

Abstract
To capitalize on the immunogenic effects of radiation in cancer treatment, we hypothesized it may be advantageous to deliver radiation to all tumor sites. Using targeted radionuclide therapy (TRT) to deliver radiation semi-selectively to tumors, we tested an approach to enhance response to immune checkpoint inhibitors (ICIs) by TRT. NM600 is a theranostic alkylphosphocholine analog that preferentially accumulates in nearly all tumor types. NM600 chelates a radioisotope and semi-selectively delivers this to tumor microenvironments (TME) for therapeutic or diagnostic applications. Using serial 86Y-NM600 PET/CT imaging, we estimate the dosimetry of 90Y-NM600 in immunologically “cold” syngeneic murine models that do not respond to ICIs alone. We observed strong therapeutic efficacy and report optimal dose (2.5 – 5 Gy) and sequencing for 90Y-NM600 in combination with ICIs. Following combined treatment, 45–66% of mice exhibited complete response and tumor-specific T cell memory, compared to 0% with 90Y-NM600 or ICIs alone. This required expression of STING in tumor cells. Combined TRT and ICI activated production of pro-inflammatory cytokines in the TME, promoted tumor infiltration by and clonal expansion of effector CD8+ T cells, and reduced spontaneous metastases. In mice bearing multiple tumors, combining TRT with moderate-dose (12 Gy) external beam radiotherapy (EBRT) targeting a single tumor further augmented response to ICIs compared to combination of ICIs with either TRT or EBRT alone. Safety of TRT was confirmed in a canine study. Low-dose TRT enables a safe and rapidly translatable approach to promoting response to ICIs for potentially any tumor type in any location.
One Sentence Summary:
Combination low dose targeted radionuclide therapy and immune checkpoint inhibition enhances complete response rates in preclinical tumors models.
Introduction
Immune checkpoint inhibitors [ICIs; e.g. anti-CTLA-4, anti-PD-1, or anti-PD-L1] modulate tumor tolerance among immune cells and are used clinically to treat a growing variety of cancers. Although a subset of patients treated with ICIs experience durable and/or complete tumor regression (1, 2), response is more limited in most patients and ICIs are not typically effective against immunologically “cold” tumors characterized by poor de novo anti-tumor immunity and limited T cell infiltration (3–7). Preclinical studies demonstrate that external beam radiotherapy (EBRT) targeting a single tumor can enhance response to ICIs at non-radiated tumor sites (8–13). In this setting, EBRT elicits an in situ tumor vaccination effect, converting the targeted tumor into a nidus for enhanced tumor antigen presentation and augmented T cell sensitization, resulting in greater diversity of antigen recognition by T cells (14–17). A recent clinical study demonstrated this in situ vaccine effect in patients treated with a combination of EBRT and anti-CTLA-4 (18). However, randomized clinical trials have not yet demonstrated whether locally delivered EBRT can augment the systemic response to ICIs in settings of metastatic disease (19).
The mechanisms whereby radiation elicits in situ vaccination are diverse and vary in their response to dose. Radiation triggers immunogenic tumor cell death with increasing dose as a function of the linear-quadratic model (20–22) and elicits a type I interferon (IFN) response in surviving tumor cells via the cyclic GMP-AMP Synthase (cGAS) – Stimulator of Interferon Genes (STING) pathway (23–27). Activation of this type I IFN response appears to be necessary for the in situ vaccine effect of EBRT and is optimally activated at moderate doses (e.g. 8–12 Gy) and with hypo-fractionated regimens (e.g. 8 Gy x3 fractions) (25). In addition to these high and moderate dose-dependent mechanisms, radiation also elicits immunomodulatory effects in the tumor microenvironment (TME) at lower doses. Lymphocytes are among the most radiosensitive mammalian cells, a dose of only 3 Gy triggers apoptosis in 90% of naïve lymphocytes within 2–8 hours after irradiation (28). Activated circulating T effector cells and immune suppressive regulatory T cells (Tregs) are slightly less sensitive to radiation when compared to naïve T cells but these are still quite sensitive relative to most tumor cells (29–31). On the other hand, tumor infiltrating T cells and tissue-resident memory T cells exhibit radioresistance that is influenced by the microenvironment in which they exist (32). Low dose radiation (2–5 Gy) can also trigger local release of inflammatory cytokines and damage-associated molecular patterns that increase immune cell trafficking and activation (33, 34). By activating inflammatory cytokine signaling, low dose radiation may reconstitute a TME that is more favorable to the propagation of anti-tumor immunity. Interestingly, these immunomodulatory effects of low dose radiation show little dependence on dose rate (33–36).
Progressing tumors often contain infiltrating T cells that are capable of killing tumor cells if removed from the suppressive milieu of the TME (37). Tumor progression in the face of these tumor-specific lymphocytes results from local and systemic effects of the immunosuppressive TME and the limited susceptibility of tumor cells to immune-mediated killing. While ICIs may overcome some of these barriers, they alone are not adequate in patients with immunologically cold tumors. Similarly, although the in situ vaccine effect of focal EBRT may prime a more diversified T cell response, these additional tumor-specific T cell clones will circulate to non-radiated tumors and encounter suppressive TMEs and poorly susceptible tumor cells that, in most patients with progressive cancers, are demonstrably capable of evading eradication by tumor- specific T cells. We therefore hypothesized that the response to ICIs or ICIs + an EBRT in situ vaccination could be improved by delivering low dose radiation to all tumor sites for the purpose of immunomodulating these TMEs to 1) promote systemic propagation of anti-tumor immunity and 2) facilitate tumor destruction at all sites by the actions of immune effector cells.
For most patients with metastatic cancers, it is not feasible to deliver low dose immunomodulatory radiation to all tumors using EBRT due to the inability to target radiographically occult sites and the toxicity, lymphopenia and associated systemic immune- suppression that result from large field or whole body radiation (38). Therefore, to test whether delivering low dose immunomodulatory radiation to all tumor sites might enhance response to ICIs, we utilized targeted radionuclide therapy (TRT). TRT is a growing class of cancer therapeutics that employs a vector or biologic mechanism to carry a radionuclide to tumor, where, upon decay, it deposits radiation to the TME. Because of tumor selectivity, TRT affords safe delivery of radiation to all tumor sites, even in settings of metastatic disease. TRTs are used for the treatment of various malignancies and are being developed for nearly every type of cancer (39, 40). Yet, it is unknown what effect TRT may have on the TME or response to ICIs.
We have pioneered the development of a new class of TRT using alkylphosphocholine analogs. Tumor cells contain an over-abundance of alkylphosphocholines (41) and we have shown that analogs to these display nearly universal and semi-selective uptake and retention in mammalian tumor cells, regardless of anatomic location or tumor type (40, 42, 43). NM600 is an alkylphosphocholine analog that can chelate a variety of radiometals including yttrium (Y). Among available TRT agents, NM600 is uniquely well suited for highly translational preclinical studies, in part because its robust uptake across mammalian tumor types enables testing of generalizable mechanisms. In contrast to most TRT applications in cancer, which aim to deliver a tumoricidal dose of radiation (44, 45), we set out to evaluate immunomodulatory effects of low dose radiation (~2–5 Gy) from TRT. Prior studies demonstrate that 90Y-NM600 can deliver this dose range to tumor sites in mice bearing syngeneic tumors without causing bone marrow suppression or systemic lymphopenia (46, 47). Here, we use 90Y-NM600 to test the hypothesis that low dose TRT can enhance response at all tumor sites to ICIs alone or in combination with an EBRT in situ vaccine to a single site. We evaluate the impact of TRT dose and timing in this therapeutic interaction and determine the immunologic effects of TRT on tumor cells and the TME as well as the impact of these on the anti-tumor immune response.
Results
90Y-NM600 dosimetry and toxicity studies.
NM600 is a “theranostic” TRT capable of chelating radiometals and delivering these to tumor for either diagnostic imaging (e.g. from positron-emitting 86Y-NM600) or therapeutic delivery of radiation (e.g. from beta-particle emitting 90Y-NM600). This dual capacity enables calculation of host- and tumor-specific dose distribution that is resolved over space and time for a given radionuclide (42). For this, we used a Monte Carlo-based dosimetry calculation platform that we developed to determine the distribution and timing of radiation dose delivery to tumor and normal tissues following TRT administration in preclinical or clinical settings (39, 41, 46–48). To begin, we evaluated the uptake of NM600 in tumors and normal tissues using serial positron emission tomography/computed tomography (PET/CT) imaging at 3, 24, 48 h after IV injection of 86Y-NM600. Maximum intensity projection PET images demonstrated semi-selective uptake and retention of NM600 in tumor for B78, NXS2 and 4T1 murine cancer models (Figure 1A). Pharmacokinetic biodistribution studies (Figure 1B) revealed increased uptake and retention over time in tumor compared with other normal tissues, with the exception of somewhat greater non-specific uptake by the liver (due to hepatic excretion of NM600) in all 3 models, and in the kidney for NXS2. Dosimetry calculations (Figure 1C) revealed significantly increased (p < 0.05) dose delivered to tumor compared to lymphatic normal tissue organs (spleen, bone marrow). The total delivered dose was dependent on tumor size with greater dose per injected activity delivered to larger compared to smaller tumors (Figure S1).
Figure 1. Imaging, uptake, and dosimetry of 86/90Y-NM600.

(A) Mice with a single (~150–250 mm3) B78, NXS2, or 4T1 flank tumor (arrow) were serially imaged using PET/CT following IV injection of 86Y-NM600. (B) Image analysis was performed to determine uptake of injected activity per gram of tissue (%IA/g) over time (at 3, 24, and 48 hr) and shows progressive accumulation of NM600 in tumor and progressive washout from normal tissues. (C) Tissue dosimetry was performed using Monte Carlo methods. (N = 3. Significance determined with one-way ANOVA with Tukey correction for multiple comparisons testing for tissue dosimetry. *P < 0.05, **P < 0.01, ***P<0.001)
Prior toxicity studies by our group demonstrated that there was no difference in white blood cell (WBC) or lymphocyte counts in C57Bl/6 mice with 50 μCi of 90Y-NM600 compared to cold NM600 compound that is not radiolabeled (45). However, with higher activity of 90Y-NM600, 250 μCi, there was a temporary depletion of WBCs and lymphocytes with rebound by 28 days (45). For our studies we examined the effect of the addition of anti-CTLA-4 to 50 μCi of 90Y-NM600 on systemic toxicity in C57Bl/6 mice with B78 melanoma tumors using serial CBC measurements from pretreatment to 35 days post TRT administration. In contrast to our prior studies, we did find modest but statistically significant (p < 0.05) WBC and lymphocyte count reduction at day 7 post TRT administration compared to cold compound, but this difference disappeared by day 14 post TRT administration (Figure S2A.B). No differences in hemoglobin, red blood cell counts, platelets, or animal weight were noted between mice treated cold NM600, anti-CTLA4 + cold NM600, or 50 μCi of 90Y-NM600 + anti-CTLA4 (Figure S2C–F). Histological analysis of liver, spleen, marrow, small intestine, and kidney at day 35 did not show any gross pathologic evidence of toxicity in any treatment group (Figure S3)
Low dose 90Y-NM600 enhances response to ICIs.
To begin testing whether low dose TRT can enhance response to ICI therapies, C57Bl/6 mice bearing B78 melanoma tumors were randomized to receive 0, 25, 50, or 100 μCi of 90Y-NM600 (delivering 0, 1.25, 2.5, or 5.0 Gy to the B78 tumor, respectively) either alone or in combination with anti-CTLA-4 alone, or with dual ICI (anti-CTLA-4 in combination with anti-PD-L1) in some experiments (Figure 2A). Tumor growth curves demonstrated significantly improved (p < 0.05) tumor response and increased survival with the combination of 50 or 100 μCi 90Y-NM600 and anti-CTLA-4, compared with either 90Y-NM600 or anti-CTLA-4 alone (Figure 2B–D). However, there was no significant (p =ns) benefit from increasing the injected TRT activity from 50 to 100 μCi; thus for our subsequent studies we used 50 μCi as our standard injected activity. No mice receiving the combination of 90Y-NM600 and anti-CTLA-4 exhibited weight loss, lethargy, hunched posture, or other visible toxicity and no mice required euthanasia for treatment-related toxicity.
Figure 2. Low-dose TRT enhances response to ICI.

(A) Mice with a single (~70–150 mm3) B78 flank tumors were treated with combinations of TRT (90Y-NM600), anti-CTLA-4 (C4), anti-PD-L1 (PDL1), or vehicle-only (VO) control injections. (B-D) Tumor response and animal survival after treatment with TRT and ICI are shown as a function of TRT dose. The mean tumor growth for each group is shown in B, the growth curves for each individual mouse in each group are shown in C and the survival for all groups in D. (E-G) Tumor response and survival based on ICI timing in relation to TRT are shown for tumor growth by group (E), by individual mouse (F), and survival by group (G). (H-J). While these data suggest comparable effect of day 4 single dose and our three-dose regimen, we have used the three-dose regimen as a standard so that our data can be evaluated in the context of prior studies that have used this regimen together with EBRT (8, 48). Tumor growth and survival for single vs dual ICI are shown for tumor growth by group (H), by individual mouse (I), and survival by group (J). In this tumor model, response and survival after treatment with anti-PD-L1 or combined anti-CTLA-4 and anti-PD-L1 is not significantly (p = ns) different from that with VO. In mice with CR to treatment in the TRT dose response studies (K), C4 timing response studies (L), and dual checkpoint studies (M), we tested for immunologic memory to B78 re-challenge and for those mice that rejected the B78 re-challenge, we then tested for rejection of subsequent B16 and Panc02 challenge as shown (K-M, Ø indicates no mice rejected challenge with symbol color representing challenge type). (N = 6 per replicate, 2 replicates). Significance determined by linear mixed effects regression analysis and two-way ANOVA with Tukey multiple comparisons testing for tumor growth [significant differences, p < 0.05, demarcated by * with the color of the asterisk representing which group from which the sample is significantly different], Kaplan–Meier with Log-rank testing for survival analysis, Chi-square contingency testing for immune memory shown in Table S1–3. * P < 0.05)
We examined the optimal timing of anti-CTLA-4 administration relative to 90Y-NM600 with single doses of anti-CTLA-4 administered from 6 days prior to 12 days after 90Y-NM600 injection. We also compared single dosing to three adjuvant anti-CTLA-4 doses, a regimen commonly used in preclinical studies (8, 48). All anti-CTLA-4 sequencing regimens led to increased tumor response and improved survival compared to vehicle only control groups (Figure 2E–F). Next, we tested the capacity of low dose 90Y-NM600 to augment the efficacy of dual checkpoint blockade with anti-CTLA-4 and anti-PD-L1, which is the current clinical standard of care for metastatic melanoma. Addition of anti-PD-L1 did not have any apparent therapeutic benefit on tumor growth (Fig. 2H–J) or on survival (Figure 2I) in this tumor model. However, in mice bearing B78 melanoma, treatment with 90Y-NM600 + anti- CTLA-4 + anti-PD-L1 led to a significant improvement (p < 0.05) in survival compared to the group receiving dual checkpoint blockade alone (Figure 2I).
Only mice receiving combination TRT + ICI treatment exhibited durable complete responses (CR). Among mice treated with either 50 or 100 μCi in combination with anti-CTLA-4 in the TRT dose study, we observed complete and durable tumor response (cure) in 66% and 50%, respectively (Figure 2K). All mice exhibiting CR subsequently demonstrated tumor-specific immune memory by rejecting re-engraftment with the same B78 tumor line they had cleared as well as the parental B16 melanoma tumor line (which shares common tumor antigens) but not the unrelated syngeneic Panc02 tumor line (Figure 2K). CRs and tumor specific immune memory were observed in all groups receiving 50 μCi TRT + anti-CTLA-4 independent of sequencing (Figure 2L). Therefore, we chose to continue with the previously established three- dose adjuvant regimen for subsequent studies. CRs and tumor-specific immune memory were also observed in TRT + single or dual checkpoint blockade (Figure 2M).
To evaluate the generalizability of our observations in B78 melanoma and confirm the impact of low dose TRT on response to ICIs, we tested these treatments in additional syngeneic tumor models from different murine strains. We examined the efficacy of 50 μCi 90Y-NM600 + anti- CTLA-4 in BALB/c or A/J mice bearing 4T1 breast cancer or NXS2 neuroblastoma, respectively. The combination of 90Y-NM600 + anti-CTLA-4 treatment led to decreased tumor growth and significantly improved survival (p < 0.0001) in both NXS2 and 4T1 compared to cohorts of mice receiving either treatment alone (Figure 3A–F). Moreover, a significantly increased CR rate (p < 0.05) was seen with TRT + anti-CTLA-4 treatment in both the NXS2 (45%) and 4T1 (60%) models compared to 0% in all other treatment groups (Figure 3G). In both models, mice with CRs, were re-challenged at day 90 following treatment initiation with the same tumor line they had eradicated. Consistent with the development of immunologic memory, these mice rejected re-engraftment compared to age matched naïve controls engrafted with the same cells (Figure 3H). Mice bearing 4T1 breast tumors consistently and spontaneously develop metastatic disease. In a cohort of mice bearing 4T1 breast cancer tumors, we quantified the number of metastatic lung nodules at day 30 following treatment with 90Y-NM600 and/or anti-CTLA-4. In these mice, the combination of 90Y-NM600 + anti-CTLA-4 resulted in significantly (p < 0.01) reduced metastases compared to either treatment alone (Figure 3I–J). In addition to examining the efficacy of 50 μCi 90Y-NM600 + anti- CTLA-4 in other tumor models, we also tested for potential gender differences in male mice with B78 tumors. Similar to results in female mice with B78 melanoma, 50 μCi 90Y-NM600 + anti- CTLA-4 + anti- CTLA-4 resulted in improved tumor growth delay and survival compared to 50 μCi 90Y-NM600 + IgG2a isotype control, cold NM600 + anti-CTLA4, or cold NM600 + IgG2a isotype control (Figure S4).
Figure 3. TRT enhances efficacy of ICI in 4T1 breast cancer and NXS2 neuroblastoma models.
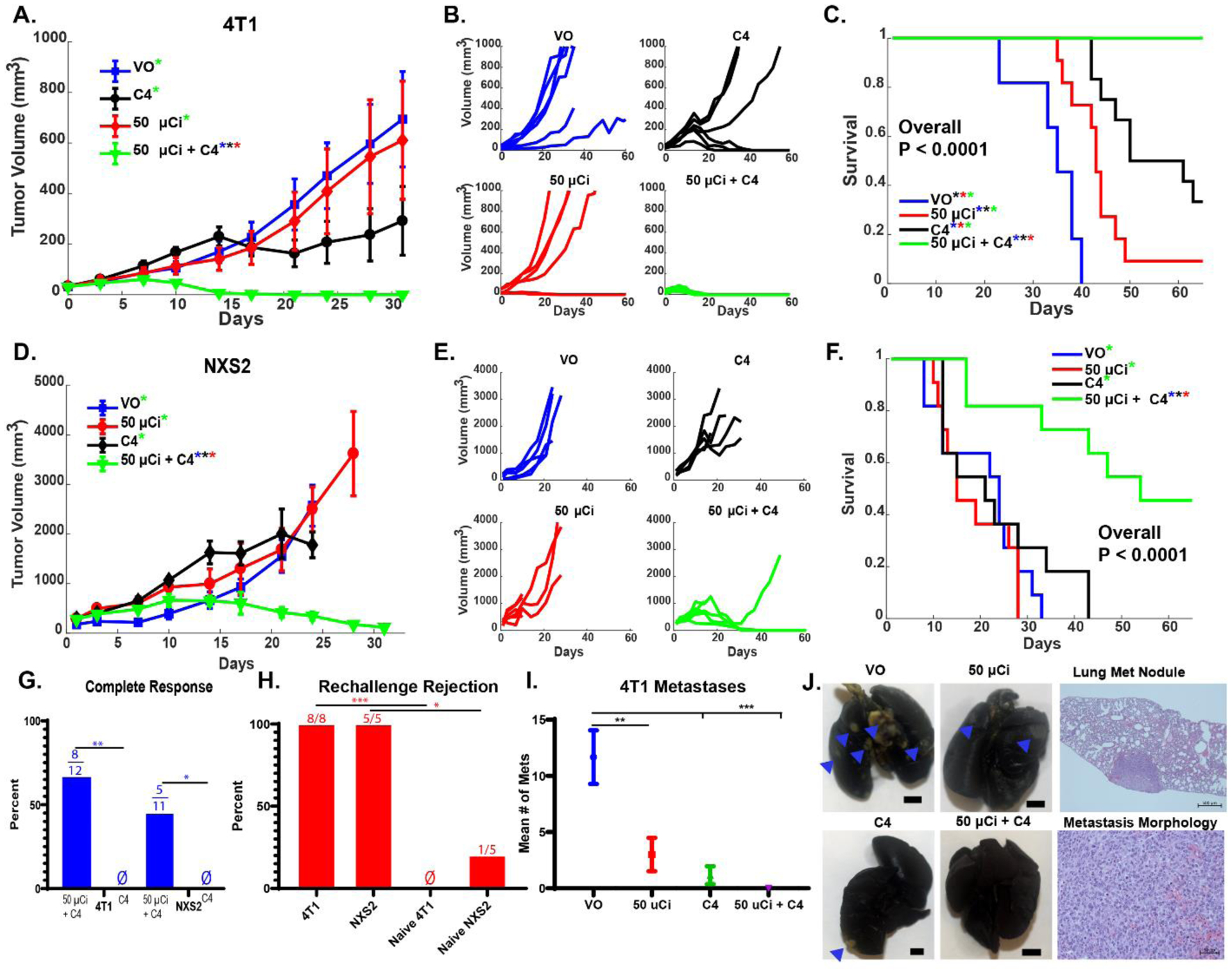
Mice with a single (~70–150 mm3) 4T1 or NXS2 flank tumor were treated with VO, 50 μCi TRT (90Y-NM600), anti-CTLA-4 (C4), or 50 μCi TRT + C4. Tumor response, by group, by individual animal, and animal survival are shown for 4T1 (in A, B and C) and for NXS2 (in D, E and F). Mice with complete response to treatment with TRT + C4 (G, Ø indicates no complete responders) to initial treatment were rechallenged with the same tumor they initially rejected, and rejection rates were compared to those for naïve controls (H, Ø indicates no rejection). The 4T1 model develops spontaneous lung metastases and the number of discrete metastases at Day 25 was compared by treatment group (I). (N = 6 per replicate, 12 total). Representative lungs are shown by gross examination for the 4 treatment groups shown in I, to demonstrate what the 4T1 metastases look like in India-ink injected lungs (with blue arrows indicating 4T1 nodules that are visible by gross inspection; along with a low power photomicrograph (power of 50X) to show a lung metastases at low power, and a higher power photomicrograph (power of 400X), to show the tumor morphology within one such 4T1 nodule (J). [Significance determined by linear mixed effects regression analysis with Tukey multiple comparisons testing for tumor growth (significant differences, p < 0.05, demarcated by * with the color of the asterisk representing which group from which the sample is significantly different), Kaplan–Meier with Log-rank testing and Cox Regression for survival analysis, Chi-square contingency testing for complete response rate and immune memory, and a two-way ANOVA for number of metastases. *P < 0.05, **P < 0.01, ***P<0.001]
Response to 90Y-NM600 + anti-CTLA-4 is radiation, and T cell dependent.
One advantage of radiation delivery via TRT is the ability to treat all sites of metastatic disease including those that are radiographically occult. While low doses of radiation (2–3 Gy) can be safely delivered with EBRT to large fields or to the whole-body, this would risk systemic lymphocyte depletion and systemic immune suppression that can hinder the development of anti- tumor immunity (38). Therefore, we compared the therapeutic efficacy of 2.5 Gy of radiation delivered to a single tumor via targeted EBRT, whole mouse EBRT, or systemically delivered 90Y-NM600, either alone or in combination with anti-CTLA-4 (Figure 4A, Figure S5). Low dose tumor targeted EBRT + anti-CTLA-4 significantly (p = 0.0026) increased tumor response compared to tumor targeted EBRT alone (Figure 4B). This effect was lost when whole mouse EBRT was delivered at the same dose (Figure 4C). This is consistent with a systemic immunosuppressive effect of whole-body radiation eliminating the potential for cooperative therapeutic interaction with ICI. On the other hand, systemically administered 90Y-NM600 (Figure 4D) maintained a similar response profile to tumor targeted EBRT (Figure 4B) in this single tumor model. Tumor response to this combination of TRT and ICI was dependent upon T cells and was abrogated by T cell depletion (Figure 4E, Figure S6 [depletion confirmation]), consistent with a therapeutic effect that is immune mediated, at least in part. In contrast to dose equivalent whole mouse EBRT, we have previously reported that systemically administered 90Y-NM600 does not result in systemic lymphopenia until moderate dose ranges are reached (~12.5 Gy tumor dose) (46, 47). Importantly, nonradioactive NM600 alone did not elicit antitumor activity either alone or in combination with anti-CTLA-4 (Figure 4F).
Figure 4. Tumor targeted radiation is necessary for enhanced efficacy of RT + ICI.

(A) C57Bl/6 mice with a single (~90 mm3) B78 tumor were treated with ~2.5 Gy RT delivered via tumor directed EBRT, whole mouse (WM) EBRT, or TRT (90Y-NM600) either alone or with anti-CTLA-4 (C4). (B-D) Tumor response was tracked for 30 days. (E) To test the effects of T cell depletion on tumor response, anti-CD4 and anti-CD8 depleting antibodies or control rat IgG were administered during TRT + C4 treatment and compared to VO (no TRT or C4 or depleting antibody) control. (F) Tumor response to 90Y-NM600 + C4 vs cold (non-radioactive) NM600 + C4 compared to VO (no NM600 or C4) control is shown. (N = 6 per replicate, 12 total. Significance determined by linear mixed effects regression analysis with Tukey multiple comparisons [significant differences, p < 0.05, demarcated by * with the color of the asterisk representing which group from which the sample is significantly different].
Immunomodulatory effects of low dose TRT.
These therapeutic studies suggest that low dose radiation delivered by TRT can modulate the TME of tumors and render these more responsive to ICIs. To elucidate mechanisms by which low dose TRT may modulate the TME and to compare these to effects of low and moderate dose EBRT, we began by examining differences in tumor immune cell infiltrate at days 1, 7, and 14 after radiation. Syngeneic C57Bl/6 mice bearing B78 flank tumors were treated with either 2.5 Gy or 12 Gy tumor-directed EBRT, 50 μCi 90Y-NM600 delivering 2.5 Gy to the tumor, or sham/vehicle only control treatments. Pre-determined cohorts of mice were euthanized at indicated times and flow cytometry was performed on disaggregated tumors. No significant (p =ns) difference was observed between radiation groups with regard to total immune cell infiltrate (CD45+), however moderate dose EBRT (12 Gy) resulted in a trend towards a decrease in tumor infiltrating immune cells at day 1 post radiation with a rebound effect seen at days 7 and 14 (Figure 5A–F). Innate myeloid (CD11b+) and natural killer (NK) cells were significantly (p < 0.05) increased compared to vehicle only controls at day 7 after low dose TRT (Figure 5A–F). The ratio of effector T cells (CD8+) to suppressor Tregs (CD4+CD25+FOXP3+) was increased in the low dose TRT treated group at day 1 post treatment compared to either low dose or high dose EBRT treated groups. Overall, alterations in tumor immune cell infiltrates were modest following these radiation treatments alone.
Figure 5. Dose and time dependent radiation effects on the TME.

Flow cytometry analyses of tumor immune cell infiltrates [total immune cells (CD45+), myeloid cells (CD11b+), T effector cells (CD8+), Tregs (CD4+CD25+FOXP3+), and NK cells (NK1.1+)] as a percent of total live cells normalized to mean of control (VO) is shown at 1, 7, 14 days after RT administration in B78 melanoma (A-F). Gene expression of chemokines, type I IFN pathway genes, acute phase inflammatory cytokines, cell adhesion and immune activation genes, and apoptosis/DNA damage repair genes in harvested tumors is displayed as the log of the fold change in expression at 1, 7, and 14 days after RT administration in comparison for values of tumors in tumor-bearing VO treated mice, obtained at the same times (days 1, 7, and 14) (G). RT effects on D7 expression of select STING pathway genes is shown (H). 50 μCi of TRT (90Y-NM600) + anti-CTLA-4 (C4) improves survival compared to single treatment controls in a B16 WT tumors but not in B16 Tmem173−/− (STING KO) tumors (I-J). (N = 5 for flow cytometry and gene expression studies, Significance determined by two-way ANOVA; N=4–6 per replicate, 8–12 total for survival studies, Kaplan–Meier with Log-rank testing and Cox Regression for survival analysis: *P < 0.05, **P < 0.01, ***P<0.001, ****P<0.0001).
We next examined the effect of TRT or EBRT on the expression of immune-related genes in the TME at days 1, 7, and 14 after treatment of mice bearing B78 melanoma tumors (Figure 5G). At day 1, overall differences in chemokine or type I Ifn genes in the 3 treated groups were modest relative to gene expression in vehicle only (VO) treated controls. Notable differences in gene expression at day 1 included reduced expression of the acute phase cytokine interleukin-6 (Il-6) with TRT treatment, increased expression of vascular cell adhesion protein 1 (Vcam1) in low dose TRT and EBRT groups, and increased expression of apoptosis antigen 1 (Fas) across all groups. Differences in immune-related gene expression peaked at day 7 post radiation with increased expression of Ifn-β, a marker of activation of the cGAS/STING pathway, seen in all groups. However, treatment with moderate dose EBRT (12 Gy) resulted in significantly (p < 0.05) greater expression of downstream type I IFN response genes, such as Oas2, Oas3, and Mx1, and of the major histocompatibility complex I (Mhc1) (Figure 5G, H). By day 14 post treatment, expression of all investigated genes was trending back to baseline. Notably, activation of a type I IFN response by cGAS/STING is critical to the in situ vaccine effect of moderate dose EBRT (25) and here we tested whether STING was also necessary for the cooperative therapeutic interaction between 90Y-NM600 and ICIs. To test this hypothesis, we first confirmed 86Y-NM600 uptake in both B16 wild-type and STING KO lines via PET imaging (Figure S7). Then, in mice bearing B16 melanoma STING KO (Tmem173 −/−) tumors we observed a loss of cooperative therapeutic efficacy that was otherwise detected between 50 μCi 90Y-NM600 and anti-CTLA-4 in wild-type B16 tumors (Figure 5 I, J, Figure S8 [tumor growth curves]).
Combining low dose TRT and ICIs renders immunologically “cold” tumors “hot”.
To investigate the therapeutic effects of combined TRT + ICI on the TME, we performed flow cytometry to evaluate differences in B78 tumor immune cell infiltrates at day 25 after treatment with vehicle only, 50 μCi 90Y-NM600, anti-CTLA-4, or 50 μCi 90Y-NM600 + anti-CTLA-4. The combination of 90Y-NM600 + anti-CTLA-4 treatment significantly (p < 0.05) increased the percentage of total immune cells (CD45+), tumor infiltrating lymphocytes (CD3+), effector T cells (CD8+), tissue resident effector memory T cells (CD8+CD103+), and innate γδ T cells (Figure 6A, Figure S9). In addition to stimulating an increased effector T cell infiltration of tumors, 90Y-NM600 + anti-CTLA-4 also reduced T cell exhaustion, as determined by reduction of PD1 expression (by MFI) on CD8+ T cells (Figure 6A) and on the breadth of CD45+ immune cells (by tSNE plot in Figure 6B).
Figure 6. TRT + ICI enhances immune infiltrates in the TME.

C57Bl/6 mice with B78 tumors were treated with VO, 50 μCi TRT (90Y-NM600), anti-CTLA-4 (C4), or 50 μCi TRT + C4. (A)Tumors were harvested on Day 25 post radiation (after radioisotope reached background) and tumor immune cell infiltrates [total immune cells (CD45+), lymphocytes (CD3), T effector cells (CD8+), Tregs (CD4+CD25+FOXP3+), NK cells (NK1.1+), γδ Tcells (CD3+γδTCR+), and resident memory effector cells (CD8+CD103+)] were quantitated as a percent of total live cells via flow cytometric analysis. In addition, PD1 expression on CD8 cells was quantified by MFI. (B)A tSNE transformation was calculated for a pooled sample of all CD45+ immune cells across all treatment groups and differential staining of Pd1 staining by treatment group is shown. (C) A separate cohort of mice bearing B78 tumors were treated with the same treatment groups with double the mice in the 50 μCi TRT + C4 group. Mice in the TRT + ICI group (μCi + C4) with tumors growing at Day 25 were labeled as non-responders (NR). Cytokine and chemokine concentrations in tumor lysates were measured by multiplex immunoassay. Hierarchical clustering analysis was performed, and the assay results were displayed as a Z-score for each cytokine. (D) TCRβ sequencing of TILs demonstrated increased total and unique CDR3 chains as well as decreased D50 with TRT + ICI, but no increase in Shannon diversity index. (N = 5 for flow cytometry, multiplex, and TCR sequencing studies, Significance determined by two-way ANOVA,*P < 0.05, **P < 0.01, ***P<0.001).
To more directly evaluate functional differences among immune cells in the TME we performed multiplex cytokine profiling of B78 tumors at day 25 after treatment with vehicle only, 50 μCi 90Y-NM600, anti-CTLA-4, or 50 μCi 90Y-NM600 + anti-CTLA-4. In addition to treatment group, specimens were annotated based on whether or not tumors were responding (decreasing in size) or progressing at day 25 after treatment initiation. Unsupervised clustering analysis was performed to group all treated mice according to the tumor cytokine profile. The combination of 90Y-NM600 + anti-CTLA-4 increased the expression of most cytokines in the TME compared to single or control treatment cohorts (Figure 6C), particularly in the 5 mice that showed a therapeutic response, but less so in 3 of the 4 mice shown in this group that were non responders (NR). One notable exception to this observation was the cytokine, IL10, which was reduced in most tumors receiving combination treatment. On the other hand, IL-10 was increased in most tumors from mice treated with 90Y-NM600 alone. This is consistent with prior studies that show a rebound increase in tumor infiltration by IL-10-secreting lineages including Tregs and suppressive monocyte populations at late time points following radiation (49, 50).
To further elucidate functional differences among immune cells in the TME, we isolated tumor infiltrating lymphocytes (TILs) from the TME on day 11 after treatment in mice treated with vehicle only, 50 μCi 90Y-NM600, anti-CTLA-4, or 50 μCi 90Y-NM600 + anti-CTLA-4. TILs were then stimulated with varying concentration of CD3 in the presence of CD28 and we found that TILs isolated from tumors treated with 50 μCi 90Y-NM600 + anti-CTLA-4 had significantly (p < 0.05) greater interferon-γ production compared to VO or monotherapy groups (Figure S10).
In preclinical and clinical studies, a signature of the cooperative therapeutic interaction between moderate dose, single tumor-directed EBRT and ICIs has been the diversification of the anti- tumor T cell response, consistent with an in situ vaccine effect of EBRT (8, 18). To evaluate the mechanisms of interaction between low dose TRT and ICIs, we performed next-generation deep sequencing of the T cell receptor (TCR) β subunit in B78 tumors collected at day 25 after treatment with vehicle only, 50 μCi 90Y-NM600, anti-CTLA-4, or 50 μCi 90Y-NM600 + anti- CTLA-4. The combination of 90Y-NM600 + anti-CTLA-4 treatment significantly (p < 0.05) increased the total number of complementary determining region 3 (CDR3) and of unique CDR3 in the TME, but did not affect the Shannon diversity index of the TCR repertoire in these tumors (Figure 6D). Rather, the addition of 90Y-NM600 to anti-CTLA-4 significantly (p < 0.05) reduced the percentage of unique CDR3 sequences that make up 50% of the total tumor infiltrating CDR3 sequences (D50) (Figure 6D). This is consistent with an increase in the clonal expansion of T cells resulting from the combination of 90Y-NM600 and anti-CTLA-4, compared to anti-CTLA-4 alone.
TRT enhances local and distant tumor response in combination with local EBRT and ICI.
In most clinical trials currently investigating combinations of radiotherapy and immunotherapy in patients with metastatic cancer, a single tumor site is treated with moderate dose EBRT in conjunction with ICI administration with a goal of eliciting an in situ vaccine effect that primes abscopal responses at non-radiated tumor sites. Given the distinct mechanistic effects of moderate dose EBRT and low dose TRT in priming diversification and propagating clonal expansion of the tumor infiltrating TCR repertoire, respectively, we hypothesized that these two forms of radiotherapy might be complementary in augmenting response to ICIs. In syngeneic mice with two established B78 melanoma tumors, we tested whether the addition of 90Y-NM600 to single-tumor directed EBRT might enhance response to anti-CTLA-4 more than either form of radiotherapy alone (Figure 7A). We observed that the combination of anti-CTLA-4 with both low dose 90Y-NM600 and moderate dose single-tumor directed EBRT significantly (p < 0.05) improved tumor response at the “secondary tumor” that was not targeted by EBRT and improved overall survival compared to combinations of anti-CTLA-4 with either TRT or EBRT alone (Figure 7B–D). All two-tumor mice experiencing CR to the combination of anti-CTLA-4 + EBRT + 90Y- NM600 demonstrated tumor-specific immune memory to B78 rechallenge and all except one mouse additionally rejected the parental B16 melanoma, which shares common antigens with B78 (Figure 7E).
Figure 7. Addition of TRT to EBRT + anti-CTLA-4 enhances primary and distant tumor response.

C57Bl/6 mice with equal sized (~120 mm3) bilateral B78 tumors were treated with 50 μCi TRT (90Y-NM600) + 12 Gy to a primary tumor (P), 12 Gy P + anti-CTLA-4 (C4), 50 μCi TRT + C4, or 12 Gy P + 50 μCi TRT +C4 (A). Tumor growth at both the primary and secondary (S) tumor site was tracked for 30 days, survival for 60 days, and all mice with CR were rechallenged at D90 with B78 and on D120 with B16/Panco2 (B-E). (N = 6 mice per group in each of 2 replicate experiments, 12 mice total per treatment]. (B) shows mean tumor growth for the primary and secondary tumors in all 4 treatment groups. (C) shows overall survival for all 4 groups. (D) primary and secondary tumor growth for all mice in one of 2 replicate experiments. (E) All mice that had CR in Figure 7D were rechallenged with B78, and those that rejected it were then rechallenged with B16 and Panc02 (Ø indicates no mice rejected challenge with symbol color representing challenge type), Significance determined by linear mixed effects regression analysis with Tukey multiple comparisons testing for tumor growth [significant differences, p < 0.05, demarcated by * with the color of the asterisk representing which group from which the sample is significantly different], Kaplan–Meier with Log-rank testing and Cox Regression for survival analysis, and Chi-square contingency testing for immune memory shown in supplemental Table S4. *P < 0.05, **P < 0.01, ***P<0.001)
Delivery of low dose TRT and moderate dose EBRT in a companion canine with cancer.
As a proof of concept, in companion canines with spontaneous, metastatic cancer we tested the feasibility and safety of performing real-time tumor- and host-specific dosimetry from serial 86Y- NM600 PET/CT imaging and delivering low dose radiation to all tumor sites using 90Y-NM600 together with single-tumor-directed moderate dose EBRT. Because the range of radiation emitted from TRT agents is constant regardless of tumor context, these large animals more closely mimic the setting that will be encountered upon clinical translation of this combined modality approach in humans.
The first two companion dogs enrolled for this study presented with widespread metastatic osteosarcoma (OSA) and metastatic melanoma. Semi-selective uptake and retention of 86Y-NM600 in tumor was observed in primary and all documented metastatic lesions on serial PET/CT images (Figure 8A,B). Blood circulation and hepatobiliary excretion of the radiotracer was evidenced by the initial elevation of radioactivity within the vascular compartment and gradual accumulation of radioactivity in the gallbladder and feces. Compared to mice, 86Y-NM600 uptake in the liver was lower and comparable to that of tumor, reducing concerns about possible liver toxicity. Monte Carlo dose calculations provided a voxel-based estimate of patient-specific dosimetry for 90Y- NM600 with respect to tumors and pertinent normal tissues. Importantly, all identified metastases including those in lung and subcutaneous soft tissue exhibited > 2:1 tumor to bone marrow uptake (Figure 8C,D). This demonstrates a therapeutic window for using 90Y-NM600 to immunomodulate all metastatic tumor sites without inducing immunosuppression from bone marrow depletion.
Figure 8. 86Y-NM600 imaging, dosimetry, and post 86Y-NM600 treatment.

Companion canines with osteosarcoma (A) and oral melanoma (B) were imaged with 86Y-NM600. Axial images are shown at 2, 24 and 48 hours, with arrows indicating the primary (index) lesion and metastatic lesions, as well as a coronal image at 48 hours. (C,D) Dose delivered to sites of tumor vs normal tissue per GBq of injected activity was determined.
To complete the personalized theranostic paradigm, one week after completion of serial 86Y- NM600 PET/CT imaging studies in these dogs, we used patient-specific dosimetry to prescribe an activity of 90Y-NM600, which was calculated to deliver a mean tumor dose of ≥ 2 Gy while limiting mean bone marrow dose to 1 Gy in each patient. Following IV injection of 90Y- NM600, we monitored for acute and sub-acute adverse events and performed serial lab tests including complete blood counts with differentials, comprehensive metabolic panels (CMP), and urinalyses. In addition, a client-documented quality of life questionnaire was completed after treatment. No substantial clinical or pathologic adverse effects were documented as graded and attributed using the veterinary cooperative oncology group common terminology criteria for adverse events (VCOG-CTCAE v1.1). No clinically meaningful changes were observed in laboratory testing including CMP (note, the elevated baseline and post treatment Alkaline phosphatase values were due to the osteosarcoma itself) and blood counts of neutrophils, lymphocytes, platelets, and red blood cells (Table S5, S6).
Discussion
We developed and tested a strategy to deliver low dose radiation safely to all tumor sites in settings of solid tumors and metastatic disease of potentially any tumor type using 90Y-NM600 and we demonstrated that this agent can immunomodulate TMEs in a manner that promotes response to ICIs. Aided by the semi-selectivity of NM600 for nearly any tumor type, we observed generalizability for these effects across multiple murine strains and tumor types, including immunologically cold tumors that exhibit limited de novo T cell infiltration and do not respond to ICIs alone. This combination of TRT and ICIs dramatically improved tumor response, reduced spontaneously arising metastatic tumor burden, increased rates of CR, and prolonged overall survival compared to ICIs or TRT alone. Mice cured by this combined modality regimen consistently demonstrated tumor-specific memory consistent with the development of a potent adaptive anti-tumor immune response. We evaluated the influence of ICI timing relative to the TRT administration and demonstrated similar efficacy with ICI initiated before or after the TRT (Figure 2E–G). We also evaluated the influence of TRT dose and demonstrated that low dose 90Y-NM600 was adequate to achieve cooperative efficacy with ICIs such that no additional benefit was observed with dose escalation from 2.5 to 5 Gy tumor dose (Figure 2B–D). We further demonstrated that this approach was feasible to translate clinically in settings of spontaneous cancers arising in large mammals such as companion canines with metastatic disease. With these findings, we are now planning to test this approach in early phase clinical trials in humans with metastatic cancers.
To our knowledge, we present the first comparative evaluation of the effects of TRT versus EBRT in immunomodulating the TME. Like low or moderate dose EBRT, low dose TRT activated a type I IFN response in tumors, although activation of downstream IFN response genes was greater with moderate dose compared to low dose EBRT (Figure 5G, H). In addition to these responses in tumor cell-associated markers, we observed comparable effects from EBRT and TRT in modulating markers associated with endothelial and immune components of the TME (e.g. Vcam1, Ccl3, Cxcl11; Figure 5G). In contrast to moderate dose EBRT, we did not detect a consistent effect of low dose TRT on the expression of Mhc I in tumors. These results demonstrate a clear immunomodulatory effect of low dose TRT but suggest that moderate dose radiation may be better suited for augmenting antigen presentation. The time course of these gene-expression effects has implications for the timing of combined TRT/EBRT and immunotherapy regimens.
When co-administered with ICIs, low dose TRT from 90Y-NM600 elicited a marked increase in the production of pro-inflammatory cytokines in the TME (Figure 6C) and enhanced tumor infiltration by effector T cell lineages (Figure 6A). Deep sequencing analyses of the TCR β subunit among tumor infiltrating T cell revealed that low dose TRT increased the clonal expansion of tumor infiltrating T cells without significant (p = ns) effect on diversity (Figure 6D). This contrasts with prior studies using the B16 melanoma model and demonstrating an effect of moderate dose EBRT in diversifying the TCR repertoire when combined with ICIs (8). Consistent with these distinct effects, we demonstrated that moderate dose single-tumor-targeted EBRT and low dose TRT are complementary and non-redundant in their ability to augment response to ICIs especially in the presence of multiple tumor sites (Figure 7B–D). These findings suggest a role for moderate dose single-tumor-targeted EBRT in priming diversification of antigen recognition by T cells through an in situ vaccine effect. This may be driven by the unique abilities of moderate dose EBRT to stimulate immunogenic cell death, increased expression of MHC I, and activation of type I IFN response genes in tumor cells; low dose TRT induces many of these same mechanisms, but less potently than moderate dose EBRT. On the other hand, our data suggest a critical role for low dose TRT in propagating anti-tumor T cell immunity by augmenting cytokine secretion and immunomodulating the TME at all tumor sites to enhance the trafficking and persistent activation of effector T cells therein. Interestingly, we observed that expression of STING in tumor cells was necessary for the cooperative therapeutic interaction between 90Y-NM600 and anti-CTLA-4 (Figure 5I, J). Prior studies demonstrated that this pathway was necessary for the in situ vaccine effect of moderate dose EBRT in combination with ICIs (25). These findings shed light on conserved and divergent mechanisms that contribute to the immunologic effects of low dose TRT and low or moderate dose tumor-targeted EBRT.
Immunologically “cold” tumors are those that are poorly infiltrated by T cells and these typically do not respond to ICIs alone (51). Our results suggest that both moderate dose single-tumor targeted EBRT and low dose TRT may enhance the response to ICIs in such tumors. When tumors are “functionally” cold due to inactivation or exclusion of a pre-existing anti-tumor T cell response in the TME, low dose TRT in combination with ICIs may be adequate to restore anti-tumor immunity by aiding the propagation of this pre- existing response at all tumor sites. In tumors that are “intrinsically” cold due to inadequate tumor recognition by T cells (e.g. tumors with few neoantigens due to low mutation burden) it may be beneficial to combine an EBRT in situ vaccine with low dose TRT in order to both prime and subsequently propagate an adaptive anti-tumor T cell response in conjunction with ICIs.
When tumor recognition by host T cells is not possible (e.g. tumors with very few immunologically recognizable tumor antigens and/or loss of MHC I), endogenous adaptive immunity induced by in situ vaccination may be ineffective. In these settings, “synthetic recognition” of tumor-associated germ-line encoded differentiation or cancer-testis antigens, via lab engineered/expanded immune cells (i.e. TILs) or antibody based recognition pathways (ie: bispecific T cell engagers, tumor-reactive mAbs or mAb-directed CAR T cells) may be required to achieve immune-mediated tumor destruction. In future studies, it will be intriguing to test whether immunomodulation of all tumor sites using low dose TRT may augment response to such synthetic-recognition-based immunotherapies. Notably, in clinical studies using expanded tumor infiltrating lymphocytes, delivery of low or moderate dose EBRT to the whole body enhanced overall response rates (52). Our data suggest that TRT may provide a more targeted and safer approach to achieve similar effect.
Among TRTs, NM600 has several potential advantages for studies involving combination with immunotherapies. NM600 can semi-selectively deliver radionuclides to a wide variety of tumor types in mice, canines, and humans (43). Unlike antigen-targeted TRTs, NM600 targets via lipid rafts, a highly conserved mechanism of accumulation of alkylphosphocholines in tumor cells. This limits potential for antigen-loss resistance to NM600. Moreover, NM600 crosses the blood-brain barrier and can deliver low dose radiation to metastatic or primary tumors in any location in the body (46). Yet, one disadvantage of its broad tumor uptake is that NM600 may have reduced tumor-targeting capacity compared to high selectivity antigen targeted TRT agents (44, 53). This limitation is overcome by the relatively low dose of radiation necessary for immunomodulation and NM600’s low uptake in lymph nodes, spleen, and bone marrow, thereby minimizing potential immune or hematologic toxicities of NM600-based TRT. NM600 also exhibits low albumin binding, enabling rapid clearance from blood. The ability of NM600 to chelate a variety of radiometals enables theranostic approaches to determine radionuclide-, host-, and tumor- specific dosimetry. This facilitates the study of dose-dependent mechanisms and guides the appropriate timing of TRT administration. The latter is particularly important because of the capacity of TRT to deplete radiosensitive immune cells circulating into the TME. We have focused our study on low dose 90Y-NM600 regimens because we observed that escalation above 2.5 Gy tumor dose did not improve response (Figure 2B–D). This raises the translatability of our observations by limiting concerns for radiation-related toxicities. Future studies will investigate this approach with alternative TRT vectors and using diverse radionuclides, including high linear-energy transfer, short-range alpha emitters that may exert distinct immunological effects on tumor cells and/or the TME.
Limitations of this study include the reliance on transplantable murine tumor models that do not recapitulate the tumor heterogeneity or spatial separation of tumor and immune organs in humans. Although our findings in a number of transplant tumor models suggest that radiation therapy and immune checkpoint blockade is a promising treatment approach, others have previously shown that transplanting tumor cells into syngeneic mice can create pre-existing immunity that is critical for the response to radiation therapy and immune checkpoint inhibition (54, 55). Therefore, future studies with this therapy in autochthonous tumors in mice or canine patients would further support the potential of this treatment for patients with cancer. Testing in companion canines begins to redress this limitation and demonstrated that patient-specific dosimetry can be performed to account for tumor heterogeneity with theranostics serving as their own biomarker. However, a lack of available canine ICIs currently prevents testing of our combined modality approach in dogs. A similar limitation arises from the greater response to anti-CTLA-4 compared to anti-PD1/L1 therapies in murine tumor models. This contrasts with clinical studies in patients with cancer and may reflect differences between murine and human immunity and/or differences in the immunogenicity of murine versus human tumors. To overcome this limitation and test whether and how TRT may enhance response rates to ICIs, we have focused on poorly immunogenic murine tumor models that do not respond well to ICIs alone and we have tested dual as well as single agent ICI regimens.
Our results demonstrate a cooperative therapeutic interaction between low dose TRT from 90Y- NM600 and ICIs that is further enhanced by combination with a moderate dose EBRT in situ vaccination. Future studies will evaluate approaches to enhance this response by optimizing the choice or mixture of therapeutic radionuclides, using alternative in situ vaccine approaches, and combining with alternative immunotherapies. Evaluating mechanisms of escape from combined TRT and ICI treatment will be critical. Cytokine profiling following our TRT + ICI regimen revealed that non-responding tumors had either lower concentrations of inflammatory cytokines or increased concentrations of other cytokines like IL-10 (Figure 6C). This observation provides leads for adding other immunotherapies to this TRT +ICI regimen to overcome these pathways of non-response including other checkpoint inhibitors targeting alternative exhaustion pathways including T cell immunoglobulin and mucin domain-containing protein 3 (TIM3), Lymphocyte-activation gene 3 (LAG-3), and others. Enhancing CR rates in difficult to treat immunologically cold tumors is an area of utmost interest in clinical oncology; further preclinical and clinical studies are warranted to evaluate the therapeutic potential of combinations of TRTs with immunotherapies.
Materials and Methods
Study Design.
The primary objective of this study was to test whether and how low dose TRT from 90Y-NM600 enhances the efficacy of ICIs in difficult to treat tumor models. All in vivo tumor response studies were replicated with at least six mice per experimental group. These experiments were powered utilizing effect size and variance from pilot studies to detect differences between treatment groups at predefined endpoints, either tumor growth at day 30 after TRT injection (80% power, alpha 0.05) or survival at day 60 after TRT injection (aggregate analysis of replicate studies, 90% power, alpha 0.05).
Cell lines.
The 4T1 murine breast cancer cell line was purchased from ATCC. The B78-D14 cell line was derived from B16 melanoma and was obtained from Dr. Ralph Reisfield (Scripps Research Institute). The NXS2 cell murine neuroblastoma cell line was obtained from Dr. Alice Yu. B16 Tmem173 −/− CRISPR KO and corresponding wild type (WT) melanoma cells were obtained from Dr. Samuel Bakhoum (Memorial Sloan Kettering Cancer Center). Panc02 pancreatic cancer line was obtained from NCI. B78, 4T1, NXS2, Panc02, and B16 cells were grown in RPMI 1640 or DMEM (Mediatech) media and supplemented with 10% fetal bovine serum (FBS), 2 mM L- glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells were maintained in culture below 80% confluence for all passages, and early passages after thaw (3–8) were used for all experiments. Cell authentication was performed per ATCC guidelines using morphology, growth curves, and mycoplasma testing within 6 months of use.
Tumor Models.
All animal studies were conducted under an approved University of Wisconsin institutional animal care and use committee (IACUC) protocol. Male or female mice aged 6–8 weeks were purchased from Taconic (C57Bl/6, Balb/c) or Jackson (A/J).B78 and NXS2, tumors were engrafted by subcutaneous flank injection of 2×106 cells into the right (for one tumor models) or right and left (for two tumor models) flanks. For 4T1 and B16, 1×105 and 5×105 cells respectively were inoculated in the right flank. Tumor size was tracked using caliper measurements twice weekly and volume was approximated as (width2 × length)/2. Mice were randomized at a mean size of 80–175 mm3 prior to imaging or treatment. After treatment, mice were tracked for tumor growth with biweekly caliper measurements until day 30 after treatment initiation and overall survival until day 60 after treatment initiation [endpoint/euthanasia of mice criteria included: tumor diameter > 15–20 mm, hunched posture, or veterinary recommendation]. At day 90 after treatment initiation, mice with tumor complete response (CR) were re-challenged by re-injection of the same tumor cell line they had been cured of to test for tumor-specific immune memory, as measured by rates of tumor engraftment compared to rates in age matched naïve control mice.
For some tumor lines, mice rejecting re-engraftment were challenged with a related tumor line sharing common antigens and unrelated tumor line to determine specificity of this response. To quantify effect of treatment on metastases, a cohort of mice bearing a spontaneously metastatic 4T1 tumor were euthanized by CO2 asphyxiation on day 30 after TRT injection. Lungs from these mice were stained with India ink and the number of metastatic nodules was visually quantified and analyzed by treatment group.
Radiochemistry and 86Y production.
The positron emitter 86Y (β+, t1/2 = 14.7 h) was produced as previously described (56). Briefly, 86Y was produced in a PETrace biomedical cyclotron via proton (14.1 MeV) bombardment of enriched [86Sr]SrCO3 (96.4 ± 0.1%) solid targets. Irradiated targets were dissolved in 6 N hydrochloric acid (HCl) and 86Y was quantitatively eluted from a DGA extraction resin column in ~600 μL of 0.1 M HCl. Clinical grade 90YCl3 was purchased from Perkin Elmer and 2-(trimethylammonio)ethyl(18-(4-(2-(4,7,10-tris(carboxymethyl)-1,4,7,10- tetraazacyclododecan-1-yl)acetamido)phenyl)octadecyl) phosphate (NM600) was kindly provided by Archeus Technologies (Madison, WI). Radiolabeling of NM600 with 86/90Y was performed by mixing 185–370 MBq (5 −10 mCi) of 86/90Y and 54–81 nmol/GBq (10–15 nmol/mCi) of NM600 in 0.1 M NaOAc (pH = 5.5) buffer. Following incubation at 90 °C for 30 minutes under constant shaking (500 rpm), 86/90Y-NM600 was purified by solid phase extraction using an HLB (Waters) cartridge. For in vivo use, 86/90Y-NM600 was formulated in vehicle consisting of normal saline containing 0.4% v/v Tween 20 and sodium ascorbate (0.5% w/v).
Imaging and bio-distribution.
Mice bearing B78, 4T1, NXS2, B16 WT, and B16 Tmem173 −/− (n = 3) flank tumors (~150 mm3) were administered 9.25 MBq of 86Y-NM600 intravenously (lateral tail vein) and serial CT (80 kVp; 1000 mAs; 220 angles) and 80 million coincidence events static PET scans (time window: 3.432 ns; energy window: 350–650 keV) were acquired with an Inveon microPET/microCT scanner (Siemens Medical Solutions, Knoxville, TN) at 3, 24, and 48 hours post injection of the radiotracer. For each scan, mice were anesthetized with isoflurane (4% induction; 2% maintenance) and placed on the scanner bed in a prone position. Static PET scans were reconstructed using a three- dimensional ordered subset expectation maximization (OSEM3D) algorithm. CT images were employed for attenuation correction, anatomical referencing, and to generate density maps for the dosimetry estimations. Region-of-interest analysis of the PET images was performed to determine the magnitude and kinetics of 86Y-NM600 uptake in the tumor and normal tissues of interest. Quantitative data were expressed as percent injected activity per gram of tissue (%IA/g; mean ± SD). Following PET/CT, mice were euthanized by CO2 asphyxiation. Tumors and normal tissue specimens were collected, wet-weighed, counted in an automated γ-counter (Wizard 2, Perking Elmer, MA), and the %IA/g (mean ± SD) corresponding to each tissue was calculated.
Tumor Dosimetry Calculations.
Subject-specific tumor dosimetry for 90Y-NM600 TRT was estimated using the Radiopharmaceutical Assessment Platform for Internal Dosimetry (RAPID) (46, 57, 58). The theranostic dosimetry approach in this work utilized serial 86Y-NM600 microPET/CT imaging to estimate the biodistribution of 90Y-NM600 over time, correcting for the different rates of radioactive decay. PET and CT volumes at each time point were used to define the source and geometry distributions, respectively, in Monte Carlo (Geant4 v9.6) simulations. Contours for tumor and organs of interest were delineated on the anatomic CT images and used to quantify the in vivo pharmacokinetics of NM600 as well as characterize the spatial distribution of absorbed dose imparted by 90Y-NM600 TRT. The cumulative absorbed dose in each region of interest was calculated by integrating the mean absorbed dose rate at each timepoint using a hybrid trapezoidal-exponential model of the time-dose-rate curves. The processing of PET/CT image volumes and generation of 3D cumulative absorbed dose distributions (0.42 × 0.42 × 0.42 mm) in RAPID were previously described in detail (46, 57, 58).
Toxicity Assays
Mice bearing B78 melanoma (N=5) flank tumors (~150 mm3) were randomized into treatment groups of cold NM600 (day 1), anti-CTLA4 (200 ug day 4,7,10), or anti-CTLA4 + 50 μCi 90Y‐NM600 (day 1). Blood samples were collected via tail vein nick (50 μL) on day 0 then weekly until day 35 and analyzed with a VETSCAN HM5 hematology analyzer (Abaxis). Mouse weight was also tracked weekly. On day 35 after TRT administration lymphatic organs and organs of elimination (liver, kidney, spleen, bone marrow, small bowel) were collected, fixed, stained, and imaged as previously described (47).
Treatments.
90Y‐NM600 treatments (at activities of 25, 50, or 100 μCi per dose per mouse) or injection vehicle only or unlabeled NM600 controls were administered via IV tail vein injection ontreatment “day 1”. Tumor-targeted EBRT was delivered at a dose of 12 Gy or 2.5 Gy on day 1 with an XRad320 (PXi) irradiator with the tumor exposed and the rest of the animal shielded using custom lead blocks. For whole mouse radiation, animals were anesthetized with 2% isoflurane and treated Anterior-to-Posterior and Posterior-to-Anterior (AP/PA) to a calculated midline dose of 2.5 Gy without any shielding. Anti-murineCTLA-4 (IgG2a or IgG2c, clone 9D9) was kindly provided by Bristol Myers Squibb and Anti-murine PD-L1 was purchased from BioXcell (IgG2b, Clone 10F.9G2). ICI or isotype control antibody (BioXcell)were delivered by intraperitoneal (IP) injection of 200 μg (in regimens giving three dose administrations) or 250 μg (single treatment dose). T-cell depletion was performed as previously described (59).
Flow Cytometry.
Flow cytometry studies were performed as previously described (60). Briefly, tumors were harvested and disassociated with DNAse and collagenase on a Miltenyi GentleMACS Octodissociator. Single cell suspensions were stained with innate and adaptive immune cell antibody panels (Table S7) followed by fixation in eBioscience FOXP3 fixation/permeabilization buffer. For tumors harvested from TRT-treated mice prior to day 25 after treatment initiation, cells were frozen after fixation in 10% DMSO in FBS solution and stored at −80 °C until reaching background radiation (>10 half-lives of the injected isotope). Cells were then thawed rapidly and acquired with an Attune Nxt flow cytometer (ThermoFisher, Waltham, MA) and analysis of flow data was performed on FCS Express 6.0 using a gating strategy as previously described (60). All analyses were normalized to vehicle only treated control samples at each time point to account for potential differences in cell loss due to sample processing, freezing, and thawing.
T cell stimulation Assays.
At day 11 after treatment initiation, B78 tumors (n=3) were harvested from mice treated with VO (cold NM600), anti-CTLA4 (IP, 200 ug day 4,7,10), 50 μCi 90Y‐NM600 (day 1) or anti-CTLA4 + 50 μCi 90Y‐NM600. Tumors were harvested and disassociated with DNAse and collagenase on a Miltenyi GentleMACS Octodissociator and then tumor infiltrating lymphocytes (TILs) were separated with magnetic bead separation using CD4/CD8 microbeads (Miltenyi Biotec). Isolated TILs were then spun down and diluted in media [RPMI 1640 containing 10% heat inactivated FBS and supplemented to a final concentration with L-glutamine (2 mM), penicillin (50 U/ml), streptomycin (50 μg/ml), 2-mercaptoethanol (50 μM), 150 Units/mL IL-2, and 1% ITS: insulin (1.7 μM), transferrin (68.8 μM) and sodium selenite (3.9 nM)], and placed in a 96 well plate previously coated with either 0, 1, or 5 ug/mL anti-CD3ε (Clone 145–2C11, BD). TILs were then allowed incubate for 48 hours in the presence of 5 ug/mL free anti-CD28 (clone 37.51, BD) in all wells. GolgiSTop (BD) was added 4 hours prior to collecting cells for staining. Cells were then prepped for flow cytometry as described above and differences in the Median Fluorescence Intensity (MFI) of IFN-γ in TILs was compared across treatment groups.
Gene Expression.
For gene expression analyses in tumor, freshly dissected specimens were homogenized using a Bead Mill Homogenizer (Bead Ruptor Elite, Omni International Cat # 19–040E). Total RNA was extracted using RNeasy Mini Kit (Qiagen, Germany, Cat # 74106) according to the manufacturer’s instructions. cDNA was derived using QuantiTect Reverse Transcription Kit (Qiagen, Germany, Cat # 205314) according to the manufacturer’s instructions. Quantitative polymerase chain reaction (qRT-PCR) was performed using PowerUp SYBR Green qPCR Master Mix. The reaction (5μL total volume) was prepared using Labcyte Echo 550 and MANTIS liquid handling systems. Thermal cycling conditions (Vii7A Cycler, Applied Biosystems) included the UDG activation stage at 50°C for 2 min, followed by Dual-Lock™ DNA polymerase activation stage at 95°C for 2 min followed by 40 cycles of each PCR step (denaturation) 95°C for 15s and (annealing/extension) 60°C for 1 min. A melt curve analysis was done to ensure specificity of the corresponding qRT-PCR reactions. For data analysis, the Ct values were exported to an Excel file and fold change was calculated using the ΔΔCt method.
ΔΔCt values were then imported into Matlab (version R2019b) and log and Z transformations were applied. Unsupervised hierarchical clustering was performed. HPRT, PGK1, and TBP were used as endogenous controls. Additional gene expression details are provided in the Supplemental Methods Section. A list of all targets and detailed primer information are provided (Table S8).
Cytokine Multiplex Immunoassay.
At day 25 after treatment initiation, mice with engrafted B78 tumors were classified as responders or non-responders based on whether or not their tumors were regressing or growing in size at the most recent measurement. Tumors were harvested, weighed, and 5 μl/mg of Cell Lysis Buffer with PMSF (Cell Signaling Technology) and Halt™ Phosphatase Inhibitor Cocktail (Thermo Scientific) was added to the tumor lysate. The tumors were homogenized and the lysates were stored at −80°C until use. A multiplex immunoassay was used to determine the concentration of 32 cytokines and chemokines in the tumor lysates (MILLIPLEX MAP Mouse Cytokine/Chemokine Magnetic Bead Panel, Millipore) following the manufacturer’s instructions. The multiplex plate was read on the MAGPIX System (Millipore) and the protein concentrations were interpolated from curves constructed from the protein standards and their respective median fluorescence intensity (MFI) readings (Milliplex Analyst, Millipore). Log and Z-transformation of the data was performed followed by unbiased hierarchical clustering.
T-cell Receptor (TCR) Sequencing.
At day 25 after treatment initiation, B78 flank tumors were harvested from mice treated with vehicle only, 50 μCi 90Y-NM600, anti-CTLA-4 (IgG2c), or combination 90Y-NM600 and anti- CTLA-4. Total RNA was extracted using an RNeasy Kit (Qiagen) followed by sample library preparation with two paired long read TCRβ IR-Profile kits (10 unique bar codes per kit, Irepertoire). Pooled libraries were sequenced with miSeq (Illumina). After initial filtering and alignment with the Irepertoire analysis platform the total complementary determining region 3 (CDR3) of TCR β-chains of T cells sequenced for each sample was determined along with the total number of unique CDR3 sequences, the Shannon Diversity Index, and the D50 (a metric for clonal expansion, lower values corresponding to increased clonality) (61).
Canine Trial.
A proof of concept companion canine clinical study was performed to test the bio-distribution and dosimetry of our 86/90Y-NM600 TRT agent in spontaneously metastatic tumors. All procedures were approved by the University of Wisconsin IACUC. 86Y-NM600 (5 mCi, bolus injection) was administered intravenously to a canine patient with metastatic osteosarcoma and another with metastatic oral melanoma and serial PET/CT scans were acquired at 2, 24, and 48h post-injection. The RAPID platform performed patient-specific Monte Carlo dosimetry for 90Y-NM600 using these images. One week after 86Y-NM600 injection, 90Y-NM600 was injected IV at an activity calculated to deliver a mean tumor dose of 2–5 Gy to all tumor sites and a mean bone marrow dose < 1 Gy. Complete blood count (CBC) and comprehensive metabolic panel (CMP) analyses were performed periodically after the administration of either the imaging or TRT compound to assess safety.
Statistical Analysis.
For flow cytometry, RT-PCR, TCR sequencing, and quantification of metastases the statistical significance of observed differences between groups was assessed using a two-way analysis of variance (ANOVA) followed by a Tukey’s post hoc comparisons test. For survival analysis, Kaplan–Meier curves were generated, and a logrank test was performed to test for overall differences between all survival curves. Then Cox regression analysis or individual logrank tests with Bonferroni corrections for multiple comparisons were performed to determine differences. For immune memory, a Chi-square test was performed to assess the statistical significance.
Analyses were performed on GraphPad Prism 7.04. Tumor growth curves were compared using a linear mixed-effects model, including random intercepts for subjects, followed by Tukey’s post hoc comparisons. The tumor volumes were log transformed to account for the log–linear growth pattern. Analyses were performed in R 3.5.0 using the NLME package.
Supplementary Material
Supplementary Methods
Figure S1: Delivery of TRT dose to tumor depends on tumor size
Figure S2: Evaluation for hematological toxicity
Figure S3. Histological evaluation of toxicity
Figure S4. Low-dose TRT enhances response to ICI in male mice.
Figure S5. Radiation volume/field size affects efficacy of RT + ICI
Figure S6: Confirmation of T cell depletion.
Figure S7. PET/CT imaging demonstrates uptake of 86Y-NM600 in B16F10
Figure S8. Tumor growth curves after TRT + ICI treatment in B16F10 and B16F10 Tmem173-/−.
Figure S9. Normalization of flow cytometry results in Figure 6.
Figure S10. Tumor infiltrating T cells from tumors treated with TRT + ICI are more effectively activated after TCR stimulation.
Table S1: Complete response and memory response by treatment group for the TRT dosing study
Table S2: Complete response and memory response by treatment group for the anti-CTLA-4 timing response study
Table S3: Complete response and memory response by treatment group for the TRT with single vs dual checkpoint study
Table S4: Complete response and memory response by treatment group for the B78 two tumor study
Table S5: Canine CBC values after treatment with 90Y-NM600
Table S6: Canine CMP values after treatment with 90Y-NM600
Table S7: List of flow cytometry antibody targets, clones, and fluorophores
Table S8: List of forward and reverse primers utilized for quantitative RT-PCR experiments
Acknowledgments:
We would like to thank Dr. Samuel Bakhoum for sharing his STING KO cell lines, Justin Jeffery in the UW Small Animal Imaging and Radiotherapy Facility, and the staff in the UW Flow Core for their help with this work.
Funding:
This work was supported by the National Institute of Health under the following awards 1K08CA241319-01, 1DP5OD024576-01, U01CA233102, R35CA197078, U54CA232568, TL1TR002375, T32CA009206, T32CA009206, P01CA250972, the University of Wisconsin Carbone Cancer Center Support Grant P30CA014520, A UW 2020 grant from the UWCCC and the UW Graduate School, the Waisman Center Core Grant from the National Institute of Child Health and Human Development U54 HD090256, the NIH MSTP Training Grant T32 GM008692, the Radiological Society of North America Research Fellow Grant RF1716, the American Society of Clinical Oncology Hayden Family Foundation Young Investigator Award 12805, the Midwest Athletes Against Childhood Cancer, the Hillman Cancer Center Early Career Fellowship for Innovative Cancer Research, and the Bentson Translational Research Fellowship. Bristol- Myers Squibb kindly provided all anti-CTLA-4 antibodies for these studies and Archeus Technologies provided NM600 compound.
Footnotes
This manuscript has been accepted for publication in Science Translational Medicine. This version has not undergone final editing. Please refer to the complete version of record at www.sciencetranslationalmedicine.org/. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior written permission of AAAS.”
Competing interests: ZM, JW, RH, and JG have financial interest in Archeus Technologies. ZM is a member of the Scientific Advisory Boards for Archeus Technologies and for Seneca Therapeutics. PS is an unpaid Medical Advisor for Invenra Inc. JW is a co-founder, CSO, and director of Archeus Technologies which holds the license rights to NM600 related technologies. BB and JG are co-founders of Voximetry, Inc and BB is the CSO. The following patents have been applied for or filed by the University of Wisconsin Alumni Research Foundation: Pat No. US 10,736,949 Radiohalogenated Agents for in Situ Immune Modulated Cancer Vaccination with ZM, PS, JW, BB as inventors, Pat No. US 10,751,430 Targeted Radiotherapy Chelates for in Situ Immune Modulated Cancer Vaccination with ZM, PS, JW, BB, PC as inventors, App No. 15/809,427 Using targeted radiotherapy to drive anti-tumor immune response to immunotherapies – ZM, PS, JW, PC, JG, RH as inventors, and US 2011/0060602 A1 Treatment Planning System For Radiopharmaceuticals with BB and JG as inventors.
References and Notes:
- 1.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, Ferrucci PF, Hill A, Wagstaff J, Carlino MS, Haanen JB, Maio M, Marquez-Rodas I, McArthur GA, Ascierto PA, Long GV, Callahan MK, Postow MA, Grossmann K, Sznol M, Dreno B, Bastholt L, Yang A, Rollin LM, Horak C, Hodi FS, Wolchok JD, Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma, N Engl J Med 373, 23–34 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS, Shaheen M, Ernstoff MS, Minor D, Salama AK, Taylor M, Ott PA, Rollin LM, Horak C, Gagnier P, Wolchok JD, Hodi FS, Nivolumab and Ipilimumab versus Ipilimumab in Untreated Melanoma, N Engl J Med 372, 2006–2017 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tawbi HA, Burgess M, Bolejack V, Van Tine BA, Schuetze SM, Hu J, D’Angelo S, Attia S, Riedel RF, Priebat DA, Movva S, Davis LE, Okuno SH, Reed DR, Crowley J, Butterfield LH, Salazar R, Rodriguez-Canales J, Lazar AJ, Wistuba II, Baker LH, Maki RG, Reinke D, Patel S, Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, phase 2 trial, The Lancet Oncology 18, 1493–1501 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ben-Ami E, Barysauskas CM, Solomon S, Tahlil K, Malley R, Hohos M, Polson K, Loucks M, Severgnini M, Patel T, Cunningham A, Rodig SJ, Hodi FS, Morgan JA, Merriam P, Wagner AJ, Shapiro GI, George S, Immunotherapy with single agent nivolumab for advanced leiomyosarcoma of the uterus: Results of a phase 2 study: Nivolumab for Uterine Leiomyosarcoma, Cancer 123, 3285–3290 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu Y-C, Robbins PF, Targeting neoantigens for cancer immunotherapy: Table 1., INTIMM 28, 365–370 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A, Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy, Cell 168, 707–723 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gajewski TF, The Next Hurdle in Cancer Immunotherapy: Overcoming the Non–T-Cell–Inflamed Tumor Microenvironment, Seminars in Oncology 42, 663–671 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi PM, Herati RS, Mansfield KD, Patsch D, Amaravadi RK, Schuchter LM, Ishwaran H, Mick R, Pryma DA, Xu X, Feldman MD, Gangadhar TC, Hahn SM, Wherry EJ, Vonderheide RH, Minn AJ, Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer, Nature 520, 373–377 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, Demaria S, Fractionated but Not Single-Dose Radiotherapy Induces an Immune-Mediated Abscopal Effect when Combined with Anti–CTLA-4 Antibody, Clin Cancer Res 15, 5379–5388 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Demaria S, Kawashima N, Yang AM, Devitt ML, Babb JS, Allison JP, Formenti SC, Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer, Clin Cancer Res 11, 728–734 (2005). [PubMed] [Google Scholar]
- 11.Verbrugge I, Hagekyriakou J, Sharp LL, Galli M, West A, McLaughlin NM, Duret H, Yagita H, Johnstone RW, Smyth MJ, Haynes NM, Radiotherapy Increases the Permissiveness of Established Mammary Tumors to Rejection by Immunomodulatory Antibodies, Cancer Res 72, 3163–3174 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Sharabi AB, Nirschl CJ, Kochel CM, Nirschl TR, Francica BJ, Velarde E, Deweese TL, Drake CG, Stereotactic Radiation Therapy Augments Antigen-Specific PD-1–Mediated Antitumor Immune Responses via Cross-Presentation of Tumor Antigen, Cancer Immunol Res 3, 345–355 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rudqvist N-P, Pilones KA, Lhuillier C, Wennerberg E, Sidhom J-W, Emerson RO, Robins HS, Schneck J, Formenti SC, Demaria S, Radiotherapy and CTLA-4 Blockade Shape the TCR Repertoire of Tumor-Infiltrating T Cells, Cancer Immunol Res 6, 139–150 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brody JD, Ai WZ, Czerwinski DK, Torchia JA, Levy M, Advani RH, Kim YH, Hoppe RT, Knox SJ, Shin LK, Wapnir I, Tibshirani RJ, Levy R, In Situ Vaccination With a TLR9 Agonist Induces Systemic Lymphoma Regression: A Phase I/II Study, JCO 28, 4324–4332 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marabelle A, Tselikas L, de Baere T, Houot R, Intratumoral immunotherapy: using the tumor as the remedy, Annals of Oncology 28, xii33–xii43 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Patel R, Czapar AE, Fiering S, Oleinick NL, Steinmetz NF, Radiation Therapy Combined with Cowpea Mosaic Virus Nanoparticle in Situ Vaccination Initiates Immune-Mediated Tumor Regression, ACS Omega 3, 3702–3707 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Formenti SC, Demaria S, Radiation Therapy to Convert the Tumor Into an In Situ Vaccine, International Journal of Radiation Oncology*Biology*Physics 84, 879–880 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Formenti SC, Rudqvist N-P, Golden E, Cooper B, Wennerberg E, Lhuillier C, Vanpouille-Box C, Friedman K, Ferrari de Andrade L, Wucherpfennig KW, Heguy A, Imai N, Gnjatic S, Emerson RO, Zhou XK, Zhang T, Chachoua A, Demaria S, Radiotherapy induces responses of lung cancer to CTLA-4 blockade, Nat Med 24, 1845–1851 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Theelen WSME, Peulen HMU, Lalezari F, van der Noort V, de Vries JF, Aerts JGJV, Dumoulin DW, Bahce I, Niemeijer A-LN, de Langen AJ, Monkhorst K, Baas P, Effect of Pembrolizumab After Stereotactic Body Radiotherapy vs Pembrolizumab Alone on Tumor Response in Patients With Advanced Non-Small Cell Lung Cancer: Results of the PEMBRO-RT Phase 2 Randomized Clinical Trial, JAMA Oncol (2019), doi: 10.1001/jamaoncol.2019.1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fowler JF, The linear-quadratic formula and progress in fractionated radiotherapy, BJR 62, 679–694 (1989). [DOI] [PubMed] [Google Scholar]
- 21.Golden EB, Apetoh L, Radiotherapy and Immunogenic Cell Death, Seminars in Radiation Oncology 25, 11–17 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, Bratton DL, Oldenborg P-A, Michalak M, Henson PM, Cell-Surface Calreticulin Initiates Clearance of Viable or Apoptotic Cells through trans-Activation of LRP on the Phagocyte, Cell 123, 321–334 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA, Mitotic progression following DNA damage enables pattern recognition within micronuclei, Nature 548, 466–470 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang H, Hu S, Chen X, Shi H, Chen C, Sun L, Chen ZJ, cGAS is essential for the antitumor effect of immune checkpoint blockade, Proc Natl Acad Sci USA 114, 1637–1642 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, Inghirami G, Coleman CN, Formenti SC, Demaria S, DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity, Nat Commun 8, 15618 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mackenzie KJ, Carroll P, Martin C-A, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, Osborn RT, Wheeler AP, Nowotny M, Gilbert N, Chandra T, Reijns MAM, Jackson AP, cGAS surveillance of micronuclei links genome instability to innate immunity, Nature 548, 461–465 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Q, Sun L, Chen ZJ, Regulation and function of the cGAS–STING pathway of cytosolic DNA sensing, Nature Immunology 17, 1142–1149 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Nakamura N, Kusunoki Y, Akiyama M, Radiosensitivity of CD4 or CD8 Positive Human T-Lymphocytes by an in Vitro Colony Formation Assay, Radiation Research 123, 224–227 (1990). [PubMed] [Google Scholar]
- 29.Liu R, Xiong S, Zhang L, Chu Y, Enhancement of antitumor immunity by low-dose total body irradiationis associated with selectively decreasing the proportion and number of T regulatorycells, Cellular & Molecular Immunology 7, 157–162 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu S, Sun X, Luo J, Zhu H, Yang X, Guo Q, Song Y, Sun X, Effects of radiation on T regulatory cells in normal states and cancer: mechanisms and clinical implications, Am J Cancer Res 5, 3276–3285 (2015). [PMC free article] [PubMed] [Google Scholar]
- 31.Balogh A, Persa E, Bogdándi EN, Benedek A, Hegyesi H, Sáfrány G, Lumniczky K, The effect of ionizing radiation on the homeostasis and functional integrity of murine splenic regulatory T cells, Inflamm. Res 62, 201–212 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Arina A, Beckett M, Fernandez C, Zheng W, Pitroda S, Chmura SJ, Luke JJ, Forde M, Hou Y, Burnette B, Mauceri H, Lowy I, Sims T, Khodarev N, Fu Y-X, Weichselbaum RR, Tumor-reprogrammed resident T cells resist radiation to control tumors, Nat Commun 10, 3959 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu S-Z, Nonlinear Dose-Response Relationship in the Immune System following Exposure to Ionizing Radiation: Mechanisms and Implications, Nonlinearity in Biology, Toxicology, Medicine 1, 15401420390844484 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodriguez-Ruiz ME, Garasa S, Rodriguez I, Solorzano JL, Barbes B, Yanguas A, Teijeira A, Etxeberria I, Aristu JJ, Halin C, Melero I, Rouzaut A, Intercellular Adhesion Molecule-1 and Vascular Cell Adhesion Molecule Are Induced by Ionizing Radiation on Lymphatic Endothelium, Int J Radiat Oncol Biol Phys 97, 389–400 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Ware JH, Sanzari J, Avery S, Sayers C, Krigsfeld G, Nuth M, Wan XS, Rusek A, Kennedy AR, Effects of Proton Radiation Dose, Dose Rate and Dose Fractionation on Hematopoietic Cells in Mice, rare 174, 325–330 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maks CJ, Wan XS, Ware JH, Romero-Weaver AL, Sanzari JK, Wilson JM, Rightnar S, Wroe AJ, Koss P, Gridley DS, Slater JM, Kennedy AR, Analysis of white blood cell counts in mice after gamma- or proton-radiation exposure, Radiat Res 176, 170–176 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dudley ME, Cancer Regression and Autoimmunity in Patients After Clonal Repopulation with Antitumor Lymphocytes, Science 298, 850–854 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ellsworth SG, Field size effects on the risk and severity of treatment-induced lymphopenia in patients undergoing radiation therapy for solid tumors, Advances in Radiation Oncology 3, 512–519 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gudkov S, Shilyagina N, Vodeneev V, Zvyagin A, Targeted Radionuclide Therapy of Human Tumors, IJMS 17, 33 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hernandez R, Grudzinski JJ, Aluicio-Sarduy E, Massey CF, Pinchuk AN, Bitton AN, Patel R, Zhang R, Rao AV, Iyer G, Engle JW, Weichert JP, 177Lu-NM600 Targeted Radionuclide Therapy Extends Survival in Syngeneic Murine Models of Triple-Negative Breast Cancer, J Nucl Med 61, 1187–1194 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Snyder F, Wood R, Alkyl and alk-1-enyl ethers of glycerol in lipids from normal and neoplastic human tissues, Cancer Res 29, 251–257 (1969). [PubMed] [Google Scholar]
- 42.Baiu DC, Marsh IR, Boruch AE, Shahi A, Bhattacharya S, Jeffery JJ, Zhao Q, Hall LT, Weichert JP, Bednarz BP, Otto M, Targeted Molecular Radiotherapy of Pediatric Solid Tumors Using a Radioiodinated Alkyl-Phospholipid Ether Analog, J Nucl Med 59, 244–250 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weichert JP, Clark PA, Kandela IK, Vaccaro AM, Clarke W, Longino MA, Pinchuk AN, Farhoud M, Swanson KI, Floberg JM, Grudzinski J, Titz B, Traynor AM, Chen H-E, Hall LT, Pazoles CJ, Pickhardt PJ, Kuo JS, Alkylphosphocholine analogs for broad-spectrum cancer imaging and therapy, Sci Transl Med 6, 240ra75 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheal SM, Xu H, Guo H, Patel M, Punzalan B, Fung EK, Lee S, Bell M, Singh M, Jungbluth AA, Zanzonico PB, Piersigilli A, Larson SM, Cheung N-KV, Theranostic pretargeted radioimmunotherapy of internalizing solid tumor antigens in human tumor xenografts in mice: Curative treatment of HER2-positive breast carcinoma, Theranostics 8, 5106–5125 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choi J, Beaino W, Fecek RJ, Fabian KPL, Laymon CM, Kurland BF, Storkus WJ, Anderson CJ, Combined VLA-4–Targeted Radionuclide Therapy and Immunotherapy in a Mouse Model of Melanoma, J Nucl Med 59, 1843–1849 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grudzinski JJ, Hernandez R, Marsh I, Patel RB, Aluicio-Sarduy E, Engle J, Morris Z, Bednarz B, Weichert J, Preclinical Characterization of 86/90Y-NM600 in a Variety of Murine and Human Cancer Tumor Models, Journal of Nuclear Medicine 60, 1622–1628 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hernandez R, Walker KL, Grudzinski JJ, Aluicio-Sarduy E, Patel R, Zahm CD, Pinchuk AN, Massey CF, Bitton AN, Brown RJ, Sondel PM, Morris ZS, Engle JW, Capitini CM, Weichert JP, 90 Y-NM600 targeted radionuclide therapy induces immunologic memory in syngeneic models of T-cell Non-Hodgkin’s Lymphoma, Communications Biology 2, 1–12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vanpouille-Box C, Pilones KA, Wennerberg E, Formenti SC, Demaria S, In situ vaccination by radiotherapy to improve responses to anti-CTLA-4 treatment, Vaccine 33, 7415–7422 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Muroyama Y, Nirschl TR, Kochel CM, Lopez-Bujanda Z, Theodros D, Mao W, Carrera-Haro MA, Ghasemzadeh A, Marciscano AE, Velarde E, Tam AJ, Thoburn CJ, Uddin M, Meeker AK, Anders RA, Pardoll DM, Drake CG, Stereotactic Radiotherapy Increases Functionally Suppressive Regulatory T Cells in the Tumor Microenvironment, Cancer Immunol Res 5, 992–1004 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wennerberg E, Lhuillier C, Vanpouille-Box C, Pilones KA, García-Martínez E, Rudqvist N-P, Formenti SC, Demaria S, Barriers to Radiation-Induced In Situ Tumor Vaccination, Front. Immunol 8 (2017), doi: 10.3389/fimmu.2017.00229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maleki Vareki S, High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors, j. immunotherapy cancer 6, 157, s40425–018–0479–7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, Robbins PF, Huang J, Citrin DE, Leitman SF, Wunderlich J, Restifo NP, Thomasian A, Downey SG, Smith FO, Klapper J, Morton K, Laurencot C, White DE, Rosenberg SA, Adoptive Cell Therapy for Patients With Metastatic Melanoma: Evaluation of Intensive Myeloablative Chemoradiation Preparative Regimens, JCO 26, 5233–5239 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Okamoto S, Thieme A, Allmann J, D’Alessandria C, Maurer T, Retz M, Tauber R, Heck MM, Wester H-J, Tamaki N, Fendler WP, Herrmann K, Pfob CH, Scheidhauer K, Schwaiger M, Ziegler S, Eiber M, Radiation Dosimetry for 177 Lu-PSMA I&T in Metastatic Castration-Resistant Prostate Cancer: Absorbed Dose in Normal Organs and Tumor Lesions, J Nucl Med 58, 445–450 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Crittenden MR, Zebertavage L, Kramer G, Bambina S, Friedman D, Troesch V, Blair T, Baird JR, Alice A, Gough MJ, Tumor cure by radiation therapy and checkpoint inhibitors depends on pre-existing immunity, Scientific Reports 8, 7012 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wisdom AJ, Mowery YM, Hong CS, Himes JE, Nabet BY, Qin X, Zhang D, Chen L, Fradin H, Patel R, Bassil AM, Muise ES, King DA, Xu ES, Carpenter DJ, Kent CL, Smythe KS, Williams NT, Luo L, Ma Y, Alizadeh AA, Owzar K, Diehn M, Bradley T, Kirsch DG, Single cell analysis reveals distinct immune landscapes in transplant and primary sarcomas that determine response or resistance to immunotherapy, Nature Communications 11, 6410 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aluicio-Sarduy E, Hernandez R, Valdovinos HF, Kutyreff CJ, Ellison PA, Barnhart TE, Nickles RJ, Engle JW, Simplified and automatable radiochemical separation strategy for the production of radiopharmaceutical quality 86Y using single column extraction chromatography, Applied Radiation and Isotopes 142, 28–31 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Besemer AE, Yang YM, Grudzinski JJ, Hall LT, Bednarz BP, Development and Validation of RAPID: A Patient-Specific Monte Carlo Three-Dimensional Internal Dosimetry Platform, Cancer Biotherapy and Radiopharmaceuticals 33, 155–165 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bednarz B, Grudzinski J, Marsh I, Besemer A, Baiu D, Weichert J, Otto M, Murine-specific Internal Dosimetry for Preclinical Investigations of Imaging and Therapeutic Agents:, Health Physics 114, 450–459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morris ZS, Guy EI, Francis DM, Gressett MM, Werner LR, Carmichael LL, Yang RK, Armstrong EA, Huang S, Navid F, Gillies SD, Korman A, Hank JA, Rakhmilevich AL, Harari PM, Sondel PM, In Situ Tumor Vaccination by Combining Local Radiation and Tumor-Specific Antibody or Immunocytokine Treatments, Cancer Res 76, 3929–3941 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Patel RB, Ye M, Carlson PM, Jaquish A, Zangl L, Ma B, Wang Y, Arthur I, Xie R, Brown RJ, Wang X, Sriramaneni R, Kim K, Gong S, Morris ZS, Development of an In Situ Cancer Vaccine via Combinational Radiation and Bacterial‐Membrane‐Coated Nanoparticles, Adv. Mater 31, 1902626 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chaudhary N, Wesemann DR, Analyzing Immunoglobulin Repertoires, Front. Immunol 9 (2018), doi: 10.3389/fimmu.2018.00462. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods
Figure S1: Delivery of TRT dose to tumor depends on tumor size
Figure S2: Evaluation for hematological toxicity
Figure S3. Histological evaluation of toxicity
Figure S4. Low-dose TRT enhances response to ICI in male mice.
Figure S5. Radiation volume/field size affects efficacy of RT + ICI
Figure S6: Confirmation of T cell depletion.
Figure S7. PET/CT imaging demonstrates uptake of 86Y-NM600 in B16F10
Figure S8. Tumor growth curves after TRT + ICI treatment in B16F10 and B16F10 Tmem173-/−.
Figure S9. Normalization of flow cytometry results in Figure 6.
Figure S10. Tumor infiltrating T cells from tumors treated with TRT + ICI are more effectively activated after TCR stimulation.
Table S1: Complete response and memory response by treatment group for the TRT dosing study
Table S2: Complete response and memory response by treatment group for the anti-CTLA-4 timing response study
Table S3: Complete response and memory response by treatment group for the TRT with single vs dual checkpoint study
Table S4: Complete response and memory response by treatment group for the B78 two tumor study
Table S5: Canine CBC values after treatment with 90Y-NM600
Table S6: Canine CMP values after treatment with 90Y-NM600
Table S7: List of flow cytometry antibody targets, clones, and fluorophores
Table S8: List of forward and reverse primers utilized for quantitative RT-PCR experiments