

Abstract
Purpose
To report a series of patients with occlusive retinal vasculitis associated with systemic sclerosis (SSc) and elevated antiphospholipid antibody titers.
Method
Case series. Main outcome measures included clinical and fluorescein angiographic findings at presentation and over time.
Observations
Case 1 - A 61-year-old woman initially diagnosed with idiopathic, bilateral panuveitis and retinal vasculitis causing peripheral nonperfusion was subsequently diagnosed with limited cutaneous systemic sclerosis (lcSSc). Her ocular inflammation and retinal vasculitis were controlled with topical and periocular corticosteroids, but she eventually developed peripheral retinal vascular occlusion that progressed to macular ischemia 11 years after presentation. Repeat serologic evaluation detected interval development of antiphospholipid antibodies. Case 2 – A 58-year-old woman was found to have bilateral peripheral nonperfusion and retinal neovascularization in her right eye. Given her elevated hemoglobin A1c of 8.5%, she was diagnosed with presumed proliferative diabetic retinopathy. Three years after initial presentation, she was diagnosed with lcSSc. Subsequent serum workup detected elevated B2-glycoprotein antibody titers. Her peripheral nonperfusion progressed despite adequate glycemic control, resulting in further neovascularization in each eye. Case 3 – A 40-year-old woman with diffuse cutaneous systemic sclerosis (dcSSc) and elevated titers of anti-cardiolipin antibodies developed multiple branch retinal artery occlusions with subsequent neovascularization of the retina, optic disc, and angle in the right eye.
Conclusion and Importance
Vision-threatening occlusive retinal vasculitis may develop in select patients with SSc. The presence of elevated anti-phospholipid antibody titers may confer increased risk for this vision-threatening complication.
Keywords: Cardiolipin, B2 glycoprotein, CREST syndrome, Phosphotidylserine, Retinal ischemia, Scleroderma
1. Introduction
Scleroderma describes a group of related connective tissue diseases of unknown etiology that can be localized, such as in morphea, linear Scleroderma, linear scleroderma en coup de sabre (CDS), and progressive hemifacial atrophy (PHA; Parry-Romberg syndrome), or systemic with involvement of internal organs, as seen in limited cutaneous systemic sclerosis (lcSSc; formerly CREST syndrome; comprised of Calcinosis, Raynaud's phenomenon, Esophageal dysmotility, Sclerodactyly, and Telangiectasia) and diffuse cutaneous systemic sclerosis (dcSSc).1,2 Occlusive retinal vasculitis is a rare complication of scleroderma that has been described in two patients with lcSSc3 and one patient with both PHA and CDS.4 We present three additional patients with vision-threatening occlusive retinal vasculitis who were found to have either lcSSc or dcSSc in conjunction with elevated antiphospholipid antibody titers.
2. Case 1
A 61-year-old Asian woman was found to have optic disc hyperemia and isolated peripheral intraretinal hemorrhages in her left eye. Fluorescein angiography (FA) revealed optic disc and vascular leakage with scattered areas of peripheral nonperfusion in the left eye. Extensive serum evaluation for infectious, autoimmune, and coagulopathy markers, including antiphospholipid antibodies, was unremarkable, but her hemoglobin A1c (6.5%) and cholesterol levels were elevated. On subsequent examination two years later, the patient was noted to have mild, bilateral anterior chamber and vitreous inflammation. Repeat FA showed diffuse vascular leakage with patchy peripheral areas of nonperfusion in both eyes (Fig. 1 – A, B) and formation of small vessel telangiectasias in the mid-periphery of the left eye (Fig. 1 – C, D). The patient was diagnosed with bilateral panuveitis with occlusive retinal vasculitis from an unknown etiology, and was treated with topical corticosteroid eyedrops and intermittent intravitreal sustained-release dexamethasone implants. Approximately nine years after presentation, the patient developed a non-healing ulcer on her left index finger, prompting a workup that eventually led to the diagnosis of incomplete lcSSc, based on presence of sclerodactyly, finger ischemia (Fig. 2), telangiectasias, and dysphagia. No serum rheumatologic aberrations were identified. Two years later, the patient's vision declined from 20/32 and 20/40 to 20/80 and 20/63 in the right and left eyes, respectively. She was found to have moderate anterior chamber inflammation and macular edema with cotton wool spots in each eye (Fig. 3 – A, B). Fluorescein angiography identified leakage of the vessels, optic disc, and macula, and areas of capillary nonperfusion in the nasal macula and periphery in both eyes (Fig. 3 – C, D). Extensive serum evaluation for rheumatologic markers found elevated titers of cardiolipin (IgM) and phosphotidylserine (IgM) antibodies.
Fig. 1.

Case 1 (At presentation): A: Late phase fluorescein angiogram of the right eye showing diffuse vascular leakage. B: Mid-phase fluorescein angiogram of the left eye showing mild leakage at the optic disc, and peripheral areas of capillary nonperfusion. C: Fundus photograph of the right eye showing mild epiretinal membrane formation. D. Fundus photograph of the left eye showing mild optic disc hyperemia and mild epiretinal membrane formation.
Fig. 2.
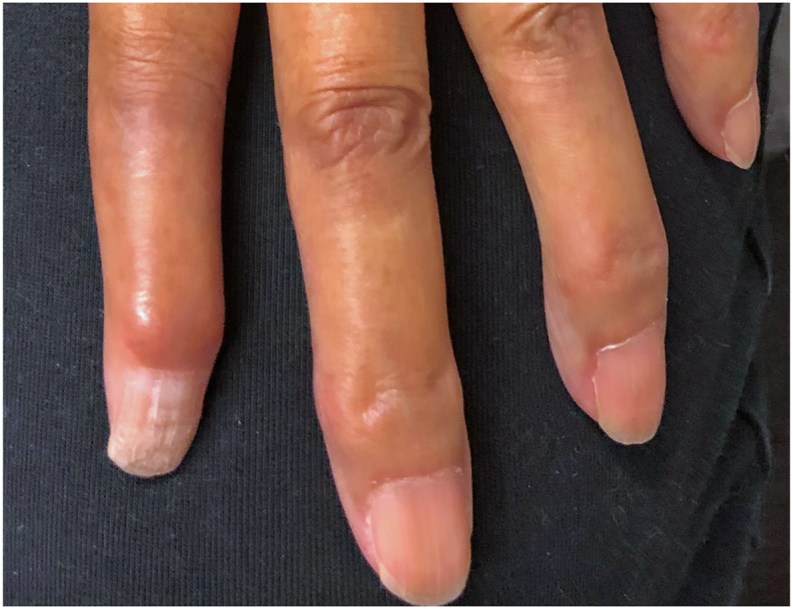
Case 1 (11 years after presentation): External photograph of the left hand showing hardening and atrophy of the distal digits, characteristic of systemic sclerosis (SSc).
Fig. 3.

Case 1 (11 years after presentation): A: Fundus photograph of the right eye showing mild epiretinal membrane. B: Fundus photograph of the left eye showing posterior vitreous detachment, mild epiretinal membrane formation. C: Mid-phase fluorescein angiogram of the right eye showing leakage from the optic disc, staining of temporal peripapillary vessels, and areas of non-perfusion. D: Mid-phase fluorescein angiogram of the left eye showing leakage from the optic disc, leakage from inferotemporal peripapillary vessels, and areas of nonperfusion.
3. Case 2
A 58-year-old Caucasian woman with systemic hypertension and a 25-year history of type II diabetes mellitus was referred for evaluation of bilateral retinal hemorrhages. Examination of her posterior segment revealed scattered microaneurysms and intraretinal hemorrhages in the periphery of each eye, and an area of neovascularization with surrounding cotton wool spots in the nasal mid-periphery of the right eye. Fluorescein angiography showed leakage consistent with neovascularization, and peripheral capillary nonperfusion on the right. Since her hemoglobin A1c was elevated (8.5%), she was presumed to have non-proliferative retinopathy of the left eye and proliferative diabetic retinopathy of the right eye, prompting panretinal photocoagulation (PRP) on the right. Three years after initial presentation, she was diagnosed with incomplete lcSSc, based on diffuse scattered telangiectasias, sclerodactyly, a history of Raynaud's phenomenon, and positive anti-centromere antibody titers. Despite maintenance of glycemic control with A1c ranging from 6.5% to 7.5% over the next seven years, she developed recurrent vitreous hemorrhages in the right eye that were treated with intravitreal bevacizumab injections, and worsening peripheral non-perfusion (Fig. 4A) and eventual neovascularization of the peripheral retina and optic disc in the left eye (Fig. 4B). On subsequent rheumatologic testing, she was found to have elevated titers of beta-2 glycoprotein 1 (IgM) antibodies.
Fig. 4.

Case 2: A: Fundus photograph of the left eye showing an intraretinal hemorrhage and sclerotic vessels in the temporal periphery. B: Mid-phase fluorescein angiogram of the left eye showing a leakage from the optic disc, a tuft of retinal neovascularization (arrow), punctate areas of hyperfluorescence, and capillary nonperfusion in the temporal periphery.
4. Case 3
A 40-year-old woman with history of systemic hypertension and dcSSc that was diagnosed based on a history of sclerodactyly, nonhealing skin ulcers, esophageal dysmotility, interstitial lung disease, and Raynaud's phenomenon was referred for decreased vision in both eyes. Her visual acuity was HM in the right eye and 20/60 in the left. Intraocular pressures were normal. Anterior segment examination revealed diffuse neovascularization of the angle in the right eye, and posterior segment examination revealed numerous branch retinal artery occlusions and a small preretinal hemorrhage in the right eye and intraretinal hemorrhages in the left eye (Fig. 5A). She denied any hearing loss. Anti-cardiolipin antibody titers were strongly positive. Fluorescein angiography confirmed the presence of multiple branch retinal artery occlusions, and also revealed optic disc neovascularization and extensive peripheral nonperfusion (Fig. 5B). She was treated with PRP on the right, which controlled her proliferative retinopathy.
Fig. 5.

Case 3: A: Fundus photography of the right eye showing multiple sclerosed retinal arterioles, vascular remodeling with telangiectasis, and intraretinal hemorrhages. B – Fluorescein angiography of the right eye showing leakage from the optic disc consistent with neovascularization, blockage from a pre-retinal hemorrhage, and arteriolar drop out with a relatively avascular periphery.
5. Discussion
We present three cases of occlusive retinal vasculitis associated with systemic sclerosis (SSc) and elevated antiphospholipid titers (see Table 1). Two of the three of patients also had elevated Hg A1c levels and an equal number had systemic hypertension. Bilateral involvement was present in two patients, resulting in five affected eyes. All eyes were found to have intraretinal hemorrhages and peripheral capillary nonperfusion, with retinal neovascularization also present in three eyes, macular non-perfusion in two eyes, and neovascularization of the disc and angle in one eye.
Table 1.
Summary of case histories, clinical findings, and treatments.
| Case | 1 | 2 | 3 |
|---|---|---|---|
| Age (years) | 61 | 58 | 40 |
| Gender | F | F | F |
| Initial VA | 20/32 OD, 20/40 OS | 20/20 OD, 20/25 OS | HM OD, 20/60 OS |
| Scleroderma subtype | lcSSC | lcSSC | dcSSC |
| Comorbidities (duration in years) | Type II Diabetes Mellitus (11) | Type II Diabetes Mellitus (32) Systemic hypertension (?) |
Systemic hypertension (?) |
| Location of Capillary Non-perfusion |
Macula and Peripheral Retina OU | Peripheral Retina OU | Peripheral Retina OD |
| Presence/Location of Neovascularization | None | Periphery OU Optic Disc OS |
Angle, Periphery, and Optic Disc OD |
| Elevated Antiphospholipid Antibodies (isotype) |
Cardiolipin (IgM) and Phosphotidylserine (IgM) | Beta-2 glycoprotein 1 (IgM) | Cardiolipin (N/A) |
| Treatment | Topical corticosteroids; Sustained-release dexamethasone implant; Oral corticosteroids; 325 mg ASA PO/day |
PRP OD; IVT Bevacizumab OU; Oral corticosteroids; 325 mg ASA PO/day |
PRP OD |
| Final VA | 20/50 OD, 20/63 OS (pseudophakic OU) | 20/25 OD, 20/16 OS | N/A |
F = female, VA = visual acuity, OD = right eye, OS = left eye, lcSSC = limited cutaneous systemic sclerosis, dcSSC = diffuse cutaneous systemic sclerosis, ? = unknown, OU = both eyes, ASA = acetylsalicyclic acid, PO = taken by mouth, PRP = panretinal photocoagulation, IVT = intravitreal, N/A = not available.
The diagnosis of SSc is established from careful clinical examination, antibody testing, and ancillary testing.1 On initial and subsequent examinations, the skin is examined for thickening, fingertip lesions, telangiectasis, abnormal nail fold capillaries, and the Raynaud phenomena. Close monitoring of blood pressure is also required as baseline or new-onset systemic hypertension may indicate renal involvement. Serial assessment of urine protein content is another means of monitoring renal function. A chest computed tomography scan should be obtained to assess for evidence of interstitial lung disease or a dilated, air-filled esophagus from long-standing esophageal dysmotility. Earlier disease can be detected by means of pulmonary function testing, endoscopy, esophageal manometry, and barium swallow studies. A transthoracic echocardiography should be obtained when there is suspicion for pulmonary arterial hypertension or pericardial effusion. More than 90% of the cases of SSc will have elevated titers of anti-nuclear antibodies.1 Patients with SSc may also possess either anti-centromere, anti-topoisomerase I (Scl-70), anti-RNA polymerase III, or anti-U3-RNP (fibrillarin) antibodies.1 Of these, a positive anti-centromere titer is specific to lcSSc, whereas the presence of the other three are associated with dcSSc.1 A patient is diagnosed with the lcSSc when the manifestations of SSc are limited to calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia.1
While SSc is a long-recognized autoimmune condition, with reported prevalence of 242–443 cases per million adults in North America,1 associated occlusive retinal vasculitis appears to be rare with only two cases reported previously by Yang and colleagues in 2018.3 Since early manifestations of SSc may be difficult to diagnose, particularly when lcSSc, and the ocular manifestations may pre-date the systemic diagnosis, it could be that some patients with SSc and occlusive retinal vasculitis go unrecognized for some time, as with our Case 1, or the retinal findings may be ascribed to more common causes, such as diabetic retinopathy, particularly when seen in the setting of elevated Hg A1c levels, as in Cases 1 and 2. Moreover, peripheral retinal non-perfusion was challenging to diagnose prior to routine imaging of the peripheral retina with wide-field fluorescein angiography.
While our cases resembled those reported by Yang et al. we show here for the first time a history of progressive retinal nonperfusion despite adequate control of retinal vascular leakage as in Case 1, development of proliferative retinopathy as in cases 2 and 3, and the presence of potentially occlusion-promoting antiphospholipid antibodies in all three cases.5, 6, 7 One of the patients described by Yang3 was contributed by a co-author (JTR) of this report, but she differed from the patients in our series as she tested negative for phospholipid antibodies and lupus anti-coagulant. These antibodies, which target cardiolipin, beta-2 glycoprotein 1, and phosphatidylserine, are common in patients with systemic lupus erythematosus (SLE), but they also occur in patients with other autoimmune diseases such as SSc or in individuals with no other apparent autoimmunity.7 Paradoxically, antibodies that cause clotting in vivo can prolong the partial thomboplastin time in vitro - a phenomenon known as the lupus anticoagulant. In a dated, but still important, longitudinal study from Stafford-Brady et al.,8 patients with SLE and the lupus anticoagulant were more likely to develop cotton wool spots. Antiphospholipid antibody testing should be considered, therefore, in any patient who develops an occlusive retinal arterial or venular vasculopathy in the absence of infection, especially if the leukocyte response in the anterior chamber or vitreous humor is mild or absent.
Our series highlights possible therapeutic approaches to the management of occlusive retinal vasculitis associated with SSc. Use of regional corticosteroids was effective in halting the vascular leakage in Case 1, but the patient's non-perfusion progressed and eventually involved the macula, raising the question of whether long-term corticosteroid-sparing immunosuppressant therapy would have prevented this complication. While no therapy exists to alter the course of scleroderma, a variety of disease-modifying agents, such as cyclophosphamide, mycophenolate mofetil, methotrexate, azathioprine, and hydroxychloroquine, have been reported to have efficacy in reducing the severity of skin and lung manifestations.1 However, little is known about the role of immunosuppressant therapy in management of scleroderma-related ocular disease. In patients with proliferative retinopathy, PRP can both treat and lower the risk of vision loss related to retinal neovascularization and neovascular glaucoma. While anti-VEGF agents may play a role in some such cases, we and others have reported catastrophic retinal vascular occlusion following intravitreal bevacizumab injection in eyes with multiple risk factors for retinal vascular occlusion, including diabetes mellitus, systemic hypertension, and SSc,9 suggesting that these agents should be used with caution in such vulnerable patients. Patients with elevated antiphospholipid titers may be at higher risk for deep vein thromboses, pulmonary embolism, and non-bacterial thrombotic endocarditis.7 The management can be complex, therefore, and may involve the use of anti-coagulants, anti-platelet agents, and biologics, especially rituximab10,11 After the retinal findings for Case 1 and 2 were relayed to rheumatology, both patients were treated with high-dose oral corticosteroids and a daily 325 mg dose of acetylsalicylic acid; more potent anticoagulation was recommended for signs of large vessel occlusion or further progression of occlusive retinal vasculitis.
In addition to lcSSc, occlusive retinal vasculitis has been reported in PHA4 - a localized form of scleroderma characterized by hemifacial degenerative changes from atrophy of the underlying subcutaneous tissue, fat, and muscle.2,12 Patients with PHA frequently have associated deformity of the tongue, teeth, and gingiva, seizures, corneal and retinal changes, enophthalmos, and eyelid and orbit abnormalities.2,12 Linear scleroderma CDS, a rare subset of linear scleroderma affecting the forehead and scalp and associated with neurological disease, often co-occurs with PHA as in the reported case with occlusive retinal vasculitis,4 and there has been debate whether these subsets of localized scleroderma are part of the same entity.12 Those with SSc developed occlusive retinal vasculitis in the fifth decade or later,3 whereas the single patient with occlusive vasculitis attributed to PHA/CDS was 17 years of age.4 Isolated reports of retinal vascular leakage, central retinal artery occlusion, and central retinal vein occlusion related to PHA have also appeared in patients within the first two decades of life, half of whom also possessed CDS.2,13
Given that occlusive retinal vasculitis associated with SSc is a rare and difficult condition to diagnosis, alternate causes of ischemic retinal vasculitis should be considered. Ischemic retinal vasculitis has been described in patients with tuberculous hypersensitivity, West Nile virus infection, Behçet's disease, sarcoidosis, multiple sclerosis, systemic lupus erythematosus, Takayasu's disease, idiopathic retinal vasculitis, dermatomyositis, Churg-Strauss syndrome, Crohn's disease, Polyarteritis nodosa, and Susac syndrome.14
In summary, vision-threatening occlusive retinal vasculitis may develop in select patients with SSc and the presence of elevated anti-phospholipid antibody titers may confer increased risk for this complication. Patient with otherwise unexplained retinal vasculitis, including nonperfusion, should be asked about signs or symptoms that might suggest SSc or the related conditions of PHA/CDS, and should be tested for the presence of anti-phospholipid antibodies. Regional corticosteroids appear to be effective at controlling both active inflammation and retinal vascular leakage, whereas PRP has been used effectively to both treat and limit the loss of vision loss related to retinal neovascularization and neovascular glaucoma. The role of longer-term non-corticosteroid immunosuppressive therapy remains to be studied.
Patient consent
Patients in case 1 and 2 consented to the publication of the case in writing. The patient in case 3 is deceased at the time of this report, and no consent was able to be obtained.
Funding
San Francisco Retina Foundation.
Authorship
All authors attest that they meet the current ICMJE criteria for Authorship.
Declaration of competing interest
None of the authors have any financial disclosures.
Acknowledgments
None.
References
- 1.Adigun R., Goyal A., Bansal P., Hariz A. 2020. Systemic Sclerosis (CREST Syndrome) StatPearls [Internet] Apr 28. [Google Scholar]
- 2.Tolkachjov S.N., Patel N.G., Tollefson M.M. Progressive hemifacial atrophy: a review. Orphanet J Rare Dis. 2015;10(1):1–3. doi: 10.1186/s13023-015-0250-9. Dec. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang S., Kopplin L.J., Rosenbaum J.T. Retinal vasculitis associated with CREST syndrome. American journal of ophthalmology case reports. 2018;10:185–188. doi: 10.1016/j.ajoc.2018.02.022. Jun 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vafa A., Gevorgyan O., De D., Hassan S. Retinal vasculitis the first clue in the diagnosis of progressive hemifacial atrophy. European journal of rheumatology. 2019;6(4):219. doi: 10.5152/eurjrheum.2019.18100. Oct. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Merashli M., Ames P.R. Clinical relevance of antiphospholipid antibodies in systemic sclerosis: a systematic review and meta-analysis. Semin Arthritis Rheum. 2017;46(5):615–624. doi: 10.1016/j.semarthrit.2016.10.004. Apr 1. WB Saunders. [DOI] [PubMed] [Google Scholar]
- 6.Sobanski V., Lemaire-Olivier A., Giovannelli J. Prevalence and clinical associations of antiphospholipid antibodies in systemic sclerosis: new data from a French cross-sectional study, systematic review, and meta-analysis. Front Immunol. 2018;9:2457. doi: 10.3389/fimmu.2018.02457. Nov 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giannakopoulos B., Krilis S.A. The pathogenesis of the antiphospholipid syndrome. N Engl J Med. 2013;368(11):1033–1044. doi: 10.1056/NEJMra1112830. Mar 14. [DOI] [PubMed] [Google Scholar]
- 8.Stafford-Brady F.J., Urowitz M.B., Gladman D.D., Easterbrook M. Lupus retinopathy. Patterns, associations, and prognosis. Arthritis Rheum. 1988;31:1105–1110. doi: 10.1002/art.1780310904. [DOI] [PubMed] [Google Scholar]
- 9.Ng C.C., Brill D., Cunningham E.T. Jr, Burckhard B.A., Jumper J.M., Heier J., Rifkin L.M., Eliott D., McDonald H.R., Sobrin L. Catastrophic, bilateral retinal vascular occlusion after intravitreal bevacizumab injection. Retin Cases Brief Rep. 2021 May 10. doi: 10.1097/ICB.0000000000001158. Epub ahead of print. PMID: 33988542. [DOI] [PubMed]
- 10.Cohen H., Cuadrado M.J., Erkan D. 16th international congress on antiphospholipid antibodies task force report on antiphospholipid syndrome treatment trends. Lupus. 2020;29(12):1571–1593. doi: 10.1177/0961203320950461. Oct. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agmon-Levin N., Berman M., Harel L. Rituximab for refractory manifestations of the antiphospholipid syndrome: a multicentre Israeli experience. Clin Exp Rheumatol. 2020 doi: 10.55563/clinexprheumatol/cc5taf. Oct 27. Epub ahead of print. PMID: 33124581. [DOI] [PubMed] [Google Scholar]
- 12.Khamaganova I. Progressive hemifacial atrophy and linear scleroderma en coup de sabre: a spectrum of the same disease? Front Med. 2018;4:258. doi: 10.3389/fmed.2017.00258. Jan 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ehmann D., Riyaz R., Greve M. Central retinal artery occlusion in a child with Parry–Romberg syndrome. Can J Ophthalmol. 2014;49(1):e9–10. doi: 10.1016/j.jcjo.2013.09.012. Feb 1. [DOI] [PubMed] [Google Scholar]
- 14.Talat L., Lightman S., Tomkins-Netzer O. Ischemic retinal vasculitis and its management. J Ophthalmol. 2014:197675. doi: 10.1155/2014/197675. 2014. Epub 2014 Apr 15. PMID: 24839552; PMCID: PMC4009272. [DOI] [PMC free article] [PubMed] [Google Scholar]