

Abstract
Tumor necrosis factor alpha (TNF) antagonists provide benefit to patients with inflammatory autoimmune disorders such as Crohn’s disease, rheumatoid arthritis, and ankylosing spondylitis. However, TNF antagonism unexplainably exacerbates CNS autoimmunity including multiple sclerosis (MS) and neuromyelitis optica (NMO). The underlying mechanisms remain enigmatic. We demonstrate that TNF receptor type 2 (TNFR2) deficiency results in female-biased spontaneous autoimmune CNS demyelination in myelin oligodendrocyte glycoprotein (MOG)-specific 2D2 T cell receptor transgenic mice. Disease in TNFR2−/− 2D2 mice associated with CNS infiltration of T and B cells as well as increased production of MOG-specific IL-17, IFN-γ, and IgG2b. Attenuated disease in TNF−/− 2D2 mice relative to TNFR2−/− 2D2 mice identified distinctive roles for TNFR1 and TNFR2. Oral antibiotic treatment eliminated spontaneous autoimmunity in TNFR2−/− 2D2 mice suggesting a role for gut microbiota. Illumina sequencing of fecal 16S rRNA identified a distinct microbiota profile in male TNFR2−/− 2D2 that associated with disease protection. Akkermansia muciniphila, Sutterella sp., Oscillospira sp., Bacteroides acidifaciens, and Anaeroplasma sp. were selectively more abundant in male TNFR2−/− 2D2 mice. In contrast, Bacteroides sp., Bacteroides uniformis, and Parabacteroides sp. were more abundant in affected female TNFR2−/− 2D2 mice suggesting a role in disease causation. Overall, TNFR2 blockade appears to disrupt commensal bacteria-host immune symbiosis to reveal autoimmune demyelination in genetically susceptible mice. Under this paradigm, microbes likely contribute to an individual’s response to anti-TNF therapy. This model provides a foundation for host immune-microbiota directed measures for the prevention and treatment of CNS-demyelinating autoimmune disorders.
Keywords: TNF, TNFR2, MS, EAE, NMO, IgG2b, Th-17, Foxp3, regulatory T cell, autoimmunity, commensal, gut microbiome, Akkermansia muciniphila, Sutterella, microbiome
Introduction
Tumor necrosis factor alpha (TNF) antagonists provide significant benefit in the treatment of autoimmune inflammatory disorders such as Crohn’s disease, rheumatoid arthritis, and ankylosing spondylitis. In contrast, despite a correlation between the level of TNF and disease severity (1–3), anti-TNF agents paradoxically exacerbate CNS demyelinating autoimmune disorders, including optic neuritis (ON), multiple sclerosis (MS), neuromyelitis optica (NMO), Guillain-Barré syndrome (GBS), and transverse myelitis (4, 5). Continuation of anti-TNF therapy after the development of neurological symptoms is associated with permanent disease, while discontinuation of the therapy usually results in complete or partial recovery from neurological deficits. The etiology of CNS demyelinating inflammatory disorders remains poorly understood. However, in accordance with clinical observations, anti-TNF therapy is currently contraindicated in patients with a pre-existing or family history of CNS demyelinating inflammation.
Optic neuritis, an acute demyelinating disease of the optic nerve, can rapidly lead to blindness within days and is often one of the first symptoms of chronic incurable CNS demyelinating autoimmune diseases including MS and NMO. MS and NMO each manifests as a heterogeneous relapsing-remitting neurodegenerative disorder in which periods of demyelination, in response to migration of activated lymphocytes across the blood-brain barrier and attack on oligodendrocytes and neuronal processes, are followed by periods of recovery and repair of the damaged myelin (6, 7). MS affects the brain, optic nerves, and spinal cord. In contrast, NMO characteristically targets optic nerves and the spinal cord. Both diseases typically present with varying degrees of sensory motor disturbances, bladder-bowel dysfunction, and visual loss. The development of MS and experimental autoimmune encephalomyelitis (EAE), an animal model of MS, has been mostly attributed to the activation of autoreactive CD4+ T cells secreting IFN-γ (Th1 cells) and IL-17 (Th17 cells) (8, 9). Although their role is poorly understood, B cells have also been implicated. Current evidence suggests that B cells are not required for myelin oligodendrocyte glycoprotein (MOG) 35–55-induced EAE (10), but are required for EAE that is induced by human, but not mouse or rat, MOG protein (11, 12). In contrast to EAE and MS, complement-activating autoantibodies against aquaporin 4 (AQP4), the predominant CNS water channel, have been implicated in the induction of inflammatory demyelination in humans with NMO (13). More recently, AQP4-specific T cells exhibiting a Th17 bias and displaying cross-reactivity to the gut commensal bacteria Clostridium perfringens have also been implicated (14, 15).
The underlying mechanism(s) for the unpredictable exacerbation of disease in response to TNF blockade remains enigmatic. However, it is now appreciated that TNF exists in two bioactive forms and signals through two distinct receptors (16, 17). TNF receptor type 1 (TNFR1) is ubiquitously expressed, activated by soluble TNF, and promotes apoptosis and inflammation. In contrast, TNF receptor type 2 (TNFR2) expression is mostly restricted to endothelial, hematopoietic, microglial, and some neuronal cells, is selectively activated in response to transmembrane TNF, and promotes cell survival and proliferation, including expansion of CD4+ Foxp3+ T regulatory cells (Tregs) (5, 18–20). The gene encoding TNFR1 has recently been identified as an MS risk allele (21), thus suggesting that the onset of autoimmune disease following non-selective TNF blockade may be a consequence TNFR signaling diversity. Consistent with this hypothesis, TNF plays dual roles during autoimmune demyelination, namely exacerbation of inflammation at the onset of disease and facilitation of the resolution of inflammation and myelin repair during ongoing disease (18, 22). In agreement, selective pharmacological inhibition of soluble TNF prevents transcriptional changes associated with the onset of CNS inflammatory demyelination and protects against MOG-induced EAE (23). Therefore, it is conceivable that selective blockade of TNFR1 may minimize undesirable side effects from anti-TNF therapy.
EAE rarely develops spontaneously in mice, but can be induced by injection of myelin antigens or by adoptive transfer of CD4+ T cells isolated from mice primed with myelin antigens. Disrupted homeostasis between encephalitogenic lymphocytes and Tregs plays a key role in the disease pathogenesis of both MS and EAE (24–26). Given that Tregs regulate MOG35–55-specific EAE and TNFR2 modulates Treg function, a genetic loss-of-function approach was used to study the influence of selective TNFR2 blockade on the control of autoreactive T cells. Accordingly, C57BL/6J TNFR2−/− mice were cross-bred with 2D2 Foxp3gfp reporter mice to generate 2D2 TNFR2−/− Foxp3gfp reporter mice (TNFR2−/− 2D2) that recognize the CD4+ T cell epitope, MOG35–55, presented by MHC class II (I-Ab). Unexpectedly, TNFR2−/− 2D2 mice demonstrated a breakdown in functional tolerance of autoreactive T and B cells with development of highly penetrant fulminant spontaneous CNS demyelinating autoimmunity with a pronounced sex-bias. While highly implicated in the pathogenesis of NMO, B cells have only recently received attention as a major contributor to EAE, and the involvement of B cells in the induction of spontaneous EAE is rare. Inflammation, demyelination, and neuronal loss in the optic nerves and spinal cord of the diseased female TNFR2−/− 2D2 mice are also consistent with NMO pathogenesis. We further correlate spontaneous demyelinating autoimmunity in female TNFR2−/− 2D2 mice with increased IL-17, IFN-γ, and IgG2b production and sex- and genotype-dependent changes in the composition of the gut microbiota.
Emerging data strongly suggest an interaction between the gastrointestinal system and CNS disorders. The composition of the gut microbiota varies between monozygotic twins (27) and influences the development of the immune system (28). The composition of the gut commensal microbiota has recently been identified as an environmental factor that can trigger CNS demyelinating autoimmune disease (14, 29, 30). Increased abundance of Bacteroides sp., Bacteroides uniformis, and Parabacteroides sp.in female TNFR2−/− 2D2 mice and increased abundance of Akkermansia muciniphila, Oscillospira sp., Bacteroides acidifaciens, Anaeroplasma sp. and Sutterella sp. in male TNFR2−/− 2D2 mice suggest possible microbial influences on disease causation and protection, respectively. Increased IL-17, IFN-γ, and IgG2b are consistent with autoreactive cell and humoral immune responses in the female TNFR2−/− 2D2 mice. Overall, these data suggest that TNFR2 potentiates autoimmune reactions that are driven by cross-reactivity between commensal microbiota and autoantigens in female mice. Understanding whether selective TNF blockade influences the composition of the microbiota in the human gut may produce a paradigm shift for anti-TNF therapy in autoimmune diseases. This model provides a foundation for host immune-gut microbiota directed measures for the prevention and treatment of CNS demyelinating autoimmune disorders.
Materials and Methods
Mice
TNF and closely linked lymphotoxin (LT)-α and LT-β genes are located within the major histocompatibility complex, thus all mice used in these studies were obtained on the C57BL/6J (H-2b) background and crossbred to avoid genetic heterogeneity introduced through backcrossing (31). Homozygous TNFR2−/− C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and bred with Foxp3gfp 2D2 TCR transgenic reporter mice to generate MOG35–55-specific TNFR2-deficient Foxp3gfp reporter mice (henceforth referred to as 2D2 TNFR2−/− mice). The 2D2 TCR mice were generated from a clone (2D2) that expresses a TCR combination of Vα3.2 and Vβ11 with specificity for the MOG35–55 peptide in the context of MHC class II (I-Ab) (32, 33). The GFP expressed in these mice is not fused with Foxp3, but is concomitantly expressed from a bicistronic mRNA, thus leaving Foxp3 intact and functional. TNF−/− C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and bred with Foxp3gfp 2D2 mice to generate homozygous Foxp3gfp 2D2 TNF−/− reporter (2D2 TNF−/−) mice. Timed-pregnancy surrogate CD-1 female mice were purchased from Charles River Laboratories (Portage, MI). These mice were housed in the Area P10 barrier at Charles River prior to purchase. After arrival at MU, they were housed in a separate limited-access quarantine room. All animals were bred and housed under specific pathogen-free conditions in MU facilities that are accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. All experimental procedures using animals were approved by the MU Institutional Animal Care and Use Committee and were performed in accordance with the Guide for the Use and Care of Laboratory Animals.
Clinical Scoring of CNS Demyelinating Autoimmune Disease
Clinical paralysis was scored using the following scale: 0, clinically normal; 1, decreased tail tone or weak tail only; 2, hind limb weakness (paraparesis); 3, hind limb paralysis (paraplegia) and/or urinary incontinence; 4,weakness of front limbs with paraparesis or paraplegia (quadriparesis) and/or atonic bladder; and 5, paralysis of all limbs (quadriplegia)
Oral Antibiotic Administration
Trimethoprim/sulfamethoxazole (TMS) (MWI, Chicago, IL) was added to drinking bottles at a targeted dosage of 80 mg/kg/day. Water bottles were changed weekly. TMS has bactericidal activity against a broad spectrum of Gram-positive and Gram-negative bacteria, including: Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus pneumonia, Staphylococcus aureus, Staphylococcus epidermidis, Listeria monocytogenes, Nocardia asteroides, Mycobacterium fortuitum, Escherichia coli, Shigella dysenteriae, Salmonella enterica, Klebsiella pneumoniae, Enterobacter cloacae, Serratia marcescens, Proteus mirabilis, Stenotrophomonas maltophilia, Haemophilus influenzae, Pasteurella multocida, Bordetella pertussis, Brucella melitensis, Neisseria gonorrhoeae, and Neisseria meningitidis. Sulfamethoxazole inhibits dihydrofolic acid synthesis, trimethoprim inhibits thymidine and DNA synthesis, and together, these two agents act synergistically to inhibit folic acid synthesis.
Cross-foster rederivation
Before, during, and after rederivation, barrier mice were housed in autoclaved microisolator cages with autoclaved compressed pelleted paper bedding and nestlets with ad libitum access to irradiated diet (breeder diet, LabDiet® 5058; stock diet, LabDiet®5053) and acidified, autoclaved water, under a 12:12 light/dark cycle. Cross-fostering was carried out in a separate limited-access quarantine room. After rederivation, mice were housed on ventilated racks (Thoren, Hazleton, PA). All cages were changed in Class II Type A biosafety cabinets. Dedicated staff were assigned to barrier and quarantine rooms. Four TNFR2+/− 2D2 C57BL/6 × TNFR2+/− 2D2 C57BL/6 breeding pairs were moved from barrier housing and mated in quarantine. Within 12 h post-delivery, pups were removed from donor cages and placed with a CD-1 surrogate recipient litter that was born within 2 days of the transfer. Within one week of cross-fostering, the recipient’s pups were removed so that the total number of pups in her cage was less than ten. At weaning, cross-fostered pups were tested for Pasteurella pneumotropica and Helicobacter spp. and each surrogate dam was submitted for a comprehensive necropsy profile. Two weeks after weaning, a second Helicobacter PCR test was performed on all cross-fostered pups before they were transferred to restricted-access barrier housing. In the barriers, sentinel testing was routinely performed to confirm that colonies remained free of opportunistic pathogens, including Bordetella bronchiseptica, cilia-associated respiratory (CAR) bacillus, Citrobacter rodentium, Clostridium piliforme, Corynebacterium bovis, Corynebacterium kutscheri, Helicobacter spp., Mycoplasma spp., Pasteurella pneumotropica, Pneumocystis carinii, Salmonella spp., Streptobacillus moniliformis, Streptococcus pneumoniae; adventitious viruses including H1, Hantaan, KRV, LCMV, MAD1, MNV, PVM, RCV/SDAV, REO3, RMV, RPV, RTV, and Sendai viruses; intestinal protozoa including Spironucleus muris, Giardia muris, Entamoeba muris, trichomonads, and other large intestinal flagellates and amoebae; intestinal parasites including pinworms and tapeworms; and external parasites including all species of lice and mites. All testing, including sentinels, was performed by Charles Rivers and IDEXX BioResearch (Columbia, MO).
Isolation of CNS mononuclear cells
Mice were euthanized by inhaled carbon dioxide and perfused with cold PBS. Brains and spinal cords were minced and incubated with 0.2 units Liberase R1 (Roche, Basel, Switzerland) and 50 μg/ml DNase I (Invitrogen™/Life Technologies, Grand Island, NY). Digested pieces were filtered through 40 μm SEFAR NITEX® (Small Parts, Inc., Logansport, IN) to yield single cell suspensions. Mononuclear cells were isolated by a discontinuous Percoll (GE Healthcare Life Sciences, Pittsburgh, PA) gradient (34).
Flow Cytometry
Cells were incubated with anti-CD16/32 prior to staining in order to prevent nonspecific Ab capture by FcRs. Cells were then stained directly with conjugated antibodies in FACS buffer (PBS containing 1% FCS, 0.05% EDTA, and 0.02% sodium azide). 7-amino actinomycin D (7-AAD) was used to exclude dead cells for data analyses. FACSCalibur™ (BD Biosciences) and FlowJo v8.8.7 (FlowJo Ashland, OR) software were used.
Cytokine and Cytokine Receptor Determinations
To determine IFN-γ and IL-17 secretion, cells isolated from CNS draining lymph nodes were adjusted to 5 × 106 cells (c)/ml in RPMI 1640 medium, containing 8.6% FCS, 4 mM glutamine, 1 mM sodium pyruvate, 0.1 mM MEM non-essential amino acids, 200 μg/ml penicillin, 200 μg/ml streptomycin, 25 μg/ml gentamicin, and 50 μM 2-ME and stimulated with MOG35–55 (MEVGWYRSPFSRVVHLYRNGK) (Ana Spec, Fremont, CA) in Costar® 96-well round-bottom culture plates (Thermo Fisher Scientific Inc., Waltham, MA) for 96 h. Cell-free supernatants were collected and cytokines were quantified by ELISA using Ready-SET-Go!® ELISA kits (eBioscience, San Diego, CA). To determine TNF, TNFR1, and TNFR2 concentrations, serum samples were collected and quantified using eBioscience Ready-SET-Go!® ELISA kits (TNF and TNFR2) or the mouse TNFR1 DuoSet® ELISA kit (R&D Systems, Minneapolis, MN). All cell-free culture supernatants and serum samples were stored at −80°C until use. The absorbance (450 nm) minus background (570 nm) of the colorimetric reactions were quantified using a Synergy™ 4 spectrophotometry microplate reader (BioTek, Winooski, VT). The lower limit of sensitivity of the TNF assay was 8 pg/ml. The recognition of TNF by the Ready-SET-Go!® assay is not affected by 1000-fold excess soluble TNFR1 or TNFR2.
Detection of MOG-specific Abs by ELISA
MOG35–55 (7 μg/ml) was coated onto 96-well plates and incubated overnight at 4°C. The plates were washed and saturated with PBS-3% BSA. Sera (1:100 dilution) was added and incubated overnight at 4°C. After extensive washes, bound Ig was detected by ELISA, consisting of biotinylated isotype-specific anti-mouse Ig and a HRP complex (eBioscience) with 3,3′,5,5′-tetramethylbenzidine (TMB) (eBioscience) as a color substrate (measured at 450 nm). Control serum was from a C57BL/6 mouse immunized with MOG35–55 plus adjuvant for 10 d. Rat anti-mouse IgM (II/41), IgG1 (A85-3), IgG2a (R11-89), and IgG2b (R9-91) capture antibodies and rat anti-mouse IgM (R6-60.2), IgG1 (A85-1), IgG2a (R19-15), and IgG2b (R12-3) detection antibodies were purchased from BD Pharmingen. The absorbance (450 nm) minus background (570 nm) of the colorimetric reactions were quantified using a BioTek Synergy™ 4 spectrophotometry microplate reader.
Necropsy and sample collection
Mice were euthanized by an inhaled overdose of carbon dioxide. Immediately after death, the thoracic cavity was opened and the heart located. A butterfly catheter was placed in the heart and mice were perfused with saline until blood draining from incised jugular veins cleared. Mice were then perfused with 4% paraformaldehyde. The brain, spinal column (containing the spinal cord), spleen, lymph nodes (cervical, inguinal, axillary and mesenteric), pancreas, liver and kidneys were removed and immersion-fixed in paraformaldehyde for 48–72 h. The spinal column was decalcified in formic acid in a sodium citrate buffer for 48–72 h. Sections of spleen, lymph nodes, pancreas, liver and kidneys were paraffin-embedded and 7 μm-thick, hematoxylin and eosin-stained sections were prepared for histologic examination. Brains were sectioned into four coronal sections and the decalcified spinal cord was sectioned into 14 sections. These sections were paraffin-embedded and slides were prepared and stained with hematoxylin and eosin. To further characterize lesions, select sections were also stained with Luxol® fast blue (myelin) and Bielschowsky (axon) stains and probed, by immunohistochemistry, for CD3-positive (T cells), CD45R (B220)-positive (B cells), and CD68-positive cells (macrophages).
Immunohistochemistry
Five-μm thick sections were mounted onto ProbeOn™ Plus microscope slides (Fisher Scientific Inc., Pittsburgh, PA). Sections were de-waxed in xylene, rehydrated through graded concentrations of ethanol, and rinsed in distilled water. Antigen retrieval was performed by immersing sections in heated 10 mM citrate buffer (pH 6.0) for 30 min and then allowed to cool to room temperature. Slides were treated with 3% hydrogen peroxide (to inactivate endogenous peroxidase activity), and rinsed prior to incubation in blocking buffer with 5% BSA for 20 min to minimize non-specific antibody binding. Sections were then incubated for 60 min at room temperature with each of the following antibodies: anti-CD3 (rabbit polyclonal anti-CD3 at a 1:200 dilution [clone G7; A0452], DAKO, Carpenteria, CA), anti-CD45R (rat monoclonal anti-CD45R/B220 at a 1:500 dilution [RA3-6B2; RM6200], Invitrogen™/Life Technologies, Grand Island, NY) and anti-CD68 (rat monoclonal anti-CD68 at a 1:200 dilution [FA-11; MCA1957], AbD Serotec, Oxford, UK). Sections of mouse spleen were similarly probed with all three antibodies for positive control samples while sections of experimental tissue were incubated with normal rabbit and rat antibodies as negative controls. Sections were rinsed and then incubated for 30 min with one of two detection protocols. Sections probed with anti-CD3 were incubated with a HRP–labeled polymer conjugated to anti-rabbit antibodies. Sections probed with anti-CD45R and anti-CD68 were incubated with a biotinylated rabbit anti-rat antibody (cat. # E0464 at 1:200 dilution; DAKO), rinsed, and incubated with HRP-labeled streptavidin (DAKO) for 30 min. Bound antibodies were visualized following incubation with 3,3’-diaminobenzidine solution (0.05% with 0.015% H2O2 in PBS; DAKO) for 3–5 min. Sections were counterstained with Meyer’s hematoxylin, dehydrated, and cover-slipped for microscopic examination.
DNA extraction
Two fresh fecal pellets were collected from each mouse directly into a 2 ml round-bottom tube containing lysis buffer adapted from (35) and a 0.5 cm diameter stainless steel bead and stored at −80°C. Following mechanical disruption using a TissueLyser II (Qiagen, Venlo, Netherlands), tubes were incubated at 70°C for 15 min, and then centrifuged at 16,000 × g for 5 min at room temperature. The supernatant was transferred to a clean 1.5 ml Eppendorf tube and mixed with ammonium acetate (10 M, 200 μl), incubated on ice for 5 min, and then centrifuged as above. Supernatant was mixed with one volume of chilled isopropanol and incubated at −20°C, while mixing, for 30 min, and then centrifuged at 16000 × g at 4°C for 15 min. The supernatant was aspirated and the DNA pellet was washed three times with 70% ethanol and resuspended in Tris-EDTA (150 μl) with Proteinase K (20 mg/ml, 15 μl) and Buffer AL (200 μl, DNeasy Blood and Tissue kit, Qiagen). Following incubation at 70°C for 10 min, 100% ethanol (200 μl) was added and the contents were transferred to a spin column (DNeasy kit) for DNA isolation according to the manufacturer’s instructions. DNA was eluted in Tris HCl (10 mM, 100 μl). The Quant-iT™ BR dsDNA reagent kit (Qubit, Life Technologies, Carlsbad, CA) and Nanodrop™ (Thermo Fisher Scientific, Waltham, MA) were used to obtain the quantity and purity, respectively.
16S rRNA library preparation and sequencing
Fecal DNA was processed at the University of Missouri DNA Core Facility. Bacterial 16S rDNA amplicons were constructed via amplification of the V4 hypervariable region of the 16S rDNA gene using universal primers (U515F/806R) previously designed against the V4 region, flanked by Illumina standard adapter sequences (36, 37); sequences are available at probeBase (38). A single forward primer and reverse primers with a unique 12-base index were used in all reactions. PCR reactions (50 μl) contained 100 ng of genomic DNA, forward and reverse primers (0.2 μM each), dNTPs (200 μM each), and Phusion High-Fidelity DNA Polymerase (1U). Thermal cycling parameters were 98°C for 3 min; 25 cycles of 98°C for 15 sec, 50°C for 30 sec, and 72°C for 30 sec; followed by 72°C for 7 min. Amplified product (5 μl) from each reaction was combined and thoroughly mixed; pooled amplicons were purified by addition of Axygen® AxyPrep™ MagPCR clean-up beads to an equal volume of 50 μl of amplicons and incubated for 15 min at room temperature. Products were washed with 80% ethanol, air dried, resuspended in 32.5 μl EB Buffer (Qiagen) for 2 min at room temperature, and placed on a magnetic stand for an additional 5 min. The final amplicon pool was evaluated using the Fragment Analyzer™ automated electrophoresis system (Advanced Analytical, Ames, IA), quantified with Qubit flourometer using the Quant-iT™ HS dsDNA reagent kit (Invitrogen™), and diluted according to MiSeq specifications (Illumina, San Diego, CA).
Informatics analysis
Assembly, binning, and annotation of DNA sequences was performed at the University of Missouri Informatics Research Core Facility. Briefly, contiguous sequences of DNA were assembled using FLASH software (39), and contigs were culled if found to be short after trimming for a base quality less than 31. Quantitative Insights into Microbial Ecology (QIIME) software v1.7 (40) was used to perform de novo and reference-based chimera detection and removal, and remaining contigs were assigned to operational taxonomic units (OTUs) using a criterion of 97% nucleotide identity. Taxonomy was assigned to selected OTUs using BLAST (41) against the Greengenes database (42) of 16S rRNA sequences and taxonomy. Samples yielding fewer than 10,000 sequences were removed from all analyses based on QIIME-generated rarefaction curves. Principal component analyses and loading plots were generated using a non-linear iterative partial least square (NIPALS) algorithm, implemented in a Macro-enabled Excel worksheet kindly provided by Hiroshi Tsugawa of the Riken Institute (Wako, Japan). Mean Chao1 indices were calculated using QIIME-generated rarefaction data, i.e., iterative subsampling of sequence data at the maximum coverage attained by all samples. All microbiome sequence data were uploaded to the National Center for Biotechnology Information (NCBI) Sequence Read Achive (SRA) database [http://www.ncbi.nlm.nih.gov/Traces/sra] under accession number PRJNA289572. For statistical analyses, a two-way analysis of variance was used to test for differences between groups in the relative abundance of taxa, overall richness, and the alpha diversity (Chao1) index, with sex and TCR (2D2 TCR+ versus 2D2 TCR−) as independent variables.
Statistical Analyses
All error bars represent mean ± SEM. For comparisons in the relative abundance of individual taxa, richness, and alpha-diversity and identification of interactions between variables, two-way analysis of variance (ANOVA) was performed using SigmaPlot 12.3 (Systat Software, Inc., San Jose, CA) with sex and TCR as independent variables. Elsewhere, means were compared using either the two-tailed unpaired Student t test for single comparisons or ANOVA followed by Student-Newman-Keuls, or in some cases Bonferroni’s, post-test correction for multiple group comparisons using GraphPad InStat v.3 (GraphPad Software, Inc., La Jolla, CA). For all comparisons, p < 0.05 was considered to be significant. *p < 0.05, **p < 0.01, and ***p < 0.001.
Results
Concentration- and time-dependent induction of TNFR2 by Foxp3gfp T regulatory cells (Tregs) and CD4+T effector cells (Teffs) in response to MOG35–55.
Previously, we determined that CD4+ T cell intrinsic TNFR2 promotes Il2 expression (43). Because IL-2 is required for the expansion and function of Tregs and impairs Th17 differentiation, we hypothesized that a loss of TNFR2 signaling may be sufficient to break functional tolerance of autoreactive T cells. MOG35–55 is a potent encephalitogenic peptide in C57BL/6 mice. Tregs have been implicated in the pathogenesis of MS and suppress MOG35–55-induced EAE. Because TNF blockade can unexplainably augment MS, we selected the MOG35–55-specific 2D2 TCR Tg mouse model to test our hypothesis. We first validated that TNFR2 expression was indeed upregulated by MOG35–55-specific CD4+ T cells in response to cognate antigen (Ag). For these experiments, T cells from 2D2 Foxp3gfp reporter mice were stimulated with increasing concentrations of MOG35–55 for 66 h and cell surface TNFR2 expression was quantified by flow cytometry (Fig. 1A). As expected, prior to activation, TNFR2 was constitutively expressed on the surface of Foxp3gfp Tregs but not CD4+ Teffs. Following MOG35–55 activation, TNFR2 expression was upregulated by both Tregs and Teffs in an Ag concentration- (Fig. 1A) and time- (Fig. 1B) dependent manner. The level of TNFR2 expression was elevated on Tregs obtained from lymph nodes (LN) in comparison to splenic Tregs (Fig. 1A and 1B). TNFR2 is readily cleaved from the cell surface by matrix metalloproteases including TNF cleavage enzyme (TACE, also called ADAM17), therefore we expanded our studies to ask whether decreased soluble TNFR2 associated with increased cell surface TNFR2 expression by Tregs in comparison to Teffs. For these experiments, LN and splenic Teffs and Foxp3gfp Tregs were FACS-purified (>95% purity) and stimulated separately with plate-bound anti-TCRβ plus soluble anti-CD28. At times thereafter, cells were harvested and processed for quantification of membrane-bound TNFR2 by flow cytometry. Analyses of cell-free culture supernatants revealed greater production of soluble TNFR2 by Foxp3gfp Tregs in comparison to Teffs (Fig. 1C) (p < 0.001). In contrast to unfractionated splenic and LN cells (Fig. 1A and 1B), TNFR2 expression did not differ between purified LN and splenic Foxp3gfp Tregs (Fig. 1C and 1D). It remains to be determined whether enhanced TACE activity augments TNFR2 shedding in spleens compared to lymph nodes. Collectively, these results show MOG35–55-specific induction of TNFR2 expression by Foxp3gfp Tregs and CD4+ Teffs. Further, impaired shedding of membrane-bound TNFR2 does not contribute to elevated cell surface TNFR2 expression on Foxp3gfp Tregs compared to CD4+ Teffs.
Figure 1. Concentration- and time-dependent induction of TNFR2 by CD4+ Foxp3+ Tregs versus CD4+ Teffs in response to MOG35–55 stimulation.
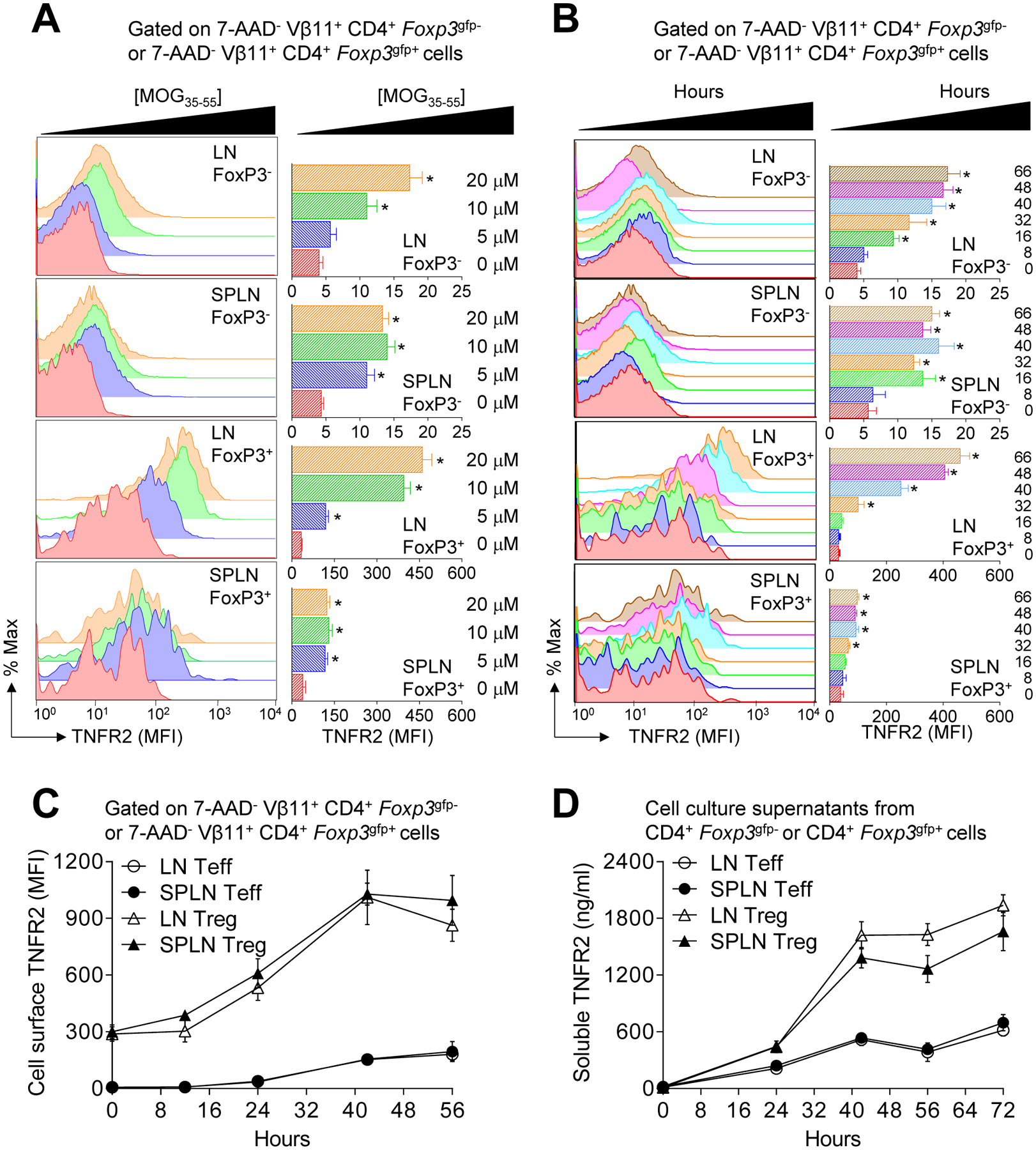
(A) Flow cytometric analysis of concentration-dependent induction of cell surface TNFR2 expression on 7-AAD− CD4+ Foxp3gfp+ Tregs and 7-AAD− CD4+ Foxp3gfp- Teffs from 2D2 Foxp3gfp reporter mice. Total lymph nodes (LN) or splenic cells isolated from three adult (6–12 wk of age) female mice were pooled and stimulated (2 × 106 c/ml) with 0, 5, 10, or 20 μM MOG35–55 peptide for 66 h. Cells were then stained for CD4, 7-AAD, and TNFR2; then 7-AAD− CD4+ Foxp3gfp+ and 7-AAD− CD4+ Foxp3gfp- cells were gated to quantify surface TNFR2 expression. Representative histograms (left) and mean MFI ± SEM (n = 3 independent experiments) (right) of TNFR2 expression are shown. (B) Flow cytometric analysis of time-dependent induction of cell surface TNFR2 expression on 7-AAD− CD4+ Foxp3gfp+ Tregs and 7-AAD− CD4+ Foxp3gfp- Teffs. Total LN or splenic cells isolated from three mice were pooled and stimulated (2 × 106 c/ml) with 10 μM MOG35–55 for 0, 8, 16, 32, 40, 48, and 66 h. Cells were then stained for CD4, 7-AAD, and TNFR2; then 7-AAD− CD4+ Foxp3gfp+ and 7-AAD− CD4+ Foxp3gfp- cells were gated to quantify surface TNFR2 expression. A representative histogram of each time-point for each population of gated-cells is shown (left). The TNFR2 MFI (mean ± SEM) of three independent experiments are shown (right). (C) Flow cytometric analysis of time-dependent induction of cell surface TNFR2 expression on FACS purified CD4+ Foxp3gfp+ Tregs and CD4+ Foxp3gfp- Teffs. Total LN or splenic cells were isolated from three mice, FACS-purified (purity >95%), and then stimulated (2 × 106 c/ml) with 10 μg/ml plate-bound anti-TCRβ (H57) and soluble anti-CD28 for 0, 12, 24, 40, and 56 h. Cells were collected, stained for CD4 and TNFR2, and 7-AAD− CD4+ Foxp3gfp+ and 7-AAD− CD4+ Foxp3gfp- cells were gated for analyses. The TNFR2 MFI (mean ± SEM) from three independent experiments is shown. (D) Time-dependent secretion of soluble TNFR2 by FACS purified CD4+ Foxp3gfp+ Tregs and CD4+ Foxp3gfp- Teffs. Soluble TNFR2 was quantified by ELISA. Secreted TNFR2 (ng/ml; mean ± SEM) from three independent experiments is shown.
Female, but not male, MOG35–55-specific TNFR2−/− 2D2 mice develop fulminant spontaneous inflammatory CNS demyelination
Previous studies have demonstrated attenuated or exacerbated MOG-induced EAE with TNFR1 or TNFR2 deficiency, respectively (44–46). Here, we use a genetic loss-of-function approach to determine whether selective ablation of TNFR2 signaling is sufficient to augment MOG35–55-specific CD4+ T cell autoreactivity. TNFR2−/− C57BL/6J mice were crossbred with MOG35–55-specific Foxp3gfp 2D2 reporter mice to generate TNFR2−/− 2D2 Foxp3gfp mice (henceforth referred to as TNFR2−/− 2D2) with the expectation of exacerbated disease upon active induction of EAE.
Unexpectedly, we observed spontaneous disease development in most female, but few male, TNFR2−/− 2D2 mice. Following this initial observation, mice were monitored, beginning at 14 days of age, for clinical signs of spontaneous disease development. A striking difference between female and male TNFR2−/− 2D2 mice was observed. At birth, female and male TNFR2−/− 2D2 mice were indistinguishable from TNFR2+/+ 2D2 and TNFR2−/− 2D2 littermates (Fig. 2A). However, 92% (59/64) of the female TNFR2−/− 2D2 mice eventually developed severe, non-remitting paralyses between 29–79 days of age (Figs. 2B and 2C, Table I). It is not clear why disease developed earlier in some female mice than others, but a difference in the frequency of MOG35–55-specific CD4+ T cells or 2D2 TCR expression by individual CD4+ T cells are possible contributing factors. Regardless of the age of the mouse at disease onset, the severity of the disease in affected female mice rapidly progressed to hind- and front-limb paralyses, without remission, and mice were euthanized once they reached this stage. In contrast, only a few male TNFR2−/− 2D2 littermates developed clinical signs (Fig. 2C). Except for a delay in disease onset and decreased disease severity, as determined by the mean peak score, in the few male TNFR2−/− 2D2 mice that did exhibit clinical signs (Table I), the disease was similar to female TNFR2−/− 2D2 littermates. The incidence of spontaneous disease in female TNFR2+/− 2D2 littermates (7.5%; n = 40) did not differ from TNFR2+/+ 2D2 controls (6.8%; n = 73) showing a lack of a gene dosage effect. Collectively, these results demonstrate that genetic ablation of TNFR2 predisposes female, but not male, 2D2 mice to develop a greater incidence of spontaneous disease.
FIGURE 2. Female 2D2 TNFR2−/− mice develop fulminant spontaneous inflammatory demyelination.
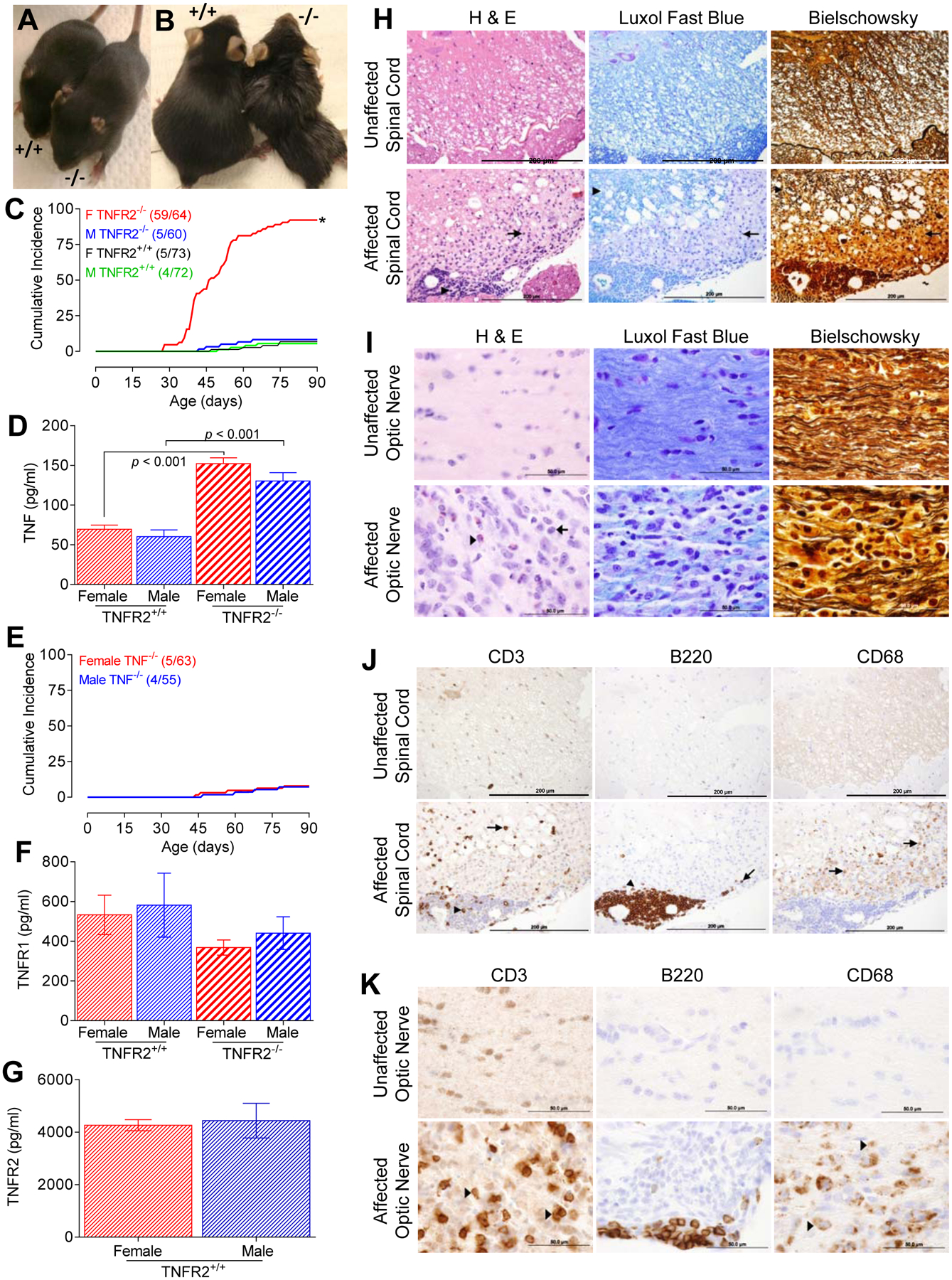
(A) Female C57BL6/J 2D2 TNFR2−/− mice appear normal at birth and at early age; shown are representative 14 day old female TNFR2−/− and TNFR2+/+ mice. (B) 92% of female 2D2 TNFR2−/− mice spontaneously developed disease between 28–79 d of age. Representative affected 65 d old female 2D2 TNFR2−/− and unaffected age- and gender-matched 2D2 TNFR2+/+ mice are shown. (C) The cumulative incidence of 2D2 TNFR2−/− mice with clinical manifestations of disease with a score ≥ 2 (female 2D2 TNFR2−/−, (59/64); male 2D2 TNFR2−/−, (5/60); female 2D2 TNFR2+/+, (5/73); male 2D2 TNFR2+/+; 4/72). Remission of disease severity was not observed in any of the mice studied. (D) Soluble TNF (pg/ml) was determined in sera collected from 4–5 wk old, unaffected male and female 2D2 TNFR2+/+ and 2D2 TNFR2−/− mice by ELISA. The data are reported as mean ± SEM; n = 3 per group. (E) The cumulative incidence of 2D2 TNF−/− mice with a score ≥ 2 (female, 5/63; male, 4/55). Remission of disease severity was not observed in any of the mice studied. (F) Soluble TNFR1 (pg/ml) was determined by ELISA from sera collected from the unaffected mice described in D. The data are reported as mean ± SEM; n = 3. (G) Soluble TNFR2 (pg/ml) was determined by ELISA from sera collected from the unaffected 2D2 TNFR2+/+ mice described in D. The data are reported as mean ± SEM; n = 3. (H) Representative paraffin-embedded spinal cord sections from unaffected female 2D2 TNFR2−/− mice (top) or age and gender-matched affected 2D2 TNFR2−/− mice (bottom) stained with H&E, luxol fast blue (LFB), or Bielschowsky staining to visualize inflammatory cell infiltration, demyelination, and axonal damage, respectively. H&E stained infiltrates are evident in perivascular cuffs (arrow head) and the parenchyma (arrow). LFB show limited loss of myelin in the central white matter (arrow head), but extensive demyelination of peripheral white matter (arrow). Bielschowsky staining shows mild (arrow head) to severe (arrow) axonal loss. Scale bar = 200 μm. (I) Representative paraffin-embedded optic nerve sections from unaffected female 2D2 TNFR2−/− mice (top) or age and gender-matched affected 2D2 TNFR2−/− mice (bottom) stained with H&E, LFB, or Bielschowsky staining demonstrate profound inflammation (left) and demyelination (middle) and moderate axonal loss (right), respectively. A representative monocyte (arrow) and eosinophil infiltrate (arrowhead) are indicated. Scale bar = 50 μm. (J) T cell (anti-CD3), B cell (anti-B220), and macrophage/microglia (anti-CD68) infiltration into the spinal cord of unaffected (top) or age and gender-match affected (bottom) female 2D2 TNFR2−/− mice. Representative paraffin-embedded spinal cord sections are shown. Localization of cells in the perivascular cuffs of the spinal cord (arrowheads) and localization to the parenchyma (arrows). Scale bar = 200 μm. (K) T cell (anti-CD3), B cell (anti-B220), and macrophage/microglia (anti-CD68) infiltration into the optic nerves of unaffected (top) or age and gender-match affected (bottom) female 2D2 TNFR2−/− mice. Representative paraffin-embedded optic nerve sections are shown with representative T cells and macrophages/microglia indicated (arrowheads). Scale bar = 50 μm.
Table I.
Summary of spontaneous clinical paralyses in C57BL/6 2D2 TNFR2−/− and TNF−/− mice.
| Genotype | Gender | % Incidencea | Mean Day of Onsetb | Mean Peak Scorec |
|---|---|---|---|---|
| 2D2 TNFR−/− | ♀ | 92(59/64) | 48.6 ± 1.5 | 3.88 ± 0.04 |
| 2D2 TNFR−/− | ♂ | 8.3 (5/60) | 75.2 ± 2.6 | 3.00 ± 0.09 |
| 2D2 TNF−/− | ♀ | 7.9 (5/63) | 86.6 ± 2.4 | 2.80 ± 0.11 |
| 2D2 TNF−/− | ♂ | 7.2 (4/55) | 85.8 ± 2.9 | 2.50 ± 0.08 |
| 2D2 TNFR−/− | ♀ | 7.5 (3/40) | 87.7 ± 1.1 | 2.33 ± 0.09 |
| 2D2 TNFR−/− | ♂ | 4.1 (1/24) | 79.0 | 2 |
| 2D2 TNFR−/− | ♀ | 6.8 (5/73) | 87.0 ± 1.7 | 2.40 ± 0.06 |
| 2D2 TNFR−/− | ♂ | 5.5 (4/72) | N.D. | N.D. |
The % incidence was calculated by dividing the number of mice that developed clinical paralyses within 6-mo-of-age by the total number of mice scored in the group.
The mean day of onset (mean ± SEM) was calculated by adding the first day of clinical signs of individual mice and dividing with the number of mice in the group, not including mice that did not develop EAE.
The mean peak score (mean ± SEM) was calculated by adding the peak scores of individual mice and dividing by the number of mice in each group. Mice that did not develop clinical symptoms of paralyses were not included in the analyses.
Since circulating levels of TNF are elevated in asymptomatic TNFR2−/− mice showing no signs of disease (Fig. 2D), and increased TNF has been linked to autoimmunity, we next bred C57BL/6J TNF−/− mice with 2D2 Foxp3gfp mice to generate MOG35–55-specific mice that completely lacked TNF signaling. These mice were monitored for disease development for up to six months after birth. As shown in Fig. 2E and Table I, the cumulative incidence of spontaneous disease in TNF−/− 2D2 mice did not increase above the background incidence observed in TNF+/+ 2D2 mice. A low incidence (4%) of spontaneous EAE has previously been reported for 2D2 mice (32), but this is remarkably different than the sex-biased 92% incidence observed in female TNFR2−/− 2D2 mice. Spontaneous NMO-like disease has also been seen in 2D2 mice crossed with MOG35–55-specific Ig heavy-chain knock-in mice (IgHMOG), in which 20% of the peripheral B cells recognize the same autoantigen as 95% of the CD4+ T cells (33, 47). However, TNFR2−/− 2D2 mice differ strikingly from these models with respect to sex-bias, cumulative incidence, and severity of disease. The mortality rate of the female TNFR2−/− 2D2 mice that develop disease is 100%.
Soluble TNF (sTNF), but not TNFR1, concentrations were elevated in TNFR2−/− mice (Fig. 2D and 2F), but elevated sTNF levels did not differ between male and female TNFR2−/− mice. In agreement with our previous studies (48), the levels of sTNFR2 were consistently greater than those of sTNFR1 (p < 0.001). Collectively these data suggest that increased TNFR1 signaling alone does not account for the sex-bias spontaneous disease in female 2D2−/− mice. Taken together, our studies unmask a profound and previously unrecognized sex-bias regulatory role for TNFR2 in the development of autoimmune disease.
We next examined the optic nerves, brain, and spinal cord of affected female 2D2 TNFR2−/− mice for pathological lesions consistent with EAE. No gross lesions were observed upon visual examination of any tissues, with the exception of the optic nerves. In all cases, either one or both nerves were smaller in size compared to asymptomatic 2D2 TNFR2−/− and 2D2 TNFR2+/+ mice but similar to symptomatic 2D2 TNFR2+/+ mice (32). Histological examination identified extensive inflammation, demyelination, and axonal loss in the spinal cord (Fig. 2H) and optic nerves (Fig. 2I). The disease presented unilaterally in the optic nerves of some 2D2 TNFR2−/− mice but was bilateral in others. Consistent with a predominant localization of MOG-mediated immune responses to the optic nerve and spinal cord (32, 49), we did not observe lesions in the brain. The pathological lesions in the spinal cord were multifocal and segmental, and varied from mild perivascular lymphocyte cuffing to severe demyelination. Eosinophils were scattered throughout the parenchyma of the optic nerve, but were rarely found in the spinal cord, and were undetected in the brain (data not shown). CD3+ T cells and B220+ B cells were identified as the dominant inflammatory infiltrates in the spinal cord and optic nerves of the symptomatic 2D2 TNFR2−/− mice (Figs. 2J and 2K, respectively). B220+ B cell infiltrates were mostly confined to the perivascular space with fewer of these cells infiltrating into the parenchyma. CD68+ macrophages/microglia were also found scattered throughout the parenchyma (Figs. 2J and 2K). Overall, these results suggest that TNFR2 deficiency does not appear to alter the pattern of inflammatory demyelination observed in the 2D2 transgenic mice (7, 50), but rather, potentiates a remarkable sex-bias increase in disease incidence (23-fold) and severity. Given the similar phenotypes of TNFR2−/− 2D2 and double-transgenic mice that express MOG-specific receptors on T and B cells (33, 47), genetic deficiency of TNFR2 in 2D2 mice appears to be sufficient to overcome the requirement for the MOG-specific B cell receptor (BCR) transgene for spontaneous autoimmune disease development (33, 47). Despite similarities between TNFR2−/− 2D2 mice and the 2D2 × MOGBCR mice, these two experimental models differ markedly with regard to sexual dimorphic disease incidence and severity. Overall, these results suggest that TNFR2 deletion is sufficient to break functional peripheral tolerance of autoreactive MOG-specific lymphocytes to yield spontaneous disease in female TCR 2D2 transgenic mice.
Ex vivo analysis of encephalitogenic T and B cells in TNFR2−/− 2D2 mice
Infiltration of T and B cells into the CNS of TNFR2−/− 2D2 mice was confirmed ex vivo by flow cytometry. Cells were isolated from the spinal cord, lymph nodes, and spleen of unaffected (healthy) TNFR2−/− 2D2 mice and compared with affected TNFR2−/− 2D2 mice. Consistent with immunohistochemistry results, T and B cells were undetected in the brain (data not shown). Optic nerves were not studied ex vivo. CD4+ T and B cells were present in the spinal cords of unaffected as well as affected TNFR2−/− 2D2 mice (Figs. 3A and 3F), but increased in the affected mice (p < 0.05). The ratio of spinal cord B cells:T cells was also increased in affected mice (unaffected, 0.8:1 and affected, 1.4:1) (Fig. 3A). The low number of spinal cord infiltrates in unaffected TNFR2−/− 2D2 mice likely contributed to their lack of detection by immunohistochemistry. In contrast to TNFR2−/− 2D2 mice, B220+ B cells were not detected in the spinal cords of unaffected 2D2 mice (data not shown). The frequency of spinal cord CD4+ T cells expressing the Vβ11 TCR (p < 0.05) declined from 97% in unaffected mice to 69% in affected mice (Figs. 3C and 3F). This reduction in Vβ11+ TCR frequency, and presumably MOG35–55 specificity, was evident in Teffs as well as Foxp3gfp Tregs. The frequency of Tregs, regardless of Vβ11 specificity, increased in affected TNFR2−/− 2D2 mice compared to unaffected healthy cohorts.
FIGURE 3. Ex vivo analysis of encephalitogenic T and B cells in TNFR2−/− 2D2 mice.
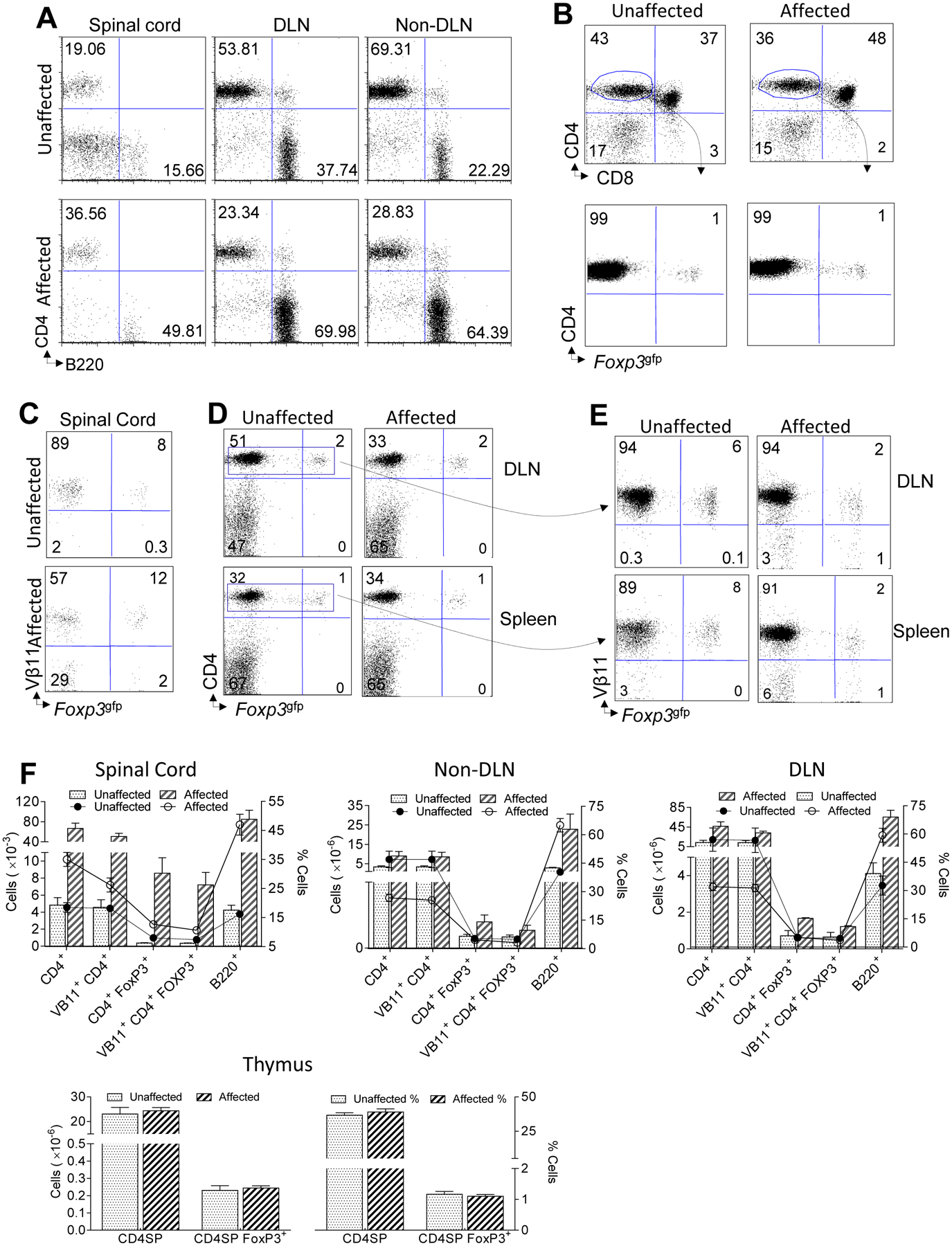
Freshly isolated mononuclear cells from the spinal cord, CNS draining lymph nodes (DLN), or CNS non-DLNs (pooled axillary and brachial) of unaffected (top) or age- and gender-matched affected (bottom) 2D2 TNFR2−/− mice are compared. FACS plots of CD4+ T cells and B220+ B cells from 7-AAD− lymphocyte-gated cells are shown. (B) Freshly isolated cells from the thymus of unaffected 2D2 TNFR2−/− mice (left) are compared with age-matched affected 2D2 TNFR2−/− mice (right). FACS plots of CD4+ T cells versus CD8+ T cells (top) and CD4+ versus Foxp3gfp+ Tregs (bottom) are shown. The cells shown in top panels are gated on 7-AAD− lymphocytes. The cells shown in the bottom panels are gated on CD4+ CD8− 7-AAD−lymphocytes. (C) Freshly isolated mononuclear cells from the spinal cords of unaffected (top) or age- and gender-matched affected (bottom) 2D2 TNFR2−/− mice are indicated. FACS plots of Vβ11 TCR and GFP expression from 7-AAD− lymphocytes are shown. (D) Comparison of CD4 Foxp3gfp+ Tregs isolated form the DLNs (top) or spleen (bottom) of unaffected (left) and age-matched affected (right) 2D2 TNFR2−/− mice. FACS plots of CD4 and Foxp3 expression from 7-AAD− lymphocytes are shown. (E) CD4+ 7-AAD− gated T cells from panel D were further gated for Vβ11 TCR expression. Shown are the frequencies of Vβ11+ and Vβ11− CD4+ Foxp3gfp- Teffs and CD4+ Foxp3gfp+ Tregs representative of DLNs (top) or spleens (bottom) from unaffected (left) and age-matched affected (right) 2D2 TNFR2−/− mice. Plots shown are representative of 3 mice per genotype. (F) The graphs depict the numbers and frequencies of cells shown in Figs A-E. Mean values ± SEM are indicated for each group.
Within the CNS draining and non-draining lymph nodes, the numbers of CD4+ Teffs (Vβ11− and Vβ11+) and B220+ B cells as well as the B:T cell ratio increased in affected versus unaffected TNFR2−/− 2D2 mice (Figs. 3A and 3F). Although the frequency of Foxp3gfp Tregs were comparable, the total number of lymph nodal Tregs were elevated in affected versus unaffected TNFR2−/− 2D2 mice (Figs. 3D, 3E, and 3F). The frequency of splenic Foxp3gfp Tregs did not differ between affected and unaffected TNFR2−/− 2D2 mice (Figs. 3D and 3F). In the thymus, the frequency and number of CD4+CD8−Foxp3gfp-, CD4+CD8−Foxp3gfp+, and CD4+CD8+ double positive T cells were comparable between affected and unaffected TNFR2−/− 2D2 mice (p > 0.05) (Figs. 3B and 3F).
Elevated ex vivo MOG35–55-specific cytokine production and circulating antibodies
To gain further insight into the mechanism(s) underlying the sex-biased spontaneous CNS-demyelinating autoimmunity in female TNFR2−/− 2D2 mice, we next examined ex vivo CD4+ T cell responsiveness to MOG35–55 and circulating antibodies. Female TNFR2−/− 2D2 mice were compared with healthy male TNFR2−/− 2D2, female TNFR2+/+ 2D2 mice, and male TNFR2+/+ 2D2 mice. Cells isolated from female TNFR2−/− 2D2 mice produced significantly more MOG35–55-dependent IFN-γ and IL-17 (Fig. 4A). We next investigated the differentiation state of B cells from the affected TNFR2−/− 2D2 mice. For these studies, MOG35–55-specific antibodies were quantified from sera collected from female and male TNFR2−/− 2D2 or 2D2 mice. Without immunization, female TNFR2−/− 2D2 mice, regardless of whether they were affected or unaffected, had elevated titers of MOG35–55 IgG2b, MOG35–55 IgM, and total IgG2b (p < 0.05) (Figs. 4B and 4C). In contrast, MOG35–55 IgG1 titers were unaltered in female TNFR2−/− 2D2 mice. MOG35–55 IgG2a was not detected in any of the mice. Anti-IgG2b sera titers were comparable between 4-wk-old and 12-wk-old TNFR2−/− 2D2 mice (data not shown). Taken together, elevated IFN-γ, IL-17, and MOG35–55-specific IgG2b titers associate with spontaneous CNS demyelination in female TNFR2−/− 2D2 mice.
FIGURE 4. Elevated ex vivo MOG35–55-specific cytokine production and circulating antibodies.
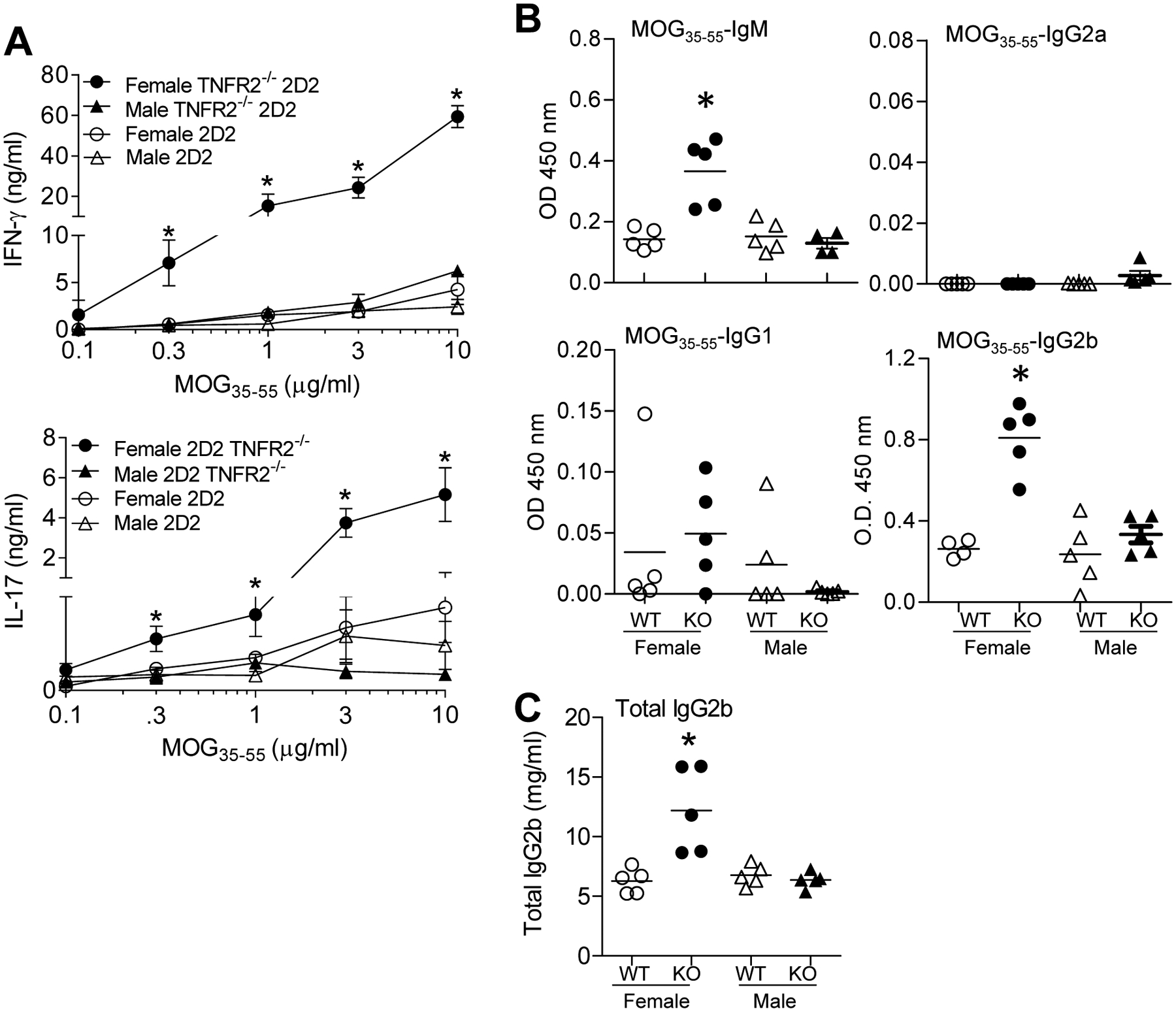
(A) MOG35–35-specific Th1 and Th17 responses. 1 × 106 splenic or cervical LN cells from affected 2D2 TNFR2−/− mice (n = 3) compared with unaffected age-matched 2D2 TNFR2+/+ mice (n = 4) were cultured with increasing concentrations (0, 0.1, 0.3, 1, 3, or 10 μg/ml) of MOG35–55 for 96 h. Cell-free supernatants were collected and IFN-γ and IL-17 production were determined by ELISA. Shown are the mean ± SEM. (B) MOG35–35-specific B cell responses were determined by ELISA. Relative concentrations of MOG35–55-specific sera Ig antibodies (1:100 diluted serum) from female affected 2D2 TNFR2−/− mice (indicated as Female KO; n = 5) and age-matched unaffected female TNFR2+/+ mice (indicated as Female 2D2; n = 4 or 5) and unaffected male 2D2 TNFR2−/− (indicated as Male KO; n = 5) control mice as determined by isotype specific sandwich ELISA. Individual values and mean absorbance at OD 450 nm are shown. Sera from MOG35–55-immunized C57BL/6 mice was used as a control: IgM, 0.27 ± 0.05; IgG1, 0.88 ± 0.11; IgG2a, 0.82 ± 0. 02; and IgG2b, 1.97 ± 0.04 (mean ± SEM). (C) Total serum IgG2b was determined by ELISA. Sera collected from affected 2D2 TNFR2−/− mice was compared with unaffected female TNFR2+/+ mice and unaffected female and male 2D2 TNFR2−/− mice. Purified IgG2b was used in the ELISA to generate a curve for quantification. Mean values ± SEM are indicated for each group.
Antibiotic treatment reduces the incidence and severity of spontaneous CNS demyelination in TNFR2−/− 2D2 mice
In response to an unexplainable production decline and onset of early mortality in our barrier-housed breeding colonies, we initiated a 6 wk. course of therapeutic trimethoprim/sulfamethoxazole (TMS) for opportunistic pathogen prophylaxis and observed a loss of disease in offspring born over a 10-month span (Fig. 5A) which was reversed by cross-fostering to surrogate Harlan CD-1 recipients. Given that the microbial community within the gut is established early in life, we hypothesized a role for gut microbiota in the precipitation of sex-biased autoimmunity in TNFR2−/− 2D2 mice. To determine whether disease susceptibility in female TNFR2−/− 2D2 mice associated with a change in gut microbiota composition, fecal samples were collected from 4–8 wk old male and female TNFR2−/− and TNFR2−/− 2D2 mice for Illumina-based sequencing of the hypervariable V4 region of bacterial 16S rRNA genes. Male and female mice were housed separately, but 2D2 TCR+ and 2D2 TCR− littermates were co-housed. At fecal collection, none of the female TNFR2−/− 2D2 mice had clinical signs of disease, but all of them eventually developed paralyses. No other mice in the study developed clinical signs of disease. After filtering, a total of 4,846,601 quality reads comprising 18 different bacterial classes from nine phyla (Actinobacteria, Bacteroidetes, Cyanobacteria, Deferribacteres, Firmicutes, Proteobacteria, TM7, Tenericutes, and Verrucomicrobia) were detected in samples from 46 mice (13 female TNFR2−/− 2D2, 14 female TNFR2−/−, 9 male TNFR2−/− 2D2, and 10 male TNFR2−/− mice) with an average coverage of 105,360 reads per sample. Microbial richness, as determined by the total number of OTUs detected, differed, in a sex-independent manner, between TNFR2−/− (42.00 ± 0.65, mean ± SEM) and TNFR2−/− 2D2 (45.74 ± 0.73, mean ± SEM) mice. Conversely, α-diversity, estimated using the Chao1 index, revealed a restricted microbial diversity in female TNFR2−/− mice compared to TNFR2−/− 2D2 mice (Fig. 5B).
FIGURE 5. Sex-specific microbiota profiles define disease-resistant male TNFR2−/− 2D2 mice and disease susceptible female TNFR2−/− 2D2 mice.
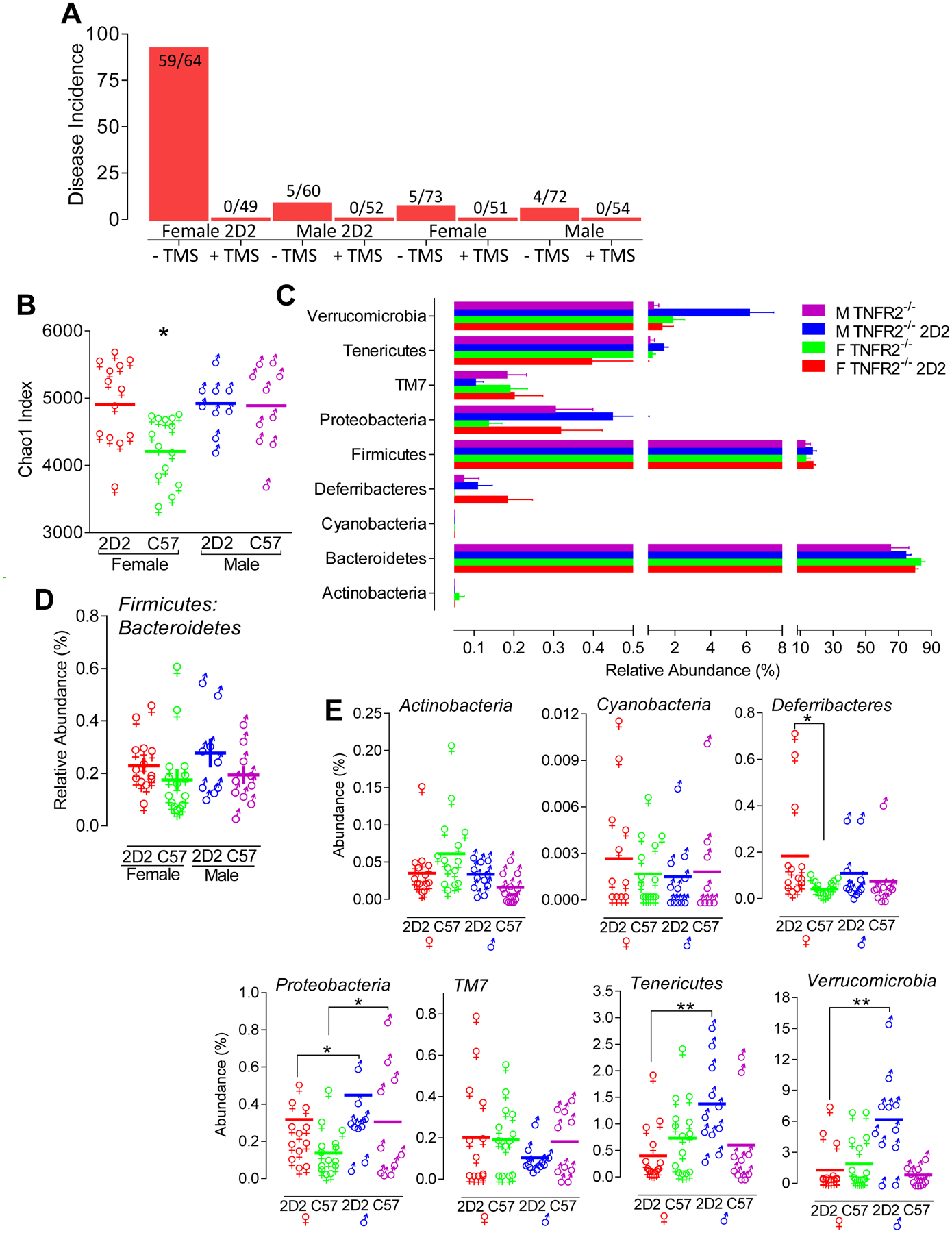
(A) The spontaneous development of paralyses in TNFR2−/− 2D2 and 2D2 mice is lost following antibiotic (oral TMS) treatment. Both males and females were affected. The bar graph indicates the percent of disease incidence. Shown are the number of mice that succumb to disease relative to the total number of mice studied. All mice were monitored for disease onset for a minimum of 90 days of age. (B) Bacterial 16S rRNA amplicon sequencing of fecal samples was used to define the microbiome profiles of female TNFR2−/− 2D2 (red, n = 13), female TNFR2−/− (green, n = 14), male TNFR2−/− 2D2 (blue, n = 9), and male TNFR2−/− 2D2 (purple, n = 10) mice. Chao1 indices in fecal microbiota. Statistical analysis was performed using Mann-Whitney rank sum tests. (C) Relative abundance at the taxonomic level of phylum. Bar charts compare the fecal bacterial composition between unaffected male and female TNFR2−/− and TNFR2−/− 2D2 mice. (D) Firmicutes:Bacteroidetes ratios were determined for each individual mouse by dividing % abundance of Firmicutes by % abundance of Bacteroidetes. The mean ± SEM of these determinations are shown. (E) Annotation at the level of phylum for each individual mouse is shown to compare the difference of bacterial abundance between groups. The solid line depicts the mean % abundance. *p < 0.05 and **p < 0.01.
Sex- and TCR-dependent differences in the relative abundance of microbial taxa
We next characterized the taxonomic variation across groups at the phylum level. For all four experimental groups, Bacteroidetes and Firmicutes were the predominant phyla, and ranged in abundance from 65% to 84% and 13% to 29%, respectively (Fig. 5C). Neither the abundance of Bacteroidetes or Firmicutes, nor the ratio of Firmicutes:Bacteroidetes differed between any of the groups (p > 0.05) (Fig. 5D). Sex-dependent differences were detected in three of the remaining seven phyla, Proteobacteria, Tenericutes, and Verrucomicrobia, with male mice harboring significantly greater proportions of microbes than females in all three phyla (p < .05), regardless of TCR genotype (Fig. 5E).
Relative abundance at taxonomic level of family, genus, and species.
When resolved to the OTU level, in which all sequences met a threshold of 97% nucleotide identity, several taxa differed between groups (Table II). There were also several interactions detected suggesting that the influence of one variable was dependent on the status of the other variable. With regard to sex-dependent differences, microbes in the genus Oscillospira and genus Sutterella were present at greater relative abundance in males (p < 0.05) (Fig. 6A). Similarly, B. acidifaciens, B. ovatus, Anaeroplasma sp. and Akkermansia muciniphila were more abundant in male TNFR2−/− 2D2 mice compared to female TNFR2−/− 2D2 mice (Fig. 6B). Anaeroplasma sp. and A. muciniphila were also more abundance in male TNFR2−/− 2D2 mice compared to male TNFR2−/− mice (p < 0.001) but no TCR-dependent difference was detected in female mice (Fig. 6B). B. acidifaciens and B. ovatus were present at greater levels in both male and female TNFR2−/− 2D2 mice relative to TNFR2−/− mice. Bacteroides sp., Bacteroides uniformis, and Parabacteroides sp. were more abundant in female mice compared to males (p < 0.05) (Fig. 6C). Turicibacter sp. and family Clostridiaceae were more abundant in female TNFR2−/− mice relative to male TNFR2−/− mice (p < 0.01), but no difference was detected between female and male TNFR2−/− 2D2 mice (Fig. 6D).
Table II.
Sex-biased taxa dominating the bacterial microbiota of TNFR2−/− 2D2 mice.
| Phyla | Class | Order | Family | Genus | Species | Fold Increase |
|---|---|---|---|---|---|---|
| Increased in Males | ||||||
| Bacteroidetes | Bacteroidia | Bacteroidales | Bacteroidaceae | Bacteroides | acidifaciens | 1.81 |
| Bacteroidetes | Bacteroidia | Bacteroidales | Bacteroidaceae | Bacteroides | ovatus | 1.52 |
| Firmicutes | Clostridia | Clostridiales | Ruminococcaceae | Oscillospira | 1.53 | |
| Verrucomicrobia | Verrucomicrobiae | Verrucomicrobiales | Verrucomicrobiaceae | Akkermansia | muciniphila | 5.87 |
| Tenericutes | Mollicutes | Anaeroplasmatales | Anaeroplasmataceae | Anaeroplasma | 13.31 | |
| Proteobacteria | Betaproteobacteria | Burkholderiales | Alcaligenaceae | Sutterella | 3.45 | |
| Increased in Females | ||||||
| Bacteroidetes | Bacteroidia | Bacteroidales | Porphyromonadaceae | Parabacteroides | 3.69 | |
| Bacteroidetes | Bacteroidia | Bacteroidales | Bacteroidaceae | Bacteroides | uniformis | 7.65 |
| Bacteroidetes | Bacteroidia | Bacteroidales | Bacteroidaceae | Bacteroides | 1.38 | |
FIGURE 6. Relative abundance at taxonomic level of family, genus, and species.
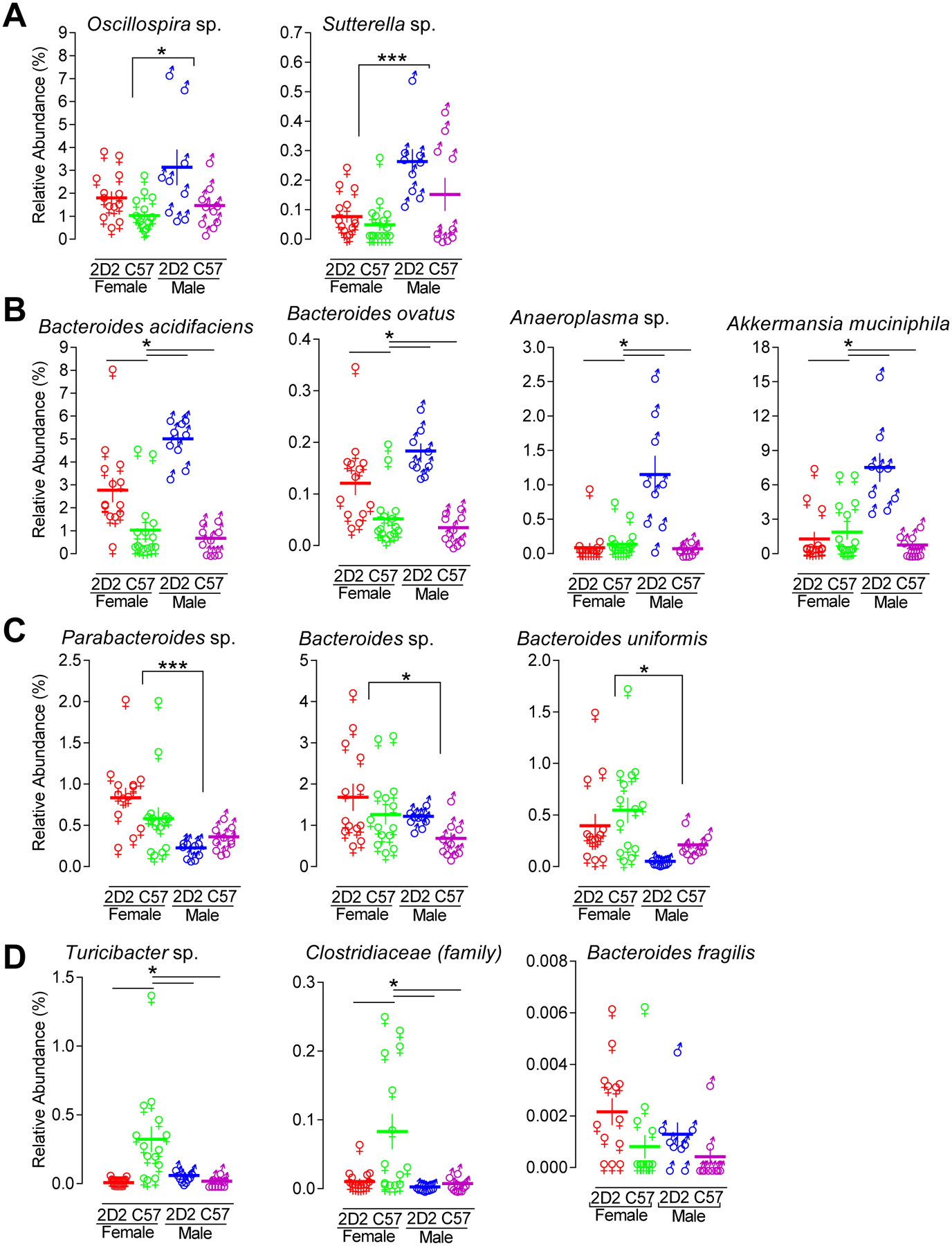
16S bacterial rRNA sequencing of fecal samples was used to define the microbiome profiles of female TNFR2−/− 2D2 (red, n = 13), female TNFR2−/− (green, n = 14), male TNFR2−/− 2D2 (blue, n = 9), and male TNFR2−/− 2D2 (purple, n = 10) mice. (A) The microbes that were significantly more abundant in male mice relative to female mice are shown. (B) The microbes that were significantly more abundant in unaffected disease-resistant male TNFR2−/− 2D2 mice relative to unaffected disease susceptible female TNFR2−/− 2D2 mice are shown. (C) The microbes that were significantly more abundant in female mice relative to male mice are shown. (D) The microbes that were significantly less abundant in unaffected disease resistant male TNFR2−/− 2D2 mice relative to unaffected disease susceptible female TNFR2−/− 2D2 mice are shown. Each data point represents an individual mouse. The horizontal line indicates the mean of each group. The vertical line represents the SEM of each group. *p < 0.05 and ***p < 0.001.
Relative abundance of key bacterial players in intestinal immunity and host health
Over the past decade, select gut commensal bacteria, including Candidatus arthromitus, Lactobacillus, Clostridium, B. fragilis, and Bifidobacterium have been identified as key regulators of immunity and host health (51). None of these bacteria differed (p > 0.05) with respect to sex or genotype between male or female TNFR2−/− and TNFR2−/− 2D2 mice. It is well accepted that interactions between commensal bacteria is important for overall robustness. Therefore, it is possible that non-significant (p > 0.05) trends of these individual bacteria may contribute to a larger collective effect. Candidatus arthromitus, commonly called segmented filamentous bacteria (SFB), was detected in all groups of mice, but its abundance was neither sex- or TCR-dependent (Fig. 7A). B. fragilis was detected in the majority of TNFR2−/− 2D2 mice but in few TNFR2−/− mice, and was more abundant in female compared to male TNFR2−/− 2D2 mice (Fig. 7B). Clostridium sp. was detected in all groups with greater abundance in TNFR2−/− 2D2 mice without sex-dependence (Fig. 7C). Clostridium perfringens was detected in only one of the 46 mice studied (Fig. 7C). Bifidobacterium was not detected in any of the male TNFR2−/− mice, but was expressed in all male TNFR2−/− 2D2 and most female mice (Fig. 7D). Lactobacillus was detected in all mice with greater abundance in TNFR2−/− male mice (Fig. 7E). Coprococcus, previously associated with MS, was detected in all mice without either TCR- or sex-dependence (Fig. 7F). Dorea, another member of the Lachnospiraceae family, was found in greater abundance in female TNFR2−/− 2D2 mice (Fig. 7F).
FIGURE 7. Relative abundance of key bacterial players in intestinal immunity and host health.
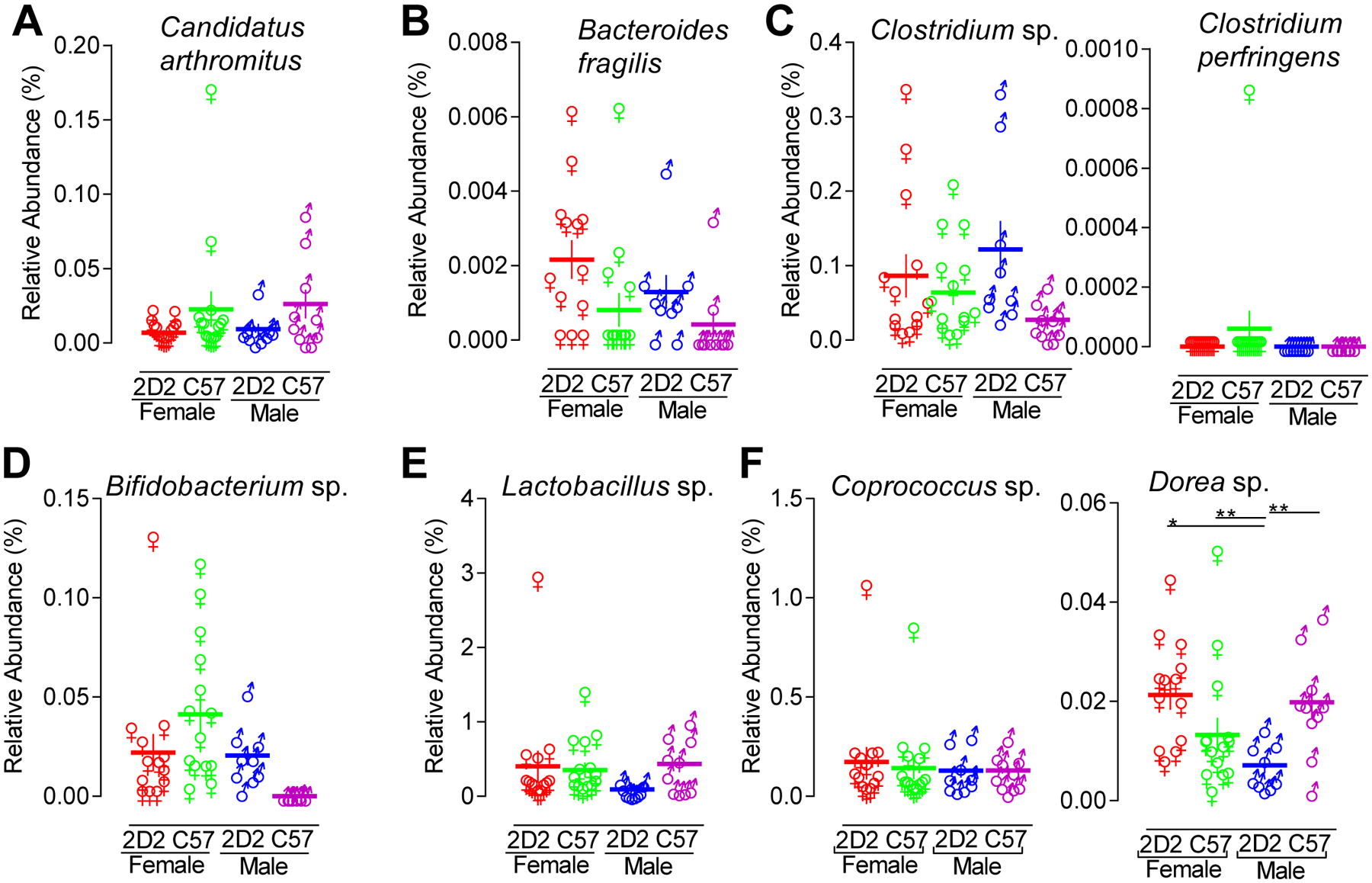
16S bacterial rRNA sequencing of fecal samples was used to define the microbiome profiles of female TNFR2−/− 2D2 (red, n = 13), female TNFR2−/− (green, n = 14), male TNFR2−/− 2D2 (blue, n = 9), and male TNFR2−/− 2D2 (purple, n = 10) mice. The relative abundance of (A) Candidatus arthromitus, (B) B. fragilis, (C) Clostridium sp. and Clostridium perfringens, (D) Bifidobacterium sp., (E) Lactobacillus sp., and (F) Coprococcus sp. and Dorea sp. are shown. Each data point represents an individual mouse. The horizontal line indicates the mean of each group. The vertical line represents the SEM of each group. *p < 0.05 and **p < 0.01.
Gut microbiota in TNFR2−/− and TNFR2−/− 2D2 mice clusters according to sex and genotype
To assess β-diversity among the gut microbiota and characterize the differences between experimental groups, taking into account all taxa detected, principal component analysis (PCA) was performed using a non-linear iterative partial least squares algorithm. Comparison of principal component (PC) 1 and PC2, explaining 21.40% and 18.52% variation respectively, demonstrated a distinct separation of male TNFR2−/− 2D2 and TNFR2−/− mice (Fig. 8A) but not female TNFR2−/− 2D2 and TNFR2−/− mice. In contrast, when PC1 was visualized against PC3, explaining 13.07% variation, female TNFR2−/− 2D2 and TNFR2−/− mice did separate along PC3 (Fig. 8B). Overall, these results identify a distinct composition of the microbiota in the male TNFR2−/− 2D2 that relates to disease protection.
FIGURE 8. Sex-specific microbiota profiles define disease-resistant male TNFR2−/− 2D2 mice.
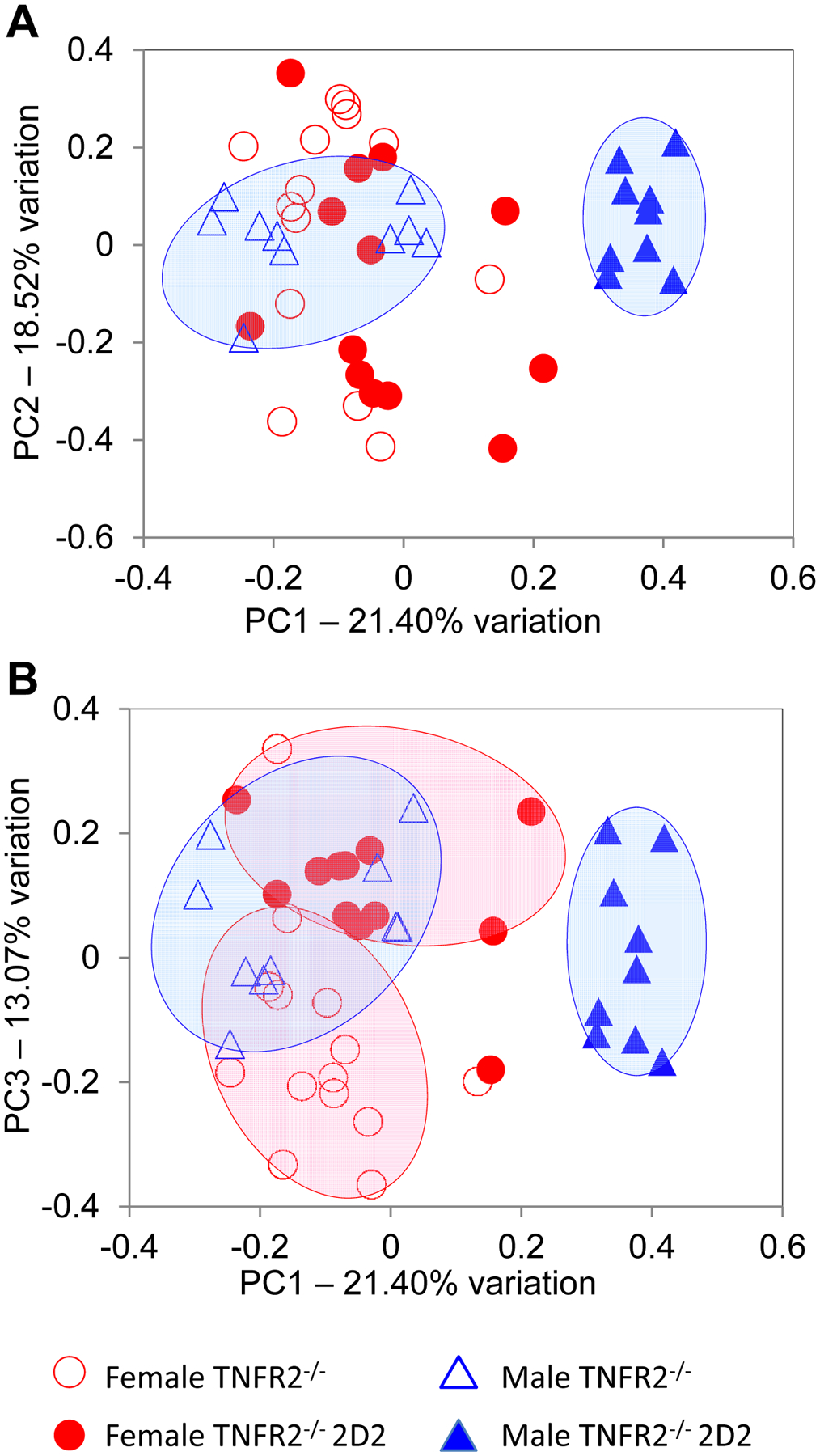
Principal component analysis (PCA) of gut microbial communities detected in the fecal samples are shown from female TNFR2−/− (open red circles); female TNFR2−/− 2D2 mice (closed red circles); male TNFR2−/− (open blue triangles); and male TNFR2−/− 2D2 (closed blue triangles) mice are shown. (A) Depiction of PC1 vs. PC2 separates male TNFR2−/− and TNFR2−/− 2D2 mice. (B) Depiction of PC1 vs. PC3 separates female TNFR2−/− and TNFR2−/− 2D2 mice as well as male TNFR2−/− and TNFR2−/− 2D2 mice.
Discussion
TNF blockade has become a mainstay of therapy for inflammatory bowel disease (IBD) and rheumatoid arthritis (RA). However, TNF antagonists, including infliximab (Remicade®), etanercept (Enbrel®), and adalimumab (Humira®), sometimes precipitate CNS autoimmune demyelinating diseases such as neuromyelitis optica (NMO) and multiple sclerosis (MS). Why? Various theories, including failed antagonist entry into the CNS (52), downregulated TNFR2-mediated immune suppression (53), and abrogated TNFR2-mediated re-myelination (54) have been proposed. While these postulates provide plausible explanations for the failure to treat demyelinating autoimmunity, they do not explain the onset of new or worsened CNS demyelination that occurs in IBD or RA patients. Here, we provide one possible explanation. We present evidence to suggest that TNFR2 is key to maintain immunological tolerance against commensal bacteria in mice that are at high genetic risk for autoimmune disease. Taking into account the interactions between the immune system, the gut microbiota, and the nervous system, we hypothesize that TNFR2 blockade disrupts this symbiotic relationship to unmask autoimmune demyelination in genetically susceptible individuals. Under this paradigm, individuals respond differently to anti-TNF therapy, not because of their genes, but because of the microbes they carry. There are multiple direct and indirect pathways through which TNF might modulate immune-microbial-neuro-endocrine communication to facilitate sex-biased CNS autoimmune demyelination. These include immune activation, microbiota, and endocrine and neurotransmitter signaling.
Sexual dimorphism is characteristic of many autoimmune disorders with disease incidence more often higher in females compared to males. In humans, there is a significant female predominance in both NMO and MS, with the female-to-male ratio being even greater in NMO, ranging from 5–11: 1 (55). The reason(s) for differences between females and males in autoimmune disease susceptibility remains an enigma. Sex hormones, the presence or absence of a second X chromosome, microbiota, and disruption at the blood-spinal cord and brain barriers have been implicated as contributing factors (56–58). While progress has been made in identifying the genetic determinants of autoimmunity (59), advances in identifying how sex hormones, microbiota, and their interactions with the immune system influence gender-bias autoimmunity remain limited. Realization that the environment is an even greater factor than genes in an immune response (60) exemplifies the importance of unraveling these complex interactions. Our current work does not explain fully the mechanism(s) for the sex differences in TNFR2−/− mice. However, our studies do provide a foundation to explore sexual dimorphism in microbiota-targeted approaches to treat CNS autoimmunity. In particular, we show that TNFR2 deficiency alone is not responsible for the observed sex-related differences, as TNFR2−/− males and females both have a low incidence of spontaneous disease. Rather, TNFR2 deficiency in combination with autoreactive MOG35–55-specific T cells precipitates a nearly complete penetrance of disease in female mice. The high disease incidence in sexual dimorphic TNFR2−/− 2D2 mice exceeds that of double-transgenic 2D2 × IgHMOG mice which produce MOG35–55-specific B and T cells (33, 61). Resistance to disease development in male TNFR2−/− 2D2, both sexes of TNFR2−/−, and TNF−/− 2D2 mice implicates sex, CNS antigen-reactive CD4+ T cells, and TNFR2 specificity as important factors in disease development. A role for microbiota is implicated by the loss of disease with antibiotic treatment and segregation of microbiota profiles that interact with sex and genotype. Disease protection in male TNFR2−/− 2D2 mice relates to increased abundance of A. muciniphila, B. acidifaciens, B. ovatus, Anaeroplasma sp., Sutterella sp., and Oscillospira sp.. Disease susceptibility in female TNFR2−/− 2D2 mice correlates with increased Parabacteroides sp., B. uniformis, and Bacteroides sp. A distinct gut microbiota profile in TNFR2−/− 2D2 mice seems to parallel male microbiome-mediated protection similar to that previously reported in the NOD T1D model (62, 63). Elevated production of MOG35–55-specific IFN-γ, IL-17, and IgG2b suggests the loss of functional T as well as B cell tolerance. Ongoing studies aim to explain the unexpectedly increased frequency of CD4+ Foxp3+ Tregs with loss of functional immune tolerance.
Infectious pathogenic viruses and bacteria have long been speculated to contribute to human CNS autoimmune pathogenesis (64, 65). Yet, no etiologic or causal effect has been firmly established. In contrast, our results are consistent with numerous other more recent studies indicating that commensal, rather than pathogenic, bacteria facilitate CNS autoimmune demyelination (29, 30, 66–68). Attenuated MOG-induced EAE development in antibiotic-treated and germ-free mice has linked gut microbiota to CNS demyelination (29, 30). SFB has been positively correlated with EAE (29). In contrast, B. fragilis, Bifidobacterium sp, and some strains of Lactobacillus negatively correlate with EAE (69–72). Evidence is now also emerging for a role by commensal microbiota in the etiology of human MS. Faecalibacterium (phylum Firmicutes) have been found in low abundance in untreated MS patients and this was reversed by glatimer acetate (GA) treatment (73). GA treatment was also shown to increase Bacteroidaceae, Ruminococcus, Lactobacillaceae, and Clostridium in these MS patients. In contrast, Vitamin D supplementation increased Faecalibacterium, A. muciniphilia, and Coprococcus in untreated, but not GA-treated MS patients. In considering these microbes in disease etiology, Faecalibacterium prausnitzii, a highly abundant butyrate producing commensal, has been found in low abundance in IBD patients and has been shown to ameliorate experimental colitis, at least in part, by promoting Treg differentiation and enhancing gut barrier integrity (74, 75). Bacteroidaceae, Ruminococcus, Lactobacillaceae, Clostridium, Coprococcus, and A. muciniphilia all share a similar capacity to promote immune tolerance by targeting T cell differentiation and gut barrier integrity. An association between commensal bacteria and host immunity is less well established in NMO patients, but a link is evident. For example, aquaporin-4 (AQP4) is considered a primary autoimmune target in NMO (76) and AQP4-specific T cells show cross-reactivity to a protein product of Clostridium perfringens (14).
In our studies, A. muciniphilia, was the most abundant bacteria that was selectively increased in protected male TNFR2−/− 2D2 mice. A. muciniphilia, a gram-negative bacteria, constitutes 3–5% of the microbiome in healthy humans, is characterized as a mucin-degrader and also enhances gut barrier integrity (77). It remains to be determined whether A. muciniphilia provides similar protection at blood-brain or spinal cord barriers. A. muciniphilia has also been implicated in the regulation of genes involved in host lipid metabolism and inflammation, including histone deacetylases and peroxisome proliferator-activated receptor gamma (PPAR-γ) (78, 79). Further, A. muciniphilia abundance has been inversely correlated with MS (73), T1D (80), T2D (81), and metabolic disorders associated with obesity (82), but positively correlated with the neurological disorder, autism (83). It is not known how TNFR2 deficiency impacts A. muciniphilia abundance, but intracellular cross-talk between SCFA and sex-hormone signaling and gene regulation is one possibility. Sutterella, also selectively more abundant in male TNFR2−/− 2D2 mice, has been positively correlated with autism and Down syndrome (84), and degradation of secretory IgA (sIgA) (85). In addition to providing a first line of defense against enteric toxins and pathogens, intestinal sIgA, promotes antigen clearance, quenches bacterial virulence factors, and changes the composition of the intestinal microbiota (86). Therefore, Sutterella-mediated protection in male TNFR2−/− 2D2 mice is plausible. Less in known about how other bacteria found in high abundance in the male TNFR2−/− 2D2 mice may benefit health to provide protection against autoimmunity. These bacteria include, in order of increasing abundance, B. acidifaciens, Oscillospira sp., Anaeroplasma sp., and B. ovatus. With that said, carbohydrate fermentation by Bacteroides and other bacteria provides a significant proportion of the host’s daily energy requirements. Moreover, different colonization likely reflect survival and functional interactions between commensal bacteria within the intestine. For example, interactions among acetate or butyrate producers, poly- or oligosaccharide degraders, or even sulfate-reducing bacteria and other microbes can be affected by the carbon sources available. In contrast to the males, the bacteria that were selectively more abundant in female TNFR2−/− 2D2 mice were all Bacteroidetes of the genera Parabacteroid and Bacteroides, and studies to define their contribution to disease are ongoing. Previous studies have positively correlated F. prausnitzii, a potent butyrate producer and HDAC inhibitor, with Crohn’s disease (87) and T2D (81). Although Faecalibacterium spp. was undetected in our study, Dorea, another Lachnospiraceae family member, was found in high abundance in female TNFR2−/− 2D2 mice. Dorea has also been linked to autoimmune and metabolic disorders (81, 88).
Towards defining a mechanism, the intestine is an important site for peripheral lymphocyte development. Exposure to different gut microbiota products, including SCFA and PSA, may differentiate MOG-reactive CD4+ T cells into an auto-reactive phenotype through molecular mimicry (89). It is also plausible that the induction of autoimmunity in the TNFR2−/− 2D2 mice relies on a direct effect of gut microbiota on B cell function. B cell development in the intestinal mucosa is influenced by commensal bacteria, including Sutterella, and alterations to the gut microbiome may modulate B cell function (30, 70). B cell activation might be also be explained by the recruitment of MOG-specific B cells from the naïve repertoire by autoreactive CD4+ T cells (61). MOG expression in the periphery, perhaps as debris from trafficking of activated CD4+ T cells, might also promote MOG-specific B cell expansion (90). In considering a mechanism for the unexpected Treg expansion, increased abundance of B. fragilis and Lactobacillus sp. in female TNFR2−/− 2D2 mice is consistent microbial-mediated CD4+ Foxp3+ Treg differentiation (70, 91, 92). It remains to be determined whether Foxp3+ Treg function differs between male and female TNFR2−/− 2D2 mice.
Changes to the gut microbiome may be a consequence, and not a cause, of autoimmunity (93). For instance, it is plausible that female TNFR2−/− 2D2 mice have increased disease susceptibility as a direct consequence of estrogen-induced IFN-γ expression and Th1-mediated IgG2b class switching (94–96). Estrogen has been also reported to suppress TNF production (97). Yet, we observed comparable levels of TNF in male and female mice to suggest that elevated concentrations of TNF alone is insufficient to link gut microbiota and spontaneous sexual dimorphic autoimmunity in 2D2 TNFR2−/− mice. Interestingly, male rheumatoid arthritis patients respond better than females to anti-TNF therapy (98–101) and female mice respond better than male mice to TNFR2-dependent cardioprotection (102). Therefore, a similar sex-bias role of TNFR2-mediated immune modulation may be applicable to other diseases such as myocarditis, colitis, and arthritis (103–105).
Regardless of whether males are protected or females are susceptible, our data demonstrate that genetic loss of TNFR2 in 2D2 TCR mice is sufficient to break functional tolerance of both T cells and B cells. Taken together, we propose disruption of immune modulation of the CNS-gut-microbe axis as a plausible explanation for some of the dichotomous effects of nonselective TNF blockade in patients. Our results shed new light on a growing body of evidence suggesting that TNFR1-selective antagonism may enhance treatment of CNS demyelinating autoimmune disorders. While the mechanistic basis remain unclear, the TNFR2−/− 2D2 model provides a unique experimental paradigm to investigate the involvement of microbiotia on T and B cell tolerance in sexual dimorphic CNS autoimmune disorders. Lastly, gender should be considered as a variable in the design and implementation of microbiota directed therapeutic measures to treat CNS autoimmunity.
Acknowledgements
We sincerely thank Dr. Nancy H. Ruddle (Yale Univ.) for the many enlightening discussions and numerous manuscript critiques; Drs. Lon Dixon and Erin O’Connor and Dana Weir, from the MU Office of Animal Resources, for assistance with cross-fostering and animal care; Dr. Habib Zaghouani for sharing of mice and reagents; Nathan Bivens, from the MU DNA Core Facility, for Illumina sequencing; Scott Givan, Christopher Bottoms, and Bill Spollen, from the MU Bioinformatics Core Facility, for assistance with bioinformatics; Jacob Lucas, Azeem Khan, Wesley Reznicek, and Giedre Turner for excellent technical assistance; and the staff of the MU Cell & Immunobiology Core Facility for flow cytometry and cell sorting.
This work was supported by NIH R01 ES022966 (S.C.M) and a University of Missouri Richard Wallace Faculty Incentive Grant Award (S.C.M.).
Abbreviations used in this article:
- 2D2
a TCR transgenic mouse (2D2) specific for the MOG 35–55 peptide
- 7-AAD
7-aminoactinomycin D
- ANOVA
analysis of variance
- APC
allophycocyanin
- AQP4
aquaporin 4
- c
cells
- CAR
cilia-associated respiratory
- DLN
draining lymph node
- EAE
experimental autoimmune encephalomyelitis
- Foxp3
forkhead box P3
- GA
glatirmer acetate
- GABA
γ-aminobutyric acid
- GBS
Guillain-Barre syndrome
- KO
knockout
- LFB
luxol fast blue
- LN
lymph node
- LT
lymphotoxin
- LTα3
lymphotoxin-α homotrimer
- MHC II
major histocompatibility complex II
- MHV
murine hepatitis virus
- MOG
myelin oligodendrocyte glycoprotein
- MOG35–55
MOG peptide 35–55
- MOG92–106
MOG peptide 92–106
- MS
multiple sclerosis
- MU
University of Missouri
- NIPALS
non-linear iterative partial least square
- NMO
neuromyelitis optica
- ON
optic neuritis
- OSE
opticospinal encephalomyelitis
- OTU
operational taxonomic units
- PC
principal component
- PCA
principal component analysis
- QIIME
Quantitative Insights into Microbial Ecology
- SCFA
short chain fatty acid
- SFB
segmented filamentous bacteria
- SRA
short read archive
- TACE
TNF cleavage enzyme
- Tconv
CD4 T conventional
- Teff
T effector cell
- Tg
transgenic
- TMB
3,3′,5,5′-tertamethylbenzidine
- TMS
trimethoprim/sulfamethoxazole
- TNF
tumor necrosis factor-α
- TNFR1
TNF receptor type 1
- TNFR2
TNF receptor type 2
- Treg
T regulatory cell
- VitD
vitamin D
Footnotes
Conflict of interest disclosure
The authors declare no competing financial interests.
References
- 1.Selmaj K, Raine CS, Cannella B, and Brosnan CF. 1991. Identification of lymphotoxin and tumor necrosis factor in multiple sclerosis lesions. J Clin Invest 87: 949–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Begolka WS, Vanderlugt CL, Rahbe SM, and Miller SD. 1998. Differential expression of inflammatory cytokines parallels progression of central nervous system pathology in two clinically distinct models of multiple sclerosis. J Immunol 161: 4437–4446. [PubMed] [Google Scholar]
- 3.Powell MB, Mitchell D, Lederman J, Buckmeier J, Zamvil SS, Graham M, Ruddle NH, and Steinman L. 1990. Lymphotoxin and tumor necrosis factor-alpha production by myelin basic protein-specific T cell clones correlates with encephalitogenicity. Int Immunol 2: 539–544. [DOI] [PubMed] [Google Scholar]
- 4.Lenercept-Group. 1999. TNF neutralization in MS: results of a randomized, placebo-controlled multicenter study. The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. Neurology 53: 457–465. [PubMed] [Google Scholar]
- 5.Feldmann M, and Steinman L. 2005. Design of effective immunotherapy for human autoimmunity. Nature 435: 612–619. [DOI] [PubMed] [Google Scholar]
- 6.Blakemore WF, and Irvine KA. 2008. Endogenous or exogenous oligodendrocytes for remyelination. Journal of the Neurological Sciences 265: 43–46. [DOI] [PubMed] [Google Scholar]
- 7.Wingerchuk DM 2006. Neuromyelitis optica. Int MS J 13: 42–50. [PubMed] [Google Scholar]
- 8.Bettelli E, Korn T, Oukka M, and Kuchroo VK. 2008. Induction and effector functions of T(H)17 cells. Nature 453: 1051–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lovett-Racke AE, Yang Y, and Racke MK. 2010. Th1 versus Th17: are T cell cytokines relevant in multiple sclerosis? Biochimica et Biophysica Acta 1812: 246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hjelmstrom P, Juedes AE, Fjell J, and Ruddle NH. 1998. B-cell-deficient mice develop experimental allergic encephalomyelitis with demyelination after myelin oligodendrocyte glycoprotein sensitization. J Immunol 161: 4480–4483. [PubMed] [Google Scholar]
- 11.Lyons JA, San M, Happ MP, and Cross AH. 1999. B cells are critical to induction of experimental allergic encephalomyelitis by protein but not by a short encephalitogenic peptide. Eur J Immunol 29: 3432–3439. [DOI] [PubMed] [Google Scholar]
- 12.Oliver AR, Lyon GM, and Ruddle NH. 2003. Rat and human myelin oligodendrocyte glycoproteins induce experimental autoimmune encephalomyelitis by different mechanisms in C57BL/6 mice. J Immunol 171: 462–468. [DOI] [PubMed] [Google Scholar]
- 13.Jarius S, Paul F, Franciotta D, Waters P, Zipp F, Hohlfeld R, Vincent A, and Wildemann B. 2008. Mechanisms of disease: aquaporin-4 antibodies in neuromyelitis optica. Nature clinical practice. Neurology 4: 202–214. [DOI] [PubMed] [Google Scholar]
- 14.Varrin-Doyer M, Spencer CM, Schulze-Topphoff U, Nelson PA, Stroud RM, Cree BA, and Zamvil SS. 2012. Aquaporin 4-specific T cells in neuromyelitis optica exhibit a Th17 bias and recognize Clostridium ABC transporter. Ann Neurol 72: 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones MV, Huang H, Calabresi PA, and Levy M. 2015. Pathogenic aquaporin-4 reactive T cells are sufficient to induce mouse model of neuromyelitis optica. Acta Neuropathol Commun 3: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vandenabeele P, Declercq W, Beyaert R, and Fiers W. 1995. Two tumour necrosis factor receptors: structure and function. Trends Cell Biol 5: 392–399. [DOI] [PubMed] [Google Scholar]
- 17.Vinay DS, and Kwon BS. 2011. The tumour necrosis factor/TNF receptor superfamily: therapeutic targets in autoimmune diseases. Clinical and Experimental Immunology 164: 145–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Faustman D, and Davis M. 2010. TNF receptor 2 pathway: drug target for autoimmune diseases. Nature Reviews. Drug Discovery 9: 482–493. [DOI] [PubMed] [Google Scholar]
- 19.Chen X, Baumel M, Mannel DN, Howard OM, and Oppenheim JJ. 2007. Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+CD25+ T regulatory cells. J Immunol 179: 154–161. [DOI] [PubMed] [Google Scholar]
- 20.Chen X, Subleski JJ, Hamano R, Howard OM, Wiltrout RH, and Oppenheim JJ. 2010. Co-expression of TNFR2 and CD25 identifies more of the functional CD4+FOXP3+ regulatory T cells in human peripheral blood. Eur J Immunol 40: 1099–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gregory AP, Dendrou CA, Attfield KE, Haghikia A, Xifara DK, Butter F, Poschmann G, Kaur G, Lambert L, Leach OA, Promel S, Punwani D, Felce JH, Davis SJ, Gold R, Nielsen FC, Siegel RM, Mann M, Bell JI, McVean G, and Fugger L. 2012. TNF receptor 1 genetic risk mirrors outcome of anti-TNF therapy in multiple sclerosis. Nature 488: 508–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eugster HP, Frei K, Bachmann R, Bluethmann H, Lassmann H, and Fontana A. 1999. Severity of symptoms and demyelination in MOG-induced EAE depends on TNFR1. Eur J Immunol 29: 626–632. [DOI] [PubMed] [Google Scholar]
- 23.Evangelidou M, Karamita M, Vamvakas SS, Szymkowski DE, and Probert L. 2014. Altered expression of oligodendrocyte and neuronal marker genes predicts the clinical onset of autoimmune encephalomyelitis and indicates the effectiveness of multiple sclerosis-directed therapeutics. J Immunol 192: 4122–4133. [DOI] [PubMed] [Google Scholar]
- 24.Kohm AP, Carpentier PA, Anger HA, and Miller SD. 2002. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol 169: 4712–4716. [DOI] [PubMed] [Google Scholar]
- 25.Zhang X, Koldzic DN, Izikson L, Reddy J, Nazareno RF, Sakaguchi S, Kuchroo VK, and Weiner HL. 2004. IL-10 is involved in the suppression of experimental autoimmune encephalomyelitis by CD25+CD4+ regulatory T cells. Int Immunol 16: 249–256. [DOI] [PubMed] [Google Scholar]
- 26.McGeachy MJ, Stephens LA, and Anderton SM. 2005. Natural recovery and protection from autoimmune encephalomyelitis: contribution of CD4+CD25+ regulatory cells within the central nervous system. J Immunol 175: 3025–3032. [DOI] [PubMed] [Google Scholar]
- 27.Hyttinen V, Kaprio J, Kinnunen L, Koskenvuo M, and Tuomilehto J. 2003. Genetic liability of type 1 diabetes and the onset age among 22,650 young Finnish twin pairs: a nationwide follow-up study. Diabetes 52: 1052–1055. [DOI] [PubMed] [Google Scholar]
- 28.Gaboriau-Routhiau V, Rakotobe S, Lecuyer E, Mulder I, Lan A, Bridonneau C, Rochet V, Pisi A, De Paepe M, Brandi G, Eberl G, Snel J, Kelly D, and Cerf-Bensussan N. 2009. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity 31: 677–689. [DOI] [PubMed] [Google Scholar]
- 29.Lee YK, Menezes JS, Umesaki Y, and Mazmanian SK. 2011. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A 108Suppl 1: 4615–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berer K, Mues M, Koutrolos M, Rasbi ZA, Boziki M, Johner C, Wekerle H, and Krishnamoorthy G. 2011. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature 479: 538–541. [DOI] [PubMed] [Google Scholar]
- 31.Muller U, Jongeneel CV, Nedospasov SA, Lindahl KF, and Steinmetz M. 1987. Tumour necrosis factor and lymphotoxin genes map close to H-2D in the mouse major histocompatibility complex. Nature 325: 265–267. [DOI] [PubMed] [Google Scholar]
- 32.Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, and Kuchroo VK. 2003. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med 197: 1073–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bettelli E, Baeten D, Jager A, Sobel RA, and Kuchroo VK. 2006. Myelin oligodendrocyte glycoprotein-specific T and B cells cooperate to induce a Devic-like disease in mice. J Clin Invest 116: 2393–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bailey SL, Schreiner B, McMahon EJ, and Miller SD. 2007. CNS myeloid DCs presenting endogenous myelin peptides ‘preferentially’ polarize CD4+ T(H)-17 cells in relapsing EAE. Nat Immunol 8: 172–180. [DOI] [PubMed] [Google Scholar]
- 35.Yu Z, and Morrison M. 2004. Improved extraction of PCR-quality community DNA from digesta and fecal samples. BioTechniques 36: 808–812. [DOI] [PubMed] [Google Scholar]
- 36.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, and Knight R. 2011. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A 108Suppl 1: 4516–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walters WA, Caporaso JG, Lauber CL, Berg-Lyons D, Fierer N, and Knight R. 2011. PrimerProspector: de novo design and taxonomic analysis of barcoded polymerase chain reaction primers. Bioinformatics 27: 1159–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Loy A, Maixner F, Wagner M, and Horn M. 2007. probeBase--an online resource for rRNA-targeted oligonucleotide probes: new features 2007. Nucleic Acids Res 35: D800–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Magoc T, and Salzberg SL. 2011. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27: 2957–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuczynski J, Stombaugh J, Walters WA, Gonzalez A, Caporaso JG, and Knight R. 2011. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr Protoc Bioinformatics Chapter 10: Unit 10 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, and Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, and Andersen GL. 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72: 5069–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller PG, Bonn MB, and McKarns SC. 2015. Transmembrane TNF-TNFR2 Impairs Th17 Differentiation by Promoting Il2 Expression. J Immunol 195: 2633–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suvannavejh GC, Lee HO, Padilla J, Dal Canto MC, Barrett TA, and Miller SD. 2000. Divergent roles for p55 and p75 tumor necrosis factor receptors in the pathogenesis of MOG(35–55)-induced experimental autoimmune encephalomyelitis. Cell Immunol 205: 24–33. [DOI] [PubMed] [Google Scholar]
- 45.Kassiotis G, and Kollias G. 2001. Uncoupling the proinflammatory from the immunosuppressive properties of tumor necrosis factor (TNF) at the p55 TNF receptor level: implications for pathogenesis and therapy of autoimmune demyelination. J Exp Med 193: 427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Williams SK, Maier O, Fischer R, Fairless R, Hochmeister S, Stojic A, Pick L, Haar D, Musiol S, Storch MK, Pfizenmaier K, and Diem R. 2014. Antibody-mediated inhibition of TNFR1 attenuates disease in a mouse model of multiple sclerosis. PLoS One 9: e90117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krishnamoorthy G, Lassmann H, Wekerle H, and Holz A. 2006. Spontaneous opticospinal encephalomyelitis in a double-transgenic mouse model of autoimmune T cell/B cell cooperation. J Clin Invest 116: 2385–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McKarns SC, and Schwartz RH. 2008. Biphasic regulation of Il2 transcription in CD4+ T cells: roles for TNF-alpha receptor signaling and chromatin structure. J Immunol 181: 1272–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berger T, Weerth S, Kojima K, Linington C, Wekerle H, and Lassmann H. 1997. Experimental autoimmune encephalomyelitis: the antigen specificity of T lymphocytes determines the topography of lesions in the central and peripheral nervous system. Laboratory Investigation; A Journal of Technical Methods and Pathology 76: 355–364. [PubMed] [Google Scholar]
- 50.O’Riordan JI, Gallagher HL, Thompson AJ, Howard RS, Kingsley DP, Thompson EJ, McDonald WI, and Miller DH. 1996. Clinical, CSF, and MRI findings in Devic’s neuromyelitis optica. Journal of Neurology, Neurosurgery, and Psychiatry 60: 382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reading NC, and Kasper DL. 2011. The starting lineup: key microbial players in intestinal immunity and homeostasis. Front Microbiol 2: 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robinson WH, Genovese MC, and Moreland LW. 2001. Demyelinating and neurologic events reported in association with tumor necrosis factor alpha antagonism: by what mechanisms could tumor necrosis factor alpha antagonists improve rheumatoid arthritis but exacerbate multiple sclerosis? Arthritis and Rheumatism 44: 1977–1983. [DOI] [PubMed] [Google Scholar]
- 53.Sotgiu S, Pugliatti M, Fois ML, Arru G, Sanna A, Sotgiu MA, and Rosati G. 2004. Genes, environment, and susceptibility to multiple sclerosis. Neurobiol Dis 17: 131–143. [DOI] [PubMed] [Google Scholar]
- 54.Rovira A, Swanton J, Tintore M, Huerga E, Barkhof F, Filippi M, Frederiksen JL, Langkilde A, Miszkiel K, Polman C, Rovaris M, Sastre-Garriga J, Miller D, and Montalban X. 2009. A single, early magnetic resonance imaging study in the diagnosis of multiple sclerosis. Arch Neurol 66: 587–592. [DOI] [PubMed] [Google Scholar]
- 55.Oh J, and Levy M. 2012. Neuromyelitis optica: an antibody-mediated disorder of the central nervous system. Neurol Res Int 2012: 460825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rubtsova K, Marrack P, and Rubtsov AV. 2015. Sexual dimorphism in autoimmunity. J Clin Invest 125: 2187–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cruz-Orengo L, Daniels BP, Dorsey D, Basak SA, Grajales-Reyes JG, McCandless EE, Piccio L, Schmidt RE, Cross AH, Crosby SD, and Klein RS. 2014. Enhanced sphingosine-1-phosphate receptor 2 expression underlies female CNS autoimmunity susceptibility. J Clin Invest 124: 2571–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burek M, Arias-Loza PA, Roewer N, and Forster CY. 2010. Claudin-5 as a novel estrogen target in vascular endothelium. Arterioscler Thromb Vasc Biol 30: 298–304. [DOI] [PubMed] [Google Scholar]
- 59.Becker KG, Simon RM, Bailey-Wilson JE, Freidlin B, Biddison WE, McFarland HF, and Trent JM. 1998. Clustering of non-major histocompatibility complex susceptibility candidate loci in human autoimmune diseases. Proc Natl Acad Sci U S A 95: 9979–9984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brodin P, Jojic V, Gao T, Bhattacharya S, Angel CJ, Furman D, Shen-Orr S, Dekker CL, Swan GE, Butte AJ, Maecker HT, and Davis MM. 2015. Variation in the human immune system is largely driven by non-heritable influences. Cell 160: 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pollinger B, Krishnamoorthy G, Berer K, Lassmann H, Bosl MR, Dunn R, Domingues HS, Holz A, Kurschus FC, and Wekerle H. 2009. Spontaneous relapsing-remitting EAE in the SJL/J mouse: MOG-reactive transgenic T cells recruit endogenous MOG-specific B cells. J Exp Med 206: 1303–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Markle JG, Frank DN, Mortin-Toth S, Robertson CE, Feazel LM, Rolle-Kampczyk U, von Bergen M, McCoy KD, Macpherson AJ, and Danska JS. 2013. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science 339: 1084–1088. [DOI] [PubMed] [Google Scholar]
- 63.Yurkovetskiy L, Burrows M, Khan AA, Graham L, Volchkov P, Becker L, Antonopoulos D, Umesaki Y, and Chervonsky AV. 2013. Gender bias in autoimmunity is influenced by microbiota. Immunity 39: 400–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cohen JI 2000. Epstein-Barr virus infection. N Engl J Med 343: 481–492. [DOI] [PubMed] [Google Scholar]
- 65.Thacker EL, Mirzaei F, and Ascherio A. 2006. Infectious mononucleosis and risk for multiple sclerosis: a meta-analysis. Ann Neurol 59: 499–503. [DOI] [PubMed] [Google Scholar]
- 66.Westall FC 2006. Molecular mimicry revisited: gut bacteria and multiple sclerosis. J Clin Microbiol 44: 2099–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Berer K, and Krishnamoorthy G. 2012. Commensal gut flora and brain autoimmunity: a love or hate affair? Acta Neuropathol 123: 639–651. [DOI] [PubMed] [Google Scholar]
- 68.Mazmanian SK, Liu CH, Tzianabos AO, and Kasper DL. 2005. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 122: 107–118. [DOI] [PubMed] [Google Scholar]
- 69.Wang Y, Telesford KM, Ochoa-Reparaz J, Haque-Begum S, Christy M, Kasper EJ, Wang L, Wu Y, Robson SC, Kasper DL, and Kasper LH. 2014. An intestinal commensal symbiosis factor controls neuroinflammation via TLR2-mediated CD39 signalling. Nat Commun 5: 4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ochoa-Reparaz J, Mielcarz DW, Haque-Begum S, and Kasper LH. 2010. Induction of a regulatory B cell population in experimental allergic encephalomyelitis by alteration of the gut commensal microflora. Gut Microbes 1: 103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ezendam J, de Klerk A, Gremmer ER, and van Loveren H. 2008. Effects of Bifidobacterium animalis administered during lactation on allergic and autoimmune responses in rodents. Clin Exp Immunol 154: 424–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lavasani S, Dzhambazov B, Nouri M, Fak F, Buske S, Molin G, Thorlacius H, Alenfall J, Jeppsson B, and Westrom B. 2010. A novel probiotic mixture exerts a therapeutic effect on experimental autoimmune encephalomyelitis mediated by IL-10 producing regulatory T cells. PLoS One 5: e9009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cantarel BL, Waubant E, Chehoud C, Kuczynski J, DeSantis TZ, Warrington J, Venkatesan A, Fraser CM, and Mowry EM. 2015. Gut microbiota in multiple sclerosis: possible influence of immunomodulators. J Investig Med 63: 729–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thorkildsen LT, Nwosu FC, Avershina E, Ricanek P, Perminow G, Brackmann S, Vatn MH, and Rudi K. 2013. Dominant fecal microbiota in newly diagnosed untreated inflammatory bowel disease patients. Gastroenterol Res Pract 2013: 636785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Carlsson AH, Yakymenko O, Olivier I, Hakansson F, Postma E, Keita AV, and Soderholm JD. 2013. Faecalibacterium prausnitzii supernatant improves intestinal barrier function in mice DSS colitis. Scand J Gastroenterol 48: 1136–1144. [DOI] [PubMed] [Google Scholar]
- 76.Kitley J, Waters P, Woodhall M, Leite MI, Murchison A, George J, Kuker W, Chandratre S, Vincent A, and Palace J. 2014. Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: a comparative study. JAMA Neurol 71: 276–283. [DOI] [PubMed] [Google Scholar]
- 77.Derrien M, Van Baarlen P, Hooiveld G, Norin E, Muller M, and de Vos WM. 2011. Modulation of Mucosal Immune Response, Tolerance, and Proliferation in Mice Colonized by the Mucin-Degrader Akkermansia muciniphila. Front Microbiol 2: 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lukovac S, Belzer C, Pellis L, Keijser BJ, de Vos WM, Montijn RC, and Roeselers G. 2014. Differential modulation by Akkermansia muciniphila and Faecalibacterium prausnitzii of host peripheral lipid metabolism and histone acetylation in mouse gut organoids. MBio 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, Benoist C, and Mathis D. 2012. PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 486: 549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hansen CH, Krych L, Nielsen DS, Vogensen FK, Hansen LH, Sorensen SJ, Buschard K, and Hansen AK. 2012. Early life treatment with vancomycin propagates Akkermansia muciniphila and reduces diabetes incidence in the NOD mouse. Diabetologia 55: 2285–2294. [DOI] [PubMed] [Google Scholar]
- 81.Zhang X, Shen D, Fang Z, Jie Z, Qiu X, Zhang C, Chen Y, and Ji L. 2013. Human gut microbiota changes reveal the progression of glucose intolerance. PLoS One 8: e71108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, Guiot Y, Derrien M, Muccioli GG, Delzenne NM, de Vos WM, and Cani PD. 2013. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci U S A 110: 9066–9071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.De Angelis M, Piccolo M, Vannini L, Siragusa S, De Giacomo A, Serrazzanetti DI, Cristofori F, Guerzoni ME, Gobbetti M, and Francavilla R. 2013. Fecal microbiota and metabolome of children with autism and pervasive developmental disorder not otherwise specified. PLoS One 8: e76993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Biagi E, Candela M, Centanni M, Consolandi C, Rampelli S, Turroni S, Severgnini M, Peano C, Ghezzo A, Scurti M, Salvioli S, Franceschi C, and Brigidi P. 2014. Gut microbiome in Down syndrome. PLoS One 9: e112023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Moon C, Baldridge MT, Wallace MA, Burnham CA, Virgin HW, and Stappenbeck TS. 2015. Vertically transmitted faecal IgA levels determine extra-chromosomal phenotypic variation. Nature 521: 90–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mantis NJ, Rol N, and Corthesy B. 2011. Secretory IgA’s complex roles in immunity and mucosal homeostasis in the gut. Mucosal Immunol 4: 603–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fujimoto T, Imaeda H, Takahashi K, Kasumi E, Bamba S, Fujiyama Y, and Andoh A. 2013. Decreased abundance of Faecalibacterium prausnitzii in the gut microbiota of Crohn’s disease. J Gastroenterol Hepatol 28: 613–619. [DOI] [PubMed] [Google Scholar]
- 88.Brahe LK, Le Chatelier E, Prifti E, Pons N, Kennedy S, Hansen T, Pedersen O, Astrup A, Ehrlich SD, and Larsen LH. 2015. Specific gut microbiota features and metabolic markers in postmenopausal women with obesity. Nutr Diabetes 5: e159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ercolini AM, and Miller SD. 2007. Molecular mimics can induce novel self peptide-reactive CD4+ T cell clonotypes in autoimmune disease. J Immunol 179: 6604–6612. [DOI] [PubMed] [Google Scholar]
- 90.de Vos AF, van Meurs M, Brok HP, Boven LA, Hintzen RQ, van der Valk P, Ravid R, Rensing S, Boon L, t Hart BA, and Laman JD. 2002. Transfer of central nervous system autoantigens and presentation in secondary lymphoid organs. J Immunol 169: 5415–5423. [DOI] [PubMed] [Google Scholar]
- 91.Round JL, and Mazmanian SK. 2010. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc Natl Acad Sci U S A 107: 12204–12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mohamadzadeh M, and Klaenhammer TR. 2008. Specific Lactobacillus species differentially activate Toll-like receptors and downstream signals in dendritic cells. Expert Rev Vaccines 7: 1155–1164. [DOI] [PubMed] [Google Scholar]
- 93.Shulzhenko N, Morgun A, Hsiao W, Battle M, Yao M, Gavrilova O, Orandle M, Mayer L, Macpherson AJ, McCoy KD, Fraser-Liggett C, and Matzinger P. 2011. Crosstalk between B lymphocytes, microbiota and the intestinal epithelium governs immunity versus metabolism in the gut. Nat Med 17: 1585–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fox HS, Bond BL, and Parslow TG. 1991. Estrogen regulates the IFN-gamma promoter. J Immunol 146: 4362–4367. [PubMed] [Google Scholar]
- 95.Johnson-Leger C, Hasbold J, Holman M, and Klaus GG. 1997. The effects of IFN-gamma on CD40-mediated activation of B cells from X-linked immunodeficient or normal mice. J Immunol 159: 1150–1159. [PubMed] [Google Scholar]
- 96.Finkelman FD, Holmes J, Katona IM, Urban JF Jr., Beckmann MP, Park LS, Schooley KA, Coffman RL, Mosmann TR, and Paul WE. 1990. Lymphokine control of in vivo immunoglobulin isotype selection. Annu Rev Immunol 8: 303–333. [DOI] [PubMed] [Google Scholar]
- 97.Ito A, Bebo BF Jr., Matejuk A, Zamora A, Silverman M, Fyfe-Johnson A, and Offner H. 2001. Estrogen treatment down-regulates TNF-alpha production and reduces the severity of experimental autoimmune encephalomyelitis in cytokine knockout mice. J Immunol 167: 542–552. [DOI] [PubMed] [Google Scholar]
- 98.Jawaheer D, Olsen J, and Hetland ML. 2011. Sex differences in response to anti-tumor necrosis factor therapy in early and established rheumatoid arthritis -- results from the DANBIO registry. The Journal of Rheumatology 39: 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Saevarsdottir S, Wallin H, Seddighzadeh M, Ernestam S, Geborek P, Petersson IF, Bratt J, and van Vollenhoven RF. 2010. Predictors of response to methotrexate in early DMARD naive rheumatoid arthritis: results from the initial open-label phase of the SWEFOT trial. Annals of the Rheumatic Diseases 70: 469–475. [DOI] [PubMed] [Google Scholar]
- 100.Makinen H, Hannonen P, and Sokka T. 2008. Sex: a major predictor of remission as measured by 28-joint Disease Activity Score (DAS28) in early rheumatoid arthritis? Annals of the Rheumatic Diseases 67: 1052–1053. [DOI] [PubMed] [Google Scholar]
- 101.Burmester GR, Ferraccioli G, Flipo RM, Monteagudo-Saez I, Unnebrink K, Kary S, and Kupper H. 2008. Clinical remission and/or minimal disease activity in patients receiving adalimumab treatment in a multinational, open-label, twelve-week study. Arthritis and Rheumatism 59: 32–41. [DOI] [PubMed] [Google Scholar]
- 102.Wang M, Crisostomo PR, Markel TA, Wang Y, and Meldrum DR. 2008. Mechanisms of sex differences in TNFR2-mediated cardioprotection. Circulation 118: S38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dayer Schneider J, Seibold I, Saxer-Sekulic N, Paredes BE, Saurer L, and Mueller C. 2009. Lack of TNFR2 expression by CD4(+) T cells exacerbates experimental colitis. Eur J Immunol 39: 1743–1753. [DOI] [PubMed] [Google Scholar]
- 104.Housley WJ, Adams CO, Nichols FC, Puddington L, Lingenheld EG, Zhu L, Rajan TV, and Clark RB. 2011. Natural but not inducible regulatory T cells require TNF-alpha signaling for in vivo function. J Immunol 186: 6779–6787. [DOI] [PubMed] [Google Scholar]
- 105.Peschon JJ, Torrance DS, Stocking KL, Glaccum MB, Otten C, Willis CR, Charrier K, Morrissey PJ, Ware CB, and Mohler KM. 1998. TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J Immunol 160: 943–952. [PubMed] [Google Scholar]