type dependent manner, and ERK1/2 activation has been associated with anti-apoptotic effects (49, 50). Our identification of their negative regulator DUSP3 as an OCN and our surprising findings that cotreatment with venetoclax and ruxolitinib or dasatinib also decreased phosphorylated ERK1/2 and JNK levels suggest that MAP kinases are involved in critical Ph-like ALL signaling crosstalk.
Our studies reveal in an unbiased manner that both CRLF2-rearranged and ABL1-rearranged Ph-like ALL appear to have previously unknown synergistic dependencies between STAT5 and BCL-2 pathways, among others. Our additional demonstration of superior in vivo antileukemia effects of combining ruxolitinib or dasatinib and venetoclax in six PDX models comprised of various genetic backgrounds provides strong preclinical rationale for bench-to-bedside development of dual BCL-2 and kinase inhibition strategies in next-generation clinical trials for patients with Ph-like ALL. Further elucidation of key genetic and molecular factors that may contribute to the observed heterogeneity of inhibitor treatment responses in our Ph-like ALL PDX models will facilitate potential future clinical translation of these findings. Importantly, we also observed effective combinatorial in vivo leukemia burden reduction when venetoclax and dasatinib were administered at subtherapeutic dosing, suggesting the ability to achieve effective antileukemia activity while potentially reducing therapy-associated toxicity. This observation lends additional support for our network-based approach which takes pathway crosstalk into account to spare unperturbed pathways to minimize potential toxicity.
Collectively, our application of a systems biology framework to a high-risk leukemia subtype provides critical new insights regarding cancer gene network controllability and the ability to facilitate unbiased discovery of novel target pairings. Although our results certainly provide compelling rationale for clinical investigation of dual venetoclax and ruxolitinib or dasatinib strategies for patients with Ph-like ALL, our network controllability-based methodology for inferring synergistic gene regulatory nodes also provides an innovative paradigm for rational design of combination therapy approaches that has wide applicability to other human cancers and diseases.
Authors’ Disclosures
S.K. Tasian reports grants and nonfinancial support from Incyte Corporation during the conduct of the study, as well as grants from Gilead Sciences and personal fees from Aleta Biotherapeutics and Kura Oncology outside the submitted work. No disclosures were reported by the other authors.
Supplementary Material
Supplementary Legend
Gene expression of Ph-like cell lines recapitulates that of primary Ph-like ALL patient sample datasets. (A) Principal component analysis (PCA) of TARGET patient samples based on microarray gene expression show that their transcriptomes cluster separately by B-ALL subtype. Ph-like cell line gene expression by RNA-Seq (MUTZ5 and MHH-cALL-4 data from Cancer Cell Line Encyclopedia, TVA-1 data generated by authors) appropriately clusters with Ph+ and Ph-like B-ALL primary patient samples. (B) Expression of predicted OCN genes in Ph-like patient samples as compared to non-Ph-like favorable-risk B-ALL subtypes (defined here as ETV6-RUNX1, ERG-dysregulated, and hyperdiploid patient B-ALL samples).
CRISPR/Cas9 single and double-knockout gene editing efficiency of STAT5B and BAG1 estimated by TIDE assay. Top (MUTZ5) and bottom (TVA-1) panels show spectrum and frequency of small (0-10 bp) insertions and deletions (indels) 4 days after nucleofection of single and double gene targeting RNPs (cell viability data shown in Figure 2A). DKO: double knockout; SKO: single knockout; X-axis shows insertion or deletion at given position; y-axis represents percentage of sequences with that particular edit. Editing efficacy (defined by small indel frequency with p<0.001) was high and similar between single and double knockout conditions.
Ph-like ALL cell viability with in vitro venetoclax and TKI exposure. (A) Cell viability at 72 hours after treatment with specified dose combinations of drugs tested and shown on isobolograms in Figure 2. Viability is depicted relative to 0.1% DMSO vehicle control, measured by CellTiter-Glo absorbance. V=venetoclax, R=ruxolitinib, D=dasatinib. (B) Leukemia cell viability at 72 hours of the specified drug dose combination chosen to investigate mechanisms of synergy. Ven=venetoclax, Rux=ruxolitinib, Das=dasatinib. Each bar represents the average of triplicate technical replicates, error bars represent standard deviation. (C) Ruxolitinib and venetoclax individual dose response curves and IC50 values for the non-Ph-like B-ALL cell line REH. Viability was assayed at 72 hours using the CellTiter-Glo assay with data normalization to 0.1% DMSO vehicle treatment. Each data point represents the mean of three replicate measures +/- SD. (D) Isobologram for combination of ruxolitinib (rux) and venetoclax (ven) at various dose combinations in the REH cell line, along with the effect size (percent reduction in cell viability relative to the DMSO control) observed at each dose combination and the Combination Index (CI) values calculated using Compusyn. One dose combination had moderate synergistic effect (CI = 0.57), but all other combinations had antagonistic effects (CI >1.0) in this non-Ph-like cell line.
Gene expression changes in Ph-like ALL cells treated with combination therapy. (A) PCA plot showing replicate samples for each treatment condition for the three Ph-like cell lines. Inter-cell line differences predominate. DMSO- (vehicle control) and venetoclax-treated samples of each cell line cluster closely, whereas single-agent TKI and dual TKI/venetoclax-treated samples cluster together. (B) Gene expression changes during monotherapy and combination therapy in select apoptosis pathway and cytokine/kinase signaling pathway genes including those in SRC-family and MAPK-family pathways, as well as in 11 of the 14 OCNs. Expression changes in 3 of the 14 OCNs (STAT5B, BAG1, DUSP3) are shown in Figure 3E.
Apoptosis assessment of drug-treated Ph-like ALL cell lines. MUTZ5, MHH-cALL-4, and TVA-1 cells were treated with 0.1% DMSO vehicle control, venetoclax, TKI, or both drugs and assessed for apoptosis via flow cytometry assays with annexin-V and PI co-staining. Early apoptotic cells are defined as the percentage of cells in Annexin V-FITC+/ PI- quadrant, and late apoptotic/necrotic cells are defined as the percentage of cells in Annexin V-FITC+/ PI+ quadrant. Flow cytometric analyses (FACS) were performed after 4, 24, 48, and 72 hours of treatment. Plots shown are representative data at 72 hours. Triplicate samples were analyzed for each condition at the separate timepoints for each cell line, and each individual experiment was repeated twice. Gating and analyses were performed using FlowJo software.
Weights of patient-derived leukemia xenografted mice remained stable during combination therapy. Mice weights (in grams, g) during treatment with TKI, venetoclax, or combination treatment were monitored as a proxy for drug toxicity and clinical status. Combination therapy did not result in more significant body weight loss (>20% of initial weight) as compared to vehicle or monotherapy in any of the six PDX models. N= 5 mice per treatment group. Each point represents mean +/- SD. The six leukemia PDX models correspond to those shown in Figure 5.
Sources of gene expression data used in this study. Patient data were stratified based on their leukemia subtypes (column header).
List of gene fusions identified and used in this study. Fusions were called using TARGET B-ALL datasets and the methods from He et al. (21). COG, Children's Oncology Group. USI, unique specimen identifier.
List of recurrently mutated genes in B-ALL used in this study. Mutation calls were obtained from studies by Roberts et al. (4) and Ma et al. (34).
List of differentially expressed genes in Ph-like versus non-Ph-like ALL. Ph-like ALL patient microarray data was compared to that of prognostically-favorable B-ALL subtypes ETV6-RUNX1, hyperdiploidy, and DUX4-rearrangements/ERG-altered subtypes. Genes are ranked by multiple-testing adjusted p-value (using the Benjamini-Hochberg method), and considered differentially expressed for adjusted p-value <0.05.
The gene regulatory network used in this study. The network consists of 5,959 genes and 108,281 regulatory interactions as we previously detailed (13). Interactions were collected from three public pathway databases that were manually curated by experts: Reactome (19) (1597 pathways), KEGG (18) (195 pathways) and NCI-Nature Pathway Interaction database (20) (745 pathways).
OCN pairs nominated to be synergistic, along with small molecule inhibitors identified from querying Therapeutic Target Database (TTD), DrugBank, DGIdb, and PubChem. BH, Benjamini-Hochberg method for multiple testing correction.
Optimal Control Nodes (OCNs) identified using the OptiCon algorithm and Ph-like B-ALL specific gene regulatory network, and genes in the optimal control regions (OCRs) of each OCN.
List of genetic lesions in Ph-like cell lines and patient-derived xenograft models used in this study.
Acknowledgments
This work was supported by the NIH/National Child Health and Human Development award T32HD043021 to the Children's Hospital of Philadelphia (to Y.-Y. Ding), NIH/NCI awards K12CA076931 to the University of Pennsylvania (to Y.-Y. Ding), K08CA184418 (to S.K. Tasian), U01CA232486 (to S.K. Tasian), and U01CA243072 (to S.K. Tasian, K. Tan), Department of Defense Translational Team Science award CA180683P1 (to S.K. Tasian), the V Foundation for Cancer Research (to S.K. Tasian), Alex's Lemonade Stand Foundation (to Y.-Y. Ding), Canadian Institutes of Health Research Doctoral Foreign Study Award #433117 (to G.M. Chen), and institutional support from the Children's Hospital of Philadelphia Center for Childhood Cancer Research (CHOP CCCR; to S.K. Tasian, K. Tan) and from the Children's Hospital of Philadelphia Research Institute (to Y.-Y. Ding). We acknowledge the Children's Oncology Group and the CHOP CCCR biorepositories for provision of primary patient leukemia specimens. We also gratefully thank Dr. David Fruman at the University of California, Irvine, for sharing the TVA-1 PDX model; Dr. Matthew Stubbs at the Incyte Corporation for generous provision of ruxolitinib rodent chow for animal studies; and Dr. Michael Hogarty at the Children's Hospital of Philadelphia and Dr. Christian Hurtz at the University of Pennsylvania for helpful scientific discussions.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
This article is featured in Highlights of This Issue, p. 4945
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Contributions
Y.-Y. Ding: Conceptualization, data curation, formal analysis, validation, investigation, visualization, writing–original draft, writing–review and editing. H. Kim: Data curation, software, formal analysis. K. Madden: Validation, investigation. J.P. Loftus: Validation, investigation. G.M. Chen: Software, formal analysis. D.H. Allen: Validation, investigation. R. Zhang: Data curation, software, formal analysis. J. Xu: Data curation, software, formal analysis. C.-H. Chen: Investigation. Y. Hu: Data curation, software, formal analysis. S.K. Tasian: Conceptualization, resources, supervision, funding acquisition, validation, writing–original draft, project administration, writing–review and editing. K. Tan: Conceptualization, resources, data curation, formal analysis, supervision, funding acquisition, investigation, writing–original draft, project administration, writing–review and editing.
References
- 1. Huang A, Garraway LA, Ashworth A, Weber B. Synthetic lethality as an engine for cancer drug target discovery. Nat Rev Drug Discov 2020;19:23–38. [DOI] [PubMed] [Google Scholar]
- 2. Laufer C, Fischer B, Billmann M, Huber W, Boutros M. Mapping genetic interactions in human cancer cells with RNAi and multiparametric phenotyping. Nat Methods 2013;10:427–31. [DOI] [PubMed] [Google Scholar]
- 3. Du D, Roguev A, Gordon DE, Chen M, Chen S-H, Shales M, et al. Genetic interaction mapping in mammalian cells using CRISPR interference. Nat Methods 2017;14:577–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang Y-L, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med 2014;371:1005–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reshmi SC, Harvey RC, Roberts KG, Stonerock E, Smith A, Jenkins H, et al. Targetable kinase gene fusions in high-risk B-ALL: a study from the Children's Oncology Group. Blood 2017;129:3352–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jain N, Roberts KG, Jabbour E, Patel K, Eterovic AK, Chen K, et al. Ph-like acute lymphoblastic leukemia: a high-risk subtype in adults. Blood 2017;129:572–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tasian SK, Loh ML, Hunger SP. Philadelphia chromosome-like acute lymphoblastic leukemia. Blood 2017;130:2064–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Harvey RC, Mullighan CG, Wang X, Dobbin KK, Davidson GS, Bedrick EJ, et al. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood 2010;116:4874–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maude SL, Tasian SK, Vincent T, Hall JW, Sheen C, Roberts KG, et al. Targeting JAK1/2 and mTOR in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood 2012;120:3510–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tasian SK, Teachey DT, Li Y, Shen F, Harvey RC, Chen I-M, et al. Potent efficacy of combined PI3K/mTOR and JAK or ABL inhibition in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood 2016;129:177–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ding YY, Stern JW, Jubelirer TF, Wertheim GB, Lin F, Chang F, et al. Clinical efficacy of ruxolitinib and chemotherapy in a child with Philadelphia chromosome-like acute lymphoblastic leukemia with GOLGA5-JAK2 fusion and induction failure. Haematologica 2018;103:e427–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tasian SK, Doral MY, Borowitz MJ, Wood BL, Chen I-M, Harvey RC, et al. Aberrant STAT5 and PI3K/mTOR pathway signaling occurs in human CRLF2-rearranged B-precursor acute lymphoblastic leukemia. Blood 2012;120:833–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hu Y, Chen C-H, Ding Y-Y, Wen X, Wang B, Gao L, et al. Optimal control nodes in disease-perturbed networks as targets for combination therapy. Nat Commun 2019;10:2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu Y-Y, Slotine J-J, Barabási A-L. Controllability of complex networks. Nature 2011;473:167–73. [DOI] [PubMed] [Google Scholar]
- 15. Zhu F, Shi Z, Qin C, Tao L, Liu X, Xu F, et al. Therapeutic target database update 2012: a resource for facilitating target-oriented drug discovery. Nucleic Acids Res 2012;40:D1128–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 2018;46:D1074–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cotto KC, Wagner AH, Feng Y-Y, Kiwala S, Coffman AC, Spies G, et al. DGIdb 3.0: a redesign and expansion of the drug–gene interaction database. Nucleic Acids Res 2018;46:D1068–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 2017;45:D353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fabregat A, Sidiropoulos K, Garapati P, Gillespie M, Hausmann K, Haw R, et al. The reactome pathway knowledgebase. Nucleic Acids Res 2016;44:D481–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schaefer CF, Anthony K, Krupa S, Buchoff J, Day M, Hannay T, et al. PID: the pathway interaction database. Nucleic Acids Res 2009;37:D674–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. He B, Gao P, Ding Y-Y, Chen C-H, Chen G, Chen C, et al. Diverse noncoding mutations contribute to deregulation of cis-regulatory landscape in pediatric cancers. Sci Adv 2020;6:eaba3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bhojwani D, Pei D, Sandlund JT, Jeha S, Ribeiro RC, Rubnitz JE, et al. ETV6-RUNX1-positive childhood acute lymphoblastic leukemia: improved outcome with contemporary therapy. Leukemia 2012;26:265–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang J, McCastlain K, Yoshihara H, Xu B, Chang Y, Churchman ML, et al. Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat Genet 2016;48:1481–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Heerema NA, Sather HN, Sensel MG, Zhang T, Hutchinson RJ, Nachman JB, et al. Prognostic impact of trisomies of chromosomes 10, 17, and 5 among children with acute lymphoblastic leukemia and high hyperdiploidy (>50 chromosomes). J Clin Oncol 2000;18:1876–87. [DOI] [PubMed] [Google Scholar]
- 25. Gotesman M, Vo T-TT, Herzog L-O, Tea T, Mallya S, Tasian SK, et al. mTOR inhibition enhances efficacy of dasatinib in ABL-rearranged Ph-like B-ALL. Oncotarget 2018;9:6562–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brinkman EK, Chen T, Amendola M, van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 2014;42:e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chou TC. Drug combination studies and their synergy quantification using the chou-talalay method. Cancer Res 2010;70:440–6. [DOI] [PubMed] [Google Scholar]
- 28. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009;4:44–57. [DOI] [PubMed] [Google Scholar]
- 29. Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009;462:108–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hurtz C, Wertheim GB, Loftus JP, Blumenthal D, Lehman A, Li Y, et al. Oncogene-independent BCR-like signaling adaptation confers drug resistance in Ph-like ALL. J Clin Invest 2020;130:3637–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Leonard JT, Rowley JSJ, Eide CA, Traer E, Hayes-Lattin B, Loriaux M, et al. Targeting BCL-2 and ABL/LYN in Philadelphia chromosome–positive acute lymphoblastic leukemia. Sci Transl Med 2016;8:354ra114. [DOI] [PubMed] [Google Scholar]
- 32. Fischer U, Forster M, Rinaldi A, Risch T, Sungalee S, Warnatz H-J, et al. Genomics and drug profiling of fatal TCF3-HLF−positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat Genet 2015;47:ng.3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gill S, Tasian SK, Ruella M, Shestova O, Li Y, Porter DL, et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood. 2014;123:2343–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ma X, Liu Y, Liu Y, Alexandrov LB, Edmonson MN, Gawad C, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 2018;555:371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Matsumoto A, Masuhara M, Mitsui K, Yokouchi M, Ohtsubo M, Misawa H, et al. CIS, a cytokine inducible SH2 protein, is a target of the JAK-STAT5 pathway and modulates STAT5 activation. Blood 1997;89:3148–54. [PubMed] [Google Scholar]
- 36. Amand M, Erpicum C, Bajou K, Cerignoli F, Blacher S, Martin M, et al. DUSP3/VHR is a pro-angiogenic atypical dual-specificity phosphatase. Mol Cancer 2014;13:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bride KL, Vincent TL, Im S-Y, Aplenc R, Barrett DM, Carroll WL, et al. Preclinical efficacy of daratumumab in T-cell acute lymphoblastic leukemia. Blood 2018;131:995–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jee HJ, Kim AJ, Song N, Kim H-J, Kim M, Koh H, et al. Nek6 overexpression antagonizes p53-induced senescence in human cancer cells. Cell Cycle 2010;9:4703–10. [DOI] [PubMed] [Google Scholar]
- 39. Aveic S, Pigazzi M, Basso G. BAG1: the guardian of anti-apoptotic proteins in acute myeloid leukemia. PLoS One 2011;6:e26097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Donato MD, Righino B, Filippetti F, Battaglia A, Petrillo M, Pirolli D, et al. Identification and antitumor activity of a novel inhibitor of the NIMA-related kinase NEK6. Sci Rep 2018;8:16047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Takayama S, Bimston DN, Matsuzawa S, Freeman BC, Aime-Sempe C, Xie Z, et al. BAG-1 modulates the chaperone activity of Hsp70/Hsc70. EMBO J 1997;16:4887–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Weigert O, Lane AA, Bird L, Kopp N, Chapuy B, van Bodegom D, et al. Genetic resistance to JAK2 enzymatic inhibitors is overcome by HSP90 inhibition. J Exp Med 2012;209:259–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhou T, Georgeon S, Moser R, Moore DJ, Caflisch A, Hantschel O. Specificity and mechanism-of-action of the JAK2 tyrosine kinase inhibitors ruxolitinib and SAR302503 (TG101348). Leukemia 2014;28:404–7. [DOI] [PubMed] [Google Scholar]
- 44. Andreeff M, Jiang S, Zhang X, Konopleva M, Estrov Z, Snell VE, et al. Expression of Bcl-2-related genes in normal and AML progenitors: changes induced by chemotherapy and retinoic acid. Leukemia 1999;13:1881–92. [DOI] [PubMed] [Google Scholar]
- 45. Tessoulin B, Papin A, Gomez-Bougie P, Bellanger C, Amiot M, Pellat-Deceunynck C, et al. BCL2-family dysregulation in B-cell malignancies: from gene expression regulation to a targeted therapy biomarker. Front Oncol. 2019;8:645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yang-Yen H-F. Mcl-1: a highly regulated cell death and survival controller. J Biomed Sci 2006;13:201–4. [DOI] [PubMed] [Google Scholar]
- 47. Moujalled DM, Hanna DT, Hediyeh-Zadeh S, Pomilio G, Brown L, Litalien V, et al. Cotargeting BCL-2 and MCL-1 in high-risk B-ALL. Blood advances. American Society of Hematology 2020;4:2762–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Senkevitch E, Li W, Hixon JA, Andrews C, Cramer SD, Pauly GT, et al. Inhibiting janus kinase 1 and BCL-2 to treat T cell acute lymphoblastic leukemia with IL7-Rα mutations. Oncotarget 2018;9:22605–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yue J, López JM. Understanding MAPK signaling pathways in apoptosis. Int J Mol Sci 2020;21:2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dhanasekaran DN, Reddy EP. JNK-signaling: a multiplexing hub in programmed cell death. Genes Cancer 2017;8:682–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Legend
Gene expression of Ph-like cell lines recapitulates that of primary Ph-like ALL patient sample datasets. (A) Principal component analysis (PCA) of TARGET patient samples based on microarray gene expression show that their transcriptomes cluster separately by B-ALL subtype. Ph-like cell line gene expression by RNA-Seq (MUTZ5 and MHH-cALL-4 data from Cancer Cell Line Encyclopedia, TVA-1 data generated by authors) appropriately clusters with Ph+ and Ph-like B-ALL primary patient samples. (B) Expression of predicted OCN genes in Ph-like patient samples as compared to non-Ph-like favorable-risk B-ALL subtypes (defined here as ETV6-RUNX1, ERG-dysregulated, and hyperdiploid patient B-ALL samples).
CRISPR/Cas9 single and double-knockout gene editing efficiency of STAT5B and BAG1 estimated by TIDE assay. Top (MUTZ5) and bottom (TVA-1) panels show spectrum and frequency of small (0-10 bp) insertions and deletions (indels) 4 days after nucleofection of single and double gene targeting RNPs (cell viability data shown in Figure 2A). DKO: double knockout; SKO: single knockout; X-axis shows insertion or deletion at given position; y-axis represents percentage of sequences with that particular edit. Editing efficacy (defined by small indel frequency with p<0.001) was high and similar between single and double knockout conditions.
Ph-like ALL cell viability with in vitro venetoclax and TKI exposure. (A) Cell viability at 72 hours after treatment with specified dose combinations of drugs tested and shown on isobolograms in Figure 2. Viability is depicted relative to 0.1% DMSO vehicle control, measured by CellTiter-Glo absorbance. V=venetoclax, R=ruxolitinib, D=dasatinib. (B) Leukemia cell viability at 72 hours of the specified drug dose combination chosen to investigate mechanisms of synergy. Ven=venetoclax, Rux=ruxolitinib, Das=dasatinib. Each bar represents the average of triplicate technical replicates, error bars represent standard deviation. (C) Ruxolitinib and venetoclax individual dose response curves and IC50 values for the non-Ph-like B-ALL cell line REH. Viability was assayed at 72 hours using the CellTiter-Glo assay with data normalization to 0.1% DMSO vehicle treatment. Each data point represents the mean of three replicate measures +/- SD. (D) Isobologram for combination of ruxolitinib (rux) and venetoclax (ven) at various dose combinations in the REH cell line, along with the effect size (percent reduction in cell viability relative to the DMSO control) observed at each dose combination and the Combination Index (CI) values calculated using Compusyn. One dose combination had moderate synergistic effect (CI = 0.57), but all other combinations had antagonistic effects (CI >1.0) in this non-Ph-like cell line.
Gene expression changes in Ph-like ALL cells treated with combination therapy. (A) PCA plot showing replicate samples for each treatment condition for the three Ph-like cell lines. Inter-cell line differences predominate. DMSO- (vehicle control) and venetoclax-treated samples of each cell line cluster closely, whereas single-agent TKI and dual TKI/venetoclax-treated samples cluster together. (B) Gene expression changes during monotherapy and combination therapy in select apoptosis pathway and cytokine/kinase signaling pathway genes including those in SRC-family and MAPK-family pathways, as well as in 11 of the 14 OCNs. Expression changes in 3 of the 14 OCNs (STAT5B, BAG1, DUSP3) are shown in Figure 3E.
Apoptosis assessment of drug-treated Ph-like ALL cell lines. MUTZ5, MHH-cALL-4, and TVA-1 cells were treated with 0.1% DMSO vehicle control, venetoclax, TKI, or both drugs and assessed for apoptosis via flow cytometry assays with annexin-V and PI co-staining. Early apoptotic cells are defined as the percentage of cells in Annexin V-FITC+/ PI- quadrant, and late apoptotic/necrotic cells are defined as the percentage of cells in Annexin V-FITC+/ PI+ quadrant. Flow cytometric analyses (FACS) were performed after 4, 24, 48, and 72 hours of treatment. Plots shown are representative data at 72 hours. Triplicate samples were analyzed for each condition at the separate timepoints for each cell line, and each individual experiment was repeated twice. Gating and analyses were performed using FlowJo software.
Weights of patient-derived leukemia xenografted mice remained stable during combination therapy. Mice weights (in grams, g) during treatment with TKI, venetoclax, or combination treatment were monitored as a proxy for drug toxicity and clinical status. Combination therapy did not result in more significant body weight loss (>20% of initial weight) as compared to vehicle or monotherapy in any of the six PDX models. N= 5 mice per treatment group. Each point represents mean +/- SD. The six leukemia PDX models correspond to those shown in Figure 5.
Sources of gene expression data used in this study. Patient data were stratified based on their leukemia subtypes (column header).
List of gene fusions identified and used in this study. Fusions were called using TARGET B-ALL datasets and the methods from He et al. (21). COG, Children's Oncology Group. USI, unique specimen identifier.
List of recurrently mutated genes in B-ALL used in this study. Mutation calls were obtained from studies by Roberts et al. (4) and Ma et al. (34).
List of differentially expressed genes in Ph-like versus non-Ph-like ALL. Ph-like ALL patient microarray data was compared to that of prognostically-favorable B-ALL subtypes ETV6-RUNX1, hyperdiploidy, and DUX4-rearrangements/ERG-altered subtypes. Genes are ranked by multiple-testing adjusted p-value (using the Benjamini-Hochberg method), and considered differentially expressed for adjusted p-value <0.05.
The gene regulatory network used in this study. The network consists of 5,959 genes and 108,281 regulatory interactions as we previously detailed (13). Interactions were collected from three public pathway databases that were manually curated by experts: Reactome (19) (1597 pathways), KEGG (18) (195 pathways) and NCI-Nature Pathway Interaction database (20) (745 pathways).
OCN pairs nominated to be synergistic, along with small molecule inhibitors identified from querying Therapeutic Target Database (TTD), DrugBank, DGIdb, and PubChem. BH, Benjamini-Hochberg method for multiple testing correction.
Optimal Control Nodes (OCNs) identified using the OptiCon algorithm and Ph-like B-ALL specific gene regulatory network, and genes in the optimal control regions (OCRs) of each OCN.
List of genetic lesions in Ph-like cell lines and patient-derived xenograft models used in this study.
Data Availability Statement
The results published here are in part based upon data generated by the Therapeutically Applicable Research to Generate Effective Treatments (TARGET) initiative (https://ocg.cancer.gov/programs/target). Whole genome sequencing (WGS), whole exome sequencing (WES), and microarray expression data were downloaded from dbGaP with study identifier phs000218 (including phs000463, phs000464). The data used for these analyses are available at https://portal.gdc.cancer.gov/projects. Transcriptomic datasets of childhood B-ALL samples were downloaded from publicly available databases as specified in “Source Details” of Supplementary Table S1. The drug/gene databases were downloaded from the Therapeutic Target Database (TTD; ref. 15), DrugBank (16), and DGIdb (17). The RNA sequencing data of untreated human B-ALL cell lines (except TVA-1) were downloaded from the Broad Institute Cancer Cell Line Encyclopedia (CCLE) available at https://portals.broadinstitute.org/ccle. The RNA sequencing data generated in this study is deposited at Gene Expression Ominbus (GEO) under the accession number GSE161939. All software supporting the analysis in this study can be found in public repositories. Software package implementing the OptiCon algorithm has been deposited at GitHub (https://github.com/tanlabcode/OptiCon).