

Summary
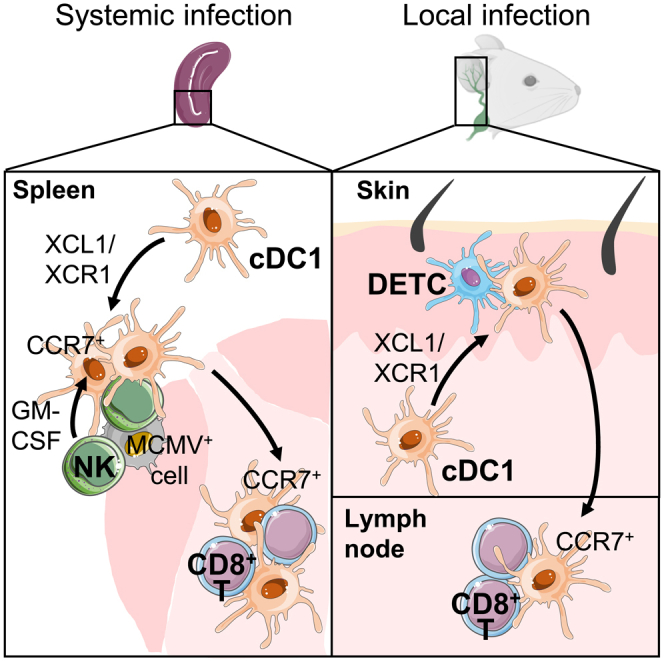
Successful immune responses rely on a regulated delivery of the right signals to the right cells at the right time. Here we show that natural killer (NK) and dendritic epidermal γδ T cells (DETCs) use similar mechanisms to spatiotemporally orchestrate conventional type 1 dendritic cell (cDC1) functions in the spleen, skin, and its draining lymph nodes (dLNs). Upon MCMV infection in the spleen, cDC1 clusterize with activated NK cells in marginal zones. This XCR1-dependent repositioning of cDC1 toward NK cells allows contact delivery of IL-12 and IL-15/IL-15Rα by cDC1, which is critical for NK cell responses. NK cells deliver granulocyte-macrophage colony-stimulating factor (GM-CSF) to cDC1, guiding their CCR7-dependent relocalization into the T cell zone. In MCMV-infected skin, XCL1-secreting DETCs promote cDC1 migration from the skin to the dLNs. This XCR1-dependent licensing of cDC1 both in the spleen and skin accelerates antiviral CD8+ T cell responses, revealing an additional mechanism through which cDC1 bridge innate and adaptive immunity.
Subject areas: Biological sciences, Immune system, Cell biology
Graphical abstract
Highlights
-
•
Upon viral infection in the spleen, NK cells clusterize with cDC1 in the marginal zone
-
•
This XCL1/XCR1-dependent interaction allows mutual delivery of activating signals
-
•
NK cell GM-CSF directs cDC1 migration to T cell zone boosting CD8+ T cell priming
-
•
In the skin, DETCs contact cDC1 via XCL1/XCR1 to promote antiviral CD8+ T cell priming
Biological sciences; Immune system; Cell biology
Introduction
The success of an immune response against infections depends on timely coordinated actions of both innate and adaptive immunity. In lymphoid organs, the delivery of the right signals to the right cells at the right time guarantees a rapid and efficient relay from innate cells to T cells, boosting specific and durable protective immunity. Dendritic cells (DCs) are key in the activation of innate lymphoid cells (ILCs) and in the priming of T lymphocytes. How DCs deliver the help from innate immune cells to naive T cells remains largely unknown.
Type 1 conventional DCs (cDC1) are critical in mounting cytotoxic CD8+ T lymphocyte (CTL) responses against intracellular pathogens (Merad et al., 2013) and cancers (Cancel et al., 2019; Wculek et al., 2020), in part due to their high efficacy at cross-presenting exogenous antigens (Alexandre et al., 2014). Through their capacity to rapidly produce large amounts of IL-12 (Dalod et al., 2002; Reis e Sousa et al., 1997), cDC1 efficiently activate ILCs, such as natural killer (NK) cells, and of T cell subsets with innate-like functions, such as memory CD8+ T cells (Mashayekhi et al., 2011; Miyake et al., 2009; Alexandre et al., 2016). cDC1 encompass the XCR1+CD8α+ cDC1 of lymphoid organs and the XCR1+CD103+CD11b- cDC1 residing in nonlymphoid tissues. Regardless of their tissues of residency, cDC1 are phenotypically defined by their selective expression of the chemokine receptor XCR1 (Bachem et al., 2012; Crozat et al, 2010, 2011; Dorner et al., 2009). The unique known ligand for XCR1 in mice is XCL1. NK cells and memory T cells express Xcl1 transcript (http://www.immgen.org) but do not translate it at steady state, which poises them for rapid secretion of XCL1 upon proper stimulation (Dorner et al., 2004; Crozat et al., 2010; Yamazaki et al., 2010). XCR1 expression on cDC1 potentiates CD8+ T cell priming and effector functions against infection and immunization (Crozat et al., 2010; Dorner et al., 2009), in part by orchestrating in lymphoid organs the formation of clusters between cDC1 and naive CD8+ T cells which rapidly produce XCL1 upon activation (Brewitz et al., 2017). XCR1 was also suggested to help cDC1 interpreting guiding cues provided in tumors by NK1.1+ or AsialoGM1+ cells (Böttcher et al., 2018), which encompass NK, natural killer T (NKT), and subsets of T cells (Iigo et al., 1997; Trambley et al., 1999; Slifka et al., 2000; Yamanokuchi et al., 2005; Kosaka et al., 2007; Moore et al., 2008; Nour-Eldine et al., 2018). Ectopic overexpression of XCL1 by inflammatory tumors increases cDC1 infiltration (Böttcher et al., 2018). However, the physiological contribution of the XCL1/XCR1 axis in NK/cDC1 interactions has not been determined. In addition, the mode of action of XCL1/XCR1 is not well understood: the XCL1/XCR1 axis could act by stabilizing cDC1/lymphocyte conjugates or by promoting cDC1 microanatomical repositioning toward XCL1-producing cells.
In the spleen, cDC1 are scattered in the red pulp and the white pulp with the exception of B cell follicles (Alexandre et al., 2016; Calabro et al., 2016; Dorner et al., 2009). The spleen filters circulating blood for blood borne pathogens. The red pulp region is the first entry point and hosts cells with innate immune functions such as monocytes, macrophages, NK cells, and NKT cells. The white pulps are embedded in the red pulp and delineated by MOMA-1+ marginal areas. They contain B cell follicles encircling T cell zones, where naïve T cells are confined. The bridging channel links the red pulp to the T cell zones, facilitating cell passage between the two compartments. Their distribution in the spleen raises the question of the division of labor between cDC1 subsets: the red pulp cDC1 subset could be more prone to interact with innate cells, whereas T cell zone cDC1 could favor adaptive responses. This potential division of labor might not occur between distinct cDC1 subsets but rather over time in the same cell during its activation trajectory because cDC1 are proposed to migrate from the red pulp to the T cell zone during their maturation, with CCR7 being a master regulator of this process (Calabro et al., 2016). Yet, the precise migration pattern of cDC1 in the spleen, the critical time window during which cDC1 may acquire guiding cues, and the molecular mechanisms regulating their distribution are still unknown.
To address these questions, we used mouse cytomegalovirus (MCMV) because its infection induces a robust immune response of both splenic NK and CD8+ T cells, which reside in the red pulp and the T cell zone, respectively, and whose activation strongly relies on cDC1 including their IL-12 production (Dalod et al., 2002; Krug et al., 2004), and their cross-presentations of viral antigens (Busche et al., 2013; Snyder et al., 2010; Torti et al., 2011). Moreover, NK cells rapidly produce XCL1 as early as one day after infection, before they reach full activation (Dorner et al., 2004; Bezman et al., 2012). Hence, MCMV infection is well suited to assess the specific contribution of the XCL1/XCR1 axis in NK/cDC1 interactions in situ.
Results
XCR1 on cDC1 promotes NK cell activation, microanatomical repositioning, and proliferation
To address the contribution of the XCL1/XCR1 axis in promoting NK/cDC1 interactions, we used the polyinosinic-polycytidylic acid (poly(I:C)), a synthetic analogue of double-stranded RNA which triggers TLR3 signaling including in cDC1 (McCartney et al., 2009; Lauterbach et al., 2010). Poly(I:C) administration induces a strong NK cell activation (Akazawa et al., 2007; McCartney et al., 2009), in part through direct contacts with DCs (Akazawa et al., 2007; Beuneu et al., 2009; Ebihara et al., 2010; Kasamatsu et al., 2014). Depletion of cDC1 or NK cells before poly(I:C) administration nearly abrogated NK cell IFN-γ production (Figure S1A) and cDC1 maturation (Figure S1B), respectively. Xcr1 deficiency also decreased cDC1 and NK cell responses to poly(I:C) (Figures S1C–S1E), showing the critical contribution of the XCL1/XCR1 axis to the cooperation between cDC1 and NK cells. We wondered whether a similar phenomenon occurs during productive viral infection. At 40 h after infection, Xcr1 deficiency altered NK cell activation and effector responses, as assessed by their CD69 expression (Figures 1A and S1F), IFN-γ production (Figure 1B), and upregulation of granzyme B (GzmB) (Figure 1C). We then investigated the migratory behavior of NK cells in the spleen upon infection. At steady state, NK cells mostly reside in the red pulp (Figure S1G) (Gregoire et al., 2008; Jaeger et al., 2012; Walzer et al., 2007). Upon MCMV infection, NK cells relocalized in clusters in the marginal zone, around infected cells expressing the MCMV immediate early gene 1 (IE-1+ cells), and produced IFN-γ (Figure 1D; arrowheads). We defined NK cell clusters as the gathering of at least 10 IFN-γ+ cells, including NKp46+ cells, around at least one IE-1+ cell, in the marginal zone. Xcr1 deficiency significantly decreased the numbers of activated NK cell clusters (Figures 1E and 1F), but did not render mice more susceptible to MCMV than controls, at 40 h and 48 h after infection (Figure S1H). The absence of XCR1 also impaired proliferation of MCMV-specific Ly49H+ NK cells (Figures 1G and 1H). Altogether, these results showed that XCR1 expression on cDC1 promoted NK cell antiviral responses against MCMV and most likely through specific NK/cDC1 interactions.
Figure 1.

XCR1 promotes NK cell activation and redistribution in the spleen upon MCMV infection
(A–C) Spleens of Xcr1−/− mice and Wt controls were harvested 40 h (A–F), 4 days (G), and 5 days (H) after MCMV infection. Splenic NK cells (NK1.1+TCRβ−) were stained for CD69 (A) and intracellularly for IFN-γ (B) and GzmB (C) directly ex vivo.
(D) Visualization of activated NK cell clusters in the spleens of MCMV-infected mice. Scale bar: 50 μm.
(E) Analysis of NK cell clusters in Xcr1−/− vs littermate controls upon MCMV infection. Spleen sections were prepared from KarmaCre;Rosa26tdRFP;Xcr1−/− mice and KarmaCre;Rosa26tdRFP;Xcr1+/− controls and stained for NKp46 (green), MOMA-1 (purple), IFN-γ (red), and IE-1 (cyan). Inserts show examples of clusters areas as defined in the material and methods. NK cell clusters were identified as groups of at least 10 IFN-γ+ cells, encompassing NKp46+ cells and gathered around MCMV-infected cells (IE-1+) in the marginal zone (MOMA-1+). Scale bar: 50 μm.
(F) Quantification of NK cell clusters in the spleens of MCMV-infected KarmaCre;Rosa26tdRFP;Xcr1−/− mice and their littermate Xcr1+/− control 40 h after infection.
(G) Splenic NK cells (NK1.1+TCRβ−) were stained intracellularly for KI67 4 days after infection.
(H) Proportion and absolute number of Ly49H+ NK cells in Wt vs Xcr1−/− mice 5 days after infection. One experiment representative of at least 4 independent ones with at least 4 mice per infected group is shown, except for B-E, where two experiments with at least 3 mice per group were pooled. NI, noninfected; ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001.
See also Figure S1.
XCR1 participates in cDC1 redistribution toward NK cells, promoting cDC1production of IL-12
We sought to determine to what extent XCR1 regulated cDC1 responses to MCMV infection. cDC1 were the major source of IL-12 among DC subsets (Figures 2A and S2A). Xcr1 deficiency impaired IL-12 production by cDC1 40 h after infection (Figure 2A), likely explaining the lower proportion of IFN-γ+ NK cells (Figure 1B), but did not alter the overall absolute number of cDC1 (Figure S2B), nor their maturation 48 h after infection (Figure S2C). We wondered whether XCR1 induced the relocalization of splenic cDC1 to the NK cell islets at the sites of viral replication. To track cDC1 in situ in spleen, we bred Xcr1-deficient animals to KarmaCre;Rosa26tdRFP mice in which the tandem dimer red fluorescent protein (tdRFP) expression selectively traces cDC1 (Mattiuz et al., 2018), and we compared Xcr1−/− animals with Xcr1+/− littermate controls. At steady state, splenic cDC1 are scattered in the red pulp and concentrated in the T cell area of the white pulp (Figure S2D) (Alexandre et al., 2016; Yamazaki et al., 2013). Although dispensable for the microanatomical distribution of splenic cDC1 at steady state (Figure S2D), XCR1 was instrumental to orchestrate their attraction inside NK cell clusters upon MCMV infection (Figures 2B and 2C), without affecting their survival (Figure S2B). Inside these clusters, cDC1 adopted dendritic morphologies, which were also dependent on XCR1 expression because the Xcr1-deficient cDC1 that had reached these clusters remained mostly round shaped (Figures 2B–2D). Depleting NK1.1-expressing cells abrogated cDC1 redistribution (Figure 2E), suggesting that NK cells were central in providing guiding cues to attract cDC1 in these marginal zone clusters and to engage them into tripartite interactions with MCMV-infected IE-1+ cells leading to IFN-γ production by NK cells (Figure 2F).
Figure 2.

XCR1 regulates cDC1 repositioning into NK cell clusters where IL-12-expressing cDC1 contact IFN-γ-producing NK cells
(A) Spleens were harvested at 40 h (A–I) or 4 days (J) after MCMV infection. (A) Analysis of IL-12p40/p70 production by splenic DCs of Xcr1−/− and Wt animals. One experiment representative of at least 4 independent ones with at least 4 mice per infected group is shown. NI, noninfected; n.s., nonsignificant; ∗, p < 0.05.
(B) Visualization of cDC1 in individual NK cell cluster in KarmaCre;Rosa26tdRFP;Xcr1−/− mice and their Xcr1+/− littermate controls. Spleen sections (scale bar: 20 μm) were stained for NKp46 (green), IFN-γ (yellow), tdRFP (cDC1, red), and IE-1 (cyan).
(C) Quantification of the dRFP intensity per NK cell cluster area (μm2) as a mean to evaluate cDC1 presence in individual NK cell cluster. For B and C, two independent experiments with at least 2-3 mice per group were pooled. An average of two clusters was analyzed in each mouse. ∗∗∗∗, p < 0.0001.
(D) Analysis of cDC1 sphericity when in NK cell clusters. We used the HK-means tool (Dufour et al., 2008) from Icy software to segment the tdRFP signal in each cluster, to computationally define and assess the sphericity of individual cDC1.
(E) Quantification of the dRFP intensity per μm2 as a mean to evaluate cDC1 relocalization in clusters in mice depleted of NK1.1+ cells upon MCMV infection. Clusters were defined as >10 IFN-γ+ cells around IE-1+ cells situated in the MZ.
(F) Visualization of tripartite interactions between cDC1, MCMV-infected cells (IE-1+) and activated IFN-γ-producing NK cells in marginal clusters. A representative image from 4 distinct MCMV-infected control spleens is shown (scale bar: 10 μm).
(G) Analysis of IL-12-producing cDC1 in NK cell clusters in KarmaCre;Rosa26tdRFP;Il12eYFP+ mice. The color obtained from overlaying green IL-12-eYFP+ on purple tdRFP-expressing cDC1 is white. Scale bar: 20 μm.
(H) Visualization of physical contacts between IL-12-eYFP-expressing cDC1 and IFN-γ-producing NK cells (arrows) in marginal ring clusters. The micrographs shown (G and H) are representative of the analyses of 6 mice from two independent experiments.
(I) Proportion of tdRFP+ eYFP+ cDC1-contacting NK cells within individual marginal zone cluster in KarmaCre;Rosa26tdRFP;Il12eYFP+;Xcr1−/− mice and littermate controls. Two independent experiments with 2-3 mice per infected group were pooled. An average of 3.5 and 2.2 clusters was analyzed in Xcr1+/− and Xcr1−/−KarmaCre;Rosa26tdRFP;Il12eYFP+ mice, respectively. ∗∗∗, p < 0.001.
(J) Analysis of NK cell absolute number 4 days after infection in the spleen of Xcr1Cre/wt;Il15raKO/flox mice and their respective controls (Il15raKO/flox). One experiment representative of at least 2 independent ones with at least 4 mice per infected group is shown. NI, noninfected; n.s., nonsignificant; ∗, p < 0.05.
See also Figure S2.
To determine whether those cDC1 could be delivering IL-12 to NK cells, we bred the KarmaCre;Rosa26tdRFP mice with Il12eYFP reporter animals (Reinhardt et al., 2006). Inside NK cell clusters, IL-12+ cDC1 were in contact with NK cells (Figure 2G), many of which were polarizing their IFN-γ at the interface with cDC1 (Figure 2H, arrows, and Video S1). This observation confirmed the formation of stimulatory synapses between DCs and NK cells, as previously described in vitro (Borg et al., 2004). The absence of XCR1 reduced the proportion of IL-12+ cDC1 interacting with NK cells in marginal zone clusters (Figure 2I), demonstrating that XCR1-mediated cDC1 relocalization allowed cDC1 to deliver bioactive IL-12 to NK cells. In addition, genetic inactivation of IL-15Rα selectively in cDC1, abrogating their ability to transpresent IL-15, also reduced NK cell expansion (Figure 2J), confirming that NK cell responses against MCMV depended on NK/cDC1 physical contacts. Altogether, these results showed that the XCL1/XCR1 axis reinforced the attraction of cDC1 toward NK cell clusters at 40 h after infection, promoting a feedforward loop relying on physical interactions between these two cell types and allowing local delivery of IL-12 and IL-15/IL-15Rα by cDC1 to NK cells.
XCL1 from NK cells is not necessary to promote NK/cDC1 physical interactions
To identify the major source of XCL1 upon MCMV infection, we examined Xcl1 expression in the total spleen of infected mice. Commercially available anti-XCL1 antibodies did not reliably stain XCL1 ex vivo in our hands. Hence, we generated an Xcl1mTfp1-flox mouse model by knocking a LoxP-exon3-IRES-mTfp1-LoxP cassette in frame in the Xcl1 gene (Figure S3A) to 1) trace Xcl1 mRNA-expressing cells as positive for the monomeric Teal fluorescent protein 1 (mTFP1) and 2) genetically inactivate Xcl1 and mTfp1 selectively in Cre-expressing cells. At steady state, mTFP1 expression was the strongest in ILC1, NK, and NKT cells (Figure 3A), with NK cells being the major cell subset expressing Xcl1 (49.9% of all mTFP1+ splenocytes), followed by memory αβ T cells (CD44+CD8+, 13.1% and CD44+CD4+, 12.4%, Figure 3C). Although NK cells remained the major source of Xcl1 upon MCMV infection (44.3%) (Figures 3B and 3D), Xcl1 inactivation in NK cells and partly in ILC1 (Figure S3B) did not decrease their capacity to produce IFN-γ, contrary to what happened in Xcr1−/− animals (Figure 3E). These results implied that memory/activated T cells, NKT cells, or ILC1 could compensate for the lack of XCL1 production by NK cells during MCMV infection, with all these XCL1-producing cells likely acting together to attract cDC1 in specific foci around infected cells in the splenic marginal zone.
Figure 3.

NK cells are a main source of XCL1 but not a critical one upon MCMV infection
(A and B) Analysis of mTFP1 expression in splenocytes of Wt (dark grey) and Xcl1-mTfp1fl/fl (white) mice at steady state (A) or 40 h after infection (B). Cells were gated as follow: NK cells (TCRβ- CD19- NK1.1+), ILC1 (NK1.1+ TCRβ- CD19- CD127+), NKT cells (CD19- CD1d+), CD8+ T (CD1d- NK1.1- TCRβ+ CD19- CD8+), CD4+ T (CD1d- NK1.1- TCRβ+ CD19- CD4+), γδ T cells (CD1d- NK1.1- CD3ε+ TCRβ- TCRγδ+) and B cells (NK1.1- TCRβ- CD19+). One representative experiment of 4 independent ones with at least three mice per group is shown.
(C and D) Proportion of immune populations within mTFP1-positive cells at steady state (C) and 40 h after MCMV infection (D). Others: sum of all the other cell subsets not detailed in the pie charts. One representative experiment of 4 independent ones with at least three mice per group is shown.
(E) IFN-γ production by NK cells in Wt, Xcr1−/−, Xcl1fl/fl, and Nkp46Cre;Xcl1fl/fl mice 40 h after infection. One representative experiment of two independent ones with at least four mice per group is shown. A one-way ANOVA statistical analysis with a Tukey's multiple comparison test was applied. ∗, p < 0.05; ∗∗, p < 0.01, n.s., nonsignificant; ANOVA, analysis of variance.
See also Figure S3.
NK/cDC1 interactions promote cDC1 redistribution from the red pulp to the T cell zone via bridging channels
Upon MCMV infection, after moving in the marginal zone at 40 h after infection, cDC1 then localized in the bridging channel at 44 h to reach the T cell zone at 48 h (Figures 4A and S4). XCR1 inactivation compromised this migratory pattern of cDC1, as Xcr1−/− cDC1 accumulated in the marginal zone at 48 h after infection, rather than into the T cell area (Figures 4A and S4). We further investigated the molecular mechanisms providing guidance cues for marginal cDC1 to enter the T cell zone.
Figure 4.

The spatiotemporal relocalization of cDC1 in the different regions of the spleen is associated with the regulation of EBI2 and CCR7 expression
(A) Quantification of tdRFP intensity as a readout of cDC1 anatomical location in marginal areas, bridging channels and T cell zones of KarmaCre;Rosa26tdRFP;Xcr1−/− and their Xcr1+/− littermate controls, during the course of MCMV infection. MOMA-1+ ring defined marginal areas, B220 staining delineated T cell areas, and breaks in both B220 and MOMA-1 stainings defined bridging channels (see Figure S4). Each dot (mean ± SD) represents a pool of two experiments with 2-3 mice per group. Statistical test: 2-way ANOVA; ∗, p < 0.05; ∗∗∗, p < 0.001. ANOVA, analysis of variance.
(B) Relative expression of the Gpr183/Ebi2 gene from transcriptomic analysis of splenic cDC1 sorted from Wt vs Xcr1−/− 38 h after infection. Data are represented as mean (+/− SEM). NI, noninfected; SEM, standard error of mean.
(C) Transwell migration assay of cDC1 in response to the EBI2 dihydroxycholesterol ligand, 7α25OH. DCs from Wt and Xcr1−/− spleens were enriched by optiprep gradient 40 h after infection. Data are represented as mean (+/− SEM). NI, noninfected; SEM, standard error of mean.
(D) Analysis of CCR7 expression on cDC1 in Wt and Xcr1−/− mice 48 h after infection. Two independent experiments with at least 3 mice per infected group were pooled. NI, noninfected. ∗, p < 0.05; ∗∗∗, p < 0.001.
See also Figure S4.
The Epstein-Barr virus–induced protein 2 (EBI2; also known as Gpr183) is a Gαi-coupled chemoattractant receptor required for cDC2 positioning in the bridging channel and T cell zone in response to 7α,25- (7α,25-HC) and 7α,27- (7α,27-HC) dihydroxycholesterols (Lu et al., 2017; Yi and Cyster, 2013). Gpr183 induction in cDC1 upon MCMV infection was strongly decreased in Xcr1−/− mice as compared with Wt animals (Figure 4B). As a consequence, the migration of cDC1 isolated from MCMV-infected Xcr1-deficient animals was completely abrogated in response to 7α,25-HC (Figure 4C). These data supported the impaired cDC1 relocalization in bridging channels in Xcr1-deficient mice at 44 h after infection (Figure 4A).
The chemokine receptor CCR7 also partly orchestrates DC movement into the T cell area in the spleen (Calabro et al., 2016; Gunn et al., 1999; Umemoto et al., 2012; Yi and Cyster, 2013), by binding CCL19 and CCL21 expressed on fibroblast reticular cells (FRC) in the bridging channel and T cell zone (Luther et al., 2000; Bajénoff et al., 2008). In Xcr1−/− mice, cDC1 showed a reduced expression of CCR7 at 48h post-infection (Figure 4D), when cDC1 relocalization peaked in the T cell zone in control mice (Figure 4A).
Thus, during MCMV infection, their XCR1-dependent repositioning within NK cell clusters in the marginal zone licensed cDC1 to express the membrane receptors with guiding functions Gpr183 and CCR7, instructing their further migration through bridging channels into the T cell zone.
NK cells induce CCR7 on cDC1 by delivering them granulocyte-macrophage colony-stimulating factor through cell-cell contacts
We next thought to determine whether and how NK cells were directly inducing CCR7 on cDC1. NK cells isolated from MCMV-infected animals efficiently induced CCR7 expression on cDC1 in an in vitro coculture experiment, without changing their phenotypic maturation as assessed by CD86 expression (Figures 5A and S5A–S5D). This response was significantly reduced when cDC1 and NK cells were physically separated by a transwell (Figure 5B), indicating that it required cell-cell contacts. In situ in NK cell clusters, the CCR7 antibody mostly stained cDC1 that touched IFN-γ-producing NK cells (Figure 5C, yellow on overlay). The analysis of pairwise correlation between CCR7 and tdRFP expression in marginal zone clusters showed that CCR7 was less expressed by Xcr1−/− cDC1 than by control cDC1 (Figure 5D). Hence, to be licensed for migration to the T cell area early during MCMV infection, cDC1 required first to establish physical contacts with activated antiviral NK cells by relocalizing in immune cell clusters around MCMV-infected cells in the marginal zone.
Figure 5.

NK cell delivery of GM-CSF to cDC1 requires physical contacts to trigger CCR7 upregulation on cDC1
(A and B) Analysis of CCR7 and CD86 expression on cDC1 when in coculture for 7 h with NK cells from spleens of Wt mice infected or not for 40 h with MCMV. cDC1 and NK cells were cocultured either in the same well (A) or separated by a 0.4-um-diameter pore-bearing transwell apparatus (B). One representative experiment of two independent ones with at least four mice per group is shown. n.s., nonsignificant; ∗, p < 0.05.
(C) Analysis of CCR7 expression on cDC1 in NK cell clusters 40 h after MCMV infection. Spleen sections of KarmaCre;Rosa26tdRFP mice were stained for IFN-γ, NKp46, CCR7, and tdRFP (cDC1). This cluster was imaged in the marginal area. The micrograph shown is representative of the analyses of 4 mice, from two independent experiments. Scale bar: 10 μm.
(D) Coefficients of Pearson correlation between CCR7 staining and tdRFP expression as detected in clusters (C) of KarmaCre;Rosa26tdRFP;Xcr1−/− mice and their Xcr1+/− littermate controls. Coefficients of Pearson correlation between CCR7 and NKp46 stainings are shown as controls. Two independent experiments with at least 2-3 mice per group were pooled. ∗∗, p < 0.01; n.s., nonsignificant.
(E) Kinetics of induction of Csf2 (Gmcsf) expression in total spleen during MCMV infection. Total RNA was extracted from spleens from Wt mice at the indicated times after MCMV infection, and RT-qPCR was performed using Hprt as a housekeeping gene. Data are represented as mean (+/− SD). One representative experiment of two independent ones with at least 4 mice per group is shown.
(F) Analysis of GM-CSF production by different lymphocyte populations 40 h after MCMV infection. Splenocytes were intracellularly stained for GM-CSF. Data are represented as mean (+/− SEM).
(G) RT-qPCR analysis of Csf2, Xcl1 and Ifng expression in purified NK cells activated in vitro with different stimuli. Data are represented as mean (+/− SEM). Isot, Isotype; med, medium.
(H) Analysis of CCR7 upregulation on Csf2rb-deficient eqcDC1 by activated NK cells. Wt and Csf2rb-deficient immature eqcDC1 were enriched from FLT3-L BM cultures and cocultured for 7 h with NK cells purified from spleens of mice infected or not for 40 h with MCMV. eqcDC1 were gated as CD11c+SiglecH−SIRPα−CD24+ cells. Data are represented as mean (+/− SEM).
(I) Analysis of CCR7 expression on Wt vs Csf2rb−/− cDC1 from Wt:Csf2rb−/− mouse chimeras 48 h after infection. ∗∗, p < 0.01. Error bars represent standard deviations.
See also Figure S5.
Neutralization of IFN-γ in vivo did not impair CCR7 upregulation on cDC1 (Figure S5E). CCR7 upregulation on cDC1 48h after MCMV infection was correlated with a decrease in XCR1 expression (Figure S5F). However, recombinant XCL1 did not induce CCR7 on cDC1s derived in vitro in FLT3-L bone marrow (BM) cultures (eqcDC1) (Figure S5G). Next, we focused on granulocyte-macrophage colony-stimulating factor (GM-CSF, encoded by the Csf2 gene) because it was reported to improve eqcDC1 survival, differentiation, and functions (Sathe et al., 2011; Zhan et al., 2011; Mayer et al., 2014). Low concentrations of GM-CSF induced a significant upregulation of CCR7 expression on eqcDC1 (Figure S5H). During MCMV infection, Csf2 transcription followed a kinetic similar to that of Ifng and Xcl1 in the spleen, reaching a peak at 40 h (Figure 5E), when cDC1 encountered NK cells in the marginal zone. At that time, GM-CSF was mainly produced by NK cells (Figure 5F). In vitro, Csf2 transcription in NK cells was triggered most efficiently by IL-12 (Figure 5G), which is highly produced by cDC1 upon MCMV infection (Figure 2A). This suggested that, in the marginal zone cell clusters, local delivery of IL-12 by cDC1 may drive GM-CSF production by NK cells, which could in turn promote CCR7 upregulation on cDC1. Therefore, we analyzed whether GM-CSF signaling on cDC1 induced CCR7. In vitro, only NK cells isolated from MCMV-infected mice induced CCR7 on eqcDC1 (Figure 5H), as observed with splenic cDC1 (Figure 5A), and this response was abrogated in GM-CSFR-deficient (Csf2rb−/−) eqcDC1 (Figure 5H). In vivo, CCR7 induction was also impaired in Csf2rb−/− cDC1 as compared with their Wt counterparts, in the spleen of Wt:Csfr2b−/− mixed BM chimera mice infected with MCMV (Figure 5I). Thus, early after MCMV infection, in marginal zone cell clusters, GM-CSF delivery by activated antiviral NK cells to cDC1 licenses them for further migration within the spleen, to the T cell zone, by inducing their upregulation of CCR7.
NK-cell-mediated cDC1 relocalization to the T cell area in the spleen promotes MCMV-specific CD8+ T cell responses and control of MCMV infection
We investigated whether the impeded migration of Xcr1−/− cDC1 to the T cell area of the spleen affected the induction of antiviral adaptive immunity. The expansion of CD8+ T cells specific for the MCMV peptide m45 was significantly reduced 6 days after infection in the absence of XCR1 (Figure 6A) and in the absence of CCR7 expression specifically on cDC1 (Figure 6B). This observation confirmed the critical role of cDC1 in cross-priming antiviral CD8+ T cells during an acute MCMV infection (Figure S6) (Busche et al., 2013; Snyder et al., 2010; Torti et al., 2011).
Figure 6.

CCR7-mediated cDC1 relocalization into the T cell zone promotes MCMV-specific CD8+ T cell responses and host resistance to MCMV infection
(A) Analysis of m45-specific CD8+ T cell response in Xcr1−/− mice and littermate controls 6 days after MCMV infection. One representative experiment of two independent ones with at least 4 mice per group is shown. ∗, p < 0.05.
(B) Analysis of m45-specific CD8+ T cell response in Wt:Xcr1-Dta, Wt:Ccr7−/− and Xcr1-Dta:Ccr7−/− BM chimera mice 6 days after MCMV infection (104 PFU). Two independent experiments with at least 4 mice per infected group were pooled. A one-way ANOVA statistical analysis with a Tukey's multiple comparison test was applied. ∗∗, p < 0.01; ∗, p < 0.05, n.s., nonsignificant. Statistical analysis between all other groups is nonsignificant.
(C) MCMV titers in spleens and livers of Wt and Xcr1−/− mice 5 days after MCMV infection (104 PFU). Two independent experiments with at least 3 mice per infected group were pooled. NI, noninfected. ∗∗∗, p < 0.001.
See also Figure S6.
Because the absence of XCR1 hampered both NK and CD8+ T cell antiviral responses, we investigated whether this compromised viral control. Viral loads were still high in the spleen and liver of Xcr1-deficient animals at 5 days after infection, whereas they had become very low to undetectable in control mice as expected (Figure 6C). Altogether, these results showed that the XCR1-dependent repositioning of cDC1 close to NK cells at 40 h and CCR7-dependent migration of cDC1 into the T cell zone both promoted faster downstream antiviral CD8+ T cell responses strongly enhancing host resistance.
Dendritic epidermal T cells control dermal cDC1 migration from the skin to the draining lymph nodes in an XCR1-dependent mechanism
We then assessed whether the spatiotemporal dynamic of cDC1 we described in the spleen upon MCMV infection also occurred under a similar regulation by innate immune mechanisms in the periphery. We focused on the skin because biting is a natural route of transmission for salivary-gland-persistent MCMV. We found that dendritic epidermal T cells (DETCs) defined as CD3ε+ TCRγδ+ lymphocytes accounted for the vast majority of Xcl1-expressing cells in the skin at steady state (Figures 7A and S7). DETCs resided exclusively in the epidermis (Figures 7B and 7C), whereas cDC1 remained in the dermis (Figure 7C). As Karma is also expressed by mast cells in the skin (Mattiuz et al., 2018), we used Avidin to exclude any Avidin+ tdRFP+ mast cells from our analysis (Tharp et al., 1985). Upon MCMV infection of the skin, dermal cDC1 underwent a local repositioning from the dermis to the epidermis to reach DETCs (Figure 7D) and to engage into interactions with them (Figure 7F). In the absence of XCR1, cDC1 remained in the dermis (Figure 7E) and did not physically contact DETCs (Figure 7G). These cDC1/DETC interactions occurred 40 h after MCMV infection of the skin (Figure 7G) and required the migration of dermis-resident cDC1 into the epidermal layer (Figure 7H). The XCL1/XCR1 axis directed this cDC1 local redistribution in the skin (Figures 7G and 7H) and participated in the licensing of cDC1 to migrate from the infected skin to the draining lymph nodes (LNs) (Figure 7I). The XCR1-dependent interactions between cDC1 and DETCs in the skin also promoted CCR7 upregulation on skin-derived migratory cDC1 (migcDC1) (Figure 7J) and was required for efficient priming of MCMV-specific CD8+ T cells in the LN draining the infected skin (Figures 7K and 7L), akin to the mechanism described in the spleen upon systemic infection. Altogether, these results showed that XCR1 finely tuned cDC1 local dynamics in the periphery to place them in contact with XCL1-expressing lymphocytes that licensed them to migrate into the draining LNs to prime naïve T cells.
Figure 7.

Dermal cDC1 make cell/cell contacts with Xcl1-expressing DETCs
(A) Analysis of the lymphocyte heterogeneity within mTFP1+ cells in the skin of Xcl1mTfp1fl/fl mice at steady state. One representative experiment of 2 independent ones with at least three mice per group is shown.
(B) DETC staining in the skin at steady state. DETCs are CD3ε+TCRγδ+ lymphocytes localized in the epidermis. The dotted line delineates the basal layer separating the dermis (D) from the epidermis (EP). C, cartilage. Scale bar: 50 μm
(C–E) Analysis of cDC1 localization and contacts with DETCs in the skin, at steady state in KarmaCre;Rosa26tdRFP;Xcr1+/− mice (C), and at 40 h after MCMV infection in KarmaCre;Rosa26tdRFP;Xcr1+/− (D) and KarmaCre;Rosa26tdRFP;Xcr1−/− (E) mice. The dotted line delineates the basal layer separating the dermis from the epidermis. Skin sections (scale bar: 50 μm) were stained for tdRFP (red; cDC1), CD3ε (cyan), TCRγδ (yellow), IE-1 (purple), and Avidin (green; mast cell). EP, epidermis; D, dermis; C, cartilage.
(F) 3D visualization of DETC/cDC1 contacts identified in (D).
(G) Kinetics of cDC1/DETC contacts occurring in the epidermis and through the epidermis-dermis border per mm of skin length, during MCMV infection. Data are represented as mean (+/− SEM). p.i., postinfection.
(H) Quantification of cDC1 nuclear bodies inside the epidermis per mm of skin length 40 h after skin infection. For (G-H), two independent experiments with at least 4 mice per infected group were pooled. n.s., nonsignificant; ∗∗, p < 0.01; ∗∗∗, p < 0.001.
(I) Proportion of CD103+ migcDCs in the ear-draining LN of Xcr1−/− and littermate controls 48 h after skin infection.
(J) Analysis of CCR7 expression on migcDCs in ear-draining LN 48 h after skin infection of Xcr1−/− and Wt mice. Two independent experiments with at least 3 mice per infected group were pooled. ∗∗, p < 0.01.
(K and L) Analysis of m45-specific CD8+ T cell response in Xcl1−/− mice (K), Xcr1−/− mice (L) and their respective controls 6 days after MCMV infection of the ear. CD8+ T cells from ear-draining LN were stimulated in vitro for 4 h with m45 peptide. Two independent experiments with at least 3 mice per infected group were pooled for (K), and one experiment with at least 4 mice per group is shown for (L). ∗, p < 0.05∗∗, p < 0.01.
See also Figure S7.
Discussion
Here we use MCMV infection to describe the molecular mechanisms underpinning the protective crosstalk between NK cells and cDC1 promoting efficient antiviral responses, including cDC1 spatiotemporal repositioning in the spleen toward CD8+ T cell to fast-forward their priming. This licensing of cDC1 migration under the instruction of lymphocytes with innate functions, from the red pulp to the T cell zone within the spleen was preserved in the periphery from the skin to the draining LN. We found that the XCL1/XCR1 axis constitutes the foremost step that triggers the attraction of cDC1 close to NK cells in the spleen and to DETCs in the epidermis. In the spleen in particular, the failure of cDC1 to reposition themselves in contact with NK cells curtails the antiviral defenses by limiting NK cell and antiviral CD8+ T cell responses.
We demonstrate for the first time that NK cells tune the spatiotemporal redistribution of cDC1, within a lymphoid organ, during a viral infection, by providing them guiding cues and other signals (Figure S8). Although NK cells are the main source of XCL1, we found that XCL1 genetic inactivation selectively in NK cells does not phenocopy XCR1 deficiency (Figure 3E). This indicates that other cells can compensate for the loss of NK cell ability to produce XCL1. Early during MCMV infection, plasmacytoid DCs (pDCs) migrate close to infected cells in the marginal zone of the spleen and upregulate Xcl1 expression (Tomasello et al., 2018; Abbas et al., 2020). In other infectious models, activated and memory CD8+ T cells and NKT cells relocalized in cell clusters at the edge of the marginal zone (Alexandre et al., 2016; Bajenoff et al., 2010; Barral et al., 2012), together with NK cells (Alexandre et al., 2016). Therefore, clusterized memory αβ T cells, NKT cells, or even pDCs may act collectively with NK cells to attract cDC1 in their vicinity through XCL1 production. Although γδ T cells represent only a minute fraction of XCL1-producing cells in the spleen upon MCMV infection (Figures 3B and 3D), this is not the case in other organs such as the skin where they are more prominent than NK cells and take the lead by attracting efficiently cDC1 (Figure 7A). XCR1 is necessary to endow cDC1 with migratory features during immune responses against viruses, at least with MCMV (this study) or Vaccinia (Brewitz et al., 2017). In contrast, in mouse cancer models, XCR1 was individually dispensable in promoting the recruitment of cDC1 for the activation of protective effector lymphocytes, even in highly inflammatory tumors (Böttcher et al., 2018). Hence, it is possible that the XCL1/XCR1 axis is critical to promote a rapid defense against acute threats but redundant for resistance to chronic pathologies.
Lymphoid cells, in particular NK cells or ILC3, are known to secrete large amounts of GM-CSF (Dougan et al., 2019). MCMV infection triggers GM-CSF production by NK cells (Figure 5F) most likely through IL-12 secretion by DCs (Figure 5G). Similarly, the IL-12 family member IL-23 triggers GM-CSF production by ILC3 in colon upon CD40-induced colitis (Pearson et al., 2016). The role of NK cell GM-CSF has been largely overlooked until recently. NK cell GM-CSF production was reported to feed inflammation during autoantibody-mediated inflammatory arthritis, by increasing the life span and functions of neutrophils and macrophages (Louis et al., 2020), and to promote neutrophil antifungal activity in a model of Candida albicans infection (Bär et al., 2014). For a long time, studies on GM-CSF function focused mostly on myeloid cell ontogeny (Dougan et al., 2019). GM-CSF promotes the differentiation of cDC1 only in peripheral tissues that are constantly under microbiota stimulation, such as the skin or intestine, but has no major impact on lymphoid-tissue-resident cDC1 (King et al., 2010; Greter et al., 2012). Our study goes further in describing the critical function for GM-CSF for licensing cDC1 migration within the spleen during a systemic viral infection. We showed that NK-cell-derived GM-CSF drives CCR7 expression on cDC1 through cell/cell contacts, thus instructing their migration to the T cell zone. Interestingly, GM-CSF is not required for the upregulation of the costimulatory molecule CD86 on cDC1 in our experimental setting, contrary to what has been reported before in vitro with eqcDC1 (Mayer et al., 2014). This may be explained by differential DC exposures to GM-CSF concentrations and durations or because activated NK cells provide other signals able to promote costimulatory molecule upregulation on cDC1, such as IFN-γ and TNFα (Walzer et al., 2005; Crouse et al., 2015).
NK cells can functionally affect the magnitude and quality of adaptive immune responses through direct and indirect pathways (Su et al., 2001; Robbins et al., 2007; Waggoner et al., 2011). NK cell secretion of cytokines, such as IFN-γ, or NK cell cytotoxicity, can directly regulate T cell responses (Crouse et al., 2015). NK cells also indirectly modulate T cell priming by promoting DC-specific functions. Through killing target cells, NK cells provide cDC1 with apoptotic-cell-derived antigens, which are cross-presented to naïve CD8+ T cells (Krebs et al., 2009; Iyoda et al., 2002; Deauvieau et al., 2015; Dao et al., 2005). NK cells also induce DC maturation, although the specific molecular mechanisms that came into play remained in part elusive (Gerosa et al., 2002; Walzer et al., 2005; Crouse et al., 2015). Here we unravel how NK cells orchestrate the spatiotemporal repositioning of cDC1 in regions where they have more chance to prime naive CD8+ T cells. This aspect has never been described before, although the regulation of DC migration by ILCs has been recently suggested (Böttcher et al., 2018).
Upon local infection, we show that cDC1 residing in the dermis reach the epidermis by responding to DETCs secreting XCL1. Once in the epidermis, cDC1 engage into physical contacts with DETCs, which could promote acquisition of CCR7 on cDC1 and their migration into skin-draining LNs, akin to NK cells in the spleen. So far, skin cDC1 have always been described as residing exclusively in the dermis, and to the best of our knowledge, the ability of cDC1 to reposition themselves in the epidermis in response to an acute inflammatory stimulation of the skin has never been reported before. Interestingly, in the buccal and gingival mucosa that are continuously challenged by microbiota, a small proportion of the Langerin+ EpCam+ DCs present in the epithelium express CD103 and XCR1 (Capucha et al., 2015), which are exclusively coexpressed on bona fide cDC1 in all other tissues examined. These cells might thus be cDC1 originating from lamina propria and migrating in the oral epithelium. There, they may acquire the epithelial cell adhesion molecule EpCAM, a marker associated to Langerhans cells within DCs but more generally regulating cell motility and adhesion (Gaiser et al., 2012; Ouchi et al., 2016). The ontogeny of these cells remains to be fully investigated.
γδ T cells can confer protection to systemic MCMV infection in the absence of any other conventional adaptive immune responses (Sell et al., 2015; Khairallah et al., 2015). This antiviral response was attributed to the expansion and activation of the Vγ1 T subset, which resides in the spleen, LNs, and liver (Khairallah et al., 2015; Sell et al., 2015). Our results suggest that MCMV infection of the skin activates the epidermal Vγ5+ DETCs to secrete XCL1, but the modalities of this activation remain poorly defined. DETCs express the Xcl1 transcript (Figure 7A) at steady state and rapidly upregulate its expression upon TCR stimulation (Boismenu et al., 1996; Rezende et al., 2019). Because DETCs acquire cytotoxic functions and IFN-γ production through engagement of the NK cell receptor NKG2D, which recognizes MHC-I–related ligands (Ibusuki et al., 2014; Nitahara et al., 2006; Girardi et al., 2001), it is also possible that NKG2D signaling regulates XCL1 secretion by DETCs. Indeed, MCMV-encoded protein m18 triggers the expression of the NKG2D ligand RAE-1 in infected cells (Greene et al., 2016). More generally, MCMV infection can upregulate the expression of other NKG2D ligands that belong to the stress-induced, “empty” or nonpresenting, MHC-like molecules such as the histocompatibility 60 (H60) and the murine UL16-binding protein-like transcript (MULT-1) (Slavuljica et al., 2011).
Our study identified a mechanism through which activated NK cells boost adaptive immune responses to MCMV, which had remained in part elusive (Robbins et al., 2007). Here, we find that XCL1/XCR1-dependent cellular interactions orchestrate cDC1 migration, in a conserved manner within the spleen during a systemic infection and from peripheral tissues to their draining LNs during a local infection. Indeed, XCL1-producing lymphoid cells surrounding infected cells, NK cells in the spleen and DETCs in the skin, attract cDC1 and instructing them locally for activation and further migration to the lymphoid structures where naïve CD8+ T cells are primed. This cDC1 relocalization optimizes the relay between innate and adaptive immunity, by accelerating and potentiating T cell expansion and effector functions, therefore yielding a more robust resistance to infections.
Limitations of the study
Here we defined how NK cells guide the delivery of specific functions of cDC1 to the right cells at the right time in the spleen in response to a systemic viral infection. Mechanistically, NK cells attracted red-pulp-resident cDC1 in the marginal zone of the spleen to establish physical interactions. This cDC1 repositioning depended on the XCL1/XCR1 axis and led to tripartite interactions between MCMV-infected cells, cDC1, and NK cells at the marginal zone in the spleen. We showed that several contact-dependent signals were delivered by cDC1 to NK cells, triggering their full activation and the expansion of their Ly49H+ subset known to specifically recognize MCMV-infected cells that express the viral protein m157 (Brown et al., 2001; Lee et al., 2001). Whether the recognition of MCMV-infected m157-expressing cells by Ly49H+ NK cells constitutes the early step of a cascade of cellular events leading to cDC1 recruitment, their licensing, and the formation of a “ménage à trois” with MCMV-infected cells remain to be determined. Interestingly, by impeding cDC1 repositioning toward NK cells, the absence of XCL1/XCR1 not only impaired NK cell responses but also decreased drastically the CCR7-dependent migration of cDC1 into the T cell zone and therefore the early priming of antiviral CD8+ T cell responses by cDC1. Further studies are required to determine the extent to which the decrease in NK cell and/or CD8+ T cell antiviral responses was responsible for the viral persistence observed in Xcr1-deficient animals at 5 days after infection. It is likely that XCL1/XCR1 was critical for both NK and CD8+ T cell protective functions. Our report and its follow-up studies would thus highlight the nonredundant role of XCL1/XCR1 in innate and adaptive immune defenses against infections or cancers and would explain why the XCL1/XCR1 axis has been conserved across evolution among potentially all warm-blooded vertebrates (Crozat et al., 2010; Vu Manh et al., 2015).
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti B220/CD45R A647 (Clone RA3-6B2) | BD Biosciences | #557683 |
| Anti CCR6 PerCP-Cy5.5 (Clone 29-2L17) | BioLegend | #129809 |
| Anti CD103 biotin (Clone 2E7) | Fischer Scientific eBioscience | #15239669 |
| Anti CD103 PE (Clone 2E7) | ThermoFisher eBioscience | #12-1031-82 |
| Anti CD11b PerCP-Cy5.5 (Clone M1/70) | BD Biosciences | #561114 |
| Anti CD11c PECy7 (Clone N418) | ThermoFisher eBioscience | #25-01114-82 |
| Anti CD169 FITC (Clone MOMA-1) | AbD Serotec | #MCA947F |
| Anti CD19 FITC (Clone 1D3) | BD Biosciences | #553785 |
| Anti CD19 PE (Clone 1D3) | BD Biosciences | #553786 |
| Anti CD19 A700 (Clone 6D5) | Fischer Scientific eBioscience | #115528 |
| Anti CD19 APC-Cy7 (Clone 6D5) | BioLegend | #115529 |
| Anti CD197 (CCR7) Purified (Clone 4B12) | BioLegend | #120101 |
| Anti CD197 (CCR7) Biotin (Clone 4B12) | ThermoFisher eBioscience | #13-1971-80 |
| Anti CD24 Pacific Blue (Clone M1/69) | BioLegend | #101820 |
| Ani CD26 BV786 (Clone H194-112) | BD Biosciences | #740868 |
| Anti-CD3 complex eF450 (Clone 17A2) | ThermoFisher eBioscience | #48-0032-82 |
| Anti CD3ε A700 (Clone eBio500A2) | Fischer Scientific eBioscience | #15217989 |
| Anti CD3ε PE (Clone 145-2C11) | BD Biosciences | #553064 |
| Anti CD4 APC-H7 (Clone GK1.5) | BD Biosciences | #560246 |
| Anti CD40 PE (Clone 3/23) | BD Biosciences | #553791 |
| Anti CD44 PE-Cy7 (Clone IM7) | ThermoFisher eBioscience | #25-0441-82 |
| Anti CD45.2 BUV395 (Clone 104) | BD Biosciences | #564616 |
| Anti CD69 FITC (Clone H1.2F3) | ThermoFisher eBioscience | #11-0691-82 |
| Anti CD80 APC (Clone 16-10A1) | BD Biosciences | #560016 |
| Anti CD86 PerCP/Cy5.5 (Clone GL-1) | BioLegend | #105028 |
| Anti CD8α BV711 (Clone 53-6.7) | BD Biosciences | #563046 |
| Anti CD8α APC (Clone 53-6.7) | BD Biosciences | #553035 |
| Anti CD8α Pacific Blue (Clone 53-6.7) | BD Biosciences | #558106 |
| Anti dsRED, dsRFP Purified (Rabbit Polyclonal IgG) | Rockland | #600-401-379 |
| Anti Fluorescein/Oregon Green A488 (Polyclonal) | ThermoFisher | #A11090 |
| Anti-Goat IgG A594 (Polyclonal Donkey Fab) | Jackson Immunoresearch | #705-587-003 |
| Anti GzmB APC (Clone GB11) | ThermoFisher | #GRB05 |
| Anti I-A/I-E (MHC-II) A700 (Clone M5/114.15.2) | ThermoFisher eBioscience | #56-5321-82 |
| Anti IL-12(p40/p70) APC (Clone C15.6) | BD Biosciences | #554480 |
| Anti IFNγ A700 (Clone XMG1.2) | BD Biosciences | #557998 |
| Anti IFNγ Biotin (Clone XMG1.2) | BD Biosciences | #554410 |
| Anti IFNγ Purified (Clone XMG1.2) | BioXCell | #BE0055 |
| Anti Ly49H biotin (Clone 3D10) | Fischer Scientific eBioscience | #15219899 |
| Anti MCMV IE1/m123 Purified (Clone IE1.01) | Capri/MEDRI | #IE1.01 |
| Anti mouse IgG2a A633 (Polyclonal Goat) | ThermoFisher | #A-21136 |
| Anti NK1.1 A700 (Clone PK136) | BD Biosciences | #560515 |
| Anti NK1.1 PE (Clone PK136) | BD Biosciences | #557391 |
| Anti NK1.1 PE-Cy7 (Clone PK136) | BD Biosciences | #552878 |
| Anti NK1.1 Purified (Clone PK136) | Purified in house | N/A |
| Anti NKp46/CD335 PE (Clone 29A1.4) | Fischer Scientific eBioscience | #15228749 |
| Anti NKp46/CD335 Purified (Polyclonal Goat IgG) | RnD Systems | #AF2225 |
| Anti rabbit IgG A546 (Polyclonal Donkey) | ThermoFisher | #A10040 |
| Anti rat IgG A594 (Polyclonal Donkey F(ab')₂) | Jackson Immunoresearch | #712-586-153 |
| Anti Siglec-H FITC (Clone 551) | BioLegend | #129604 |
| Anti SIRPα (CD172a) APC-Cy7 (Clone P84) | BioLegend | #144018 |
| Anti TCRβ FITC (Clone H57-597) | BD Biosciences | #553171 |
| Anti TCRβ PerCP/Cy5.5 (Clone H57-597) | BioLegend | #109228 |
| Anti TCRγδ BV711 (Clone GL3) | BD Biosciences | #563994 |
| Anti XCR1 APC (Clone ZET) | BioLegend | #148206 |
| Avidin A488 | ThermoFisher | #A21370 |
| Streptavidin APC | BD Biosciences | #554067 |
| Streptavidin eFluor 450 | ThermoFisher eBiosciences | #48-4317-82 |
| Streptavidin PE | BD Biosciences | #554061 |
| Streptavidin PE-Cy7 | BD Biosciences | #557598 |
| Virus strains | ||
| MCMV smith strain | ATCC | ATCC #VR-1399 |
| Chemicals, peptides, and recombinant proteins | ||
| 7α,25-Dihydroxycholesterol | Sigma-Aldrich | #SML0541-5MG |
| AntigenFix | DiaPath | #P0016 |
| m45 peptide HGIRNASFI | ProImmune | N/A |
| Poly(I:C) High Molecular Weight | InvivoGen | #tlrl-pic |
| Critical commercial assays | ||
| NK Cell Isolation Kit, mouse | Miltenyi Biotec | #130-115-818 |
| CD8+ Dendritic Cell Isolation Kit, mouse | Miltenyi Biotec | #130-091-169 |
| Deposited data | ||
| microarray data GSE142402 | GEO database | https://www.ncbi.nlm.nih.gov/geo/ |
| Experimental models: mouse strains | ||
| Ccr7−/− (B6.129P2(C)-Ccr7tm1Rfor/J) | The Jackson Laboratory | JAX #006621 |
| Cd45.1+/+ (B6.SJL-PtprcaPepcb/BoyJ) | The Jackson Laboratory | JAX #002014 |
| Csf2rb−/− (B6.129S1-Csf2rbtm1Cgb/J) | The Jackson Laboratory | JAX #005940 |
| Il12eYFP (B6.129-Il12btm1.1Lky/J) | The Jackson Laboratory | JAX #006412 |
| Il15rako/ko (Il15ratm1Ama) | The Jackson Laboratory | JAX #003723 |
| Il15raflox/flox (Il15ratm2.1Ama) | The Jackson Laboratory | JAX #022365 |
| KarmaCre (B6-Gpr141btm2Ciphe) | Mattiuz et al., 2018 | N/A |
| KarmatdTomato-hDtr (Gp141btm1Ciphe) | Alexandre et al., 2016 | N/A |
| Nkp46Cre (Ncr1tm1.1(iCre)Viv) | Narni-Mancinelli et al., 2011 | N/A |
| Rosa26lox−stop−lox−DTA (B6.129P2-Gt(ROSA)26Sortm1(DTA)Lky/J) | The Jackson Laboratory | JAX #009669 |
| Rosa26lox−stop−lox−tdRFP (Gt(ROSA)26Sortm1Hjf) | Luche et al., 2007 | N/A |
| Xcl1mTfp1-fl (Xcl1tm1Ciphe) | This paper | N/A |
| Xcr1−/− (B6.129P2-Xcr1tm1Dgen) | The Jackson Laboratory | JAX #005791 |
| Xcr1Cre (B6-Xcr1tm1Ciphe) | Mattiuz et al., 2018 | N/A |
| Oligonucleotides | ||
|
Csf2 forward GGCCTTGGAAGCATGTAGAGG |
This paper | N/A |
|
Csf2 reverse GGAGAACTCGTTAGAGACGACTT |
This paper | N/A |
|
Hprt forward TCAGTCAACGGGGGACATAAA |
This paper | N/A |
|
Hprt reverse GGGGCTGTACTGCTTAACCAG |
This paper | N/A |
|
Ifng forward CAACAGCAAGGCGAAAAAGG |
This paper | N/A |
|
Ifng reverse CCTGTGGGTTGTTGACCTCAA |
This paper | N/A |
|
Xcl1 forward AGAAGCCAAATGGGTGAAAGC |
This paper | N/A |
|
Xcl1 reverse TCAGCCATGTTCTTTCTGGTACTG |
This paper | N/A |
| Software and algorithms | ||
| BD FACSDiva™ Software v8.0 | BD Biosciences | https://www.bdbiosciences.com/en-us/instruments/research-instruments/research-software/flow-cytometry-acquisition/facsdiva-software |
| FlowJo v10.4 | Tree Star | https://www.flowjo.com |
| GraphPad Prism 7 | GraphPad | https://www.graphpad.com/scientific-software/prism |
| Icy Bioimage analysis | Icy Bioimage analysis | http://icy.bioimageanalysis.org |
| ImageJ | NIH | https://imagej.nih.gov/ij/download.html |
| Zen | Zeiss | https://www.zeiss.fr/microscopie/produits/microscope-software/zen-lite/zen-2-lite-download.html |
| Imaris | Oxford Instruments | https://imaris.oxinst.com |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Karine Crozat (karine.crozat@univ-rennes1.fr). Inquiries to access to mutant mice targeting cDC1 or Xcl1 must be addressed to Marc Dalod (dalod@ciml.univ-mrs.fr).
Materials availability
Materials generated in this study are available upon reasonable request from Karine Crozat (karine.crozat@univ-rennes1.fr).
Data and code availability
The microarray data generated for this study have been deposited in the GEO database (http://www.ncbi.nlm.nih.gov/geo/) under the series accession number: GSE142402.
This paper does not report original codes.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and subject details
Mice
C57BL/6J and BALB/cByJ mice were purchased from Charles River Laboratories. Xcr1−/− (B6.129P2-Xcr1tm1Dgen; MGI:3604538) were backcrossed >10 times into C57BL/6J background before use. C57BL/6J mice or, when possible, littermates were used as controls. To deplete cDC1, KarmatdTomato-hDtr (Gp141btm1Ciphe) mice (Alexandre et al., 2016) received 32 ng/g of diphtheria toxin (DT) (Merck) 12h before MCMV infection, then injections of 16 ng/g every 60 h. KarmaCre (B6-Gpr141btm2Ciphe) mice (Mattiuz et al., 2018) were first bred with Rosa26lox−stop−lox−tdRFP (Gt(ROSA)26Sortm1Hjf) (Luche et al., 2007) to give KarmaCre;Rosa26tdRFP, then with Xcr1−/− mice to allow tracking of Xcr1-deficient cDC1. Xcr1+/− littermate controls were used. For some experiments, KarmaCre;Rosa26tdRFP mice were bred to Il12eYFP (B6.129-Il12btm1.1Lky/J) (Reinhardt et al., 2006) backcrossed >10 times to C57BL/6J). To selectively inactivate Il15ra gene in cDC1, we followed a specific breeding schema, which prevents germline recombination, using Xcr1Cre (B6-Xcr1tm1Ciphe) (Mattiuz et al., 2018), Il15rako/ko (Il15ratm1Ama) and Il15raflox/flox (Il15ratm2.1Ama) mice (Mortier et al., 2009) . We bred Xcr1cre/cre;Il15Rako/ko mice to Il15raflox/flox to obtain Xcr1Cre/wt;Il15Rafl/ko mice that we compared to Il15Rafl/ko controls. Xcl1mTfp1-flox (Xcl1tm1Ciphe) mice were generated according to a standard gene targeting approach in C57BL/6N-derived ES cells. A LoxP-exon3-IRES-mTfp1-LoxP cassette was inserted into the 3′-UTR of the Xcl1 gene (Figure S3A). Xcl1mTfp1-flox mice were backcrossed for >3 generations with C57BL/6J mice, then with Nkp46Cre (Ncr1tm1.1(iCre)Viv) mice (Narni-Mancinelli et al., 2011) to inactivate the Xcl1 gene in NK cells. Csf2rb−/− (B6.129S1-Csf2rbtm1Cgb/J) (Robb et al., 1995) were bred to CD45.1+ congenic C57BL/6J (B6.SJL-PtprcaPepcb/BoyJ) mice to generate Cd45.1+Csf2rb−/− mice. cDC1-less mice (Xcr1-Dta) were generated by breeding Xcr1Cre to Rosa26lox−stop−lox−DTA (B6.129P2-Gt(ROSA)26Sortm1(DTA)Lky/J) (Voehringer et al., 2008) . To generate BM chimera mice, recipient CD45.2+ C57BL/6J mice were 8 Gy irradiated, and then reconstituted with equal proportions of BM cells derived from CD45.1+Csf2rb−/− and CD45.2+ C57BL/6J (Wt) mice for Figure 5I, and from Xcr1-Dta and Ccr7−/− (B6.129P2(C)-Ccr7tm1Rfor/J) mice for Figure 6B. Mice were used at least 8 weeks after BM reconstitution. All mice were bred under pathogen-free conditions at the Centre d’Immunophénomique de Marseille-Luminy (CIPHE). Seven to 12 weeks-old mice, females or males, were used for experiments, in which we compared groups of animals of same gender and same age. The animal care and use protocols (APAFIS #1212 and #16547) were designed in accordance with national and international laws for laboratory animal welfare and experimentation (EEC Council Directive, 2010/63/EU, September 2010), and approved by the Marseille Ethical Committee for Animal Experimentation (registered by the Comité National de Réflexion Ethique sur l’Expérimentation Animale under no. 14).
MCMV infection and viral loads
MCMV infections were performed ip with 1x105 plaques forming units (PFU) of salivary gland-extracted MCMV (Smith strain; ATCC; VR-1399). At this dose, all C57BL/6J mice survived the infection whereas all BALB/c died between 5 and 6 dpi (not shown). For viral loads, 5 days after infection, total spleens and pieces of livers were harvested. Pieces of livers were weighted before proceeding to conventional plaque assay on organ homogenates (Crozat et al., 2007). For skin infection, 1.3 × 105 PFU in 20 μl were inoculated intradermally in one ear. RPMI was injected in control mice.
Method details
Poly(I:C) and in vivo antibody administration
50ug of poly(I:C) (high molecular weight, InvivoGen) was administrated iv. For in vivo depletion of NK1.1-expressing cells, mice were injected ip with the anti-NK1.1 antibody clone PK136 (200ug) 12h or 1 day prior to poly(IC) or MCMV administration respectively. In vivo IFN-γ blocking was achieved by ip administration of the anti-IFN-γ clone XMG1.2 (1mg) 7h prior to infection. Both PK136 and XMG1.2 were produced in house.
Organ preparation for flow cytometry analysis
For lymphocytes analysis, spleens were smashed through nylon meshes in complete RPMI medium, red blood cells lysed, and filtered again through a nylon mesh before proceeding to antibody staining. Splenocytes were intracellularly stained directly ex vivo, except for GM-CSF for which an incubation with Brefeldin A (10 μg/ml) in complete RPMI medium for 3h at 37°C was performed. NKT cells were stained using CD1d tetramers. MCMV-specific CD8+ T cell responses were evaluated by incubating splenocytes or cell suspension from ear-draining LN in complete medium with the MCMV H-2Db-restricted epitope m45 (HGIRNASFI; 1μg/ml) and Brefeldin A (10μg/ml) for 4h prior to intracellular staining. For skin, ears were separated into ventral and dorsal layers and incubated in liberase LD solution (0.5 mg/mL) for 60 min at 37°C. The epidermis was separated from the dermis then chopped in collagenase D (0.5 mg/mL) for a further 25 min, then filtered through a 70μm-nylon mesh. For DC analysis, spleens and LNs were prepared as follow: Spleens were digested with an enzymatic cocktail made of Collagenase D (1 mg/ml) and DNase I (70 μg/ml, both Roche) in plain RPMI 1640 for 25 min at 37°C. Ice cold EDTA (2 mM) was added for additional 5 min. Cells were filtered through a 70-μm nylon sieve, and exposed to 0.155 M NH4Cl, 10 mM KHCO3, 0.127 M EDTA to lyse red blood cells. Auricular LNs were cut into small pieces and digested for 25 min at 37°C with a mixture of type II collagenase (Worthington Biochemical) and DNase I (Sigma-Aldrich) in plain RPMI 1640. The resulting cell suspension was treated with 2 mM EDTA and filtered through a 70 μm nylon sieve (BD Falcon). For the analysis of cytokine induction in DCs, 10 μg/ml Brefeldin A was added to the enzymatic cocktail during the incubation at 37°C. Antibodies used for flow cytometry are listed in the key resource table. Stained cell acquisition was performed on a FACSCANTO II or a LSR II flow cytometer (BD Biosciences). cDCs were defined as CD3e−, NK1.1−, CD19−, CD11chigh, SiglecH−, and further split into cDC1 and cDC2 which were defined as CD11b−CD8α+ versus CD11b+, CD8α−, since XCR1 as a defining marker could not be used in experiments involving Xcr1−/− mice. pDCs were defined as CD3e−, NK1.1−, CD19−, CD11cmed, SiglecH+.
Immunohistofluorescence
Spleens and ears were fixed for at least 2h in Antigenfix (DiaPath) at 4°C, then washed in phosphate buffer (PB1X: 0.02 M NaH2PO4 and 0.08 M Na2HPO4) for 1h. Finally, they were dehydrated in 30% sucrose overnight at 4°C, before embedding in optimal cutting temperature (OCT) freezing media (Sakura Fineteck). 8-12-μm spleen and transversal skin sections were cut with a Leica CM3050 S cryostat. For staining, sections were blocked in PB1X containing 0.2% saponin, 1% Protein Block (Abcam), and 2% 2.4G2 supernatant, and stained in PB1X, 0.2% saponin. Antibodies used for flow cytometry are listed in the Key resources table. We used an anti-RFP antibody (Rockland) and a secondary donkey anti-rabbit antibody (Jackson Immuno Research) to amplify the tdRFP signal. NK cells and ILC1 were stained using a goat polyclonal anti-NKp46/Ncr1 antibody (RnD Systems) followed by a donkey anti-goat polyclonal Fab fragment antibody (Jackson Immuno Research). The MCMV immediate-early protein IE-1 was detected using the monoclonal mouse IgG2a,k anti-IE1 (Clone IE1.01, CapRI, Center for Proteomics, University of Rijeka, Rijeka, Croatia), followed by a polyclonal goat anti-mouse IgG2a antibody (Molecular Probes). When the staining required further blocking steps, normal goat, mouse or rabbit sera were added. Stained sections were mounted in ProLong Gold antifade reagent (Invitrogen), and were acquired using the spectral mode on a LSM780 confocal microscope (Carl Zeiss).
Co-culture experiments and migration assays
For co-culture experiments, splenic cDC1 and NK cells were enriched using Miltenyi Biotec CD8+ Dendritic Cell isolation kit and NK cell isolation kit respectively. NK cells were isolated from non-infected and 40h-infected mice. For co-culture using FLT3-L-differentiated BM-derived eqcDC1, BM cell suspensions were prepared from Wt and Csf2rb−/− mice, and red blood cells were lysed. After washing in complete RPMI 1640 medium, cells were cultured at 3 × 106 cells/ml in 24 well plates, with 10% FBS, RPMI 1640 medium containing murine FLT3-L (in house supernatant from B16-Flt3l cells, used at 1/20 final) at 37°C in 5% CO2. Four days after, half of the culture medium was replaced by fresh FLT3-L. 14 days later, cells were harvested and enriched for eqcDC1 by removing magnetically SIRPα-expressing cells using the Dynabeads system (ThermoFisher). The enriched cell populations were co-cultured for 6h at a ratio of 1DC:3NK, stained and live cells were analyzed by flow cytometry. We used 0.4um pore Corning Transwells to evaluate the contribution of NK/cDC1 physical contacts to CCR7 upregulation. For migration assay, splenic DCs were enriched by OptiPrep (Sigma-Aldrich), resuspended in migration medium (0.5% BSA in RPMI) and distributed in the upper chamber of a 24 Transwell system insert (8-μm pore size; Corning). The lower chamber was filled with migration medium alone or containing 10nM of the 7α,25-dihydroxycholesterol (7α,25-OH) (Sigma-Aldrich). Cells were incubated for 3–4 h at 37°C in 5% CO2. A constant number of polybead polystyrene microspheres (Polyscience) was added in each lower chamber before harvesting migrated cells. Migrated cells were then stained and analyzed by flow cytometry. A constant event of microspheres for each well was acquired to evaluate the absolute number of cells recovered after migration. The percentage of migrated cells was calculated by the following formula for each subset: [number of cells having migrated in response to XCL-1 − number of cells having migrated spontaneously]/[total number of input cells] × 100.
Microarray experiments and analysis
Each cell type studied was FACS sorted from spleen to over 98% purity in independent duplicate samples. Microarray experiments were performed as follow (Robbins et al., 2008). Briefly, RNA was extracted from between 105 and 3 × 106 cells for each cell subset with the Qiagen (Courtaboeuf, France) micro RNAeasy kit, yielding between 100 and 300 ng of total RNA for each sample. Quality and absence of genomic DNA contamination were assessed with a Bioanalyser (Agilent, Massy, France). RNA (100 ng) from each sample was used to synthesize probes, and hybridized to the GeneChip Mouse Gene 1.0ST Array (Affymetrix, Santa Clara, CA, USA). Affymetrix DAT files were processed using Affymetrix Gene Chip Operating System (GCOS) to generate. CEL files. Quality control of array hybridization, and data normalization and analysis were performed using the affy and oligo packages in R (version 3.2.2) (Baranek et al., 2012). Differentially expressed genes were selected using linear models from Limma package in Bioconductor. The microarray data have been deposited in the GEO database under the series accession number GSE142402.
Quantification and statistical analysis
Analysis of cell distribution and cell contacts
For cDC1 distribution analysis and quantification of clusters in the spleen, we imaged half spleen sections, and randomly mixed them using the random number generator (www.dcode.fr). Three independent manipulators blindly counted clusters using Zen software with a fixed zoom of 30 and 40%. Cluster quantifications were normalized to the section area (mm2). We quantified tdRFP intensity as a read-out of cDC1 presence. Each cluster was isolated by drawing a Region of Interest (ROI), which was then exported as unmodified uncompressed TIFF files. The tdRFP intensity was quantified in each ROI using ImageJ, and normalized to the ROI area (mm2). To follow cDC1 relocalization in the three splenic areas marginal zone, bridging channel and T cell regions, we applied a high throughput acquisition of whole spleen sections using the Pannoramic Scan slide scanner (3dHistec), and quantified tdRFP intensity minus background in each these areas. We identified them using MOMA-1 and B220 staining as shown in Figure S4. Between 6 and 14 white pulp follicles were analyzed per mouse. The 3-dimensional (3D) reconstruction and the Video S1 were generated using the Imaris software (Oxford Instruments). cDC1/DETC contacts were quantified in the skin and normalized to the length of the skin analyzed in the section. The Icy software (http://icy.bioimageanalysis.org/) was used for some analysis as detailed in the figure legend. Other software used for image analysis are listed in the Key resources table.
Statistical analyses
Statistical analyses were performed using a nonparametric Mann-Whitney test performed with Prism 8 (GraphPad Software) for all experiments unless specified otherwise. n.s., nonsignificant (p > 0.05); ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Acknowledgments
We acknowledge France Bio lmaging infrastructure supported by the Agence Nationale de la Recherche (ANR-10-INBS-04-01, call “Investissement d’Avenir”), in particular Sébastien Mailfert, Roxanne Fabre, Mathieu Fallet and Lionel Chasson from the CIML Immagimm and histology core facilities for technical help. We thank Eric Vivier (CIML) for the Nkp46-Cre mice, Sandrine Sarrazin and Michael Sieweke (CIML) for Csfr2b-deficient mice, and Mauro Gaya (CIML) for CD1d tetramers. This work benefited from institutional supports from CNRS and Inserm and was funded by the I2HD CIML-SANOFI project, by grants from Fondation pour la Recherche Médicale (“Equipe FRM 2011”, #DEQ20110421284 to MD) and from the European Research Council under the European Community's Seventh Framework Programme (FP7/2007-2013 grant agreement 281225, to M.D.), by the XCR1DirectingCells ANR Grant (KC), and by the Fondation ARC pour la Recherche sur le Cancer (KC). S.G. was supported by a doctoral fellowship from the French Ministère de l’Enseignement Supérieur et de la Recherche and from Fondation ARC pour la Recherche sur le Cancer. J.C.C. was supported by a fellowship from La Ligue Nationale Contre le Cancer.
Author contributions
S.G. designed and performed experiments, analyzed and interpreted data, and contributed to manuscript preparation; M.A. designed and performed experiments, quantified and analyzed microscopy data, and contributed to manuscript preparation; J.C.C. performed experiments and analyzed data; M.M. analyzed microarray data; H.L. provided critical technical support for imaging; M.D. directed and contributed to funding the project, and edited the manuscript. K.C. contributed to conceptualization, directed and contributed to funding the project, designed and performed experiments, analyzed and interpreted data, and wrote the manuscript.
Declaration of interests
The authors declare no conflict of interests.
Published: September 24, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103059.
Supplemental information
Color code: yellow, IL-12eYFP; green, NKp46; purple, IFN-g; red, tdRFP (cDC1). Scale bar: 10μm.
References
- Abbas A., Vu Manh T.-P., Valente M., Collinet N., Attaf N., Dong C., Naciri K., Chelbi R., Brelurut G., Cervera-Marzal I. The activation trajectory of plasmacytoid dendritic cells in vivo during a viral infection. Nat. Immunol. 2020;21:983–997. doi: 10.1038/s41590-020-0731-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akazawa T., Ebihara T., Okuno M., Okuda Y., Shingai M., Tsujimura K., Takahashi T., Ikawa M., Okabe M., Inoue N. Antitumor NK activation induced by the Toll-like receptor 3-TICAM-1 (TRIF) pathway in myeloid dendritic cells. Proc. Natl. Acad. Sci. U S A. 2007;104:252–257. doi: 10.1073/pnas.0605978104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandre Y.O., Cocita C.D., Ghilas S., Dalod M. Deciphering the role of DC subsets in MCMV infection to better understand immune protection against viral infections. Front Microbiol. 2014;5:378. doi: 10.3389/fmicb.2014.00378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandre Y.O., Ghilas S., Sanchez C., Le Bon A., Crozat K., Dalod M. XCR1+ dendritic cells promote memory CD8+ T cell recall upon secondary infections with Listeria monocytogenes or certain viruses. J. Exp. Med. 2016;213:75–92. doi: 10.1084/jem.20142350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachem A., Hartung E., Guttler S., Mora A., Zhou X., Hegemann A., Plantinga M., Mazzini E., Stoitzner P., Gurka S. Expression of XCR1 characterizes the batf3-dependent lineage of dendritic cells capable of antigen cross-presentation. Front Immunol. 2012;3:214. doi: 10.3389/fimmu.2012.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajénoff M., Glaichenhaus N., Germain R.N. Fibroblastic reticular cells guide T lymphocyte entry into and migration within the splenic T cell zone. J. Immunol. Baltim. Md 1950. 2008;181:3947–3954. doi: 10.4049/jimmunol.181.6.3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajenoff M., Narni-Mancinelli E., Brau F., Lauvau G. Visualizing early splenic memory CD8+ T cells reactivation against intracellular bacteria in the mouse. PLoS ONE. 2010;5:e11524. doi: 10.1371/journal.pone.0011524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bär E., Whitney P.G., Moor K., Reis e Sousa C., LeibundGut-Landmann S. IL-17 regulates systemic fungal immunity by controlling the functional competence of NK cells. Immunity. 2014;40:117–127. doi: 10.1016/j.immuni.2013.12.002. [DOI] [PubMed] [Google Scholar]
- Baranek T., Vu Manh T.P., Alexandre Y., Maqbool M.A., Cabeza J.Z., Tomasello E., Crozat K., Bessou G., Zucchini N., Robbins S.H. Differential responses of immune cells to type I interferon contribute to host resistance to viral infection. Cell Host Microbe. 2012;12:571–584. doi: 10.1016/j.chom.2012.09.002. [DOI] [PubMed] [Google Scholar]
- Barral P., Sánchez-Niño M.D., van Rooijen N., Cerundolo V., Batista F.D. The location of splenic NKT cells favours their rapid activation by blood-borne antigen. EMBO J. 2012;31:2378–2390. doi: 10.1038/emboj.2012.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuneu H., Deguine J., Breart B., Mandelboim O., Di Santo J.P., Bousso P. Dynamic behavior of NK cells during activation in lymph nodes. Blood. 2009;114:3227–3234. doi: 10.1182/blood-2009-06-228759. [DOI] [PubMed] [Google Scholar]
- Bezman N.A., Kim C.C., Sun J.C., Min-Oo G., Hendricks D.W., Kamimura Y., Best J.A., Goldrath A.W., Lanier L.L., Immunological Genome Project C. Molecular definition of the identity and activation of natural killer cells. Nat. Immunol. 2012;13:1000–1009. doi: 10.1038/ni.2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boismenu R., Feng L., Xia Y.Y., Chang J.C., Havran W.L. Chemokine expression by intraepithelial gamma delta T cells. Implications for the recruitment of inflammatory cells to damaged epithelia. J. Immunol. Baltim. Md 1950. 1996;157:985–992. [PubMed] [Google Scholar]
- Borg C., Jalil A., Laderach D., Maruyama K., Wakasugi H., Charrier S., Ryffel B., Cambi A., Figdor C., Vainchenker W. NK cell activation by dendritic cells (DCs) requires the formation of a synapse leading to IL-12 polarization in DCs. Blood. 2004;104:3267–3275. doi: 10.1182/blood-2004-01-0380. [DOI] [PubMed] [Google Scholar]
- Böttcher J.P., Bonavita E., Chakravarty P., Blees H., Cabeza-Cabrerizo M., Sammicheli S., Rogers N.C., Sahai E., Zelenay S., e Sousa C.R. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. 2018;172:1022–1037.e14. doi: 10.1016/j.cell.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewitz A., Eickhoff S., Dahling S., Quast T., Bedoui S., Kroczek R.A., Kurts C., Garbi N., Barchet W., Iannacone M. CD8+ T cells orchestrate pDC-XCR1+ dendritic cell spatial and functional cooperativity to optimize priming. Immunity. 2017;46:205–219. doi: 10.1016/j.immuni.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M.G., Dokun A.O., Heusel J.W., Smith H.R., Beckman D.L., Blattenberger E.A., Dubbelde C.E., Stone L.R., Scalzo A.A., Yokoyama W.M. Vital involvement of a natural killer cell activation receptor in resistance to viral infection. Science. 2001;292:934–937. doi: 10.1126/science.1060042. [DOI] [PubMed] [Google Scholar]
- Busche A., Jirmo A.C., Welten S.P.M., Zischke J., Noack J., Constabel H., Gatzke A.-K., Keyser K.A., Arens R., Behrens G.M.N., Messerle M. Priming of CD8+ T cells against cytomegalovirus-encoded antigens is dominated by cross-presentation. J. Immunol. 2013;190:2767–2777. doi: 10.4049/jimmunol.1200966. [DOI] [PubMed] [Google Scholar]
- Calabro S., Liu D., Gallman A., Nascimento M.S., Yu Z., Zhang T.T., Chen P., Zhang B., Xu L., Gowthaman U. Differential intrasplenic migration of dendritic cell subsets tailors adaptive immunity. Cell Rep. 2016;16:2472–2485. doi: 10.1016/j.celrep.2016.07.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancel J.-C., Crozat K., Dalod M., Mattiuz R. Are conventional type 1 dendritic cells critical for protective antitumor immunity and how? Front. Immunol. 2019;10:9. doi: 10.3389/fimmu.2019.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capucha T., Mizraji G., Segev H., Blecher-Gonen R., Winter D., Khalaileh A., Tabib Y., Attal T., Nassar M., Zelentsova K. Distinct murine mucosal Langerhans cell subsets develop from pre-dendritic cells and monocytes. Immunity. 2015;43:369–381. doi: 10.1016/j.immuni.2015.06.017. [DOI] [PubMed] [Google Scholar]
- Crouse J., Xu H.C., Lang P.A., Oxenius A. NK cells regulating T cell responses: mechanisms and outcome. Trends Immunol. 2015;36:49–58. doi: 10.1016/j.it.2014.11.001. [DOI] [PubMed] [Google Scholar]
- Crozat K., Guiton R., Contreras V., Feuillet V., Dutertre C.A., Ventre E., Vu Manh T.P., Baranek T., Storset A.K., Marvel J. The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8alpha+ dendritic cells. J. Exp. Med. 2010;207:1283–1292. doi: 10.1084/jem.20100223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozat K., Hoebe K., Ugolini S., Hong N.A., Janssen E., Rutschmann S., Mudd S., Sovath S., Vivier E., Beutler B. Jinx, an MCMV susceptibility phenotype caused by disruption of Unc13d: a mouse model of type 3 familial hemophagocytic lymphohistiocytosis. J. Exp. Med. 2007;204:853–863. doi: 10.1084/jem.20062447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozat K., Tamoutounour S., Vu Manh T.P., Fossum E., Luche H., Ardouin L., Guilliams M., Azukizawa H., Bogen B., Malissen B. Cutting edge: expression of XCR1 defines mouse lymphoid-tissue resident and migratory dendritic cells of the CD8alpha+ type. J. Immunol. 2011;187:4411–4415. doi: 10.4049/jimmunol.1101717. [DOI] [PubMed] [Google Scholar]
- Dalod M., Salazar-Mather T.P., Malmgaard L., Lewis C., Asselin-Paturel C., Briere F., Trinchieri G., Biron C.A. Interferon alpha/beta and interleukin 12 responses to viral infections: pathways regulating dendritic cell cytokine expression in vivo. J. Exp. Med. 2002;195:517–528. doi: 10.1084/jem.20011672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao T., Gomez-Nunez M., Antczak C., Kappel B., Jaggi J.S., Korontsvit T., Zakhaleva V., Scheinberg D.A. Natural killer cells license dendritic cell cross-presentation of B lymphoma cell--associated antigens. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005;11:8763–8772. doi: 10.1158/1078-0432.CCR-05-0975. [DOI] [PubMed] [Google Scholar]
- Deauvieau F., Ollion V., Doffin A.-C., Achard C., Fonteneau J.-F., Verronese E., Durand I., Ghittoni R., Marvel J., Dezutter-Dambuyant C. Human natural killer cells promote cross-presentation of tumor cell-derived antigens by dendritic cells. Int. J. Cancer. 2015;136:1085–1094. doi: 10.1002/ijc.29087. [DOI] [PubMed] [Google Scholar]
- Dorner B.G., Dorner M.B., Zhou X., Opitz C., Mora A., Guttler S., Hutloff A., Mages H.W., Ranke K., Schaefer M. Selective expression of the chemokine receptor XCR1 on cross-presenting dendritic cells determines cooperation with CD8+ T cells. Immunity. 2009;31:823–833. doi: 10.1016/j.immuni.2009.08.027. [DOI] [PubMed] [Google Scholar]
- Dorner B.G., Smith H.R., French A.R., Kim S., Poursine-Laurent J., Beckman D.L., Pingel J.T., Kroczek R.A., Yokoyama W.M. Coordinate expression of cytokines and chemokines by NK cells during murine cytomegalovirus infection. J. Immunol. 2004;172:3119–3131. doi: 10.4049/jimmunol.172.5.3119. [DOI] [PubMed] [Google Scholar]
- Dougan M., Dranoff G., Dougan S.K. GM-CSF, IL-3, and IL-5 family of cytokines: regulators of inflammation. Immunity. 2019;50:796–811. doi: 10.1016/j.immuni.2019.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour A., Meas-Yedid V., Grassart A., Olivo-Marin J.-C. 2008 19th International Conference on Pattern Recognition. 2008. Automated quantification of cell endocytosis using active contours and wavelets; pp. 1–4. [Google Scholar]
- Ebihara T., Azuma M., Oshiumi H., Kasamatsu J., Iwabuchi K., Matsumoto K., Saito H., Taniguchi T., Matsumoto M., Seya T. Identification of a polyI:C-inducible membrane protein that participates in dendritic cell-mediated natural killer cell activation. J. Exp. Med. 2010;207:2675–2687. doi: 10.1084/jem.20091573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaiser M.R., Lämmermann T., Feng X., Igyarto B.Z., Kaplan D.H., Tessarollo L., Germain R.N., Udey M.C. Cancer-associated epithelial cell adhesion molecule (EpCAM; CD326) enables epidermal Langerhans cell motility and migration in vivo. Proc. Natl. Acad. Sci. U S A. 2012;109:5563–5564. doi: 10.1073/pnas.1117674109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerosa F., Baldani-Guerra B., Nisii C., Marchesini V., Carra G., Trinchieri G. Reciprocal activating interaction between natural killer cells and dendritic cells. J. Exp. Med. 2002;195:327–333. doi: 10.1084/jem.20010938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girardi M., Oppenheim D.E., Steele C.R., Lewis J.M., Glusac E., Filler R., Hobby P., Sutton B., Tigelaar R.E., Hayday A.C. Regulation of cutaneous malignancy by gammadelta T cells. Science. 2001;294:605–609. doi: 10.1126/science.1063916. [DOI] [PubMed] [Google Scholar]
- Greene T.T., Tokuyama M., Knudsen G.M., Kunz M., Lin J., Greninger A.L., DeFilippis V.R., DeRisi J.L., Raulet D.H., Coscoy L. A Herpesviral induction of RAE-1 NKG2D ligand expression occurs through release of HDAC mediated repression. eLife. 2016;5:e14749. doi: 10.7554/eLife.14749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregoire C., Cognet C., Chasson L., Coupet C.A., Dalod M., Reboldi A., Marvel J., Sallusto F., Vivier E., Walzer T. Intrasplenic trafficking of natural killer cells is redirected by chemokines upon inflammation. Eur. J. Immunol. 2008;38:2076–2084. doi: 10.1002/eji.200838550. [DOI] [PubMed] [Google Scholar]
- Greter M., Helft J., Chow A., Hashimoto D., Mortha A., Agudo-Cantero J., Bogunovic M., Gautier E.L., Miller J., Leboeuf M. GM-CSF controls nonlymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity. 2012;36:1031–1046. doi: 10.1016/j.immuni.2012.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn M.D., Kyuwa S., Tam C., Kakiuchi T., Matsuzawa A., Williams L.T., Nakano H. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J. Exp. Med. 1999;189:451–460. doi: 10.1084/jem.189.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibusuki A., Kawai K., Yoshida S., Uchida Y., Nitahara-Takeuchi A., Kuroki K., Kajikawa M., Ose T., Maenaka K., Kasahara M., Kanekura T. NKG2D triggers cytotoxicity in murine epidermal γδ T cells via PI3K-dependent, Syk/ZAP70-independent signaling pathway. J. Invest. Dermatol. 2014;134:396–404. doi: 10.1038/jid.2013.353. [DOI] [PubMed] [Google Scholar]
- Iigo M., Moriyama M., Suzuki I., Tsuda H. Markedly induced asialoGM1+CD8+ T cell production and enhancement of antimetastatic activity by interferon beta with folic or folinic acid. Cancer Immunol. Immunother. CII. 1997;44:65–69. doi: 10.1007/s002620050356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyoda T., Shimoyama S., Liu K., Omatsu Y., Akiyama Y., Maeda Y., Takahara K., Steinman R.M., Inaba K. The CD8+ dendritic cell subset selectively endocytoses dying cells in culture and in vivo. J. Exp. Med. 2002;195:1289–1302. doi: 10.1084/jem.20020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger B.N., Donadieu J., Cognet C., Bernat C., Ordoñez-Rueda D., Barlogis V., Mahlaoui N., Fenis A., Narni-Mancinelli E., Beaupain B. Neutrophil depletion impairs natural killer cell maturation, function, and homeostasis. J. Exp. Med. 2012;209:565–580. doi: 10.1084/jem.20111908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasamatsu J., Azuma M., Oshiumi H., Morioka Y., Okabe M., Ebihara T., Matsumoto M., Seya T. INAM plays a critical role in IFN-γ production by NK cells interacting with polyinosinic-polycytidylic acid–stimulated accessory cells. J. Immunol. 2014;193:5199–5207. doi: 10.4049/jimmunol.1400924. [DOI] [PubMed] [Google Scholar]
- Khairallah C., Netzer S., Villacreces A., Juzan M., Rousseau B., Dulanto S., Giese A., Costet P., Praloran V., Moreau J.-F. γδ T cells confer protection against murine cytomegalovirus (MCMV) Plos Pathog. 2015;11 doi: 10.1371/journal.ppat.1004702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King I.L., Kroenke M.A., Segal B.M. GM-CSF-dependent, CD103+ dermal dendritic cells play a critical role in Th effector cell differentiation after subcutaneous immunization. J. Exp. Med. 2010;207:953–961. doi: 10.1084/jem.20091844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka A., Wakita D., Matsubara N., Togashi Y., Nishimura S.-I., Kitamura H., Nishimura T. AsialoGM1+CD8+ central memory-type T cells in unimmunized mice as novel immunomodulator of IFN-gamma-dependent type 1 immunity. Int. Immunol. 2007;19:249–256. doi: 10.1093/intimm/dxl140. [DOI] [PubMed] [Google Scholar]
- Krebs P., Barnes M.J., Lampe K., Whitley K., Bahjat K.S., Beutler B., Janssen E., Hoebe K. NK-cell-mediated killing of target cells triggers robust antigen-specific T-cell-mediated and humoral responses. Blood. 2009;113:6593–6602. doi: 10.1182/blood-2009-01-201467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug A., French A.R., Barchet W., Fischer J.A., Dzionek A., Pingel J.T., Orihuela M.M., Akira S., Yokoyama W.M., Colonna M. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity. 2004;21:107–119. doi: 10.1016/j.immuni.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Lauterbach H., Bathke B., Gilles S., Traidl-Hoffmann C., Luber C.A., Fejer G., Freudenberg M.A., Davey G.M., Vremec D., Kallies A. Mouse CD8alpha+ DCs and human BDCA3+ DCs are major producers of IFN-lambda in response to poly IC. J. Exp. Med. 2010;207:2703–2717. doi: 10.1084/jem.20092720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.H., Girard S., Macina D., Busa M., Zafer A., Belouchi A., Gros P., Vidal S.M. Susceptibility to mouse cytomegalovirus is associated with deletion of an activating natural killer cell receptor of the C-type lectin superfamily. Nat. Genet. 2001;28:42–45. doi: 10.1038/88247. [DOI] [PubMed] [Google Scholar]
- Louis C., Guimaraes F., Yang Y., D’Silva D., Kratina T., Dagley L., Hediyeh-Zadeh S., Rautela J., Masters S.L., Davis M.J. NK cell-derived GM-CSF potentiates inflammatory arthritis and is negatively regulated by CIS. J. Exp. Med. 2020;217 doi: 10.1084/jem.20191421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu E., Dang E.V., McDonald J.G., Cyster J.G. Distinct oxysterol requirements for positioning naïve and activated dendritic cells in the spleen. Sci. Immunol. 2017;2 doi: 10.1126/sciimmunol.aal5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luche H., Weber O., Nageswara Rao T., Blum C., Fehling H.J. Faithful activation of an extra-bright red fluorescent protein in “knock-in” Cre-reporter mice ideally suited for lineage tracing studies. Eur. J. Immunol. 2007;37:43–53. doi: 10.1002/eji.200636745. [DOI] [PubMed] [Google Scholar]
- Luther S.A., Tang H.L., Hyman P.L., Farr A.G., Cyster J.G. Coexpression of the chemokines ELC and SLC by T zone stromal cells and deletion of the ELC gene in the plt/plt mouse. Proc. Natl. Acad. Sci. U S A. 2000;97:12694–12699. doi: 10.1073/pnas.97.23.12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashayekhi M., Sandau M.M., Dunay I.R., Frickel E.M., Khan A., Goldszmid R.S., Sher A., Ploegh H.L., Murphy T.L., Sibley L.D., Murphy K.M. CD8alpha(+) dendritic cells are the critical source of interleukin-12 that controls acute infection by Toxoplasma gondii tachyzoites. Immunity. 2011;35:249–259. doi: 10.1016/j.immuni.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiuz R., Wohn C., Ghilas S., Ambrosini M., Alexandre Y.O., Sanchez C., Fries A., Vu Manh T.-P., Malissen B., Dalod M., Crozat K. Novel cre-expressing mouse strains permitting to selectively track and edit type 1 conventional dendritic cells facilitate disentangling their complexity in vivo. Front. Immunol. 2018;9:2805. doi: 10.3389/fimmu.2018.02805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer C.T., Ghorbani P., Nandan A., Dudek M., Arnold-Schrauf C., Hesse C., Berod L., Stüve P., Puttur F., Merad M., Sparwasser T. Selective and efficient generation of functional Batf3-dependent CD103+ dendritic cells from mouse bone marrow. Blood. 2014;124:3081–3091. doi: 10.1182/blood-2013-12-545772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCartney S., Vermi W., Gilfillan S., Cella M., Murphy T.L., Schreiber R.D., Murphy K.M., Colonna M. Distinct and complementary functions of MDA5 and TLR3 in poly(I:C)-mediated activation of mouse NK cells. J. Exp. Med. 2009;206:2967–2976. doi: 10.1084/jem.20091181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merad M., Sathe P., Helft J., Miller J., Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol. 2013;31:563–604. doi: 10.1146/annurev-immunol-020711-074950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake T., Kumagai Y., Kato H., Guo Z., Matsushita K., Satoh T., Kawagoe T., Kumar H., Jang M.H., Kawai T. Poly I:C-induced activation of NK cells by CD8 + dendritic cells via the IPS-1 and TRIF-dependent pathways. J. Immunol. 2009;183:2522–2528. doi: 10.4049/jimmunol.0901500. [DOI] [PubMed] [Google Scholar]
- Moore M.L., Chi M.H., Goleniewska K., Durbin J.E., Peebles R.S. Differential regulation of GM1 and asialo-GM1 expression by T cells and natural killer (NK) cells in respiratory syncytial virus infection. Viral Immunol. 2008;21:327–339. doi: 10.1089/vim.2008.0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortier E., Advincula R., Kim L., Chmura S., Barrera J., Reizis B., Malynn B., Ma A. Macrophage- and dendritic-cell-derived interleukin-15 receptor alpha supports homeostasis of distinct CD8+ T cell subsets. Immunity. 2009;31:811–822. doi: 10.1016/j.immuni.2009.09.017. [DOI] [PubMed] [Google Scholar]
- Narni-Mancinelli E., Chaix J., Fenis A., Kerdiles Y.M., Yessaad N., Reynders A., Grégoire C., Luche H., Ugolini S., Tomasello E., Walzer T., Vivier E. Fate mapping analysis of lymphoid cells expressing the NKp46 cell surface receptor. Proc. Natl. Acad Sci. U S A. 2011;108:18324–18329. doi: 10.1073/pnas.1112064108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitahara A., Shimura H., Ito A., Tomiyama K., Ito M., Kawai K. NKG2D ligation without T cell receptor engagement triggers both cytotoxicity and cytokine production in dendritic epidermal T cells. J. Invest. Dermatol. 2006;126:1052–1058. doi: 10.1038/sj.jid.5700112. [DOI] [PubMed] [Google Scholar]
- Nour-Eldine W., Joffre J., Zibara K., Esposito B., Giraud A., Zeboudj L., Vilar J., Terada M., Bruneval P., Vivier E. Genetic depletion or hyperresponsiveness of natural killer cells do not affect atherosclerosis development. Circ. Res. 2018;122:47–57. doi: 10.1161/CIRCRESAHA.117.311743. [DOI] [PubMed] [Google Scholar]
- Ouchi T., Nakato G., Udey M.C. EpCAM expressed by murine epidermal Langerhans cells modulates immunization to an epicutaneously applied protein antigen. J. Invest. Dermatol. 2016;136:1627–1635. doi: 10.1016/j.jid.2016.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson C., Thornton E.E., McKenzie B., Schaupp A.-L., Huskens N., Griseri T., West N., Tung S., Seddon B.P., Uhlig H.H., Powrie F. ILC3 GM-CSF production and mobilisation orchestrate acute intestinal inflammation. eLife. 2016;5:e10066. doi: 10.7554/eLife.10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt R.L., Hong S., Kang S.J., Wang Z.E., Locksley R.M. Visualization of IL-12/23p40 in vivo reveals immunostimulatory dendritic cell migrants that promote Th1 differentiation. J. Immunol. 2006;177:1618–1627. doi: 10.4049/jimmunol.177.3.1618. [DOI] [PubMed] [Google Scholar]
- Reis e Sousa C., Hieny S., Scharton-Kersten T., Jankovic D., Charest H., Germain R.N., Sher A. In vivo microbial stimulation induces rapid CD40 ligand-independent production of interleukin 12 by dendritic cells and their redistribution to T cell areas. J. Exp. Med. 1997;186:1819–1829. doi: 10.1084/jem.186.11.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezende R.M., Nakagaki B.N., Moreira T.G., Lopes J.R., Kuhn C., Tatematsu B.K., Boulenouar S., Maghzi A.-H., Rubino S., Menezes G.B. Γδ T cell-secreted XCL1 mediates anti-CD3-induced oral tolerance. J. Immunol. Baltim. Md 1950. 2019 doi: 10.4049/jimmunol.1900784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robb L., Drinkwater C.C., Metcalf D., Li R., Köntgen F., Nicola N.A., Begley C.G. Hematopoietic and lung abnormalities in mice with a null mutation of the common beta subunit of the receptors for granulocyte-macrophage colony-stimulating factor and interleukins 3 and 5. Proc. Natl. Acad. Sci. U.S.A. 1995;92:9565–9569. doi: 10.1073/pnas.92.21.9565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins S.H., Bessou G., Cornillon A., Zucchini N., Rupp B., Ruzsics Z., Sacher T., Tomasello E., Vivier E., Koszinowski U.H., Dalod M. Natural killer cells promote early CD8 T cell responses against cytomegalovirus. Plos Pathog. 2007;3:e123. doi: 10.1371/journal.ppat.0030123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins S.H., Walzer T., Dembele D., Thilbault C., Defays A., Bessou G., Xu H., Vivier E., Sellars M., Pierre P. Novel insights into the relationships between dendritic cell subsets in human and mouse revealed by genome-wide expression profiling. Genome Biol. 2008;9 doi: 10.1186/Gb-2008-9-1-R17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathe P., Pooley J., Vremec D., Mintern J., Jin J.-O., Wu L., Kwak J.-Y., Villadangos J.A., Shortman K. The acquisition of antigen cross-presentation function by newly formed dendritic cells. J. Immunol. Baltim. Md 1950. 2011;186:5184–5192. doi: 10.4049/jimmunol.1002683. [DOI] [PubMed] [Google Scholar]
- Sell S., Dietz M., Schneider A., Holtappels R., Mach M., Winkler T.H. Control of murine cytomegalovirus infection by γδ T cells. PLOS Pathog. 2015;11:e1004481. doi: 10.1371/journal.ppat.1004481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavuljica I., Krmpotić A., Jonjić S. Manipulation of NKG2D ligands by cytomegaloviruses: impact on innate and adaptive immune response. Front. Immunol. 2011;2:85. doi: 10.3389/fimmu.2011.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slifka M.K., Pagarigan R.R., Whitton J.L. NK markers are expressed on a high percentage of virus-specific CD8+ and CD4+ T cells. J. Immunol. Baltim. Md 1950. 2000;164:2009–2015. doi: 10.4049/jimmunol.164.4.2009. [DOI] [PubMed] [Google Scholar]
- Snyder C.M., Allan J.E., Bonnett E.L., Doom C.M., Hill A.B. Cross-Presentation of a spread-defective MCMV is sufficient to prime the majority of virus-specific CD8+ T cells. PLoS ONE. 2010;5:e9681. doi: 10.1371/journal.pone.0009681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H.C., Nguyen K.B., Salazar-Mather T.P., Ruzek M.C., Dalod M.Y., Biron C.A. NK cell functions restrain T cell responses during viral infections. Eur. J. Immunol. 2001;31:3048–3055. doi: 10.1002/1521-4141(2001010)31:10<3048::aid-immu3048>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Tharp M.D., Seelig L.L., Tigelaar R.E., Bergstresser P.R. Conjugated avidin binds to mast cell granules. J. Histochem. Cytochem. Off. J. Histochem. Soc. 1985;33:27–32. doi: 10.1177/33.1.2578142. [DOI] [PubMed] [Google Scholar]
- Tomasello E., Naciri K., Chelbi R., Bessou G., Fries A., Gressier E., Abbas A., Pollet E., Pierre P., Lawrence T. Molecular dissection of plasmacytoid dendritic cell activation in vivo during a viral infection. EMBO J. 2018;37 doi: 10.15252/embj.201798836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torti N., Walton S.M., Murphy K.M., Oxenius A. Batf3 transcription factor-dependent DC subsets in murine CMV infection: differential impact on T-cell priming and memory inflation. Eur. J. Immunol. 2011;41:2612–2618. doi: 10.1002/eji.201041075. [DOI] [PubMed] [Google Scholar]
- Trambley J., Bingaman A.W., Lin A., Elwood E.T., Waitze S.Y., Ha J., Durham M.M., Corbascio M., Cowan S.R., Pearson T.C., Larsen C.P. Asialo GM1(+) CD8(+) T cells play a critical role in costimulation blockade-resistant allograft rejection. J. Clin. Invest. 1999;104:1715–1722. doi: 10.1172/JCI8082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umemoto E., Otani K., Ikeno T., Verjan Garcia N., Hayasaka H., Bai Z., Jang M.H., Tanaka T., Nagasawa T., Ueda K., Miyasaka M. Constitutive plasmacytoid dendritic cell migration to the splenic white pulp is cooperatively regulated by CCR7- and CXCR4-mediated signaling. J. Immunol. 2012;189:191–199. doi: 10.4049/jimmunol.1200802. [DOI] [PubMed] [Google Scholar]
- Voehringer D., Liang H.E., Locksley R.M. Homeostasis and effector function of lymphopenia-induced “memory-like” T cells in constitutively T cell-depleted mice. J. Immunol. 2008;180:4742–4753. doi: 10.4049/jimmunol.180.7.4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu Manh T.P., Elhmouzi-Younes J., Urien C., Ruscanu S., Jouneau L., Bourge M., Moroldo M., Foucras G., Salmon H., Marty H. Defining mononuclear phagocyte subset homology across several distant warm-blooded vertebrates through comparative transcriptomics. Front Immunol. 2015;6:299. doi: 10.3389/fimmu.2015.00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waggoner S.N., Cornberg M., Selin L.K., Welsh R.M. Natural killer cells act as rheostats modulating antiviral T cells. Nature. 2011;481:394–398. doi: 10.1038/nature10624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walzer T., Chiossone L., Chaix J., Calver A., Carozzo C., Garrigue-Antar L., Jacques Y., Baratin M., Tomasello E., Vivier E. Natural killer cell trafficking in vivo requires a dedicated sphingosine 1-phosphate receptor. Nat. Immunol. 2007;8:1337–1344. doi: 10.1038/ni1523. [DOI] [PubMed] [Google Scholar]
- Walzer T., Dalod M., Robbins S.H., Zitvogel L., Vivier E. Natural-killer cells and dendritic cells: “l’union fait la force. Blood. 2005;106:2252–2258. doi: 10.1182/blood-2005-03-1154. [DOI] [PubMed] [Google Scholar]
- Wculek S.K., Cueto F.J., Mujal A.M., Melero I., Krummel M.F., Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020;20:7–24. doi: 10.1038/s41577-019-0210-z. [DOI] [PubMed] [Google Scholar]
- Yamanokuchi S., Ikai I., Nishitai R., Matsushita T., Sugimoto S., Shiotani T., Yamaoka Y. Asialo GM1 positive CD8+ T cells induce skin allograft rejection in the absence of the secondary lymphoid organs. J. Surg. Res. 2005;129:57–63. doi: 10.1016/j.jss.2005.07.009. [DOI] [PubMed] [Google Scholar]
- Yamazaki C., Miyamoto R., Hoshino K., Fukuda Y., Sasaki I., Saito M., Ishiguchi H., Yano T., Sugiyama T., Hemmi H. Conservation of a chemokine system, XCR1 and its ligand, XCL1, between human and mice. Biochem. Biophys. Res. Commun. 2010;397:756–761. doi: 10.1016/j.bbrc.2010.06.029. [DOI] [PubMed] [Google Scholar]
- Yamazaki C., Sugiyama M., Ohta T., Hemmi H., Hamada E., Sasaki I., Fukuda Y., Yano T., Nobuoka M., Hirashima T. Critical roles of a dendritic cell subset expressing a chemokine receptor, XCR1. J. Immunol. 2013;190:6071–6082. doi: 10.4049/jimmunol.1202798. [DOI] [PubMed] [Google Scholar]
- Yi T., Cyster J.G. EBI2-mediated bridging channel positioning supports splenic dendritic cell homeostasis and particulate antigen capture. eLife. 2013;2:e00757. doi: 10.7554/eLife.00757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Y., Carrington E.M., van Nieuwenhuijze A., Bedoui S., Seah S., Xu Y., Wang N., Mintern J.D., Villadangos J.A., Wicks I.P., Lew A.M. GM-CSF increases cross-presentation and CD103 expression by mouse CD8+ spleen dendritic cells. Eur. J. Immunol. 2011;41:2585–2595. doi: 10.1002/eji.201141540. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Color code: yellow, IL-12eYFP; green, NKp46; purple, IFN-g; red, tdRFP (cDC1). Scale bar: 10μm.
Data Availability Statement
The microarray data generated for this study have been deposited in the GEO database (http://www.ncbi.nlm.nih.gov/geo/) under the series accession number: GSE142402.
This paper does not report original codes.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.