

Abstract
The magnitude and functional patterns of intraspecific transcriptional variation in the anophelines, including those of sex-biased genes underlying sex-specific traits relevant for malaria transmission, remain understudied. As a result, how changes in expression levels drive adaptation in these species is poorly understood. We sequenced the female, male, and larval transcriptomes of three populations of Anopheles arabiensis from Burkina Faso. One-third of the genes were differentially expressed between populations, often involving insecticide resistance-related genes in a sample type-specific manner, and with the females showing the largest number of differentially expressed genes. At the genomic level, the X chromosome appears depleted of differentially expressed genes compared with the autosomes, chromosomes harboring inversions do not exhibit evidence for enrichment of such genes, and genes that are top contributors to functional enrichment patterns of population differentiation tend to be clustered in the genome. Further, the magnitude of variation for the sex expression ratio across populations did not substantially differ between male- and female-biased genes, except for some populations in which male-limited expressed genes showed more variation than their female counterparts. In fact, female-biased genes exhibited a larger level of interpopulation variation than male-biased genes, both when assayed in males and females. Beyond uncovering the extensive adaptive potential of transcriptional variation in An. Arabiensis, our findings suggest that the evolutionary rate of changes in expression levels on the X chromosome exceeds that on the autosomes, while pointing to female-biased genes as the most variable component of the An. Arabiensis transcriptome.
Keywords: transcriptome variation, functional diversification, sex-biased gene expression, faster-X effect, Anopheles arabiensis
Significance
Transcriptome evolution in the anophelines has been primarily studied at the interspecific level, paying little attention to the patterns of interpopulation differentiation. As a result, our understanding about intraspecific variation in expression levels, particularly at different life stages, in relation to chromosome organization, and in relation to sex-biased expression, remains limited. We present a portrait of the transcriptome variation in Anopheles arabiensis, an increasingly concerning malaria vector. Our results uncover the patterns of functional differentiation underlying expression variation at different life stages, expose a nonrandom distribution of gene expression polymorphism at the chromosome and fine scales, and reveal substantial differences in the patterns of interpopulation expression polymorphism between female- and male-biased genes.
Introduction
In Africa, three members of the species complex Anopheles gambiae sensu lato (s.l.), An. gambiae s.s., Anopheles coluzzii, and Anopheles arabiensis, are considered major vectors of transmission of the malaria parasite Plasmodium (White et al. 2011; Barron et al. 2019). As disease vectors, these species impose a substantial burden on human health with ∼215 million cases and ∼384,000 casualties just in 2019 (WHO 2020). Malaria transmission is shaped by multiple genetic and ecological factors, including the immune response of these vectors against the Plasmodium parasite, which in turn affects vector reproduction and longevity (Mitchell et al. 2015); standing genetic variation in insecticide resistance (Main et al. 2016; Lucas et al. 2019) and differential survival during the larval and adult stages (Roux and Robert 2019); and access to feeding resources (Barredo and DeGennaro 2020).
Organismal traits relevant for understanding malaria transmission in the anophelines, such as reproduction and longevity, are interrelated through a partially shared genetic basis (Lawniczak et al. 2007; Rono et al. 2010; Mitchell and Catteruccia 2017). Large titers of the hormone 20-hydroxecdysone (20E) generated by the male accessory glands are transferred to the female during mating, inducing oogenesis, egg-laying, and preventing further mating (Baldini et al. 2013; Gabrieli et al. 2014). This monandrous behavior enhances female longevity (Dao et al. 2010), favoring indirectly the completion of the Plasmodium sporogony, that is, the parasite multiplication within the mosquito. This lipid-transporter mediated male-enhanced oogenesis reduces the mosquito immune response against Plasmodium, further facilitating its development in the female (Rono et al. 2010). Equally important, in the anthropophilic anopheline species, blood-feeding triggers two temporally and physiologically tied process: oogenesis and Plasmodium sporogony (Mitchell and Catteruccia 2017). Further, longevity in the anophelines exhibits a complex interplay with insecticide resistance (Rivero et al. 2010), whereas survival is strongly associated with desiccation resistance (Ayala et al. 2019), in both cases affecting malaria transmission. All these organismal traits are susceptible to both inter- and intraspecific adaptive evolution as abundant naturally occurring genetic variation exists (Neafsey et al. 2010; Main et al. 2016; Ayala et al. 2019). Nevertheless, the functional consequences of this intraspecific genetic variation in relation to organismal traits that impact malaria transmission remain poorly characterized in the anophelines.
An important aspect of the anopheline biology is its unique sex-specific component as females are not only mainly monandrous (Tripet et al. 2003; Yuval 2006), but are also the only blood-feeding sex (Yuval 2006; Carnevale and Robert 2009; Barredo and DeGennaro 2020). This could impact the rate of evolution of the above indicated organismal traits as they could be targeted by sex-specific natural and sexual selection (Darwin 1871; Ellegren and Parsch 2007). For example, in An. Arabiensis, all the 20E content generated by the male accessory glands is transferred to the female during copulation whereas only half of the total 20E is transferred by other anophelines males (Mitchell et al. 2015). The genomic signature of this sex-specific fast evolution has been explored at the interspecific level, finding that the magnitude of divergence in expression levels, the rate of evolution of protein-coding genes, and the rate of gene turnover, are substantially higher for genes with female-biased expression than for male- and nonsex-biased genes (Papa et al. 2017). These patterns are in stark contrast to previous findings in other Diptera and animal lineages (Grath and Parsch 2016), being this difference interpreted as the outcome of an enhanced selection intensity operating on females coupled with ameliorated male–male competition at the postcopulatory level as a result of female monandry. Whether similar patterns are reproduced at the intraspecific level has not been explored.
Anopheles arabiensis is the most common malaria vector in the driest parts of Africa (White et al. 2011; Main et al. 2016), and it is expanding its range due to climate change (Dabire et al. 2014; Bimbile Somda et al. 2018). In this species, our understanding of patterns of naturally occurring genetic variation is primarily confined to SNP and chromosomal inversion surveys (Fontaine et al. 2015; Main et al. 2016; Ayala et al. 2017), whereas functional genomic information derives primarily from interspecific studies based on a single laboratory strain from Dongola, Sudan (Neafsey et al. 2015; Papa et al. 2017; Izquierdo et al. 2019). To our knowledge, only one study has tried to decipher the genome-wide transcriptional patterns that arises from interpopulation genetic variation (Simma et al. 2019), and thus far no study has tried to investigate the functional patterns associated with this genetic variation while taking into account sex-specific aspects of the anopheline biology. As a result, we lack a basic understanding about the magnitude of this subset of functional variation upon which selective mechanisms can operate and how it might be linked to the evolution of key traits that contribute to the role of An. Arabiensis as a malaria vector and its ability to thrive in the most arid areas of Africa.
Using RNA-sequencing, we dissected the patterns of transcriptome variation in females, males, and larvae across three populations from Burkina Faso exposed to different types of savanna climate. Notably, in Burkina Faso, one of the countries most affected by malaria burden and deaths (WHO 2020), insecticide resistance in An. Arabiensis has been increasingly documented (Jones et al. 2012a). This resistance can arise in different ways (Ranson et al. 2002; Feyereisen et al. 2015), in some cases in association with gene expression changes, typically in the form of overexpression of detoxifying enzyme-encoding genes leading to metabolic resistance (Ibrahim et al. 2016), and expression changes in genes relevant for cuticle composition leading to penetration resistance (Jones et al. 2013). Here, we aimed at 1) determining the magnitude and patterns of interpopulation variation in transcript abundance for An. Arabiensis in Western Africa; 2) uncovering the functional attributes of differentially expressed genes most strongly impacted by population differentiation, particularly in relation to metabolic and penetrance insecticide resistance; 3) testing for the existence of hot spots of transcriptional differentiation in the An. Arabiensis genome, for example, in regards to inversion polymorphism, which could favor the accumulation of transcriptional differences in particular chromosomes or genomic regions (Marques-Bonet et al. 2004); and 4) characterizing how genes with different sex biases might differ in their level of expression polymorphism such that we can determine the extent to which interspecific patterns of divergence (Papa et al. 2017) are reproduced at the intraspecific level and explore how gene expression variation is impacted by sex in the anophelines. Through this population framework, we have generated a comprehensive portrait of the patterns of interpopulation functional variation in An. Arabiensis, which should accelerate our understanding about the potential for adaptation and fast evolution in this vector species (Powell 2018; Lai et al. 2019), while providing a more nuanced picture of anopheline transcriptome evolution.
Results
Field Work and Colony Construction
We collected gravid females from rural areas of Nanoro, Bobo, and Kodeni not overly exposed to insecticides. These three sites are located across two different ecological and climate zones of Burkina Faso (West Africa; fig. 1; Materials and Methods). Subsequently, we constructed colonies by first molecularly excluding the presence of A. coluzzi and An. gambiae (supplementary fig. S1, Supplementary Material online), and then by enriching through particular mating schemes for the presence of the most frequent chromosomal inversions according to cytological squashes, that is, the 2Rb in all the colonies and the 2Ra and 3Ra in Kodeni and Nanoro, respectively (supplementary fig. S2 and table S1, Supplementary Material online; Materials and Methods). This approach resulted in three stable colonies, one per collection site, virtually fixed for particular combinations of chromosomal inversions (supplementary table S1, Supplementary Material online). Hereafter, we will refer to these colonies as populations.
Fig. 1.

Collection sites of gravid females An. Arabiensis in Burkina Faso. Nanoro is located in the Sudano-Sahelian zone, which extends throughout much of the central part of the country. In this zone, vegetation is constituted of an arid savannah and the climate is characterized by a short rainy season from July to September. Bobo Dioulasso, or Bobo, and Kodeni are situated in the Sudano-Guinean zone at the Southwest of the country. In this zone, the type of vegetation is humid savannah, and the rainy season extends from June to October.
Sequencing Results
To ameliorate any unintended effect from long-term laboratory adaptation (Bonizzoni et al. 2012), RNA-seq data were generated shortly after colony construction. We sequenced in triplicates the transcriptome of L4 larvae, naïve males, and virgin nonblood-fed females. In the case of the larvae, their sex was unknown, nevertheless, the probability of including skewed biological replicates due to sex bias was low and therefore the impact in downstream analysis negligible (supplementary note S1, Supplementary Material online). In the case of adult individuals, the choice of using only virgins was to minimize allometric differences between the sexes (Baker and Russell 2011; Jiang et al. 2015). Approximately 523 million sequencing reads (58.2 ± 10.58; mean ± SD per sample type and population) were used in subsequent downstream analyses. From those, 88% ± 15.6% per sample type and population mapped to the genome assembly of An. Arabiensis. Mapping efficiency did not differ by collection site (Wilcoxon rank-sum test, P = 0.701), and was strongly correlated between pairs of biological replicates for the sample type and collection site (Pearson’s r = 0.929–0.997). Upon discarding any batch effect (supplementary fig. S3, Supplementary Material online), multidimensional scaling analysis additionally showed a perfect clustering of the biological replicates per population for each of the sequenced sample types, confirming the reliability and reproducibility of the sequencing results (supplementary fig. S4, Supplementary Material online).
Interpopulation Transcriptional Variation
The total number of genes with nonzero read counts in at least one sample was 13,657 (99.8%). However, a gene was considered to be expressed only if there was evidence of expression (≥1 cpm) in ≥2 biological replicates of at least one of the particular sample type by population combinations, that is, a biological group, surveyed. Under these criteria, 12,575 genes (91.9% of all the genes surveyed) were considered in further analyses (supplementary table S2, Supplementary Material online). First, we performed three pairwise population contrasts per sample type (supplementary table S3, Supplementary Material online). At a 1% false discovery rate (FDR), fold-change (FC) ≥2, a minimum of 1,150 (9.1%) and a maximum of 2,304 (18.3%) genes showed differential expression in larvae and females, respectively. In total, across sample types, 4,035 genes were differentially expressed between populations, that is, 32.1% of all annotated genes, of which, 76.2% showed sample type-specific expression (supplementary fig. S5 and table S4, Supplementary Material online).
We next examined the commonalities and differences in expression differentiation across population contrasts, which were reproducible at different minimum expression thresholds (fig. 2A; supplementary figs. S6 and S7, Supplementary Material online). Females exhibit the largest magnitude of interpopulation differentiation, which is clearly linked to the Kodeni population. This population shows the largest number of differentially expressed genes (1,813) in relation to Bobo. The differentiation of the Kodeni female transcriptome from those from Bobo and Nanoro involves 1,063 common genes, which represent more than 50% of the subtotal in each pairwise contrast (58.6% and 68.3% for the Kodeni–Bobo contrast and the Kodeni–Nanoro contrasts, respectively; fig. 2B). For the male and larval transcriptomes, the contrasts involving Nanoro entail the most differentially expressed genes. According to these levels of expression polymorphism, for males and larvae but not for females, the mode of interpopulation differentiation in mRNA abundance seems to mirror more closely the ecotype zone to which the collection site belongs (fig. 2C). Lastly, we examined whether there was any global bias in the directionality of the differential expression, that is, toward over- or underexpression, across population contrasts and sample types. We found evidence of such bias at Padj < 0.01 in six out of the nine contrasts (supplementary fig. S7, Supplementary Material online), although no consistency in the directionality of the trend was found for the same population contrast across sample types. In sum, we documented an overall lack of consistency in magnitude, mode, and directionality in the patterns of differentiation in mRNA abundance across sample types and population contrasts.
Fig. 2.

Magnitude and patterns of interpopulation differentiation of the transcriptome of An. Arabiensis across sample types. (A) Number of differentially expressed genes at a 1% FDR and fold-change ≥2 for females, males, and larvae. The total number of unique differentially expressed genes across contrasts for each sample type is indicated in parenthesis. (B) Venn diagrams showing the degree of overlap among genes differentially expressed across the three contrasts performed for each sample type. (C) Hierarchical clustering of differentially expressed genes for different sample types. The genes included correspond to those in the union set of the Venn diagrams in (B). B, Bobo; K, Kodeni; and N, Nanoro. Unlike Nanoro, Bobo, and Kodeni are associated with the same ecotype zone.
Nonuniform Interpopulation Functional Diversification across Sample Types
To understand the patterns of functional differentiation associated with changes in gene expression across populations and sample types, we tested for enrichment in Gene Ontology (GO) terms and pathways among the differentially expressed genes in each population contrast (supplementary table S6 and note S2, Supplementary Material online). For females, the populations from Bobo and Kodeni showed the largest level of differentiation across functional rubrics (supplementary table S7, Supplementary Material online). In contrast, for males, Nanoro exhibited the most pronounced pattern of functional differentiation, both in relation to Kodeni and Bobo. For larvae, no consistent pattern of functional differentiation emerged across population contrasts. Overall, we found very limited overlap in trends of functional differentiation both among sample types within the same population and among populations for the same sample type (fig. 3A). These patterns were essentially reproduced when performing a complementary analysis focused on genes found overexpressed in one population in relation to the other two (supplementary table S5, Supplementary Material online), and when considering only the 161 genes that contribute to the statistical enrichment across multiple functional terms, hereafter top contributors (fig. 3B; supplementary table S8 and note S3, Supplementary Material online; Materials and Methods).
Fig. 3.

Commonalities in functional enrichment patterns across different functional rubrics are limited among sample types. (A) Degree of overlap among sets of differentially expressed genes in at least one population contrast at a 1% FDR across the three types of samples assayed for GO terms and pathways. GO term and pathway enrichment was assessed using g:Profiler (Raudvere et al. 2019) and VectorBase (Giraldo-Calderon et al. 2015), respectively. (B) Degree of overlap among sets of differentially expressed genes tagged as top contributors to patterns of functional enrichment for GO terms (without distinguishing among Biological Process, Molecular Function, and Cellular Component) and pathways.
When specific GO terms and pathways were considered, several related to protein metabolism stood out as being enriched across two or more population contrasts both in males and larvae (supplementary figs. S8 and S9, Supplementary Material online). Also in males, enrichment for pathways and GO terms related to multiple detoxification processes underlying metabolic and penetration resistance was very common across population contrasts. For example, we found multiple genes encoding cytochrome P450s such as AARA015789 (CYP6P4), which has been shown to underlie An. Arabiensis resistance against several pyrethroids in Central Africa (Ibrahim et al. 2016), and glutathione S-transferases, such as AARA015764 (Gstd3), which has been shown to underlie resistance of An. Arabiensis to DDT in Burkina Faso (Jones et al. 2012b). We also detected genes encoding proteins relevant for cuticle composition such as AARA003376. This gene is involved in the detoxification of bisphenol, a compound known for interfering in the tanning and sclerotization reaction in mosquito (Sacher 1971), and also in that of repellents such as nicotine and limonene (Huong et al. 2019). Although not part of enriched functional terms, we also found genes recently proposed to be candidates of insecticide resistance in An. gambiae such as AARA016114 (hexamerin) and AARA002269 (α-crystallin; Ingham et al. 2018). Notably as well, 31.4% (126/401) of the differentially expressed genes at 1% FDR and FC > 2 between Ethiopian resistant populations of An. Arabiensis and other populations not resistant to DDT and the pyrethroid deltamethrin were also found to be polymorphic in expression across Burkina Faso populations (Simma et al. 2019). In good agreement with the general patterns described above, most of these differentially expressed genes were not consistent across sample types (supplementary fig. S10, Supplementary Material online).
Transcriptional Variation across the Genome
For each sample type, we sought to establish whether genes polymorphic in expression were randomly distributed across the five chromosomal elements of An. Arabiensis the X chromosome, and the left and right arms of the autosomes 2 and 3 (2L and 2R; 3L and 3R; fig. 4A). We found a significant deficit of differentially expressed genes on the X chromosome across sample types, being more robust in larvae as such deficit was reproduced across all population contrasts (supplementary table S9, Supplementary Material online); for females, two population contrasts, and for males, one population contrast showed this reduction. Additionally, in the case of females, we also detected an enrichment for differentially expressed genes on chromosome 3R (supplementary table S9, Supplementary Material online).
Fig. 4.

Differentially expressed genes are nonrandomly distributed across the An. Arabiensis chromosomal element. (A) Per sample type. (B) Main contributors to functional enrichment patterns. Top, different chromosomes, with L and R denoting the left and right arms of the metacentric chromosomes 2 and 3, respectively. Only genes with FC ≥ 2 in expression at a 1% FDR in at least one of the population contrasts performed are considered. Each of these genes is indicated with a vertical solid line. Significant enrichment (↑) or depletion (↓) at 1% FDR-corrected P-value (*) or at 5% FDR-corrected P-value (•) is shown.
Alternative chromosomal arrangements generated by inversions are known to facilitate functional divergence (Marques-Bonet et al. 2004). The populations of An. Arabiensis analyzed carry the inversions 2Ra and 3Ra (supplementary table S1, Supplementary Material online), allowing us to investigate any possible transcriptional effect of such inversions. Although we could not test precisely whether expression polymorphism is associated with the indicated inversions, as their breakpoints have not been mapped molecularly, none of our analyses identified the chromosomes carrying the indicated inversions as a preferential source of interpopulation polymorphism in gene expression. This result differs markedly from what has been reported in other Diptera (Fuller et al. 2016; Lavington and Kern 2017). Our results provide strong evidence for a chromosome effect on the genome-wide expression polymorphism documented here, but not one mediated by chromosome inversions.
Expression polymorphism can also be nonrandomly distributed at a finer scale, and not only at the chromosome-wide level. We tested this possibility across scaffolds, adjusting for their size in number of included genes. Only for females, in the case of the contrast between Kodeni and Nanoro, is there evidence of enrichment in the 1,254 kb long scaffold KB704718, which is located on the chromosome 2L. Out of the 57 expressed genes harbored by this scaffold, 16 (30.4%) are differentially expressed in the indicated contrast, although there is no consistency in the population directionality of the expression changes. Therefore, we discarded the possibility of local chromatin-level regulatory differentiation in the genomic region represented by the scaffold KB7047818.
Subsequently, we examined how the 165 top contributor genes were organized in the genome (fig. 4B). Of them, 154 genes were mapped to chromosomes, showing a small but significant underrepresentation on the X and 3R (two-sided Fisher’s exact test, FDR-corrected; X, Padj = 0.0132; 3R, Padj = 0.0175) and a larger deficit on the 3L (two-sided Fisher’s exact test, FDR-corrected, Padj = 9.53 × 10−5). Notably, 60 of these top contributors (i.e., 36.4%) form part of 22 clusters, defined as groups of at least two differentially expressed genes located less than 10 kb away (supplementary table S8, Supplementary Material online). Collectively, these findings support the conclusion that polymorphic genes underlying multiple functionally enriched trends of population differentiation are not randomly distributed across the An. Arabiensis genome, but rather they tend to form clusters.
Sex-Biased Gene Expression
We examined the expression ratio between the sexes, that is, log2(male/female), from a population perspective. We considered increasing expression differences between the sexes in our gene categorization: moderate sex bias (2≤FC < 4); strong sex bias (FC ≥ 4); and sex-limited expression (supplementary table S10, Supplementary Material online; Materials and Methods). We discovered overt interpopulation differences in the distribution of sex-biased expression (fig. 5A). These differences are well reflected in the disparity among the overall proportion of genes showing sex bias in gene expression across populations (table 1; three-sample test for equality of proportions without continuity correction, X2 = 1,303, d.f. = 2, Padj = 8.8 × 10−16), which is also apparent when examined in a pairwise fashion (supplementary table S11, Supplementary Material online). Thus, while 41.0% of the genes showed some degree of significant sex bias in Kodeni, only 20.4% and 27.8% showed such pattern in Bobo and Nanoro, respectively. These percentages are similar to those in another An. Arabiensis strain from Dongola, Sudan, in which 34.6% (3,911/11,300) of the genes were also found to be sex-biased in expression (Papa et al. 2017). In addition, the proportion of female-biased genes was greater than that of male-biased genes (three-sample test for equality of proportions without continuity correction, X2 = 33.81, d.f. = 2, Padj = 7.36 × 10−8), which is significant in two of the three population contrasts (supplementary table S11, Supplementary Material online). This overrepresentation of female-biased genes has also been reported for the Dongola strain, with 2,025 genes being female-biased and 1,886 genes being male-biased (Papa et al. 2017). Overall, Eastern and Western African populations of An. Arabiensis show strong commonalities relative to sex-biased gene expression.
Fig. 5.

Abundant and inconsistent sex-biased gene expression across populations of An. Arabiensis. (A) The violin plots show the proportion and range of fold-change values of differentially expressed genes between males and females in the three surveyed populations. (B) For sex-biased gene expression, female (red) or male (blue) bias might not be found in all three populations (no consistently category). In fact, the percentage of genes showing the same sex bias across populations (consistently category) is only 12.54% (4.32% + 8.22%). (C) Sankey charts showing the lability in the sex-biased expression between populations. The seven delineated categories in table 1 are color-coded. Bobo and Nanoro are more closely related to each other based on the patterns of sex-biased expression than either of them to Kodeni. See supplementary table S12, Supplementary Material online for the precise number of genes associated with different pairwise combinations of categories based on the magnitude and type of sex-biased expression between the two populations involved in each case. F, female-biased; M, male-biased; No DE, no differentially expressed between the sexes, that is, nonsex-biased.
Table 1.
Population Patterns of Sex-Biased Gene Expression
| Female-Biased* |
Male-Biased* |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Population | Nonsex-Biased | Moderate | Strong | Limited | Subtotal | Moderate | Strong | Limited | Subtotal | Sex-Biased |
| Bobo | 10,006 | 676 | 262 | 587 | 1,525 | 572 | 199 | 273 | 1,044 | 2569 |
| Kodeni | 7,422 | 1,465 | 662 | 594 | 2,721 | 1,403 | 475 | 554 | 2,432 | 5153 |
| Nanoro | 9,073 | 911 | 395 | 690 | 1,996 | 935 | 264 | 307 | 1,506 | 3502 |
Note.—Fold-change differences: Moderate (2 ≥ FC ≤ 4); Strong (FC > 4); Limited (expression evidence in only one sex). See supplementary table S10, Supplementary Material online.
Given the known plasticity of the transcriptome and sex-biased expression (Grath and Parsch 2016), we inspected the degree of consistency of different expression trends across our populations (fig. 5B; supplementary table S12, Supplementary Material online). We found that 48.8% of the expressed genes consistently show no sex bias, whereas the rest show evidence of significant sex bias in at least one of the populations. Within this last group of genes, 25.9% are a female-biased in at least one of the populations and unbiased in the rest, 23.3% are male-biased in at least one of the populations and unbiased in the rest, and 2% show a mixed bias pattern (i.e., reversal of sex bias across populations). When requiring consistency in the trend, that is, the same sex bias pattern across the three populations, the fraction of genes showing such biases was reduced to 8.2% and 4.3%, for female- and male-biased expression, respectively. This lack of consistency affects even well-known fitness-increasing genes such as AARA017140 or MISO (Mating-induced stimulator of oogenesis; Mitchell et al. 2015), which is categorized as moderately female-biased in Bobo and Kodeni but nonsex-biased in Nanoro.
At the interpopulation level, Kodeni showed the most differentiated pattern of categorization of sex-biased expression relative to the other two populations (fig. 5C). This interpopulation lability was further underscored when we parsed our categorization against that in the Dongola population (Papa et al. 2017; (supplementary fig. S11, table S13, and note S4, Supplementary Material online). Further, this lability in the categorization of sex-biased genes is even reflected in the differential enrichment for functional rubrics of the different sex-biased gene categories across Burkina Faso populations. Only in the case of genes with sex-limited expression, were more than 50% of the enriched functional rubrics in each population shared by at least two of the surveyed populations (supplementary table S14, Supplementary Material online).
The interspecific comparison of the sex-biased expression across four Anopheles species, including An. Arabiensis, revealed that male-biased genes showed a higher magnitude of sex bias compared with female-biased genes, whereas female-biased genes exhibited a higher rate of interspecific differentiation compared with male-biased genes (Papa et al. 2017). We examined the reproducibility of both patterns in the Burkina Faso populations. In the case of the first pattern, we found no differences in the magnitude of sex bias between the male- and female-biased gene categories when a combination of P < 0.01 and effect size (i.e., log2 FC) ≥1 was considered (supplementary fig. S12 and table S15, Supplementary Material online). Subsequently, we tested for an increased expression variation of female-biased relative to male-biased genes. For that, we calculated the standard deviation of the log2 sex expression ratio (SD, Materials and Methods), a common metric (Zhang et al. 2007; Papa et al. 2017). At a Padj < 0.01, and an effect size >0.3 to capture truly meaningful biological effects (Cohen 1992), only 2 out of the 12 contrasts showed differences in the SD of the sex expression between the sexes (Fig. 6; supplementary table S15, Supplementary Material online). The differences found were consistent with higher expression variation for male-biased genes in Bobo and Nanoro, being primarily associated with the category of sex-limited expression. Repeating the analysis with only genes showing consistently the same pattern of sex bias across populations did not change the result (supplementary table S16, Supplementary Material online). Collectively, these results indicate a lack of reproducibility of the patterns previously documented at the interspecific level in our Burkina Faso populations.
Fig. 6.
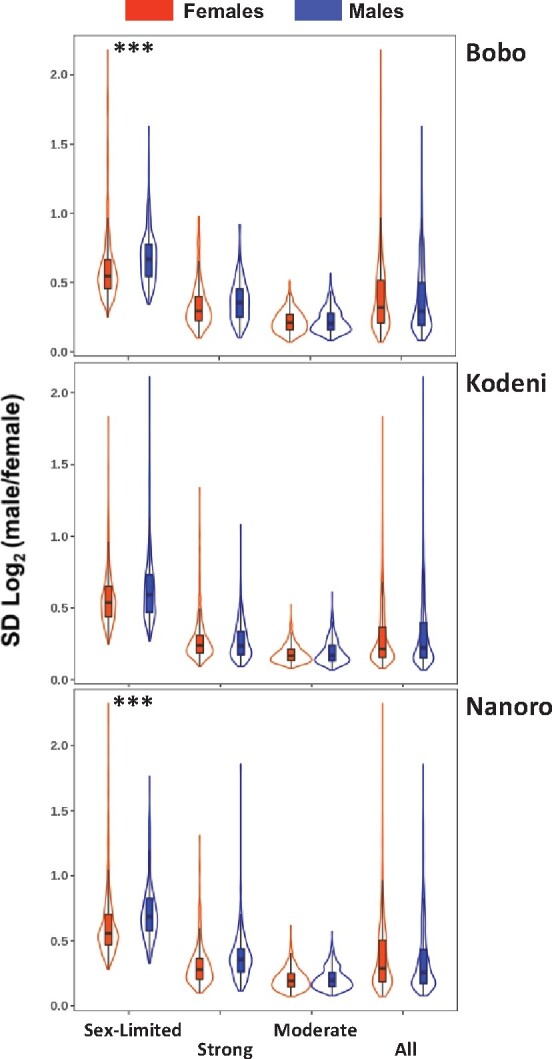
Magnitude of variation in sex expression ratio of sex-biased genes. The violin plots show the proportion and range of the variation in sex expression ratio across the three populations of An. Arabiensis investigated. For each population, all sex-biased genes, or subsets of them based on the magnitude of the sex bias, are plotted side by side. The magnitude of variation in sex expression ratio was calculated as the standard deviation (SD) of log2(male/female) expression. Only in 2 out of 12 contrasts is there evidence of significantly higher variation between the sexes; in all cases male-biased genes show significantly higher variation than female-biased genes with an effect size >0.3 and statistical significance (***) at Padj < 0.001 (supplementary table S15, Supplementary Material online).
We also investigated whether there is a positive association between the magnitude of sex-biased expression and the degree of variation in mRNA abundance in Burkina Faso populations. Compared with nonsex-biased genes, we found evidence for a higher magnitude of variation for both sex-limited and strongly sex-biased genes, but not for moderately sex-biased genes (supplementary fig. S13 and table S17, Supplementary Material online). We saw no obvious difference between male- and female-biased genes in terms of magnitude of variation relative to nonsex-biased genes. Further, in other Diptera, male-biased genes are more polymorphic than female- or nonsex-biased genes, and generally, any given gene category based on its sex bias (i.e., female-, male-, or nonsex-biased) shows more expression variability when assayed in males compared with females (Meiklejohn et al. 2003; Hutter et al. 2008; Muller et al. 2011). Contrary to this expectation, when examining the degree of interpopulation variation of different sex-biased gene categories within the same sex, we detected an overall higher level of expression polymorphism for female- compared with male-biased genes. This was obvious in males, particularly for strongly sex-biased genes and most comparisons including the nonsex-biased category (supplementary table S18, Supplementary Material online). The same trend was reproduced in females, but in some cases, the effect size was not as large as in males. Based on this trend, we predicted that the strongly sex-biased gene categories should display larger interpopulation variation when expressed in males compared with when expressed in females. Upon adjusting for population-level differences, we found support to our prediction (supplementary fig. S13 and table S19, Supplementary Material online). Altogether, the patterns of sex-biased expression described here in An. Arabiensis share limited similarity with those previously reported in Drosophila melanogaster (Meiklejohn et al. 2003; Hutter et al. 2008; Muller et al. 2011).
Discussion
Patterns of Interpopulation Variation in Gene Expression
Genome-wide analyses of molecular variation in An. Arabiensis and other anopheline species have primarily focused on the sequence evolution of protein-coding genes (Neafsey et al. 2010; Fontaine et al. 2015; Neafsey et al. 2015). In contrast, equivalent analysis on expression variability have been scarce (Bonizzoni et al. 2012) and largely limited to the response to insecticides (Bonizzoni et al. 2015; Ingham et al. 2018; Simma et al. 2019). As the anophelines are subject to strong selection pressures including climate change, habitat disturbance, and human-mediated interventions to control malaria, transcriptome studies are arguably essential to understand the adaptive response of these species.
Our portrait of the expression polymorphism in An. Arabiensis has revealed remarkable levels of mRNA variation among females, males, and larvae from three populations of Burkina Faso located within ∼350 km2, with larvae showing the lowest level of expression polymorphism. This is largely expected due to several factors. The first is the enhanced selection intensity that operates on this developmental stage, as it requires an aquatic environment and is highly susceptible to predation, which results in higher mortality rates compared with adulthood (Awono-Ambene and Robert 1999; Carnevale and Robert 2009). The second factor is the greater developmental constraints associated with the insect larval stage, constraints that decrease as development progresses (Artieri and Singh 2010). And the third is the reduced power to detect interpopulation expression differences when sequencing a mix of male and female larvae as a gene can be differentially expressed between two populations only in one sex, or in both sexes but in opposite directions, that is, overexpressed in one sex and underexpressed in the other.
Notably, we observed discordant patterns of transcriptome variation among populations across sample types. Using the fraction of differentially expressed genes between two populations as a distance metric, the Nanoro population, which is the only one of the three located at the Sudano-Sahelian ecotype zone, shows the most variable transcriptome for males and larvae, whereas the Kodeni population does so for the female transcriptome. Particularly noticeable is the relatively low number of differentially expressed genes between the females of Bobo and Nanoro, a pattern for which its biological underpinnings are not apparent at this time and should be further explored. Overall, these differential patterns of expression polymorphism could be explained by asymmetrical functional constraints and selective pressures among sample types, which underscores the need of performing expression analyses with different sample types to obtain a more accurate view of the genome-wide patterns of population differentiation in the anophelines.
From a functional perspective, the patterns of transcriptome differentiation, based on the number of GO terms and pathways that showed significant enrichment among differentially expressed genes, only mirror the ecotype location of the three analyzed populations in the case of the males but not in the case of females and larvae. This is particularly evident for the females from Bobo and Kodeni, which exhibit the most marked patterns of functional differentiation despite belonging to the same ecotype zone. Overall, the disparate patterns of functional differentiation shown by different sample types across populations could be explained by uneven effects of local variation in abiotic and biotic factors although experiments directly testing this possibility remain to be done. We also documented very little overlap among enriched functional terms across sample types, which, to some extent, is unsurprising given the different ontogenetic constraints and selection pressures to which larvae, males, and females are exposed.
Expression Polymorphism Is Abundant among Insecticide Resistance Genes
We detected that many polymorphic genes are associated with functional terms relevant for insecticide resistance. For example, 251 (∼2/3) of the differentially expressed genes in females in response to DDT and the pyrethroid deltamethrin in Ethiopian populations (Simma et al. 2019) were found polymorphic across our Burkina Faso populations in not only females (126 or 31.4%) but also males (162 or 40.4%) and larvae (91 or 22.7%). Although tentatively some of these differentiated genes could be used as markers for tracking resistance in Western and Eastern African populations of An. Arabiensis, analyses of the genome-wide transcriptional response to insecticides are yet to be done in Burkina Faso populations. Furthermore, we note that among these genes, we have identified not only those typically known to mediate metabolic resistance in anopheline species but also others previously overlooked but recently proposed to contribute to alternative pathways and biological mechanisms of resistance (Ingham et al. 2018). An overall concerning aspect is the abundant naturally occurring variation associated with metabolic and penetration resistance across sample types, which might hinder long-term vector control strategies that rely primarily on insecticide-based interventions.
The X Chromosome Exhibits Reduced Levels of Transcriptional Variation Relative to the Autosomes
Genes showing interpopulation expression polymorphism are nonrandomly distributed between the X chromosome and the autosomes of An. Arabiensis. The X chromosome appeared significantly depleted of these genes across all sample types investigated, particularly in larvae, which should be studied more precisely in this developmental stage by analyzing each sex separately. This decreased level of expression polymorphism of X-linked genes is reminiscent of observations at the intraspecific level in embryos and adult males of different D. melanogaster inbred strains (Hutter et al. 2008; Kayserili et al. 2012), potentially reflecting a faster-X effect, that is, a rate of evolution on the X that exceeds that on the autosomes. This effect can result from two nonmutually exclusive mechanisms. First, it could result from a more efficient fixation of beneficial recessive mutations via positive selection when hemizygous (Charlesworth et al. 1987). Second, it could reflect a higher fixation of dominant slightly deleterious mutations through genetic drift due to a reduced effective population size of the X chromosome relative to the autosomes (Charlesworth et al. 1987; Vicoso and Charlesworth 2009)—the X chromosome but not the autosomes is in lower number in males relative to females, that is, NeX = 3/4NeA. Upward alterations of the relationship between the Ne of the X and autosomes due to intense male–male competition as in D. melanogaster, that is, NeX > 3/4NeA (Laporte and Charlesworth 2002; Vicoso and Charlesworth 2009), would not apply to the same extent to An. Arabiensis as females are essentially monandrous (Tripet et al. 2003; Yuval 2006; Carnevale and Robert 2009).
A faster-X effect on expression has been often interpreted as a result of an adaptive scenario driven by X-linked recessive cis-regulatory mutations (Kayserili et al. 2012; Meisel et al. 2012; Coolon et al. 2015; Llopart 2012, 2018), particularly in association with male-biased genes. Male-biased, as opposed to female-biased genes, are preferentially located on the autosomes of Drosophila species, are the gene category showing the largest divergence between species, and, in some reports, have the highest levels of gene expression polymorphism (Meiklejohn et al. 2003; Ranz et al. 2003; Zhang et al. 2007; Hutter et al. 2008). As reported at the species level in the anophelines (Baker and Russell 2011; Magnusson et al. 2012; Jiang et al. 2015; Papa et al. 2017), and here for the Burkina Faso populations of An. Arabiensis, female-biased genes are also located in excess on the X chromosome whereas male-biased genes are preferentially located on the autosomes (supplementary fig. S15, Supplementary Material online). However, it is not clear whether male-biased genes are driving a faster-X effect in An. Arabiensis, as the strongest evidence for depletion of polymorphic genes is found in larvae—a developmental stage more moderately impacted by sexual dimorphism—followed by females, with males showing the weakest evidence for depletion of X-linked polymorphic genes. Additionally, some categories of female-, but not male-biased genes, are in fact the most polymorphic in expression in An. Arabiensis (supplementary Table S18, Supplementary Material online). Further, adopting the rationale that a high correlation between male and female gene expression reflects strong selective constraints, that is, weak relaxed selection (Meisel et al. 2012), we found that the X chromosome only exhibits higher levels of selective constraints relative to the 3R arm (supplementary figure S16, Supplementary Material online). Therefore, a faster-X effect in An. Arabiensis seems, if anything, more influenced by nonadaptive than adaptive causes, showing substantial general differences with previous findings in Drosophila.
Genomic Hot Spots of Transcriptome Variation
Inversion polymorphism might favor the accumulation of transcriptional differences in particular chromosomes or genomic regions (Marques-Bonet et al. 2004). The lack of information about the location of the breakpoints of the inversions 2Ra and 3Ra precluded a precise assessment of whether expression differences were particularly associated with these inversions or whether there was any position effect at their breakpoints. Nevertheless, we did not find that the chromosomes carrying such inversions, the 2R and 3R, were particularly enriched in differentially expressed genes as it has been found in other chromosome inversion polymorphism systems in Drosophila species (Fuller et al. 2016; Lavington and Kern 2017). One possible explanation is that these inversions had arisen very recently and therefore not enough time has elapsed as for detecting the expected signature of expression differentiation between populations in association with such inversions. Additional work deciphering the evolutionary origin of these inversions will be necessary to test this possibility.
Further, we found evidence of a nonrandom distribution of the top contributing genes to functional enrichment patterns of population differentiation across the genome. This unequal distribution is not only happening at the whole-chromosome scale, but more importantly, it is consistent with an incipient emergence of hot spots of transcriptome differentiation, possibly denoting effects of population divergence due to genetic changes affecting cis-regulatory sequences or local chromatin properties.
The Lability of Sex-Biased Expression Requires a Population Framework
Our analysis of sex-biased gene expression stresses the need for adopting a population framework relative to the categorization of genes according to their sex bias trend. We found that 2,227 (68%) of the female-biased genes and 2,381 (81%) of the male-biased genes in at least one population showed transitions between nonsex bias and sex bias in gene expression—or vice versa—across Burkina Faso populations. This intraspecific lability of sex-biased expression is even more evident when we compared our sets of genes consistently showing male or female bias in relation to a population from Dongola, Sudan (Papa et al. 2017). We found that 0.63% (2/318) and 14.29% (60/420) of the genes in these two categories, respectively, which also show sex-biased expression in the Dongola population, reverse the direction of their bias between studies. Similarly, when we considered jointly genes that show consistent and nonconsistent sex bias, we detected 248 genes exhibiting reversals of the sex bias across Burkina Faso populations, a number significantly higher than the only seven equivalent cases documented between the Dongola population of An. Arabiensis and another of An. gambiae (two-tailed Fisher’s exact test; P = 1.18 × 10−18; Papa et al. 2017). The lability of sex-biased gene expression featured by the Burkina Faso populations is not surprising as different populations might be exposed not only to different abiotic factors but also to very different regimes of sexual selection, which has been proven to be a main factor in the evolution of sex-biased expression in experimentally evolved populations of Drosophila species over very short timespans (Hollis et al. 2014).
Patterns of Sex-Biased Transcriptome Evolution
A previous comparative analysis of four Anopheles species, including An. Arabiensis, found evidence that female-biased genes were more rapidly evolving interspecifically than male-biased genes, in sharp contrast to former findings in other eukaryotes (Ranz et al. 2003; Khaitovich et al. 2005; Voolstra et al. 2007). This peculiarity of the anophelines was interpreted to be the consequence of two factors. First, females in these species are under strong selection pressures, as only this sex blood-feeds, which is essential for oogenesis (Attardo et al. 2003; Mitchell and Catteruccia 2017). Second, anopheline males would be exposed to decreased postcopulatory sexual selection compared with other Diptera, as females typically mate only once during their lifetime (Tripet et al. 2003; Yuval 2006; Carnevale and Robert 2009), which is due to a male-induced mating plug and the subsequent refractory response (Rogers et al. 2009; Mitchell et al. 2015). Nevertheless, an increased rate of evolution at the interspecific level could also result from nonadaptive processes. Finding evidence of genes with female-biased expression being more influenced by positive selection compared with male-biased genes will ultimately require determining whether a significantly higher proportion of between-species divergence to within-species polymorphism exists that is associated with female-biased genes (Meiklejohn et al. 2003; Nuzhdin et al. 2004; Harrison et al. 2012; Yeh et al. 2014).
At the intraspecific level in An. Arabiensis, we have found that overall female-biased genes show similar magnitude of sex-expression ratio variation to that of male-biased genes, although male-limited expressed genes in Bobo and Nanoro showed a significantly higher expression variation than their female-limited counterparts. This overall lack of differences between female- and male-biased genes at the intraspecific level does not refute nor confirm whether intense positive selection is the primary factor driving the fast evolution of female-biased genes in anopheline species. Nevertheless, several observations are not in line with the factors considered as evidence for the preferential action of positive selection on the female transcriptome, which call for additional studies to more reliably discard relaxed selection as a relevant contributing factor to the fast evolution of female- relative to male-biased genes at the interspecific level. First, we have found unambiguous evidence that female-biased genes are more variable than male-biased genes at the interpopulation level when assayed both in males and females, which is compatible with female-biased genes being subject to less stringent selective constraints. Note that this last observation does not contradict the notion reported in other Diptera that gene expression is more variable in males than in females (Meiklejohn et al. 2003; Muller et al. 2011), due to an increased susceptibility to genetic or environmental modifiers (Jiang et al. 2010). In fact, we do observe a larger magnitude of interpopulation differentiation for the same gene category, regardless of its sex bias, when expressed in males compared with in females.
Additionally, there is evidence for accelerated evolution of male-related traits at the postcopulatory level, which can have adaptive potential. In An. Arabiensis, the entire 20E content generated by the male accessory glands is transferred to the female during copulation, whereas other anophelines only transfer half (Mitchell et al. 2015). As this 20E content elicits vital physiological responses in the females, impacting their remating ability (Baldini et al. 2013; Gabrieli et al. 2014), and ultimately their vectorial capacity (Rono et al. 2010), male–male competition could still take place, however, not by minimizing the direct fertilization success of the rival sperm but by impacting indirectly some sperm traits such as longevity and motility, or by modulating the reproductive fitness of the female, in part through its endocrine system (Rogers et al. 2008). In fact, we found significant differences in the magnitude of sex bias among genes expressed in the female spermathecae (AARA002778 and AARA017412), whose orthologs in An. gambiae (an ABC transporter and a peroxidase encoding genes) regulate the lipid reserves for sperm storage and acquisition of sperm motility, respectively (Aitken and Baker 2006; Rogers et al. 2008). Our results add to the increasingly accepted view that the strength of sexual selection operating on males might be higher than previously recognized (Cator et al. 2021).
Conclusions
Intraspecific expression surveys in the anophelines are fundamental to understand the potential of these species for adaptation and trait diversification, which is crucial for vector control. These studies should interrogate the transcriptome response to environmental cues such as insecticides and increasing temperatures while considering sex-specific aspects of the anopheline biology. Equally important, these studies should also address what genetic changes are responsible for differential expression. While expression differences between the sexes primarily reflect their distinct reproductive systems (Assis et al. 2012; Jiang et al. 2015; Grath and Parsch 2016; Papa et al. 2017), intraspecific expression differences for the same sample type under controlled environmental conditions can result from cis- and trans-regulatory changes as well as from copy number variation (Itokawa et al. 2010; Meiklejohn et al. 2014; Faucon et al. 2015; Cardoso-Moreira et al. 2016). Therefore, genome sequencing of these, or similar, populations should help gauge the relative importance of these different types of changes in their contribution to the transcriptome diversification of An. Arabiensis, and consequently to the expansion of its geographic range while transmitting malaria across Africa.
Materials and Methods
Mosquito Collection and Identification
Mosquitoes were collected from July to November 2012. No specific permissions were required for the locations where the collection was performed. Blood-fed and unfed resting females were collected using manual aspirators in human dwellings. Subsequently, An. gambiae s.l. mosquitoes were sorted from other species using morphological identification criteria (Gillies and De Meillon 1968; Gillies and Coetzee 1987). Once in the laboratory, unfed mosquitoes were provided a blood meal on a rabbit. Field collected or laboratory blood-fed females were kept in cages for 4 days until they became gravid and ready to lay eggs. Then, females were individually placed in plastic cups containing ∼25 ml of water for oviposition. Once having laid eggs, they were killed and genomic DNA extracted from legs, followed by species identification by PCR (Santolamazza et al. 2008) as the member species from the An. gambiae s.l. are morphologically indistinguishable. (In Burkina Faso, three members of this complex are present: An. gambiae, A. coluzzii, and An. arabiensis [Costantini et al. 2009].) The PCR is based on the amplification of a ∼200 nt long Short Interpersed Element (SINE 200) locus on the X chromosome (locus S200 X 6.1.) using the flanking primers SINE 200X6.1F (5′-TCGCCTTAGACCTTGGGTTA-3′) and SINE 200X6.1R (5′-CGCTTC AAGAATTCGAGATAC-3′). The reaction mixture contained 2 µl of 10x PCR buffer (Promega, Madison, WI, USA), 1 mM MgCl2, 0.2 mM of each dNTP, 0.3 µM of each primer (Eurogentec, Angers, France), 0.35 unit of Taq DNA Polymerase (Promega) and 2 μl of template DNA, in a total volume of 20 μl. Amplification runs were performed under the following conditions: an initial denaturation step at 94 °C for 10 min followed by 35 cycles of 30 s at 94 °C, 30 s at 54 °C, 60 s at 72 °C, and a final elongation step of 10 min at 72 °C. The amplification products were run in 2% agarose gels (supplementary fig. S1, Supplementary Material online). Progeny of all mosquitoes molecularly identified as An. Arabiensis were pooled according to the study sites and reared under common insectary conditions. Larval offspring were reared at 30 °C and fed with Baby Tetramine fish food. Adults were maintained at 28 °C and 80–85% relative humidity with ad libitum supply of 10% sucrose solution.
Chromosome Karyotyping
Ovaries were extracted from 20 to 22 h post blood-feeding females under a stereoscope (Leica Microsystem GmbH, Wetzlar, Germany) and preserved in Carnoy’s fixative solution (three parts of absolute ethanol and one part of glacial acetic acid). After one night at room temperature, ovaries were kept at −20 °C until chromosomal analysis was performed. Ovaries were first removed from the Carnoy’s fixative solution and placed in a drop of 50% propionic acid on a microscope slide. Then, follicles were carefully separated from each other using fine needles under a stereoscope. After 3–5 min, a cover slip was placed over the preparation and a gentle pressure applied by tapping with a pencil to squash the follicles allowing polytene chromosomes to spread out. The slides were examined under a contrast phase microscope (Motic BA410, Motic China group co., Ltd., Xiamen China) and chromosome arrangements identified using a reference of the cytogenetic map for An. Arabiensis (Coluzzi et al. 2002).
Construction of Isofemale Lines
Naïve males and virgin females were obtained by sexing pupae prior to emergence (Jones 1957), and placing them into separate cages. Individuals were supplied with 10% sucrose during the first 2 days to allow complete reproductive maturation. On the third day after emergence, females were blood-fed and subsequently forced to mate with naïve males (one male for one female). Four days after insemination, females were provided a blood meal and subsequently allowed to lay eggs as described above. Isofemale progenies were then carefully reared separately until F4–F6.
Construction of Homokaryotypic Colonies
Subsets of mosquitoes were blood-fed and allowed to lay eggs individually. Once having laid eggs, mosquitoes were given a second blood meal and their ovaries were extracted 20–22 h later for karyotyping. Specimens carrying the inversion 2Ra, 2Rb, or 3Ra were then identified and progeny of females sharing identical chromosome arrangement were pooled together for mating. After mating and blood-feeding, F1 mosquitoes were placed in individual cups for oviposition, reblood-fed, and their ovaries extracted for karyotyping. As previously, F2 mosquitoes were sorted by their karyotype. The process was repeated until each inversion was completely fixed in the colony as tested by further karyotyping. The resulting colonies were maintained in the insectary of the Institute de Recherche en Sciences de la Santé (IRSS) at Bobo Dioulasso, Burkina Faso, under controlled conditions of 28 °C, 80–85% relative humidity and a 12 h/12 h light–dark cycle without crepuscular transitions. Larvae were fed using TetraMin baby fish food whereas adults were provided ad libitum 10% sucrose solution. Females were allowed to feed on a rabbit for production of eggs, following the approved UCI IACUC protocol number 2012–3035.
RNA Extraction and Sequencing
Stage 4 larvae and 3- to 5-day-old naïve males and virgin nonblood-fed females from homokaryotypic colonies were frozen in TRIzol (Invitrogen) for subsequent total RNA extraction. Specimens were macerated using motorized pestles and the resulting total RNA samples were purified with RNeasy Mini kit (Qiagen). Concentration, quality, and integrity of the RNA samples were assessed using a Qubit 2.0 Fluorometer, a NanoDrop 8000 Spectrophotometer, and with the RNA 6000 Pico kit in a BioAnalyzer 2100 (Agilent Technologies), respectively. Unstranded (female) and stranded (male and larvae), non-polyA, ribodepleted libraries were prepared using the TruSeq Total RNA (Illumina) and the Ribo-Zero (Epicentre) kits. cDNAs were sequenced using 100-bp paired-end reads on an Illumina HiSeq 2500. Size selection was performed using 2% agarose gels on the Sage Sciences Blue Pippin, resulting in a library insert size of ∼480 nt. Three biological replicates were sequenced per biological group, that is, a particular sample type by population combination, being 27 the total number of libraries (i.e., 3 samples × 3 populations × 3 replicates). The set of nine libraries corresponding to a particular sample type was sequenced jointly in two lanes.
Transcriptome Analyses
Quality control and processing of RNA-seq outputs were performed using the pipeline HTStream (https://github.com/s4hts/HTStream; last accessed August 1, 2020), including removal of known Anopheles rRNA-related sequences as present in NCBI, PCR duplicates, adapter sequences, reads shorter than 50 nt, and filtered for low-quality bases using a sliding window approach that required a window size of 10 nt and a minimum quality Qscore of 20. Reads were mapped using the splice-aware STAR Aligner v2.7.3a (Dobin et al. 2013) to the AaraD1 (release 47; April 2020) genome assembly, which was softmasked using RepeatMasker, Dust, and TRF. The AaraD1.11 gene annotation was used. Both annotation and genome assembly were downloaded from VectorBase (as of July 31, 2020). Scaffold location at the chromosomal level was taken as reported (Waterhouse et al. 2020). SAM alignment files were sorted and converted into BAM files using SAMtools v1.10 (Li et al. 2009) and subsequently into count matrices based on the gene annotation used.
Differential expression analysis was performed on those genes deemed as expressed, which implied the detection of at least 1 count per million (cpm) in at least two of the three biological replicates of at least one biological group. Firstly, read counts across sample types for the genes deemed as expressed were scaled using TMM normalization (Robinson and Oshlack 2010) and log2 transformed. Secondly, precision weights were estimated using the voom method (Law et al. 2014) and provided as input for linear models. For each sample type, three linear models were considered corresponding to the three pairwise comparisons between populations. Mathematically, each model was of the form yij=α+ βi+ϵij where, yij denoted the normalized and log2 transformed gene expression in the jth replicate from the ith population, α denoted the baseline expression, βi denoted the average increase (or decrease) in expression with respect to baseline in replicates from the ith population, and ϵij∼ N(0, σ2) denoted an independent and identically distributed error term with mean 0 and variance σ2. The above model was implemented in limma (Ritchie et al. 2015) to test the null hypothesis that β1=β2, that is, for a given sample type, the average expression of a gene was the same in the two populations labeled 1 and 2. We rejected the null hypothesis and considered a gene to be differentially expressed if the FDR (Benjamini and Hochberg 1995) adjusted P-value was < 0.01 and FC≥ 2, that is, more strict criteria than those conventionally used to ensure that the same genes will be identified as differentially expressed even in the case that the number of biological replicates per biological group was increased. Lastly, to determine whether the differentially expressed genes in a given contrast, that is, between two populations of the same sample type were preferentially over- or underexpressed, a goodness-of-fit test was performed under the null hypothesis that p1= p2, where p1 and p2 denote the proportion of differentially expressed genes that are over- and underexpressed, respectively. We rejected the null hypothesis if the FDR-adjusted P-value < 0.01.
Functional Enrichment Analysis
Lists of differentially expressed genes were used to identify enriched pathways using VectorBase’s Metabolic Pathway Enrichment tool (Giraldo-Calderon et al. 2015), and enriched categories of GO terms using g:Profiler (Raudvere et al. 2019), in both cases as of October 2020. For any relevant set of genes, a particular pathway was considered to be enriched for differentially expressed genes if the FDR adjusted P-value was < 0.01. In the case of the GO terms, the g:SCS multiple test correction was applied also at a P-value < 0.01. This more conservative correction does not assume independence among the multiple tests that have to be done as it considers the dependencies of GO terms within the GO term hierarchy.
For the identification of top contributor genes in terms of biological relevance, that is, those genes that contribute to the statistical enrichment across multiple functional terms, we determined whether the joint probability of the gene being differentially expressed and an associated pathway (or GO term) being enriched was greater than or equal to an arbitrary threshold value Qthresh. For a given sample type, the joint probability was defined as Q(g, w) = P(g)×P(w|g). Here, g and w denote a differentially expressed gene and an enriched pathway, respectively. Because there are only three pairwise comparisons of interest, P(g) = N/3, where 0 ≤ N ≤ 3 and P(w|g) = M/N, where 0 ≤ M ≤ N. We set Qthresh to 0.33 or 0.66 depending on whether the gene was differentially expressed in only one of the pairwise comparisons or more. If a gene is differentially expressed in only one comparison, then the maximum permissible value for Q(g, w) = 1 3×1 1 = 0.33. In contrast, if a gene is differentially expressed in ≥2 comparisons, then Q(g, w) = 0.66, which ensures that the pathway is enriched in more than 50% of such cases. For each gene examined, we counted X, that is, the number of pathways (or GO terms) where Q(g, w) ≥ Qthresh and if XY>0.25, where Y denotes the total number of enriched pathways (or GO terms), then the gene was deemed a top contributor gene. The threshold of 0.25 was selected based on the empirical observation that, for most pairwise comparisons, it resulted in a small number of genes (<30) being included in the list of top contributors, thereby facilitating the downstream examination of their properties.
Sex-Biased Expression Analysis
Population-specific linear models, similar to those described above, were developed to identify differentially expressed genes (FC ≥ 2 and FDR-adjusted P-value < 0.01) between males and females. The differentially expressed genes were divided into six groups based on the type (overexpressed in male or female) and magnitude of sex bias. Unlike genes showing sex-limited expression, those preferentially expressed in one of the sexes were further subdivided according to the fold-change (2 ≤ FC <4, moderate sex bias; and FC ≥4, strong sex bias). A FC of 4 was selected based on the empirical observation that, in each of the three populations, the third quartile of fold-changes was ∼4 implying that only 25% of the genes had fold-change exceeding this threshold.
Subsequently, a Wilcoxon rank-sum test was performed to determine whether male- and female-biased genes showed differences in expression variability. First, the six groups were mapped to three categories: exclusive (expressed only in male or female); nonexclusive with strong bias (overexpressed in male or female with FC ≥4); and nonexclusive with moderate bias (overexpressed in male or female and 2 ≤ FC <4). Second, for each differentially expressed gene, the standard deviation for the expression ratio between the sexes, that is, the standard deviation for log2(male/female), was estimated using the empirical Bayes method in limma. Third, for each of the three categories, a two-sided Wilcoxon rank-sum test was performed to test the null hypothesis (H0) that the standard deviation was the same in male- and female-biased genes. Fourth, the U-statistic for Wilcoxon rank-sum test was converted into rank-biserial correlation (Kerby 2014) to estimate the “effect size” or f-s where f and s denote the probability of a male- or female-biased gene, respectively, having a higher standard deviation. Lastly, for a given category, we rejected H0 if the FDR-adjusted P-value (Wilcoxon rank-sum test) <0.01 and effect size ≥0.3. For the effect size, the cutoff value used was in accordance with reported guidelines (Cohen 1992) for considering an effect size to be biologically meaningful rather than just statistically significant, as the latter can simply result from an increase in the power of the test associated with a large sample size. It should be noted that the use of the effect size for Wilcoxon rank-sum test is equivalent to the use of a FC in gene expression analysis in addition to the P-value for identifying genes with nontrivial changes.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
The authors are grateful to Elizabeth Hemming-Schroeder, Anthony James, Colin Meiklejohn, Igor Sharakhov, and Guiyun Yan for providing useful comments on early versions of the manuscript, and to the staff at the IRSS for technical assistance at different stages in establishing and maintaining the mosquito populations. This work was supported by an award from the National Institutes of Health (R21AI096084) and intramural funds from the University of California Irvine to J.M.R.
Data Availability
All raw sequencing data were deposited as part of the NCBI BioProject PRJNA707074.
Contributor Information
Vivek Jayaswal, School of Mathematics and Statistics, The University of Sydney, Sydney, NSW, Australia.
Cyrille Ndo, Institut de Recherche en Sciences de la Santé, Bobo-Dioulasso, Burkina Faso.
Hsiu-Ching Ma, Department of Ecology and Evolutionary Biology, University of California Irvine, Irvine, California, USA.
Bryan D Clifton, Department of Ecology and Evolutionary Biology, University of California Irvine, Irvine, California, USA.
Marco Pombi, Dipartimento di Sanità Pubblica e Malattie Infettive, University of Rome “Sapienza”, Rome, Italy.
Kevin Cabrera, Department of Ecology and Evolutionary Biology, University of California Irvine, Irvine, California, USA.
Anna Cohuet, Unité Mixte de Recherche Maladies Infectieuses et Vecteurs: Ecologie, Génétique, Evolution et Contrôle, University of Montpellier, CNRS, Montpellier, IRD, France.
Karine Mouline, Unité Mixte de Recherche Maladies Infectieuses et Vecteurs: Ecologie, Génétique, Evolution et Contrôle, University of Montpellier, CNRS, Montpellier, IRD, France.
Abdoulaye Diabaté, Institut de Recherche en Sciences de la Santé, Bobo-Dioulasso, Burkina Faso.
Roch Dabiré, Institut de Recherche en Sciences de la Santé, Bobo-Dioulasso, Burkina Faso.
Diego Ayala, Unité Mixte de Recherche Maladies Infectieuses et Vecteurs: Ecologie, Génétique, Evolution et Contrôle, University of Montpellier, CNRS, Montpellier, IRD, France.
José M Ranz, Department of Ecology and Evolutionary Biology, University of California Irvine, Irvine, California, USA.
References
- Aitken RJ, Baker MA. 2006. Oxidative stress, sperm survival and fertility control. Mol Cell Endocrinol. 250(1–2):66–69. [DOI] [PubMed] [Google Scholar]
- Artieri CG, Singh RS. 2010. Demystifying phenotypes: the comparative genomics of evo-devo. Fly (Austin) 4(1):18–20. [DOI] [PubMed] [Google Scholar]
- Assis R, Zhou Q, Bachtrog D. 2012. Sex-biased transcriptome evolution in Drosophila. Genome Biol Evol. 4(11):1189–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attardo GM, Higgs S, Klingler KA, Vanlandingham DL, Raikhel AS. 2003. RNA interference-mediated knockdown of a GATA factor reveals a link to anautogeny in the mosquito Aedes aegypti. Proc Natl Acad Sci USA. 100(23):13374–13379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awono-Ambene HP, Robert V. 1999. Survival and emergence of immature Anopheles arabiensis mosquitoes in market-gardener wells in Dakar, Senegal. Parasite 6(2):179–184. [DOI] [PubMed] [Google Scholar]
- Ayala D, et al. 2017. Chromosome inversions and ecological plasticity in the main African malaria mosquitoes. Evolution 71(3):686–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala D, et al. 2019. Association mapping desiccation resistance within chromosomal inversions in the African malaria vector Anopheles gambiae. Mol Ecol. 28(6):1333–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DA, Russell S. 2011. Role of testis-specific gene expression in sex-chromosome evolution of Anopheles gambiae. Genetics 189(3):1117–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldini F, et al. 2013. The interaction between a sexually transferred steroid hormone and a female protein regulates oogenesis in the malaria mosquito Anopheles gambiae. PLoS Biol. 11(10):e1001695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barredo E, DeGennaro M. 2020. Not just from blood: mosquito nutrient acquisition from nectar sources. Trends Parasitol. 36(5):473–484. [DOI] [PubMed] [Google Scholar]
- Barron MG, et al. 2019. A new species in the major malaria vector complex sheds light on reticulated species evolution. Sci Rep. 9(1):14753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate—a practical and powerful approach to multiple testing. J R Stat Soc B Methodol. 57(1):289–300. [Google Scholar]
- Bimbile Somda NS, et al. 2018. Ecology of reproduction of Anopheles arabiensis in an urban area of Bobo-Dioulasso, Burkina Faso (West Africa): monthly swarming and mating frequency and their relation to environmental factors. PLoS One. 13(11):e0205966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonizzoni M, et al. 2012. Strain variation in the transcriptome of the dengue fever vector, Aedes aegypti. G3 (Bethesda) 2:103–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonizzoni M, et al. 2015. RNA-seq analyses of changes in the Anopheles gambiae transcriptome associated with resistance to pyrethroids in Kenya: identification of candidate-resistance genes and candidate-resistance SNPs. Parasit Vectors. 8:474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso-Moreira M, et al. 2016. Evidence for the fixation of gene duplications by positive selection in Drosophila. Genome Res. 26(6):787–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnevale P, Robert V. 2009. Les Anophèles: Biologie, transmission du Plasmodium et lutte antivectorielle. Marseille: IRD Éditions. [Google Scholar]
- Cator LJ, Wyer CAS, Harrington LC. 2021. Mosquito sexual selection and reproductive control programs. Trends Parasitol. 37(4):330–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ΘCharlesworth B, Coyne JA, Barton NH. 1987. The relative rates of evolution of sex chromosomes and autosomes. Am Nat. 130(1):113–146. [Google Scholar]
- Cohen J. 1992. Statistical power analysis. Curr Dir Psychol Sci. 1(3):98–101. [Google Scholar]
- Coluzzi M, Sabatini A, della Torre A, Di Deco MA, Petrarca V. 2002. A polytene chromosome analysis of the Anopheles gambiae species complex. Science 298(5597):1415–1418. [DOI] [PubMed] [Google Scholar]
- Coolon JD, et al. 2015. Molecular mechanisms and evolutionary processes contributing to accelerated divergence of gene expression on the Drosophila X chromosome. Mol Biol Evol. 32(10):2605–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini C, et al. 2009. Living at the edge: biogeographic patterns of habitat segregation conform to speciation by niche expansion in Anopheles gambiae. BMC Ecol. 9:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabire KR, et al. 2014. Occurrence of natural Anopheles arabiensis swarms in an urban area of Bobo-Dioulasso City, Burkina Faso, West Africa. Acta Trop. 132(Suppl):S35–S41. [DOI] [PubMed] [Google Scholar]
- Dao A, et al. 2010. Reproduction-longevity trade-off in Anopheles gambiae (Diptera: Culicidae). J Med Entomol. 47(5):769–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darwin C. 1871. The descent of man and selection in relation to sex. London: John Murray. [Google Scholar]
- Dobin A, et al. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren H, Parsch J. 2007. The evolution of sex-biased genes and sex-biased gene expression. Nat Rev Genet. 8(9):689–698. [DOI] [PubMed] [Google Scholar]
- Faucon F, et al. 2015. Identifying genomic changes associated with insecticide resistance in the dengue mosquito Aedes aegypti by deep targeted sequencing. Genome Res. 25(9):1347–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feyereisen R, Dermauw W, Van Leeuwen T. 2015. Genotype to phenotype, the molecular and physiological dimensions of resistance in arthropods. Pestic Biochem Physiol. 121:61–77. [DOI] [PubMed] [Google Scholar]
- Fontaine MC, et al. 2015. Mosquito genomics. Extensive introgression in a malaria vector species complex revealed by phylogenomics. Science 347(6217):1258524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller ZL, Haynes GD, Richards S, Schaeffer SW. 2016. Genomics of natural populations: how differentially expressed genes shape the evolution of chromosomal inversions in Drosophila pseudoobscura. Genetics 204(1):287–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrieli P, et al. 2014. Sexual transfer of the steroid hormone 20E induces the postmating switch in Anopheles gambiae. Proc Natl Acad Sci USA. 111(46):16353–16358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillies MT, Coetzee M. 1987. A supplement to the Anophelinae of Africa south of the Sahara (Afrotropical Region). Johannesburg: The South African Institute for Medical Research.
- Gillies MT, De Meillon B. 1968. The Anophelinae of Africa south of the Sahara (Ethiopian Zoogeographical Region). Johannesburg: The South African Institute for Medical Research.
- Giraldo-Calderon GI, et al. ; VectorBase Consortium. 2015. VectorBase: an updated bioinformatics resource for invertebrate vectors and other organisms related with human diseases. Nucleic Acids Res. 43(Database issue):D707–D713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grath S, Parsch J. 2016. Sex-biased gene expression. Annu Rev Genet. 50:29–44. [DOI] [PubMed] [Google Scholar]
- Harrison PW, Wright AE, Mank JE. 2012. The evolution of gene expression and the transcriptome-phenotype relationship. Semin Cell Dev Biol. 23(2):222–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollis B, Houle D, Yan Z, Kawecki TJ, Keller L. 2014. Evolution under monogamy feminizes gene expression in Drosophila melanogaster. Nat Commun. 5:3482. [DOI] [PubMed] [Google Scholar]
- Huong LT, et al. 2019. Chemical compositions and mosquito larvicidal activities of essential oils from piper species growing wild in Central Vietnam. Molecules 24(21):3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutter S, Saminadin-Peter SS, Stephan W, Parsch J. 2008. Gene expression variation in African and European populations of Drosophila melanogaster. Genome Biol. 9(1):R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim SS, Riveron JM, Stott R, Irving H, Wondji CS. 2016. The cytochrome P450 CYP6P4 is responsible for the high pyrethroid resistance in knockdown resistance-free Anopheles arabiensis. Insect Biochem Mol Biol. 68:23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingham VA, Wagstaff S, Ranson H. 2018. Transcriptomic meta-signatures identified in Anopheles gambiae populations reveal previously undetected insecticide resistance mechanisms. Nat Commun. 9(1):5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itokawa K, et al. 2010. Genomic structures of Cyp9m10 in pyrethroid resistant and susceptible strains of Culex quinquefasciatus. Insect Biochem Mol Biol. 40(9):631–640. [DOI] [PubMed] [Google Scholar]
- Izquierdo A, et al. 2019. Evolution of gene expression levels in the male reproductive organs of Anopheles mosquitoes. Life Sci Alliance. 2(1):e201800191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang PP, Hartl DL, Lemos B. 2010. Y not a dead end: epistatic interactions between Y-linked regulatory polymorphisms and genetic background affect global gene expression in Drosophila melanogaster. Genetics 186(1):109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Biedler JK, Qi Y, Hall AB, Tu Z. 2015. Complete dosage compensation in Anopheles stephensi and the evolution of sex-biased genes in mosquitoes. Genome Biol Evol. 7(7):1914–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CM, et al. 2013. The dynamics of pyrethroid resistance in Anopheles arabiensis from Zanzibar and an assessment of the underlying genetic basis. Parasit Vectors. 6:343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CM, et al. 2012a. Additional selection for insecticide resistance in urban malaria vectors: DDT resistance in Anopheles arabiensis from Bobo-Dioulasso, Burkina Faso. PLoS One 7(9):e45995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones FC, et al. ; Broad Institute Genome Sequencing Platform & Whole Genome Assembly Team. 2012b. The genomic basis of adaptive evolution in threespine sticklebacks. Nature 484(7392):55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JC. 1957. A simple method for sexing living Anopheles larvae (Diptera, Culicidae). Ann Entomol Soc Am. 50(1):104–106. [Google Scholar]
- Kayserili MA, Gerrard DT, Tomancak P, Kalinka AT. 2012. An excess of gene expression divergence on the X chromosome in Drosophila embryos: implications for the faster-X hypothesis. PLoS Genet. 8(12):e1003200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerby DS. 2014. The simple difference formula: an approach to teaching nonparametric correlation. Compr Psychol. 3:1. doi:10.2466/11.It.3.1. [Google Scholar]
- Khaitovich P, et al. 2005. Parallel patterns of evolution in the genomes and transcriptomes of humans and chimpanzees. Science 309(5742):1850–1854. [DOI] [PubMed] [Google Scholar]
- Lai YT, et al. 2019. Standing genetic variation as the predominant source for adaptation of a songbird. Proc Natl Acad Sci USA. 116(6):2152–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laporte V, Charlesworth B. 2002. Effective population size and population subdivision in demographically structured populations. Genetics 162(1):501–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavington E, Kern AD. 2017. The effect of common inversion polymorphisms In(2L)t and In(3R)Mo on patterns of transcriptional variation in Drosophila melanogaster. G3 (Bethesda) 7(11):3659–3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, Smyth GK. 2014. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15(2):R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawniczak MK, et al. 2007. Mating and immunity in invertebrates. Trends Ecol Evol. 22(1):48–55. [DOI] [PubMed] [Google Scholar]
- Li H, et al. ; 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16):2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llopart A. 2012. The rapid evolution of X-linked male-biased gene expression and the large-X effect in Drosophila yakuba, D. santomea, and their hybrids. Mol Biol Evol. 29(12):3873–3886. [DOI] [PubMed] [Google Scholar]
- Llopart A. 2018. Faster-X evolution of gene expression is driven by recessive adaptive cis-regulatory variation in Drosophila. Mol Ecol. 27(19):3811–3821. [DOI] [PubMed] [Google Scholar]
- Lucas ER, et al. ; Anopheles gambiae 1000 Genomes Consortium. 2019. Whole-genome sequencing reveals high complexity of copy number variation at insecticide resistance loci in malaria mosquitoes. Genome Res. 29(8):1250–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusson K, et al. 2012. Demasculinization of the Anopheles gambiae X chromosome. BMC Evol Biol. 12:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Main BJ, et al. 2016. The genetic basis of host preference and resting behavior in the major African Malaria Vector, Anopheles arabiensis. PLoS Genet. 12(9):e1006303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques-Bonet T, et al. 2004. Chromosomal rearrangements and the genomic distribution of gene-expression divergence in humans and chimpanzees. Trends Genet. 20(11):524–529. [DOI] [PubMed] [Google Scholar]
- Meiklejohn CD, Coolon JD, Hartl DL, Wittkopp PJ. 2014. The roles of cis- and trans-regulation in the evolution of regulatory incompatibilities and sexually dimorphic gene expression. Genome Res. 24(1):84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiklejohn CD, Parsch J, Ranz JM, Hartl DL. 2003. Rapid evolution of male-biased gene expression in Drosophila. Proc Natl Acad Sci USA. 100(17):9894–9899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel RP, Malone JH, Clark AG. 2012. Faster-X evolution of gene expression in Drosophila. PLoS Genet. 8(10):e1003013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell SN, Catteruccia F. 2017. Anopheline reproductive biology: impacts on vectorial capacity and potential avenues for malaria control. Cold Spring Harb Perspect Med. 7(12):a025593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell SN, et al. 2015. Mosquito biology. Evolution of sexual traits influencing vectorial capacity in Anopheline mosquitoes. Science 347(6225):985–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller L, et al. 2011. Population transcriptomics of Drosophila melanogaster females. BMC Genomics 12:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neafsey DE, et al. 2010. SNP genotyping defines complex gene-flow boundaries among African malaria vector mosquitoes. Science 330(6003):514–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neafsey DE, et al. 2015. Mosquito genomics. Highly evolvable malaria vectors: the genomes of 16 Anopheles mosquitoes. Science 347(6217):1258522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ΘNuzhdin SV, Wayne ML, Harmon KL, McIntyre LM. 2004. Common pattern of evolution of gene expression level and protein sequence in Drosophila. Mol Biol Evol. 21(7):1308–1317. [DOI] [PubMed] [Google Scholar]
- Papa F, et al. 2017. Rapid evolution of female-biased genes among four species of Anopheles malaria mosquitoes. Genome Res. 27(9):1536–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ΘPowell JR. 2018. Genetic variation in insect vectors: death of typology? Insects 9(4):139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranson H, et al. 2002. Evolution of supergene families associated with insecticide resistance. Science 298(5591):179–181. [DOI] [PubMed] [Google Scholar]
- Ranz JM, Castillo-Davis CI, Meiklejohn CD, Hartl DL. 2003. Sex-dependent gene expression and evolution of the Drosophila transcriptome. Science 300(5626):1742–1745. [DOI] [PubMed] [Google Scholar]
- Raudvere U, et al. 2019. g: profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47(W1):W191–W198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, et al. 2015. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43(7):e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivero A, Vézilier J, Weill M, Read AF, Gandon S. 2010. Insecticide control of vector-borne diseases: when is insecticide resistance a problem? PLoS Pathog. 6(8):e1001000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, Oshlack A. 2010. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11(3):R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers DW, et al. 2008. Molecular and cellular components of the mating machinery in Anopheles gambiae females. Proc Natl Acad Sci USA. 105(49):19390–19395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers DW, et al. 2009. Transglutaminase-mediated semen coagulation controls sperm storage in the malaria mosquito. PLoS Biol. 7(12):e1000272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rono MK, Whitten MM, Oulad-Abdelghani M, Levashina EA, Marois E. 2010. The major yolk protein vitellogenin interferes with the anti-plasmodium response in the malaria mosquito Anopheles gambiae. PLoS Biol. 8(7):e1000434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux O, Robert V. 2019. Larval predation in malaria vectors and its potential implication in malaria transmission: an overlooked ecosystem service? Parasit Vectors. 12(1):217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ΘSacher RM. 1971. A mosquito larvicide with favorable environmental properties. Mosquito News 31:513–516. [Google Scholar]
- Santolamazza F, et al. 2008. Insertion polymorphisms of SINE200 retrotransposons within speciation islands of Anopheles gambiae molecular forms. Malar J. 7(1):163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simma EA, et al. 2019. Genome-wide gene expression profiling reveals that cuticle alterations and P450 detoxification are associated with deltamethrin and DDT resistance in Anopheles arabiensis populations from Ethiopia. Pest Manag Sci. 75(7):1808–1818. [DOI] [PubMed] [Google Scholar]
- Tripet F, Toure YT, Dolo G, Lanzaro GC. 2003. Frequency of multiple inseminations in field-collected Anopheles gambiae females revealed by DNA analysis of transferred sperm. Am J Trop Med Hyg. 68(1):1–5. [PubMed] [Google Scholar]
- Vicoso B, Charlesworth B. 2009. Effective population size and the faster-X effect: an extended model. Evolution 63(9):2413–2426. [DOI] [PubMed] [Google Scholar]
- Voolstra C, Tautz D, Farbrother P, Eichinger L, Harr B. 2007. Contrasting evolution of expression differences in the testis between species and subspecies of the house mouse. Genome Res. 17(1):42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse RM, et al. 2020. Evolutionary superscaffolding and chromosome anchoring to improve Anopheles genome assemblies. BMC Biol. 18(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White BJ, Collins FH, Besansky NJ. 2011. Evolution of Anopheles gambiae in relation to humans and malaria. Annu Rev Ecol Evol Syst. 42(1):111–132. [Google Scholar]
- WHO. 2020. World malaria report 2020: 20 years of global progress and challenges. Geneva: World Health Organization.
- Yeh SD, et al. 2014. Functional divergence of the miRNA transcriptome at the onset of Drosophila metamorphosis. Mol Biol Evol. 31(10):2557–2572. [DOI] [PubMed] [Google Scholar]
- Yuval B. 2006. Mating systems of blood-feeding flies. Annu Rev Entomol. 51:413–440. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Sturgill D, Parisi M, Kumar S, Oliver B. 2007. Constraint and turnover in sex-biased gene expression in the genus Drosophila. Nature 450(7167):233–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw sequencing data were deposited as part of the NCBI BioProject PRJNA707074.