

Abstract
High‐dimensional analyses of tissue samples from patients with rheumatic diseases are providing increasingly detailed descriptions of the immune cell populations that infiltrate tissues in different rheumatic diseases. Here we review key observations emerging from high‐dimensional analyses of T cells within tissues in different rheumatic diseases, highlighting common themes across diseases as well as distinguishing features. Single‐cell RNA sequencing analyses capture several dimensions of T‐cell states, yet surprisingly, these analyses generally have not demonstrated distinct clusters of paradigmatic T‐cell effector subsets, such as T helper (Th) 1, Th2, and Th17 cells. Rather, global transcriptomics robustly identify both proliferating T cells and regulatory T cells and have also helped to reveal new effector subsets in inflamed tissues, including T peripheral helper cells and granzyme K+ T cells. Further characterization of the T‐cell populations that accumulate within target tissues should enable more precise targeting of biologic therapies and accelerate development of more specific biomarkers to track activity of relevant immune pathways in patients with rheumatic diseases.
Characterizing T‐cell phenotypes in autoimmunity
A core principle of prototypical autoimmune diseases is that autoreactive T cells recognize and respond to self‐antigens. These autoreactive T cells can execute a varying set of functions, which may include killing target cells and producing cytokines to recruit, activate, or enhance functions of other immune cell populations, such as macrophages, B cells, natural killer (NK) cells, and neutrophils, as well as local stromal and parenchymal cells.
The central importance of antigen presentation to T cells as a cause of autoimmunity is demonstrated by the strong genetic association with HLA antigen alleles for many autoimmune rheumatic diseases, including rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), systemic sclerosis (SSc), psoriatic arthritis (PsA), and others (1). Human leukocyte antigen (HLA) genes encode major histocompatibility complex (MHC) molecules, whose primary role is to present antigens to T cells, indicating that antigen presentation to T cells is a critical factor in the development of autoimmune rheumatic diseases. T cells infiltrate the target tissue in multiple autoimmune rheumatic diseases, supporting their likely role in instigating tissue damage. In some diseases, direct targeting of T‐cell activation with abatacept (CTLA4‐Ig) has proven effective, at least in some patients (2).
In addition to abatacept, many disease‐modifying antirheumatic drugs can inhibit T‐cell activation and function; however, almost all of these therapies are nonspecific, broadly inhibiting all T cells instead of only the specific T‐cell populations that drive pathology. A more detailed understanding of the specific T‐cell phenotypes and functions that induce pathology in different rheumatic diseases may enable more specific targeting of the relevant T‐cell populations, while leaving the rest of the T‐cell compartment unaffected. However, defining the relevant T‐cell subsets and functions in different rheumatic diseases has been challenging because of the highly diverse, overlapping, and malleable functions of activated T cells.
T cells can acquire the ability to perform a wide range of effector functions. This is particularly clear for CD4 T cells, although CD8 T cells also vary in their functions. Depending on the conditions at the time of activation, T cells can differentiate into discrete subsets that perform different functions, such as production of cytokines such as interferon‐γ (IFN‐γ), interleukin (IL)‐17A, IL‐4, IL‐10, IL‐21, and many others. Often, patterns of production of these individual cytokines are used to classify T cells into subsets (eg, T helper [Th] 1, Th17, Th2, Type 1 regulatory, and T follicular helper [Tfh] cells). However, it has not generally been possible to measure all of the potential factors that individual T cells might be making when trying to classify T cells into categories. In addition, T cells make different combinations of these factors, often crossing theoretical lines of differentiated subsets. For example, IFN‐γ/IL‐17A coproducing T cells are found in many conditions (3). T cells that produce both IFN‐γ and IL‐10 or IL‐17A and IL‐10 are well appreciated, such that IL‐10 production is not clearly restricted to any T‐cell subset (4). Furthermore, T‐cell production of cytokines can change over time depending on the local stimuli. This polyfunctionality and plasticity makes it challenging to easily summarize T‐cell phenotypes within tissues of patients with autoimmune diseases.
The tools that immunologists have relied on for decades to study T‐cell populations, including quantitative polymerase chain reaction, enzyme‐linked immunosorbent assay, immunofluorescence microscopy, and even multicolor flow cytometry, often struggle to measure a sufficient number of variables to fully describe T‐cell populations in patient samples. The advent of even higher‐dimensional analyses, including mass cytometry and single‐cell transcriptomics, has brought us closer to a comprehensive understanding of the T cells in inflamed tissues, offering complementary views of the T‐cell states in chronically inflamed tissues (Figure 1).
Figure 1.
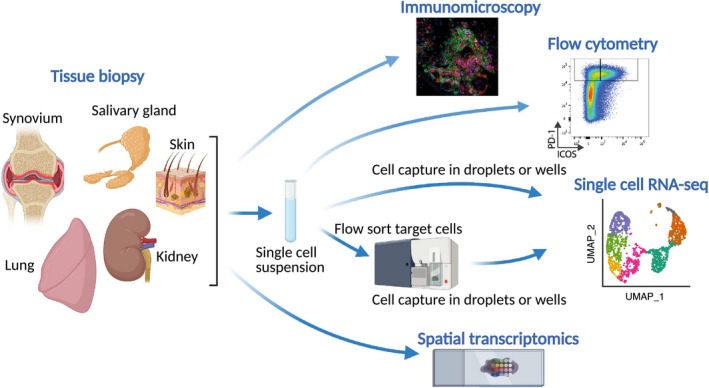
Approaches to studying tissue biopsies in rheumatic diseases. Schematic of strategies to analyze tissue biopsy samples, including well‐established techniques, such as microscopy and flow cytometry, as well as higher‐dimensional analyses, such as single‐cell RNA sequencing (RNA‐seq) and spatial transcriptomics.
High‐dimensional analyses of T‐cell phenotypes with single‐cell resolution
The past decade has provided an explosion of technologies to perform detailed descriptive analyses of patient samples, providing a progressively higher‐resolution view of the cells, pathways, and gene signatures active in disease. Mass cytometry can measure greater than 40 proteins on or inside cells simultaneously with single‐cell resolution (5). This level of resolution helped to discover a unique T peripheral helper (Tph) cell population abundant in RA synovial fluid and tissue and also highly expanded in the circulation of patients with SLE (6, 7). However, even this dimensionality becomes limiting when attempting to simultaneously assess surface markers, transcription factors, cytokines, differentiation states, migratory programs, and other features in diverse T‐cell populations.
The advent of whole transcriptomics of single cells has now enabled quantification of broad gene expression patterns in single cells, allowing investigators to evaluate T‐cell phenotypes without having to select specific markers to measure. Initial methods relied on fluorescence‐activated cell sorting (FACS) or micromanipulation to separate cells into the individual wells of a plate and could profile on the order of hundreds to thousands of cells per study. Advances in approaches, including microfluidic‐ and droplet‐based methodologies, can now accommodate tens to hundreds of thousands of cells at a time, allowing for a global interrogation of the cell states present in a sample (8). In parallel to developments in hardware components, advances in analytical methods have similarly made great strides, with computational approaches to address issues of batch correction and data integration (9, 10, 11). As a result, comparative analyses across patients, experimental conditions, tissues, and techniques are feasible, for example, allowing for the evaluation of the effects of a particular disease across multiple tissues in the body or across multiple patients or the examination of cell state dynamics across multiple associated diseases.
T cells from tissue samples from several rheumatic diseases have now been evaluated by single‐cell transcriptomics. Here we will review some of the key observations emerging from these analyses, which have yielded several surprises and highlighted new axes of T‐cell functions not previously well appreciated. As we will discuss, single‐cell RNA sequencing (scRNA‐seq) analyses appear particularly well suited to identify distinct populations of proliferating cells, which stand out as transcriptomically unique given the host of genes associated with cell cycle. Perhaps unexpectedly, T regulatory cells (Tregs), B‐cell‐helper T cells (such as Tph cells and Tfh cells), and distinct CD8 effector subsets are well resolved in scRNA‐seq data sets across multiple diseases, whereas distinct subsets of Th1, Th17, or Th2 cells are less apparent. Here we do not comprehensively review all T‐cell subsets that have been associated with each rheumatic disease, nor do we review every rheumatic disease; rather, we focus on recent examples that help to illustrate common themes and key differences in the T‐cell phenotypes captured by high‐dimensional analyses, with a specific emphasis on T cells within the target tissues from patients with rheumatic diseases.
High‐dimensional views of T‐cell infiltrates in rheumatic diseases
SLE
Several high‐dimensional tissue analyses have been performed by the Accelerating Medicines Partnership (AMP) RA/SLE Network, a multisite collaborative effort that has focused on single‐cell analyses of tissue biopsies from patients with SLE and RA (12, 13, 14, 15, 16). The AMP Network prioritized lupus nephritis as a primary target tissue in SLE, and protocols were developed to collect, store, and dissociate kidney biopsy tissue to isolate both leukocytes and parenchymal cells for scRNA‐seq analyses (15). As part of AMP Network phase 1 analyses, CD45+ leukocytes were flow sorted from lupus nephritis kidney biopsy specimens into individual wells of 384‐well plates and analyzed by scRNA‐seq via the CelSeq2 pipeline (12, 17). Lupus nephritis kidney biopsy specimens from 24 patients were processed to generate scRNA‐seq transcriptomes on 2736 leukocytes. The T cells and NK cells from this analysis, represented by 1764 cells, clustered into 10 distinct populations (eight T‐cell clusters and two NK cell clusters) (12).
CD4 T cells
Among CD4+ T cells within lupus nephritis kidneys, the largest cluster of cells contained few distinguishing effector functions but could be separated into two subclusters, one with early central memory‐like features (CCL7, SELL) and a second with more effector memory‐like features (CXCR6, CCR5). A distinct Tfh‐like population, which may include Tph and/or Tfh cells, clustered separately from the main CD4 cluster (12). This cluster displayed the highest expression of PDCD1 (encoding PD‐1), the transcription factor MAF, and the B‐cell chemoattractant CXCL13. Flow cytometric assessment of these same kidney biopsy specimens confirmed the presence of a PD‐1hi CD4 T‐cell population that correlated with the presence of B cells in kidney tissue (6). Immunofluorescence microscopy analyses have also demonstrated the presence of PD‐1+ B‐cell‐helper T cells with lupus nephritis kidneys (18, 19). Notably, CXCR5− PD‐1hi Tph cells and a similar population of Th10 cells, which produce IL‐10 and succinate to stimulate B‐cell responses, are also highly expanded in the circulation of patients with lupus (6, 18). scRNA‐seq analyses also revealed a Treg population with high expression of FOXP3 and IKZF2, which clustered closely with Tfh‐like cells (12). In addition, whereas most cell clusters in lupus nephritis kidneys show an elevation of interferon‐inducible genes compared with control kidneys, a small population of CD4 T cells with very high expression of interferon‐inducible genes was also identified as a discrete cluster (12). Single‐cell analyses of cells from blood have further helped to identify specific cell subsets with distinctively upregulated interferon signatures in patients with SLE (20).
CD8 T cells
The CD8 T cells in lupus nephritis samples also separated into three distinct clusters (12). The largest of these clusters showed high expression of cytotoxicity‐associated genes (GZMB, GNLY, PRF1) and appeared transcriptomically similar to cytotoxic NK cells found in the same samples. The presence of both a large cytotoxic CD8 T‐cell population and cytotoxic NK cell population suggests that cytotoxic T cells may play important roles in lupus nephritis, particularly in the interstitial space (21). Interestingly, a separate cluster of CD8 T cells showed a stark difference in the expression of granzymes, with almost no expression of GZMB (encoding granzyme B), low expression of PRF1, and yet high expression of GZMK (encoding granzyme K, a granzyme with cleavage specificity that differs from that of granzyme B) (22). The localization and function of this granzyme K+ CD8 population has not yet been determined. A third CD8 T‐cell population that expressed markers of T resident memory cells, including XCL1, ZNF683 (encoding Hobit), and ITGA1, also appeared (23, 24). This cell subset was the most abundant among the CD8 T cells obtained from healthy kidney tissue (12, 25), suggesting that a population of resident memory CD8 T cells exists in healthy kidney tissue, perhaps analogous to T resident memory cell populations well appreciated at epithelial barriers (25).
RA
High‐dimensional analyses of synovial biopsy tissue from patients with RA has been a major focus of the AMP Network (14, 16). In phase 1 of the AMP effort, 36 RA synovial samples were analyzed by a combination of mass cytometry, bulk RNA sequencing of sorted subsets (T cells, B cells, fibroblasts, and macrophages), and scRNA‐seq of the same sorted subsets (16). CD3 T cells were sorted as single cells into individual wells of 384‐well plates, and global transcriptomes were generated through the CelSeq2 platform as for SLE. A total of 1529 T cells, derived from 18 samples from patients with RA , were clustered on the basis of their scRNA‐seq transcriptomes, with the aid of the bulk RNA sequencing data, through a canonical correlation analysis approach; this analysis yielded three CD4 T‐cell clusters and three CD8 T‐cell clusters (16).
CD4 T cells
As in lupus nephritis kidneys, the major CD4 T‐cell population showed few defining effector functions and contained both cells with central memory features and cells with effector memory features. A population of Tph and Tfh cells clustered separately from the main cluster, displaying high expression of PDCD1, MAF, and CXCL13, consistent with the prior description of Tph cells in RA synovium (7, 16). Comparison of the T‐cell clusters between RA and osteoarthritis (OA) samples demonstrated that the Tph/Tfh cell cluster was significantly expanded in RA samples compared with OA samples (16). The Tph cell cluster in RA synovium was the only cell cluster across all cell populations with detectable expression of CXCL13, suggesting that Tph cells may play a critical role in recruiting B cells into RA synovium through production of this potent B‐cell chemoattractant (26). An independent analysis of RA synovial tissue by droplet‐based scRNA‐seq also revealed a prominent Tph cell population, which again appeared as the sole source of CXCL13 messenger RNA (mRNA) expression in RA synovium (27). These results, coupled with the recognition that CXCL13 is most the highly upregulated gene in T cells from RA synovium compared with T cells from OA synovium (16), underscore that the accumulation of CXCL13+ Tph cells is a highly characteristic feature of the T‐cell infiltrate in RA synovium. The AMP scRNA‐seq analysis also revealed a Treg cluster with FOXP3 and IKZF2, which again clustered closely to Tph/Tfh cells, similar to the pattern seen in lupus nephritis (12, 16).
CD8 T cells
Representing approximately 40% of synovial T cells, the CD8 T cells in RA synovium demonstrated a surprising pattern, with only a small number of CD8 cells grouping together into a cluster with a clear set of cytotoxic features (GZMB, PRF1, GNLY) (16). In contrast, a much larger population of CD8 T cells clustered separately, with low or intermediate expression of GZMB but clear expression of GZMK. In addition, a third population expressed both GZMB and GZMK; this population with dual GZMK/GZMB expression was not observed in lupus nephritis kidneys (12, 16). The large population of granzyme K+ T cells has gone largely unappreciated prior to these scRNA‐seq analyses; understanding the functions of this population and its relationship to granzyme B+ CD8 T cells is now of major interest.
PsA
High‐dimensional analyses of T cells in PsA synovial fluid and tissue have been recently described as using a combination of mass cytometry and scRNA‐seq, with varied scRNA‐seq methods used to analyze 41 453 T cells obtained from samples from eight patients (28).
CD4 T cells
Among the CD4 T cells, large clusters could be distinguished with higher or lower expression of central memory–associated features (CCR7, SELL), although no clear effector functions were identified in these clusters (28). A substantial cluster (14% of T cells) was identified that expressed some features of Th17 cells, including increased average expression of KLRB1, CCR6, and RORC; however, expression of key effector cytokines IL‐17A and IL‐17F was not reported. Intracellular cytokine‐staining analyses of T cells from PsA synovial fluid and tissue samples have generally indicated that less than 5% of CD4 T cells in PsA synovial fluid make IL‐17A after strong stimulation, with similar frequencies observed in RA synovial fluid (29). Interestingly, cytometric studies of synovial fluid cells have indicated that CD8 T cells can produce IL‐17, and these IL‐17+ CD8 T cells are more abundant in PsA than in RA (30). IL‐17+ CD8 T cells differ in the transcriptomes from other CD8 T cells and from Th17 CD4 cells, with increased expression of RORC, IL23R, and CCR6, as well as GZMA, GZMB, and PRF1 (31). In addition, the frequency of circulating IL‐17+ CD8 T cells was positively correlated with disease activity, the C‐reactive protein level, and the erythrocyte sedimentation rate (30). However, the frequency of IL‐17A+ CD8 T cells appears lower than the frequency of IL‐17A+ CD4 T cells, and scRNA‐seq analyses have not yet clearly visualized this IL‐17+ CD8 population.
scRNA‐seq clustering identified several additional CD4 T‐cell clusters, including one small cluster characterized by high expression of interferon‐induced genes, such as MX1 (28). In addition, as in other tissues, Tregs in PsA synovial samples appeared as a distinct cluster, with high expression of FOXP3 and IL2RA, consistent with prior cytometric studies indicating an abundance of FoxP3+ Tregs (32, 33). Additional clusters also appeared in the PsA analyses, yet no clear effector functions were determined, in part, because the low sensitivity of scRNA‐seq makes it is challenging to detect many cytokines. This weakness can make it challenging to map previously described populations to the scRNA‐seq data. For example, polyfunctional CD4 T cells previously reported in PsA synovial fluid, which produce multiple cytokines, including granulocyte‐macrophage colony‐stimulating factor (GM‐CSF), tumor necrosis factor (TNF), and IL‐17A or IFN‐γ, are not easily mapped to the scRNA‐seq clusters (34). These cells are positively correlated with disease activity of PsA and likely contribute to pathology through production of multiple inflammatory cytokines (34, 35); however, dissecting their presence in scRNA‐seq data sets will take further analysis.
CD8 T cells
CD8 T cells were clustered into seven populations, which included a mucosal associated invariant T (MAIT) cell cluster and a cluster of central memory‐like CD8 cells (CCR7, SELL) that did not express granzymes or cytokines (28). As in RA synovium, distinct effector CD8 T‐cell clusters appeared with varying patterns of GZMB and GZMK. The largest of these clusters, labeled HLA antigen‐DRlow, predominantly expressed GZMK but not GZMB. A moderately sized cluster labeled HLA antigen‐DRhigh, expressed both GZMK and GZMB, whereas a smaller cluster characterized by expression of ZNF683, a transcription factor associated with tissue residency (23), expressed GZMB and GNLY without GZMK, characteristic of a cytotoxic effector CD8 T cell.
Notably, scRNA‐seq revealed an upregulation of the chemokine receptor CXCR3 broadly among PsA T cells (28). Consistent with this observation, the interferon‐inducible chemokines CXCL9 and CXCL10, both ligands for CXCR3, were abundant in PsA synovial fluid, likely contributing to the recruitment of CXCR3+ CD8 and CD4 T cells to affected joints (28, 36, 37). The high frequency of CXCR3 expression on T cells in PsA synovial fluid (as well as RA synovial fluid) is consistent with prior flow cytometry assessments that indicated that the majority of T cells in PsA and RA synovial fluid express CXCR3 (37, 38, 39, 40).
Juvenile idiopathic arthritis
scRNA‐seq analysis of synovial fluid T cells from seven patients with oligoarticular juvenile idiopathic arthritis (JIA) was recently reported (41). T cells were flow cytometrically sorted, and 74 891 scRNA‐seq transcriptomes were generated by the 10X Genomics platform.
CD4 T cells
CD4 T cells separated into six clusters, the largest of which was a central memory‐like cluster (CCR7, SELL), similar to findings from RA and PsA samples (16, 28, 41). The next largest cluster appeared as a Tph cluster, with high expression of PDCD1, IL21, CXCL13, ENTPD1, and CD40LG, as well as increased expression of cytokines CSF2 (encoding GM‐CSF), TNF, and IFNG. These cells lacked CXCR5 at the mRNA and protein level and instead expressed transcriptomic signatures of peripheral tissue localization, including expression of the chemokine receptor CXCR6 (41). A second highly activated cluster displayed a different set of functional properties, characterized by high expression of IL10, PRF1, GZMK, and the transcription factor EOMES. This cluster of CD4 T cells was also present in peripheral blood from patients with JIA, as indicated by an overlap in both T‐cell receptor (TCR) repertoire and expression of cytotoxicity‐associated genes (41).
In addition, a cluster with increased expression of Th17‐associated genes RORC, IL23R, and CCR6 was also identified and comprised 13% of the synovial fluid CD4 T cells (41). Although comparisons across studies are limited by technical and data processing differences, these cells appear roughly similar to the Th17 cluster identified in PsA samples (28). However, detection of Th17 effector cytokines, such as IL‐17A and IL‐17F, was not reported; thus, the extent to which these cells are IL‐17‐producing T cells remains to be demonstrated. IL‐17+ CD4 T cells have been identified in synovial fluid from patients with JIA, with approximately 1% to 5% of CD4 T cells producing IL‐17A after strong in vitro stimulation (42).
scRNA‐seq data were also generated on Tregs sorted from synovial fluid of patients with JIA, which complements prior detailed analyses of JIA synovial Treg phenotypes (43). Cytometric, imaging, and functional studies have demonstrated substantial heterogeneity of Tregs in JIA synovial fluid, including a subset of functional CD25hi CD127− Tregs that have downregulated FoxP3 expression (43) and subpopulations that differ in CD161 expression (44). Whether these Treg subpopulations can be discriminated transcriptomically remains to be demonstrated. Notably, high‐dimensional mass cytometry has also been used to characterize Tregs in the synovial fluid and blood of patients with systemic JIA (45). In these studies, Tregs from JIA synovial fluid and blood were shown to possess Th17‐associated features, which could be reversed by IL‐1 blockade (45).
CD8 T cells
Clustering of CD8 T cells from JIA synovial fluid yielded 10 clusters, although the defining features of these clusters were not described in detail (41). Interestingly, GZMK and GZMH expression was observed throughout the vast majority of CD8 T cells. The patterns of GZMB and GNLY were not demonstrated, yet the expression of these markers appears to have been most frequent in some of the smaller clusters. Overall, this pattern seems consistent with CD8 T cells from RA and PsA, in which there is broad GZMK expression among infiltrating T cells (16, 28).
Among synovial CD8 T cells in JIA analyzed by scRNA‐seq, one cluster stood out with little expression of GZMK; this cluster instead expressed several markers resembling CD4 Tph cells, including CXCL13, PDCD1, ENTPD1, and CXCR6 (41). These findings are consistent with prior work that evaluated an activated clonally expanded population of CD8 T cells marked by surface expression of PD‐1 in JIA synovial fluid (46). Functional and transcriptomic analyses indicated that this PD‐1+ CD8 T‐cell subset was metabolically active and functional, despite expression of several genes and proteins associated with exhaustion, such as LAG3, HAVCR2, TIGIT, and CTLA‐4. Elevated expression of CXCL13 in the PD‐1+ CD8 T cell was noted in this study as well (46). The presence of a CXCL13+ CD8 T‐cell population seems to be a distinct difference from the pattern seen in RA synovial T cells, in which CXCL13 expression has been observed exclusively in CD4 T cells (16, 27), and raises the possibility that CD8 T cells may play a unique B‐cell‐helper role in JIA.
SSc
The majority of tissue analyses in SSc have focused on the skin. Although analyses of T cells in SSc skin is likely to vary with the disease course, for example, with increased detection of CD4 and CD8 T cells in early diffuse disease compared with later stages (47, 48), cytometric, histologic, and bulk transcriptomic approaches have demonstrated key features of the T‐cell infiltrate in SSc. In addition, a recent report has provided the first scRNA‐seq assessment of skin T cells in SSc, evaluating 2862 T cells obtained from 27 skin biopsies using the 10X Genomics platform (49).
CD4 T cells
scRNA‐seq analyses identified several subpopulations of CD4 T cells with both central memory and resident memory features, yet specific functions for the majority of these cells were not clearly evident from the transcriptomes (49). Prior analyses of skin T cells have emphasized the increased frequency of Th2 cells in SSc skin, which produce factors such IL‐4, IL‐5, and IL‐13 and may contribute to tissue fibrosis (50, 51, 52, 53, 54, 55, 56, 57, 58). T cells with expression of both CD4 and CD8 (“double positive” cells) have been isolated from SSc skin and have been shown to produce large amounts of IL‐4 (58). However, although increased in SSc skin compared with control skin, GATA3+ Th2 cells do not appear to be the most abundant T‐cell subset in SSc skin (although they were shown to be abundant in skin from patients with bullous pemphigoid) (59). Rather, by immunofluorescence microscopy analyses, the predominant CD4 T‐cell population infiltrating the skin of patients with early diffuse cutaneous SSc was a granzyme A+ cytotoxic CD4 T‐cell population (59). This microscopy analysis also detected Rorgt+ Th17 cells, CXCR5+ Tfh cells, and FoxP3+ Tregs in decreasing order of abundance.
Cytotoxic CD4 T cells are marked by surface expression of SLAMF7 and produce both granzymes and cytokines, such as IL‐1β and transforming growth factor β (TGF‐β) (59). It was hypothesized that they may promote fibrosis by selectively killing endothelial cells. Cytotoxic CD4 T cells were also detectable in blood, where they are marked by loss of CD28 and expression of CD57 and CX3CR1 (59). TCR repertoire analysis on CD28− CD57+ cytotoxic CD4 T cells from the blood of patients with SSc showed marked clonal expansion, and transcriptomic analysis showed upregulation of cytotoxicity‐associated genes, including GZMA, GZMB, GZMH, GZMM, PRF1, GNLY, SLAMF7, and KLRG1 (59). scRNA‐seq analysis of SSc skin identified a prominent cluster of T cells with cytotoxic features; this cluster contained most of the CD8 T cells captured, as well as some cells that lacked CD8A expression and were potentially cytotoxic CD4 T cells (49). Cytotoxicity‐associated genes were also found in a portion of T cells in other clusters predominantly composed of CD4 T cells; these cells also represented potential cytotoxic CD4 T cells. Integrating analyses from scRNA‐seq and microscopy will be of interest to further define this T‐cell population.
A separate study used microscopy to demonstrate an increased frequency of PD‐1+ ICOS+ CD4 T cells in the skin of patients with SSc, which were hypothesized to contribute to fibrosis through production of IL‐21 (60). High expression of PD‐1 and ICOS might suggest a B‐cell‐helper phenotype, such as Tph or Tfh cells. mRNA expression of PDCD1 and ICOS in SSc skin samples was positively correlated with expression of CXCR5, which can be detected on T cells in SSc skin (59); however, the cell source of CXCR5 expression (ie, on B cells or T cells) that correlated with PDCD1 and ICOS expression was not determined (61). scRNA‐seq analyses have provided new insight on this B‐cell‐helper T‐cell population by highlighting the presence of a CXCL13 + T‐cell cluster with expression of ICOS and TIGIT (49). These cells lacked detectable BCL6 and CXCR5 expression by scRNA‐seq (which may miss expression because of low sensitivity) as well as by immunofluorescence microscopy (49), strongly suggesting that CXCL13+ CXCR5− Tph cells are present in SSc skin. Microscopy analyses have also detected CXCR5+ T cells in SSc skin (59); thus, further evaluation of the relative proportions of Tph cells and Tfh cells in SSc skin will be of interest.
Both microscopy and scRNA‐seq analyses have indicated the presence of Tregs in SSc skin (49, 59). Surprisingly, Tregs from SSc skin, but not blood, can produce IL‐4 and IL‐13, raising the possibility that Tregs in SSc skin may contribute to the fibrotic process (62). IL‐33 acting through ST2 was identified as a candidate signal to induce this set of Th2 features in skin Tregs (62).
CD8 T cells
Microscopy and flow cytometric analyses have demonstrated increased numbers of CD28− CD8 T cells in the skin of patients with early diffuse cutaneous SSc (63, 64). These cells frequently display multiple features of cytotoxicity, including expression of granzyme A and granzyme B, such that by microscopy, the numbers of granzyme A+ CD8 T cells and granzyme A+ CD4 T cells are roughly comparable in SSc skin (59). scRNA‐seq analysis also clearly illustrated a CD8 T‐cell population with a cytotoxic gene expression program (49). CD28− CD8 T cells in SSc skin frequently express migratory receptors for skin homing, including CCR10 and cutaneous lymphocyte antigen (CLA) (63), which may promote their accumulation in skin. Interestingly, the CD28− CD8 T‐cell population appears to be one source of the Th2‐associated factor IL‐13, which can promote skin fibrosis (63, 64, 65, 66); however, most of the detectable IL13 expression in T cells by scRNA‐seq appears to come from CD8A− cells (likely CD4 T cells) and from a potential γδ T‐cell population (49). Defining whether there is a discrete subset of cytotoxic CD8 T cells that acquire an IL‐13‐producing phenotype and what distinguishes these cells from other cytotoxic cells will be of substantial interest.
A recently published analysis of scRNA‐seq of cells from lungs of patients with interstitial lung disease (ILD) due to SSc has also provided an initial look at the transcriptomes of T cells that infiltrate the lung in SSc. Lung tissue samples from eight patients with SSc were analyzed by scRNA‐seq via the 10X Genomics platform, and populations of CD8 T cells, CD4 T cells, and Tregs emerged from the clustering of total lung cells (67). Although a detailed dissection of T‐cell subpopulations was not described in this work, pathway analyses suggested that CD8 T cells from SSc‐ILD lungs showed an increased expression of interferon‐inducible genes and IL‐6 pathway–associated genes compared with T cells from idiopathic pulmonary fibrosis and control samples (67).
Immunoglobulin G4–related disease
High‐dimensional analyses have not yet been reported for T cells from immunoglobulin G4–related disease (IgG4‐RD) lesions; however, flow cytometric and microscopy analyses have demonstrated abundant cell phenotypes that are valuable to put in the context of other diseases, including Tfh cells and cytotoxic T cells.
Given the defining feature of accumulation of polyclonal IgG4+ B cells in tissue lesions, several studies have evaluated potential B‐cell‐helper T cells within IgG4‐RD‐affected tissues. Flow cytometry analyses have demonstrated accumulation of a large population of T cells with typical Tfh features, including high expression of PD‐1, ICOS, CXCR5, and BCL6, within submandibular gland lesions (68, 69, 70). The Tfh cells in submandibular glands of IgG4‐RD show a distinct phenotype with the ability to produce IL‐4 but not IL‐5 or IL‐13 (68). These IL‐4+ Tfh cells express high levels of both BCL6 and BATF but not the Th2‐associated transcription factor GATA3 (68). IL‐4+ Tfh cells were associated with AID+ B cells predominantly outside germinal centers, and their frequency in blood correlated with serum IgG4 levels (68). Consistent with other rheumatic diseases, Tregs are found in IgG4‐RD lesions as well (71, 72, 73, 74).
Microscopic analyses similar to those described above in SSc have also demonstrated accumulation of a cytotoxic CD4 T‐cell population within IgG4‐RD lesions, which are identified by expression of SLAMF7 and granzyme A (75, 76). These cells, which are clonally expanded and found in numerous tissue sites in IgG4‐RD, can contribute to fibrosis through multiple mechanisms, including direct killing of stromal or mesenchymal cells and production of cytokines, such as IL‐1β and TGF‐β (75). Cytotoxic CD8 T cells can also be identified in these lesions and may also contribute to mesenchymal cell death (76).
Themes and distinctions across rheumatic diseases
Detailed assessments of the T‐cell phenotypes within tissues across multiple diseases have begun to yield some initial patterns. Trying to compare across studies that use different technologies and analysis methods is risky, in that apparent differences may be due to differences in the technologic platform or analysis methods rather than the underlying biology. In addition, high‐dimensional analyses are often applied to only a small number of patients with a given disease, which may yield a skewed view of overall disease patterns. Furthermore, cell composition may vary on the basis of the tissue analyzed, for example, skin versus kidney in SLE or skin versus lung in SSc or synovial fluid versus synovial tissue in inflammatory arthritides. Nonetheless, it is valuable to begin to summarize patterns that are emerging from current reports, recognizing that analyses in the near future may revise these initial impressions (Figure 2).
Figure 2.
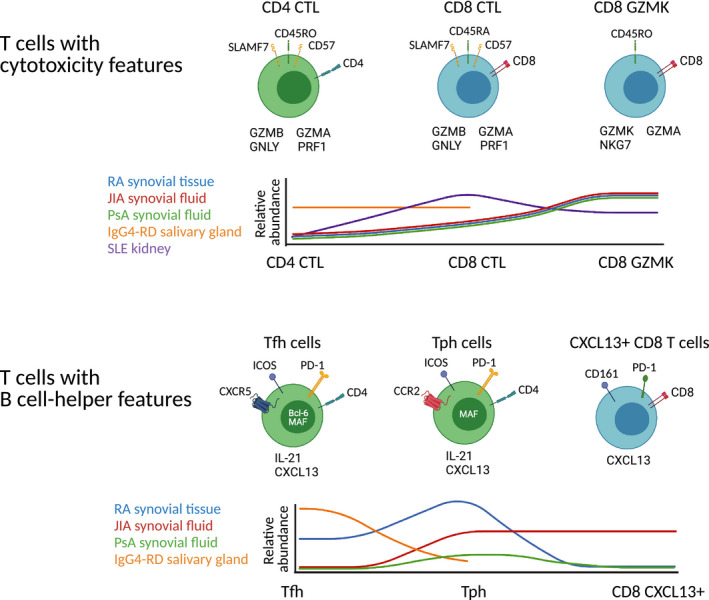
Varied cell phenotypes highlighted by single‐cell RNA sequencing (scRNA‐seq). scRNA‐seq analyses of rheumatic disease samples have illustrated different versions of cells with cytotoxicity‐associated features (top row), including paradigmatic cytotoxic CD8 T cells, CD8 T cells that express granzyme K and granzyme A with little granzyme B, and cytotoxic CD4 T cells. scRNA‐seq analyses have also illustrated a range of cell types expressing features associated with B‐cell‐helper function (bottom row), including T follicular helper (Tfh) cells, T peripheral helper (Tph) cells, and CXCL13+ CD8 T cells. Histogram plots show preliminary estimates of the relative abundances of the different cell states listed in the plots across diseases based on data currently available. CTL, cytotoxic T lymphocytes; IgG4‐RD, immunoglobulin G4–related disease; JIA, juvenile idiopathic arthritis; PsA, psoriatic arthritis; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus.
Perhaps the biggest surprise to T‐cell immunologists from the analyses described above is that CD4 T cells from inflamed tissues tend not to cluster into expected Th1, Th2, and Th17 subsets. At the outset of many of these studies, we frequently asked whether a given disease should be considered a Th1 disease or a Th17 disease. In some cases, this question may capture the pathophysiology well; for example, the dramatic efficacy of IL‐17A blockade in psoriasis strongly argues that psoriasis is a Th17 disease (77). Yet the CD4 T cells in RA and SLE do not easily separate into clean Th1 and Th17 clusters on the basis of their transcriptomics. The scRNA‐seq data from JIA and PsA do identify clusters with Th17‐associated features, including some CCR6 and RORC expression, yet the extent to which these cells represent IL‐17A/IL‐17F‐producing effector T cells remains to be determined. In addition, the majority of CD4 T cells in both RA and PsA express CXCR3 (28, 37, 38, 39, 40), a Th1‐associated factor, suggesting that the axis of Th1 versus Th17 is unlikely to cleanly define many rheumatic diseases. Careful flow cytometric studies of T cells from inflammatory arthritides have highlighted the presence of polyfunctional T cells that produce multiple cytokines, including TNF, GM‐CSF, and IFN‐γ or IL‐17A (34). This polyfunctionality is likely the rule rather than the exception for T cells because in vitro stimulated T cells transition through states of making different combinations of cytokines (78).
Although current scRNA‐seq often does not robustly detect low abundance transcripts of cytokines, chemokine receptors, and transcription factors (the typical classification markers of T‐cell subsets), global transcriptomics has provided a new view of T‐cell states with a different set of axes complementary to the patterns defined through traditional T‐cell immunology. For example, proliferating cells are unambiguously separated by scRNA‐seq given the dramatic shifts in gene expression associated with cell cycle (12, 16, 28, 41, 67). This can enable interrogation of the identities and clonal relationships of the proliferating cells in a tissue. In addition, scRNA‐seq has revealed unexpected patterns of T‐cell effector functions, for example, the more frequent expression of IFN‐γ among CD8 T cells, rather than CD4 T cells, in both RA synovium and lupus nephritis (12, 16). Furthermore, scRNA‐seq has helped to highlight certain effector functions not previously well appreciated in T cells, such as the production of chemokines (eg, CXCL13, CCL4, CCL5) (12, 16, 27), likely because chemokine mRNA transcripts tend to be expressed at very high levels, which enables detection by scRNA‐seq.
Single‐cell analyses have identified a transcriptomically distinct Treg cluster across essentially all scRNA‐seq analyses of rheumatic disease tissues (12, 16, 28, 41, 67). Rather than being buried or intermixed with other subsets, Tregs robustly cluster apart from most other T‐cell populations. Although it can be challenging to distinguish Tregs from activated CD4 effector cells on the basis of cytometric markers (activated human T cells can upregulate CD25, downregulate CD127, and express intermediate FoxP3 levels) (79), scRNA‐seq clearly identifies these cells in multiple data sets. This argues strongly that Tregs are present in inflamed tissues in rheumatic diseases and raises some questions: Are the Tregs in tissues functional, and if so, why do they not successfully suppress the immune response?
T cells with a B‐cell‐helper phenotype, including Tph cells and Tfh cells, also cluster out separately from the rest of the CD4 T cells in both SLE and RA data sets. Interestingly, Tph and Tfh cells cluster closely with Tregs, suggesting some shared transcriptomic programs. Across diseases, a Tph/Tfh cluster appeared in scRNA‐seq data from RA synovium, JIA synovial fluid, and lupus nephritis kidneys but not in PsA synovial fluid (Figure 2) (16, 28, 41). Although this may be related to technological differences, the observation is consistent with prior cytometric analyses indicating a higher frequency of Tph cells in RA joints compared with seronegative arthritides, such as PsA (7). In this context, the identification of a CXCL13+ Tph population in JIA is somewhat surprising. Even more surprisingly, a CXCL13+ CD8 T‐cell population with some resemblance to Tph cells was also identified in JIA synovial fluid samples (41). This appears to differ from the pattern in RA synovium, in which CXCL13 expression is highly restricted to CD4 T cells (5, 10, 21). Direct comparisons of these cell types from RA and JIA will be informative to help define functional similarities and differences.
Distinct populations of B‐cell‐helper T cells, such as Tph and Tfh cells, were initially distinguished by distinct expression of chemokine receptors, with Tfh cells defined, in part, on the basis of expression of CXCR5, a chemokine receptor that allows migration to follicles, and Tph cells defined, in part, on the basis of expression of chemokine receptors that confer migration to inflamed tissues (26). scRNA‐seq does not detect chemokine receptors robustly enough to distinguish Tph cells from Tfh cells on the basis of these features alone; thus, further analyses will be needed to define the B‐cell‐helper cells that infiltrate different tissues, such as kidneys in lupus nephritis. Cytometric analyses demonstrate abundant Tph cells, but not Tfh cells, in RA synovial fluid and both Tph and Tfh cells in RA synovial tissue, with the Tfh cells located primarily within T‐cell/B‐cell aggregates (5). In contrast, cytometric analyses of IgG4‐RD demonstrate that the majority of B‐cell‐helper T cells in salivary gland tissue are BCL6+ CXCR5+ Tfh cells, with fewer Tph cells present. Extrafollicular helper T cells, such as Tph cells (5), Th10 cells (12), and CXCR4+ extrafollicular helper cells (22), can express different migratory receptors, which may allow localization to different sites. Thus, it seems likely that B‐cell‐helper T cells may exist in many flavors, with a B‐cell‐helper program coupled to different migratory programs (26). In total, scRNA‐seq appears well suited to identify the overall B‐cell‐helper phenotype and distinguish these cells, including Tph and Tfh cells, from other infiltrating CD4 T‐cell populations.
In addition to Tregs and B‐cell‐helper populations, T‐cell populations with distinct expression of granzymes have emerged as an exciting new dimension in T‐cell immunology revealed by scRNA‐seq (Figure 2). In lupus nephritis kidneys, a large population of cytotoxic CD8 T cells emerged, with high expression of cytotoxicity genes, such as GZMB, PRF1, and GNLY, similar to that seen in NK cells (6). In contrast, a similarly appearing cytotoxic CD8 population was relatively underrepresented in RA synovium. Microscopy and cytometry analyses have highlighted both cytotoxic CD8 T cells and, more recently appreciated, cytotoxic CD4 T cells within skin in SSc and salivary glands in IgG4‐RD. Cytotoxic CD4 T cells may target different tissue cell types that express MHC class II, with microscopy evidence suggesting that they may target endothelium in SSc and mesenchymal cells, such as fibroblasts, in IgG4‐RD. Cytotoxic CD4 T cells have also been recognized in RA synovium, although their roles in RA pathology are not yet defined (80).
Although CD8 T cells compose almost of the T‐cell infiltrate in RA synovium, transcriptomics suggests that most of the CD8 T cells in RA synovium are not highly cytotoxic cells but rather show a characteristic elevation of a distinct granzyme called granzyme K, which does not appear to be a highly cytotoxic granzyme (22), suggesting that granzyme K+ CD8 T cells may have functions distinct from cytotoxic CD8 T cells. Granzyme K is also widely expressed in T cells from PsA (28) and JIA synovial samples (41), with partially overlapping expression with granzyme B, suggesting that granzyme K+ cells may have common functions in these inflammatory arthritides. A distinct granzyme K+ cluster was also observed in lupus nephritis kidneys, in this case, with almost no overlap with granzyme B expression. Understanding the roles of granzyme K+ cells in inflamed tissues will be of major interest.
Putting the pieces back together and making new connections
scRNA‐seq of dissociated tissues has proven enormously informative in describing the immune cell types that accumulate within target tissues. However, the process of generating a single‐cell suspension from intact tissues destroys a key set of information about the localization of the cells in situ: Where were they in the tissue? With which other cell types do they interact? Microscopy analyses of cytotoxic CD4 T cells have provided intriguing new hypotheses about the targets of the cytotoxic T‐cell response in SSc and IgG4‐RD. Asking similar questions about the localization of Tregs, Tph cells, granzyme K–expressing cells, and others is a critical next step. For specific hypotheses or cell types, localization can be achieved through immunofluorescence microscopy or immunohistochemistry. Higher‐dimensional imaging through techniques such as co‐detection by indexing (CODEX) (81) and imaging mass cytometry (82) allows for the interrogation of many more parameters in the same tissue section and begins to enable cellular network analyses.
Advances in spatial transcriptomics are making it increasingly feasible to evaluate tissue sections with resolution approaching genomic scale. Multiple in situ hybridization‐based techniques have allowed for the highly multiplexed detection of transcriptomic targets at subcellular levels (83, 84). These technologies often require specialized equipment, and the cost of an experiment can become significant with an increased number of targets. Transcriptome‐wide approaches have aimed to address the biased nature of such targeted in situ methods, allowing for an analysis of the complete transcriptome of a tissue of interest (85, 86, 87). However, the RNA detection rates of these technologies fall behind their targeted counterparts. Similarly, these whole‐transcriptome methods often suffer from lower spatial resolution, in which current techniques cannot resolve cell‐specific transcriptomics, instead providing information about multiple cells within a region of detection. Further technological development will no doubt bring about whole‐transcriptome spatial analysis at a subcellular level, allowing for high‐resolution, unbiased examination of spatial dynamics implicated in homeostasis and disease.
Advances in single‐cell RNA‐seq technologies are also expanding the ways in which T cells and other cells can be characterized. The addition of oligo‐tagged antibodies targeting surface molecules now allows for interrogation of surface protein expression in parallel to transcriptomics (88, 89); this information is helpful to translate between single‐cell transcriptomic clusters and the well‐established patterns of T‐cell phenotypes defined over decades on the basis surface markers. In addition, single‐cell assay for transposase‐accessible chromatin using sequencing (ATAC‐seq), evaluating epigenetics at the single‐cell level, provides a complementary view of T‐cell states relative to scRNA‐seq profilers (90).
Simultaneous capture of the paired α and β chains of the TCR from the same T cells profiled by scRNA‐seq has provided a unique new dimension with which to understand T‐cell populations. Because each naïve T cell emerging from the thymus generates its own unique TCR, all progeny of that single T cell can be traced back to a common precursor, with the TCRs essentially functioning as barcodes for different T‐cell clones. This allows one to ask multiple questions about the pathologically expanded T cells in autoimmune tissues. Can a single ‐ cell clone give rise to both T effector cells and Tregs in the tissue, suggesting cross‐differentiation one way or the other? Are the same T‐cell clones found in different tissues in the same patients, for example, in two different joints in patients with RA or in both the skin and kidneys in patients with SLE? Are pathologically expanded T‐cell clones retained over time, or do they continually turn over? Connecting TCRs to T‐cell phenotypes at the single‐cell level enables this kind of dissection of the T‐cell response.
In addition, TCR sequencing methods allow for the potential discovery or prediction of antigens associated with identified TCRs. With a growth in TCR sequencing use at both the bulk and single‐cell levels, experimental and computational techniques to discover antigens have been put forth. Experimental approaches largely rely on detection of TCR‐epitope pairing using antigen libraries, such as those displayed on the surface of yeast (91) or through the detection of T‐cell killing activity (92). Although these methodologies can give evidence to the physical interaction of TCR‐epitope pairs, they mainly rely on antigen discovery in a low‐throughput manner, only examining one or a few TCRs simultaneously. Recently, multiple computational efforts have sought to use machine learning techniques to predict pairing in a high‐throughput manner (93, 94, 95).
Clinical utility of understanding T‐cell phenotypes
A clear definition of the T‐cell phenotypes that accumulate within target tissues of patients with autoimmune rheumatic diseases has several practical implications for clinical use. Because of the diverse range of phenotypes that T cells can acquire, they are particularly informative to identifying pathways activated in a specific disease or specific patient. It is not sufficient to simply know that T cells are present; it is essential to determine what kinds of T cells are present and what their most prominent effector programs are because the treatment implications of finding a large IL‐17A+ population versus a large Tph cell population or a large granzyme B+ cytotoxic population will differ. Defining T‐cell subsets has become increasingly nuanced as our experimental resolution improves, yet a robust description of the effector functions expressed (or not expressed) by tissue‐infiltrating T cells should help to more efficiently select relevant therapeutic strategies. As one example, the discovery of a large Tph cell population raises the possibility that therapies specifically directed against this population may be effective in RA; one might predict that a Tph‐directed therapy would be less likely to succeed in PsA, in which fewer Tph cells are found.
In addition to helping to guide use of existing therapies and potentially reveal new therapeutic targets, understanding the phenotypes of T cells in the synovium will help with developing more specific biomarkers of disease activity. Once Tph cells were identified in RA synovium, it became feasible to track this cell population in the circulation, revealing an increased frequency of Tph cells in patients with active arthritis, even in early disease, and a decrease with effective therapy (7, 96). The ability to compare TCRs of T cells in blood and tissues allows for further validation that the T cells being tracked in the blood are likely to reflect the pathologic cell types in blood. The extent to which T‐cell phenotypes in blood can predict what is happening in the tissue requires further investigation. For example, it will be of major interest to determine whether circulating T‐cell phenotypes, particularly those that reflect tissue phenotypes, correlate with the patterns of immune infiltration in synovium in RA, such as lymphoid, myeloid, and fibroid pathotypes (97, 98).
Conclusions
Efforts to catalogue the T‐cell phenotypes within tissues in rheumatic diseases are revealing unappreciated aspects of T‐cell effector states in chronically inflamed tissues and are starting to provide benchmarks for the types of T‐cell phenotypes that are shared across diseases as well as those that are selectively enriched in certain diseases. We are optimistic that these features can be used to design new biomarkers to track specific T‐cell effector pathways and to more efficiently tailor therapy toward the relevant pathways active in specific diseases or in individual patients.
AUTHOR CONTRIBUTIONS
All authors drafted the article, revised it critically for important intellectual content, and approved the final version to be published.
Dr. Rao’s work was supported by NIH National Institute of Arthritis and Musculoskeletal and Skin Diseases grants K08‐AR‐072791, P30‐AR‐070253, and UH2‐AR‐067688, as well as grants from the Lupus Research Alliance, Burroughs Wellcome Fund Career Award for Medical Scientists, and Doris Duke Charitable Foundation Clinical Scientist Development Award. Dr. Elahee’s work was supported by NIH grant T32‐AR‐007530.
Yidan Gao, BS, BA, Garrett Dunlap, BS, Mehreen Elahee, MD, Deepak A. Rao, MD, PhD: Brigham and Women's Hospital and Harvard Medical School, Boston, Massachusetts.
Dr. Rao reports personal fees from Pfizer, Janssen, Merck, Scipher Medicine, GlaxoSmithKline, and Bristol‐Myers Squibb and grant support from Janssen and Bristol‐Myers Squibb, outside the submitted w ork. In addition, Dr. Rao is a coinventor on a patent submitted on T peripheral helper cells. No other disclosures relevant to this article were reported.
REFERENCES
- 1. Gutierrez‐Arcelus M, Rich SS, Raychaudhuri S. Autoimmune diseases ‐ connecting risk alleles with molecular traits of the immune system. Nat Rev Genet 2016;17:160–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Genovese MC, Becker JC, Schiff M, Luggen M, Sherrer Y, Kremer J, et al. Abatacept for rheumatoid arthritis refractory to tumor necrosis factor α inhibition. N Engl J Med 2005;353:1114–23. [DOI] [PubMed] [Google Scholar]
- 3. Kamali AN, Noorbakhsh SM, Hamedifar H, Jadidi‐Niaragh F, Yazdani R, Bautista JM, et al. A role for Th1‐like Th17 cells in the pathogenesis of inflammatory and autoimmune disorders. Mol Immunol 2019;105:1407–15. [DOI] [PubMed] [Google Scholar]
- 4. Sabat R, Grütz G, Warszawska K, Kirsch S, Witte E, Wolk K, et al. Biology of interleukin‐10. Cytokine Growth Factor Rev 2010;21:331–44. [DOI] [PubMed] [Google Scholar]
- 5. Spitzer MH, Nolan GP. Mass cytometry: single cells, many features. Cell 2016;165:780–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bocharnikov AV, Keegan J, Wacleche VS, Cao Y, Fonseka CY, Wang G, et al. PD‐1hiCXCR5: T peripheral helper cells promote B cell responses in lupus via MAF and IL‐21. JCI Insight 2019;4:e130062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature 2017;542:110–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Valihrach L, Androvic P, Kubista M. Platforms for single‐cell collection and analysis. Int J Mol Sci 2018;19:807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, et al. Fast, sensitive and accurate integration of single‐cell data with Harmony. Nat Methods 2019;16:1289–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single‐cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 2018;36:411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tran HT, Ang KS, Chevrier M, Zhang X, Lee NY, Goh M, et al. A benchmark of batch‐effect correction methods for single‐cell RNA sequencing data. Genome Biol 2020;21:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Arazi A, Rao DA, Berthier CC, Davidson A, Liu Y, Hoover PJ, et al. The immune cell landscape in kidneys of patients with lupus nephritis. Nat Immunol 2019;20:902–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Der E, Suryawanshi H, Morozov P, Kustagi M, Goilav B, Ranabothu S, et al. Tubular cell and keratinocyte single‐cell transcriptomics applied to lupus nephritis reveal type I IFN and fibrosis relevant pathways. Nat Immunol 2019;20:915–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Donlin LT, Rao DA, Wei K, Slowikowski K, McGeachy MJ, Turner JD, et al. Methods for high‐dimensional analysis of cells dissociated from cryopreserved synovial tissue. Arthritis Res Ther 2018;20:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rao DA, Arazi A, Wofsy D, Diamond B. Design and application of single‐cell RNA sequencing to study kidney immune cells in lupus nephritis. Nat Rev Nephrol 2020;16:238–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang F, Wei K, Slowikowski K, Fonseka CY, Rao DA, Kelly S, et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single‐cell transcriptomics and mass cytometry. Nat Immunol 2019;20:928–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hashimshony T, Senderovich N, Avital G, Klochendler A, de Leeuw Y , Anavy L, et al. CEL‐Seq2: sensitive highly‐multiplexed single‐cell RNA‐Seq. Genome Biol 2016;17:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Caielli S, Veiga DT, Balasubramanian P, Athale S, Domic B, Murat E, et al. A CD4+ T cell population expanded in lupus blood provides B cell help through interleukin‐10 and succinate. Nat Med 2019;25:75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liarski VM, Kaverina N, Chang A, Brandt D, Yanez D, Talasnik L, et al. Cell distance mapping identifies functional T follicular helper cells in inflamed human renal tissue. Sci Transl Med 2014;6:230ra46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nehar‐Belaid D, Hong S, Marches R, Chen G, Bolisetty M, Baisch J, et al. Mapping systemic lupus erythematosus heterogeneity at the single‐cell level. Nat Immunol 2020;21:1094–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Winchester R, Wiesendanger M, Zhang HZ, Steshenko V, Peterson K, Geraldino‐Pardilla L, et al. Immunologic characteristics of intrarenal T cells: trafficking of expanded CD8+ T cell β‐chain clonotypes in progressive lupus nephritis. Arthritis Rheum 2012;64:1589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wensink AC, Hack CE, Bovenschen N. Granzymes regulate proinflammatory cytokine responses. J Immunol 2015;194:491–7. [DOI] [PubMed] [Google Scholar]
- 23. Mackay LK, Minnich M, Kragten NA, Liao Y, Nota B, Seillet C, et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 2016;352:459–63. [DOI] [PubMed] [Google Scholar]
- 24. Bray NL, Pimentel H, Melsted P, Pachter L. Near‐optimal probabilistic RNA‐seq quantification. Nat Biotechnol 2016;34:525–7. [DOI] [PubMed] [Google Scholar]
- 25. Mueller SN, Gebhardt T, Carbone FR, Heath WR. Memory T cell subsets, migration patterns, and tissue residence. Annu Rev Immunol 2013;31:137–61. [DOI] [PubMed] [Google Scholar]
- 26. Rao DA. T cells that help B cells in chronically inflamed tissues. Front Immunol 2018;9:1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stephenson W, Donlin LT, Butler A, Rozo C, Bracken B, Rashidfarrokhi A, et al. Single‐cell RNA‐seq of rheumatoid arthritis synovial tissue using low‐cost microfluidic instrumentation. Nat Commun 2018;9:791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Penkava F, Velasco‐Herrera MD, Young MD, Yager N, Nwosu LN, Pratt AG, et al. Single‐cell sequencing reveals clonal expansions of pro‐inflammatory synovial CD8 T cells expressing tissue‐homing receptors in psoriatic arthritis. Nat Commun 2020;11:4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Benham H, Norris P, Goodall J, Wechalekar MD, FitzGerald O, Szentpetery A, et al. Th17 and Th22 cells in psoriatic arthritis and psoriasis. Arthritis Res Ther 2013;15:R136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Menon B, Gullick NJ, Walter GJ, Rajasekhar M, Garrood T, Evans HG, et al. Interleukin‐17+CD8+ T cells are enriched in the joints of patients with psoriatic arthritis and correlate with disease activity and joint damage progression. Arthritis Rheumatol 2014;66:1272–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Steel KJ, Srenathan U, Ridley M, Durham LE, Wu SY, Ryan SE, et al. Polyfunctional, proinflammatory, tissue‐resident memory phenotype and function of synovial interleukin‐17A+CD8+ T cells in psoriatic arthritis. Arthritis Rheumatol 2020;72:435–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fiocco U, Martini V, Accordi B, Caso F, Costa L, Oliviero F, et al. Transcriptional network profile on synovial fluid T cells in psoriatic arthritis. Clin Rheumatol 2015;34:1571–80. [DOI] [PubMed] [Google Scholar]
- 33. Fiocco U, Martini V, Accordi B, Caso F, Costa L, Oliviero F, et al. Ex vivo signaling protein mapping in T lymphocytes in the psoriatic arthritis joints. J Rheumatol Suppl 2015;93:48–52. [DOI] [PubMed] [Google Scholar]
- 34. Wade SM, Canavan M, McGarry T, Low C, Wade SC, Mullan RH, et al. Association of synovial tissue polyfunctional T‐cells with DAPSA in psoriatic arthritis. Ann Rheum Dis 2019;78:350–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Livshits G, Kalinkovich A. Hierarchical, imbalanced pro‐inflammatory cytokine networks govern the pathogenesis of chronic arthropathies. Osteoarthritis Cartilage 2018;26:7–17. [DOI] [PubMed] [Google Scholar]
- 36. Groom JR, Luster AD. CXCR3 in T cell function. Exp Cell Res 2011;317:620–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Diani M, Casciano F, Marongiu L, Longhi M, Altomare A, Pigatto PD, et al. Increased frequency of activated CD8+ T cell effectors in patients with psoriatic arthritis. Sci Rep 2019;9:10870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Patel DD, Zachariah JP, Whichard LP. CXCR3 and CCR5 ligands in rheumatoid arthritis synovium. Clin Immunol 2001;98:39–45. [DOI] [PubMed] [Google Scholar]
- 39. Lee J‐H, Kim B, Jin WJ, Kim H‐H, Ha H, Lee ZH. Pathogenic roles of CXCL10 signaling through CXCR3 and TLR4 in macrophages and T cells: relevance for arthritis. Arthritis Res Ther 2017;19:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Aldridge J, Ekwall AH, Mark L, Bergström B, Andersson K, Gjertsson I, et al. T helper cells in synovial fluid of patients with rheumatoid arthritis primarily have a Th1 and a CXCR3+Th2 phenotype. Arthritis Res Ther 2020;22:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Maschmeyer P, Heinz GA, Skopnik CM, Lutter L, Mazzoni A, Heinrich F, et al. Antigen‐driven PD‐1+TOX+BHLHE40+ and PD‐1+TOX+EOMES+ T lymphocytes regulate juvenile idiopathic arthritis in situ. Eur J Immunol 2021;51:915–29. [DOI] [PubMed] [Google Scholar]
- 42. Nistala K, Moncrieffe H, Newton KR, Varsani H, Hunter P, Wedderburn LR, et al. Interleukin‐17‐producing T cells are enriched in the joints of children with arthritis, but have a reciprocal relationship to regulatory T cell numbers. Arthritis Rheum 2008;58:875–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Copland A, Bending D. Foxp3 molecular dynamics in Treg in juvenile idiopathic arthritis. Front Immunol 2018;9:2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Duurland CL, Brown CC, O’Shaughnessy RF, Wedderburn LR. CD161+ Tconv and CD161+ Treg share a transcriptional and functional phenotype despite limited overlap in TCRβ repertoire. Front Immunol 2017;8:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Henderson LA, Hoyt KJ, Lee PY, Rao DA, Jonsson AH, Nguyen JP, et al. Th17 reprogramming of T cells in systemic juvenile idiopathic arthritis. JCI Insight 2020;5:e132508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Petrelli A, Mijnheer G, Hoytema van Konijnenburg DP, van der Wal MM , Giovannone B, Mocholi E, et al. PD‐1+CD8+ T cells are clonally expanding effectors in human chronic inflammation. J Clin Invest 2018;128:4669–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Skaug B, Khanna D, Swindell WR, Hinchcliff ME, Frech TM, Steen VD, et al. Global skin gene expression analysis of early diffuse cutaneous systemic sclerosis shows a prominent innate and adaptive inflammatory profile. Ann Rheum Dis 2020;79:379–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Assassi S, Swindell WR, Wu M, Tan FD, Khanna D, Furst DE, et al. Dissecting the heterogeneity of skin gene expression patterns in systemic sclerosis. Arthritis Rheumatol 2015;67:3016–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gaydosik AM, Tabib T, Domsic R, Khanna D, Lafyatis R, Fuschiotti P. Single‐cell transcriptome analysis identifies skin‐specific T‐cell responses in systemic sclerosis. Ann Rheum Dis. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chizzolini C, Boin F. The role of the acquired immune response in systemic sclerosis. Semin Immunopathol 2015;37:519–28. [DOI] [PubMed] [Google Scholar]
- 51. Meloni F, Solari N, Cavagna L, Morosini M, Montecucco CM, Fietta AM. Frequency of TH1, TH2 and TH17 producing T lymphocytes in bronchoalveolar lavage of patients with systemic sclerosis. Clin Exp Rheumatol 2009;27:765–72. [PubMed] [Google Scholar]
- 52. Mavilia C, Scaletti C, Romagnani P, Carossino AM, Pignone A, Emmi L, et al. Type 2 helper T‐cell predominance and high CD30 expression in systemic sclerosis. Am J Pathol 1997;151:1751–8. [PMC free article] [PubMed] [Google Scholar]
- 53. Salmon‐Ehr V, Serpier H, Nawrocki B, Gillery P, Clavel C, Kalis B, et al. Expression of interleukin‐4 in scleroderma skin specimens and scleroderma fibroblast cultures: potential role in fibrosis. Arch Dermatol 1996;132:802–6. [PubMed] [Google Scholar]
- 54. Scaletti C, Vultaggio A, Bonifacio S, Emmi L, Torricelli F, Maggi E, et al. Th2‐oriented profile of male offspring T cells present in women with systemic sclerosis and reactive with maternal major histocompatibility complex antigens. Arthritis Rheum 2002;46:445–50. [DOI] [PubMed] [Google Scholar]
- 55. Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol 2008;214:199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med 2011;208:1339–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wynn TA. Fibrotic disease and the TH1/TH2 paradigm. Nat Rev Immunol 2004;4:583–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Parel Y, Aurrand‐Lions M, Scheja A, Dayer JM, Roosnek E, Chizzolini C. Presence of CD4+CD8+ double‐positive T cells with very high interleukin‐4 production potential in lesional skin of patients with systemic sclerosis. Arthritis Rheum 2007;56:3459–67. [DOI] [PubMed] [Google Scholar]
- 59. Maehara T, Kaneko N, Perugino CA, Mattoo H, Kers J, Allard‐Chamard H, et al. Cytotoxic CD4+ T lymphocytes may induce endothelial cell apoptosis in systemic sclerosis. J Clin Invest 2020;130:2451–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hasegawa M, Fujimoto M, Matsushita T, Hamaguchi Y, Takehara K. Augmented ICOS expression in patients with early diffuse cutaneous systemic sclerosis. Rheumatology (Oxford) 2013;52:242–51. [DOI] [PubMed] [Google Scholar]
- 61. Taylor DK, Mittereder N, Kuta E, Delaney T, Burwell T, Dacosta K, et al. T follicular helper‐like cells contribute to skin fibrosis. Sci Transl Med 2018;10:eaaf5307. [DOI] [PubMed] [Google Scholar]
- 62. MacDonald KG, Dawson NAJ, Huang Q, Dunne JV, Levings MK, Broady R. Regulatory T cells produce profibrotic cytokines in the skin of patients with systemic sclerosis. J Allergy Clin Immunol 2015;135:946–55. [DOI] [PubMed] [Google Scholar]
- 63. Li G, Larregina AT, Domsic RT, Stolz DB, Medsger TA Jr, Lafyatis R, et al. Skin‐resident effector memory CD8+CD282212− T cells exhibit a profibrotic phenotype in patients with systemic sclerosis. J Invest Dermatol 2017;137:1042–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fuschiotti P, Larregina AT, Ho J, Feghali‐Bostwick C, Medsger TA Jr. Interleukin‐13‐producing CD8+ T cells mediate dermal fibrosis in patients with systemic sclerosis. Arthritis Rheum 2013;65:236–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Medsger TA Jr, Ivanco DE, Kardava L, Morel PA, Lucas MR, Fuschiotti P. GATA‐3 up‐regulation in CD8+ T cells as a biomarker of immune dysfunction in systemic sclerosis, resulting in excessive interleukin‐13 production. Arthritis Rheum 2011;63:1738–47. [DOI] [PubMed] [Google Scholar]
- 66. Fuschiotti P, Medsger TA Jr, Morel PA. Effector CD8+ T cells in systemic sclerosis patients produce abnormally high levels of interleukin‐13 associated with increased skin fibrosis. Arthritis Rheum 2009;60:1119–28. [DOI] [PubMed] [Google Scholar]
- 67. Valenzi E, Tabib T, Papazoglou A, Sembrat J, Trejo Bittar HE, Rojas M, et al. Disparate interferon signaling and shared aberrant basaloid cells in single‐cell profiling of idiopathic pulmonary fibrosis and systemic sclerosis‐associated interstitial lung disease. Front Immunol 2021;12:595811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Maehara T, Mattoo H, Mahajan VS, Murphy SJ, Yuen GJ, Ishiguro N, et al. The expansion in lymphoid organs of IL‐4+ BATF+ T follicular helper cells is linked to IgG4 class switching in vivo. Life Sci Alliance 2018;1:e201800050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chen Y, Lin W, Yang H, Wang M, Zhang P, Feng R, et al. Aberrant expansion and function of follicular helper T cell subsets in IgG4‐related disease. Arthritis Rheumatol 2018;70:1853–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kamekura R, Takano K, Yamamoto M, Kawata K, Shigehara K, Jitsukawa S, et al. Cutting edge: a critical role of lesional T follicular helper cells in the pathogenesis of IgG4‐related disease. J Immunol 2017;199:2624–9. [DOI] [PubMed] [Google Scholar]
- 71. Kamekura R, Takahashi H, Ichimiya S. New insights into IgG4‐related disease: emerging new CD4+ T‐cell subsets. Curr Opin Rheumatol 2019;31:9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Koyabu M, Uchida K, Miyoshi H, Sakaguchi Y, Fukui T, Ikeda H, et al. Analysis of regulatory T cells and IgG4‐positive plasma cells among patients of IgG4‐related sclerosing cholangitis and autoimmune liver diseases. J Gastroenterol 2010;45:732–41. [DOI] [PubMed] [Google Scholar]
- 73. Heeringa JJ, Karim AF, van Laar JA , Verdijk RM, Paridaens D, van Hagen PM , et al. Expansion of blood IgG4 + B, TH2, and regulatory T cells in patients with IgG4‐related disease. J Allergy Clin Immunol 2018;141:1831–43. [DOI] [PubMed] [Google Scholar]
- 74. Zen Y, Fujii T, Harada K, Kawano M, Yamada K, Takahira M, et al. Th2 and regulatory immune reactions are increased in immunoglobin G4‐related sclerosing pancreatitis and cholangitis. Hepatology 2007;45:1538–46. [DOI] [PubMed] [Google Scholar]
- 75. Mattoo H, Mahajan VS, Maehara T, Deshpande V, Della‐Torre E, Wallace ZS, et al. Clonal expansion of CD4(+) cytotoxic T lymphocytes in patients with IgG4‐related disease. J Allergy Clin Immunol 2016;138:825–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Perugino CA, Kaneko N, Maehara T, Mattoo H, Kers J, Allard‐Chamard H, et al. CD4+ and CD8+ cytotoxic T lymphocytes may induce mesenchymal cell apoptosis in IgG4‐related disease. J Allergy Clin Immunol 2021;147:368–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Green L, Weinberg JM, Menter A, Soung J, Lain E, Jacobson A. Clinical and molecular effects of interleukin‐17 pathway blockade in psoriasis. J Drugs Dermatol 2020;19:138–43. [DOI] [PubMed] [Google Scholar]
- 78. Han Q, Bagheri N, Bradshaw EM, Hafler DA, Lauffenburger DA, Love JC. Polyfunctional responses by human T cells result from sequential release of cytokines. Proc Natl Acad Sci U S A 2012;109:1607–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kleinewietfeld M, Starke M, Di Mitri D, Borsellino G, Battistini L, Rötzschke O, et al. CD49d provides access to “untouched” human Foxp3+ Treg free of contaminating effector cells. Blood 2009;113:827–36. [DOI] [PubMed] [Google Scholar]
- 80. Fonseka CY, Rao DA, Teslovich NC, Korsunsky I, Hannes SK, Slowikowski K, et al. Mixed‐effects association of single cells identifies an expanded effector CD4+ T cell subset in rheumatoid arthritis. Sci Transl Med 2018;10:eaaq0305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Goltsev Y, Samusik N, Kennedy‐Darling J, Bhate S, Hale M, Vazquez G, et al. Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell 2018;174:968–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Giesen C, Wang HA, Schapiro D, Zivanovic N, Jacobs A, Hattendorf B, et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat Methods 2014;11:417–22. [DOI] [PubMed] [Google Scholar]
- 83. Eng CL, Lawson M, Zhu Q, Dries R, Koulena N, Takei Y, et al. Transcriptome‐scale super‐resolved imaging in tissues by RNA seqFISH. Nature 2019;568:235–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Chen KH, Boettiger AN, Moffitt JR, Wang S, Zhuang X. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 2015;348:aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR, et al. Slide‐seq: a scalable technology for measuring genome‐wide expression at high spatial resolution. Science 2019;363:1463–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016;353:78–82. [DOI] [PubMed] [Google Scholar]
- 87. Vickovic S, Eraslan G, Salmén F, Klughammer J, Stenbeck L, Schapiro D, et al. High‐definition spatial transcriptomics for in situ tissue profiling. Nat Methods 2019;16:987–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Stoeckius M, Hafemeister C, Stephenson W, Houck‐Loomis B, Chattopadhyay PK, Swerdlow H, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods 2017;14:865–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Peterson VM, Zhang KX, Kumar N, Wong J, Li L, Wilson DC, et al. Multiplexed quantification of proteins and transcripts in single cells. Nat Biotechnol 2017;35:936–9. [DOI] [PubMed] [Google Scholar]
- 90. Satpathy AT, Saligrama N, Buenrostro JD, Wei Y, Wu B, Rubin AJ, et al. Transcript‐indexed ATAC‐seq for precision immune profiling. Nat Med 2018;24:580–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Birnbaum ME, Mendoza JL, Sethi DK, Dong S, Glanville J, Dobbins J, et al. Deconstructing the peptide‐MHC specificity of T cell recognition. Cell 2014;157:1073–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kula T, Dezfulian MH, Wang CI, Abdelfattah NS, Hartman ZC, Wucherpfennig KW, et al. T‐scan: a genome‐wide method for the systematic discovery of T cell epitopes. Cell 2019;178:1016–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Fischer DS, Wu Y, Schubert B, Theis FJ. Predicting antigen specificity of single T cells based on TCR CDR3 regions. Mol Syst Biol 2020;16:e9416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Gielis S, Moris P, Bittremieux W, De Neuter N, Ogunjimi B, Laukens K, et al. Detection of enriched T cell epitope specificity in full T cell receptor sequence repertoires. Front Immunol 2019;10:2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Jurtz VI, Jessen LE, Bentzen AK, Jespersen MC, Mahajan S, Via R, et al. NetTCR: sequence‐based prediction of TCR binding to peptide‐MHC complexes using convolutional neural networks. bioRxiv Preprint posted online October 3, 2018. 10.1101/433706. [DOI] [Google Scholar]
- 96. Fortea‐Gordo P, Nuño L, Villalba A, Peiteado D, Monjo I, Sánchez‐Mateos P, et al. Two populations of circulating PD‐1hiCD4 T cells with distinct B cell helping capacity are elevated in early rheumatoid arthritis. Rheumatology (Oxford) 2019;58:1662–73. [DOI] [PubMed] [Google Scholar]
- 97. Lewis MJ, Barnes MR, Blighe K, Goldmann K, Rana S, Hackney JA, et al. Molecular portraits of early rheumatoid arthritis identify clinical and treatment response phenotypes. Cell Rep 2019;28:2455–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Pitzalis C, Kelly S, Humby F. New learnings on the pathophysiology of RA from synovial biopsies. Curr Opin Rheumatol 2013;25:334–44. [DOI] [PubMed] [Google Scholar]