

Abstract
A series of adenosine and 2′-deoxyadenosine pairs modified with a 1,12-dicarba-closo-dodecaborane cluster or alternatively with a phenyl group at the same position was synthesized, and their affinity was determined at A1, A2A, A2B and A3 adenosine receptors (ARs). While AR affinity differences were noted, a general tendency to preferentially bind A3 AR over other ARs was observed for most tested ligands. In particular, 5′-ethylcarbamoyl-N6-(3-phenylpropyl)adenosine (18), N6-(3-phenylpropyl)-2-chloroadenosine (24) and N6-(3-phenylpropyl)adenosine (40) showed nanomolar A3 affinity (Ki 4.5, 6.4 and 7.5 nM, respectively). Among the boron cluster-containing compounds, the highest A3 affinity (Ki 206 nM) was for adenosine derivative 41 modified at C2. In the matched molecular pairs, analogs bearing boron clusters were found to show lower binding affinity for adenosine receptors than the corresponding phenyl analogs. Nevertheless, interestingly, several boron cluster modified adenosine ligands showed significantly higher A3 receptor selectivity than the corresponding phenyl analogs: 7 vs. 8, 15 vs. 16, 17 vs. 18.
Keywords: Adenosine, Boron cluster, Agonists, Antagonists, Selectivity, Nucleosides, Purinergic receptors
1. Introduction
Adenosine is a naturally occurring nucleoside that is ubiquitously present throughout the body as a metabolic intermediate. In addition to its metabolic role within cells as a building block of RNA and many small biomolecules, such as the chemical energy source ATP, adenosine is released into the extracellular space. Extracellular adenosine, through interaction with the extracellular domain of G-protein-coupled receptors (A1 AR, A2A AR, A2B AR and A3 AR) exerts its regulatory role in the nervous, circulatory, endocrine, and immunological systems [1]. Adenosine levels are markedly increased in response to hypoxia, inflammation, or ischemia, and serve as a protectant under these conditions [2]. The prospect of harnessing these effects specifically for therapeutic purposes is attractive [3].
The availability of selective AR (adenosine receptor) ligands facilitates research on therapeutic applications of strategies modulating AR activity and, in some cases, has already provided clinical candidates [4]. Currently, the A2A AR agonist regadenoson is being evaluated as an agent that reduces COVID-19-induced lung injury through inhibition of hyperinflammation (ClinicalTrials.gov Identifier: NCT04606069).
In the search for potent and selective AR ligands, a plethora of adenosine and nonnucleoside ligands have been synthesized and tested [1,3–6]. Although selective AR modulators have promise for numerous therapeutic applications in practice, this goal, as in the case of many other targets, is not easy to achieve. The difficulties in the use of AR ligands in clinical practice result mainly from: i) low tissue specificity of the adenosine receptors in the body, and ii) cross-activation of nontarget AR receptors by highly potent selective ligands. One of the possible approaches to solve this problem is to develop ligands that do not cause 100% activation of the receptors but activate only a smaller percentage. In this way, not all adenosine receptors are targeted, but only those essential for achieving desirable clinical outcomes.
The development of nucleoside-boron cluster conjugates, including adenosine derivatives, is a new avenue of research in this field that is attracting considerable attention. Medicinal chemists are increasingly utilizing boron clusters (polyhedral boron cages) as a new generation of 3-dimensional (3D), abiotic privileged scaffolds, modifiers and pharmacophores in diverse bioactive molecular designs [7–10]. As we show in this article, the use of boron clusters as adenosine-modifying units may be one of the possible solutions enabling the construction of more selective AR ligands with sufficient potency at the same time.
The properties of boron clusters that are useful in drug design are listed below. (1) These clusters can form unique noncovalent interactions such as dihydrogen bonds due to the hydride character of H atoms, σ-hole bonding or ionic interactions; as a result, the types of interactions of boron cluster-containing compounds with biological targets may differ from purely organic molecules. (2) Boron clusters exhibit a spherical or ellipsoidal geometry and rigid 3D arrangement, which offer versatile platforms for 3D molecular construction. (3) The lipophilicity, amphiphilicity or hydrophilicity depend on the type of boron cluster used and allow the tuning of pharmacokinetics and bioavailability. (4) Boron clusters exhibit chemical stability and simultaneous susceptibility to functionalization, as well as (5) bioorthogonality, stability in biological environments and a decreased susceptibility to metabolism [7,9,11]. Due to the above reasons, the interactions of boron cluster drugs with their protein targets may be slightly different from the lock and key system that has evolved for purely organic ligands. Therefore, the use of boron cluster modification can be expected to allow us to challenge known drug targets in a new way and test new targets that have not been accessible thus far.
All ARs belong to the same class of purinergic G protein-coupled receptors; however, although they build on the same molecular blueprint, they differ in structural details and function. Due to the unique 3D structure of the boron cluster and the hydric character of hydrogen atoms of B–H bonds allowing for the formation of unconventional hydrogen bonds, the boron cluster modifier seems to allow for better sensing of these subtle differences between A3 and A1, A2A, A2B ARs than in the case of derivatives bearing purely organic groups.
We previously reported the chemical synthesis of a range of nucleoside-boron cluster conjugates, including those formed from adenosine, and evaluated their activity as blood platelet aggregation inhibitors [12], reactive oxygen species inhibitors [13], antivirals [14,15] and antitumor agents [16]. Some of these compounds are potential ligands for purinergic receptors and the observed biological activities might be attributed to the modulation of AR activity [13].
Here, a series of 30 adenosine derivatives that we designed modified with either inorganic, boron clusters (carborane groups) or organic phenyl groups for comparison were synthesized, and the effects of organic and inorganic modifications on the affinity of the ligands for the A1, A2A, A2B and A3 ARs were evaluated and compared. While no chemical modification can guarantee the transformation of any compound into drug candidates, we believe that our investigation of the chemistry and biology of innovative adenosine derivatives containing boron clusters expands the range of AR ligand types available for testing. Moreover, the effect of modification with a boron cluster on the increased selectivity of adenosine ligands towards A3 AR demonstrated herein is a finding of practical importance.
2. Results
2.1. The synthesis of boron cluster donors
The synthesis of 1-(3-azidopropyl)-1,12-dicarba-closo-dodecaborane (2) [13] was performed using a method analogous to the method described previously for 1,7-dicarba-closo-dodecaborane isomer [17] by treating bromide 1 [18] with sodium azide in a dimethylformamide (DMF) solution to provide compound 2 with a 93% yield (Scheme 1). Alternatively, a tosyl derivative instead of bromide can be used analogously, as described for 1-(3-azidopropyl)-1,2-dicarba-closo-dodecaborane synthesis [19]. The 1-(3-aminopropyl)-1,12-dicarba-closo-dodecaborane (4) [13] was obtained from azide 2 via catalytic hydrogenation according to the procedure published for isomeric 1,2- or 1,7-dicarba-closo-dodecaborane [17,19].
Scheme 1.

Methods of amine 4 synthesis: i. NaN3, DMF, rt; ii. QHSO4 in 2 M NaOH, HN(Boc)2 in CH2Cl2, reflux; iii. H2/10% Pd/C, CH3OH, rt; iv. HCl in CH3OH, rt.
Another method used to synthesize amine 4 is based on the conversion of bromide 1 to di-Boc-protected amine 3, as originally described for the synthesis of an analogous derivative of the 1,2-dicarba-closo-dodecaborane isomer. Amine 4, in the form of a hydrochloride, was obtained by removing the Boc groups with methanolic hydrogen chloride (Scheme 1) [20,21]. Next, 1-(4-aminobutyl)-1,12-dicarba-closo-dodecaborane (6), an aminoalkyl derivative of 1,12-dicarba-closo-dodecaborane with an extended butyl linker, was synthesized as described in a previous study [22] using a method based on phthalimide chemistry [19,22].
2.2. Synthesis of adenosine derivatives bearing boron clusters
Previously described compounds (34 [23], 37 [24], 38 [25], 39 [12], 40 [26], 41 [13], 42 [27], 43 [12], 47 [16] and 49 [16]), new adenosine derivatives containing 1,12-dicarba-closo-dodecaborane clusters (45 and 51) or phenyl groups (44, 46, 48, 50 and 52, Table 1) are described elsewhere [28], and a series of adenosine derivative pairs containing inorganic boron cluster modifications or alternative organic phenyl modifications (7, 8, 15–18, 23, 24, 27, 28 and 33, 35, 36) described here were synthesized (Table 1 and Schemes 2–5).
Table 1.
Binding affinity of compounds 7, 8, 15–18, 23, 24, 27, 28, 33–52, and unmodified adenosine and 2’-deoxyadenosine at the human A1, A2A, A2B and A3 adenosine receptors.
| Compound/reference | Structure | Ki [nM] or percent of inhibition [10 μM] | |||
|---|---|---|---|---|---|
| A1 AR | A2A AR | A2B AR | A3 AR | ||
| 7 |

|
29 ± 1 %a | 2 ± 1 %a | 1 ± 1 %a | 444a |
| 8 26 |

|
864a | 11 ± 2 %a | 19 ± 3%a | 32.9a |
| 15 |

|
40 ± 2 %a | 9 ± 1 %a | 7 ± 2 %a | 437a |
| 16 |

|
803a | 7100a | 3330a | 9.2a |
| 17 |

|
18 ± 3 %a | 1 ± 1 %a | 4 ± 4 %a | 34 ± 4 %a |
| 18 |

|
49 ± 2 %a | 42 ± 1 %a | 1380a | 4.5a |
| 23 |

|
38 ± 2 %a | 1 ± 1 %a | 5940a,b | 2200a |
| 24 |

|
3340a | 27 ± 5 %a | 24 ± 1 %a | 6.44a |
| 27 |

|
25 ± 1 %a | 1 ± 1 %a | 4670a | 10 ± 3 %a |
| 28 |

|
13,300a,b | 14 ± 2 %a | 1 ± 1 %a | 8770a,b |
| 33 |

|
29 ± 7 % | 17.1 ± 8 % | nd | 48 ± 12 % |
| 34 24 |

|
11 ± 3 %c | 3 ± 2 %c | n.d. | 856 ± 291c |
| 35 |

|
16 ± 7 % | 18 ± 4 % | n.d. | 9 ± 13 % |
| 36 |

|
11 ± 11 % | 15 ± 10 % | n.d. | 8 ± 13 % |
| 37 25 |

|
6 ± 10 % | 7 ± 2 % | n.d. | 19 ± 14 % |
| 38 26 |
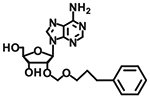
|
4 ± 10 % | 3 ± 0 % | n.d. | 29 ± 12 % |
| 39 14 |

|
32 ± 1 % | 0 ± 1 % | n.d. | 0 ± 12 % |
| 40 27 |

|
78.8 ± 34.3 | 2050 ± 40 | n.d. | 54.6 ± 5.8 |
| 41 14 |

|
2870 ± 1690 | 3070 ± 530 | n.d. | 206 ± 31 |
| 42 28 |

|
646 ± 4d | 1040 ± 200d | n.d. | 42 ± 2d |
| 43 13 |
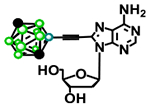
|
6 ± 6 % | 10 ± 5 % | n.d. | 1 ± 8 % |
| 44 29 |
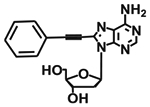
|
40 ± 9 % | 1990 ± 180 | n.d. | 879 ± 151 |
| 45 29 |

|
20 ± 6 % | 7080 ± 240 | n.d. | 2780 ± 800 |
| 46 29 |

|
1940 ± 1340 | 22 ± 9 % | n.d. | 8610 ± 1390 |
| 47 17 |

|
15 ± 1 % | 17 ± 2.7 % | n.d. | 4390 ± 2020 |
| 48 29 |

|
27.6 ± 7.2 % | 15 ± 22 % | n.d. | 1230 ± 220 |
| 49 17 |

|
27 ± 9 % | 12 ± 14 % | n.d. | 25 ± 8 % |
| 50 29 |

|
15 ± 20 % | −5 ± 6 % | n.d. | 54 ± 6 % |
| 51 29 |

|
16 ± 4 % | 7 ± 2 % | n.d. | 34 ± 7 % |
| 52 29 |

|
28 ± 28 % | 20 ± 6 % | n.d. | 44 ± 9 % |
| Ade |

|
13 ± 1 % | 3 ± 1 % | 4 ± 2 % | 10 ± 1 % |
| dAde |

|
5 ± 1 % | 2 ± 1 % | 12 ± 3% | 5 ± 1 % |
The radioligand replacement assay of compounds 7, 8, 15–18, 23, 24, 27 and 28 was performed under a contractual service agreement with Plataforma de Screening de Farmacos (USEF), 15782 Santiago de Compostela, Spain; Binding affinity assays of compounds 33–52 were performed in authors’ laboratory. Compounds 37–40 were double tested, for USEF data please see Supplementary Information, Table S2.
Compound 28 for A1 AR and A3 AR and 23 for A2B AR does not completely displace the radioligand. Ki value extrapolated by the analysis software might be not accurately estimated. n.d. = not determined.
Compound 34: A1 Ki > 100 μM (human), A2A Ki > 100 μM (human), A2B Ki > 100 μM (human), A3 Ki = 790 nM (human).[23]
Scheme 2.

Synthesis of adenosine derivatives with boron cluster 7 and phenyl 8 modifications, with an extended linker between the modification and exo-amino group of adenosine: i. Et3N in n-BuOH was used as the solvent.
Scheme 5.

Synthesis of adenosine derivatives with a boron cluster (33, 35) or a phenyl group (34, 36) attached to the carbon in the eight position of adenine nucleobase: i. Et3N/CuI/Pd(PPh3)2Cl2 (29), ii. H2/10% Pd/C, CH3OH.
Based on the results of an in silico virtual screen (data not shown), adenosine derivatives with an extended butyl linker between the boron cluster or phenyl modification and the exocyclic amino group of adenosine were synthesized. Adenosine was modified at position 6 via the substitution of the chlorine in commercially available 6-chloroadenosine (5) with a 1-(4-aminobutyl)-1,12-dicarba-closo-dodecaborane (6), which was used as the boron cluster donor, or alternatively, with its fully organic counterpart, 4-phenylbutylamine (9).
The substitution of chlorine at position 6 of compound 5 with a boron cluster containing amine 6 was performed using a method developed in a previous study for the synthesis of a homolog of compound 7 bearing a propyl linker [13]. N6-(4-Phenylbutyl) adenosine (8) was synthesized using a published procedure (Scheme 3) [26,29]. 5′-N-Ethylcarboxamidoadenosine (NECA) derivatives (Scheme 3) modified at position 6 with a boron cluster or phenyl group were synthesized by substituting chlorine at position 6 using a method analogous to the procedure described for compounds 7 and 8 above. The key intermediate 5′-ethylamino-5′-oxo-6-chloroadenosine (13) was obtained using published procedures with some modifications.
Scheme 3.

Synthesis of adenosine derivatives with boron cluster (15, 17) or phenyl (16, 17) modification, with a propyl (17, 18) or butyl (15, 16) linker between the modification and exo-amino group of adenosine, and with 5′-ethylamino group: i. Et3N in n-BuOH; ii. 2 M HCl in H2O/1,4-dioxane (1:1); iii. DMP, PTSA in DMF; iv. TEMPO, BAIB in CH3CN/H2O; v. SOCl2 in DMF/CHCl3, vi. EtNH2/THF.
First, 6-chloroadenosine (5) was treated with 2,2-dimethoxypropane in the presence of catalytic amounts of p-toluenesulfonic acid [30] to produce 2′,3′-O,O-isopropylidene-6-chloroadenosine (10) [31]. The remaining 5′-hydroxyl functional group of compound 10 was oxidized upon treatment with TEMPO/BAIB (2,2,6,6-tetramethyl-1-piperidinyloxyl/bisacetoxyiodobenzene) providing 2′,3′-O,O-isopropylidene-6-chloro-5′-oxoadenosine (11) [32]. The 4′-carboxylic group in compound 11 was amidated by first transforming it into C4′-acid chloride and then performing in situ ethylamine-mediated aminolysis to produce compound 12 [33]. The final removal of the isopropylidene protection in compound 12 was achieved under acidic conditions using 2 M hydrochloric acid, providing 5′-ethylamino-5′-oxo-6-chloroadenosine adenosine (13) [33]. The introduction of various amino substituents into the purine 6-position was achieved by treating compound 13 with boron cluster-containing amines 4 or 6 or alternatively with their fully organic counterparts, amines 9 or 14, yielding modified NECA derivatives 15–18.
The synthesis of 2-chloroadenosine or 2-chloro-2′-deoxyadenosine modified at the 6-position with a 1-(3-aminopropyl)-1,12-dicarba-closo-dodecaborane (4) or fully organic counterpart, 3-phenylpropylamine (14) (Scheme 4), was performed using the methods described above for compounds 7, 8 and 15–18. The much higher reactivity of the chlorine substituent at position 6 compared to position 2 was exploited [34,35]. Sonogashira-type cross coupling [36] between 8-bromo-2′-deoxyadenosine (30) and 2-ethynyl-1,12-dicarba-closo-dodecaborane (31) [37], a boron cluster donor, or phenylacetylene (32), its organic counterpart containing a triple bond, in the presence of bis(triphenylphosphine) palladium(II) dichloride (Pd(PPh3)2Cl2; 29) was used to tether the modification to nucleoside acceptor 30 (Scheme 5).
Scheme 4.

Synthesis of 2-chloro-adenosine and 2-chloro-2′-deoxyadenosine derivatives with boron cluster (23, 27) or phenyl (24, 28) modification: i. DMF/Et3N; ii. NH3 in CH3OH.
Although almost all adenosine derivatives described in the present study are modified at different positions of the adenine nucleobase, compounds 37 and 38 containing boron cluster modification or alternative organic phenyl modifications at the 2′-position of the sugar residue were also obtained (Scheme S1).
Compound 37 bearing a boron cluster was synthesized via the formation of a formacetal linkage using a previously described method [24]. Compound 38 containing a phenyl group at the 2’ position was obtained analogously using 3-phenyl-1-propanol instead of 1-(3-hydroxypropyl)-1,12-dicarba-closo-dodecaborane (Scheme S1 showing the synthesis of compounds 37 and 38 is available in Supplementary Information) [24,25,38].
2.3. Affinity and selectivity for adenosine receptors
The affinity and selectivity of the compounds were assayed by the radioligand binding method with the use of transfected cell lines overexpressing only one AR subtype. The radioligand replacement assay based on the competitive binding of the tested compound and a ligand with known affinity to the receptor was performed under a contractual service agreement with Plataforma de Screening de Farmacos (USEF), 15782 Santiago de Compostela, Spain (compounds 7, 8, 15–18, 23, 24, 27, and 28) or in the NIH laboratory (compounds 33–52) (Table 1). Compounds 37–40 were double tested; for USEF data, please see Supplementary Information, Table S2. Adenosine and 2′-deoxyadenosine were tested under the same conditions for comparison.
For the assessment of compounds 7, 8, 15–18, 23, 24, 27 and 28 as radioligands for A1 and A2B receptors, 1,3-[3H]-dipropyl-8-cyclopentylxanthine ([3H]-DPCPX), a selective antagonist of A1, was administered at different concentrations, 1 nM and 25 nM. An adenine isostere and a high-affinity selective antagonist, 2,8-substituted [1,2,4]triazolo[1,5-a] [1,3,5]triazine ([3H]-ZM241385), were selected for the A2A receptor. For the A3 receptor, 1-[3H]-(6-amino-9H-purin-9-yl)-1-deoxy-N-ethyl-β-d-ribofuronamide ([3H]-NECA) was used as a radioligand in the replacement assay. Nonspecific binding was determined in the presence of 6-N-R-phenylisopropyladenosine (R-PIA) for A1 and A3, and NECA for A2A and A2B receptors. CGS15943, ZM241385, MRS1220 and XAC were used as the standard controls for the A1, A2A, A2B and A3 receptors, respectively. CHO-A1, HeLa-A2A, HEK-A2B and HeLa-A3 cells expressing human A1, A2A, A2B or A3 receptors were used.
Radioligand binding assays for compounds 33–52 were performed using HEK293 cells expressing human A1, A2A, or A3 receptors and standard radioligands, as previously described [39]. Unlike the binding performed by USEF, which utilized antagonist radioligands for A1 AR and A2A AR, these determinations at human A1, A2A and A3 ARs used agonist radioligands (concentration, KD value; nM): [3H]-N6-R-phenylisopropyladenosine (1.0, 1.5), [3H]-2-[p-(2-carboxyethyl)phenyl-ethylamino]-5′-N-ethylcarboxamidoadenosine (10, 16.2), [125I]-N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide (0.2, 1.22), respectively. Nonspecific binding was determined using adenosine 5′-N-ethyluronamide (10 μM) [39].
2.4. Assessment of the functions of compounds 7, 8, 15, 16, 23 and 24 as ligands for the human A3 receptor
Among compounds that interact with the A3 receptor, a set of three pairs of boron cluster- or phenyl-modified adenosine derivatives with the highest affinity towards the A3 AR in their categories (“boronic” compounds vs. “phenylic” compounds) was selected for the functional study. The set of boronic compounds included compounds 7 (A3 Ki = 444 nM), 15 (A3 Ki = 437 nM) and 23 (A3 Ki = 2200 nM), and the phenylic compounds included compounds 8 (A3 Ki = 33 nM), 16 (A3 Ki = 9.2 nM) and 24 (A3 Ki = 6.4 nM). The interaction of agonists with the A3 receptor induces cAMP accumulation in the CHO-A3 cell line. The cAMP level was determined in CHO-A3 cells incubated with increasing concentrations of selected compounds to describe the A3 AR agonistic profiles of the compounds. Subsequently, the ability of the compounds to inhibit NECA-induced cAMP accumulation was evaluated to determine their antagonistic features. The values of the equilibrium dissociation constant (KB) for antagonists and agonist potency (50% maximal response, EC50) and maximal agonist effects (Emax) were determined. Based on the results of the functional study, all tested compounds exclusively exhibited A3 receptor agonist characteristics, and none inhibited the function of the A3 receptor (Table 2). Phenyl derivatives were the most potent agonists of the A3 receptor compared to boronic agonists. Among the boronic compounds, compound 7 was distinguished as the most effective activator of A3 receptors. The assessments of the affinity and biological activity of the phenylic compounds produced consistent results. Compound 24 displayed the highest A3 receptor affinity (Ki = 6.4 nM) and the highest potency in inducing cAMP production in CHO-A3 cells (EC50 = 10.4 nM), whereas the lowest affinity compound, 8, was the least potent among the analyzed phenylic compounds (EC50 = 54.3 nM).
Table 2.
Results of the functional assessment of the tested compounds (cAMP, EC50, nM) and the relationship of cAMP levels and A3 receptor binding.
| Compound | KB | EC50 [nM] | % Emax |
|---|---|---|---|
| 7 | no antagonist | 271 | 103 |
| 8 | no antagonist | 54.3 | 102 |
| 15 | no antagonist | 1010 | 99 |
| 16 | no antagonist | 16.3 | 102 |
| 23 | no antagonist | 3760 | 104 |
| 24 | no antagonist | 10.4 | 93 |
2.5. Assessment of the affinity of the selected compounds towards the A2A receptor
Based on our previous observations of the activity of some adenosine and 2′-deoxyadenosine modified with boron clusters as platelet function modulators [12], and the known role of A2A AR activation in antiplatelet activity, the compound library obtained in the present study was evaluated as an A2A ligand and in addition to Ki, as shown in Table 1.
The IC50 values for selected boron cluster/phenyl-modified pairs, 7, 8; 15, 16; 17, 18; 23, 24; 39, 40, with preferential affinity for A3 but also binding to the same extent as A2A, were compared (Table 3). The results obtained prompted us to test the ability of phenyl and boron cluster-modified adenosine derivative pairs to influence platelet activation in a more complex model of human blood.
Table 3.
IC50 values of radioligand displacement calculated for A2A receptor.
| Compound | IC50 [μM] | Compound | IC50 [μM] |
|---|---|---|---|
| 7 | >100 | 23 | >100 |
| 8 | 74.1 ± 9.7 | 24 | 22.0 ± 4.4 |
| 15 | >100 | 35 | n.d |
| 16 | 26.5 ± 5.1 | 37 | >100 |
| 17 | >100 | 39 | >100 |
| 18 | 14.6 ± 0.6 | 40 | 20.3 ± 0.5 |
IC50 – Compound concentration that inhibits radioligand binding to the receptor by 50%. n.d. – not determined.
2.6. Inhibition of platelet aggregation in human blood
The ability of the synthesized compounds to decrease platelet activation was tested in a stepwise manner. As a first approach, measurements in platelet-rich plasma were conducted. In this method, platelet aggregation was induced by ADP, and the formation of platelet aggregates in the presence of the tested compounds was monitored. Red blood cells (RBCs), which are known to accumulate and metabolize adenosine, are not present in this experimental setup.
The tendency of the tested compounds to be taken up by RBCs was not known; therefore, we decided to utilize simplified conditions in the preliminary tests. If a particular compound was not effective in these RBC-free conditions (platelet-rich plasma), the compound was unlikely to possess inhibitory activity in whole blood, where RBCs are present. These ineffective compounds were not tested in whole blood. As shown in Table 4, compounds 18 and 40 that are modified with a phenyl group significantly decreased platelet activation induced by ADP by approximately 50%, while other compounds did not possess an inhibitory effect or inhibited activation by less than 50%. Compound 24 inhibited platelet aggregation by 20%.
Table 4.
ADP-induced platelet aggregation in platelet-rich plasma in the presence of the compounds 7, 8, 15–18, 23, 24, 35, 37, 39 and 40.
| Compound | 0 | 10 μM | 50 μM | ||
|---|---|---|---|---|---|
| 7 | 656.2 ± 55.8 | 686.5 ± 66.1 | n.s. | 642.8 ± 45.4 | n.s. |
| 8 | 654.9 ± 123.1 | 695.3 ± 133.4 | n.s. | 623.4 ± 165.6 | n.s. |
| 15 | 654.8 ± 57.5 | 698.7 ± 74.4 | n.s. | 657.1 ± 70.7 | n.s. |
| 16 | 648.5 ± 45.0 | 662.3 ± 67.0 | n.s. | 641.1 ± 35.1 | n.s. |
| 17 | 677.7 ± 65.5 | 646.2 ± 58.6 | P < 0.001 | 639.5 ± 64.7 | n.s. |
| 18 | 708.9 ± 64.5 | 630.0 ± 80.3 | P < 0.01 | 336.8 ± 108.5 | P < 0.001 |
| 23 | 657.1 ± 57.8 | 715.3 ± 85.8 | n.s. | 657.0 ± 67.2 | n.s. |
| 24 | 678.9 ± 70.9 | 649.5 ± 70.2 | P < 0.001 | 548.6 ± 72.2 | P < 0.01 |
| 35 | 655.6 ± 38.8 | 665.7 ± 86.8 | n.s. | 682.3 ± 17.4 | n.s. |
| 37 | 651.4 ± 59.9 | 661.1 ± 61.3 | n.s. | 616.0 ± 24.7 | P< 0.05 |
| 39 | 663.0 ± 54.6 | 678.4 ± 55.7 | n.s. | 658.0 ± 46.8 | n.s. |
| 40 | 641.1 ± 85.5 | 566.4 ± 112.9 | P < 0.05 | 289.5 ± 158.9 | P < 0.001 |
Data are presented as the means ± SD, n = 6–9 samples. The results are shown as the area under an aggregation curve measured in arbitrary units and was calculated as the extent of aggregation (a.u.) × time [min]. The significance of the inhibitory effect was tested in a pairwise manner (control vs. compound) using the bootstrap-boosted (10000 iterations) paired one-tailed Student’s t-test. Inhibitory effects greater than 50% are marked in bold. n.s. = nonsignificant.
Additional analyzes of the receptor-ligand binding curves, as well as the percentage changes in platelet activity revealed a clear relationship between the ability of the active compounds to interact with the A2A receptor and inhibit platelet aggregation.
Analysis of the percentage values of both receptor binding and inhibition of platelet aggregation showed that the only relevant relationship between these two compound features is valid for the A2A receptor and not for the other receptors (rs_A2A = 0.746, P < 0.01, rs_A2B = 0.378, n.s., rs_A1 and rs_A3 < 0.1). Of the four compounds interacting with the A2A receptor, compound 16 did not affect platelet aggregation. Characteristically, the active compounds are N6-(3-phenylpropyl) adenosine derivatives 18, 24 and 40 which simultaneously preferentially bind to A3 AR. In summary, regardless of the receptor preference (mainly in relation to A3), compounds interacting with the A2A receptor, modified with a phenyl group, but not a boron cluster, showed antiplatelet activity.
Compounds 18 and 40, which possessed the highest inhibitory activity, were further tested in a whole blood aggregometry assay. In this experiment, platelet aggregation in whole blood was induced with ADP, collagen or arachidonic acid. As shown in Fig. 2, compound 40 significantly decreased ADP-stimulated platelet aggregation at all tested concentrations but did not inhibit aggregation induced by collagen or arachidonic acid (data not shown). In contrast, compound 18 significantly inhibited aggregation induced by either ADP (5 μM) or collagen (1 μg/mL), but it had no effect on platelet aggregation induced by arachidonic acid (data not shown).
Fig. 2.

Effects of compounds 18 and 40 on platelet aggregation in whole blood. The results are presented as the area under an aggregation curve. a) Effect of compound 40 on platelet aggregation induced by ADP. b) Effect of compound 18 on platelet aggregation induced by ADP. c) Effect of compound 18 on platelet aggregation induced by collagen. The significance of differences was determined using a paired Student’s t-test: P < 0.005 for 10 μM compound 40, P < 0.0001 for 30 μM compound 40 and for 50 μM compound 40 ZL-1098; P < 0.0001 for all the tested concentrations of compound 18 (both for ADP- and collagen-induced aggregation), n = 13.
As a final step, the ability of compounds 18 and 40 to inhibit thrombus formation under flow conditions in the presence of collagen was evaluated. Whole blood was perfused over a collagen-coated surface under flow conditions that emulate the conditions in a human artery. The contact of platelets with collagen in combination with shear forces induces the formation of thrombi. As shown in Fig. 3, the thrombi in samples treated with compound 40 were smaller than the thrombi in the untreated samples. Surprisingly, compound 18 did not exert an inhibitory effect under these conditions (data not shown).
Fig. 3.

Effects of compound 40 on the formation of thrombi deposited on collagen under flow conditions. A) Representative images of fluorescently labelled thrombi formed in blood treated with a vehicle (left panel) or 50 μM compound 40 (right panel). B) Comparison of the sizes of thrombi. The sizes of thrombi visualized in the channels were quantified using dedicated software. Each dot represents the summarized area of thrombi larger than 50 μM2 formed in the blood of a particular donor treated either with vehicle or compound 40. Significant differences were determined using a paired Student’s t-test, P < 0.01, n = 9.
2.7. Structure-activity relationships (SARs)
The strength of interactions of compounds and adenosine receptors varied depending on the receptor type, and for A3 AR, the strength of interactions was significantly higher than the strength of interactions for other receptors (ANOVA, p = 0.0001; logKiA3 vs. logKiA1, p = 0.0026; logKiA3 vs. logKiA2A, p = 0.0003, Tukey’s test; Kruskal–Wallis test, p = 0.0001; KiA3 vs. KiA1, p = 0.043; KiA3 vs. KiA2A, p = 0.0001). The analysis of categorical data (active versus inactive compounds) showed that the number of active compounds was clearly higher for the A3 receptor (n = 17) than for the A2A (n = 6) and A1 receptors (p = 0.0018, chi-square statistic). Therefore, the receptors were analyzed separately for the SAR model. Among substructure descriptors, the best predictors for A3 receptor affinity selected using the GRM method (general regression model, GRM, Wald chi-square statistics) were as follows: modifier (carborane or phenyl, p = 0.0008), position of modification (N6, C2, or C8; p = 0.028), and sugar type (R and not R; p = 0.018); for A1 receptor: modifier (carborane or phenyl, p = 0.007), and sugar type (R and not R; p = 0.03); results for A2A were not statistically significant due to insufficient number of active compounds per group (6 active versus 22 inactive). Structure-activity analysis was performed only for statistically significant classes/groups of descriptors. The regression approach of classification was implemented from the PLS and C&RT modules in Statistica software. The data were divided into a training set (n = 18 compounds) and a test set (n = 10 compounds, 7, 8, 23, 24, 36, 39, 43, 44, 47, 48). The C&RT tree method was used for the prediction of compound classification (Supplementary Material, Figure S129 A and B). The analysis enabled formulation of some general rules. The most important predictor of compound affinity to the A3 receptor was modification position (relative importance 100%) over modifier type (85%) and sugar type (53%). The analysis showed that active compounds with log Ki ≤ 1 are adenosine modified at the N6 position with a phenyl group (n = 3 compounds in the training set); moderately active compounds with log Ki 2–3 are adenosine modified at the C2 and N6 positions with boron clusters or at the C2 and C8 positions with phenyl groups; and low active or inactive (log Ki ≥ 4) compounds are C8 adenosine modified with carborane and 2′-deoxyadenosine derivatives. Compounds with arabinoadenosine and 2′-deoxyadenosine scaffolds are less active or inactive. In general, the most active compounds among phenyl derivatives are adenosine modified at the N6 position, whereas among carborane derivatives, adenosine is modified at the C2 and N6 positions. The boosted tree method gave similar results (importance of modification site 100%, modifier 75%, and sugar type 67%; details of the calculations are shown in Supplementary Material, Figure S129C). Evaluation of the model on numerous newly synthesized compounds in the future may verify the described principles as general for adenosine phenyl and carboranyl derivatives as adenosine receptor ligands, with higher or lower affinity to the receptor.
Additional detailed analysis showed the importance of ligand rigidity in the group of phenyl-modified compounds at both the C2 and C8 positions, which was a statistically significant feature of the compound interaction with the A3 AR (active compounds 34, 42, 44, and 48 with rigid linkers versus inactive 36, 46, 50, and 52; p = 0.002; GRM). A similar relationship was observed for the corresponding carboranyl analogs at C2 positions, among which the active derivatives contained a rigid linker (compounds 41 and 47 versus 45 and 49).
2.8. Receptor selectivity analysis
Detailed in-depth analysis of the structural similarities shows some specific relationships in receptor selectivity for individual compounds. In the group of N6 adenosine derivatives, receptor selectivity was analyzed for individual cases in relation to the linker length and the presence of specific additional groups such as N-ethyl carboxamide (in the C5′ position) and chlorine (Cl in the C2 position of purine ring). Among adenosine derivatives bearing carborane groups at the N6 position, linker length decided on receptor selectivity (C4 – active, A3 AR selective compounds 7 and 15, C3 – inactive compound 17 or compound 39 slightly active on A1 AR), regardless of the presence of the 5′ N-ethyl carboxamide substituent. However, chlorine substitution in the N6 carboranyl derivative with a C3 linker resulted in activity against A3 AR, but was poor (compound 23). In the group of phenyl N6 adenosine derivatives, no compound was found to be selective against one type of receptor; in the group of C3 linkers, the lowest selectivity was found for unsubstituted phenyl derivatives (40, all receptor types, including A2A AR). The presence of chlorine at C2 or N-ethyl carboxamide at C5′ made this type of compound more selective for A3 AR (compounds 18 and 24). Linker extension (C4 derivative, 8) resulted in a relatively better interaction with A3 AR than with the other receptor types; however, substitution with N-ethyl carboxamide at C5’ resulted in a loss of selectivity of compound (16). In summary: 1) C3 linker and 2) long C4 linker in combination with N-ethyl carboxamide substituent determined lack of receptor selectivity of N6-phenyl adenosine derivatives. However, 3) a long C4 linker and 4) a C3 linker in combination with N-ethyl carboxamide or chlorine substituents increased the selectivity for A3 AR.
The group of compounds with phenyl modification at the C8 position with the flexible linker were inactive (compounds 36 and 52), regardless of sugar structure. The rigidity of the linker determined the interaction with adenosine receptors in this group of compounds; however, receptor selectivity depended on the sugar moiety. Ribose (R)-containing compound 34 was A3 selective, but compound 44 with deoxyribose (dR) was not selective. However, the activity of a group of C8-modified compounds depended on the presence of a phenyl substituent since all analogous derivatives containing the carboranyl modifier were inactive (33, 35, 43, and 51). Interesting structure-selectivity dependencies were observed for C2 derivatives. Whereas the structure of the sugar moiety decided on the receptor selectivity, all these compounds were active, regardless of the linker or modifier structure. R-containing compounds were not selective (41, 42, 45, and 46), and arabinose (Ara)-containing compounds were A3 selective (compounds 47 and 48).
2.9. Docking results
A homology model of the A3 receptor was generated from the A2A receptor (PDB code: 4UG2), as described in the Materials and Methods, and was subsequently used for docking studies. A docking study showed that carborane and phenyl derivatives 15 and 16 are located at analogous positions inside the A3 AR, i.e., ribose downward and modifier towards the “entrance” of the receptor interior, identified as a classic A3 AR ligand-binding pocket (Fig. 4 A). This docking position was energetically preferred for compounds 15 and 16 (binding free energy, ΔG, −9.6 ± 0.076 kcal/mol and −10.4 ± 0.11 kcal/mol, respectively, see Table S1 in Supplementary Information). The binding free energy showed a higher affinity of compound 16 to the A3 AR than the affinity of compound 15, although the calculated Ki_calc values for both compounds were in the nanomolar range (26 ± 4.3 nM and 93 ± 8.4 nM, respectively; Fig. 4 A and B; Table S1 in Supplementary Information). The docking results revealed a fourfold lower affinity of compound 15 than the affinity of 16 towards the A3 receptor. Although qualitatively similar, quantitatively, these results differ from those in vitro in which compound 16 revealed a 47-fold higher affinity towards the A3 AR than compound 15 (see Table 1).
Fig. 4.

Binding modes of compounds 15 and 16 at A3 AR binding site. (A and B) Overall look on topography of 15 and 16 preferred docking position: “ribose down” (A), and alternative poses: “ribose up” (B); affinity for A3 AR was expressed as Ki calc values (nM). (C and D) Details of compounds interaction with A3 AR binding site for preferred docking positions of compounds 15 (C) and 16 (D). Structures were visualized and analyzed using the PyMOL Molecular Graphics System, Version 2.3.4, Schrödinger, LLC (Figures A and B), and The Protein-Ligand Interaction Profiler (PLIP, Figures C and D).
Docking experiments also revealed another interesting property of compounds 15 and 16. The position of ribose directed towards the binding pocket was apparently a preferred docking position for compound 15. A reverse docking position of this compound, i.e., carborane downward, was also observed (regarding the number of docking events), albeit this reverse docking position requires much higher energetic effort (ΔG −9.4 kcal/mol, Ki_calc 129 nM; Table S1 in Supplementary Information). Therefore, the carborane moiety seems to stabilize the ribose directed towards the binding pocket. However, unlike derivative 15 bearing a boron cluster, for compound 16, the position of the phenyl residue pointing towards the binding pocket seems possible; the ΔG of such binding was −9.9 kcal/mol (Ki calc 53 nM; Table S1 in Supplementary Information). Both docking positions of compounds 15 and 16, ribose directed towards the binding pocket (“ribose down”) and ribose located towards the “entrance” of the receptor (“ribose up”), are stabilized by Phe168 interacting with the purine ring through π-stacking (Table S1 in Supplementary Information). Nevertheless, the position “ribose down” seems preferable, considering the energy cost and the number of interactions with neighboring amino acids (Table S1 in Supplementary Information and Fig. 4C and D). Positions “ribose down” of both compounds were stabilized by hydrogen bonds between ribose and Ser271 and His272 and between the N-ethyl carboxamide group (in the C5 ′position) and Thr94 (Figures C and D). This position is also stabilized by hydrophobic interactions of the carboxamide group and Ile98 or Leu91; in the case of compound 16, an additional hydrophobic interaction of the carboxamide group with Trp185 is formed (Fig. 4 D). Asn250 forms two hydrogen bonds with purine nitrogen atoms (6 N and 7 N), and Phe168 forms π-stacking with purine rings in compounds 15 and 16 (Figures C and D).
Apparently, the nucleoside scaffold of both compounds interacts with similar amino acids of the binding pocket without any steric hindrance resulting from carborane or phenyl groups.
The most interesting parts of the conjugates are carboranyl or phenyl modifiers together with long, fourth-carbon linkers. The flexible linker participates in hydrophobic interactions of compounds 15 and 16 with two (Val169 and Leu264) or three (Val169, Leu264, and Ile268) amino acids, respectively. Significantly, Phe168 forms a parallel interaction with two subfragments of compound 15: π-stacking with purine ring and hydrophobic with a boron vertex of the cluster (Fig. 4C). By these interactions, Phe168 could stabilize the position of compound 15 “in the electrostatic holder” in the binding pocket, including Val169, Leu264, Tyr265 and Ile268 (Fig. 4C). The results obtained partially explain the possible interactions of carboranyl- and phenyl-modified adenosine derivatives with the A3 receptor at the molecular level.
3. Discussion
Two major types of chemical transformations were used to obtain adenosine derivative pairs containing inorganic boron cluster modifications or alternative organic phenyl modifications:1) Sonogashira-type cross coupling between 8-halogen-adenosine and a boron cluster or phenyl group donor containing a terminal triple bond (compounds 33 and 34), and 2) the substitution of chlorine at carbon 6 of adenosine with an alkylamine containing a boron cluster or a phenyl group (compounds 7, 8, 15–18, 23, 24, 27, and 28).
Catalytic hydrogenation of the triple bond in the rigid alkyne linker (compounds 33 and 34) provided adenosine derivatives 35 and 36 (Table 1) bearing a flexible ethyl tether between the nucleobase and modification. Compound 38 modified at the 2’ position was obtained analogously to the method described for compound 37 using 3-phenyl-1-propanol instead of 3-(1,12-dicarba-closo-dodecaboran-1-yl)-1-propanol via the formation of a formacetal linkage [24].
A modular approach based on the coupling of a modification acceptor (suitably functionalized adenosine component) and modification donor (boron cluster or phenyl group) was applied to synthesize all compounds described in the present study. Commercially available phenyl derivatives, 4-phenylbutylamine (9), 3-phenylpropylamine (14), 3-phenyl-1-propanol and phenyl acetylene (32), were used.
The boron cluster-containing counterparts 1-(4-aminobutyl)-1,12-dicarba-closo-dodecaborane (6), 1-(3-aminopropyl)-1,12-dicarba-closo-dodecaborane (4), 1-(3-hydroxypropyl)-1,12-dicarba-closo-dodecaborane and 2-ethynyl-1,12-dicarba-closo-dodecaborane (31) were synthesized using different chemistries. Methods for preparing azido- and aminoalkylcarboranes and derivatives of isomeric 1,2- and 1,7-dicarba-closo-dodecaborane, as well as their applications in the synthesis of various carborane derivatives have been reported [17–19,21]. Additionally, we previously described the synthesis of azido- and amino-derivatives of 1,12-dicarba-closo-dodecaboranes 2 and 4 [13], although to our knowledge, detailed procedures for their synthesis have not yet been described. Here, we fill the gap (Scheme 1). Three different methods were employed and compared to obtain amines 4 or 6: 1) catalytic hydrogenation of azides, 2) deprotection of Boc-protected amines obtained from suitable bromoalkyl carborane and bis-Bocamine (Scheme 1), or 3) deprotection of phthalimide-protected amines obtained via the alkylation of carborane with N-bromoalkyl phthalimide. Methods 1 and 2 are best suited for the synthesis of 1-(3-aminopropyl)carboranes due to the accessibility of 1-(3-bromopropyl)carboranes prepared conveniently from suitable 1-(3-hydroxypropyl)carboranes available via oxetane ring opening with lithiated carboranes [10]. The phthalimide methodology seems more versatile due to the possible use of alkyl tethers with varying lengths in the N-bromoalkyl phthalimide component. 2-Ethynyl-1,12-dicarba-closo-dodecaborane (31) was synthesized by performing a Pd-catalyzed Kumada-type cross coupling reaction of the corresponding iodinated carborane with Me3SiCCMgBr, followed by desilylation of the trimethylsilylalkynyl-substituted cluster [37].
The adenosine derivative pairs obtained modified with inorganic (boron cluster) or organic (phenyl) groups at the same location within the nucleoside unit were screened for affinity towards A1, A2A, A2B and A3 adenosine receptors using a radioligand replacement assay. Pronounced, albeit varying, effects of the modifications on the ligand affinity and selectivity towards the studied receptors were observed. In general, most of the adenosine modifications studied here, regardless of whether a boron cluster or phenyl group was used as the modifying unit, displayed higher affinity for the A3 AR than for A1, A2A or A2B, which was also confirmed by statistical calculations, including both the classification approach and the factor analysis. An exception is compound 40 with a 3-phenylpropyl modification at N6 or, to a lesser extent, compound 39 with a (1,12-dicarba-closo-dodecaboran-1-yl)propyl at the same position, which showed significant interaction with all AR types (A3, A1, and A2A, 40) or tendency to bind to A1 AR (39).
Interestingly, the transformation of the 5′-OH group in these derivatives into a 5′-N-ethylcarboxamide group (compounds 17 and 18) or an extension of the linker between N6 and phenyl or (1,12-dicarba-closo-dodecaboran-1-yl)propyl-group (compounds 7 and 8) switches this preference from A1/A2A to A3 AR (Table 1). Notably, the ability of compounds 18 and 40 to bind to A1/A2A AR, even with moderate affinity and preference for A3, appears to be important for their activity as inhibitors of platelet aggregation in blood (vide infra).
The analogs with the highest selectivity towards A3 AR include compounds: 16 (A3 Ki = 9.2 nM): A1/A3 = 87, A2A/A3 = 772, A2B/A3 = 359), 18 (A3 Ki = 4.5 nM): A1/A3 = 49% (at 10 μM) vs. Ki = 4.5 nM, A2A/A3 = 42% (at 10 μM) vs. Ki 4.5 nM, A2B/A3 = 307; 24 (A3 Ki = 6.4 nM): A1/A3 = 519, A2A/A3 = 27% (at 10 μM) vs. Ki = 6.4 nM, A2B/A3 = 24% (at 10 μM) vs. Ki = 6.4 nM, 40 (A3 Ki = 7.5 nM): A1/A3 = 54% (at 10 μM) vs. Ki = 7.5 nM, A1/A2A = 32% (at 10 μM) vs. Ki = 7.5 nM, A1/A2B = 264, 41 (A3 Ki = 206 nM): A1/A3 = 14, A2A/A3 = 15, 42 (A3 Ki = 42.3 nM): A1/A3 = 15, A2A/A3 = 25. Based on the aforementioned observations supported by the=statistical analysis of SAR, the preferential binding to the A3 AR of the ligands evaluated in the present study seems to be determined by the presence of the modification at N6 (or 2-C in the case of compounds 41 and 42), which is consistent with previous reports [5].
Importantly, a rigid ethynyl linker between the modification and nucleobase at position C2 increases the ability of the ligand to bind A3 tenfold compared with a more flexible ethyl linker for both the boron cluster and phenyl modification: compound 41 A3 Ki = 206 nM (boron cluster modification attached through an ethynyl linker) vs. compound 45 A3 Ki = 2780 nM (boron cluster modification attached through an ethyl linker) and compound 42 A3 Ki = 42.3 nM (phenyl modification attached through an ethynyl linker) vs. compound 46 A3 Ki = 8610 nM (phenyl modification attached through an ethyl linker). Interestingly, the aforementioned selectivity of the studied adenosine derivatives towards A3 AR is also valid for carborane modifications, which are better accommodated by A3 than by A1, A2A or A2B ARs.
Furthermore, for reasons that are currently unclear, regardless of the location of the modification and receptor type, in each pair of counterparts bearing a phenyl group or boron cluster modification the ligand bearing a phenyl group usually showed higher affinity for the specific receptor. According to preliminary findings based on in silico calculations, the observed difference may be due to the 50% higher van der Waals volume of the carborane cage than the rotating phenyl group and different electronic properties of both modifying units [25]. The replacement of the phenyl group with the boron cluster seems to weaken the ligand interactions with the receptor binding pocket.
Analyses of the affinity, selectivity and biological activity (intracellular cAMP production) revealed that the phenyl modifications at the exo-amino group of adenosine (compounds 8, 16 and 24) efficiently interacted with the A3 receptor. Moreover, the affinity and biological activity of the phenylic compounds were consistent. A comparison of the biological activity of phenylic compounds with their respective boronic analogs (IC50 boronic vs IC50 phenylic) indicated the significance of the phenyl substituent at a particular site of the compound to obtain the best affinity for the A3 receptor. The replacement of the phenyl group with a boronic moiety resulted in the most dramatic decrease in the biological activity of the receptor treated with compounds 23 and 24. Because a phenylic substitution of 2-chloroadenosine (compound 24) results in an approximately 300-fold increase in affinity for the A3 receptor compared with 2-chloroadenosine (Ki of 6.4 nM (this work) and 1900 nM [40]), the phenylic base functional group appears to be desirable.
However, higher affinity may not only have a beneficial effect on the compound activity but also, at the expense of selectivity, a negative effect. This problem can be approached by designing selective agonists that, although they activate a given receptor at a relatively high concentration of compound, still activate the remaining receptors to a small extent. One example may be the activity of compounds 15 and 16 towards the A3 receptor, one boron cluster modified and the other a phenyl group. Analysis of receptor selectivity showed that compound 16, with a higher affinity for the A3 receptor (Ki = 9.2 nM), binds 100% of the A1, A2A and A2B receptors at micromolar concentrations.
Compound 16 displays relatively low selectivity over A1 AR (87-fold, A1 AR Ki value is 803 nM), 771-fold selectivity over A2A AR (Ki value is 7098 nM), and 362-fold selectivity over A2B (AR Ki value is 3331 nM). Compound 15 modified with a boron cluster with affinity Ki = 437 nM to A3 AR activates 100% of the A3 receptor at micromolar concentrations, but in contrast to 16, Compound 15 does not interact noticeably with A1, A2A, or A2B receptors, showing clearly significant A3 AR selectivity. An illustrative comparison of the binding of 15 and 16 to the receptors at nanomolar and micromolar concentrations is shown in Fig. 5. Docking results revealed a good dynamic fit of carborane derivative 15 to the interior of the A3 AR binding pocket without steric hindrance. Experiments in silico have shown that the carborane moiety linked to a nucleoside scaffold via a four-carbon flexible linker significantly interacts with the surrounding hydrophobic amino acids, engaging them to stabilize the compound at the active site. The length and flexibility of the linker may be crucial for the functionality of the carborane as a modifier of A3 AR ligands based on adenosine scaffolds. An example of the influence of linker structure on A3 AR affinity can be compound 17, which differs from compound 15 in the length of the linker “only” by one methylene group. This small change, however, turns out to be sufficient for 17 to lose its affinity to this receptor (Table 1). Whether the linker length and its structure could be a paradigm of the effectiveness of adenosine ligands modified with boron clusters requires further research in terms of both chemical synthesis and biological and in silico studies (see Fig. 6).
Fig. 5.

Dose-response curves of binding compounds 15 and 16 to adenosine receptors A1 ( ), A2A (
), A2A ( ),A2B (
),A2B ( ), A3 (
), A3 ( ), evaluated in radioligand inhibition assay (details in Experimental Section). Values are mean ± SEM.
), evaluated in radioligand inhibition assay (details in Experimental Section). Values are mean ± SEM.
Fig. 6.

Biological activity of N6 derivatives of adenosine, modified with boron cluster (7, 15, and 23) or phenyl group (8, 16, and 24); median values of Ki are shown. Both groups of derivatives show activity towards A3 AR. Phenyl compounds interact significantly more strongly with A3 AR, but also activate A2A AR, interfering with the activity of blood platelets.
Although ARs are promising therapeutic targets under a wide range of conditions, one difficulty when developing active analogs is that each adenosine receptor affects not just one but a large number of body functions. The development of sufficiently selective AR modulators with the required pharmacological characteristics is an attractive but challenging goal.
The use of a boron cluster modifier instead of ordinary organic modifications such as phenyl groups provides a new perspective and can help to overcome these difficulties, as shown for A3 AR in the above example. The higher selectivity of some adenosine ligands modified with boron clusters than phenyl-modified derivatives towards the A3 receptor can be of special interest in light of the A3 AR-mediated protective response in heart diseases and skeletal muscle injury and other conditions related to the functional role of this receptor [41]. The modifications proposed in this work offer a wide range of specific structural features related to receptor specificity. Among the compounds with lower specificity, there may be those interacting with the A2A receptor (compounds 18, 24, or 40), an important therapeutic target in the hemostatic system. Even if they interact with the A2A AR in micromolar concentrations, such a low affinity turns out to be sufficient to influence the activity of components of the hemostatic system.
Antiplatelet therapies, which are administered to patients at risk of myocardial infarction or ischemic stroke, are based on the inhibition of platelet aggregation in the blood. These interventions are always associated with a risk of bleeding. At the same time, some patients respond poorly to antiplatelet therapy. An increase in the dose of the drug in these patients is not the preferred method to bypass this ‘resistance’ due to the risk of bleeding. In turn, combined therapies are applied in which at least two independent pathways regulating platelet activation are targeted simultaneously to limit platelet activation. One potential target is the activation of adenosine receptors on platelets. This approach has not yet been tested in the clinic but has been extensively studied in vitro and in vivo [42–44]. Agonists of adenosine receptors have recently been shown to enhance the inhibitory activity of P2Y12 antagonists – antiplatelet drugs used widely as antiplatelet therapy [45]. Blood platelets express A2A and A2B subtypes of adenosine receptors [46,47]. The activation of these receptors increases the intraplatelet concentration of cAMP, which decreases the ability of platelets to aggregate [48]. One of the disadvantages of using AR agonists as antiplatelet agents is their relatively short-term effect following in vivo application [49]. This limitation may be partially attributed to the effective uptake of the drug by red blood cells, which has been shown for adenosine [50]. Thus, novel agonists of ARs with more persistent effects in vivo should be developed.
Two of the newly synthesized compounds described in the present study exerted a significant inhibitory effect on platelet aggregation in whole blood stimulated with ADP or collagen. In the thrombus formation test, only one of the compounds proved to be effective. The latter test emulates pathophysiological phenomena occurring in a diseased artery. The formation of a thrombus under flow conditions involves complex interactions between factors that activate platelets, such as shear forces, thromboxane, ADP or thrombin [51]. Therefore, the thrombus formation test differs from a classical aggregation test in which only one platelet activator is used as a trigger. Thus, the confirmation of the antiplatelet activity of a compound in a classical aggregation test does not unambiguously determine its effectiveness in the thrombus formation test. This finding may also explain why compound 18 effectively inhibited collagen-induced aggregation but did not inhibit thrombus formation on collagen. In contrast, compound 40, which was not effective as an inhibitor of collagen-induced aggregation in the classical test, was a potent inhibitor in the thrombus formation test. Notably, in the thrombus formation test, collagen activated platelets, but thrombus expansion was further driven by the other factors mentioned above. Thus, we postulated that compound 40 is more effective than compound 18 at inhibiting one of these pathways of platelet activation that play a major role in thrombus growth. Since both compounds inhibited ADP-induced aggregation to a similar extent and failed to inhibit arachidonic acid-induced aggregation, another mechanism is likely involved in thrombus formation that is affected by compound 40 to a greater extent than compound 18. Each of the stimuli inducing platelet aggregation activates separate receptors, but the downstream pathways intercept at some points. These points include, among many others, a decrease in intracellular cAMP and an increase in intracellular calcium. Adenosine receptor agonists reverse these effects with varying effectiveness [52]; therefore, their ability to inhibit platelet aggregation induced by these stimuli may also differ between compounds.
The antiaggregation activity of compounds 18 and 40 and their parallel A3 AR affinity should not be surprising in light of their clear although moderate affinity towards A2A AR (Tables 1 and 3, Fig. 1). These compounds significantly interact with the A2A receptor at micromolar concentrations (IC50 for radioligand displacement 14.6 ± 0.6 μM and 20.3 ± 0.5 μM, respectively) (Table 3). However, nanomolar affinity for the A3 receptor is irrelevant for platelet activation given the absence of this receptor on the platelet surface. Furthermore, aggregation studies were conducted at 10 μM and 50 μM ligand concentrations, which seems to be sufficient for A2A AR activation, leading to inhibition of aggregation. Compound 40 was an effective inhibitor of thrombus formation under conditions that approximate the rupture of an atherosclerotic plaque. Thus, this compound might represent an interesting candidate for the development of a potential antiplatelet compound. Moreover, compound 40 may be considered a dual-acting A2A and A3 AR ligand interacting with a given target depending on the concentration.
Fig. 1.

The effect of compounds 7, 8, 15–18, 23, 24, 35, 37, 39 and 40. (50 μM) on ADP-induced platelet aggregation in platelet-rich plasma in comparison with the effect on the radioligand binding to A2A receptor. The values are expressed as percentages of maximal control values of radioligand binding and platelet aggregation (AUC parameter). Data points marked with a red line indicate compounds 18, 24 and 40 having both properties: interaction with A2A receptor (at least 50% of ligand displacement) and effect on the platelet aggregation (at least 20% inhibition). Data points marked with a black line indicate inactive compounds. White triangle - compound 16, that interact with the receptor, but is inactive on blood platelets. Spearman’s correlation coefficient calculated for all data points is rs = 0.746, P (2-tailed) = 0.0085.
However, developing potent ARs targeting drugs is a difficult and challenging task. One of the factors that must be considered, which is often overlooked, is cross-binding for the new adenosine derivatives with other adenosine-like recognition sites and non-adenosine receptor drug interactions. Discussing details of this issue goes beyond the scope of this publication. Here, we would like to draw attention to two phenomena related only to the first part of that concern. Both compounds 18 and 40, due to the activation of A2A AR, show desired antiplatelet activity while simultaneously being A3 AR ligands (Table 1). These properties may at first glance be of concern due to the side effects of A3 AR activation, but in this case, a side effect may be cardioprotection [41], which for antithrombotics could be an added value.
Another issue is the possible phosphorylation of adenosine ligands possessing free 5′-hydroxy groups. Most adenosine-based A3 ligands are modified at the 5′-position and are not vulnerable to phosphorylation. However, other derivatives with a free 5′-hydroxyl group, as we showed earlier, can be phosphorylated quite efficiently by adenosine kinase (ADK) [28] and probably also by other kinases. One of the possible paths of metabolic transformation of these derivatives resulting from ADK action, as in the case of cladribine, is conversion in subsequent phosphorylation steps into the corresponding triphosphate. The triphosphates formed interact with DNA/RNA polymerases by competition with natural nucleoside triphosphates, disturbing DNA replication, which in turn may cause cytotoxicity and other adverse effects. Awareness of possible, although not necessary, already recognized activities of potential AR-targeting therapeutics may help minimize side effects, sometimes achieve synergism and optimize therapeutic activities.
4. Conclusions
We described a series of adenosine and 2′-deoxyadenosine pairs modified with a 1,12-dicarba-closo-dodecaborane cluster or alternatively with a phenyl group at the same position and studied their affinity for the A1, A2A, A2B and A3 adenosine receptors. Regardless of whether a boron cluster or phenyl group was used as the modifying unit and the location of the modification within the nucleobase (C2, C8 or N6), most of the studied adenosine derivatives tended to exhibit higher affinity for A3 AR than for A1, A2A or A2B ARs. This finding expands previous observations of the preferential binding of adenosine ligands modified at N6 or C8 to A3 AR [24,51].
In our study, the highest selectivity for A3 was observed for adenosine derivatives modified at N6, followed by derivatives modified at C2. For adenosine ligands modified at N6, the replacement of a hydroxymethyl group at C2’ with an ethylcarbamoyl group or the incorporation of chlorine at C2 increased the selective affinity for A3 AR. In addition, phenyl modification increased the general affinity of adenosine derivatives for ARs to a greater extent than their boron cluster counterparts. At the same time, the observed higher selectivity of some AR ligands modified with boron clusters compared to the corresponding phenyl derivatives towards the A3 receptor may open the way for the development of molecules activating A3 without causing unwanted antiplatelet effects.
All selected adenosine ligands with the highest affinity for A3 displayed agonist characteristics in the functional study, regardless of whether they were modified with a phenyl group or boron cluster. Finally, the tests of inhibition of blood platelet aggregation allowed us to identify two ligands, 18 and 40, with clear but moderate antiaggregatory properties, most likely due to A2A AR activation. Both compounds are modified with phenyl groups at the N6 position.
Despite the low level of A3 AR in most cells and tissues, A3 AR expression is upregulated in blood cells in subjects with some pathological conditions. Indeed, preclinical studies have reported anti-inflammatory, anticancer and cytoprotective effects of A3 receptor agonists [41,53]. In addition, the cardioprotective effect triggered by A3 AR activation can be an added value for the identified phenyl-modified compounds with A2A AR-related antiplatelet activities. The results described here expand the range of uses of adenosine derivatives for further testing in this promising area of the therapeutic application of selective AR ligands.
5. Experimental section
5.1. Synthesis of adenosine derivatives bearing a boron cluster or phenyl group modification
5.1.1. Materials
Commercially available chemicals were of reagent grade and used as received. N-(4-Bromobutyl)phthalimide and 3-phenylpropylamine (14) were purchased from Acros Organics (Geel, Belgium). 4-Phenylbutylamine (9) and phenylacetylene (32) and 10% Pd on activated charcoal were purchased from Sigma-Aldrich (Darmstadt, Germany). 6-Chloroadenosine (5), 8-bromoadenosine (30), 2,6-dichloropurine, 1-O-acetyl-2,3,5-tri-O-benzoyl-β-d-ribofuranose and 2-deoxy-3,5-di-O-(4-toluoyl)-α-d-erythropentofuranosyl chloride (Hoffer’s chlorosugar) were purchased from Carbosynth (Compton - Berkshire, United Kingdom). Silica gel column chromatography was performed on silica gel 60 (230–400 mesh), Sigma-Aldrich (Steinheim, Germany) or E. Merck (Darmstadt, Germany). Rf values refer to analytical TLC performed using pre-coated silica gel 60 F254 plates purchased from Sigma-Aldrich (Steinheim, Germany) or E. Merck (Darmstadt, Germany) and developed in the solvent system indicated. Compounds were visualized with UV light (254 nm). Melting points are uncorrected and were measured on a Büchi (New Castle, DE, USA) Melting Point B-540 apparatus. The yields are not optimized.
Multiplate® test cuvettes for whole blood aggregometry were obtained from Roche Diagnostics GmbH (Mannheim, Germany). Multiplate test cells for turbidimetric aggregometry were purchased from Chronolog Co (Havertown, PA). ADP was purchased from Sigma-Aldrich (Steinheim, Germany). Collagen was from Chronolog Co. (Havertown, PA), arachidonic acid was from Dynabyte (Munich, Germany), Biochips for the assays of thrombi formation were purchased from Cellix (Dublin, Ireland), collagen for chip coating was from Chronolog Co. (Havertown, PA). Anti-CD61 PE-conjugated antibodies for the staining of thrombi were from Becton Dickinson (San Diego, CA).
Melting points are uncorrected and were measured on a Kofler apparatus. The purity of all tested compounds was established by HPLC and was 95–100% with the exception of compound 33, 34, 35 and 51 whose purity was higher than 90%. (for HPLC details please see the experimental section, and for HPLC traces of newly synthesized compounds 7, 8, 15–18, 23, 24, 27, 28, 33–35 see Figures S1–S13 in Supplementary Information).
5.1.2. Methods
Nuclear Magnetic Resonance (NMR).
The 1H, 13C, dept135 and 11B NMR spectra in CDCl3 or CD3OD were recorded using a Bruker AVANCE III HD (Billerica, MA, USA) 500 MHz or a BrukerAvance DPX 250 MHz spectrometer; shift values in parts per million are relative to the SiMe4 internal reference. Multiplets were assigned as s (singlet), bs (broad singlet), d (doublet), t (triplet), m (multiplet), bm (broad multiplet), dd (doublet of doublets), dt (doublet of triplets), td (triplet of doublets), tt (triplet of triplets), ddd (doublet of doublet of doublet). Coupling constants are reported in Hertz (Hz).
Mass spectrometry.
Fast atom bombardment (FAB) mass spectra were recorded on a Finnigan MAT (Bremen, Germany). The m/z was measured in positive and negative modes. The MALDI-TOF spectra were recorded on a Voyager Elite mass spectrometer (PerSeptive Biosystems Inc., Framingham, MA) in linear negative ion mode. The ESI spectra were recorded on a Waters HPLC/MS (Milford, MA, USA). High-resolution mass spectra were recorded on an LTQ Orbitrap Velos instrument (Thermo Scientific, Waltham, MA, USA). The calculated m/z corresponds to calculated values based on the average mass of the elements consisting of natural isotopes.
Ultraviolet spectroscopy measurements (UV).
UV measurements were performed on a GBC Cintra10 UV-VIS spectrometer (Dandenong, Australia). Samples for UV experiments, ca. 0.5 A260 OD of each compound were dissolved in 96% C2H5OH. The measurement was performed at ambient temperature.
Attenuated total reflectance Fourier transform infrared (ATR–FTIR) spectroscopy measurements.
Infrared absorption spectra were recorded using a Smart iTR diamond attenuated total reflectance (ATR) attachment on a Nicolet 6700 FTIR spectrometer (Thermo Scientific) equipped with an ETC EverGlo* source for the IR range, a Ge-on-KBr beam splitter, and a DLaTGS detector. Samples to be analyzed were placed on a diamond ATR element in the solid form. The Omnic 8.1 software program was used for data acquisition and processing. Alternatively, a Jasco 6200 (Easton, MD, USA) FTIR spectrometer was used, and spectra were run in a KBr pellet.
HPLC analysis.
Analyses were performed on a Hewlett-Packard 1050 system equipped with a UV detector. A Hypersil Gold C18 RP, 250 × 4.6 mm column, 5 μM particle size (Thermo Scientific, Runcorn, UK) was used, and the flow rate was 1 mL/min. All analyses were run at ambient temperature. The gradient elution profile was as follows: 20 min from 0% B to 100% B, 20 min 100% B, and 5 min from 100% B to 0% B. Buffer A contained 0.1 M TEAB (pH 7.0) in a mixture of acetonitrile and water (2:98, v/v), and buffer B contained 0.1 M TEAB (pH 7.0) in a mixture of acetonitrile and water (60:40, v/v). UV detection was performed at λ = 262 nm.
Synthesis of 1-(3-bromopropyl)-1,12-dicarba-closo-dodecaborane (1) was performed as described [18].
Synthesis of 1-(4-aminobutyl)-1,12-dicarba-closo-dodecaborane (6) was performed as described [22].
Synthesis of 2′,3′-O,O-isopropylidene-6-chloro-5′-oxoadenosine (11) was performed according to the literature procedure [32].
2′,3′,5′-tris-O-Benzoyl-2,6-dichloroadenosine (20) [54] 3′,5′-bis-O-(4-methylbenzoyl)-2,6-dichloro-2′-deoxyadenosine (19) [55] were synthesized as described.
2′-O-(1,12-dicarba-closo-dodecaboran-1-yl)propyleneoxymethyl]-adenosine (37) [24] and 2′-O-(3-phenylpropyleneoxymethyl]-adenosine (38) [25] were synthesized as described.
N6-[3-(1,12-Dicarba-closo-dodecaboran-1-yl)propyl]adenosine (39) [13] was obtained as described previously.
N6-(3-phenylpropyl)adenosine (40) [26] and N6-(4-phenylbutyl) adenosine (8) [26] were obtained according to the literature methods.
2-Ethynyl-(1,12-dicarba-closo-dodecaboran-1-yl)adenosine (41) was synthesized as described [13].
2-Phenylethynyladenosine (42) was synthesized as described [27].
Synthesis of 8-[2-(1,12-dicarba-closo-dodecaboran-1-yl)ethynyl]-2′-deoxyadenosine (43) was performed as previously described [12].
Synthesis of 2-ethynyl-(1,12-dicarba-closo-dodecaboran-2-yl) arabinoadenosine (47) [16] and 2-ethyl-(1,12-dicarba-closo-dodecaboran-2-yl)arabinoadenosine (49) [16] was performed as described earlier.
Synthesis of 1-(3-azidopropyl)-1,12-dicarba-closo-dodecaborane (2).
1-(3-Bromopropyl)-1,12-dicarba-closo-dodecaborane (1) (250 mg, 0.94 mmol) was dissolved in anhydrous DMF (3 mL) under argon atmosphere then sodium azide (71.7 mg, 1.1 mmol) was added. The reaction mixture was stirred at room temperature until TLC monitoring (CH2Cl2/hexane, 1:1) showed complete conversion of the starting material (ca. 2 h). Then the solvent was evaporated to dryness under vacuum yielding crude product 2 which was purified by silica gel column chromatography using a mixture of CH2Cl2/hexane (1:1) as an eluent. Yield: 207 mg, 93%, colourless oil. TLC (CH2Cl2/hexane, 1:1): Rf = 0.69; FT-IR (ATR) νmax, cm−1: 3065, 2960, 2933, 2865 (C–H), 2600 (B–H), 2090 (N3); 1H NMR {11B BB} (CDCl3, 600.25 MHz, 25 °C, TMS) δ, ppm: 1.41–1.47 (2H, m, CH2CH2CH2N3),1.67–1.71 (2H, m, CH2CH2CH2N3), 1.67–2.56 (10H, m, BH), 2.65 (1H, m, CHcarborane), 3.13 (2H, t, J = 6.6 Hz, CH2CH2CH2N3); 13C NMR (CDCl3, 250.13 MHz, 25 °C, TMS) δ, ppm: 28.73, 36.03, 50.72, 58.45,83.63; 11B NMR {1H BB} (CDCl3, 80.25 MHz, 25 °C, BF3/(C2H5)2O) δ, ppm: 12.56 (5B, s), −14.96 (5B, s); 11B NMR (CDCl3, 80.25 MHz, 25 °C, BF3/(C2H5)2O) δ, ppm: 14.52 (d, 5B, 2JBH = 164.8 Hz), −12.15 (d, 5B, 2JBH = 161.7 Hz); MS (APCI, +Ve): m/z (%) calculated for C5B10H17N3: 227.32, found 200.25 [M − N2]+.
Synthesis of N,N-di-tert-butyloxycarbonyl-l-(3-aminopropyl)-1,12-dicarba-closo-dodecaborane (3).
Tetrabutylammonium hydrogensulfate (494.6 mg) was dissolved in 2 M NaOH aqueous solution(1.5 mL), and then a solution of di-tert-butyl iminodicarboxylate (289.7 mg) in CH2Cl2 (7.2 mL) was added. To the resultant mixture a solution of 1-(3-bromopropyl)-1,12-dicarba-closo-dodecaborane (1, 340.0 mg, 1.28 mmol) in CH2Cl2 (5 mL) was added dropwise. The reaction mixture was stirred under reflux for 3.5 h, then the reaction was quenched with water (16.5 mL). The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (2 × 7 mL). The combined organic layers were dried over anhydrous MgSO4 and the organic solvent was evaporated to dryness. The resulting crude product was purified by silica gel column chromatography (230–400 mesh) using hexane/CH2Cl2 (1:1) as an eluting solvent system. Yield: 463.2 mg (90%); TLC (CH2Cl2): Rf = 0.37; FT-IR (ATR): v, cm−1 = 3054 (CHcarborane), 2975 (), 2923 (), 2872 (), 2852 (), 2595 (BH), 1768 (C=O), 1466 (CH2), 1363 (CH3), 1099 (CO); 1H NMR (CDCl3, 500.13 MHz, 25 °C) δ, ppm: 1.39–1.44 (2H, m, CH2), 1.48 (18H, s, 6 × CH3), 1.59 (2H, dd, J = 11.9, 5.5 Hz, CH2), 1.64–2.57 (10H, m, BH), 2.62 (1H, s, CHcarborane), 3.36 (2H, t. J = 7.1 Hz, CH2); 13C NMR (CDCl3, 125.76 MHz, 25 °C) δ, ppm: 28.17 (CH3), 28.88 (CH3), 29.83 (CH3), 36.21 (CH2),45.56 (C–N(Boc)2), 58.20 (Ccarborane), 82.50 (O–C(CH3)3), 152.49 (C=O); 11B NMR {1H BB} (CDCl3, 160.46 MHz, 25 °C) δ, ppm: 15.04 (5B, s), −12.56 (5B, s); 11B NMR (CDCl3, 160.46 MHz, 25 °C) δ, ppm: 15.54 (5B, d, J = 165.3 Hz), −13.06 (5B, d, J = 162.1 Hz); MS (ESI): m/z (%) calculated for C15H35B10O4N: 401.3569, found: 424.3475 [M + Na]+.
Synthesis of 1-(3-aminopropyl)-1,12-dicarba-closo-dodecaborane (4).
Method A. 1-(3-Azidopropyl)-1,12-dicarba-closo-dodecaborane (2, 150 mg, 0.66 mmol) was suspended in dry methanol (2 mL) under argon atmosphere, next 10% Pd/C (86 mg) was added. Reaction mixture was stirred at room temperature under H2 atmosphere (balloon reservoir). After 2 h the solvent was evaporated and the residue was resuspended in diethyl ether. Suspension was filtered through a pad of Celite and the filtrate was evaporated to dryness. Oily residue was purified by silica gel column chromatography using a mixture of hexane/Et2O/Et3N (1:1:0.02) as an eluent. Yield: 81 mg, 61%, white solid. Method B. To N,N-tert-butyloxycarbonyl-1-(3-aminopropyl)-1,12-dicarba-closo-dodecaborane (3, 475.7 mg, 1.18 mmol) a solution of hydrogen chloride in methanol (9.48 mL) was added. The reaction mixture was stirred for 18 h at room temperature, then solvent was evaporated to dryness under reduced pressure. The resulting crude product was purified by silica gel column chromatography (230–400 mesh) using a linear gradient of CH3OH in CH2Cl2 (5%–14%) as an eluting solvent system. Yield: 214 mg (76%) of free amine containing a trace of hydrochloride salt. TLC (CH2Cl2/CH3OH 95:5): Rf = 0.38 (free amine), (CH2Cl2/CH3OH, 9:1): Rf = 0.09 (hydrochloride); FT-IR (ATR) ν, cm−1: 3250–2800 (), 3061 (CHcarborane), 2921 (), 2851 (), 2601 (BH), 1603 (NH); 1H NMR {11B BB} (CDCl3, 250.13 MHz, 25 °C, TMS) δ, ppm: 1.26–1.32 (2H, m, CH2CH2CH2NH2), 1.60–1.63 (2H, m, CH2CH2CH2NH2), 1.56–2.53 (10H, m, BH), 2.33 (2H, t, J = 7.1 Hz, CH2CH2CH2NH2), 2.62 (1H, m, CHcarborane); 13C NMR (CDCl3, 250.13 MHz, 25 °C, TMS) δ, ppm:29.84 (CH3), 36.75 (CH2-carborane), 49.03 (CH2–N), 58.23 (Ccarborane), 84.50 (C(CH3)3); 11B NMR {1H BB} (CDCl3, 80.25 MHz, 25 °C, BF3/(C2H5)2O) δ, ppm: 12.53 (s, 5B), −15.04 (s, 5B), 11B NMR (CDCl3, 80.25 MHz, 25 °C, BF3/(C2H5)2O) δ, ppm: 12.53 (d, 5B, 2JBH = 162.7 Hz), −15.04 (d, 5B, 2JBH = 165.8 Hz); MS (ESI): m/z calculated for C5B10H19N: 201.252, found: 202.259 (100) [M+H].
Synthesis of N6-[(1,12-dicarba-closo-dodecaboran-1-yl)but-4-yl]adenosine (7).
The synthesis was performed according to a modified literature procedure30.6-Chloroadenosine (5, 50 mg, 0.15 mmol) and 1-(4-aminobutyl)-1,12-dicarba-closo-dodecaborane (6) (39 mg, 0.18 mmol) was dissolved in n-BuOH (0.88 mL) then Et3N (0.07 mL, 0.52 mmol) was added. The resultant solution was refluxed for 2 h then evaporated to dryness. The crude product was purified by silica gel column chromatography using linear gradient of CH3OH in CH2Cl2 (0–6%) as an eluent. Yield: 56 mg (80%), yellowish solid. TLC (CH2Cl2/CH3OH, 9:1): Rf = 0.40; UV–Vis (95% C2H5OH) λ, nm: 228 (min), 267 (max); FT-IR (ATR) ν, cm−1: 3232 (O–H), 2926 (C–H), 2861 (C–H), 2601 (B–H), 1620 (C=N), 1077 (C–O); RP-HPLC, tR = 23.34 min; 1H NMR (CD3OD, 600.13 MHz, 25 °C) δ, ppm:1.25–1.31 (4H, m, butyl CH2), 1.48–1.53 (2H, m, butyl CH2),1.70–1.73 (2H, m, CH2–CH2–NH), 1.85–2.53 (10H, m, BH), 3.10 (1H, bs, CHcarborane) 3.49 (1H, bs, NH), 3.74 (1H, dd, J = 2.4, 10.2 Hz, 5′bH), 3.88 (1H, dd, J = 2.4, 10.2 Hz, 5′aH), 4.17 (1H, d, J = 2.4 Hz, 3′H), 4.32 (1H, d, J = 2.4 Hz, 4′H), 4.73–4,75 (1H, m, 2′H), 5.94 (1H, d, J = 6.6 Hz, 1′H), 8.21 (1H, s, 2-H), 8.24 (1H, s, 8-H); 13C NMR (CD3OD, 150.90 MHz, 25 °C) δ, ppm: 26.38 (CH2), 28.60 (CH2), 29.36 (CH2),38.27 (CH2), 58.41 (CHcarborane), 62.31 (C5′), 71.32 (C-3′), 74.04 (C-2′), 86.83 (C-4′), 89.91 (C-1′),140.07 (C-8),152.11 (C-2), 154.94 (C-6), 11B NMR {1H BB} (CD3OD, 192.55 MHz, 25 °C) δ ppm: 12.65 (s, 5B), −15.12 (s, 5B); MS (ESI, +3 kV, 300 °C): m/z calculated for C16H31B10N5O4: 465.56, found: 466.35 [M+H]+.
Synthesis of 2′,3′-O,O-isopropylidene-6-chloroadenosine (10).
The synthesis was performed according to the modified literature procedure [30]. 6-Chloroadenosine (5, 1000 mg, 3.49 mmol) was dissolved in dry DMF (13 mL) then p-toluenesulfonic acid monohydrate (130 mg, 0.70 mmol) and 2,2-dimethoxypropane (0.93 mL,10.47 mmol) was added. The resultant solution was stirred overnight at ambient temperature and then evaporated to dryness. The crude product was purified by column chromatography using linear gradient of CH3OH in CH2Cl2 (0–6%). Yield: 1110 mg (97%), white solid. TLC (CH2Cl2/CH3OH, 9:1): Rf = 0.63; UV–Vis (95% C2H5OH) λ, nm: 228 (min), 264 (max); FT-IR (ATR) ν, cm−1: 3289 (O–H), 2922 (C–H), 2854 (C–H), 1663 (C=N), 1081 (C–O); 1H NMR (CDCl3, 500.13 MHz, 25 °C) δ, ppm: 1.38 (3H, s, CH3), 1.65 (3H, s, CH3), 3.82 (1H, d, J = 11 Hz, 5′aH), 3.98 (1H, d, J = 11 Hz, 5′bH), 4.56 (1H, s, 4′H), 5.11 (1H, s, 3′H), 5.20 (1H, s, 2′H), 6.00 (1H, s, 1′H), 8.30 (1H, s, 8-H),8.77 (1H, s, 2-H); 13C NMR (CDCl3, 125.76 MHz, 25 °C) δ, ppm: 25.20 (CH3), 25.36 (CH3), 63.11 (CH2) 81.54, 83.21 (C-2′, C-3′), 86.36 (C-4′),94.14 (C-1′), 114.28 (CH3CCH3), 133.00 (C-4), 145.04 (C-8), 150.40 (C-2), 151.91 (C-6); MS (ESI, −3 kV, 300 °C): m/z calculated for C12H14ClN5O4: 326.74, found: 361.1 [M+Cl]−.
Synthesis of 2′,3′-O,O-isopropylidene-5′-ethylamino-5′-oxo-6-chloroadenosine (12).
The synthesis was performed according to a modified literature procedure [33]. 2′,3′-O,O-Isopropylidene-5′-oxo-6-chloroadenosine (11, 475 mg, 1.39 mmol) was dissolved in dry CHCl3 (7 mL) then dry DMF (0.27 mL, 3.49 mmol) and SOCl2 (0.51 mL, 6.97 mmol) was added. The resulting solution was stirred under reflux for 4 h then evaporated to dryness. The brown residue was dissolved in dry THF (9.3 mL), cooled to 5 °C then 2 M solution in THF ethylamine was added (1.75 mL, 3.49 mmol). The solution was stirred for 20 min and evaporated to dryness. The crude product was purified by silica gel column chromatography using linear gradient of CH3OH in CH2Cl2 (0–10%) as an eluent. Yield: 293 mg (57%), brown oil. TLC (CH2Cl2/CH3OH, 9:1): Rf = 0.70; UV–Vis (95% C2H5OH) λ, nm: 229 (min), 265 (max); FT-IR (ATR) ν, cm−1: 3309 (N–H), 3073 (C–H), 2983 (C–H), 2936 (C–H), 1662 (C=O), 1592 (C=N), 1200 (C–O), 1093 (C–O); 1H NMR (CD3OD, 500.13 MHz, 25 °C) δ, ppm: 0.53 (3H, t, J = 7 Hz, CH3), 1.41 (3H, s, CH3), 1.59 (3H, s, CH3), 2,76 (2H, q, J = 7.0 Hz, N–CH2–C), 4.69 (1H, d, J = 1.5 Hz, 4′H), 5.59 (1H, d, J = 1.5 Hz, 3′H), 5.69 (1H, dd, J = 4.5, 1.5 Hz, 2′H), 6.50 (1H, s, 1′H), 7.65=(1H, bs, NH), 8.69–8.70 (2H,=m, 8-H, 2-H); 13C NMR (CD3OD, 125.76 MHz, 25 °C) δ, ppm: 14.50 (CH3CH2N), 25.76, 27.50 (isopropylidene CH3), 35.13 (CH3CH2N) 85.75 (C-3′), 89.90 (C-4′), 93.36 (C-1′), 115.39 (CH3CCH3), 133.26 (C-4), 149.06 (C-8), 152.06 (C-2), 153.46 (C-6), 171.85 (C=O); MS (ESI, −3 kV, 300 °C): m/z calculated for C12H14ClN5O4: 367.79, found: 402.2 [M+Cl]−.
Synthesis of 5′-ethylamino-5′-oxo-6-chloroadenosine adenosine (13).
The synthesis was performed according to the modified literature procedure [33]. 2′,3′-O,O-Isopropylidene-5′-ethylamino-5′-oxo-6-chloroadenosine (12, 150 mg, 0.41 mmol) was dissolved in a mixture of 2 M HCl solution in water and 1,4-dioxane (1:1, 3.5 mL), the resultant solution was stirred for 1 h at 70 °C then was evaporated to dryness. The crude product was purified by reverse phase flash chromatography using a gradient of CH3OH in H2O (5–50%) as an eluent. Yield: 54 mg, (41%), yellowish solid. TLC (CH2Cl2/CH3OH, 9:1): Rf = 0.27; UV–Vis (95% C2H5OH) λ, nm: 231 (min), 265 (max); FT-IR (ATR) ν, cm−1: 3251 (O–H), 3079 (N–H), 2987 (C–H), 2941 (C–H), 2683 (C–H), 1645 (C=O), 1059 (C–O); 1H NMR (acetone-d6, 600.26 MHz, 25 °C) δ, ppm: 1.16 (3H, t, J = 6 Hz, CH3), 3.31–3.35 (2H, m, N–CH2–C), 4.51–4.54 (2H, m, 2′OH,=3′OH),4.82 (1H, d, J = 6 Hz, 4′H), 4.97–5.0 (2H, m, 2′H, 3′H), 6.26 (1H, d, J = 6 Hz, 1′H), 7.99 (1H, bs, NH), 8.81 (1H, s, 8-H), 8.91 (1H, s, 2-H); 13C NMR (acetone-d6, 150.94 MHz, 25 °C) δ, ppm: 14.22 (CH3), 33.62 (CH2–N), 73.12 (C-2′), 73.64 (C-3′), 84.98 (C-4′), 89.26 (C-1′), 132.59 (C-4), 146.23 (C-8), 150.44 (C-2), 151.57 (C-6), 168.82 (C=O); MS (FAB, Gly, +Ve): m/z calculated for C12H14ClN5O4: 327.72, found: 328.2 [M+H]+.
Synthesis of N6-[(1,12-dicarba-closo-dodecaboran-1-yl)but-4-yl]-5′-ethylcarbamoyl-adenosine (15).
The synthesis was performed analogously as described for 6-chloroadenosine adenosine.30 5′-Ethylamino-5′-oxo-6-chloroadenosine adenosine (13, 50 mg,0.15 mmol) and 1-(4-aminobutyl)-1,12-dicarba-closo-dodecaborane hydrochloride (6) (36 mg, 0.17 mmol) was dissolved in n-BuOH (0.76 mL) then Et3N (0.07 mL, 0.65 mmol) was added. The resulting solution was refluxed for 2 h then evaporated to dryness. The crude product was purified by silica gel column chromatography using linear gradient of CH3OH in CH2Cl2 (0–6%) as an eluent. Yield: 39 mg, 51%, orange solid. TLC (CH2Cl2/CH3OH, 9:1): Rf = 0.43; UV–Vis (95% C2H5OH) λ, nm: 230 (min), 267 (max); FT-IR (ATR) ν, cm−1: 3364 (N–H), 3231 (O–H), 3063 (C-Hcarborane), 2928 (C–H), 2855 (C–H), 2602 (B–H), 1647 (C=O), 1614 (C=N), 1049 (C–O); RP-HPLC, tR = 24.93 min; 1H NMR (CD3OD, 600.13 MHz, 25 °C) δ, ppm: 1.21 (3H, t, J = 7.2 Hz, CH3), 1.24–1.34 (4H, m, CH2-(CH2–CH2)–CH2), 1.49–1,54 (2H, m, CH2–CH2-carborane), 1.74 (2H, m, CH2–CH2–NH), 1.98–2.52 (10H, m, BH), 3.10 (1H, s, CHcarborane),3.37 (2H, q, J = 7.2 Hz, N–CH2–CH3), 3.50 (1H, bs, NH-Bu), 4.31 (1H, d, J = 4.8 Hz, 3′H), 4.46 (1H, s, 4′H), 4.75–4,77 (1H, m, 2′H), 6.00 (1H, d, J = 7.2 Hz, 1′H), 8.24 (1H, s, 2-H), 8.27 (1H, s, 8-H); 13C NMR (CD3OD, 150.90 MHz, 25 °C) δ, ppm: 13.68 (CH3), 26.37, 28.62, 29.37,33.67 (CH2), 38.26 (CH2–N), 58.41 (C-5′), 71.95 (C-3′), 73.59 (C-2′),85.03 (C-4′), 89.11 (C-1′), 140.68 (C-8), 152.39 (C-2), 154.95 (C-6), 170.66 (C=O); 11B NMR {1H BB} (CD3OD, 192.55 MHz, 25 °C) δ, ppm: 12.67 (s, 5B), −15.13 (s, 5B); MS (ESI, +3 kV, 300 °C): m/z calculated for C18H33B10N6O4: 506.61, found: 507.37 [M+H]+.
Synthesis of 5′-ethylcarbamoyl-N6-(4-phenylbutyl)adenosine (16).
The synthesis was performed analogously as described for 6-chloroadenosine [29]. 5′-Ethylamino-5′-oxo-6-chloroadenosine adenosine (13, 96 mg, 0.29 mmol) and 4-phenylbutylamine (9) (0.056 mL, 0.35 mmol) was dissolved in n-BuOH (1.5 mL) then Et3N (0.15 mL, 1.06 mmol) was added. The resulting solution was refluxed for 2 h then was evaporated to dryness. The crude product was purified by flash chromatography using a gradient of CH3OH in CH2Cl2 (0–10%). Yield: 48 mg, 37%, white solid. TLC (CH2Cl2/MeOH, 9:1): Rf = 0.38; UV–Vis (95% C2H5OH) λ, nm: 232 (min), 268 (max); FT- IR (ATR) ν, cm−1: 3233 (O–H), 3083 (N–H), 3027 (C–H), 2975 (C–H), 2932 (C–H), 2858 (C–H), 1646 (C=O), 1615 (C=N), 1049 (C–O), 745 (Phe), 698 (Phe); RP-HPLC, tR = 18.94 min; 1H NMR (CD3OD, 500.13 MHz, 25 °C) δ, ppm: 1.20 (3H, t, J = 7.5 Hz, CH3),1.70–1.75 (4H, m, (CH2)2), 2.63–2.68 (2H, m, CH2-Phe), 3.30–3.38 (4H, m, 2xCH2NH, overlapped by methanol signal), 3.60 (1H, bs, NH-Bu-Phe), 4.31–4.47 (2H, m, 2′H, 3′H), 4.75 (1H, m, 4′H), 6.01 (1H, d, J = 7.5 Hz, 1′H), 7.11–7.24 (5H, m, Phe-H), 8.23 (1H, s, 2-H), 8.27 (1H, s, 8-H); 13C NMR (CD3OD, 125.76 MHz, 25 °C) δ, ppm:13.56 (CH3), 30.43, 37,95, 41,84 (3x CH2), 35.56, 37.05 (2x CH2N),73.85 (C-3′), 75.49 (C-2′), 86.93 (C-4′), 91.00 (C-1′), 122.07, 127.25, 129.79, 129.93, 142.53, 144.06 (5x Phe), 154.28 (C-8), 156.84 (C-2), 172.56 (C=O); MS (ESI, −3 kV, 300 °C): m/z calculated for C22H28N6O4: 440.50, found: 475.3 [M+Cl]−.
Synthesis of N6-[(1,12-dicarba-closo-dodecaboran-1-yl)prop-3-yl]-5′-ethylcarbamoyl adenosine (17).
The synthesis was performed according to the modified literature procedure [28]. 5′-Ethylamino-5′-oxo-6-chloroadenosine adenosine (13, 59 mg, 0.18 mmol) and 1-(3-aminopropyl)-1,12-dicarba-closo-dodecaborane (4, 43 mg, 0.18 mmol) was dissolved in n-BuOH (0.90 mL) then Et3N (0.09 mL,0.65 mmol) was added. The resulting solution was refluxed for 2 h, then evaporated to dryness. The crude product was purified by silica gel column chromatography using linear gradient of CH3OH in CH2Cl2 (0–7%) as an eluent. Yield: 52 mg, (59%) white solid. TLC (CH2Cl2/CH3OH, 85:15): Rf = 0.58; UV–Vis (95% C2H5OH) λ, nm: 228 (min), 270 (max); FT-IR (ATR) ν, cm−1: 3247 (N–H), 3069 (CHcarborane), 2954 (C–H), 2928 (C–H), 2857 (C–H), 2603 (B–H), 1729 (C=O), 1615 (C=N), 1125 (C–O), 1049 (C–O); RP-HPLC, tR = 23.94 min; 1H NMR (CD3OD, 600.13 MHz, 25 °C) δ, ppm: 1.20 (3H, t, J = 6 Hz, CH3, overlapped by t-BuOH signal), 1.29 (2H, m, CH2–CH2–CH2), 1.57 (2H, m, CH2-carborane), 1.78 (2H, m, CH2–CH2–NH), 1.81–2.63 (10H, m, BH), 3.13 (1H, s, CHcarborane),3.37 (2H, q, J = 6 Hz, N–CH2–CH3), 3.42 (1H, bs, NH-Pr), 4.31 (1H, m, 3′H), 4.46 (1H, m, 4′H), 4.75 (1H, m, 2′H), 6.00 (1H, d, J = 6 Hz, 1′H),8.24 (1H, s, 2-H), 8.27 (1H, s, 8-H); 13C NMR (CD3OD, 150.90 MHz, 25 °C) δ, ppm: 15.51 (CH3), 30.72, 31.58 (2xCH2), 35.53, 37.81 (2xCH2-N), 60.12, 60.55 (2xCHcarborane), 73.46 (C-3′), 75.44 (C-2′), 86.88 (C-4′), 90.96 (C-1′), 142.61 (C-8), 153.91 (C-2), 171.96 (C=O); 11B NMR {1H BB} CD3OD, 160.46 MHz, 25 °C) δ, ppm: 12.55 (s, 5B), −14.99 (s, 5B); MS (ESI, −3 kV, 300 °C): m/z calculated for C17H32B10N6O4: 492.58, found: 493 [M+H]+.
Synthesis of 5′-ethylcarbamoyl-N6-(3-phenylpropyl)adenosine (18).
The substitution of the chlorine atom was performed analogously as described for the synthesis of N6-(3-(phenylpropyl) adenosine [29]. 5′-Ethylamino-5′-oxo-6-chloroadenosine adenosine (13, 26 mg, 0.08 mmol) was dissolved in n-BuOH (0.4 mL) under argon atmosphere then 3-phenylpropylamine (14) (0.014 mL, 0.10 mmol) and Et3N (0.04 mL, 0.29 mmol) was added. The resulting solution was refluxed for 1.5 h, then evaporated to dryness. The crude product was purified by reverse phase flash chromatography using gradient of CH3OH in H2O (5–70%) as an eluent. Yield: 31 mg, 73%, white solid. TLC (CH2Cl2/CH3OH, 9:1): Rf = 0.48; UV–Vis (95% C2H5OH) λ, nm: 231 (min), 267 (max); FT-IR (ATR) ν, cm−1: 3228 (N–H), 3085 (O–H), 3029 (C–H), 2929 (C–H), 2857 (C–H), 1729 (C=O), 1614 (C=N), 1049 (C–O), 744 (Phe), 698 (Phe); RP-HPLC, tR = 19.36 min; 1H NMR (CD3OD, 600.26 MHz, 25 °C) δ, ppm: 1.23 (3H, t, J = 6 Hz, CH3), 1.31–1.37 (2H, m, CH2-Phe), 2.04 (2H, kw, J = 6 Hz, C–CH2–C), 2.77 (2H, t, J = 6 Hz, N–CH2–C2H5-Phe), 3.38 (2H, q, J = 6 Hz, N–CH2–CH3), 3.64 (1H, bs, NH-Pr), 4.34–4.35 (1H, m, 4′H), 4.49 (1H, d, J = 6 Hz, 3′H), 4.77–4.81 (1H, m, 2′H, overlapped by water signal), 6.03 (1H, d, J = 6 Hz, 1′H), 7.15–7.18 (1H, m, Phe-H), 7.23–7.28 (4H, m, Phe-H), 8.26 (1H, s, 2-H), 8.30 (1H, s, 8-H); 13C NMR (CD3OD, 150.94 MHz, 25 °C) δ, ppm: 13.62 (CH3), 32.74, 33.67 (2xCH2), 72.03 (C-3′), 73.59 (C-2′),85.04 (C-4′), 89.16 (C-1′), 125.48 (Phe), 128.01 (Phe), 140.59 (C-8), 141.59 (Phe), 152.39 (C-2), 155.06 (C-6), 170.64 (C=O); MS (FAB, Gly, +VE): m/z calculated for C21H26N6O4: 426.47, found: 427.3 [M+H]+.
Synthesis of 2′,3′,5′-tris-O-benzoyl-6-N-[3-(1,12-dicarba-closododecaboran-1-yl)propyl]-2-chloro-2′-adenosine (21) and 2′,3′,5′−tris-O-benzoyl-6-N-(3-phenylpropyl)-2-chloro-2′-adenosine (22).
To the 1 eq. of 2′,3′,5′-tris-O-benzoyl-2,6-dichloroadenosine (20) dissolved in dry DMF (10 mL/0.5 mmol of nucleoside), 3 eq. of 3-phenyl-1-propylamine (14) hydrochloride or 1.2 eq. of 3-(1,12-dikarba-closo-dodecaboran-1-yl)propylamine (4) hydrochloride followed by 3 eq. of triethylamine were added and reaction mixture was stirred at room temperature for 20 h. Obtained solution was diluted with ethyl acetate and washed twice with water and once with brine. The organic phase was dried over magnesium sulfate, filtrated and evaporated with silica gel. Compounds 21 and 22 were purified by silica gel column chromatography using a gradient of AcOEt in hexane (20–30%) as an eluent.
Yield 21: 154 mg, 78%, starting from 158 mg of dichloropurine nucleoside; TLC (CH2Cl2/CH3OH 9:1): Rf = 0.88; UV–Vis (95% C2H5OH) λ, nm: 227, 250 (min), 230, 271 (max); FT-IR (KBr) ν, cm−1: 3365, 3063, 2927, 2861, 2605, 1968, 1912, 1728, 1620, 1582, 1532, 1477, 1451, 1350, 1314, 1266, 1177, 1120, 1092, 1068, 1025; M.P., °C:94.6–97.1; 1H NMR (CDCl3, 500 MHz, 25 °C) δ, ppm: 1.49–1.56 (2H, m, CH2), 1.60- ~2.80 (10H, bm, BH) – very broad multiplet which superposes with three other peaks within the range (CH-carborane, 2 × CH2), 1.75–1.69 (2H, m, CH2), 2.63 (1H, bs, CH-carborane), 3.43 (2H, bs, CH2(NH)), 4.72 (1H, dd, J = 4.0, 12.0 Hz, 5′bH), 4.78–4.83 (1H, m, 4′H), 4.89 (1H, dd, J = 3.0, 12.0 Hz, 5′aH), 5.73 (1H, bs, NH), 6.10–6.17 (2H, m, 2′H + 3′H), 6.43 (1H, d, J = 5.5 Hz, 1′H), 7.35–7.49 (6H, m, Phe-H), 7.52–7.62 (3H, m, Phe-H), 7.86 (1H, s, 8-H),7.93–7.97 (2H, m, Phe-H), 7.99–8.02 (2H, m, Phe-H), 8.07–8.11 (2H, m, Phe-H); 13C NMR (CDCl3, 125 MHz, 25 °C): 29.4 (CH2), 36.1 (CH2),40.1 (CH2), 58.4 (CH-carborane), 63.9 (C-5′), 71.7 (C-2′), 74.5 (C-3′),81.2 (C-4′), 83.8 (CIV-carborane), 86.3 (C-1′), 119.2 (CIV–Cl), 128.5 (CIV), 128.7 (2 × Phe), 128.8 (Phe), 129.4 (CIV), 129.8 (Phe), 130.0 (Phe), 130.1 (Phe), 133.6 (Phe), 133.9 (Phe), 134.0 (Phe), 138.2 (C8), 150.0 (CIV), 155.2 (CIV), 155.4 (CIV), 165.3 (CIV), 165.5 (CIV), 166.3 (CIV) (1 (CIV) carbon is not observed, most probably it is shielded by the intense Phe signal at 128.8 ppm); 11B NMR {1H BB} (CDCl3, 160 MHz, 25 °C): 12.95 (s, 5B), −15.40 (s, 5B); HRMS (ESI, + 3 kV, 300 °C): m/z calcd for C36H41ClN5B10O7: 798.36922, found: 798.36991 [M+H]+.
Yield 22: 529 mg, 92%, starting from 500 mg of dichloropurine nucleoside; TLC (CH2Cl2/CH3OH 9:1): Rf = 0.84; UV–Vis (95% C2H5OH) λ, nm: 248 (min), 270 (max); FT-IR (KBr) ν, cm−1: 3368, 3061, 3028, 2935, 2858, 1968, 1914, 1727, 1620, 1581, 1533, 1477, 1451,1350, 1314, 1267,1177,1092,1067,1025; M.P.: 63.2–66.3 °C; 1H NMR (CDCl3, 500 MHz, 25 °C): 1.95–2.04 (2H, m, CH2), 2.72 (2H, t, J = 7.0 Hz, CH2(Ph)), 3.64 (2H, bs, CH2(NH)), 4.72 (1H, dd, J = 4.0, 12.0 Hz, 5′bH), 4.81 (1H, dt, J = 3.0, 4.0 Hz, 4′H), 4.89 (1H, dd, J = 3.0, 12.0 Hz, 5′aH), 5.96 (1H, bs, NH), 6.12–6.19 (2H, m, 2′H + 3′H),=6.44 (1H, d, J = 5.0 Hz, 1′H), 7.14–7.22 (3H, m, HAr), 7.23–7.30 (2H, m, HAr), 7.30–7.50 (6H, m, HAr), 7.50–7.65 (3H, m, HAr), 7.87 (1H, s, 8-H), 7.92–7.98 (2H, m, HAr), 7.98–8.04 (2H, m, HAr), 8.06–8.14 (2H, m, HAr); 13C NMR (CDCl3, 125 MHz, 25 °C): 31.1 (CH2), 33.1 (CH2),40.4 (CH2), 64.0 (C-5′), 71.7 (C-2′), 74.5 (C-3′), 81.1 (C-4′), 86.3 (C-1′), 119.2 (CIV–Cl), 126.1 (Phe), 128.47 (CIV), 128.48 (Phe), 128.57 (Phe), 128.65 (Phe), 128.67 (Phe), 128.79 (Phe), 128.80 (CIV), 129.4 (CIV), 129.8 (Phe), 129.99 (Phe), 130.05 (Phe), 133.6 (Phe), 133.85 (Phe), 133.93 (Phe), 138.1 (C-8), 141.3 (CIV), 149.9 (CIV), 155.2 (CIV), 155.3 (CIV), 165.3 (CIV), 165.5 (CIV), 166.2 (CIV); HRMS (ESI, + 3 kV, 300 °C): m/z calcd for C40H35ClN5O7: 732.22195, found: 732.22285 [M+H]+.
Synthesis of N6-[3-(1,12-dicarba-closo-dodecaboran-1-yl)propyl]-2-chloroadenosine (23) and 6-N-(3-phenylpropyl)-2-chloroadenosine (24).
For the removal of benzoyl groups, compounds 21 (154 mg,0.19 mmol) and 22 (448 mg, 0.61 mmol) were treated with 7 N methanolic solution of ammonia (25 mL) at room temperature for 20 h. Next, the silica gel was added directly to the post-reaction mixture, solvent was evaporated under the reduced pressure and the crude products were purified by silica gel column chromatography using a gradient of CH3OH in CHCl3 (5–10%) as an eluent.
Yield 23: 78 mg, 82%; TLC (CH2Cl2/CH3OH 9:1): Rf = 0.47; UV–Vis (95% C2H5OH) λ, nm: 236 (min), 273 (max); FT-IR=(KBr) ν, cm−1: 3334, 2926, 2605, 1624, 1581, 1534, 1476, 1355, 1309, 1229, 1113, 1078; M.P.: 183.2–184.1 °C; RP-HPLC, tR = 29.05 min; 1H NMR (CD3OD, 500 MHz, 25 °C): 1.55 – ~2.80 (10H, bm, BH) (very broad multiplet which superposes with two CH2 peaks within the range),1.48–1.58 (2H, m, CH2), 1.72–1.82 (2H, m, CH2), 3.10 (1H, bs, CH-carborane), 3.38 (1H, t, J = 7.0 Hz, CH2(NH)), 3.75 (1H, dd, J = 3.0, 12.5 Hz, 5′b-H), 3.89 (1H, dd, J = 2.5, 12.5 Hz, 5′a-H), 5.15 (1H, td, J = 2.5, 3.0 Hz, 4′-H), 4.31 (1H, dd, J = 3.0, 5.0 Hz, 3′-H), 4.66 (1H, dd, J = 5.0, 6.0 Hz, 2′-H), 5.89 (1H, d, J = 6.0 Hz, 1′-H), 8.21 (1H, s, 8-H); 13C NMR (CD3OD, 125 MHz, 25 °C): 30.3 (CH2), 37.3 (CH2), 40.8 (CH2), 59.9 (CH-carborane), 63.3 (C-5′), 72.4 (C-3′), 75.4 (C-2′), 85.3 (CIV-carborane), 87.9 (C-4′), 91.0 (C-1′), 120.2 (CIV–Cl), 141.6 (8-C), 150.3 (CIV), 156.6 (CIV), 255.3 (CIV); 11B NMR {1H BB} (CDCl3, 160 MHz, 25 °C): 12.92 (5B s), −15.36 (5B s); HRMS (ESI, + 3 kV, 300 °C): m/z calcd for C15H29ClN5B10O4: 486.29058, found: 486.29060 [M+H]+.
Yield 24: 205 mg, 80%; TLC (CH2Cl2/CH3OH 9:1): Rf = 0.65; UV–Vis (95% C2H5OH) λ, nm: 235 (min), 272 (max); FT-IR=(KBr) ν, cm−1: 3316, 2925, 2858, 1624,1581, 1536, 1479, 1453, 1355, 1310, 1226, 1200, 1111, 1078; M.P.: 140.2–140.6 °C; RP-HPLC, tR = 20.62 min; 1H NMR (CD3OD, 500 MHz, 25 °C): 1.98 (2H, tt, J = 7.0, 7.5 Hz, CH2), 2.71 (2H, t, J = 7.5 Hz, CH2(Phe)), 3.57 (2H, t, J = 7.0 Hz, CH2(NH)), 3.72 (1H, dd, J = 3.0, 12.0 Hz, 5′a-H), 3.89 (1H, dd, J = 2.5, 12.5 Hz, 5′b-H), 4.15 (1H, td, J = 2.5, 3.0 Hz, 4′-H), 4.32 (1H, dd, J = 3.0, 5.0 Hz, 3′-H), 4.67 (1H, dd, J = 5.0, 6.0 Hz, 2′-H), 5.90 (1H, d, J = 6.0 Hz, 1′-H), 7.10–7.16 (1H, m, Phe-H), 7.17–7.27 (4H, m, Phe-H), 8.21 (1H, s, 8-H); 13C NMR (CD3OD, 125 MHz, 25 °C): 32.1 (CH2), 34.1 (CH2), 41.2 (CH2), 63.4 (C-5′), 72.4 (C-3′), 75.5 (C-2′),87.9, (C-4′), 91.1 (C-1′), 120.3 (CIV–Cl), 126.9 (Phe), 129.36 (Phe), 129.42 (Phe), 141.5 (C-8), 142.9 (CIV), 150.2 (CIV), 155.4 (CIV), 156.7 (CIV); HRMS (ESI, +3 kV, 300 °C): m/z calcd for C19H23ClN5O4: 420.14331, found: 420.14323 [M+H]+.
Synthesis of 3′,5′-bis-O-(4-methylbenzoyl)-N6-[3-(1,12-dicarbacloso-dodecaboran-1-yl)propyl]-2-chloro-2′-deoxyadenosine (25) and 3′,5′-bis-O-(4-methylbenzoyl)-N6-(3-phenylpropyl)-2-chloro-2′−deoxyadenosine (26).
To the 1 eq. of 3′,5′-bis-O-(4-methylbenzoyl)-2,6-dichloro-2′-deoxyadenosine (19), dissolved in dry DMF (10 mL/0.5 mmol of nucleoside), 3 eq. of 3-phenyl-1-propylamine hydrochloride (14) or 1.2 eq. of 3-(1,12-dicarba-closo-dodecaboran-1-yl) propylamine (4) hydrochloride followed by 3 eq. of triethylamine were added and reaction mixture was stirred at room temperature for 20 h. Obtained solution was diluted with ethyl acetate and washed twice with water and once with brine. The organic phase was dried over magnesium sulfate, filtrated and evaporated with silica gel. Compounds 25 and 26 were purified by silica gel column chromatography using a gradient of ethyl acetate in hexane (20–30%) as an eluent.
Yield 25: 140 mg (80%, starting from 135 mg of dichloropurine nucleoside); TLC (CH2Cl2/CH3OH 9:1): Rf = 0.86; UV–Vis (95% C2H5OH) λ, nm: 227, 261 (min), 242, 271 (max); FT-IR (KBr) ν, cm−1: 3355, 3063, 2925, 2867, 2608, 1930, 1721, 1615, 1578, 1533, 1503, 1409, 1351, 1309, 1270, 1176, 1099, 1019; M.P.: 80.2–80.9 °C; 1H NMR (CDCl3, 500 MHz, 25 °C): 1.48–1.58 (2H, m, CH2), 1.60 – ~2.80-(10H, bm, BH) – very broad multiplet which superposes with five other peaks within the range (CH-carborane, 2xCH3, 2xCH2),1.66–1.78 (2H, m, CH2), 2.41 (3H, s, CH3), 2.45 (3H, s, CH3), 2.64 (1H, bs, CH-carborane), 2.81–2.91 (2H, m, 2′-H), 3.45 (2H, bs, CH2(NH)),4.61 (1H, td, J = 2.5, 4.0 Hz, 4′-H), 4.66 (1H, dd, J = 4.0, 12.0 Hz, 5′b-H), 4.72 (1H, dd, J = 4.0, 12.0 Hz, 5′a-H), 5.65–5.85 (2H, m, 3′-H + NH), 6.51 (1H, dd, J = 6.0, 8.0 Hz, 1′-H), 7.20–7.25 (2H, m, Phe-H), 7.27–7.31 (2H, m, Phe-H), 7.86–7.93 (3H, m, Phe-H + 8-H), 7.93–7.94 (2H, m, Phe-H); 13C NMR (CDCl3, 125 MHz, 25 °C): 21.88 (CH3), 21.92 (CH3), 29.5 (CH2), 36.1 (CH2), 38.8 (C-2′), 40.1 (CH2),58.4 (CH-carborane), 64.3 (C-5′), 75.2 (C-3′), 83.2 (C-4′), 83.8 (CIV-carborane), 84.6 (C-1′), 119.0 (CIV–Cl), 126.5 (CIV), 126.8 (CIV), 129.4 (Phe), 129.5 (Phe), 129.8 (Phe), 130.0 (Phe), 138.0 (C-8), 144.4 (CIV), 144.7 (CIV),149.7 (CIV), 154.9 (CIV), 155.3 (CIV), 166.1 (CIV), 166.3 (CIV); 11B NMR {1H BB} (CDCl3, 160 MHz, 25 °C): 12.94 (5B, s), −15.39 (5B,s); HRMS (ESI): m/z calcd for C31H41ClN5B10O5: 706.37939, found: 706.37978 [M+H]+.
Yield 26: 276 mg (78%, starting from 300 mg of dichloropurine nucleoside); TLC (CH2Cl2/CH3OH 9:1): Rf = 0.86; UV–Vis (95% C2H5OH) λ, nm: 228, 259 (min), 243, 270 (max); FT-IR (KBr) ν, cm−1: 3247, 3108, 3063, 3029, 2933, 2853, 1936, 1721, 1621, 1574, 1498, 1453, 1429, 1408, 1383, 1331, 1279, 1221, 1176, 1140, 1104, 1087, 1048, 1017; M.P.: 156.8–157.5 °C; 1H NMR (CDCl3, 500 MHz, 25 °C):1.95–2.04 (2H, m, CH2), 2.39 (3H, s, CH3), 2.73 (2H, t, J = 7.0 Hz, CH2(Phe)), 2.81–2.91 (2H, m, 2′-H), 3.65 (2H, bs, CH2(NH)), 4.61 (1H, td, J = 2.5, 4.0 Hz, 4′-H), 4.66 (1H, dd, J = 4.0, 12.0 Hz, 5′a-H), 4.73 (1H, dd, J = 4.0, 12.0 Hz, 5′b-H), 2.44 (3H, s, CH3), 5.78–5.72 (1H, m, 3′-H), 5.91 (1H, bs, NH), 6.51 (1H, dd, J = 6.0, 8.0 Hz, 1′-H),7.15–7.25 (5H, m, Phe-H), 7.25–7.31 (4H, m, Phe-H), 7.93–7.87 (3H, m, Phe-H + 8-H), 7.94–7.99 (2H, m, Phe-H); 13C NMR (CDCl3, 125 MHz, 25 °C): 21.8 (CH3), 21.9 (CH3), 31.1 (CH2), 33.1 (CH2), 38.7 (C-2′), 40.4 (CH2), 64.3 (C-5′), 75.2 (C-3′), 83.2 (C-4′), 84.5 (C-1′), 119.1 (CIV–Cl), 126.2 (Phe), 126.5 (CIV), 126.8 (CIV), 128.5 (Phe), 128.6 (Phe), 129.40 (Phe), 129.43 (Phe), 129.7 (Phe), 130.0 (Phe), 137.8 (C-8), 141.3 (CIV), 144.3 (CIV), 144.7 (CIV), 149.6 (CIV), 154.9 (CIV), 155.5 (CIV), 166.1 (CIV), 166.2 (CIV); HRMS (ESI): m/z calcd for C35H35ClN5O5: 640.23223, found: 640.23212 [M+H]+.
Synthesis of N6-[3-(1,12-dicarba-closo-dodecaboran-1-yl)propyl]-2-chloro-2′-deoxyadenosine (27) and N6-(3-phenylpropyl)-2-chloro-2′-deoxyadenosine (28).
For the removal of 4-methylbenzoyl group, compounds 25 (140 mg, 0.20 mmol) or 26 (200 mg, 0.31 mmol) were treated with 7 N methanolic solution of ammonia (25 mL) at room temperature for 20 h. Next, the silica gel was added directly to the reaction mixture, solvent was evaporated under the reduced pressure and crude the products were purified by silica gel column chromatography using a gradient of CH3OH in CHCl3 (5–10%) as an eluent. Yield of 27: 69 mg, 74%; TLC (CH2Cl2/CH3OH, 9:1): Rf = 0.49; UV–Vis (95% C2H5OH) λ, nm: 235 (min), 271 (max); FT-IR (KBr) ν, cm−1: 3324, 2928, 2605, 1622, 1581, 1535, 1476, 1352, 1308, 1231, 1182, 1094, 1067, 1005; M.P.: 114.1–120.0 °C; RP-HPLC, tR = 30.56 min; 1H NMR (CD3OD, 500 MHz, 25 °C): 1.50- ~2.80 (10H, bm, BH) – very broad multiplet which superposes with three other peaks within the range (H2’a, H2’b, 2 × CH2), 1.49–1.57 (2H, m, CH2),1.73–1.82 (2H, m, CH2), 2.40 (1H, ddd, J = 3.0, 6.0, 13.5 Hz, 2′a-H), 2.77 (1H, ddd, J = 6.0, 7.5, 13.5 Hz, 2′b-H), 3.11 (1H, bs, CH-carborane), 3.39 (1H, t, J = 7.0 Hz, CH2(NH)), 3.75 (1H, dd, J = 3.5, 12.0 Hz, 5′a-H), 3.85 (1H, dd, J = 3.0, 12.0 Hz, 5′b-H), 4.05 (1H, dt, J = 3.0, 3.5 Hz, 4′-H), 4.57 (1H, dt, J = 3.0, 3.5 Hz, 3′-H), 6.35 (1H, dd, J = 6.0, 7.5 Hz, 1′-H), 8.22 (1H, s, 8-H); 13C NMR (CD3OD, 125 MHz, 25 °C): 30.3 (CH2), 37.3 (CH2), 40.8 (CH2), 41.4 (C-2′), 59.9 (CH-carborane), 63.5 (C-5′), 72.8 (C-3′), 85.3 (CIV-carborane), 86.9 (C-1′),89.7 (C-4′), 120.0 (CIV–Cl), 141.2 (C-8), 150.3 (CIV), 155.3 (CIV), 156.5 (CIV); 11B NMR {1H BB} (CDCl3, 160 MHz, 25 °C): 12.92 (5B,s), −15.37 (5B, s); HRMS (ESI): m/z calcd for C15H29ClN5B10O3: 470.29566, found: 470.29552 [M+H]+.
Yield 28: 105 mg, 83%; TLC (CH2Cl2/CH3OH, 9:1): Rf = 49; UV–Vis (95% C2H5OH) λ, nm: 232 (min), 271 (max); FT-IR (KBr) ν, cm−1: 3316, 3026, 2925, 2857, 1706, 1623, 1578, 1536, 1477, 1453, 1354, 1309, 1227, 1093, 1065; M.P.: 58.0–58.4 °C; RP-HPLC, tR = 21.31 min; 1H NMR (CD3OD, 500 MHz, 25 °C): 1.98 (2H, tt, J = 7.0, 7.5 Hz, CH2), 2.41 (1H, ddd, J = 3.0, 6.0, 13.5 Hz 2′a-H), 2.72 (2H, t, J = 7.5 Hz, CH2(Ph)), 2.77 (1H, ddd, J = 6.0, 7.5, 13.5 Hz 2′b-H), 3.57 (2H, t, J = 7.0 Hz, CH2(NH)), 3.75 (1H, dd, J = 3.5, 12.5 Hz, 5′b-H), 3.85 (1H, dd, J = 3.0, 12.5 Hz, 5′a-H), 4.06 (1H, dt, J = 3.0, 3.5 Hz, 4′-H), 4.57 (1H, dt, J = 3.0, 6.0 Hz, 3′-H), 6.36 (1H, dd, J = 6.0, 7.5 Hz, 1′-H), 7.08–7.17 (1H, m, Phe-H), 7.17–7.32 (4H, m, Phe-H), 8.22 (1H, s, 8-H); 13C NMR (CD3OD, 125 MHz, 25 °C): 32.1 (CH2), 34.0 (CH2), 41.2 (CH2), 41.4 (C-2′), 63.6 (C-5′), 72.9 (C-3′), 86.9 (C-1′), 89.7 (C-4′), 120.1 (CIV–Cl), 126.8 (Phe), 129.3 (Phe), 129.4 (Phe), 141.1 (C-8), 142.9 (CIV), 150.2 (CIV), 155.4 (CIV), 156.6 (CIV); HRMS (ESI): m/z calcd for C19H23ClN5O3: 404.14839, found: 404.14845 [M+H] +.
Synthesis of 8-[2-(1,12-dicarba-closo-dodecaboran-1-yl)ethynyl] adenosine (33) and 8-(2-phenylethynyl)adenosine (34) [23].
The synthesis was performed under anhydrous conditions in an argon atmosphere. To the 1 eq. of 8-bromoadenosine (30), dissolved in dry DMF (10 mL/0.5 mmol of nucleoside), 1.5 eq. of appropriate acetylene derivative, 3 eq. of triethylamine were added, followed by 0.2 eq. of CuI and 0.1 eq. of Pd(PPh3)2Cl2. The reaction mixture was stirred at room temperature for 20 h. Next day, volatiles were evaporated under reduced pressure, obtained residue was redissolved in methanol and evaporated with silica gel, then purified by column chromatography using 10% methanol in chloroform as an eluent.
Yield 33: 71 mg (from 94 mg of 30), 63%; TLC (CH2Cl2/CH3OH, 9:1): Rf = 0.30; UV–Vis (95% C2H5OH) λ, nm: 252 (min), 236, 301 (max); FT-IR (KBr) ν, cm−1: 3214, 3047, 2932, 2874, 2614, 2207, 1911, 1648, 1600, 1576, 1477, 1450, 1414, 1370, 1333, 1297, 1238, 1193, 1131, 1078, 1062; M.P.: 201–202 °C; RP-HPLC, tR = 24.60 min; 1H NMR (CD3OD, 500 MHz, 25 °C) δ, ppm: 1.30–3.20 (9H, bm, BH); 3.44 (1H, bs, CHcarborane), 3.70–3.80 (2H, m, CHcarborane +5′a-H), 3.89 (1H, dd, J = 2.5, 12.5 Hz, 5′b-H), 4.18 (1H, ddd, J = 2.0, 2.5, 3.0 Hz, 4′-H), 4.38 (1H, dd, J = 2.0, J = 5.5 Hz, 3′-H); 5.01 (1H, dd, J = 5.5, 7.0 Hz, 2′-H); 8.19 (1H, s, 2-H); 13C NMR (CD3OD, 125 MHz, 25 °C) δ, ppm: 64.1 (C-5′); 65.4 (CHcarborane), 68.0 (CHcarborane), 73.1 (C-3′), 74.2 (C-2′), 85.2 (C≡C), 88.8 (C-4′), 91.9 (C-1′), ~98.5 (C≡C, very broad), 120.7 (CIV), 135.7 (CIV), 149.5 (CIV), 154.4 (C-2), 157.5 (CIV); 11B NMR {1H BB} (CD3OD3, 160 MHz, 25 °C) δ, ppm: 13.53, −14.86; HRMS (ESI): m/z calcd for C14H24B10N5O4: 434.28260, found: 434.28284 [M+H]+.
Yield 34: 814 mg (from 900 mg of 30), 85%; TLC (CH2Cl2/CH3OH, 9:1): Rf = 0.28; UV–Vis (95% C2H5OH) λ, nm: 241, 251, 267 (min), 247, 254, 314 (max); FT-IR (KBr) ν, cm−1: 3333, 2935, 2219, 1649, 1603, 1569, 1503, 1442, 1395, 1332, 1308, 1255, 1227, 1196, 1092, 1045; M.P.: 211–212 °C; RP-HPLC, tR = 17.16 min; 1H NMR (DMSO-d6, 500 MHz, 25 °C) δ, ppm: 3.55 (1H, dd, J = 4.0, 12.0 Hz, 5′a-H), 3.70 (1H, dd, J = 4.0, 12.0 Hz, 5′a-H), 3.92–4.08 (1H, m, 4′-H), 4.14–4.34 (1H, m, 3′-H), 5.03 (1H, t, J = 6.0 Hz, 2′-H), 5.06–5.30 (1H, bs, OH), 5.30–5.82 (2H, bm, 2 × OH), 6.06 (1H, d, J = 6.0 Hz, 1′-H),7.47–7.60 (3H, m, Phe-H), 7.60–7.95 (4H, m, Phe-H + NH2), 8.19 (1H, s, 2-H); 13C NMR (DMSO-d6, 125 MHz, 25 °C) δ, ppm: 62.2 (C-5′), 71.0 (C-3′), 71.8 (C-2′), 78.5 (C≡C), 86.8 (C-4′), 89.5 (C1’), 94.4 (C≡C), 119.7 (CIV), 119.9 (CIV), 129.2 (Phe), 130.5 (Phe), 131.9 (Phe), 133.4 (CIV), 148.6 (CIV), 153.4 (C-2), 156.1 (CIV); HRMS (ESI): m/z calcd for C18H18N5O4: 368.13533, found: 368.13530 [M+H]+.
Synthesis of 8-[2-(1,12-dicarba-closo-dodecaboran-1-yl)ethyl]-adenosine (35) and 8-(2-phenylethyl)adenosine (36).
Compound 33 (35 mg, 0.08 mmol) or compound 34 (400 mg, 1.09 mmol) were dissolved in methanol (20 mL/1 mmol of nucleoside) then 30 mg of palladium on activated charcoal (10% Pd basis) was added to the solution. The reaction vessel was connected to a balloon filled with hydrogen and the reaction mixture was stirred at room temperature for 20 h. The resultant solution was filtered through Celite, evaporated and the crude product was purified by silica gel column chromatography using 10% methanol in chloroform as an eluent.
Yield 35: 30 mg (85%); TLC (CH2Cl2/CH3OH, 9:1): Rf = 0.29; UV–Vis (95% C2H5OH) λ, nm: 231 (min), 262 (max); IR (KBr) ν, cm−1: 3584, 3433, 3340, 3223, 3092, 3050, 2924, 2867, 2604, 1651, 1606, 1578, 1515, 1483, 1449, 1423, 1376, 1334, 1295, 1272, 1234, 1181, 1122, 1070, 1043; M.P.: 204–205 °C; RP-HPLC, tR = 24.02 min; 1H NMR (CD3OD, 500 MHz, 25 °C) δ, ppm: 1.53–1.60 (2H, m, CH2–CH2-carborane), 1.62–2.97 (9H, m, BH), 3.00–3.13 (2H, m, CH2–CH2-carborane), 3.26 (1H, bs, CHcarborane), 3.46 (1H, bs, CHcarborane), 3.73 (1H, dd, J = 2.5, 12.5 Hz, 5′a-H), 3.89 (1H, dd, J = 2.0, 12.5 Hz, 5′b-H), 4.18–4.22 (1H, m, 4′-H), 4.34 (1H, dd, J = 1.5, 5.0 Hz, 3′-H), 5.00 (1H, dd, J = 5.0, 7.5 Hz, 2′-H), 5.92 (1H, d, J = 7.5 Hz, 1′-H), 8.11 (1H, s, 2-H); 13C NMR (CD3OD, 125 MHz, 25 °C) δ, ppm: ~14.1 (CH2–CH2-carborane, very broad); 28.1 (CH2–CH2-carborane), 64.1 (C-5′), 65.2 (CHcarborane), 67.9 (CHcarborane), 73.3 (C-3′), 74.2 (C-2′), 88.9 (C-4′), 90.6 (C-1′), 119.5 (CIV), 150.9 (CIV), 152.5 (C-2), 156.3 (CIV), 156.8 (CIV); 11B NMR {1H BB} (CD3OD3, 160 MHz, 25 °C) δ, ppm: 4.80, −13.58, −14.56, −15.93; HRMS (ESI): m/z calcd for C14H28B10N5O4: 438.31390, found: 438.31397 [M+H]+.
Yield 36: 390 mg (97%); TLC (CH2Cl2/CH3OH, 9:1): Rf = 0.25; UV–Vis (95% C2H5OH) λ, nm: 232 (min), 262 (max); IR (KBr) ν, cm−1: 3435, 3348, 3228, 2930, 2894, 1658, 1606, 1547, 1496, 1452, 1386, 1333, 1230,1092, 1044; M.P.:183–184 °C; 1H NMR (DMSO-d6, 500 MHz, 25 °C) δ, ppm: 3.02–3.25 (4H, m, ); 3.55 (1H, ddd, J = 3.0, 9.5, 12.5 Hz, 5′a-H), 3.69 (1H, ddd, J = 3.0, 3.5, 12.5 Hz, 5′b-H), 4.02 (1H, td, J = 2.0, 3.0, 4′-H), 4.15 (1H, ddd, J = 2.0, 4.5, 5.5 Hz, 3′-H), 4.91 (1H, ddd, J = 5.5, 7.0, 7.5 Hz, 2′-H), 5.25 (1H, d, J = 4.5 Hz, 3′-OH), 5.42 (1H, d, J = 7.5 Hz, 2′-OH), 5.85 (1H, d, J = 7.0 Hz, 1′-H), 5.95 (1H, dd, J = 3.5, 9.5 Hz, 5′-OH), 7.15–7.25 (1H, m, Phe-H), 7.25–7.40 (6H, m, Phe-H + NH2), 8.07 (s, 1H, 2-H); 13C NMR (DMSO-d6, 125 MHz, 25 °C) δ, ppm: 29.3 (CH2CH2Phe); 33.1 (CH2CH2Phe), 62.3 (C-5′), 71.1 (C-3′), 72.0 (C-2′), 86.9 (C-4′), 88.4 (C-1′), 118.3 (CIV), 126.2 (Phe), 128.4 (Phe), 140.7 (CIV), 149.6 (CIV), 151.4 (C-2), 151.6 (CIV), 155.6 (CIV); HRMS (ESI): m/z calcd for C18H22N5O4: 372.16663, found: 372.16643 [M+H]+.
5.2. Radioligand inhibition assay for compounds 7, 8, 15–18, 23, 24, 27–28
Competition Binding in Human A1 receptors.
Adenosine A1 receptor competition binding experiments were carried out in a multiscreen GF/C 96-well plate (Millipore, Madrid, Spain) pre-treated with binding buffer (Hepes 20 mM, NaCl 100 mM, MgCl2 10 mM, 2 U/ml adenosine deaminase, pH = 7.4). In each well was incubated 5 μg of membranes from Euroscreen CHO-A1 cell line and prepared in Plataforma de Screening de Farmacos (USEF) (Lot: A002/13-04-2011, protein concentration = 5864 μg/mL), 1 nM [3H]-DPCPX (120 Ci/mmol, 1 mCi/mL, PerkinElmer NET974001MC) and compounds studied and standard. Non-specific binding was determined in the presence of R-PIA 10 μM (Sigma P4532). The reaction mixture (Vt: 200 μl/well) was incubated at 25 °C for 60 min, after was filtered and washed four times with 250 μl wash buffer (Hepes 20 mM, NaCl 100 mM, MgCl2 10 mM pH = 7.4), before measuring in a microplate beta scintillation counter (Microbeta Trilux, PerkinElmer, Madrid, Spain).
Competition Binding in Human A2A receptors.
Adenosine A2A receptor competition binding experiments were carried out in a multiscreen GF/C 96-well plate (Millipore, Madrid, Spain) pre-treated with binding buffer (Tris-HCl 50 mM, EDTA 1 mM, MgCl2 10 mM, 2 U/ml adenosine deaminase, pH = 7.4). In each well was incubated 5 μg of membranes from HeLa-A2A cell line and prepared in Plataforma de Screening de Farmacos (USEF) (Lot: A001/18-10-2010, protein concentration = 4288 μg/mL), 3 nM [3H]-ZM241385 (50 Ci/mmol, 1 mCi/mL, ARC-ITISA 0884) and compounds studied and standard. Non-specific binding was determined in the presence of NECA 50 μM (Sigma E2387). The reaction mixture (Vt: 200 μl/well) was incubated at 25 °C for 30 min, after was filtered and washed four times with 250 μl wash buffer (Tris-HCl 50 mM, EDTA 1 mM, MgCl2 10 mM, pH = 7.4), before measuring in a microplate beta scintillation counter (Microbeta Trilux, PerkinElmer, Madrid, Spain).
Competition Binding in Human A2B receptors.
Adenosine A2B receptor competition binding experiments were carried out in a polypropylene 96-well plate. In each well was incubated 20 μg of membranes from Euroscreen HEK-A2B cell line and prepared in Plataforma de Screening de Farmacos (USEF) (Lot: A007/22-02-2016, protein concentration = 3211 μg/mL), 25 nM [3H]-DPCPX (164 Ci/mmol, 1 mCi/mL, PerkinElmer NET974001MC) and compounds studied and standard. Non-specific binding was determined in the presence of NECA 1000 μM (Sigma E2397). The reaction mixture (Vt: 250 μl/well) was incubated at 25 °C for 30 min, 200 μL was transferred to GF/C 96-well plate (Millipore, Madrid, Spain) pre-treated with binding buffer (Tris-HCl 50 mM, EDTA 1 mM, MgCl2 mM, Bacitracin 100 μg/μl, adenosine deaminase 2 U/mL, pH = 6.5), after was filtered and washed four times with 250 μl wash buffer (Tris-HCl 50 mM, EDTA 1 mM, MgCl2 5 mM, pH = 6.5), before measuring in a microplate beta scintillation counter (Microbeta Trilux, PerkinElmer, Madrid, Spain).
Competition Binding in Human A3 receptors.
Adenosine A3 receptor competition binding experiments were carried out in a multiscreen GF/B 96-well plate (Millipore, Madrid, Spain) pre-treated with binding buffer (Tris-HCl 50 mM, EDTA 1 mM, MgCl2 5 mM, 2 U/ml adenosine deaminase, pH = 7.4). In each well was incubated 70 μg of membranes from HeLa-A3 cell line and prepared in our laboratory (Lot: A003/13-04-2016, protein concentration = 2449 μg/mL), 10 nM [3H]-NECA (27.6 Ci/mmol, 1 mCi/mL, PerkinElmer NET811250UC) and compounds studied and standard. Non-specific binding was determined in the presence of R-PIA 100 μM (Sigma P4532). The reaction mixture (Vt: 200 μl/well) was incubated at 25 °C for 180 min, after was filtered and washed six times with 250 μl wash buffer (Tris-HCl 50 mM pH = 7.4), before measuring in a microplate beta scintillation counter (Microbeta Trilux, PerkinElmer, Madrid, Spain).
Radioligand inhibition assay for compounds 33–55.
Radioligand binding assays for compounds 33–52 were performed using HEK293 cells expressing human A1, A2A, or A3 receptors and standard radioligands, as previously described [39].
5.3. Functional cAMP assay for compounds 7, 8, 15, 16, 23 and 24 in human A3 receptor
Chinese hamster ovary (CHO) cells transfected with the human A3 receptor (CHO-A3, Centro de Investigación CIMUS, Barcelona) were cultured in Dulbecco’s modified eagle’s medium nutrient mixture F-12 ham (Sigma Aldrich; D8062) pH = 7.4, containing 25 mM Hepes and 30 μM rolipram. As the wash buffer Dulbecco’s modified eagle’s medium nutrient mixture F-12 ham pH = 7.4 containing 25 mM Hepes was used. Initially, the CHO-A3 cells were incubated with the vehicle (for an agonists mode) or with the increasing concentrations of compounds (for an antagonist mode) at 37 °C, 5% CO2, for 15 min. Next, the increasing concentrations of compounds and 0.1 μM of NECA (for agonists mode), or vehicle and 0.1 μM of NECA (for antagonist mode) were added for the next 15 min (37 °C, 5% CO2). Subsequently, 10 μМ οf forskolin was added and the incubation was continued for next 5 min (37 °C, 5% CO2). Determination of the cAMP amount was performed using Amersham cAMP Enzyme immunoassay (Amersham, RPN 225). Absorbance measurement was measured at 450 nm using a Tecan Infinite M1000 Pro Microplate Reader. The MRS1220 and NECA (agonist mode) were used as the standard controls for validation of the assay (antagonist and agonist mode, respectively). The affinity values obtained for standard controls were as follow: NECA, EC50 = 4 nM (described in the literature 20 nM [39]); MRS1220, KB = 4.0 nM (described in the literature 1.7 nM [56]).
5.4. Patients and biological materials
Blood was drawn from 57 healthy donors including 27 men and 30 women, with the average age of 30 years in the case of men and 33 years in the case of women. The samples were collected into a vacuum tube containing 25 μg/mL hirudin, for whole blood aggregometry, or 0.105 M buffered sodium citrate, for optical aggregation and thrombus formation in flow conditions. The study was performed under the guidelines of the Helsinki Declaration for human research and approved by the Medical University of Lodz committee on the Ethics of Research in Human Experimentation.
5.5. Inhibition of human blood platelets aggregation
Aggregation of blood platelets in a platelet rich plasma.
The blood samples were centrifuged for 12 min at 190×g to obtain platelet-rich plasma (PRP). The platelet count in the PRP was adjusted to 2 × 108 platelets/ml prior to its use in experiments. Five minutes prior to measurements samples were added with a tested compound (dissolved in 0.1% DMSO), control samples were added with 0.1% DMSO. ADP was added to each sample to final concentration of 10 μM to induce platelet aggregation, which was thereafter monitored for 10 min using an optical aggregometer Chrono-Log 490–2D (Chrono-Log,Havertown, PA, USA).The total area under the aggregation curve (AUC) and the maximal value of platelet aggregation (Amax,U) were recorded [57].
Aggregation of blood platelets in whole blood.
Samples were pre-incubated with the aliquot of a tested compound or DMSO as described above. Whole platelet aggregation was monitored thereafter for up to 15 min in response to 6.4 μM ADP, 1 μg/mL collagen or 0.5 mM arachidonic acid using the five-channel Multiplate analyzer (Roche Diagnostics GmbH, Mannheim, Germany). The total area under the aggregation curve (AUC) was recorded [57,58].
Formation of thrombus in flow conditions.
Effects of the tested compounds on the formation of thrombi were assayed with the use of Venaflux platform (Cellix, Dublin, Ireland). The channels of Fluoro + biochips were coated with type I collagen. Citrated blood was incubated with anti-CD61 PE-conjugated antibodies to label blood platelets. Tested compounds or 0.1% DMSO were added 5 min prior to measurements. Samples were recalcified with 1 mM CaCl2 shortly before running in the system. The samples were then perfused with a shear force of 20 dyn/cm2 for 5 min. The thrombi in the channels were imaged with the use of AxioExaminer epifluorescence microscope (Zeiss, Jena, Germany) equipped with a high performance EMCCD camera Rolera em-c2 (QImaging, Surrey, Canada). Areas of individual thrombi were estimated with the use of a dedicated ZEN software.
5.6. Structure-activity relationship (SAR)
Current commercial software packages do not have built-in parameters for hexacoordinated boron atoms. Therefore, to enable comparative analysis of carboranyl and phenyl compounds, qualitative structural descriptors were generated. The following substructures/features were selected for analysis: type of sugar (ribose, R; 2′-deoxyribose, dR; and arabinose, Ara), type of modifier (carborane, phenyl), position of substitution (N6, C2, and C8), number of carbon atoms in a linker (C2, C3, C4), and linker flexibility (flexible C–C versus rigid C≡C). The activity of the compounds, i.e., receptor affinity, was expressed as log Ki values. Additionally, the receptor affinity values were transformed to categorical variables for classification analysis (active, low active, and inactive) using the k-means clustering method (Statistica software package, version 13.3, TIBCO Software Inc., Palo Alto, CA, USA). The structural features of compounds in relation to A3, A1, and A2A receptor affinity were analyzed separately. For selectivity testing, compounds were categorized into two groups and analyzed as dichotomous categorical variables (A3-selective and A3-not selective).
Combining classes/groups of descriptors was performed using feature selection and a variable screening (FSL) module (Data Mining tools in Statistica software). Significant variables for model building were selected using the general regression model module (GRM, backward stepwise and best-subset search procedures, Wald chi-square statistics). Structural features specific only for one group of compounds were subjected to regression analysis separately, e.g., ligand flexibility/rigidity in short C2-linker compounds.
The relationship of compound activity (log Ki, dependent variable) and previously selected substructural features (categorical predictors) was determined by a regression approach (PLS, or general classification and regression trees, GC&RT module, Statistica, with goodness-of–fit and predictivity evaluation and cross validation, Statistica software).
All synthesized compounds complied with Lipinski’s Rule of Five (online software chemicalize.com, ChemAxon 2020). The average MW values of carborane and phenyl derivatives were 453.86 ± 27.87 and 387.75 ± 27.87, respectively. Although the MW calculated for both derivative groups was significantly different (P < 0.0001, Student’s t-test; n = 15, normal data distribution; Shapiro-Wilk test), this parameter was not related to compound activity (P > 0.05), and it was not included in the analysis.
5.7. Docking study
Homology modeling of the A3 receptor: The sequence of the A3 AR (chain A) was collected from the Swiss-Prot Protein Database (P33765). The A3 AR model was generated by LOMETS (Local Meta-Threading-Server [59]) using a template of the 2.6 Å resolution structure of human A2A receptor with CGS21680 antagonist bound (Protein Data Bank, PDB code: 4UG2). The sequence identity was 0.43; coverage of the threading alignment was 0.88 (equal to the number of aligned residues divided by the length of the query protein). The loop segments were built with Modeler [60]; the obtained structure was optimized using Maestro Schrodinger software 11.7 (OPLS3e, Schrödinger, Inc., New York, NY, 2013). The A3 AR model contained one disulfide bridge between the cysteine residue (Cys166) in the second extracellular loop (EL2) and the cysteine residue of TM3, Cys83 (3.25). The pair of carboranyl and phenyl derivatives of NECA were docked to the A3 AR model (compounds 15 and 16). Ligands were built and initially optimized in 3D using MarvinSketch 20.14.0, 2020, ChemAxon (http://www.chemaxon.com). Partial atomic charges of compounds were calculated using semiempirical CNDO method (HyperChem 7.51 program, HyperCube Inc., Gainesville, FL, USA). The resulting partial negative charges of hydrogen atoms at BH vertices (values from −0.076 to −0.082, depending on the distance from C vertices) allowed us to retain the hydridic character of H atoms in models of carborane derivatives. The compound geometry was finally optimized in an AMBER95 force field (in vacuum, distance dependent dielectric constant 1, electrostatic interactions 0.5, van der Waals interactions 0.5, HyperChem 7.51). Boron atom parameters were implemented in AutoDock 4.2.6, and into AutoDock and AutoGrid parameter files. The following have been added [61]: vdW diameter, Rii 3.96 Å; vdW well depth, epsii 0.034 kcal/mol; atomic solvation volume 32.5281 Å3; atomic solvation parameter −0.00143; the remaining parameters were default as for carbon atom. A docking procedure was performed by preserving i) the input of partial atomic charges, and ii) BH hydridic hydrogens. Polar hydrogen atoms were added and Kollman charges were assigned to protein (A3 AR) atoms. The docking area was centered at the average position of the intra- and extracellular part of the A3 AR (grid points in xyz, 60 × 60 × 80, grid point spacing 0.375 Å), which broadly covered the entire extracellular part of the receptor, binding pocket and intracellular region. The Lamarckian genetic algorithm (LGA) was used for conformational search of the flexible ligand. Docking parameters were as follows: number of individuals in population 100, GA-LS runs 100, maximum number of energy evaluations 108 (a higher value, 2.5 × 108 did not alter results), maximum number of generations 27000. Validation of the docking method was performed using I-AB-MECA, a high affinity A3 AR agonist; the calculated I-AB-MECA Ki value was 3.5 nM; binding energy, ΔG, −11.54 kcal/mol (compared to the experimental Ki values: 1.2–2.1 nM; PubChem CID 9958472). The results were analyzed using PLIP (protein-ligand interaction profiler) open source software [62] and LigPlot+ 2.2. [63] Sequence-based GPCR residue numbering was performed according to Ballesteros-Weinstein.
Supplementary Material
Acknowledgments
This work was supported in part by the National Science Center, Poland, grant 2014/13/B/NZ1/03989, (AR, ZJL) and NIDDK Intramural Program grant ZIA DK031117 (KJ, HR, Z-GG). Contributions from the Statutory Fund of IMB PAS are also gratefully acknowledged. The equipment used (AM) was sponsored in part by the Center for Preclinical Research and Technology (CePT), a project cosponsored by the European Regional Development Fund and Innovative Economy, The National Cohesion Strategy of Poland. We thank Jacek Olędzki for recording ES-MS spectra.
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2021.113607.
References
- [1].Jacobson KA, Gao ZG, Adenosine, in: Squire LR (Ed.), Encyclopedia in Neuroscience, Academic Press, Burlington, 2009, pp. 83–96. [Google Scholar]
- [2].Burnstock G, The therapeutic potential of purinergic signalling, Biochem. Pharmacol 151 (2018) 157–165, 10.1016/j.bcp.2017.07.016. [DOI] [PubMed] [Google Scholar]
- [3].Chen JF, Eltzschig HK, Fredholm BB, Adenosine receptors as drug targets–what are the challenges? Nat. Rev. Drug Discov 12 (2013) 265–286, 10.1038/nrd3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Müller CE, Jacobson KA, Recent developments in adenosine receptor ligands and their potential as novel drugs, Biochim. Biophys. Acta Biomembr 1808 (2011) 1290–1308, 10.1016/j.bbamem.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dal Ben D, Buccioni M, Lambertucci C, Marucci G, Volpini R, Cristalli G, The importance of alkynyl chain presence for the activity of adenine nucleosides/nucleotides on purinergic receptors, Curr. Med. Chem 18 (2011) 1444–1463, 10.2174/092986711795328391. [DOI] [PubMed] [Google Scholar]
- [6].Jacobson KA, Muller CE, Medicinal chemistry of adenosine, P2Y and P2X receptors, Neuropharmacology 104 (2016) 31–49, 10.1016/j.neuropharm.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hey-Hawkins E, Viñas Teixidor C, Boron-based Compounds, Wiley Online Library, 2018. [Google Scholar]
- [8].Issa F, Kassiou M, Rendina LM, Boron in drug discovery: carboranes as unique pharmacophores in biologically active compounds, Chem. Rev 111 (2011) 5701–5722, 10.1021/cr2000866. [DOI] [PubMed] [Google Scholar]
- [9].Lesnikowski ZJ, Recent developments with boron as a platform for novel drug design, Expet Opin. Drug Discov 11 (2016) 569–578, 10.1080/17460441.2016.1174687. [DOI] [PubMed] [Google Scholar]
- [10].Scholz M, Hey-Hawkins E, Carbaboranes as pharmacophores: properties, synthesis, and application strategies, Chem. Rev 111 (2011) 7035–7062, 10.1021/cr200038x. [DOI] [PubMed] [Google Scholar]
- [11].Lesnikowski ZJ, Challenges and opportunities for the application of boron clusters in drug design, J. Med. Chem 59 (2016) 7738–7758, 10.1021/acs.jmedchem.5b01932. [DOI] [PubMed] [Google Scholar]
- [12].Bednarska K, Olejniczak AB, Wojtczak BA, Sulowska Z, Lesnikowski ZJ, Adenosine and 2’-deoxyadenosine modified with boron cluster pharmacophores as new classes of human blood platelet function modulators, Chem-MedChem 5 (2010) 749–756, 10.1002/cmdc.201000075. [DOI] [PubMed] [Google Scholar]
- [13].Bednarska K, Olejniczak AB, Piskala A, Klink M, Sulowska Z, Lesnikowski ZJ, Effect of adenosine modified with a boron cluster pharmacophore on reactive oxygen species production by human neutrophils, Bioorg. Med. Chem 20 (2012) 6621–6629, 10.1016/j.bmc.2012.09.039. [DOI] [PubMed] [Google Scholar]
- [14].Białek-Pietras M, Olejniczak AB, Paradowska E, Studzińska M, Jabłońnska A, Leśnikowski ZJ, Synthesis, susceptibility to enzymatic phosphorylation, cytotoxicity and in vitro antiviral activity of lipophilic pyrimidine nucleoside/carborane conjugates, J. Organomet. Chem 865 (2018) 166–172, 10.1016/j.jorganchem.2018.03.026. [DOI] [Google Scholar]
- [15].Białek-Pietras M, Olejniczak AB, Paradowska E, Studzińska M, Suski P, Jabłońska A, Leśnikowski ZJ, Synthesis and in vitro antiviral activity of lipophilic pyrimidine nucleoside/carborane conjugates, J. Organomet. Chem 798 (2015) 99–105, 10.1016/j.jorganchem.2015.07.002. [DOI] [Google Scholar]
- [16].Zolnierczyk JD, Olejniczak AB, Mieczkowski A, Blonski JZ, Kilianska ZM, Robak T, Lesnikowski ZJ, In vitro antileukemic activity of novel adenosine derivatives bearing boron cluster modification, Bioorg. Med. Chem 24 (2016) 5076–5087, 10.1016/j.bmc.2016.08.028. [DOI] [PubMed] [Google Scholar]
- [17].Maderna A, Herzog A, Knobler CB, Hawthorne MF, The syntheses of amphiphilic camouflaged carboranes as modules for supramolecular construction, J. Am. Chem. Soc 123 (2001) 10423–10424, 10.1021/ja016768g. [DOI] [PubMed] [Google Scholar]
- [18].Tjarks W, Ghaneolhosseini H, Henssen CL, Malmquist J, Sjöberg S, Synthesis of para-and nido-carboranyl phenanthridinium compounds for neutron capture therapy, Tetrahedron Lett. 37 (1996) 6905–6908, 10.1016/0040-4039(96)01512-2. [DOI] [Google Scholar]
- [19].Wilson JG, Anisuzzaman A, Alam F, Soloway A, Development of carborane synthons: synthesis and chemistry of (aminoalkyl) carboranes, Inorg. Chem 31 (1992) 1955–1958, 10.1021/ic00036a043. [DOI] [Google Scholar]
- [20].Ghirmai S, Malmquist J, Lundquist H, Tolmachev V, Sjöberg S, Synthesis and radioiodination of 7-(3′-ammoniopropyl)-7, 8-dicarba-nido-undecaborate (−1),(ANC), J. Label. Compd. Radiopharm 47 (2004) 557–569, 10.1002/jlcr.840. [DOI] [Google Scholar]
- [21].Malmquist J, Sjoeberg S, Synthesis of. omega.-(aminoalkyl)-1, 2-closodicarbadodecaboranes (12), Inorg. Chem 31 (1992) 2534–2537, 10.1021/ic00038a041. [DOI] [Google Scholar]
- [22].Lee MW Jr., Sevryugina YV, Khan A, Ye SQ, Carboranes increase the potency of small molecule inhibitors of nicotinamide phosphoribosyltranferase, J. Med. Chem 55 (2012) 7290–7294, 10.1021/jm300740t. [DOI] [PubMed] [Google Scholar]
- [23].Volpini R, Costanzi S, Lambertucci C, Vittori S, Klotz K-N, Lorenzen A, Cristalli G, Introduction of alkynyl chains on C-8 of adenosine led to very selective antagonists of the A3 adenosine receptor, Bioorg. Med. Chem. Lett 11 (2001) 1931–1934, 10.1016/S0960-894X(01)00347-X. [DOI] [PubMed] [Google Scholar]
- [24].Olejniczak AB, Semenuk A, Kwiatkowski M, Lesnikowski ZJ, Synthesis of adenosine containing carborane modification, J. Organomet. Chem 680 (2003) 124–126, 10.1016/S0960-894X(01)00347-X. [DOI] [Google Scholar]
- [25].Vincenzi M, Bednarska K, Lesnikowski ZJ, Comparative study of carborane- and phenyl-modified adenosine derivatives as ligands for the A2A and A3 adenosine receptors based on a rigid in silico docking and radioligand replacement assay, Molecules (2018) 23, 10.3390/molecules23081846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Nishikawa S, Kumazawa Z, Kashimura N, Nishkimi Y, Uemura S, Alternating dependency of cytokinin activity on the number of methylene units in ω-phenylalkyl derivatives of some purine cytokinins and 4-substituted pyrido [3, 4-d] pyrimidine, Agric. Biol. Chem 50 (1986) 2243–2249, 10.1080/00021369.1986.10867724. [DOI] [Google Scholar]
- [27].Abiru T, Miyashita T, Watanabe Y, Yamaguchi T, Machida H, Matsuda A, Nucleosides and nucleotides. 107. 2-(cycloalkylalkynyl) adenosines: adenosine A2 receptor agonists with potent antihypertensive effects, J. Med. Chem 35 (1992) 2253–2260, 10.1021/jm00090a017. [DOI] [PubMed] [Google Scholar]
- [28].Mieczkowski A, Kierozalska A, Bialek-Pietras M, Goszczynski TM, Janczak S, Olejniczak AB, Studzinska M, Paradowska E, Lesnikowski ZJ, Comparative study of inorganic, boron-rich cluster and organic, phenyl adenosine modifications: synthesis and properties, Future Med. Chem 11 (2019) 1267–1284, 10.4155/fmc-2018-0517. [DOI] [PubMed] [Google Scholar]
- [29].Suzuki F, Shimada J, Shiozaki S, Ichikawa S, Ishii A, Nakamura J, Nonaka H, Kobayashi H, Fuse E, Adenosine A1 antagonists. 3. Structure-activity relationships on amelioration against scopolamine- or N6-((R)-phenylisopropyl) adenosine-induced cognitive disturbance, J. Med. Chem 36 (1993) 2508–2518, 10.1021/jm00069a009. [DOI] [PubMed] [Google Scholar]
- [30].Hou X, Lee HW, Tosh DK, Zhao LX, Jeong LS, Alternative and improved syntheses of highly potent and selective A3 adenosine receptor agonists, Cl-IB-MECA and thio-Cl-IB-MECA, Arch Pharm. Res. (Seoul) 30 (2007) 1205–1209, 10.1007/BF02980260. [DOI] [PubMed] [Google Scholar]
- [31].Zhu Z, Buolamwini JK, Constrained NBMPR analogue synthesis, pharmacophore mapping and 3D-QSAR modeling of equilibrative nucleoside transporter 1 (ENT1) inhibitory activity, Bioorg. Med. Chem 16 (2008) 3848–3865, 10.1016/j.bmc.2008.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Rodenko B, Detz RJ, Pinas VA, Lambertucci C, Brun R, Wanner MJ, Koomen GJ, Solid phase synthesis and antiprotozoal evaluation of di- and trisubstituted 5’-carboxamidoadenosine analogues, Bioorg. Med. Chem 14 (2006) 1618–1629, 10.1016/j.bmc.2005.10.011. [DOI] [PubMed] [Google Scholar]
- [33].Middleton RJ, Briddon SJ, Cordeaux Y, Yates AS, Dale CL, George MW, Baker JG, Hill SJ, Kellam B, New fluorescent adenosine A1-receptor agonists that allow quantification of ligand-receptor interactions in microdomains of single living cells, J. Med. Chem 50 (2007) 782–793, 10.1021/jm061279i. [DOI] [PubMed] [Google Scholar]
- [34].Dobak I, Ostafin M, Poleshchuk OK, Milecki J, Nogaj B, Reactivity of 2, 6-Dichloropurine ribonucleoside studied by 35 Cl NQR spectroscopy, Appl. Magn. Reson 34 (2008) 47–53, 10.1007/s00723-008-0091-y. [DOI] [Google Scholar]
- [35].Schaeffer HJ, Thomas HJ, Synthesis of potential anticancer agents. XIV. Ribosides of 2, 6-disubstituted purines, J. Am. Chem. Soc 80 (1958) 3738–3742, 10.1021/ja01547a068. [DOI] [Google Scholar]
- [36].Schilz M, Plenio H, A guide to Sonogashira cross-coupling reactions: the influence of substituents in aryl bromides, acetylenes, and phosphines, J. Org. Chem 77 (2012) 2798–2807, 10.1021/jo202644g. [DOI] [PubMed] [Google Scholar]
- [37].Himmelspach A, Finze M, Dicarba-closo-dodecaboranes with one and two ethynyl groups bonded to boron, Eur. J. Inorg. Chem 13 (2010) 2012–2024, 10.1002/ejic.201000064. [DOI] [Google Scholar]
- [38].Wojtczak B, Semenyuk A, Olejniczak AB, Kwiatkowski M, Lesnikowski ZJ, General method for the synthesis of 2′-O-carboranyl-nucleosides, Tetrahedron Lett. 46 (2005) 3969–3972, 10.1016/j.tetlet.2005.04.046. [DOI] [Google Scholar]
- [39].Gao ZG, Mamedova LK, Chen P, Jacobson KA, 2-Substituted adenosine derivatives: affinity and efficacy at four subtypes of human adenosine receptors, Biochem. Pharmacol 68 (2004) 1985–1993, 10.1016/j.bcp.2004.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].https://www.tocris.com/products/2-chloroadenosine_3136, (accessed13 March 2021).
- [41].Borea PA, Varani K, Vincenzi F, Baraldi PG, Tabrizi MA, Merighi S, Gessi S, The A3 adenosine receptor: history and perspectives, Pharmacol. Rev 67 (2015) 74–102, 10.1124/pr.113.008540. [DOI] [PubMed] [Google Scholar]
- [42].Fuentes E, Caballero J, Alarcon M, Rojas A, Palomo I, Chlorogenic acid inhibits human platelet activation and thrombus formation, PloS One 9 (2014), e90699, 10.1371/journal.pone.0090699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Fuentes E, Pereira J, Mezzano D, Alarcon M, Caballero J, Palomo I, Inhibition of platelet activation and thrombus formation by adenosine and inosine: studies on their relative contribution and molecular modeling, PloS One 9 (2014), e112741, 10.1371/journal.pone.0112741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Pan C, Wei X, Ye J, Liu G, Zhang S, Zhang Y, Du H, Ding Z, BF066, a novel dual target antiplatelet agent without significant bleeding, PloS One 7 (2012), e40451, 10.1371/journal.pone.0040451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Boncler M, Wzorek J, Wolska N, Polak D, Watala C, Rozalski M, Adenosine receptor agonists deepen the inhibition of platelet aggregation by P2Y12 antagonists, Vasc. Pharmacol 113 (2018) 47–56, 10.1016/j.vph.2018.11.005. [DOI] [PubMed] [Google Scholar]
- [46].Amisten S, Braun OO, Bengtsson A, Erlinge D, Gene expression profiling for the identification of G-protein coupled receptors in human platelets, Thromb. Res 122 (2008) 47–57, 10.1016/j.thromres.2007.08.014. [DOI] [PubMed] [Google Scholar]
- [47].Ledent C, Vaugeois JM, Schiffmann SN, Pedrazzini T, El Yacoubi M, Vanderhaeghen JJ, Costentin J, Heath JK, Vassart G, Parmentier M, Aggressiveness, hypoalgesia and high blood pressure in mice lacking the adenosine A2a receptor, Nature 388 (1997) 674–678, 10.1038/41771. [DOI] [PubMed] [Google Scholar]
- [48].Cooper JA, Hill SJ, Alexander SP, Rubin PC, Horn EH, Adenosine receptor-induced cyclic AMP generation and inhibition of 5-hydroxytryptamine release in human platelets, Br. J. Clin. Pharmacol 40 (1995) 43–50, 10.1111/j.1365-2125.1995.tb04533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sandoli D, Chiu PJ, Chintala M, Dionisotti S, Ongini E, In vivo and ex vivo effects of adenosine A1 and A2 receptor agonists on platelet aggregation in the rabbit, Eur. J. Pharmacol 259 (1994) 43–49, 10.1016/0014-2999(94)90155-4. [DOI] [PubMed] [Google Scholar]
- [50].Plagemann PG, Wohlhueter RM, Kraupp M, Adenosine uptake, transport, and metabolism in human erythrocytes, J. Cell. Physiol 125 (1985) 330–336, 10.1002/jcp.1041250223. [DOI] [PubMed] [Google Scholar]
- [51].Stalker TJ, Traxler EA, Wu J, Wannemacher KM, Cermignano SL, Voronov R, Diamond SL, Brass LF, Hierarchical organization in the hemostatic response and its relationship to the platelet-signaling network, Blood 121 (2013) 1875–1885, 10.1182/blood-2012-09-457739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Wolska N, Kassassir H, Luzak B, Watala C, Rozalski M, Adenosine receptor agonists increase the inhibition of platelet function by P2Y12 antagonists in a cAMP-and calcium-dependent manner, Pharmaceuticals 13 (2020) 177, 10.3390/ph13080177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Fishman P, Bar-Yehuda S, Liang BT, Jacobson KA, Pharmacological and therapeutic effects of A3 adenosine receptor agonists, Drug Discov. Today17 (2012) 359–366, 10.1016/j.drudis.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Imai K.-i., Nohara A, Honjo M, Synthesis of purine nucleosides using iodine as catalyst, Chem. Pharm. Bull 14 (1966) 1377–1381, 10.1248/cpb.14.1377. [DOI] [PubMed] [Google Scholar]
- [55].Kazimierczuk Z, Cottam HB, Revankar GR, Robins RK, Synthesis of 2’-deoxytubercidin, 2’-deoxyadenosine, and related 2’-deoxynucleosides via a novel direct stereospecific sodium salt glycosylation procedure, J. Am. Chem. Soc 106 (1984) 6379–6382, 10.1021/ja00333a046. [DOI] [Google Scholar]
- [56].Jacobson KA, Park K-S, Jiang J-L, Kim Y-C, Olah ME, Stiles GL, Ji X-D, Pharmacological characterization of novel A3 adenosine receptor-selective antagonists, Neuropharmacology 36 (1997) 1157–1165, 10.1016/S0028-3908(97)00104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Dudzinska D, Bednarska K, Boncler M, Luzak B, Watala C, The influence of Rubus idaeus and Rubus caesius leaf extracts on platelet aggregation in whole blood, Cross-talk of platelets and neutrophils, Platelets 27 (2016) 433–439, 10.3109/09537104.2015.1131254. [DOI] [PubMed] [Google Scholar]
- [58].Peerschke EI, Castellone DD, Stroobants AK, Francis J, Reference range determination for whole-blood platelet aggregation using the Multiplate analyzer, Am. J. Clin. Pathol 142 (2014) 647–656, 10.1309/AJCPP43SEYCBJLHJ. [DOI] [PubMed] [Google Scholar]
- [59].Wu S, Zhang Y, LOMETS: a local meta-threading-server for protein structure prediction, Nucleic Acids Res. 35 (2007) 3375–3382, 10.1093/nar/gkm251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Fiser A, Šali A, Modeller: generation and refinement of homology-based protein structure models, Methods Enzymol. 374 (2003) 461–491, 10.1016/S0076-6879(03)74020-8. [DOI] [PubMed] [Google Scholar]
- [61].Otkidach D, Pletnev I, Conformational analysis of boron-containing compounds using Gillespie–Kepert version of molecular mechanics, J. Mol. Struct.: THEOCHEM 536 (2001) 65–72, 10.1016/S0076-6879(03)74020-8. [DOI] [Google Scholar]
- [62].Salentin S, Schreiber S, Haupt VJ, Adasme MF, Schroeder M, PLIP: fully automated protein–ligand interaction profiler, Nucleic Acids Res. 43 (2015) W443–W447, 10.1093/nar/gkv315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Wallace AC, Laskowski RA, Thornton JM, LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions, Protein Eng. 8 (1995) 127–134, 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.