

Summary
Cellular cholesterol is regulated by at least two transcriptional mechanisms involving sterol-regulatory-element-binding proteins (SREBPs) and liver X receptors (LXRs). Although SREBP and LXR pathways are the predominant mechanisms that sense cholesterol in the endoplasmic reticulum and nucleus to alter sterol-regulated gene expression, evidence suggests cholesterol in plasma membrane can be sensed by proteins in the Hedgehog (Hh) pathway which regulate organ self-renewal and are a morphogenic driver during embryonic development. Cholesterol interacts with the G-protein-coupled receptor Smoothened (Smo), which impacts downstream Hh signaling. Although evidence suggests cholesterol influences Hh signaling, it is not known whether Smo-dependent sterol sensing impacts cholesterol homeostasis in vivo. We examined dietary-cholesterol-induced reorganization of whole-body sterol and bile acid (BA) homeostasis in adult mice with inducible hepatocyte-specific Smo deletion. These studies demonstrate Smo in hepatocytes plays a regulatory role in sensing and feedback regulation of cholesterol balance driven by excess dietary cholesterol.
Subject areas: Lipid, Molecular physiology, Molecular biology
Graphical abstract
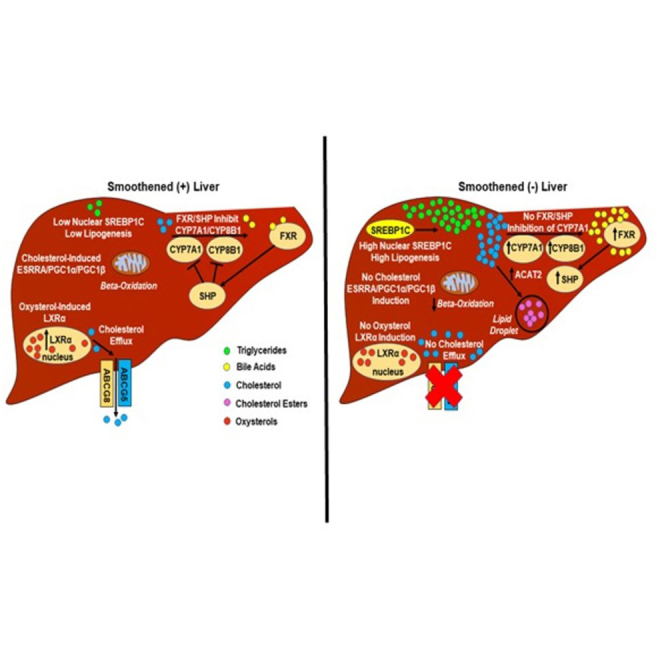
Highlights
-
•
Cholesterol and bile acid homeostasis is controlled by hepatocyte Smo activity
-
•
Smo loss blocked induction of LXRα, ABCG5, and AGCG8 that remove liver cholesterol
-
•
Smo deficiency increased hepatic CYP7A1 despite elevated SHP and bile acid levels
-
•
High-cholesterol Smo (−) livers were steatotic and did not induce ESRRA/PGC1α/PGC1β
Lipid; Molecular physiology; Molecular biology
Introduction
Strict control of cholesterol accumulation is critical for health, and dysregulated cholesterol homeostasis is a hallmark of both cancer and many degenerative diseases (Morgan et al., 2016; Xu et al., 2020). Two major signaling pathways that sense changes in the relative abundance of cholesterol and its metabolites (oxysterols) maintain cholesterol balance (Luo et al., 2020). Cholesterol accumulation inhibits proteolytic processing of sterol-regulatory-element-binding protein 2 (SREBP2) that is necessary for nuclear localization of that transcription factor. This reduces expression of HMG-CoA reductase (HMCGR), the SREBP2 target gene and rate-limiting enzyme for cholesterol biosynthesis, limiting cholesterol biosynthesis when cholesterol levels are in excess. In parallel, when cholesterol levels are high, oxysterol metabolites are readily generated which can act as direct ligands for liver X receptors (LXRα/β), thereby stimulating the expression of LXR-target genes that generally promote cellular efflux of cholesterol. The abundance of oxysterols themselves can also be influenced by the activity of farnesoid X receptor (FXR) and its target small heterodimer partner (SHP) which control the feedback regulation of de novo BA synthesis (Goodwin et al., 2000).
Cholesterol controls health in part by modulating the activity of morphogenic signaling pathways that regulate cell viability, proliferation, and differentiation. Proper functioning of the Hedgehog (Hh) pathway is particularly dependent on cholesterol (Hu and Song, 2019). All three Hh ligands, Sonic hedgehog (Shh), Indian hedgehog (Ihh), and Desert hedgehog (Dhh), are posttranslationally modified by cholesterol, as well as palmitoylation, to complete their maturation in ligand-producing cells (Banavali, 2020; Porter et al., 1996a, 1996b; Pepinsky et al., 1998). The lipid modifications enable Hh ligands to associate with exosomes, lipoproteins, and other lipid particles that control their local and systemic bioavailability (Willnow et al., 2007; Palm and Rodenfels, 2020; Prince et al., 2020). The Hh receptor is a complex consisting of the cholesterol-sensing transmembrane protein Patched (Ptc) that binds Hh ligands on Hh-responsive cells and a second transmembrane protein called Smoothened (Smo) (Zhang et al., 2018). In the absence of Hh ligand, Ptc inhibits Smo activity, and this inhibition is removed after Hh ligand binding to Ptc which allows membrane-associated cholesterol to activate Smo, thereby enabling signal transduction (Huang et al., 2016; Xiao et al., 2017; Radhakrishnan et al., 2020). Interestingly, oxysterols have also been shown to bind Smo and either inhibit or enhance its activity (Huang et al., 2016; Radhakrishnan et al., 2020; Nachtergaele et al., 2012; Nedelcu et al., 2013). Smo is a G-protein-coupled receptor, and its activation induces inhibitory G proteins that suppress the activities of protein kinase A (PKA) and glycogen synthase kinase 3 β (GSK3β), reducing the ability of those kinases to phosphorylate and degrade their targets, which include Glioma (Gli) family transcription factors. Smo-dependent inhibition of PKA and GSK3β allows Gli transcriptional activators (i.e., full-length Gli2 and Gli3) to move off primary cilia and accumulate in nuclei at the expense of Gli-transcriptional repressors (i.e., Gli3 and truncated Gli2 (Gorojankina, 2016). This turns on the canonical Hh transcriptional program which critically regulates tissue morphogenesis during embryogenesis and is essential for proper tissue repair and maintenance of tissue integrity in adulthood (Kong et al., 2019).
Although cholesterol acts at multiple levels to control Hh signaling, it is not known if the activity of this morphogenic pathway feeds back in some way to regulate cholesterol bioavailability. To address this question, we conditionally deleted Smo in hepatocytes of healthy adult mice and compared cholesterol homeostasis at baseline and after challenging the mice with a high-cholesterol (HC) diet. Remarkably, selectively deleting Smo in hepatocytes of healthy adult mice was sufficient to disrupt both hepatic and systemic homeostasis of cholesterol and BAs and dysregulated normal compensatory responses to dietary cholesterol challenge. These results reveal a novel mechanism that enables Hh, a sterol-sensitive morphogenic pathway, to titrate its own activity and the activity of other sterol-sensitive pathways by controlling the bioavailability of cholesterol.
Results
Smo deletion in hepatocytes alters hepatic cholesterol homeostasis
In chow-fed mice, total hepatic cholesterol and cholesterol ester levels were significantly higher in Smo (−) mice than in Smo (+) mice (Figures 1A and 1B), while serum levels of total cholesterol and cholesterol esters were similar in the two groups (Figures S1B and S1C). When cellular cholesterol levels are high, SREBP2 exists as an inactive membrane precursor protein in the endoplasmic reticulum (ER) and cannot enter the nucleus to activate the expression of genes involved in cholesterol synthesis such as HMGCR and acetoacetyl-CoA synthetase (AACS) (Horton et al., 2002; Hasegawa et al., 2012; Goldstein and Brown, 1990). The active form of SREBP2 was reduced approximately 2-fold in membrane and nuclear extracts from Smo (−) hepatocytes compared with Smo (+) hepatocytes (Figures 1D and 1E). Unexpectedly, despite exhibiting decreased nuclear SREBP2 protein content, Smo (−) mice expressed higher mRNA levels of the cholesterol biosynthetic genes HMGCR and AACS than Smo (+) mice (Figure 1C). However, HMGCR protein levels were reduced in Smo (−) hepatocytes compared with Smo (+) hepatocytes, while AACS protein levels were unchanged (Figures 1F and 1G). The discrepancy between mRNA and protein levels of HMGR and AACS suggests that Smo deletion impacts posttranscriptional regulatory mechanisms that control the stability, processing, and/or translation of these mRNAs and/or the turnover of the proteins they encode. In general, posttranscriptional mechanisms are major determinants of gene expression because the correlation between mRNA and protein expression is relatively poor on a genome-wide level (de Sousa Abreu et al., 2009; Vogel and Marcotte, 2012; Maier et al., 2009). Furthermore, our Gene Ontogeny (GO) analyses and Genome Set Enrichment Analyses (GSEA) of RNA sequencing data from Smo (−) and Smo (+) hepatocytes demonstrate that Smo deletion itself significantly dysregulates some of these posttranscriptional processes, including ‘regulation of mRNA stability,’ ‘regulation of translation,’ and ‘regulation of proteolysis’ (data not shown). The differences in SREBP2 nuclear accumulation and HMGCR protein content in Smo (−) versus Smo (+) hepatocytes were associated with significantly increased accumulation of INSIG1 mRNA in the Smo (−) group (Figure 1C). INSIG1 is known to facilitate HMGCR degradation and suppress activation of ER-membrane-bound SREBPs (Debose-Boyd, 2008; Yang et al., 2002). Together, these findings suggest that Smo (−) hepatocytes were attempting to suppress cholesterol biosynthesis after excessively accumulating cholesterol. Hepatocyte expression of acyl-CoA:cholesterol acyltransferase 2 (ACAT2), the major cholesterol-esterifying enzyme in the human liver (Parini et al., 2004), was also increased in Smo (−) hepatocytes (Figure 1C) and accompanied by elevated accumulation of cholesterol esters in Smo-knockout livers (Figure 1B), consistent with evidence that cholesterol esterification increases to buffer accumulation of free cholesterol when cholesterol content rises (Tabas, 2002). Excess cholesterol accumulation can also provide substrates for the generation of oxysterols, which serve as ligands for the oxysterol-sensing nuclear hormone receptors LXRα/β, thereby transcriptionally controlling cellular cholesterol efflux (Zhu et al., 2012; Repa et al., 2002). Although Smo (−) livers accumulated excess total cholesterol, they did not exhibit an increase in hepatocyte mRNA expression of LXRα/β or LXRα/β target genes ATP-binding cassette transporters A1 (ABCA1), G5 (ABCG5), and G8 (ABCG8) that facilitate hepatic cholesterol excretion (Figure 1H). On the other hand, mRNA and protein levels of scavenger receptor, class B type 1 (SRB1), a hepatic cell surface receptor that mediates high-density lipoprotein (HDL) cholesterol uptake into the liver (Shen et al., 2018), was reduced in Smo (−) hepatocytes compared with their respective Smo (+) controls (Figures 1F–1H). Therefore, the aggregate data indicate that hepatocytes in healthy adult livers rapidly accumulate excessive cholesterol when Smo is deleted and induce an array of compensatory responses to constrain further enrichment with free cholesterol. These include upregulating mechanisms that reduce cholesterol biosynthesis, increase cholesterol esterification, and limit reuptake of HDL cholesterol.
Figure 1.

Smoothened deletion in hepatocytes alters hepatic cholesterol homeostasis in chow-fed mice
Male Smo (+) and Smo (−) mice were fed a low-cholesterol chow (0.02% w/w) diet for 7 days.
(A) Total liver cholesterol.
(B) Liver cholesterol esters.
(C) Hepatocyte gene expression of SREBP2, HMGCR, LSS, AACS, INSIG1, and ACAT2.
(D and E) Western blot and densitometric analysis of hepatocyte membrane and nuclear SREBP2 (precursor, P; active A), EGFR, and TAF15 protein levels.
(F and G) Western blot and densitometric analysis of hepatocyte HMGCR, AACS, SRB1, and β-tubulin protein levels.
(H) Hepatocyte gene expression of LXRα, ABCA1, ABCG5, ABCG8, and SRB1. Results reported as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01 vs. Smo (+) chow.
See also Figure S1.
Smo deletion in hepatocytes dysregulates hepatic responses to dietary cholesterol challenge
We next investigated the effect of excess cholesterol on the ability of Smo to regulate hepatic cholesterol homeostasis by comparing responses of Smo (−) and Smo (+) mice fed HC diets for 10 days with each other and with chow-fed Smo (+) controls (Figure 2A). The HC diet induced an increase in hepatic total cholesterol and cholesterol ester levels in both Smo (+) and Smo (−) mice compared with chow-fed Smo (+) controls, but the increase was significantly greater in the Smo (−) group (Figures 2B and 2C). Compared with chow-fed Smo (+) mice, the HC diet reduced nuclear SREBP2 protein levels in both Smo (+) and Smo (−) groups (Figures 2D and 2E). The HC diet also significantly reduced Smo (+) and Smo (−) hepatic mRNA expression of cholesterol biosynthetic enzymes (HMGCR, AACS, and lanosterol synthase [LSS]), as well as HMGCR and AACS protein levels compared with chow-fed Smo (+) controls (Figures 2F–2H). However, the decreases in hepatic HMGCR protein levels were significantly less in Smo (−) livers than in Smo (+) livers (Figures 2G and 2H). Livers of HC-diet-fed Smo (−) mice also exhibited lower levels of adenosine monophosphate-activated kinase (AMPK) phosphorylation than any of the other groups (Figures 6D and 6E), and inhibited activation of AMPK increases cholesterol synthesis (Jeon, 2016). Together, these results suggest that the compensatory suppression of cholesterol biosynthesis that is typically triggered by high dietary cholesterol is blunted in mice with Smo-deficient hepatocytes and this may contribute to their higher hepatic cholesterol content. On the other hand, Smo (−) mice fed HC diets exhibited higher levels of ACAT2 protein in liver microsomes than Smo (+) mice fed HC diets (Figures 2I and 2J). This may be a compensatory mechanism, given that ACAT2-driven cholesterol esterification helps to buffer accumulation of free cholesterol in hepatocytes via facilitating cholesterol ester storage in cytosolic lipid droplets and packaging of cholesterol ester onto nascent very-low-density lipoproteins (VLDLs). Finally, cellular accumulation of cholesterol normally triggers activation of LXRα/β, leading to induction of LXRα/β target genes that encode ATP-binding cassette (ABC) transporters that export cholesterol. Hence, Smo (+) mice fed an HC diet exhibited a marked increase in hepatic mRNA expression of LXRα and the LXRα target genes ABCA1, ABCG5, and ABCG8 (Figure 2K). Induction of all but one of these transporters (ABCA1) was significantly attenuated in HC-diet-fed Smo (−) mice (Figure 2K), suggesting that reduced cholesterol efflux may also contribute to excessive hepatic cholesterol accumulation in Smo (−) mice. In contrast, Smo (−) mice exhibited lower protein expression of SRB1, which is a major transporter that facilitates cellular uptake of HDL cholesterol (Figures 2G and 2H).
Figure 2.

Smoothened deletion in hepatocytes dysregulates hepatic responses to dietary cholesterol challenge
Male Smo (+) and Smo (−) mice were fed a high-cholesterol (HC, 0.2% w/w) diet for 10 days.
(A) Hepatic SMO gene expression.
(B) Liver total cholesterol.
(C) Liver cholesterol esters.
(D and E) Western blot and densitometric analysis of hepatic membrane and nuclear SREBP2 (precursor, P; active, A), EGFR, and TAF15 protein levels.
(F) Hepatic gene expression of SREBP2, HMGCR, AACS, and LSS.
(G and H) Western blot and densitometric analysis of hepatic HMGCR, AACS, SRB1, and β-tubulin protein levels.
(I and J) Western blot and densitometric analysis of liver microsome ACAT2 and β-actin protein levels.
(K) Hepatic gene expression of LXRα, ABCA1, AGCG5, ABCG8, and SRB1. Results reported as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01 vs. Smo (+) Chow; #p < 0.05, ##p < 0.01 vs. Smo (+) HC.
Figure 6.

Smoothened deletion in hepatocytes alters activities of kinases involved in Hedgehog signaling
Male Smo (+) and Smo (−) mice were fed either a low-cholesterol chow (0.02% w/w) or high-cholesterol (HC, 0.2% w/w) diet for 10 days.
(A) Hepatic gene expression of Patched, Shh, Gli1, Gli2, and Gli3.
(B and C) Western blot analysis of hepatic cytosolic and nuclear Gli1, Gli2, Gli3, β-tubulin, and TAF15 protein levels.
(D–G) Western blot and densitometric analysis of phospho-AMPK Threonine 172, total AMPK, phospho-Akt Serine 473, total Akt, phospho-GSK3β Serine 9, and β-tubulin protein levels. Results reported as MEAN ± SEM. ∗p < 0.05, ∗∗p < 0.01 vs. Smo (+) Chow; #p < 0.05, ##p < 0.01 vs. Smo (+) HC. A, activator; R, repressor.
Smo deletion in hepatocytes increases plasma and fecal bile acids
Excessive cholesterol in hepatocytes can serve as a substrate for the production of oxysterols and bile acids (BAs), both of which perform key cellular signaling roles that help titrate cholesterol homeostasis. The generation of oxysterol can be sensed by dedicated oxysterol-binding proteins (e.g., OSBPs, LXRα/β, Smo) resulting in downstream reorganization of cholesterol synthesis and cellular export, as well as changes in the activity of cholesterol-sensitive signaling pathways. BAs are made in the liver (primary BAs) and stored in the gallbladder from which they are secreted into the proximal intestine, reabsorbed in the ileum, and returned to the liver via the enterohepatic circulation. In mice, most BAs are taurine (T) conjugated (Chiang, 2017). The primary BAs found in mice are cholic acid (TCA), chenodeoxycholic acid (TCDCA), and α- and β-muricholic acids (TMCAs) (Chiang, 2017). In the intestine, gut bacteria modify primary BAs to form secondary BAs, which include deoxycholic acid (DCA), ωMCA, hyocholic acid (HCA), hyodeoxycholic acid (HCDA), lithocholic acid (LCA), and murideoxycholic acid (MDCA). To determine if Smo-related changes in cholesterol homeostasis impacted BA balance, we compared concentrations of individual BAs in plasma and feces of Smo (−) and Smo (+) mice on chow and HC diets. In plasma, the primary BA TβMCA along with the secondary BAs taurochenodeoxycholic acid (TCDCA), CA, ωMCA, and DCA were increased in chow-fed Smo (−) mice compared with Smo (+) mice (Figures 3A and 3B). The HC diet increased the plasma levels of the primary BAs TCA, TαMCA, TβMCA, and TωMCA, as well as the secondary BA TCDCA in Smo (+) mice (Figures 3A and 3B). Plasma levels of the secondary BAs taurodeocycholic acid (TDCA), CA, ωMCA, and tauroursodeoxycholic acid (TUDCA) were even more increased in Smo (−) mice fed HC diets (Figure 3B). Fecal BA levels were consistently higher in Smo (−) mice than in Smo (+) mice, regardless of diet (Figure 3C), but Smo depletion did not influence fecal neutral sterol loss (Figure 3D). These results indicate that hepatocyte Smo activity preferentially opposes acidic sterol loss, without altering fecal disposal of neutral sterols. Together, these results link Smo-related changes in hepatic cholesterol homeostasis with systemic changes in BA balance including enhanced fecal BA excretion.
Figure 3.

Smoothened deletion in hepatocytes increases plasma and fecal bile acids
Male Smo (+) and Smo (−) mice were fed either a low-cholesterol chow (0.02% w/w) or high-cholesterol (HC, 0.2% w/w) diet for 10 days.
(A) Fold change in primary bile acids over Smo (+) chow-fed mice.
(B) Fold change in secondary bile acids over Smo (+) chow-fed mice.
(C) Fold change in total bile acids in feces over Smo (+) chow-fed mice.
(D) Fold change in fecal neutral sterol loss over Smo (+) chow-fed mice. Results reported as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01 vs. Smo (+) Chow; #p < 0.05, ##p < 0.01 vs. Smo (+) HC. TCA, taurocholic acid; TαMCA, tauro-α-murocholic acid; TβMCA, tauro-β-murocholic acid; TωMCA, tauro-ω-murocholic acid; TCDCA, taurochenoxycholic acid; TDCA, taurodeocycholic acid; CA, cholic acid; ωMCA, ω-murocholic acid; TUDCA, tauroursodeoxycholic acid; DCA, deoxycholic acid.
To begin to elucidate the mechanisms responsible for changes in plasma and fecal BA levels, we analyzed expression of receptors, enzymes, and transporters involved in BA synthesis and uptake in the liver and ileum. BA synthesis in the liver involves several enzymes that are found in either the classic or alternative BA synthetic pathways. Cholesterol 7α-hydroxylase (CYP7A1) is the rate-limiting enzyme in the classic pathway and catalyzes the formation of both CA and chenodeoxycholic acid (CDCA) from cholesterol (Myant and Mitropoulos, 1977; Russell, 2003). Sterol 12α-hydroxylase (CYP8B1) is required for the 12α-hydroxylation of 7α-hydroxy-4-cholesten-3-one which is a marker for the rate of BA synthesis by the classic pathway (Chiang, 2017). Dietary cholesterol stimulates CYP7A1 transcription via activation of LXRα (Ballatori et al., 2009). Conversely, CYP7A1 and CYP8B1 transcription are inhibited by BAs via two feedback mechanisms (Chiang, 2004). One involves the BA nuclear receptor FXR and its target gene SHP, which suppresses CYP7A1 and CYP8B1 transcription in liver cells (Goodwin et al., 2000). The other involves activation of FXR in the intestine and the induction of its target gene fibroblast growth factor 15 (FGF15), an intestinal hormone that travels to the liver where it represses the transcription of CYP7A1 and CYP8B1 (Inagaki et al., 2005). In our studies of chow-fed mice, CYP7A1, CYP8B1, and SHP mRNA and protein expression were increased in Smo (−) livers compared with Smo (+) livers (Figures 4A–4C). Thus, the higher levels of SHP protein in chow-fed Smo (−) mice were not sufficient to suppress critical SHP target genes (CYP7A1, CYP8B1) that gate the activity of the classic pathway of bile acid synthesis. The HC diet increased SHP mRNA and protein levels to comparable levels in both Smo (−) and Smo (+) livers; this diet-related increase in SHP associated with decreased expression of CYP8B1 mRNA and protein in both groups but did not reduce CYP7A1 mRNA or protein levels in either group (Figures 4A–4C). Although expression of FXR mRNA (data not shown) and protein (Figures 4E and 4F) were comparable in the two groups regardless of diet, the increased expression of SHP in Smo (−) livers of chow-fed mice suggested that Smo deletion might have increased FXR transcriptional activity when dietary cholesterol was low. However, examination of other FXR target genes (e.g., FGF15, APOC2, APOC3) revealed that only APOC2 transcript levels were significantly higher in Smo (−) mice on chow diet (Figure 4D). Furthermore, the mRNA and protein expression of these FXR target genes correlated poorly with each other in both groups on either diet (Figures 4D–4F). Intestinal mRNA levels of the FXR target gene FGF15 were similar in HC Smo (−) and Smo (+) mice (Figure 4D), although the hepatic content of FGF15 protein (Figures 4E and 4F) was somewhat lower in HC Smo (−) mice than in HC Smo (+) mice. Together, these data indicate that deleting Smo in hepatocytes differentially modulated the expression of FXR target genes and their proteins and disrupted feedback mechanisms that normally coordinate cholesterol and BA synthesis in the liver leading to systemic imbalance of BAs.
Figure 4.

Smoothened deletion in hepatocytes dysregulates pathways that critically control hepatic bile acid synthesis
Male Smo (+) and Smo (−) mice were fed either a low-cholesterol chow (0.02% w/w) or high-cholesterol (HC, 0.2% w/w) diet for 10 days.
(A) Hepatic gene expression of CYP7A1, CYP8B1, and SHP.
(B and C) Western blot and densitometric analysis of hepatic CYP7A1, CYP8B1, SHP, and β-tubulin protein levels.
(D) Gene expression of FGF15 in the small intestine and APOC2/APOC3 in the liver.
(E and F) Western blot and densitometric analysis of hepatic FXR, APOC3, APOC2, FGF15, and β-tubulin protein levels.
(G) Effect of FXR on FXR target gene expression in HC Smo (−) liver and small intestine. Results reported as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01 vs. Smo (+) Chow; #p < 0.05, ##p < 0.01 vs. Smo (+) HC.
Smo deletion in hepatocytes disrupts mechanisms that constrain liver triglyceride accumulation during dietary cholesterol challenge
Although our work reveals a role for Hh signaling in cholesterol and BA homeostasis, the Hh pathway has long been known to inhibit triglyceride accumulation (Pospisilik et al., 2010; Teperino et al., 2014), and recent studies by Matz-Soja et al revealed that Hh signaling broadly regulates liver lipid metabolism (Matz-Soja et al., 2016). Because that earlier work did not manipulate Hh activity selectively in hepatocytes, we reexamined the effect of hepatocyte-specific Smo deletion on fatty acid and triglyceride homeostasis. In chow-fed mice, we found that hepatocyte-specific deletion of Smo induced the lipogenic transcription factor SREBP1C, upregulated expression of SREBP1C-target genes that promote lipogenesis, and caused hepatic steatosis (Figures S1D–S1G). These findings are in agreement with the liver lipogenic responses that follow more global deletion of Smo in mice (Matz-Soja et al., 2016). Cholesterol is known to promote interactions between estrogen-related receptor α (ESRRA) and peroxisome-proliferator-activated receptor gamma coactivator 1α (PGC1α), thereby enhancing lipid disposal via mitochondrial beta-oxidation (Casaburi et al., 2018). Therefore, we investigated the effect of Smo deletion in hepatocytes on lipid balance in Smo (+) and Smo (−) mice fed an HC diet. Although HC diets increased hepatic TG levels in both groups, liver TG content remained significantly higher in Smo (−) mice than in Smo (+) mice after HC diet exposure (Figures 5A and 5B). These differences in hepatic TG content could no longer be explained by higher lipogenic activity in Smo (−) livers because Smo (+) and Smo (−) mice exhibited similar hepatic membrane and nuclear active SREBP1C accumulation, as well as similar mRNA expression of the lipogenic SREBP1C target-genes stearoyl-CoA desaturase 1 (SCD1) and diacylglycerol acyltransferase 2 (DGAT2) (Figures 5C–5E). In contrast, mechanisms that control lipid catabolism were differentially influenced in Smo (+) and Smo (−) mice fed HC diets. Expression of ESRRA, PGC1α, and PGC1β was induced in Smo (+) livers but not in Smo (−) liver, suggesting that suppression of mitochondrial beta-oxidation may have derailed efforts to compensate for increased lipogenesis, resulting in greater hepatic TG accumulation in Smo (−) mice during HC diet exposure (Figure 5F). These findings complement and extend the other new evidence that Smo deletion impairs cholesterol sensing mechanisms in hepatocytes.
Figure 5.

Smoothened deletion in hepatocytes disrupts mechanisms that constrain liver triglyceride accumulation during dietary cholesterol challenge
Male Smo (+) and Smo (−) mice were fed either a low-cholesterol chow (0.02% w/w) or a high-cholesterol (HC, 0.2% w/w) diet for 10 days.
(A) Hepatic triglyceride levels.
(B) Oil Red O staining in the liver. Scale bar equals 100 μm.
(C and D) Western blot and densitometric analysis of the hepatic membrane and nuclear SREBP1C (precursor, P; active, A), EGFR, and TAF15 protein levels.
(E) Hepatic SCD1 and DGAT2 gene expression.
(F) Hepatic gene expression of ESRRA, PGC1α, and PGC1β. Results reported as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01 vs. Smo (+) Chow; #p < 0.05, ##p < 0.01 vs. Smo (+) HC.
See also Figure S1.
Smo deletion in hepatocytes alters activities of kinases involved in Hh signaling
Smo is a G-protein-coupled receptor that links Hh ligand-driven activation of Ptc, the cell surface receptor for Hh ligands, to activation of Gli family transcription factors that control the expression of Hh-target genes. Recent work by Matz-Soja et al demonstrated that this canonical Hh signaling cascade can orchestrate changes in lipogenic activity by modulating the relative abundance of the three Gli family members (dubbed the ‘Gli-code’) (Matz-Soja et al., 2016). However, Smo activity is also sensitive to Ptc-independent mechanisms and Gli activity can be regulated by mechanisms that do not involve Smo. Thus, Smo and other components of the canonical Hh pathway can participate in so-called ‘noncanonical’ Hh signaling (Teperino et al., 2014; Pietrobono et al., 2019; Faria et al., 2019). To determine the relative importance of canonical versus noncanonical Hh signaling for changes that we noted after deleting Smo specifically in hepatocytes, we compared mRNA expression of Patched1, Shh, Gli1, Gli2, and Gli3 in Smo (−) and Smo (+) livers after feeding mice either chow or HC diet. In chow-fed mice, deleting Smo in hepatocytes decreased mRNA expression of Ptc, Shh, Gli1, and Gli3 (Figure 6A). HC diets decreased hepatic mRNA levels of Ptc, Shh, and Gli3 in both Smo (+) and Smo (−) livers and increased hepatic expression of Gli1 and Gli2 mRNAs in both groups (Figure 6A). Thus, although mRNAs for Hh ligand (Shh), receptor (Ptc), and two Hh-sensitive transcription factors (Gli1 and Gli3) were reduced in livers of chow-fed mice with Smo-depleted hepatocytes, hepatocyte Smo expression was not necessary for mice to upregulate either Gli1 or Gli2 transcripts during HC diets. Immunoblotting was then used to examine the hepatic nuclear distribution of the Glis. Although Gli1 and Gli3 activations were readily demonstrated and comparable in liver cell cytosols whether or not Smo was present (Figure 6B), we were unable to detect Gli1 and Gli3 in the liver nuclei of Smo (+) or Smo (−) mice fed either diet (Figure 6C). However, Gli2 was present in both strains and mainly accumulated within nuclei with the smaller Gli2 repressor form predominating in the liver nuclei of Smo (+) mice on both diets (Figures 6B and 6C). Nuclear accumulation of Gli2 repressor was generally lower in Smo (−) livers than in Smo (+) livers regardless of diet (Figure 6C). In contrast, Gli2 activator was detectable in the nuclei of chow-fed Smo (−) mice but disappeared when the Smo-depleted mice were fed HC diets (Figure 6C). Thus, deleting Smo specifically in hepatocytes appeared to have little effect on the Gli code in the liver, suggesting that the differences in cholesterol and BA homeostasis that occurred after Smo deletion were mainly mediated by noncanonical signaling activities of the Hh pathway.
Noncanonical Hh signaling is complex and involves both Smo-dependent/Gli-independent (i.e., nontranscriptional) processes, as well as Smo-independent/Gli-dependent (i.e., transcriptional) responses (Teperino et al., 2014; Pietrobono et al., 2019; Faria et al., 2019). One example of the former type of noncanonical signaling is Smo-dependent phosphorylation of AMPK-Thr172 which directly activates that enzyme (Teperino et al., 2012). Therefore, we compared hepatic levels of phosphorylated Thr172 AMPK and total AMPK in Smo (+) and Smo (−) mice. AMPK phosphorylation was significantly lower in livers of Smo (−) mice than in Smo (+) mice on both chow and HC diets (Figures 6D and 6E). Thus, depleting Smo in hepatocytes suppresses Smo-dependent/Gli-independent signaling by the Hh pathway. The net impact of Smo depletion on Smo-dependent/Gli-independent signaling is likely to be broad and context dependent, however, because many pathways interface with AMPK and other enzymes that Smo regulates. Evidence that deleting Smo in hepatocytes failed to suppress HC diet induction of Gli1 mRNAs suggests that the second type of noncanonical Hh signaling (i.e., Smo-independent/Gli-dependent signaling) may be activated in Smo (−) mice as Gli1 mRNA expression is a very reliable indicator of Gli2 transcriptional activity (Pan et al., 2006). This possibility is supported by the relative depletion of Gli2-repressor isoforms in hepatocyte nuclei of Smo (−) mice on both chow and HC diets, and the relative nuclear enrichment with Gli2 activator isoforms in Smo (−) hepatocytes of chow-fed mice (Figures 6B and 6C). Smo-independent/Gli-dependent noncanonical Hh signaling is often deployed by cancer cells to bypass Smo inhibition and involves compensatory changes in the activity of various kinases that control Gli2 stability and activity, such as GSK3β and Akt (Pietrobono et al., 2019). Akt can phosphorylate GSK3β on Ser9 to inhibit GSK3β activity. Thus, activation of Akt (by Ser473 phosphorylation) and inhibition of GSK3β (by Ser9 phosphorylation) promote stabilization/activation of Gli2 (Faria et al., 2019). Immunoblot analysis demonstrated that HC diets induced Akt activation and GSK3β inhibition in both Smo (−) and Smo (+) livers but both responses were significantly greater in the Smo (−) mice (Figures 6D, 6F and 6G), suggesting a mechanism (i.e., reduced accumulation of Gli2-repressor isoforms) that helps hepatocytes maintain Gli transcriptional activity despite Smo deficiency. Together, these findings identify previously unsuspected roles for noncanonical Hh signaling in maintaining cholesterol and BA balance.
Discussion
Dysregulated cholesterol homeostasis is a hallmark of many degenerative diseases and cancer. These disorders are also characterized by defective determination of cell fate decisions, a process that is orchestrated by a network of morphogenic signaling pathways that interact to control cell viability, proliferation, and differentiation in healthy tissues. In the present study, we uncovered cross talk that links these phenomena by demonstrating that systemic cholesterol homeostasis in healthy adult mice is determined by constitutive hepatic activity of Hh, a morphogenic pathway that is particularly cholesterol-sensitive.
The Hh pathway is very active in developing tissues but becomes restricted to small populations of cells in most healthy adult tissues where it functions to regulate fate decisions in stem/progenitor cells. Hh was thought to be silenced in adult hepatocytes because these cells failed to exhibit Hh-dependent transcriptional activity in Ptc-LacZ reporter mice (Sicklick et al., 2005). However, Matz-Soja et al recently demonstrated that deleting Smo, the obligate coreceptor for canonical Hh signaling, significantly altered lipid metabolism in healthy hepatocytes (Matz-Soja et al., 2016). The resultant lipogenic response correlated with shifts in the relative abundance of the three Gli family transcription factors in hepatocyte nuclei. Enforced manipulation of Gli factors in cultured hepatocytes and hepatoma cells also changed lipogenic gene expression, leading the authors to conclude that hepatocyte expression of genes involved in lipid metabolism is normally controlled by Smo which acts by modulating a ‘Gli-code’ that regulates the transcriptional activity of Hh target genes in hepatocytes (Schmidt-Heck et al., 2015). This important work complements and extends evidence showing that other critical morphogenic signaling pathways (e.g., Wnt, Notch) control both metabolism and fate decisions in adult hepatocytes (Russell and Monga, 2018; Siebel and Lendahl, 2017) and justifies further research to unravel the mechanisms whereby these pathways may interact to maintain tissue homeostasis (Matz-Soja et al., 2013; Kolbe et al., 2019; Kietzmann, 2019). Such work will have high clinical significance because dysregulation of these pathways promotes defective regenerative responses that result in liver cancer and cirrhosis (Siebel and Lendahl, 2017; Machado and Diehl, 2018; Russell and Monga, 2018).
Our study also shows that Smo deletion promotes hepatic lipogenesis and thus generally confirms the findings reported by Matz-Soja et al. (2016). However, our results are not identical, perhaps because our groups used different approaches to delete Smo. We relied on viral vectors that have been proven to activate Cre recombinase and delete floxed alleles specifically in adult hepatocytes (Gao et al., 2002). This enabled us to determine the consequences that ensue 7–10 days after Smo expression is selectively eliminated in healthy adult hepatocytes. Matz-Soja et al also studied healthy adult mice but deployed alternative strategies which either deleted Smo in bipotent liver epithelial progenitors and their progeny during fetal development (Matz-Soja et al., 2014), or conditionally deleted Smo in all adult cells that can activate the LAP (C/EBP β) promoter (Matz-Soja et al., 2016; Schonig et al., 2002). Earlier work from other groups shows that the first Cre recombinase driver deletes floxed alleles in both hepatocytes and cholangiocytes (Matthews et al., 2004) and that the latter Cre recombinase driver is active in the adult kidney, lung, muscle, intestine, and several other organs, as well as the liver (Schonig et al., 2002). Thus, Hh signaling may have been disrupted in nonhepatocytes in both of the other mouse models, and this might have influenced the results that were observed in hepatocytes because Hh signaling regulates metabolism and energy homeostasis in other tissues that cooperate with hepatocytes to coordinate lipid homeostasis (Teperino et al., 2014).
Importantly, in our model, targeted deletion of Smo in hepatocytes significantly influenced the expression and activity of several kinase cascades that are involved in both canonical and noncanonical Hh signaling, including AMPK, Akt, and GSK3β (Teperino et al., 2014). This provocative result merits further investigation but suggests that Smo can control hepatocyte metabolism by gating the activity of Ptc- and/or Gli-dependent ‘noncanonical’ Hh signaling mechanisms that have been thought to function independently of Smo. Therefore, Hh may not rely solely on the canonical Ptc-Smo interaction that directs the Gli-code to affect changes in hepatocyte gene expression. Rather, canonical and noncanonical Hh signaling may cooperate to modulate the activities of kinases that are downstream effectors of other morphogenic pathways, such as Wnt, Notch, TGFβ, and Hippo-Yap (Pelullo et al., 2019; Chatterjee and Sil, 2019; Zema et al., 2020), and thereby control the net output of the larger morphogenic signaling network. This has important implications for interpreting the basis for zonal differences in both metabolism and putative stem/progenitor populations that are present in healthy livers, as well as changes in these phenotypes that are required for injured livers to regenerate appropriately.
Adult livers perform a repertoire of metabolic and detoxification functions that are vital for health, as evidenced by the fact that disrupting liver function inevitably triggers systemic metabolic dysfunction, and ultimately results in multiorgan failure (Yan et al., 2014; Gu et al., 2018). However, the precise mechanisms whereby the liver maintains the health of other organs are poorly understood. To our knowledge, we are the first to demonstrate that Smo deletion in hepatocytes influences cholesterol and BA homeostasis in mice. We learned this by challenging mice with high dietary cholesterol after selectively deleting Smo in adult hepatocytes. Excess dietary cholesterol evoked the expected array of compensatory responses that operate at multiple levels to adjust cholesterol synthesis and disposal to maintain free cholesterol balance at the cellular level in wild-type mice. However, efforts to reorganize cholesterol metabolism were severely compromised in mice that lacked Smo, resulting in significant quantitative and qualitative alterations in circulating and fecal BA pools. Deletion of Smo in hepatocytes resulted in elevated hepatic cholesterol and a compensatory reduction in nuclear Srebp2 protein levels, hepatic protein levels of HMGCR, and hepatic protein levels of the HDL receptor SRB1 that transports cholesterol into the liver. However, these efforts to correct elevated hepatic cholesterol were offset by a lack of induction of LXRα and LXRα targets (e.g., ABCG5, ABCG8) that remove cholesterol from the liver, as well as persistent induction of the bile acid synthetic enzyme CYP7A1 despite high SHP levels and elevated plasma and fecal BAs. The fact that hepatocyte Smo activity controls systemic homeostasis of BAs has important implications because BAs interact with ubiquitous FXR and TGR5 receptors that critically regulate systemic energy balance (di Ciaula et al., 2017). BAs also bidirectionally integrate the metabolic activities of hosts and their intestinal microbiomes and thus help to shape the gut-liver axis (Chiang and Ferrell, 2018). The gut-liver axis both regulates and is a target of the circadian clock, and gut-liver axis disruption has been implicated in the pathogenesis/progression of various liver diseases, as well as obesity-associated disorders (Ahmad and Haeusler, 2019; Shapiro et al., 2018; Chiang and Ferrell, 2018). Therefore, our findings suggest a novel mechanism that helps to explain how Hh modulates susceptibility to cirrhosis, liver cancer, obesity, and related disorders, including nonalcoholic fatty liver disease.
Limitations of the study
This study demonstrated that Smo deletion in hepatocytes alters hepatic cholesterol homeostasis and leads to elevated hepatic cholesterol, cholesterol esters, triglycerides, and BAs. Smo deletion in hepatocytes also dysregulated hepatic responses to dietary cholesterol challenge. A limitation of this work is that it provides an overview of several cellular mechanisms Smo utilizes to regulate hepatic cholesterol balance. More work is needed to understand specifically how Smo deletion in hepatocytes results in (1) the loss of cholesterol-mediated upregulation of LXRα and LXRα target genes that function to remove excess cholesterol from the liver and (2) persistent elevation of the BA synthetic enzyme CYP7A1 despite high SHP levels and elevated plasma and fecal BAs. In addition, studies must be conducted to identify the specific canonical and noncanonical Hh signaling pathways that mediate Smo regulation of cholesterol balance. Investigating the role that the intestinal microbiome plays will also be important because evidence that Smo deletion altered fecal BAs and microbiome-derived metabolites are known to influence liver pathobiology.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-β-Tubulin | Abcam | Cat#ab6046; RRID:AB_10807712 |
| Rabbit polyclonal anti-SREBP2 | Abcam | Cat#30682; RRID:AB_779079 |
| Rabbit monoclonal anti-phospho-Akt Serine 473 (D9E) | Cell Signaling | Cat#4060; RRID:AB_2315049 |
| Rabbit polyclonal anti-Akt | Cell Signaling | Cat#9272; RRID:AB_329827 |
| Rabbit monoclonal anti-phospho-AMPKα Threonine 172 (40H9) | Cell Signaling | Cat#2535; RRID:AB_331250 |
| Rabbit polyclonal anti-AMPKα | Cell Signaling | Cat#2532; RRID:AB_330331 |
| Rabbit polyclonal anti-phospho-GSK3B Serine 9 | Cell Signaling | Cat#9336; RRID:AB_331405 |
| Mouse monoclonal Anti-TAF15 | EnCor Biotechnology | Cat#MCA-4D71; RRID:AB_2572388 |
| Mouse monoclonal anti-CYP7A1 (15B9.1) | Millipore | Cat#MABD42; RRID:AB_2756360 |
| Rabbit polyclonal anti-Gli1 | Novus Biologicals | Cat#NB600-600; RRID:AB_2111758 |
| Rabbit polyclonal anti-HMGCR (JF0981) | Novus Biologicals | Cat#NBP2-66888; RRID:AB_2860021 |
| Rabbit polyclonal anti-SRB1 | Novus Biologicals | Cat#NB400-101; RRID:AB_10107658 |
| Rabbit polyclonal anti-AACS | Proteintech | Cat#13815-1-AP; RRID:AB_2877977 |
| Rabbit polyclonal anti-ACAT2 | Proteintech | Cat#14755-1-AP; RRID:AB_2220236 |
| Mouse monoclonal anti-β-Actin HRP (C4) | Santa Cruz | Cat#sc-47778; RRID:AB_2714189 |
| Rabbit polyclonal anti-EGFR (1005) | Santa Cruz | Cat#sc-03; RRID:AB_631420 |
| Mouse monoclonal anti-FGF-15 (D-9) | Santa Cruz | Cat#sc-514647 |
| Mouse monoclonal anti-FXR/NR1H4 (D-3) | Santa Cruz | Cat#sc-25309; RRID:AB_628039 |
| Rabbit polyclonal anti-Gli2 (H-300) | Santa Cruz | Cat#sc-28674; RRID:AB_2111908 |
| Rabbit polyclonal anti-Gli3 (H-280) | Santa Cruz | Cat#sc-20688; RRID:AB_2109708 |
| Mouse monoclonal anti-SHP (H-5) | Santa Cruz | Cat#sc-271511; RRID:AB_10649803 |
| Mouse monoclonal anti-SREBP-1 (2A4) | Santa Cruz | Cat#sc-13551; RRID:AB_628282 |
| Rabbit polyclonal anti-APOC2 | Thermo Fisher | Cat#PA5-102480; RRID:AB_2851882 |
| Rabbit polyclonal anti-APOC3 | Thermo Fisher | Cat#PA5-116572 |
| Rabbit polyclonal anti-CYP8B1 | Thermo Fisher | Cat#PA5-37088; RRID:AB_2553905 |
| Anti-mouse IgG, HRP-linked antibody | Cell Signaling | Cat#7076; RRID:AB_330924 |
| Rabbit IgG HRP Linked Whole antibody | Millipore | Cat#GENA934-1ML; RRID:AB_2722659 |
| Bacterial and virus strains | ||
| AAV.TBG.Pl.Cre.rBG | James M. Wilson Lab, Penn Vector Core | Addgene AAV8; 107787-AAV8 |
| pENN.AAV.TBG.Pl.ffLuciferase.RBG | James M. Wilson Lab, Penn Vector Core | Addgene, AAV8; 105538-AAV8 |
| Chemicals, peptides, and recombinant proteins | ||
| Blotting-Grade Blocker | BIO-RAD | Cat# 1706404 |
| Clarity Western ECL Substrate | BIO-RAD | Cat# 1705060 |
| Bovine Serum Albumin ≥98% | Millipore Sigma | Cat# A7906-500G |
| Chloroform | Millipore Sigma | Cat# CX1060-1 |
| cOmplete Mini EDTA-free Protease Inhibitor Cocktail | Millipore Sigma | Cat# 4693159001 |
| Oil Red O | Millipore Sigma | Cat# O0625-25G |
| RIPA Buffer | Millipore Sigma | Cat# R0278-500ML |
| Restore Western Blot Stripping Buffer | Thermo Fisher | Cat# 21063 |
| TRIzol Reagent | Thermo Fisher | Cat# 15596018 |
| Critical commercial assays | ||
| Cholesterol/Cholesteryl Ester Quantitation Assay Kit | Abcam | Cat# ab65359 |
| Triglyceride Colorimetric Assay Kit | Cayman Chemical | Cat# 10010303 |
| Mouse Total Bile Acids Assay Kit | Crystal Chem | Cat# 80470 |
| High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher | Cat# 4368814 |
| Pierce BCA Protein Assay Kit | Thermo Fisher | Cat# 23225 |
| PowerTrack SYBR Green Master Mix | Thermo Fisher | Cat# A46111 |
| Deposited data | ||
| Raw and analyzed data from this paper | This Paper | Mendeley Data; https://doi.org/10.17632/6mcj5n2xwg.1 |
| Experimental models: Organisms/strains | ||
| Mouse Smotm2Amc/J (SMO-flox) | Jackson Laboratory | JAX stock# 004526 |
| Oligonucleotides | ||
| Primers for Mus Musculus, see Table S1 | This Paper | N/A |
| Software and algorithms | ||
| GraphPad Prism Version 5 for Windows | GraphPad Software | https://www.graphpad.com |
| Primer-Blast | NIH | https://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi?LINK_LOC=BlastHome |
| ImageJ Software | NIH | https://imagej.nih.gov/ij/ |
| Microsoft Excel | Microsoft Corporation | https://office.microsoft.com/excel |
| Other | ||
| 5053 - PicoLab Rodent Diet 20 (Chow Diet) | LabDiet | Cat# 3005740-220 |
| Standard chow diet (0.02% w/w cholesterol) | Envigo | Cat# 130104 |
| High cholesterol diet (0.2% w/w cholesterol) | Envigo | Cat# 160514 |
| Kinetex 2.6 μm C18 LC column 150 x 4.6 mm | Phenomenex | Cat# 00F-4462-E0 |
| ChemiDoc MP Imaging System | BIO-RAD | Cat# 17001402 |
| Nitrocellulose Membrane | BIO-RAD | Cat# 1620112 |
| StepOnePlus Real-Time PCR System | Applied Biosystems | Cat# 4376600 |
| Tecan Infinite M200 Pro Microplate Reader | Tecan | Model# M200 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Anna Mae Diehl (annamae.diehl@duke.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Animals
Adult male Smotm2Amc/J (Smo-flox) mice on a C57Bl6/J background (JAX stock# 004526, Jackson Laboratory, Bar Harbor, ME) were maintained in a temperature-controlled, specific pathogen-free room on 12-h light and dark cycles with ad libitum access to water and diet as indicated at Duke University (Long et al., 2001). Animal care and surgical procedures were conducted in compliance with an approved Duke University IACUC protocol, and those set forth in the “Guide for the Care and Use of Laboratory Animals” as published by the National Research Council. At 12 weeks of age, mice were injected by tail vein with 5 × 1011 genome-equivalents of AAV8-TBG-Luc (control; Smo (+)) or AAV8-TBG-Cre (Smo (-)) to selectively delete the Smo gene in hepatocytes (Nakai et al., 2005; Malato et al., 2011). Viruses were obtained from the University of Pennsylvania Viral Vector Core and Addgene. Two separate studies were conducted. In a preliminary study, Smo (+) (n = 3) and Smo (-) (n = 3) mice were fed a Purina 5053 standard chow low cholesterol diet (0.02% w/w; LabDiet, St. Louis, MO) for seven days. In the second study, Smo (+) (n = 8) and Smo (-) (n = 8) mice were fed either a standard chow low cholesterol diet (0.02% w/w; ENVIGO TD.130104) or a high cholesterol diet (0.2% w/w/; ENVIGO TD.160514) for ten days. At whole tissue harvest, slices of liver or ileum were formalin-fixed for paraffin embedding and the remainder snap frozen in liquid nitrogen for RNA and protein analysis. To obtain primary hepatocytes, liver was perfused with collagenase as described (Munker et al., 2017). Hepatocyte preparations were evaluated for viability and purity by light microscopy to assure that viability was at least 95%. Freshly isolated hepatocytes were immediately processed to obtain RNA and protein. Feces, plasma, and serum were collected at the time of sacrifice.
Methods details
Oil Red O staining and serum analysis for triglycerides and cholesterol
Lipid accumulation in liver was evaluated by Oil Red O (Cat# O0625-25G, Sigma-Aldrich, St. Louis, MO). Briefly, fresh tissue samples were fixed with 10% formalin, stained with Oil Red O (10 min), and counterstained with hematoxylin (2 min). Results were examined by light microscopy. Serum triglycerides were measured with a Triglyceride Colorimetric Assay kit (Cat# 10010303, Cayman Chemical Company, Ann Arbor, MI) with absorbance at 545 nm determined with the Tecan Infinite M200 Pro microplate reader (Tecan, Switzerland). Serum total cholesterol and cholesterol esters were measured in 96-well plates using a Cholesterol Quantification kit (Cat# ab65359, Abcam, Cambridge, UK) with absorbance at 570 nm determined with the Tecan Infinite M200 Pro microplate reader.
Liver cholesterol and triglyceride analysis
Liver tissue lipid was extracted using a modified Bligh and Dyer procedure (Bligh and Dyer, 1959). Approximately 100 mg of frozen liver tissue was placed in 2:1 CHCl3:MeOH OVN at RT. The sample was centrifuged at 1500 g for 10 min at RT and the lipid extract was evaporated at 60°C. CHCl3:MeOH (2:1) and 0.05% H2SO4 were added to the tube and tubes were centrifuged at 1500 g for 10 min. After removing the upper aqueous phase, aliquots of the bottom phase were transferred to a tube containing 2 ml 1% Triton X-100 in CHCl3, mixed, and evaporated at 60°C. Samples were reconstituted in assay diluents, heated at 60°C for 10 min, and centrifuged at 1500 g for 5 min. Total cholesterol and cholesterol esters were measured in 96-well plates using a Cholesterol Quantification kit (Cat# ab65359, Abcam) with absorbance at 570 nm determined with the Tecan Infinite M200 Pro microplate reader. Liver triglycerides were measured with a Triglyceride Colorimetric Assay kit (Cat# 10010303, Cayman Chemical Company) with absorbance at 545 nm determined with the Tecan Infinite M200 Pro microplate reader.
Quantification of fecal neutral and acidic sterol excretion
Quantitative fecal sterol excretion of both plant and animal derived sterols were analyzed by gas chromatography coupled with flame ionization detection (Warrier et al., 2015). Briefly, mice were individually housed in a cage with a wire bottom and allowed free access to low and high cholesterol diets and water for 3 consecutive days (72 hours). After a 3 day quantitative fecal collection, mice were weighed and feces were collected, dried in a 70°C vacuum oven, weighed, and crushed into a fine powder. A measured mass (50–100 mg) of feces was placed into a glass tube containing 100 μg of 5α-cholestane as an internal standard. Feces were saponified and neutral lipids were extracted into hexane, and mass analysis of extracted neutral sterols was conducted by gas chromatography flame ionization detection (Warrier et al., 2015). Total fecal neutral sterol mass represents the sum of cholesterol and the bacterial metabolite of cholesterol - coprostanol in each sample. For quantification of fecal BAs (fecal acidic sterols), dried feces were extracted (Bligh and Dyer, 1959). Approximately 100 mg of feces were placed in 3 ml of 2:1 CHCl3:MeOH OVN, and the methanolic phase was used for BA quantification (Pathak et al., 2020; Koeth et al., 2013). Fecal acidic sterol levels were measured using a mouse Total Bile Acid Assay kit (Cat# 80470, Crystal Chem, Elk Grove Village, IL).
Quantification of plasma bile acid levels
Individual plasma BA species were quantified using a quantitative stable isotope dilution LC-MS/MS analytical method (Choucair et al., 2020). Briefly, stable isotope labeled internal standards included were: D4-glycolithocholic acid, D4-glycoursodeoxycholic acid, D4-glycodeoxycholic acid, D4-glycocholic acid, D4-taurolithocholic acid, D4-tauroursodeoxycholic acid, D4-taurochenodeoxycholic acid, D4-taurodeoxycholic acid, D4-taurocholic acid, D4-lithocholic acid, D4-chenodeoxycholic acid, D4-deoxycholic acid, D4-cholic acid, and D4-glycochenodeoxycholic acid. Mouse plasma samples were mixed with ice-cold methanolic IS working solution of internal standard, vortexed for 10 min and centrifuged at 14000 g for 20 min at 4°C. Supernatants were transferred to glass HPLC vials for LC/MS/MS analysis using a 4000 Q-Trap triple quadrupole tandem mass spectrometer (SCIEX, Framingham, MA) equipped with an electrospray ionization source operating in negative ion mode. Mass spectrometry parameters were as follows: ions spray voltage – 4200 V, ion source heater temperature 500°C, source gas 1:35 psi, source gas 2:45 psi, and curtain gas 35 psi. Nitrogen gas was used for the nebulizer, curtain, and collision gas. Analyses were performed using electrospray ionization in negative-ion mode with multiple reaction monitoring (MRM) of precursor and characteristic product ions specific for each monitored BA. The HPLC system consisted of four binary pumps (LC-20 AD), autosampler operating at 10°C (Nexera X2 SIL-30AC), controller (CBM-20A) (Shimadzu Scientific Instruments, Columbia, MD) and a dual column switching valve system Rheodyne (IDEX Health & Science, Middleboro, MA). Chromatographic separations were performed on a reverse phase column (Kinetix C18, 2.6 μm, 150 mm x 4.6 mm ID; catalog # 00F-4462-E0; Phenomenex, Torrance, CA). Mobile phase A was 1 mM ammonium acetate and 0.1% acetic acid in methanol:acetonitrile:water (1:1:3; v/v/v) and mobile phase B was 0.1% acetic acid in methanol:acetonitrile:2-propanol (4.5:4.5:1; v/v/v). Samples were injected onto columns equilibrated in 100% A, and separated using a gradient as follows: 0-2 min 0% B; 2-20 min 0-100% B; 20-28.5 min 100% B. Flow rate was programmed as follows: 0.3 mL/min from 0-20 min, and 0.5 mL/min from 20-28 min. Samples were introduced to the mass spectrometer for analysis from 9-28 min. To eliminate carry over, an extensive washing step alternating between mobile phase A and B was added at the end of each run as follows: 100% A for 28-35 min, then directly switched to 100% B from 36-46 min, and equilibration step of 100% A from 46-60 min. Calibration curves were built by fitting each analyte concentration (10 different points) to peak area ratios (analyte/internal standard). The limit of detection (LOD) was defined as the lowest concentration of analyte in sample matrix (e.g. serum) that generated a signal-to-noise ratio of ≥ 3. The limit of quantification (LOQ) was defined as the lowest concentration of analyte in sample matrix that generated a signal-to-noise ratio of ≥ 10.
Quantitative real-time reverse transcription polymerase chain reaction (QRT-PCR)
Total RNAs were extracted from hepatocytes or whole livers using TRIzol reagent (Cat# 15596018, Thermo Fisher Scientific, Waltham, MA). RNA was reverse transcribed to cDNA templates using High-Capacity cDNA Reverse Transcription kit (Cat# 4368814, Thermo Fisher Scientific) according to the manufacturer’s instructions and assayed in duplicate by QRT-PCR with SYBR Green PCR Master Mix (Cat# A46111, Thermo Fisher Scientific) using a StepOnePlus Real-Time PCR System (Cat# 4376600, Applied Biosystems, Waltham, MA). Murine gene-specific primer sequences are available in Table S1. Results were normalized to mouse S9 based on the threshold cycle (Ct) and relative fold change calculated by the 2-ΔΔCt method.
Subcellular fractionation of whole liver and hepatocytes
Aliquots of frozen liver or hepatocytes (∼100 mg) were homogenized in homogenization buffer (20 mM Tris-HCl pH 7.4, 2 mM MgCl2, 250 mM sucrose, 10 mM EDTA, 10 mM EGTA, 5 mM dithiothreitol, 1 mM PMSF, 5 μg/mL pepstatin A, 10 μg/mL aprotinin, 0.5 tablet of Complete Protease Inhibitor cocktail). Following centrifugation at 1000 g at 4°C, an aliquot of the supernatant was retained as the cytosolic fraction and protein concentration was determined with a Pierce BCA Protein Assay Kit (Cat# 23225, Thermo Fisher Scientific). The rest of the supernatants were used to prepare the membrane fractions. For nuclear extracts, pellets were washed in homogenization buffer and collected by centrifugation at 1000 g at 4°C. Nuclear pellets were resuspended in nuclear buffer (20 mM HEPES-NaOH pH 7.6, 2.5% glycerol, 420 mM NaCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 0.5 tablet of Complete Protease Inhibitor cocktail), rotated at 4°C for 1 hour, and centrifuged at 100,000 g at 4°C. Supernatants were designated the nuclear extract and protein concentration was determined with a Pierce BCA Protein Assay Kit. Nuclear extracts and cytosolic fractions were subjected to SDS-PAGE and immunoblot analysis. For the membrane fraction, supernatants were centrifuged at 6800 g at 4°C. Supernatants from this spin were then centrifuged at 100,000 g at 4°C. Pellets were designated the membrane fraction and resuspended in SDS-lysis buffer (10 mM Tris-HCl pH 6.8, 1% SDS, 100 mM NaCl, 1 mM EDTA, 1 mM EGTA) and protein concentration was measured with a Pierce BCA Protein Assay Kit. Membrane fraction was mixed with an equal volume of urea buffer (150 μl, 62.5 mM Tris-HCl pH 6.8, 15% SDS, 8 M urea, 10% glycerol, 100 mM dithiothreitol). 4x SDS Loading buffer was added to this mixture and aliquots were incubated at 37°C and subjected to SDS-PAGE.
Immunoblotting
Liver tissue or isolated hepatocytes from Smo (+) and Smo (-) mice were lysed in RIPA buffer (Cat# R0278, Sigma-Aldrich) supplemented with phosphatase inhibitors and Complete Protease Inhibitor cocktail. Proteins were separated by SDS-PAGE and transferred electrophoretically to nitrocellulose membranes (Cat# 1620112, Bio-Rad, Hercules, CA). Blots were blocked, incubated OVN at 4°C with primary antibodies, probed with secondary HRP-conjugated antibodies, and visualized by ECL with detection on a Chemidoc MP Imaging system (Cat# 17001402, Bio-Rad). The details of the antibodies used are provided in the Key Resources Table and in Table S2.
Quantification and statistical analysis
Statistical analysis was performed by GraphPad Prism 5 software (GraphPad Software, San Diego, CA). All data were expressed as mean ± SEM. Statistical significance of differences was determined between two groups using unpaired Student’s t test. Differences with p ≤ 0.05 were considered to be statistically significant.
Acknowledgments
We thank Dr. Xiyou Zhou for his mouse colony management at Duke University. This work was supported by grants from the National Institutes of Health [RO1 DK077794 (A.M.D.), R01 DK120679 (J.M.B.), P01 HL 147823 (J.M.B.), P50 AA024333 (J.M.B.)] and The Florence McAlister Professorship of Medicine (A.M.D.).
Author contributions
Conceptualization, A.M.D. and J.M.B.; Methodology, G.D.D., A.M.D., and J.M.B.; Formal Analysis, Investigation, Visualization, G.D.D., S.O., L.T., S.Z., A.L.B., C.B.G., V.G., V.V., and P.P.; Writing – Original Draft, G.D.D., A.M.D., and J.M.B.; Writing – Review & Editing, G.D.D., S.O., L.T., S.Z., A.L.B., C.B.G., V.G., P.P., A.M.D., and J.M.B.; Supervision, A.M.D. and J.M.B.; Funding Acquisition, A.M.D. and J.M.B.
Declaration of interests
The authors declare no competing interests.
Published: September 24, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103089.
Supplemental information
Data and code availability
-
•
All data have been deposited at Mendeley and are publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Ahmad T.R., Haeusler R.A. Bile acids in glucose metabolism and insulin signalling - mechanisms and research needs. Nat. Rev. Endocrinol. 2019;15:701–712. doi: 10.1038/s41574-019-0266-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatori N., Li N., Fang F., Boyer J.L., Christian W.V., Hammond C.L. OST alpha-OST beta: a key membrane transporter of bile acids and conjugated steroids. Front. Biosci. (Landmark Ed) 2009;14:2829–2844. doi: 10.2741/3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banavali N.K. The mechanism of cholesterol modification of hedgehog ligand. J. Comput. Chem. 2020;41:520–527. doi: 10.1002/jcc.26097. [DOI] [PubMed] [Google Scholar]
- Bligh E.G., Dyer W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- Casaburi I., Chimento A., de Luca A., Nocito M., Sculco S., Avena P., Trotta F., Rago V., Sirianni R., Pezzi V. Cholesterol as an endogenous ERRalpha agonist: a new perspective to cancer treatment. Front. Endocrinol. 2018;9:525. doi: 10.3389/fendo.2018.00525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S., Sil P.C. Targeting the crosstalks of Wnt pathway with Hedgehog and Notch for cancer therapy. Pharmacol. Res. 2019;142:251–261. doi: 10.1016/j.phrs.2019.02.027. [DOI] [PubMed] [Google Scholar]
- Chiang J.Y. Regulation of bile acid synthesis: pathways, nuclear receptors, and mechanisms. J. Hepatol. 2004;40:539–551. doi: 10.1016/j.jhep.2003.11.006. [DOI] [PubMed] [Google Scholar]
- Chiang J.Y.L. Bile acid metabolism and signaling in liver disease and therapy. Liver Res. 2017;1:3–9. doi: 10.1016/j.livres.2017.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang J.Y.L., Ferrell J.M. Bile acid metabolism in liver pathobiology. Gene Expr. 2018;18:71–87. doi: 10.3727/105221618X15156018385515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choucair I., Nemet I., Li L., Cole M.A., Skye S.M., Kirsop J.D., Fischbach M.A., Gogonea V., Brown J.M., Tang W.H.W., Hazen S.L. Quantification of bile acids: a mass spectrometry platform for studying gut microbe connection to metabolic diseases. J. Lipid Res. 2020;61:159–177. doi: 10.1194/jlr.RA119000311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Sousa Abreu R., Penalva L.O., Marcotte E.M., Vogel C. Global signatures of protein and mRNA expression levels. Mol. Biosyst. 2009;5:1512–1526. doi: 10.1039/b908315d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debose-Boyd R.A. Feedback regulation of cholesterol synthesis: sterol-accelerated ubiquitination and degradation of HMG CoA reductase. Cell Res. 2008;18:609–621. doi: 10.1038/cr.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- di Ciaula A., Garruti G., Lunardi Baccetto R., Molina-Molina E., Bonfrate L., Wang D.Q., Portincasa P. Bile acid physiology. Ann. Hepatol. 2017;16:s4–s14. doi: 10.5604/01.3001.0010.5493. [DOI] [PubMed] [Google Scholar]
- Faria A.V.S., Akyala A.I., Parikh K., Bruggemann L.W., Spek C.A., Cao W., Bruno M.J., Bijlsma M.F., Fuhler G.M., Peppelenbosch M.P. Smoothened-dependent and -independent pathways in mammalian noncanonical Hedgehog signaling. J. Biol. Chem. 2019;294:9787–9798. doi: 10.1074/jbc.RA119.007956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao G.P., Alvira M.R., Wang L., Calcedo R., Johnston J., Wilson J.M. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. U. S. A. 2002;99:11854–11859. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein J.L., Brown M.S. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- Goodwin B., Jones S.A., Price R.R., Watson M.A., Mckee D.D., Moore L.B., Galardi C., Wilson J.G., Lewis M.C., Roth M.E. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol. Cell. 2000;6:517–526. doi: 10.1016/s1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- Gorojankina T. Hedgehog signaling pathway: a novel model and molecular mechanisms of signal transduction. Cell Mol. Life Sci. 2016;73:1317–1332. doi: 10.1007/s00018-015-2127-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C., Qiao W., Wang L., Li M., Song K. Identification of genes and pathways associated with multiple organ dysfunction syndrome by microarray analysis. Mol. Med. Rep. 2018;18:31–40. doi: 10.3892/mmr.2018.8973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa S., Noda K., Maeda A., Matsuoka M., Yamasaki M., Fukui T. Acetoacetyl-CoA synthetase, a ketone body-utilizing enzyme, is controlled by SREBP-2 and affects serum cholesterol levels. Mol. Genet. Metab. 2012;107:553–560. doi: 10.1016/j.ymgme.2012.08.017. [DOI] [PubMed] [Google Scholar]
- Horton J.D., Goldstein J.L., Brown M.S. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu A., Song B.L. The interplay of patched, smoothened and cholesterol in Hedgehog signaling. Curr. Opin. Cell Biol. 2019;61:31–38. doi: 10.1016/j.ceb.2019.06.008. [DOI] [PubMed] [Google Scholar]
- Huang P., Nedelcu D., Watanabe M., Jao C., Kim Y., Liu J., Salic A. Cellular cholesterol directly activates smoothened in Hedgehog signaling. Cell. 2016;166:1176–1187 e14. doi: 10.1016/j.cell.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki T., Choi M., Moschetta A., Peng L., Cummins C.L., Mcdonald J.G., Luo G., Jones S.A., Goodwin B., Richardson J.A. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–225. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Jeon S.M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016;48:e245. doi: 10.1038/emm.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kietzmann T. Liver zonation in health and disease: hypoxia and hypoxia-inducible transcription factors as concert masters. Int. J. Mol. Sci. 2019;20:2347. doi: 10.3390/ijms20092347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeth R.A., Wang Z., Levison B.S., Buffa J.A., Org E., Sheehy B.T., Britt E.B., Fu X., Wu Y., Li L. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013;19:576–585. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolbe E., Aleithe S., Rennert C., Spormann L., Ott F., Meierhofer D., Gajowski R., Stopel C., Hoehme S., Kucken M. Mutual zonated interactions of Wnt and Hh signaling are orchestrating the metabolism of the adult liver in mice and human. Cell Rep. 2019;29:4553–4567 e7. doi: 10.1016/j.celrep.2019.11.104. [DOI] [PubMed] [Google Scholar]
- Kong J.H., Siebold C., Rohatgi R. Biochemical mechanisms of vertebrate Hedgehog signaling. Development. 2019;146:dev166892. doi: 10.1242/dev.166892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long F., Zhang X.M., Karp S., Yang Y., Mcmahon A.P. Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development. 2001;128:5099–5108. doi: 10.1242/dev.128.24.5099. [DOI] [PubMed] [Google Scholar]
- Luo J., Yang H., Song B.L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 2020;21:225–245. doi: 10.1038/s41580-019-0190-7. [DOI] [PubMed] [Google Scholar]
- Machado M.V., Diehl A.M. Hedgehog signalling in liver pathophysiology. J. Hepatol. 2018;68:550–562. doi: 10.1016/j.jhep.2017.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier T., Guell M., Serrano L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009;583:3966–3973. doi: 10.1016/j.febslet.2009.10.036. [DOI] [PubMed] [Google Scholar]
- Malato Y., Naqvi S., Schurmann N., Ng R., Wang B., Zape J., Kay M.A., Grimm D., Willenbring H. Fate tracing of mature hepatocytes in mouse liver homeostasis and regeneration. J. Clin. Invest. 2011;121:4850–4860. doi: 10.1172/JCI59261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews V.B., Rose-John S., Yeoh G.C. Genetic manipulations utilizing albumin and alpha-fetoprotein promoter/enhancers affect both hepatocytes and oval cells. Hepatology. 2004;40:759. doi: 10.1002/hep.20396. author reply 760. [DOI] [PubMed] [Google Scholar]
- Matz-Soja M., Aleithe S., Marbach E., Bottger J., Arnold K., Schmidt-Heck W., Kratzsch J., Gebhardt R. Hepatic Hedgehog signaling contributes to the regulation of IGF1 and IGFBP1 serum levels. Cell Commun. Signal. 2014;12:11. doi: 10.1186/1478-811X-12-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matz-Soja M., Hovhannisyan A., Gebhardt R. Hedgehog signalling pathway in adult liver: a major new player in hepatocyte metabolism and zonation? Med. Hypotheses. 2013;80:589–594. doi: 10.1016/j.mehy.2013.01.032. [DOI] [PubMed] [Google Scholar]
- Matz-Soja M., Rennert C., Schonefeld K., Aleithe S., Boettger J., Schmidt-Heck W., Weiss T.S., Hovhannisyan A., Zellmer S., Kloting N. Hedgehog signaling is a potent regulator of liver lipid metabolism and reveals a GLI-code associated with steatosis. Elife. 2016;5:e13308. doi: 10.7554/eLife.13308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan A.E., Mooney K.M., Wilkinson S.J., Pickles N.A., Mc Auley M.T. Cholesterol metabolism: a review of how ageing disrupts the biological mechanisms responsible for its regulation. Ageing Res. Rev. 2016;27:108–124. doi: 10.1016/j.arr.2016.03.008. [DOI] [PubMed] [Google Scholar]
- Munker S., Wu Y.L., Ding H.G., Liebe R., Weng H.L. Can a fibrotic liver afford epithelial-mesenchymal transition? World J. Gastroenterol. 2017;23:4661–4668. doi: 10.3748/wjg.v23.i26.4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myant N.B., Mitropoulos K.A. Cholesterol 7 alpha-hydroxylase. J. Lipid Res. 1977;18:135–153. [PubMed] [Google Scholar]
- Nachtergaele S., Mydock L.K., Krishnan K., Rammohan J., Schlesinger P.H., Covey D.F., Rohatgi R. Oxysterols are allosteric activators of the oncoprotein smoothened. Nat. Chem. Biol. 2012;8:211–220. doi: 10.1038/nchembio.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai H., Fuess S., Storm T.A., Muramatsu S., Nara Y., Kay M.A. Unrestricted hepatocyte transduction with adeno-associated virus serotype 8 vectors in mice. J. Virol. 2005;79:214–224. doi: 10.1128/JVI.79.1.214-224.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedelcu D., Liu J., Xu Y., Jao C., Salic A. Oxysterol binding to the extracellular domain of smoothened in Hedgehog signaling. Nat. Chem. Biol. 2013;9:557–564. doi: 10.1038/nchembio.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm W., Rodenfels J. Understanding the role of lipids and lipoproteins in development. Development. 2020;147:dev186411. doi: 10.1242/dev.186411. [DOI] [PubMed] [Google Scholar]
- Pan Y., Bai C.B., Joyner A.L., Wang B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell Biol. 2006;26:3365–3377. doi: 10.1128/MCB.26.9.3365-3377.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parini P., Davis M., Lada A.T., Erickson S.K., Wright T.L., Gustafsson U., Sahlin S., Einarsson C., Eriksson M., Angelin B. ACAT2 is localized to hepatocytes and is the major cholesterol-esterifying enzyme in human liver. Circulation. 2004;110:2017–2023. doi: 10.1161/01.CIR.0000143163.76212.0B. [DOI] [PubMed] [Google Scholar]
- Pathak P., Helsley R.N., Brown A.L., Buffa J.A., Choucair I., Nemet I., Gogonea C.B., Gogonea V., Wang Z., Garcia-Garcia J.C. Small molecule inhibition of gut microbial choline trimethylamine lyase activity alters host cholesterol and bile acid metabolism. Am. J. Physiol. Heart Circ. Physiol. 2020;318:H1474–H1486. doi: 10.1152/ajpheart.00584.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelullo M., Zema S., Nardozza F., Checquolo S., Screpanti I., Bellavia D. Wnt, Notch, and TGF-beta pathways impinge on hedgehog signaling complexity: an open window on cancer. Front. Genet. 2019;10:711. doi: 10.3389/fgene.2019.00711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepinsky R.B., Zeng C., Wen D., Rayhorn P., Baker D.P., Williams K.P., Bixler S.A., Ambrose C.M., Garber E.A., Miatkowski K. Identification of a palmitic acid-modified form of human Sonic Hedgehog. J. Biol. Chem. 1998;273:14037–14045. doi: 10.1074/jbc.273.22.14037. [DOI] [PubMed] [Google Scholar]
- Pietrobono S., Gagliardi S., Stecca B. Non-canonical hedgehog signaling pathway in cancer: activation of GLI transcription factors beyond smoothened. Front. Genet. 2019;10:556. doi: 10.3389/fgene.2019.00556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter J.A., Ekker S.C., Park W.J., von Kessler D.P., Young K.E., Chen C.H., Ma Y., Woods A.S., Cotter R.J., Koonin E.V., Beachy P.A. Hedgehog patterning activity: role of a lipophilic modification mediated by the carboxy-terminal autoprocessing domain. Cell. 1996;86:21–34. doi: 10.1016/s0092-8674(00)80074-4. [DOI] [PubMed] [Google Scholar]
- Porter J.A., Young K.E., Beachy P.A. Cholesterol modification of hedgehog signaling proteins in animal development. Science. 1996;274:255–259. doi: 10.1126/science.274.5285.255. [DOI] [PubMed] [Google Scholar]
- Pospisilik J.A., Schramek D., Schnidar H., Cronin S.J., Nehme N.T., Zhang X., Knauf C., Cani P.D., Aumayr K., Todoric J. Drosophila genome-wide obesity screen reveals hedgehog as a determinant of brown versus white adipose cell fate. Cell. 2010;140:148–160. doi: 10.1016/j.cell.2009.12.027. [DOI] [PubMed] [Google Scholar]
- Prince E., Marcetteau J., Therond P.P. Circulating Hedgehog: a fresh view of a classic morphogen. Development. 2020;147:dev186395. doi: 10.1242/dev.186395. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan A., Rohatgi R., Siebold C. Cholesterol access in cellular membranes controls Hedgehog signaling. Nat. Chem. Biol. 2020;16:1303–1313. doi: 10.1038/s41589-020-00678-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repa J.J., Berge K.E., Pomajzl C., Richardson J.A., Hobbs H., Mangelsdorf D.J. Regulation of ATP-binding cassette sterol transporters ABCG5 and ABCG8 by the liver X receptors alpha and beta. J. Biol. Chem. 2002;277:18793–18800. doi: 10.1074/jbc.M109927200. [DOI] [PubMed] [Google Scholar]
- Russell D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 2003;72:137–174. doi: 10.1146/annurev.biochem.72.121801.161712. [DOI] [PubMed] [Google Scholar]
- Russell J.O., Monga S.P. Wnt/beta-Catenin signaling in liver development, homeostasis, and pathobiology. Annu. Rev. Pathol. 2018;13:351–378. doi: 10.1146/annurev-pathol-020117-044010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Heck W., Matz-Soja M., Aleithe S., Marbach E., Guthke R., Gebhardt R. Fuzzy modeling reveals a dynamic self-sustaining network of the GLI transcription factors controlling important metabolic regulators in adult mouse hepatocytes. Mol. Biosyst. 2015;11:2190–2197. doi: 10.1039/c5mb00129c. [DOI] [PubMed] [Google Scholar]
- Schonig K., Schwenk F., Rajewsky K., Bujard H. Stringent doxycycline dependent control of CRE recombinase in vivo. Nucleic Acids Res. 2002;30:e134. doi: 10.1093/nar/gnf134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro H., Kolodziejczyk A.A., Halstuch D., Elinav E. Bile acids in glucose metabolism in health and disease. J. Exp. Med. 2018;215:383–396. doi: 10.1084/jem.20171965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W.J., Asthana S., Kraemer F.B., Azhar S. Scavenger receptor B type 1: expression, molecular regulation, and cholesterol transport function. J. Lipid Res. 2018;59:1114–1131. doi: 10.1194/jlr.R083121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicklick J.K., Li Y.X., Choi S.S., Qi Y., Chen W., Bustamante M., Huang J., Zdanowicz M., Camp T., Torbenson M.S. Role for hedgehog signaling in hepatic stellate cell activation and viability. Lab. Invest. 2005;85:1368–1380. doi: 10.1038/labinvest.3700349. [DOI] [PubMed] [Google Scholar]
- Siebel C., Lendahl U. Notch signaling in development, tissue homeostasis, and disease. Physiol. Rev. 2017;97:1235–1294. doi: 10.1152/physrev.00005.2017. [DOI] [PubMed] [Google Scholar]
- Tabas I. Consequences of cellular cholesterol accumulation: basic concepts and physiological implications. J. Clin. Invest. 2002;110:905–911. doi: 10.1172/JCI16452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teperino R., Aberger F., Esterbauer H., Riobo N., Pospisilik J.A. Canonical and non-canonical Hedgehog signalling and the control of metabolism. Semin. Cell Dev. Biol. 2014;33:81–92. doi: 10.1016/j.semcdb.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teperino R., Amann S., Bayer M., Mcgee S.L., Loipetzberger A., Connor T., Jaeger C., Kammerer B., Winter L., Wiche G. Hedgehog partial agonism drives Warburg-like metabolism in muscle and brown fat. Cell. 2012;151:414–426. doi: 10.1016/j.cell.2012.09.021. [DOI] [PubMed] [Google Scholar]
- Vogel C., Marcotte E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012;13:227–232. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrier M., Shih D.M., Burrows A.C., Ferguson D., Gromovsky A.D., Brown A.L., Marshall S., Mcdaniel A., Schugar R.C., Wang Z. The TMAO-generating enzyme flavin monooxygenase 3 is a central regulator of cholesterol balance. Cell Rep. 2015;10:326–338. doi: 10.1016/j.celrep.2014.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willnow T.E., Hammes A., Eaton S. Lipoproteins and their receptors in embryonic development: more than cholesterol clearance. Development. 2007;134:3239–3249. doi: 10.1242/dev.004408. [DOI] [PubMed] [Google Scholar]
- Xiao X., Tang J.J., Peng C., Wang Y., Fu L., Qiu Z.P., Xiong Y., Yang L.F., Cui H.W., He X.L. Cholesterol modification of smoothened is required for hedgehog signaling. Mol. Cell. 2017;66:154–162 e10. doi: 10.1016/j.molcel.2017.02.015. [DOI] [PubMed] [Google Scholar]
- Xu H., Zhou S., Tang Q., Xia H., Bi F. Cholesterol metabolism: new functions and therapeutic approaches in cancer. Biochim. Biophys. Acta Rev. Cancer. 2020;1874:188394. doi: 10.1016/j.bbcan.2020.188394. [DOI] [PubMed] [Google Scholar]
- Yan J., Li S., Li S. The role of the liver in sepsis. Int. Rev. Immunol. 2014;33:498–510. doi: 10.3109/08830185.2014.889129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T., Espenshade P.J., Wright M.E., Yabe D., Gong Y., Aebersold R., Goldstein J.L., Brown M.S. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110:489–500. doi: 10.1016/s0092-8674(02)00872-3. [DOI] [PubMed] [Google Scholar]
- Zema S., Pelullo M., Nardozza F., Felli M.P., Screpanti I., Bellavia D. A dynamic role of Mastermind-like 1: a journey through the main (Path)ways between development and cancer. Front. Cell Dev. Biol. 2020;8:613557. doi: 10.3389/fcell.2020.613557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Bulkley D.P., Xin Y., Roberts K.J., Asarnow D.E., Sharma A., Myers B.R., Cho W., Cheng Y., Beachy P.A. Structural basis for cholesterol transport-like activity of the hedgehog receptor patched. Cell. 2018;175:1352–1364.e14. doi: 10.1016/j.cell.2018.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu R., Ou Z., Ruan X., Gong J. Role of liver X receptors in cholesterol efflux and inflammatory signaling (review) Mol. Med. Rep. 2012;5:895–900. doi: 10.3892/mmr.2012.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data have been deposited at Mendeley and are publicly available as of the date of publication. DOIs are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.