

Summary
This protocol describes the use of a biolayer interferometry platform for assessing antibody-antigen interactions. The protocol focuses on affinity determination and epitope binning, although the system can be utilized for measuring any protein-protein interaction. Readings are collected in real time, allowing the use of unlabeled molecules, and data can thus be obtained in a fast and easy manner. Experiments should be carefully designed, taking into consideration the tested interaction, available sensors, and suitable controls.
For complete details on the use and execution of this protocol, please refer to Noy-Porat et al. (2021).
Subject areas: Immunology, Antibody, Protein Biochemistry
Graphical abstract
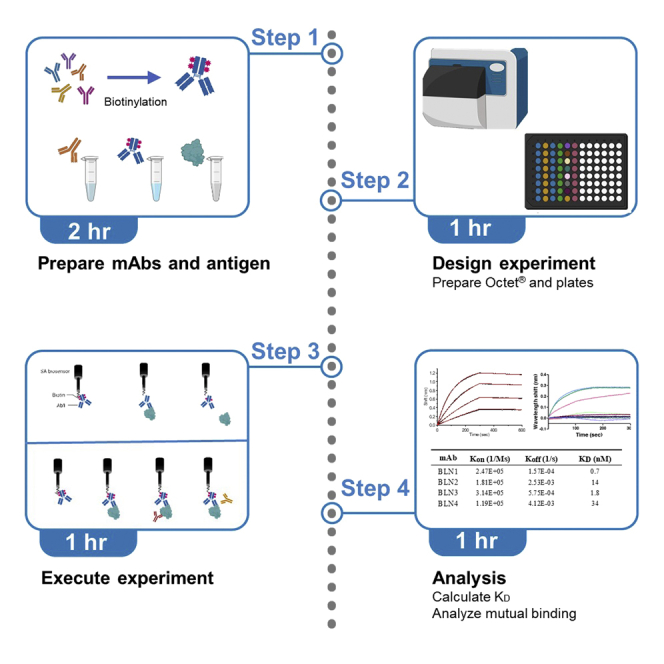
Highlights
-
•
Antibody-antigen interactions can be easily tested using Biolayer interferometry
-
•
The use of BLI for affinity measurement and epitope binning is described
-
•
Antibody and antigen concentrations should be pre-adjusted
This protocol describes the use of a biolayer interferometry platform for assessing antibody-antigen interactions. The protocol focuses on affinity determination and epitope binning, although the system can be utilized for measuring any protein-protein interaction. Readings are collected in real time, allowing the use of unlabeled molecules, and data can thus be obtained in a fast and easy manner. Experiments should be carefully designed, taking into consideration the tested interaction, available sensors, and suitable controls.
Before you begin
The development and engineering of antibodies for different purposes, such as diagnostics or therapeutics, requires comprehensive characterization to determine affinity, specificity and mechanism of action. Biolayer interferometry (BLI) is widely used for analyzing interactions between two biomolecules. Thus, it can aid in antibody characterization in a relatively easy and fast manner. In BLI, the binding between a ligand immobilized on the biosensor tip and an analyte in solution produces an increase in optical thickness at the biosensor tip, resulting in a wavelength shift proportional to the extent of binding (Azmiri and Lee, 2015; Mechaly et al., 2016). The sensor tips collect readings in real time, while immersed in the analyte solution (“dip-and-read”), without the need for continuous flow fluidics (Yang et al., 2017). The system therefore allows the measurements of different antibody-antigen interactions, using various sensors, suitable for label-free molecules or widely used tags. Here we describe the use of the Octet® RED96 BLI system, for affinity measurements and epitope binning. However, the system can also be used for a variety of other measurements involving protein-protein interactions.
When isolating antibodies from a library (such as phage/yeast display libraries) it is often required to classify the mass of resulting antibodies according to affinity (in order to identify the potentially most potent ones) and according to the epitope they bind (in order to potentially combine antibodies that bind discreet epitopes). The octet® system can be easily, rapidly and accurately applied for such classification.
Isolation of antibodies
Antibodies against a specific antigen can be isolated by a variety of means (phage or yeast display libraries or any other scaffold) and from different sources (such as immunized animals, naïve humans or synthetic libraries). These antibodies, obtained from any source, can be tested in the BLI system. Also, antibodies can be used in different formats, such as single-chain variable fragment (scFv), antigen-binding fragment (Fab), scFv-Fc (fragment crystallizable) or full-length IgG. Therefore, the antibodies should first be cloned into the desired format. Here we describe the characterization of antibodies in full-length IgG format.
Antibody expression and purification
The antibodies obtained, in full-length IgG format, can be expressed in any kind of mammalian expression system (the mammalian CHO or HEK293 cells are the most commonly used), and purified using different methods (generally based on protein-A/G). Antibodies produced in our study were expressed in ExpiCHOTM expression system (Thermoscientific, USA) and purified on HiTrap Protein-A columns (GE healthcare, UK). After purification, buffer was exchanged to PBS.
Note: BLI can also be used for quantitation of antibodies, before or after purification.
Note: Some antibody formats may require bacterial expression systems, and not mammalian, for optimal expression and folding. Therefore, the expression system used must be carefully suited to the antibody format intended to be expressed.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Human-derived antibodies | Noy-Porat et al., (2021) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| SARS-CoV-2 spike S1 subunit (antigen) | Noy-Porat et al., (2021) | N/A |
| PBS | Biological Industries | Cat#02-023-5A |
| IgG Elution Buffer (50 mM Glycine-HCl pH=2.7) | Thermo Fisher Scientific | Cat#21004 |
| BSA | Sigma-Aldrich | Cat#A9418 |
| Tween 20 | Sigma-Aldrich | Cat#P9416 |
| Critical commercial assays | ||
| EZ-Link Sulfo-NHS Biotin Kit | Thermo Fisher Scientific | Cat#21335 |
| Streptavidin (SA) biosensors | ForteBio | Cat#18-5020 |
| BCA Protein Assay Kit | Thermo Fisher Scientific | Cat#23225 |
| Software and algorithms | ||
| Octet® Data Acquisition software, version 8.1 | ForteBio | https://www.sartorius.com/en/products/protein-analysis/octet-systems-software |
| Octet® Data Analysis software, version 8.1 | ForteBio | https://www.sartorius.com/en/products/protein-analysis/octet-systems-software |
| Other | ||
| Microplate 96 well PP F-Bottom Black | Greiner | Cat#655209 |
| Amicon Ultra 0.5 mL 50,000NMWL | Merck | Cat#UFC505024 |
| Slide-A-Lizer G2 Dialysis Cassette 20,000 MWCO | Thermo Fisher Scientific | Cat#87734 |
| Biolayer Interferometry System | ForteBio | Octet® RED96 |
Materials and equipment
| Reagent | Final concentration | Amount |
|---|---|---|
| Binding buffer | PBS (pH 7.4) containing 10 mg/mL BSA and 0.1% (v/v) Tween 20 | 50 mL |
| Glycine pH 2.7 | 50mM | 3 mL |
[Store all solutions at 4°C. Maximum storage time: 1 year]
Step-by-step method details
This protocol describes two different methodologies using the BLI system. The first is affinity measurement and the second is epitope binning. Antibodies used for these assays can be obtained from diverse sources and can be used in different formats. Therefore, the protocol described here, begins after the identification and selection of antibodies, which specifically bind a certain antigen, and after the molecular cloning of these antibodies to a full-length IgG format.
Final well volume throughout all experiments should be 180–220 μL.
Biotinylation of antibodies
Timing: 2 h
In order to accurately measure the antibody affinity towards its ligand, the optimal setup is to immobilize the antibody to the sensor and incubate it with free ligand (Kumaraswamy and Tobias, 2015). Thus, it is necessary to validate that the dissociation rate of the ligand from the antibody is faster than the dissociation rate of the antibody from the sensor. Generally, immobilization of biotinylated antibody to a streptavidin coated-sensor would assure minimal dissociation drift and therefore is highly recommended for this setup.
-
1.Antibody biotinylation
-
a.Verify that antibodies are in PBS solution (or any other suitable buffer without primary amines).
-
b.Add Biotin to purified antibodies using EZ-Link Sulfo-NHS Biotin kit (ThermoScientific), according to manufacturer instructions (https://www.thermofisher.com/order/catalog/product/21217#/21217). Usually, molar ratio of 20:1 (biotin: antibody) is used.
-
c.Discard excess of free biotin. Use Amicon Ultra 50,000 NMWL (or smaller) centrifugal filters with the appropriate volume. Dialysis buffer excess of 500-fold or higher, should be obtained. Alternatively, dialysis cassettes such as Slide-A-Lizer with similar NMWL specifications can be used. Determine biotinylated antibody concentration (either by Nanodrop, BCA assay etc.) and verify that the labeled antibodies have retained their binding activity. Troubleshooting 1
-
a.
Alternatives: Other kits or methods for biotinylation of antibodies can be used. In these cases, several antibody: biotin ratios should be examined for optimal labeling.
Initial characterization of antibody-antigen binding
It is recommended to perform an initial assay using only one antigen concentration, in order to assess the integrity of the biotinylated antibody and the strength of the antibody-antigen interaction. In this assay, non-specific binding of the antigen to the sensor should also be examined, using an unloaded sensor, with no antibody.
-
2.Antibody immobilization
-
a.Rehydrate two streptavidin biosensors in a biosensor rack with a 96-well black plate containing 200 μL/well binding buffer, for at least 10 min at 20°C–25°C.
-
b.Dilute biotinylated antibody (to a final concentration of 5–10 μg/mL) in 200 μL binding buffer and use in the loading step. Add 200 μL binding buffer to a second well to test non-specific binding. Dilute antigen (50–100 nM) in 200 μL binding buffer and use in the association step.
-
c.Program the Data Acquisition software with the following steps:
-
i.Loading: 300 s
-
ii.Baseline: 60 s
-
iii.Association: 180 s
-
iv.Dissociation: 60 s
-
i.
-
d.Execute the binding assay (all steps are performed at 30°C with shaking at 1500 rpm). According to the program described in step C, the antibody is loaded on the sensor (step i), then washed (step ii) and allowed to bind the antigen (step iii). The sensor is then dipped in binding buffer (step iv) to allow dissociation.
-
e.Verify that the antibody was properly immobilized to the sensor and that it binds the antigen (Figure 1). Otherwise, please refer to the troubleshooting section (troubleshooting 1).
-
a.
Note: The loading step is mainly affected by antibody concentration and biotinylation effectivity. Optimally, a wavelength shift of ∼0.7–1.5 nm should be reached within 2–3 minutes. If such shift was not observed, antibody concentration can be adjusted accordingly.
Note: In this step, the optimal antigen concentration range for affinity measurements should be defined. The concentration used here is the one intended to be the highest concentration examined in the affinity measurements. A concentration of 50–100 nM is a good starting point and will cover most medium to low affinity antibodies. A good binding for this concentration should give ∼0.5 nm wavelength shift (in the Association step). However, if the concentration used did not give the desired wavelength shift, this step should be repeated using different concentrations (higher or lower, depending on the observed shift) until the optimal range is achieved. If the desired wavelength shift (0.5 nm) was achieved in less than 180 sec, the program can be manually switched to the next step.
Figure 1.

Initial characterization of antibody-antigen interaction
Biotinylated antibodies (mAb1-3) are loaded on separate streptavidin (SA) sensors (I; loading), washed (II; baseline) and allowed to bind the antigen (III; association). The sensors (loaded with the antibody-antigen complex) are then dipped in the binding buffer (IV) to measure dissociation. Different mAbs may exhibit different levels of loading, which may affect subsequent Rmax.
Affinity measurements
In this assay, loaded sensors are incubated in parallel with several concentrations of the antigen (Figures 2 and 3). Kon and Koff are measured and the subsequent KD is calculated, as a measure of antibody affinity towards the antigen. This assay should be carried out after validating the biotinylated antibodies’ activity and confirming that non-specific binding of antigen to the sensor is not detected.
-
3.
Rehydrate five streptavidin biosensors in a biosensor rack with a 96-well black plate containing 200 μL/well binding buffer, for at least 10 min at 20°C–25°C.
-
4.
Dilute biotinylated antibody in binding buffer to a final volume of 1000 μL.
Note: The recommended antibody concentration is 5–10 μg/ml, however, concentration should be adjusted according to the results of step 2, to obtain the desired wavelength shift of 0.7–1.5 nm, which ensures an effective presentation of the antibody for subsequent binding by the antigen, without introducing avidity effect. Since the loading level will influence binding (see Figure 1) it is important to ensure identical loading for all five sensors used for each affinity measurement.
-
5.
Dilute antigen in binding buffer (200 μL for each concentration). The antigen should be prepared in four titrated concentrations, preferably in two-fold serial dilutions (for example, in Noy-Porat et al., 2021 (Noy-Porat et al., 2021) concentrations of 12.5 nM–100 nM were used). An additional well containing only binding buffer should be used as control.
-
6.
Plate preparation and assay execution. Assays are performed in black 96-well plates. See Figure 3 for a recommended plate layout. Add the biotinylated antibody and antigen samples to the appropriate wells. Add binding buffer and Glycine solution to the appropriate wells (200 μL per well). Glycine is used for the regeneration step. Binding buffer is also used for baseline stabilization, analyte dissociation and neutralization (after sensor regeneration).
-
7.Program the Data Acquisition software with the following steps (Figure 4, phase I-VII):
-
a.Baseline: 60 s
-
b.Loading: 120 s
-
c.Baseline: 60 s
-
d.Association: 300 s
-
e.Dissociation: 600 s
-
f.Regeneration: 15 s
-
g.Neutralization: 30 s
-
a.
Note: Assay step times can be adjusted according to the strength of the interactions observed or the quality of the biotinylation. Based on this pre-assessment, the final antigen concentrations used in the assay will be determined.
CRITICAL: Plates layout can be changed. However, each step solution must be located in the same column. Make sure that the plate layout is identical to the one programmed in the Data Acquisition file.
Note: It is highly recommended to limit the antibody loading step to a wavelength shift of 0.7–1.5 nm, to avoid avidity effect that may influence the resulting KD. Therefore, the timing of the loading step (step 7b) is set to 120 sec and once the desired wavelength shift is reached, the program can be manually switched to the next step. However, if the antigen induces a subsequent low wavelength shift, the loading step can be prolonged (up to a wavelength shift of 4 nm). Troubleshooting 2
Note: In the first baseline step, a stabilization of the sensogram should be observed. Timing may be prolonged accordingly.
Note: Regeneration and neutralization steps are not mandatory and are only needed if sensors are intended for re-use.
-
8.
Execute the assay (all steps are performed at 30°C with shaking at 1500 rpm). See Figure 4 for a representative assay run.
-
9.
Analyze data with Data Analysis software (see detailed explanation in the “expected outcomes” section).
Note: Antibody-loaded sensors can be re-used in subsequent assays provided that full regeneration was accomplished (sensograms return to step 7c baseline levels). Immediately after sensor ejection to the sensor-tray, quickly collect them and immerse in binding buffer. For longer periods, sensors should be kept refrigerated while assuring that they are kept immersed in binding buffer. Some antibodies are highly sensitive to the regeneration step and thus the sensors cannot be reused. Such antibodies will not be able to bind the antigen following regeneration.
Figure 2.

Schematic representation of affinity measurement using BLI
During the antibody loading step, biotinylated mAb is simultaneously immobilized to several SA sensors. Following a wash step, each sensor is dipped in the antigen solution (containing different antigen concentrations) and the association rate between the antigen and the antibody is measured. The complex is then dipped in buffer and the dissociation rate is measured.
Figure 3.

Recommended plate layout for affinity measurements
Association is measured with four antigen concentrations (column 4). An additional sample with no antigen is added as control (well 4E). Buffer containing wells are used for initial baseline and neutralization (column 1) and for second baseline and dissociation (column 3).
Figure 4.

A representative assay run for affinity measurement
Five sensograms are presented, representing four antigen concentrations and a control (blank) sample with no antigen. Loading levels should be identical for all sensors, to ensure accurate subsequent measurements. The loading step was manually stopped once a wavelength shift of 1.5 nm was reached. Differences in association levels are expected to be dependent on antigen concentration. Control sensogram should show no wavelength shift and should be subtracted from all other sensograms.
Epitope binning
This assay allows the classification of mAbs according to different epitope clusters upon a given antigen. This method does not allow for precise epitope mapping, since two antibodies may not be able to simultaneously bind the antigen (competing antibodies) as a result of steric interference or due to targeting adjacent epitopes. However, antibodies that do bind together (non-competing), can certainly be classified as belonging to different groups. Thus, the assay allows classification of antibody binding sites on the antigen, based on the proximity of their respective epitopes.
In this assay, antibody-loaded sensors are incubated with a single concentration of antigen, washed and incubated with the unlabeled antibody counterpart (Figure 5). In each set of experiments, the background signal is obtained from a parallel sensor incubated with the same antibody as the capture antibody (unlabeled), and background signal is subtracted from all the sensograms. Such background signal may arise from non-specific binding of unlabeled mAbs to the sensor. Non-specific binding of the antigen to the sensors should also be ruled out before executing the assay, as explained in step 2.
-
10.
Rehydrate eight streptavidin biosensors in a biosensor rack with a 96-well black plate containing 200 μL/well binding buffer, for at least 10 min at 20°C–25°C.
-
11.
Dilute biotinylated antibody (b-mAb1) in binding buffer to a final volume of 200 μL. Recommended concentration: 10 μg/mL.
-
12.
Dilute each tested unlabeled antibody (mAb1 to mAb8) in binding buffer to a final volume of 200 μL. Recommended concentration: 10 μg/mL
-
13.
Dilute antigen in binding buffer to a final volume of 1600 μL. The concentration of the antigen should be pre-optimized based on the affinity of the mAbs to the desired antigen, in order to obtain a maximal saturation of the immobilized antibody. In most cases, antigen is used at a concentration of 10–20 μg/mL.
-
14.
Plate preparation and assay execution. Assays are performed in black 96-well plates. See Figure 6 for recommended plate layout. Add the antibodies and antigen samples to the appropriate wells. Add binding buffer and Glycine solution to the appropriate wells. Binding buffer is also used for baseline stabilization, analyte dissociation and neutralization.
-
15.Program the Data Acquisition software with the following steps:
-
a.Baseline: 120 s
-
b.Loading: 300 s
-
c.Baseline: 30 s
-
d.Activation: 300 s
-
e.Baseline: 30 s
-
f.Association: 300 s
-
g.Dissociation: 60 s
-
h.Regeneration: 15 s
-
i.Neutralization: 30 s
-
a.
-
16.
Execute the assay (all steps are performed at 30°C with shaking at 1500 rpm). See Figure 7 (phase I – IX) for a representative assay run.
-
17.
Analyze the data. For each run, a sample with the same antibody (e.g., mAb1) implemented as both the capture and second antibody should be used as control, to determine background drift. This sample should be subtracted from all other samples (see representative Figure 9 in the “expected outcomes” section).
Note: To achieve high signals, it is advisable to ensure maximal loading of the sensor with the immobilized antibody (3–4nm wavelength shift). This can be obtained by extending the loading step or by increasing the concentration of the biotinylated antibody.
Note: In the activation step, the antigen is allowed to bind to mAb1. Baseline time (step 15e, phase V in Figure 7) is short to avoid dissociation of the antigen from mAb1. The second, unlabeled mAb is then allowed to bind during the association step. The ability of mAb2 to bind in the presence of mAb1 will be reflected by an additional wavelength shift. Each mAb must be tested as both first (capture) and second antibody in order to achieve reliable classification.
Note: When testing two mAbs which differ substantially in their affinities towards the antigen, some wavelength shift may be observed due to the removal of the low affinity mAb by the high affinity one. Reciprocal testing of the two mAbs will thus enable a more accurate decision regarding their classification. Troubleshooting 3
Note: In cases where a decrease in wavelength is observed after the activation step (due to dissociation of the antigen), step 15e (baseline) may be omitted, to enhance subsequent association signal.
Note: If needed, diluted antigen can be reused in subsequent assays. Due to evaporation, sample volume should be re-adjusted to 200 μl with water.
Note: This method can be applied to assess mutual binding of more than two antibodies, enabling the evaluation of steric interference within antibody cocktails. Such interference may occur even if each individual antibody was assigned to a different epitope within the antigen.
Optional: This assay can also be employed in a different format: the antigen can first be immobilized on the sensor and the two mAbs are then loaded in tandem (antigen → mAb1→ mAb2). However, this setup requires that binding by mAb1 will reach saturation before applying mAb2. The setup described above, where the first mAb serves as a capture antibody, ensures that, for each antigen molecule, the binding site of mAb1 is already occupied, therefore allowing binding of mAb2 only if it does not compete with mAb1.
Figure 5.

Schematic representation of epitope binning using BLI
Biotinylated mAb1 is immobilized to a SA sensor and allows to bind the antigen. The sensor (after a wash step) is then incubated with a second, unlabeled mAb, and its ability to bind the mAb1-antigen complex is measured. If mAb2 competes with mAb1, it will not be able to bind and no additional wavelength shift will be observed. In some cases, if mAb2 has a substantially higher affinity towards the antigen than mAb1, a decrease in wavelength might be detected. A sample with the same mAb (mAb1) as both labeled and unlabeled antibodies, is used as negative (self-competing) control. The negative control sensogram should be subtracted from all other sensograms.
Figure 6.

Recommended plate layout for epitope binning
One mAb is used as capture antibody (in our assay this antibody is biotinylated: b-mAb1; column 2). A total of seven different antibodies can be measured at each run (represented by different colors in column 5) whereas an identical mAb (unlabeled mAb1) serves as control (well 5A). The antigen is used in a single concentration.
Figure 7.

Representative assay run for epitope binning
Mab1 is used as capture antibody, loaded on the sensor, and also as second antibody in the control sample (black curve). Following antigen binding to mAb1, antibodies 1–8 are added. Only mAbs 4–6 bind the antigen in the presence of mAb1, as reflected by the wavelength shift. All other mAbs do not bind, giving a flat curve, similar to the control.
Figure 9.

Representative sample before and after subtraction of background. MAb1 is used as capture antibody
(A) Before subtraction of the background signal (black curve).
(B) After subtraction of the background signal, the extent of binding of the second antibody in each sample can be viewed more clearly.
Expected outcomes
The procedures described here allow for the initial characterization of selected antibodies and aid in classification and selection of antibodies possessing desired features such as high affinity, epitope diversity or the recognition of an epitope of interest. The use of BLI is simple, fast and gives immediate results.
Affinity measurements
KD values can vary substantially, based on the strength of the antibodies’ binding. KD values in the low nano molar range, or lower, are considered high affinity.
The Octet® data analysis software is used for affinity calculations. For full analysis instructions, see the Data analysis user guide (https://www.sartorius.com).
The control (Blank) sensor should show no wavelength shift during association and dissociation steps. However, a drift in wavelength can sometimes be observed due to release of antibody from the sensor, change in buffer concentration or other reasons. The results obtained by the control sensor will be subtracted from all other results, thereby eliminating background signals. For subtraction of background signal, in the Processing tab, in step 1 choose “sensor selection” and define the reference sensor for the measurement. In step 2 – choose “reference wells” for subtraction.
For curve fitting: in the Processing tab, in step 3 – align Y axis to the association step. In step 4, choose “align to Dissociation”. In step 5, choose “Saviztky-Golay filtering” and hit “Process Data” button.
In the Analysis tab, define the curve fitting parameters. Choose all the sensors in the table on lower part of the window, right click and choose “set color by” → “sensor type”. In “step to analyze”, choose “Association and Dissociation” and choose 1:1 model. In “fitting” define “Global (Full)” and choose “Rmax Unlinked by sensor” and “group by color” and hit “Fit curves!” button.
The quality of the fit can be determined both visually, using the fitting view, and by examining the computed statistical values. The R2 value should be close to 1 and Χ2 value should be close to zero. For examples, see Figure 8. Troubleshooting 4
Figure 8.

Curves fit, using 1:1 fitting model
A “good” fit can be determined in the fitting view window and confirmed by Χ2 and R2 values. An example of. (A) a good fit. (B) a less reliable fit are presented.
Epitope binning
Epitope binning experiments result in a series of curves, representing the degree of competition between two or more mAbs. If the affinity of mAb1 to the antigen is low, a “background drift” may occur simultaneously with the binding of the other antibody to the mAb1-antigen complex. In these cases, it is necessary to subtract this background from all other sensors (see Figure 9 for a representative sample before and after the subtraction of background sensor). In the data analysis software, processing is done as described for affinity measurements, without curve fitting.
The results are generally qualitative. When assessing the relationships between a set of mAbs, the results can be summarized and presented using a matrix or heat map, to allow a more convenient view.
For example, area under curve (AUC) can be calculated and the values could be used to create a heat map (Noy-Porat et al., 2021).
Limitations
The Octet® RED96 can utilize up to eight sensors at each run (including controls). Therefore, for large screens, such as antibody library initial screens, Octet® RED384 should be considered. Alternatively, it is recommended to apply other appropriate approaches (e.g., ELISA) for the initial screen prior to evaluation by BLI.
The use of this system is most suitable for monovalent analytes. If the antigen in question has more than one epitope for a given antibody, the results obtained may not be accurate.
Due to evaporation of the samples over time, the experiment duration is limited to up to 12 h (according to manufacturer specifications).
The system limit of detection for affinity constant (KD) measurements is 1mM–10 pM. Therefore, very low affinity antibodies or ultra-potent antibodies cannot be accurately measured.
Troubleshooting
Problem 1
In rare cases, the biotinylation process may lead to a partial or full loss of the antibody ability to bind the antigen. (Step 1; 2.e)
Potential solution
Verify the absence of sodium-Azide in the antibody’s buffer prior to the biotinylation.
It is advisable to reduce the molar excess ratio of biotin in the labeling reaction (to 3–10 times molar excess of biotin compared to antibody). Alternatively, antibodies can be directly immobilized to the sensor by chemical cross-linking (e.g., Amine-reactive biosensors). Other sensors, such as Protein A/G, Ni-NTA etc. can also be used. However, substantial drift may occur during the dissociation phase and thus these options are not recommended for affinity measurements.
Problem 2
Association of the antigen to the loaded antibody is very low.(Step 7)
Potential solution
Make sure that the labeled antibody retained its binding activity. If the labeled antibody's binding activity is confirmed, try the following: increase antigen concentration, increase the length of the loading step to obtain larger amount of bound antibody, increase association time. Alternatively, immobilize the antibody on a different sensor (e.g., Protein A, FAB2G, anti Fc).
Problem 3
Non-specific binding of antigen to sensor is detected.(Step 2)
Potential solution
Use lower antigen concentration.
Use a different sensor type, if possible.
Problem 4
The concentrations used during affinity measurements did not give a clear dose response. (Step 8)
Potential solution
The concentration range used is too high. Lower concentrations should be used. Alternatively, if at least four concentrations were used, the highest concentration can be omitted from the analysis.
Problem 5
A pair of mAbs shows complete competition at one format but some mutual binding in the reciprocal format. (Step 17)
Potential solution
This phenomenon may occur when there is a substantial difference in the affinity of the antibody pair. If there is an additional antibody known to bind the same epitope as the low affinity mAb, it can be used to help evaluate the degree of epitope overlap.
Problem 6
Fitting of the affinity measurements sensograms is not optimal. (Expected outcomes)
Potential solution
The range of concentrations used should be adjusted.
Alternatively, if antibody’s affinity is beyond the system limit of detection, other methods for affinity determination should be used.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ohad Mazor (ohadm@iibr.gov.il).
Materials availability
This study did not generate new unique reagents
Acknowledgments
We wish to express our gratitude to our colleague Itai Glinert for fruitful discussions and support. Figures 2, 3, 5, and 6 were created with BioRender.com.
Author contributions
T.N.-P., R.A., A.M., E.P., E.M., R.R., and O.M. designed, carried out, and analyzed the data. T.N.-P., R.R., and O.M. wrote the manuscript. R.R. and O.M. supervised the project. All authors have approved the final manuscript.
Declaration of interests
Patent application for the described antibodies in Noy-Porat et al. (2021) was filed by the Israel Institute for Biological Research. None of the authors declared any additional competing interests.
Contributor Information
Ronit Rosenfeld, Email: ronitr@iibr.gov.il.
Ohad Mazor, Email: ohadm@iibr.gov.il.
Data and code availability
No new datasets or codes were produced during this study.
References
- Azmiri S., Lee J.E. Measuring protein-protein and protein-nucleic acid interactions by biolayer interferometry. Curr. Protoc. Protein Sci. 2015;79:19.25.1–19.25.26. doi: 10.1002/0471140864.ps1925s79. [DOI] [PubMed] [Google Scholar]
- Kumaraswamy S., Tobias R. In: Protein-Protein Interactions: Methods and Applications. Meyerkord C.L., Fu H., editors. Springer; 2015. Label-free kinetic analysis of an antibody–antigen interaction using biolayer interferometry; pp. 165–182. [DOI] [PubMed] [Google Scholar]
- Mechaly A., Cohen H., Cohen O., MazoR O. A biolayer interferometry-based assay for rapid and highly sensitive detection of biowarfare agents. Anal Biochem. 2016;506:22–27. doi: 10.1016/j.ab.2016.04.018. [DOI] [PubMed] [Google Scholar]
- Noy-Porat T., Mechaly A., Levy Y., Makdasi E., Alcalay R., Gur D., Aftalion M., Falach R., Leviatan Ben-Arye S., Lazar S. Therapeutic antibodies, targeting the SARS-CoV-2 spike N-terminal domain, protect lethally infected K18-hACE2 mice. iScience. 2021;24:102479. doi: 10.1016/j.isci.2021.102479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D., Singh A., Wu H., Kroe-Barrett R. Determination of high-affinity antibody-antigen binding kinetics using four biosensor platforms. J. Vis. Exp. 2017;122:55659. doi: 10.3791/55659. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new datasets or codes were produced during this study.