

SUMMARY
Skin is one of the most common sites of host immune response against Staphylococcus aureus infection. Here, through a combination of in vitro assays, mouse models, and intravital imaging, we find that S. aureus immune evasion in skin is controlled by a cascade composed of the ArlRS two-component regulatory system and its downstream effector, MgrA. S. aureus lacking either ArlRS or MgrA is less virulent and unable to form correct abscess structure due to de-repression of a giant surface protein, Ebh. These S. aureus mutants also have decreased expression of immune evasion factors (leukocidins, chemotaxis-inhibitory protein of S. aureus [CHIPS], staphylococcal complement inhibitor [SCIN], and nuclease) and are unable to kill neutrophils, block their chemotaxis, degrade neutrophil extracellular traps, and survive direct neutrophil attack. The combination of disrupted abscess structure and reduced immune evasion factors makes S. aureus susceptible to host defenses. ArlRS and MgrA are therefore the main regulators of S. aureus immune evasion and promising treatment targets.
In brief
Kwiecinski et al. show that Staphylococcus aureus uses the ArlRS-MgrA regulatory system to coordinate gene expression during skin infection. This cascade is required for proper abscess structuring and evasion of the host innate immune system, which together are essential for full S. aureus virulence.
Graphical Abstract
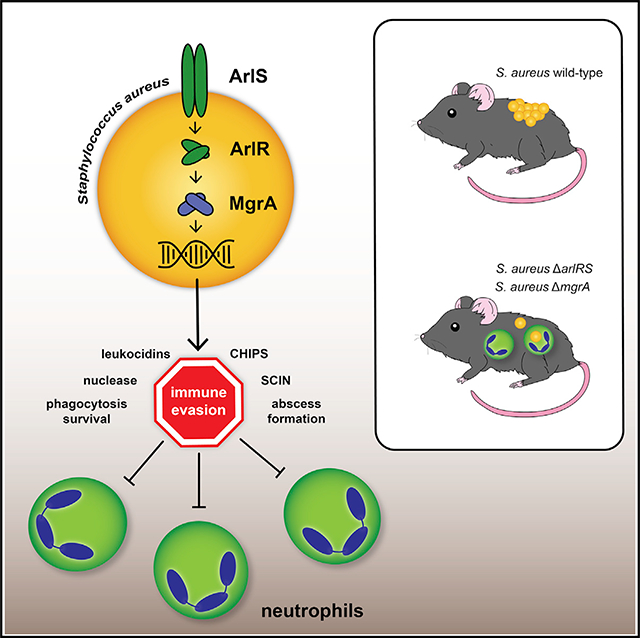
INTRODUCTION
Skin with its underlying tissues is often the first line of defense against pathogens, both as a physical barrier and as the site of the initial immune response. Staphylococcus aureus is the leading cause of the skin and soft tissue infections, with up to 80% of cases attributable to this pathogen (Ray et al., 2013). Skin infections are also the most common type of staphylococcal disease (Jacobsson et al., 2007; Landrum et al., 2012). While S. aureus skin infections usually remain self-limiting, they become recurrent in ~20% of patients (Sreeramoju et al., 2011). They can also lead to systemic spread if the immune system fails to contain the pathogen, which makes skin and soft tissue infections the main source of the S. aureus bacteremia (Wilson et al., 2011). Recently, treatment of staphylococcal skin infections has been increasingly challenging, and associated morbidity, mortality, and healthcare costs have been rising, partly due to growing prevalence of methicillin-resistant S. aureus (MRSA) (Bassetti et al., 2017). In order to design better treatments against S. aureus skin infections, an improved understanding is needed of the mechanisms for establishing infectious foci and evading the local immune response.
Neutrophils are the first immune cells recruited to the site of skin invasion by S. aureus and are essential for clearing S. aureus from the tissue and preventing its systemic spread (Kwiecinski et al., 2014; Mölne et al., 2000). Neutrophils kill the invading pathogen through phagocytosis, production of reactive oxygen species (ROS) and antimicrobial peptides, and trapping bacteria in neutrophil extracellular traps (NETs). At the same time, neutrophils actively create the structure of skin abscesses (Brandt et al., 2018; Kobayashi et al., 2015). In order to mount an effective response to these immune attacks and adapt to the novel skin niche, S. aureus must orchestrate a precise and timely production of a number of virulence factors (Balasubramanian et al., 2017). To achieve such coordination, S. aureus relies on dedicated regulatory systems (Haag and Bagnoli, 2017; Jenul and Horswill, 2019), such as the Agr quorum-sensing system, the SaeRS two-component system, and the CodY nutritional regulator, all of which have been shown to control virulence during skin infections (Cheung et al., 2011; Kobayashi et al., 2011; Montgomery et al., 2010, 2012; Nygaard et al., 2010). However, many other important regulatory systems have not been investigated in this context, including the ArlRS-MgrA regulatory cascade, a system essential for S. aureus virulence in the bloodstream and in other systemic infections (Chen et al., 2009; Crosby et al., 2016b; Gupta et al., 2013; Jonsson et al., 2008; Li et al., 2019; Liu et al., 2014; Radin et al., 2016; Walker et al., 2013). This cascade begins with the ArlRS two-component regulator, which is composed of the membrane-bound sensory kinase ArlS and the response regulator ArlR. After sensing a yet-unknown signal, ArlS phosphorylates ArlR, thus making this DNA-binding regulator active. The activated ArlR in turn drives expression of the regulators MgrA and Spx. The Spx regulator controls S. aureus response to beta-lactam antibiotics and stress, while the global regulator MgrA directly impacts virulence by controlling expression of over 100 effector genes (Bai et al., 2019; Crosby et al., 2020). The importance of this ArlRS and MgrA cascade in systemic bloodstream infections made us suspect that it might play an essential role also in the context of a localized skin infection.
In this work, we identified the ArlRS and MgrA as regulators of the virulence in MRSA skin infection, affecting both skin damage and MRSA survival in the host. These effects were largely due to ArlRS and MgrA regulating MRSA immune evasion through control of virulence factor expression. Immune evasion was also impacted by the altered spatial organization of the abscess where MRSA is shielded from host phagocytes.
RESULTS
ArlRS and MgrA regulate virulence in skin infection
When injected subcutaneously into mice, the wild-type (WT) USA300 MRSA strain LAC (hereafter referred to as MRSA WT) caused a pronounced skin infection, associated with a gradual development of skin necrosis. This virulent process was significantly attenuated when mutants lacking elements of the ArlRS regulatory cascade, either deletions of ΔarlRS or ΔmgrA, were injected. In mutants, necrotic lesions were developing slower and never reached the extent observed in the MRSA WT (Figure 1A). Decreased virulence of the ArlRS and MgrA mutants was also evident when lower dose of MRSA was injected, leading to skin abscess formation rather than to the immediate skin necrosis. In this setting, all strains managed to proliferate to a similar extent during the first day of infection (with a trend toward fewer colony-forming units [CFUs] in mutants). Afterward, the MRSA WT persisted in skin, while CFUs of the ΔarlRS and ΔmgrA mutants decreased significantly (Figure 1B). This accelerated clearance of the mutant strains by the host suggests a defect in their immune evasion mechanisms. The histopathological examination of skin showed that the ΔarlRS and ΔmgrA mutants formed slightly smaller abscesses (Figure 1C), but the most striking differences occurred in bacterial organization within the abscesses, and this was especially noticeable in Gram stains of the tissue (Figure 1C). MRSA WT formed typical abscess structure, with a densely packed bacteria in the center of the abscess, the so-called staphylococcal abscess community (SAC) (Cheng et al., 2011). The ΔarlRS and ΔmgrA mutants failed to form this type of tightly clumped community and were present dispersed across the abscess (Figure 1D). SACs are thought to protect S. aureus from host phagocytes (Cheng et al., 2011); therefore, this improper spatial organization of the cascade mutants could contribute to the accelerated clearance and the reduced virulence of mutant strains.
Figure 1. ArlRS and MgrA control S. aureus skin infection severity.
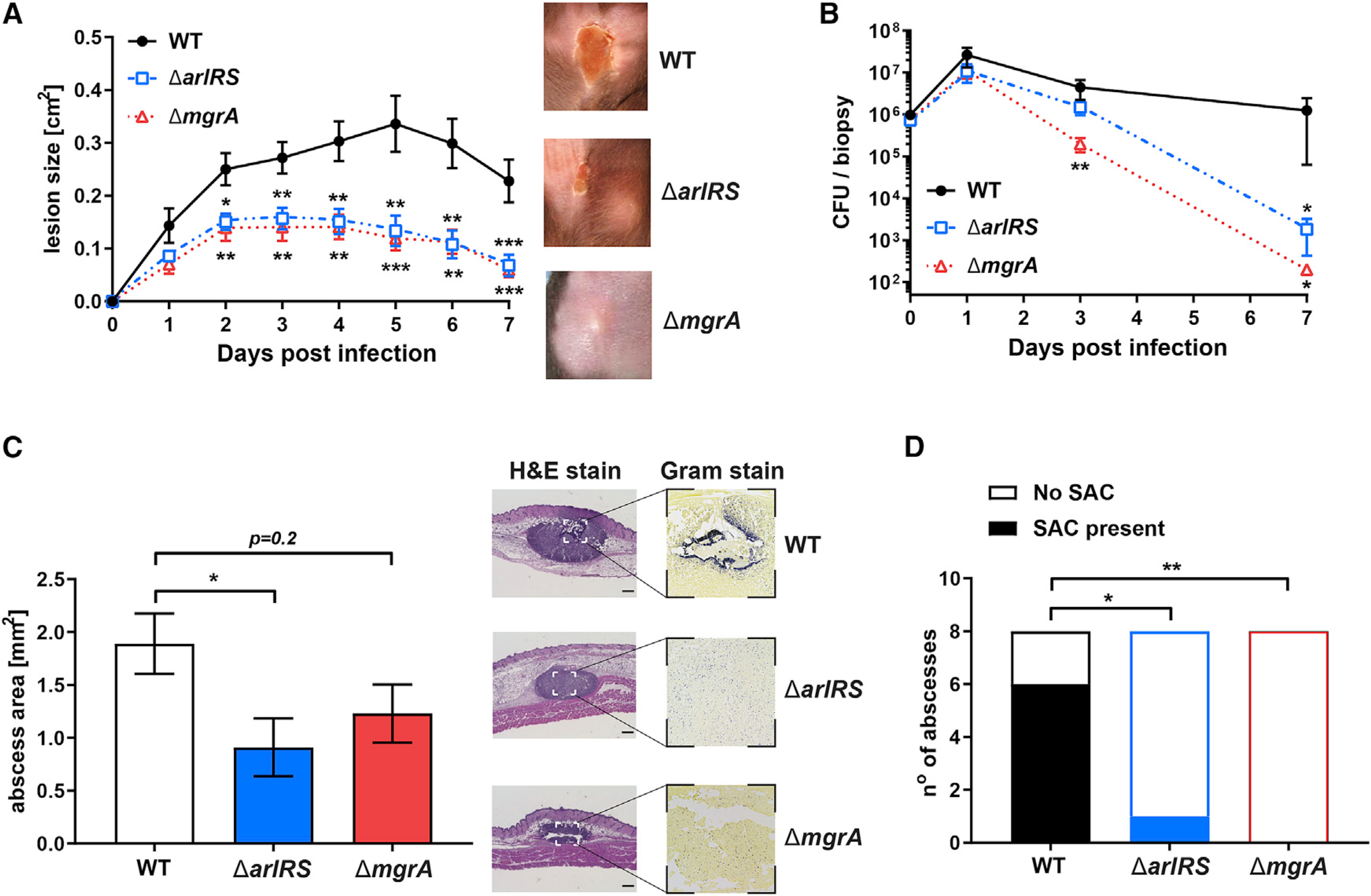
(A) C57BL/6 mice were infected with S. aureus through subcutaneous injection, and developing skin infection was observed. The size of dermonecrotic lesions was measured daily.
(B) On selected days, the infected areas were biopsied, and skin bacterial burden in homogenized biopsy specimens was measured.
(C and D) Additionally, skin biopsy specimens were taken on day 1 of infection, and histopathological sections of the biopsy specimens were used to measure the size of abscesses formed in skin (C) and the presence of tightly clumped staphylococcal abscess communities (SACs) inside these abscesses (D).
Scale bars, 300 μm. Data are shown as mean ± SEM. N = 9 (A), 5–8 (B), and 8 (C and D). *p < 0.05; **p < 0.01; ***p < 0.001. All p values are for comparisons to WT.
ArlRS-MgrA cascade regulates adhesion of S. aureus to skin cells
During infection, S. aureus adheres to and invades host cells, finding inside them a shelter from host phagocytes (Fraunholz and Sinha, 2012; Sinha et al., 1999). When adhesion to dermal fibroblasts was tested, the ΔarlRS and ΔmgrA mutants showed an adhesion defect compared to MRSA WT (Figure S1A). This adhesion deficit disappeared after chromosomal complementation of the missing genes (Figure S1A). The ΔarlRS and ΔmgrA mutants are known to de-repress the expression of the large surface proteins Ebh (extracellular-matrix-binding protein homolog) and SraP (serine-rich adhesin to platelets), which interfere with proper binding of S. aureus to host endothelial cells (Kwiecinski et al., 2019). Therefore, possible role of de-repressed Ebh and SraP in the decreased attachment to skin fibroblast was tested. As predicted, normal adhesion to skin fibroblasts was partially or fully restored when genes for SraP and Ebh were deleted in the ΔarlRS and ΔmgrA backgrounds (Figures S1B and S1C), showing that de-repression of these large proteins is responsible for the observed defect of fibroblast attachment in the cascade mutant strains. It is, however, unlikely that altered interaction with cells in dermis would alone explain either the altered spatial organization or the accelerated immune clearance of the mutant strains.
ArlRS and MgrA regulate spatial organization of S. aureus model in vitro abscesses
To understand the mechanism behind the altered spatial organization inside skin abscesses, we used a three-dimensional model of MRSA growing inside collagen gels in a presence of fibrinogen, which replicates properties of the SAC (Guggenberger et al., 2012). While growing in this dermis-like matrix, MRSA WT formed tightly clumped spherical SAC-like communities (Figure 2A). This process required presence of the fibrinogen; when fibrinogen was not added to the medium, MRSA failed to form a typical tight community and displayed instead a “starburst”-like phenotype of individual loose bacteria detaching from the central community (Figure 2A). Moreover, we could demonstrate that formation of SAC-like community requires bacteria to bind fibrinogen from the media on their surface. As S. aureus expresses a vast repertoire of different fibrinogen-binding proteins (Crosby et al., 2016a), the importance of fibrinogen binding in the in vitro abscess model was tested using Lactococcus lactis, since this bacterium lacks fibrinogen-binding proteins of its own. Only simultaneous expression of a prototypical S. aureus fibrinogen-binding protein ClfA on L. lactis surface, and the presence of fibrinogen in the medium, allowed this bacterium to form tight three-dimensional communities (Figure S2A). This emphasizes the need to bind fibrinogen on bacterial surface in order to properly form a SAC-like abscess structure.
Figure 2. ArlRS and MgrA control formation and immune evasion of model in vitro S. aureus abscess communities.
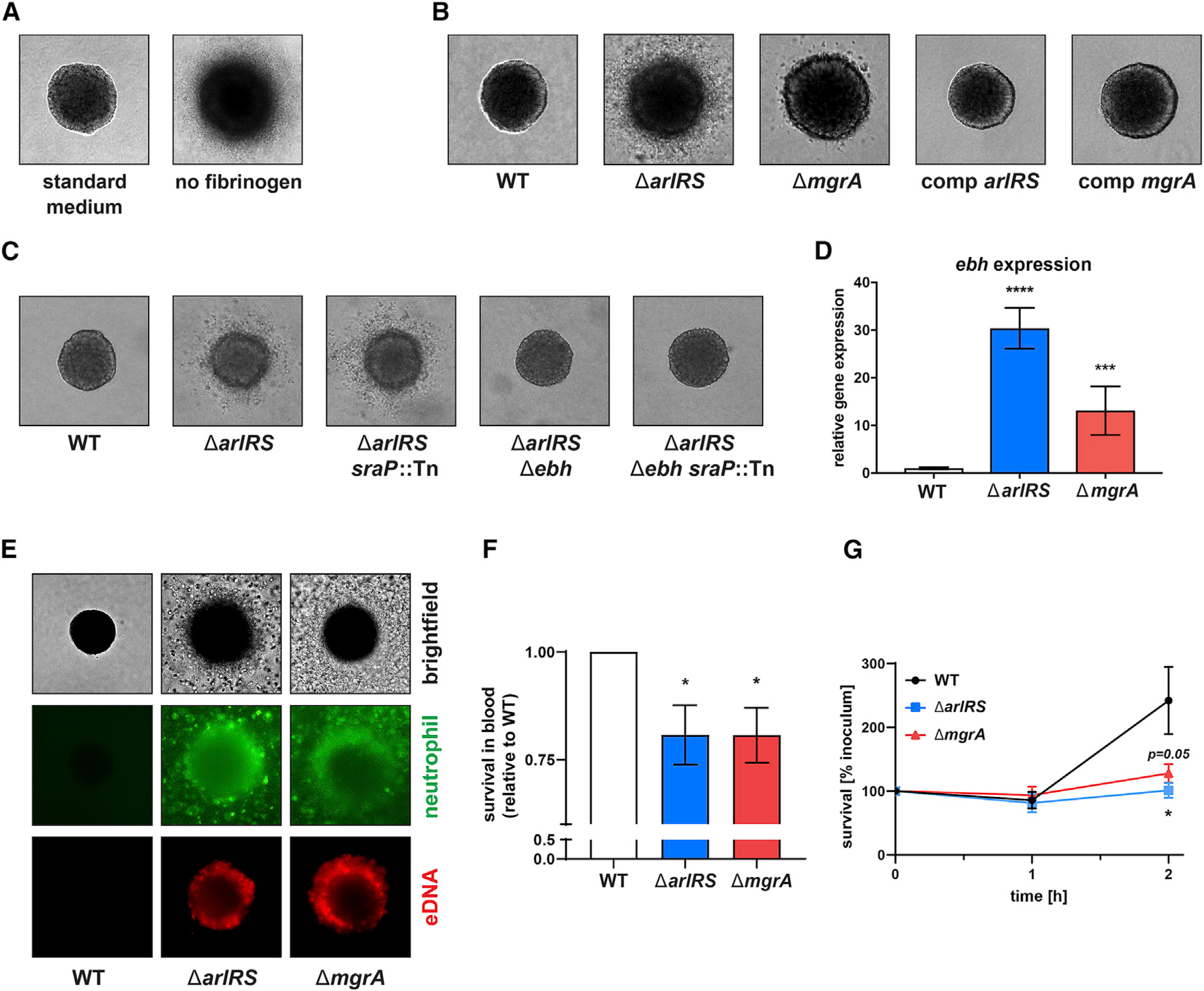
(A–C) Three-dimensional SACs formed from individual S. aureus cells after culturing in collagen/fibrinogen/RPMI gels for 16 h. These were used to determine the role of fibrinogen present in the culture medium (A), the effects of mutations in the ArlRS-MgrA signaling system (B), and the role of giant surface proteins SraP and Ebh in causing the starburst phenotype in the ΔarlRS mutant strains (C).
(D) Expression of ebh in mid-exponential S. aureus RPMI culture was measured with qPCR and normalized to gyrB expression.
(E) Behavior of human neutrophils (stained green with CFDA-SE) 3 h after addition to the in vitro three-dimensional abscess models was also visualized, with propidium iodide (PI) added before imaging to stain extracellular DNA and lysed cells.
(F) Survival of S. aureus after 1-h incubation with fresh human blood was quantified and normalized to WT survival.
(G) Survival of S. aureus co-incubated with purified human neutrophils was measured.
Representative images are shown. Image size: 350 × 350 μm. Data are shown as mean ± SEM. n = 6 (D and F) or 5 (G). *p < 0.05; ***p < 0.001; ****p < 0.0001. All p values are for comparisons to WT. See also Figures S2–S4.
In this in vitro abscess model, the ΔarlRS and ΔmgrA mutants failed to form a correct SAC-like structure and instead showed a starburst phenotype (Figure 2B) that mirrored the WT phenotype without fibrinogen in this assay (Figure 2A). Chromosomal complementation of the arlRS and mgrA genes restored the abscess phenotype to the WT appearance (Figure 2B). The observed defect of the ΔarlRS and ΔmgrA mutants paralleled the in vivo failure to form SAC in the abscess by the mutants in the mouse infection model (Figure 1D).
Previous work with this in vitro model identified Agr quorum-sensing system activation and staphylokinase (Sak) secretion as factors causing spread of bacteria from the abscess community (Guggenberger et al., 2012), but the observed “starburst” phenotype of the ΔarlRS and ΔmgrA mutants was independent from Agr and Sak. The phenotype occurred even in the strains with Δagr or Δsak backgrounds (Figure S2B). The giant surface proteins SraP and Ebh, upregulated in the absence of ArlRS or MgrA, were previously found to prevent S. aureus binding to fibrinogen and to block fibrinogen-induced clumping (Crosby et al., 2016b; Kwiecinski et al., 2019; Walker et al., 2013), making them possible mediators of the starburst phenotype. When tested, lack of SraP in ΔarlRS had no impact on the phenotype, but in the absence of Ebh, the ΔarlRS Δebh mutant regained the WT phenotype (Figure 2C). The same pattern of the starburst phenotype being caused by Ebh (despite the phenotype being overall less pronounced) was observed in the ΔmgrA background (Figure S2C). Moreover, the elevated expression of ebh, but not sraP, correlated with the degree of the phenotype in the ΔarlRS and ΔmgrA mutants (Figures 2D and S2D). This further stresses the role of the Ebh for the starburst phenotype.
As different S. aureus strains often have different repertoire of surface proteins, the effect of ΔarlRS mutation on formation of model abscesses was tested in divergent S. aureus strains. Failure to form the proper three-dimensional SAC in ΔarlRS and ΔmgrA mutants was seen in all strains that harbor the full-length ebh in the genome (strains 502A and MW2), but it did not occur in strains that express only a truncated, nonfunctional version of Ebh (strains N315 and MN8; Figure S3). Altogether, this model suggests that the overexpression of the giant surface protein Ebh, which interferes with bacterial binding of the host fibrinogen, prevents the ArlRS and MgrA mutants from forming the tightly packed SAC and thus from forming the correct spatially organized S. aureus abscesses.
ArlRS and MgrA regulate immune evasion and survival of S. aureus interacting with neutrophils
The failure to form tight SAC exposes the ΔarlRS and ΔmgrA mutants to immune attacks by the host neutrophils infiltrating the infected skin. However, S. aureus produces a vast array of immune evasion factors blocking neutrophil functions or directly killing them, which should allow S. aureus to escape host phagocytes even in the absence of protective abscess structures. It is therefore possible that altered regulation of such factors additionally accelerated clearance of the mutant strains during infection. When purified human neutrophils were added to in vitro abscess models, only a few neutrophils entered into the gel and could be observed in the vicinity of the model SAC of MRSA WT (Figure 2E). Focusing the field of view on the layer immediately above the gel revealed that neutrophils remained on the surface of the gel containing MRSA WT, many of them dead, lysed, or possibly attempting to produce NETs, as demonstrated by propidium iodide staining (Figure S4). In contrast, in the ΔarlRS and ΔmgrA mutants, neutrophils readily entered the gel, approached the model abscesses, and even penetrated the SAC, directly engaging bacteria (Figure 2E). In case of the mutant strains, staining for extracellular DNA demonstrated bright spots corresponding to individual neutrophils (presumably dead, undergoing lysis, or NETosis), as well as a large amount of diffuse staining suggestive of NETs appearing around the periphery of the SAC and ensnaring the whole community (Figure 2E).
Similarly, when different strains were added to human blood in vitro and their survival measured, the ΔarlRS and ΔmgrA mutants were more susceptible than MRSA WT to killing by blood phagocytes (Figure 2F), though this effect in suspension was not as pronounced as in the model abscesses, possibly due to additional phagocyte-independent killing mechanism present in blood. Notably, when mixed directly with isolated human neutrophils, the MRSA WT strain could evade them and began to proliferate, while the ΔarlRS and ΔmgrA mutants were kept under control by neutrophils for up to 2 h (Figure 2G). Altogether, the mutant strains lacking the ArlRS-MgrA cascade demonstrate an immune evasion defect, partly related to failure to form the protective three-dimensional structures in the infected site but apparently also due to some other mechanisms, such as possible failure to block phagocyte recruitment and/or phagocytosis and/or killing inside the phagocytes.
ArlRS and MgrA regulate expression of multiple S. aureus immune evasion genes
To understand mechanisms behind observed changes in immune evasion, we examined our earlier RNA sequencing (RNA-seq) data and noticed decreased expression of several immune evasion genes in the mutant strains (Crosby et al., 2020). We confirmed these RNA-seq data by directly measuring gene expression with qPCR. Expression of the nuclease, bicomponent leukocidins LukSF (Panton-Valentine leukocidin [PVL]) and LukAB, chemotaxis-inhibitory protein of S. aureus (CHIPS), and staphylococcal complement inhibitor (SCIN) were all dramatically reduced in the ΔarlRS and ΔmgrA mutants (Figure 3A).
Figure 3. ArlRS and MgrA control innate immune evasion of S. aureus.
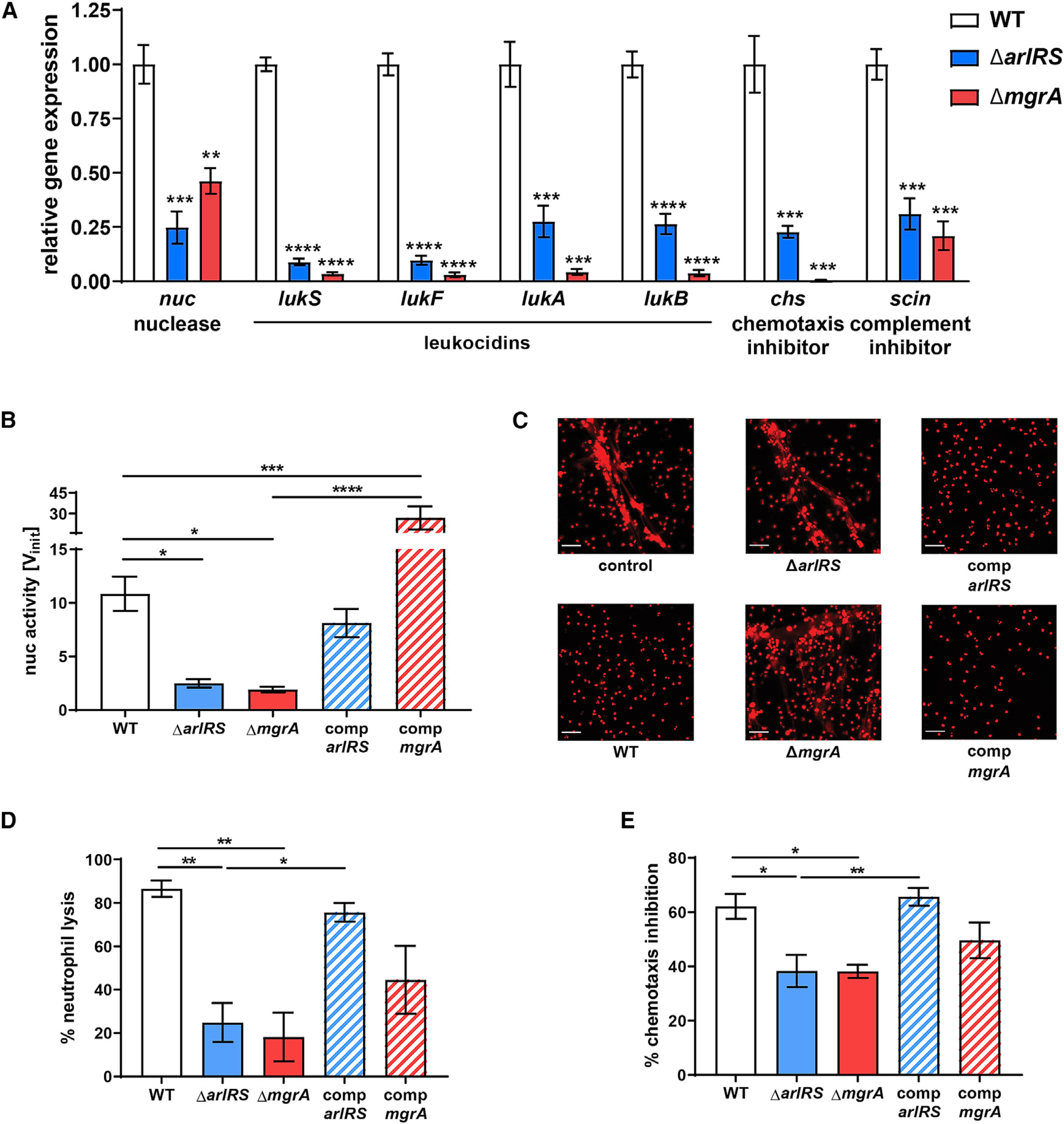
(A) Expression of immune evasion genes in mid-exponential S. aureus RPMI culture was measured with qPCR and normalized to gyrB expression.
(B and C) Nuclease activity in culture supernatants (B) and their ability to digest NETs, visualized with PI (C), were measured.
(D and E) The ability of S. aureus culture supernatants to kill human neutrophils (D) and block neutrophil chemotaxis (E) was measured.
Representative images are shown. Scale bar, 100 μm. Data are shown as mean ± SEM. n = 3 (A), 3–6 (B), 4 (D), and 6 (E). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. In (A), all p values for comparisons to WT. All significant p values between the groups are marked on graphs. See also Figure S5.
ArlRS and MgrA mutants cannot evade neutrophil NETs
Formation of NETs to ensnare the pathogens is an important host defense mechanisms in skin infections (Stephan and Fabri, 2015). Degradation of these NETs by S. aureus nuclease allows the pathogen to evade entrapment and subsequently transform degraded NET fragments into the macrophage-killing deoxyadenosine. Thus, nuclease is responsible for S. aureus immune evasion in abscesses and the spread of bacteria from the infectious foci in skin (Berends et al., 2010; Thammavongsa et al., 2013; Tseng et al., 2012; Wang et al., 2020; Yipp et al., 2012). We therefore investigated if the decreased nuc expression in the ΔarlRS and ΔmgrA mutants leads to their reduced ability to degrade NETs.
The nuclease activity in supernatants of the ΔarlRS and ΔmgrA mutants was significantly reduced (Figure 3B). This reduced activity was restored to the MRSA WT level after chromosomal complementation of arlRS and significantly exceeded the WT level when mgrA was complemented by expressing it with its native promoter from a complementing plasmid (Figure 3B). The reduced nuclease activity translated to a pronouncedly decreased ability of S. aureus to digest NETs. When stimulated human neutrophils created NETs, visible as cords and meshwork of extracellular DNA, the supernatant from MRSA WT readily destroyed these NETs, leaving behind only the nuclear DNA (Figure 3C). This activity was dependent on secretion of staphylococcal nuclease (Figure S5A). The supernatants from the ΔarlRS and ΔmgrA mutants, unlike the WT, did not destroy the NETs but regained the NET-degrading activity upon complementation of the missing genes (Figure 3C). The same pattern was confirmed by quantification of visible NETs with image-processing software (Figure S5B). Altogether, the results point to markedly decreased ability of the mutants lacking ArlRS and MgrA to evade the NETs.
ArlRS and MgrA mutants fail to kill incoming neutrophils or prevent chemotaxis
Bicomponent leukocidins, a group of toxins targeting leukocytes, are produced by S. aureus to lyse the incoming host immune cells (Lewis and Surewaard, 2018; Seilie and Bubeck Wardenburg, 2017; Spaan et al., 2017). This lytic activity can be detected in S. aureus culture supernatants and is dependent on synergistic activity of both LukAB and LukSF (PVL) (Figure S5C). Indeed, we observed that filtered spent media from MRSA WT was able to kill human neutrophils, and in contrast, spent media from the ΔarlRS and ΔmgrA mutants had significantly attenuated neutrophil killing (Figure 3D). This mutant phenotype correlated with their reduced expression of lukSF and lukAB genes seen in the qPCR analysis (Figure 3A). The ability of the ΔarlRS and ΔmgrA mutants to kill neutrophils was fully or partly restored by chromosomal complementation (Figure 3D).
As part of the immune evasion, S. aureus prevents neutrophil recruitment to the infection site (de Haas et al., 2004; de Jong et al., 2019; Lewis and Surewaard, 2018). Indeed, when we tested human neutrophil chemotaxis toward N-formyl-met-leu-phe (fMLP) chemoattractant peptide, we observed a chemotaxis-inhibiting activity of S. aureus spent media that was dependent on CHIPS (Figure S5D). The addition of supernatant from the MRSA WT strain (at a sub-lytic concentration causing no direct killing of assayed neutrophils) led to a marked inhibition of chemotaxis (Figure 3E). When ArlRS-MgrA cascade mutants were tested, they could not inhibit chemotaxis to the same extent as MRSA WT (Figure 3E), in accordance with their reduced expression of chs gene encoding the CHIPS chemotaxis-inhibitory protein (Figure 3A). Mutant strains had their chemotaxis-inhibitory ability restored upon chromosomal complementation of the missing arlRS and mgrA genes (Figure 3E).
ArlRS and MgrA mutants are more susceptible to killing by neutrophil α-defensins and oxygen radicals
Neutrophils directly kill S. aureus by producing antimicrobial peptides (like α-defensins) and ROS (mainly hypochlorite) (Brandt et al., 2018; Kobayashi et al., 2015; Lewis and Surewaard, 2018). When we measured resistance of MRSA to killing by one of human neutrophil α-defensins (HNP-1), the ΔarlRS and ΔmgrA mutants were significantly more susceptible than the WT strain, and this increased susceptibility was reversed by chromosomal complementation of the missing genes (Figure 4A). It has been previously theorized that ΔmgrA mutants might be more resistant to oxidative killing (Chen et al., 2006). Contrary to this hypothesis, we did not observe any changes in resistance to hypochlorite in the ΔarlRS and ΔmgrA mutants (Figure 4B). They were slightly but significantly more susceptible to killing by hydrogen peroxide, which could be reversed by chromosomal complementation of the missing genes (Figure 4C). In accordance with these findings, when survival inside human neutrophils was measured after phagocytosis, MRSA WT was not killed and even managed to proliferate, while the ΔarlRS and ΔmgrA mutants were killed (Figure 4D). Chromosomal complementation of the missing genes restored the ability of mutant strains to proliferate inside neutrophils (Figure 4D). This further explains the observed accelerated immune clearance of the infecting ΔarlRS and ΔmgrA mutants in infected skin.
Figure 4. ArlRS and MgrA control S. aureus survival after neutrophil phagocytosis.
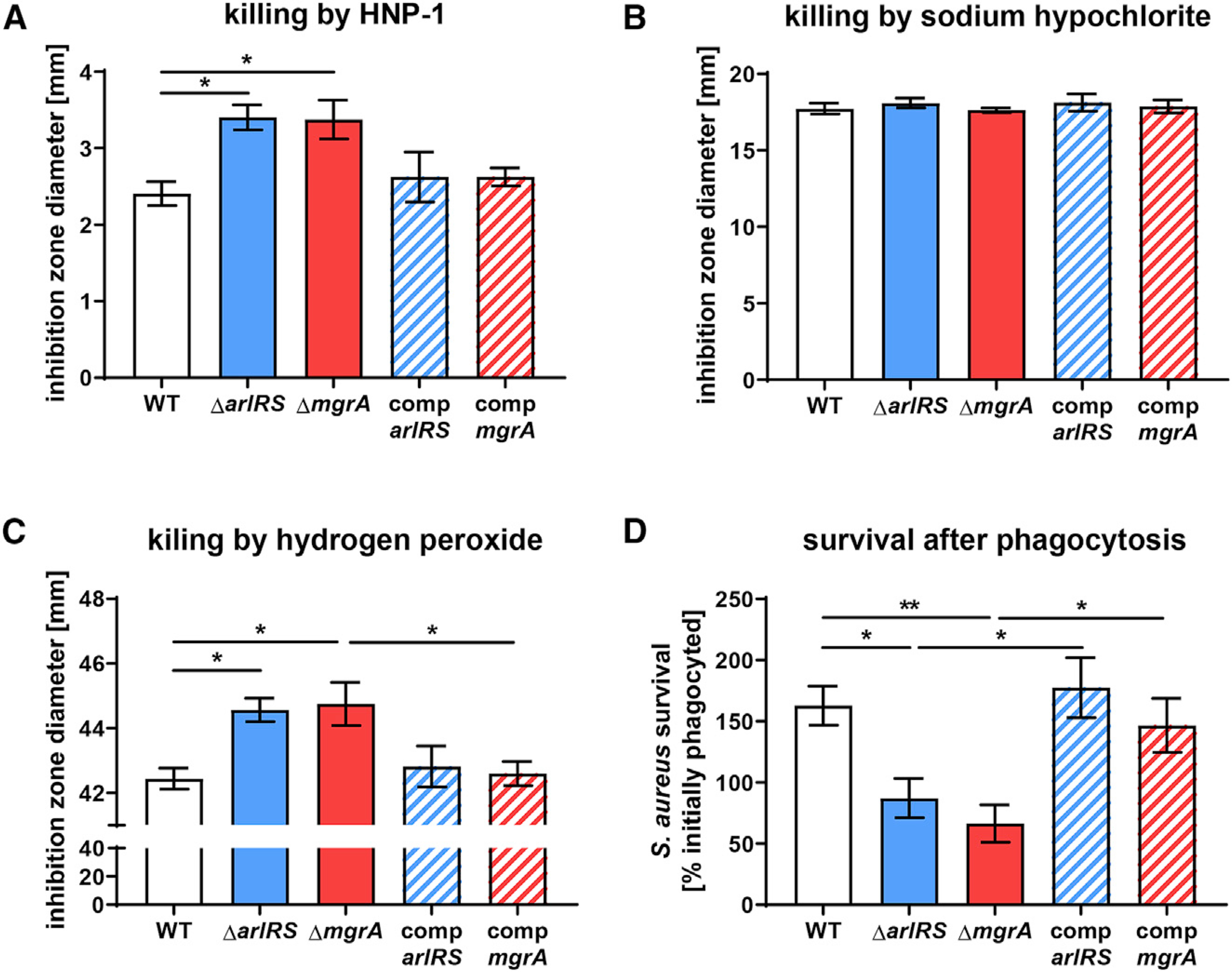
(A–C) Resistance of S. aureus to various compounds used by neutrophils to kill the bacteria, including human α-defensin HNP-1 (A), sodium hypochlorite (B), and hydrogen peroxide (C), was measured with agar diffusion assays. Additionally, survival of S. aureus 1 h after phagocytosis by human neutrophils was measured (D). Data are shown as mean ± SEM. n = 8 (A, C, and D) or 4 (B). *p < 0.05; **p < 0.01. All significant p values between the groups are marked on graphs.
ArlRS and MgrA are needed for evasion of neutrophils in skin in vivo
To observe the ongoing immune evasion during skin infection, we used multiphoton intravital microscopy to visualize interactions between S. aureus and neutrophils in vivo (Figure 5). Neutrophil behavior was tracked using the CatchupIVM-red reporter mouse after infection with MRSA WT or the ΔarlRS and ΔmgrA mutant strains at 24 h post-infection (Figure 5A). We observed the most profound phenotypic differences when the ΔmgrA mutant was used. Neutrophils from mice infected with the ΔmgrA mutant displayed longer track lengths (Figure 5B; Videos S1 and S2) and an overall increase in displacement over time (velocity) compared to neutrophils from mice infected with MRSA WT (Figure 5D). We then analyzed neutrophil 3D localization at the infection site by applying a surface reconstruction on the neutrophil and S. aureus channels (Figure 6A; Videos S3 and S4). Both MRSA WT and the mutants recruited the same number of neutrophils to the general infection site (Figures 6B and 6C), and the total surface volume of S. aureus at the visualized site did not differ between the WT and mutant strains (Figures 6D and 6E). There were, however, marked differences in the way neutrophils behaved toward bacteria within the infection site. Significantly more neutrophils directly interacted with S. aureus in the mice infected with ΔmgrA mutant (Figure 6G), resembling the earlier in vitro observations (Figure 2E). Furthermore, there was a strong trend toward neutrophils infiltrating into the S. aureus layer in ΔmgrA-infected mice (Figure 6I). The ΔarlRS mutant generally presented an intermediate phenotype between the WT and the ΔmgrA, but a significantly increased infiltration of mouse neutrophils into the S. aureus layer also occurred in the ΔarlRS strain (Figure 6H), showing that also this element of the regulatory cascade is needed for efficient evasion of neutrophil attacks. Overall, while MRSA WT was able to limit neutrophil movement and prevent them from directly engaging the growing bacterial community in skin, the strain lacking MgrA (and, to a smaller degree, the strain lacking ArlRS) was unable to induce such immune evasion. These observations from live imaging of in vivo infection confirm the findings from in vitro systems.
Figure 5. ArlRS and MgrA allow S. aureus to affect neutrophil movement during skin infection in vivo.
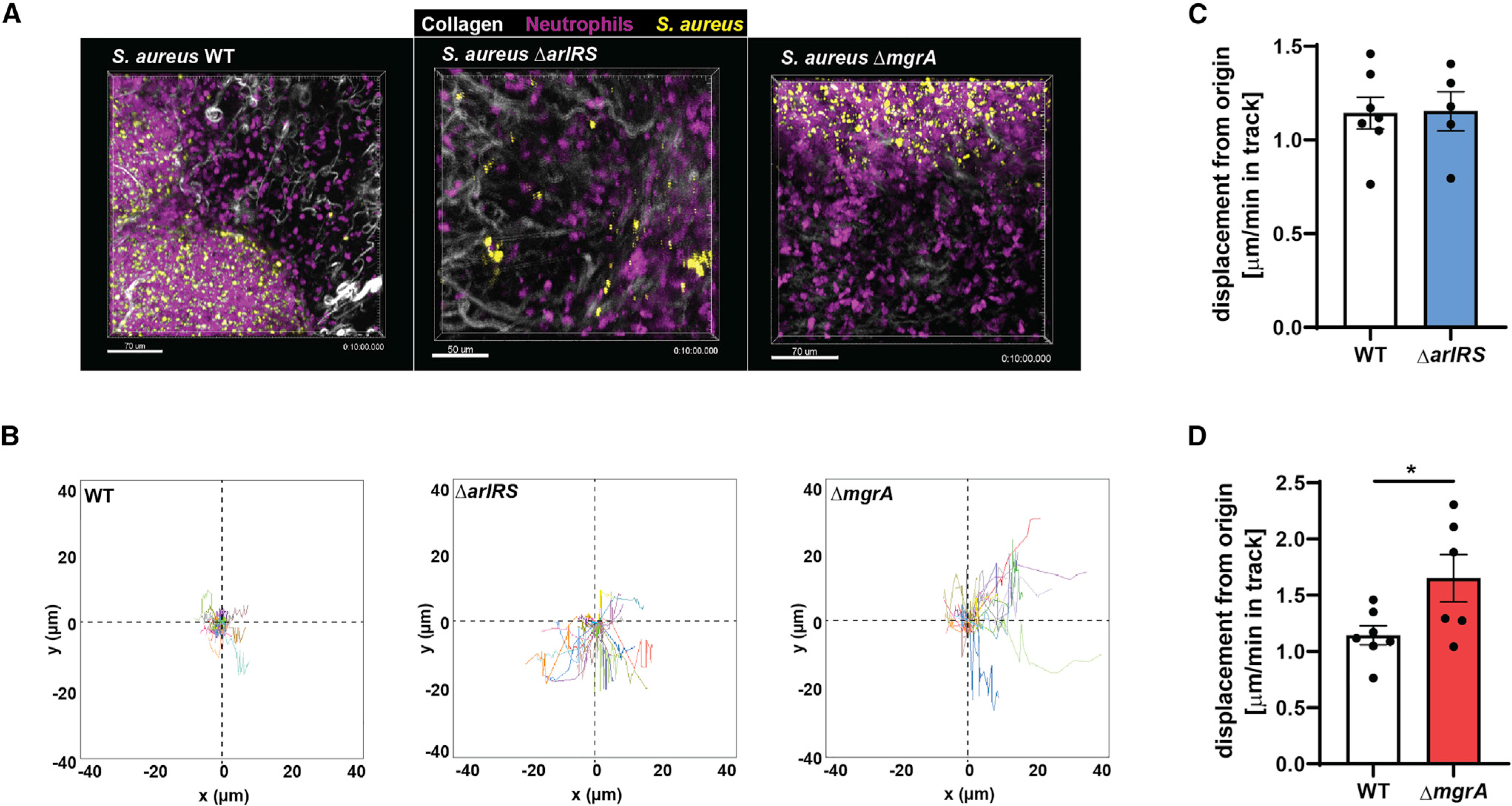
Multiphoton intravital microscopy was used to image neutrophil/S. aureus interactions in vivo for 10 min at 24 h post-infection.
(A) Representative image taken from time-lapse videos showing neutrophils at the infection site from WT, ΔarlRS, and ΔmgrA skin infections.
(B) Quantification of neutrophil track displacement length in the x-y position in S. aureus skin infections.
(C and D) Quantification of the mean displacement of neutrophils per minute (velocity).
Data are shown as mean ± SEM. n = 5–7. *p < 0.05. See also Videos S1 and S2.
Figure 6. ArlRS and MgrA are needed for immune evasion during skin infection in vivo.
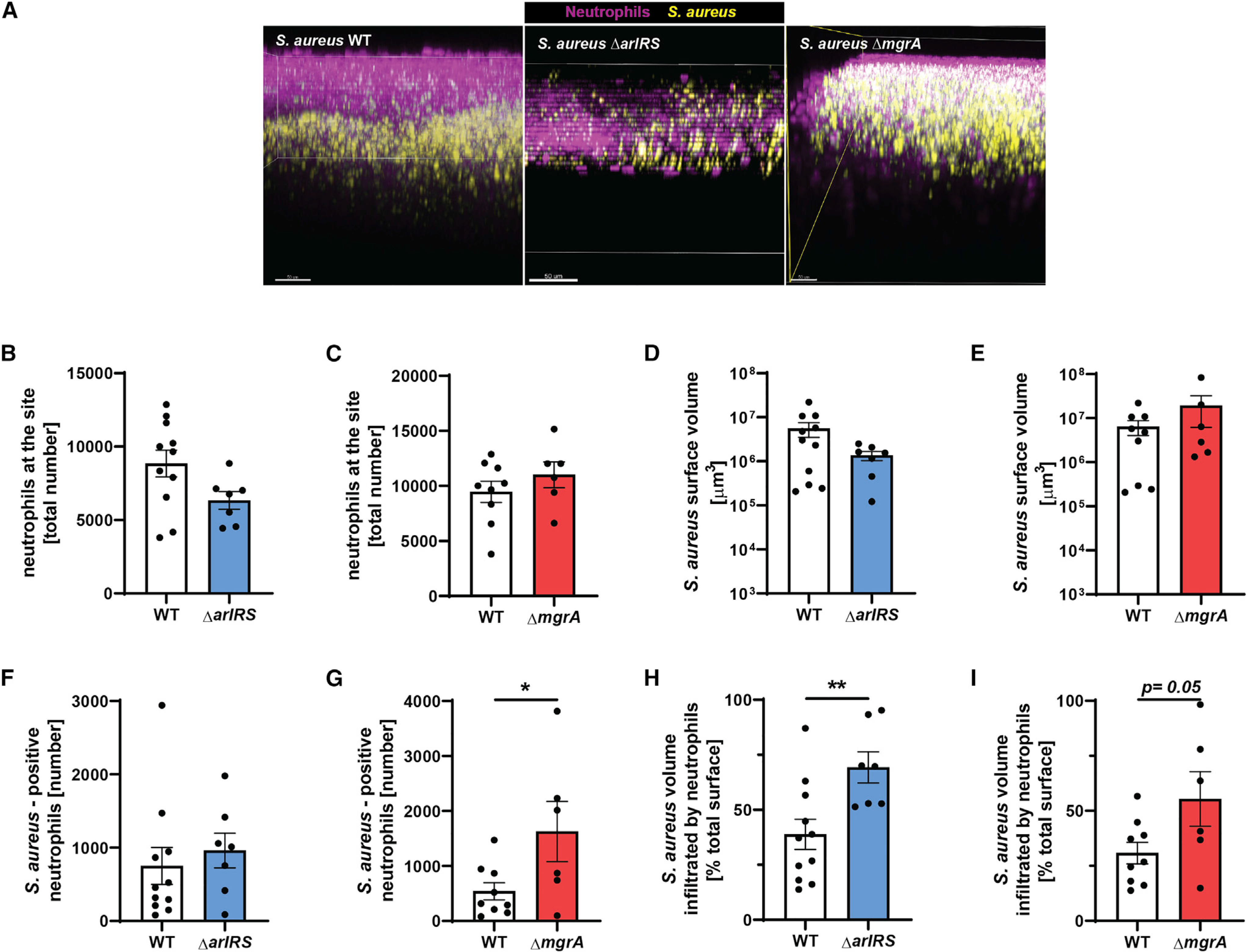
Multiphoton intravital microscopy was used to image neutrophil/S. aureus interactions in vivo at 24 h post-infection.
(A) Representative intravital image showing a three-dimensional stitched image viewed from the x-z plane (side view) showing neutrophil localization at the infection site.
(B and C) Image analysis quantification of total neutrophil spots at the infection site.
(D and E) Total S. aureus surface volume at the infection site.
(F and G) S.-aureus-positive neutrophils.
(H and I) Percentage of S. aureus volume that was infiltrated by neutrophils.
Data are shown as mean ± SEM. n = 6–11. *p < 0.05; **p < 0.01. See also Videos S3 and S4.
DISCUSSION
Skin is the most common site of S. aureus infection and the most common foci of systemic spread. The interplay of bacteria with the immune system dictates the extent of these two events. In its fight against the host’s immune system, S. aureus relies on timely and precisely regulated production of its virulence factors. In this study, we aimed to identify the regulatory system responsible for controlling these diverse direct and indirect mechanisms of immune evasion.
Using a combination of mouse infection models, intravital microscopy, and in vitro models of isolated pathogenic processes, we discovered that the ArlRS-MgrA regulatory cascade controls S. aureus virulence in skin and that this is largely due to its regulation of the staphylococcal immune evasion (outlined in Figure 7). ArlRS and MgrA directly controlled expression of immune evasion factors (nuclease, LukSF, LukAB, CHIPS, and SCIN) and affected resistance to killing by neutrophil antimicrobial peptides and ROS. It also controlled the spatial organization of the S. aureus abscesses, which affected the pathogens’ ability to hide from immune attacks. Thus, our results identify the ArlRS-MgrA regulatory cascade as being central to S. aureus skin virulence and immune evasion. This adds ArlRS and MgrA to the short list of skin virulence regulators (Agr, SaePQRS, and CodY) and expands other reports of ArlRS or MgrA being involved in systemic infections, such as staphylococcal sepsis, endocarditis, arthritis following bacteremia, and muscle infection (Benton et al., 2004; Chen et al., 2009; Crosby et al., 2016b; Gupta et al., 2013; Jonsson et al., 2008; Li et al., 2019; Liu et al., 2014; Radin et al., 2016; Walker et al., 2013). The significance of the ArlRS-MgrA cascade across so many models, both local and systemic, points to it being one of the most important S. aureus regulatory systems for survival in the host. Even though the signal activating ArlRS signaling is still unknown, our data indicate that ArlRS-MgrA is functionally active inside the host’s skin, allowing S. aureus to mount an appropriate immune evasion response.
Figure 7. Proposed model of innate immune evasion control by ArlRS and MgrA during S. aureus infection.
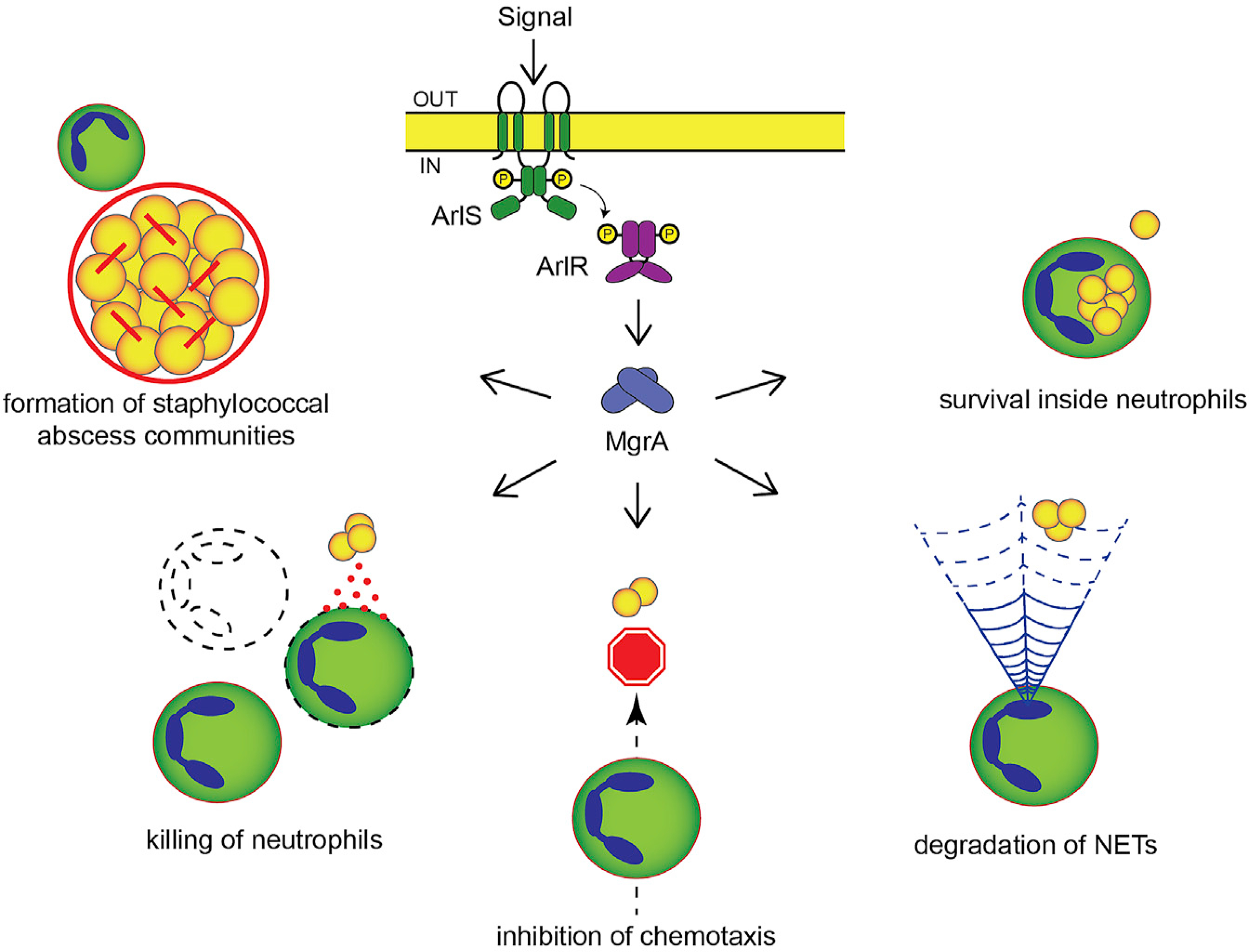
With a functional ArlRS-MgrA cascade, the initial signal detected by the ArlRS two-component system induces expression of the global regulator MgrA, which in turn controls expression of various genes involved in virulence and immune evasion. By suppressing expression of large surface proteins with anti-adhesive properties (Ebh and SraP), the active cascade allows S. aureus to bind fibrinogen and form tight three-dimensional abscess communities where bacteria are shielded from phagocytes. Active cascade also causes S. aureus to secrete various immune evasion factors, such a leukocidins (LukAB and LukSF), CHIPS, SCIN, and nuclease, which together act to kill incoming neutrophils, prevent their chemotaxis and movement, and digest NETs used by neutrophils to ensnare bacteria. Finally, due to the cascade’s involvement with S. aureus resistance to antimicrobial peptides and, to a smaller degree, oxygen radicals, active ArlRS and MgrA promote bacterial survival inside neutrophils after phagocytosis. See also Figure S6.
Abscess formation is an active process, controlled both by the host and by the invading S. aureus (Cheng et al., 2011; Kobayashi et al., 2015). The characteristic structure of S. aureus abscesses includes the SAC in the center, encased in protective layers of fibrinogen and polymerized fibrin, and surrounded by further layers of tissue debris, dead and living neutrophils, and macrophages at the periphery (Brandt et al., 2018; Cheng et al., 2011; Kobayashi et al., 2015). S. aureus virulence factors are also not randomly distributed within abscess but accumulate at distinct parts of the abscess structure (Cheng et al., 2009; Guggenberger et al., 2012). Formation of the central SAC (and the abscess in general) is thought to protect bacteria from attacks of host phagocytes and to create a niche for staphylococcal persistence. S. aureus mutants lacking ArlRS or MgrA components of the regulatory cascade failed to form the usual SAC in the center of the skin abscesses, instead producing a disordered spread of individual cells throughout the entire abscess. As shown before in in vitro models, lack of a typical spatial structure of the abscess could leave individual bacteria exposed to neutrophil phagocytosis (Guggenberger et al., 2012). This probably contributed to the accelerated clearance and the failure of ΔarlRS and ΔmgrA mutant strains to persist. When we investigated the mechanism of this altered spatial organization in in vitro model, it became apparent that it was caused by inability of bacteria to bind host fibrinogen, which acted as an organizing agent for the model SAC, crosslinking individual bacteria to form a tight, fibrinogen-encased three-dimensional structure. This is consistent with previous reports that de-repression of giant surface proteins with interfering activity in the ΔarlRS and ΔmgrA mutants leads to failed clumping and fibrinogen attachment by S. aureus (Crosby et al., 2016b; Kwiecinski et al., 2014). Indeed, the de-repression of the largest of these giant surface proteins (Ebh) prevented formation of tightly clumped S. aureus communities in our three-dimensional model of SAC. The exposure of invading S. aureus ΔarlRS and ΔmgrA mutants to immune attacks was possibly further exacerbated by mutants’ failure to adhere to dermal fibroblasts, preventing bacteria from using the intracellular niche to evade phagocytosis. We demonstrated that this phenotype, previously noted for adhesion to endothelial cells (Kwiecinski et al., 2019; Li et al., 2019; Seidl et al., 2018), was caused by the de-repression of the giant surface proteins SraP and Ebh. The overall observed anti-virulence effects of the de-repressed giant surface proteins, which interfere with typical S. aureus microbial surface components recognizing adhesive matrix molecules (MSCRAMMs) binding to their ligands (Kwiecinski et al., 2019), is consistent with the known importance of the MSCRAMMs in skin infection (Kwiecinski et al., 2014). These spatial and organizational anomalies caused by the altered regulation of giant surface proteins expose infecting S. aureus to immune attacks (Figure S6). Our findings highlight the importance of abscess three-dimensional structure for the outcome of S. aureus skin infections and identify ArlRS and MgrA as the key regulators of the abscess structuring.
At the site of infection, neutrophils kill bacteria through production of ROS and secretion of toxic compounds like antimicrobial peptides. We demonstrated that ΔarlRS and ΔmgrA mutant strains are more susceptible to neutrophil α-defensins, in accordance with earlier observation of functional MgrA being necessary for upregulation of protective mprF and dltA in response to antimicrobial peptide challenge (Li et al., 2019; Matsuo et al., 2010). We also show that lack of ArlRS and MgrA causes small but statistically significant increase in susceptibility to killing by hydrogen peroxide. This might seem counterintuitive, because MgrA was identified as oxidation-sensing molecule in S. aureus, and it was speculated that its absence in a mutant strain would lock the cell into a permanent oxidation-responsive state (Chen et al., 2006). However, other reports indicated that genes directly involved in survival of oxidative stress are regulated not by MgrA but rather by its homolog, SarZ (Chen et al., 2009). Functional MgrA was even shown to be necessary for a correct response to nitric oxide stress (Favazzo et al., 2019). A 2- to 3-fold decrease in expression of staphyloxanthin pigment synthesis genes (responsible for S. aureus resistance to ROS) was also reported in absence of ArlRS or MgrA (Crosby et al., 2020). Altogether, it appears that ArlRS-MgrA cascade is involved to some degree in protection of S. aureus from ROS, though the exact mechanism remains unknown.
In addition to direct killing, neutrophils can ensnare and kill the invading pathogens through the production of NETs, which prevent the spread of bacteria from skin (Stephan and Fabri, 2015; Tseng et al., 2012; Yipp et al., 2012). The nuclease of S. aureus allows it to evade these NETs by digesting their DNA backbone, liberating individual bacteria from the trap (Berends et al., 2010). Failure to produce nuclease in ΔarlRS and ΔmgrA mutants therefore likely contributed to their accelerated immune clearance from the infected skin in our model, adding yet another mechanism to immune evasion control by the ArlRS-MgrA cascade. Considering this increased evasion of NETs-mediated killing and direct killing by defensins and ROS, combined with the previously described involvement of the ArlRS in S. aureus resistance to the neutrophil-induced manganese starvation (Radin et al., 2016), it is evident that the ArlRS-MgrA cascade is necessary for evasion of nearly all types of neutrophil attacks.
Even more important than survival of direct killing by neutrophils is the S. aureus ability to avoid altogether attacks by host phagocytes. S. aureus achieves it by production of immune evasion molecules preventing neutrophils from approaching the infectious foci and killing phagocytes that nevertheless get too close. As we demonstrated, many of these evasion molecules, such as the neutrophil-killing bicomponent leukocidins (Lewis and Surewaard, 2018; Seilie and Bubeck Wardenburg, 2017; Spaan et al., 2017), as well as chemotaxis and complement inhibitors CHIPS and SCIN (de Haas et al., 2004; Rooijakkers et al., 2005), are all regulated by the ArlRS and MgrA system. We observed substantially decreased production of the two leukocidins (LukSF and LukAB) in the ΔarlRS and ΔmgrA mutants, leading to inability of these mutants to kill human neutrophils. This is consistent with previous suggestions that ArlRS might be involved in regulation of leukocidin expression (Crosby et al., 2020; Harper et al., 2018; Párraga Solórzano et al., 2019). Also, expression of both CHIPS and SCIN had decreased in the ΔarlRS and ΔmgrA mutant strains. All this further supports the notion of ArlRS and MgrA being central for immune evasion.
Notably, many of the mentioned immune evasion molecules are human specific. LukSF does not kill mouse neutrophils, while LukAB has only very weak killing ability (Spaan et al., 2017), though S. aureus mutants lacking these leukocidins previously show phenotypes in animal skin infection models, suggesting their limited activity might still play some role (Lacey et al., 2016; Seilie and Bubeck Wardenburg, 2017; Spaan et al., 2017). Similarly, SCIN is inactive, and CHIPS is less active in mouse than in human infections (de Haas et al., 2004; Rooijakkers et al., 2005). This indicates that the ΔarlRS and ΔmgrA mutants would present a much stronger phenotype in a real human infection than the one we observed in our mouse model. This is supported by our observation of a very profound phenotype in the in vitro abscess model with human neutrophils.
To further ascertain our findings, we visualized the real-time S. aureus interaction with neutrophils in the infected skin. Direct visualization of the infectious process inside a living organism, with all its complex multicellular interactions, provides unparalleled possibility to explore mechanistic details and confirm conclusions extrapolated from in vitro or whole-animal experiments (Scott et al., 2019). Observation of neutrophil behavior in skin infected by the WT strain and its counterparts lacking ArlRS and MgrA showed striking differences. On the large scale, both WT MRSA and the mutant strains caused similar recruitment of neutrophils to the infected area. The behavior of the neutrophils in the direct vicinity of the bacteria, however, where immune evasion strategies of S. aureus are acting, showed marked differences. While neutrophils in the WT skin infection had reduced motility and did not engage bacteria directly, in the ΔmgrA mutant infection, neutrophils presented a more actively motile phenotype and were able to access S. aureus. This failure of immune evasion was most evident in the mutant lacking MgrA, but S. aureus lacking ArlRS also could not prevent neutrophils from penetrating inside its colony. The decreased mobility and failure to enter into S. aureus community probably resulted from a combination of the protective fibrinogen layer on surface of bacterial community, decreased neutrophil chemotaxis, and neutrophil damage caused by staphylococcal toxins. As many of the immune evasion molecules of S. aureus are human specific or require high concentration to be active in mouse, it is possible that phenotypes in this model depended largely on the altered structure of bacterial community and fibrinogen deposition and that the anti-chemotactic and neutrophil-killing activities were limited to the area inside and immediately in contact with the S. aureus colony, where concentrations of the immune evasion molecules were the highest. In real-life settings of human infection, we would expect a much more striking combined effect of anti-chemotactic, anti-phagocytic, and neutrophil-killing mechanisms. Despite these reservations, the observation of an overall functional failure of immune evasion during the ongoing skin infection by the cascade mutants confirms our conclusions from the in vitro models.
The structure of the cascade (Figure 7), with MgrA being its final effector, expressed at a low basal level even in the absence of the ArlRS two-component system (Crosby et al., 2016b; 2020), indicates that phenotypes of the ΔarlRS and ΔmgrA mutants should be overlapping, but with a more pronounced phenotype in the ΔmgrA strain. This was indeed the case in majority of the experiments (including mouse infection), but in a few cases, the phenotype of the ΔarlRS strain was more pronounced. This might be due to activity of another global regulator, Spx, also controlled by the ArlRS. However, Spx is responsible for response to beta-lactams and stress and has not been linked to any of the virulent phenotypes in question (Bai et al., 2019; Crosby et al., 2020). It is more likely that the ArlRS-MgrA signaling cascade includes additional undescribed levels of signal integration. Notably, the bulk of our knowledge about the ArlRS-MgrA cascade comes from experiments conducted in rich laboratory bacteriological media under optimal conditions. Our observations indicate that under different environmental conditions, additional regulatory elements are possibly interacting with the cascade.
In conclusion, our work identified the regulatory cascade of ArlRS and MgrA as one of the main regulators involved in S. aureus skin infection, particularly in the development of abscess structure, the interaction with host cells, and evasion of the host immune response. The importance of the ArlRS and MgrA regulatory cascade for skin infections makes it a particularly promising drug target and an alternative to targeting individual virulence factors. By interfering with just this single cascade, one could block multiple and diverse immune evasion mechanisms, rendering S. aureus defenseless against host attacks. Further disentangling of different parts of S. aureus virulence regulation, identification of the relative contribution of individual virulence factors, and understanding of the overlap among host protein binding, abscess structure, and immune evasion will hopefully lead to not only a better understanding of S. aureus biology but also novel treatment strategies.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Alexander R. Horswill (alexander.horswill@cuanschutz.edu).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact and, in some instances, may require a completed materials transfer agreement.
Data and code availability
This paper does not report original datasets.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial strains and plasmids
Bacterial strains and plasmids are listed in the key resources table. S. aureus was grown in tryptic soy broth (TSB) at 37°C with shaking, or in Roswell Park Memorial Institute medium 1640 (RPMI) at 37°C and 5% CO2 with shaking. For CFU counts, samples were serially diluted, plated on tryptic soy agar (TSA), and colonies counted after incubation at 37°C. L. lactis was grown at 30°C, without shaking, in M17 broth with 0.5% glucose. When needed, antibiotics were added to media: chloramphenicol (Cm, 10 μg/ml), erythromycin (Erm, 10 μg/ml, or 5 μg/ml for L. lactis), tetracycline (Tet, 1 μg/ml), spectinomycin (Spc, 1000 μg/ml).
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Bacterial and virus strains | ||
|
| ||
| S. aureus USA300 CA-MRSA, ermS (= LAC) | Boles et al., 2010 | AH1263 |
| S. aureus LAC Δagr::tetM | Kiedrowski et al., 2011 | AH1292 |
| S. aureus LAC Δsak::tetM | This Paper | AH4990 |
| S. aureus LAC nuc::LtrB | Kiedrowski et al., 2011 | AH1680 |
| S. aureus LAC chs:: φNΣ | This Paper | AH4960 |
| S. aureus LAC lukA:: φNΣ | This Paper | AH4963 |
| S. aureus LAC lukS:: φNΣspc | This Paper | AH4987 |
| S. aureus LAC Δebh | Crosby et al., 2016b | AH3150 |
| S. aureus LAC ΔarlRS | Walker et al., 2013 | AH1975 |
| S. aureus LAC ΔarlRS φ11::LL29tet arlRS | Kwiecinski et al., 2019 | AH3244 |
| S. aureus LAC ΔarlRS Δagr::tetM | This Paper | AH3288 |
| S. aureus LAC ΔarlRS Δsak::tetM | This Paper | AH5599 |
| S. aureus LAC ΔarlRS::tetM | Crosby et al., 2016b | AH3520 |
| S. aureus LAC ΔarlRS::tetM Δebh | This Paper | AH3151 |
| S. aureus LAC ΔarlRS::tetM sraP::φNΣ | This Paper | AH3817 |
| S. aureus LAC ΔarlRS::tetM sraP::φNΣ Δebh | This Paper | AH3818 |
| S. aureus LAC ΔmgrA::tetM | Crosby et al., 2016b | AH3455 |
| S. aureus LAC ΔmgrA::tetM φ11::LL29erm mgrA | Crosby et al., 2016b | AH3485 |
| S. aureus LAC ΔmgrA::tetM Δebh | Crosby et al., 2016b | AH3481 |
| S. aureus LAC ΔmgrA::tetM sraP::φNΣ | Crosby et al., 2016b | AH3811 |
| S. aureus LAC ΔmgrA::tetM sraP::φNΣ Δebh | Crosby et al., 2016b | AH3798 |
| S. aureus LAC ΔmgrA | Crosby et al., 2016b | AH3375 |
| S. aureus LAC ΔmgrA Δagr::tetM | This Paper | AH4986 |
| S. aureus ST5 MSSA (= 502A) | Parker et al., 2014 | AH3610 |
| S. aureus 502A ΔarlRS::tetM | Crosby et al., 2016b | AH3624 |
| S. aureus 502A ΔmgrA::tetM | Crosby et al., 2016b | AH3625 |
| S. aureus USA400 MRSA (= MW2) | Baba et al., 2002 | AH843 |
| S. aureus MW2 ΔarlRS::tetM | Crosby et al., 2016b | AH3060 |
| S. aureus MW2 ΔmgrA::tetM | Crosby et al., 2016b | AH3456 |
| S. aureus USA100 MRSA (= N315) | Kuroda et al., 2001 | AH2398 |
| S. aureus N315 ΔarlRS::tetM | Crosby et al., 2016b | AH3082 |
| S. aureus N315 ΔmgrA::tetM | Crosby et al., 2016b | AH3473 |
| S. aureus USA200 MSSA (= MN8) | Schlievert and Blomster, 1983 | AH2413 |
| S. aureus MN8 ΔarlRS::tetM | Crosby et al., 2016b | AH3063 |
| S. aureus MN8 ΔmgrA::tetM | Crosby et al., 2016b | AH3480 |
| L. lactis: surrogate host for ClfA expression | O’Brien et al., 2002 | MG1363 |
|
| ||
| Biological samples | ||
|
| ||
| Whole blood, human | volunteers | N/A |
| Plasma, human | volunteers | N/A |
| Serum, human | Innovative Research | Cat# ISER10ML |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Serum albumin, human | Millipore Sigma | Cat# 12667 |
| Fibrinogen, human | Millipore Sigma | Cat# F3879 |
| Polymorphprep | Accurate Chemical | Cat# AN1114683 |
| Syto 9 stain | Thermo Fisher Scientific | Cat# S34854 |
| Propidium Iodide | Thermo Fisher Scientific | Cat# L10316 |
| Carboxyfluorescein diacetate succinimidyl ester (CFDA-SE) | BioLegend | Cat# 423801 |
| Streptokinase | Millipore Sigma | Cat# S0577 |
| Trypsin | Millipore Sigma | Cat# T4799 |
| DNase I | Millipore Sigma | Cat# DN25 |
| Phorbol 12-myristate 13-acetate (PMA) | Millipore Sigma | Cat# 524400 |
| N-Formyl-Met-Leu-Phe (fMLP) | Millipore Sigma | Cat# F3506 |
| HNP-1 (human α-defensin 1) | AnaSpec | Cat# AS-60743 |
| FRET oligonucleotide substrate | Integrated DNA Technologies (Kavanaugh et al., 2019) | Custom synthesis |
| Sodium hypochlorite | ACROS Organics | Cat# 419550250 |
| Collagen I, rat | BD Biosciences | Cat# 354236 |
|
| ||
| Critical commercial assays | ||
|
| ||
| iTaq Universal SYBR Green Supermix | Bio-Rad | Cat# 1725121 |
| LDH Cytotoxicity Detection Kit | Millipore Sigma | Cat# 11644793001 |
|
| ||
| Experimental models: cell lines | ||
|
| ||
| Dermal fibroblasts, murine | C57BL/6J female mice, isolated according to Khan and Gasser (2016) | N/A |
|
| ||
| Experimental models: organisms/strains | ||
|
| ||
| Mouse: C57BL/6J | Jackson Laboratories | RRID:IMSR_JAX:000664 |
| Mouse: CatchupIVM-red: C57BL/6-Ly6g(tm2621(Cre-tdTomato)Arte) | Hasenberg et al., 2015 | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| See Table S1 | ||
|
| ||
| Recombinant DNA | ||
|
| ||
| pJB38; mutation generation vector, CmR / AmpR | Wörmann et al., 2011 | N/A |
| pCM28; S. aureus - E. coli shuttle vector, CmR / AmpR | Pang et al., 2010 | N/A |
| pCM29; sGFP expression vector, CmR / AmpR | Pang et al., 2010 | N/A |
| pHC66; mgrA complementing vector (pCM28::mgrA), CmR / AmpR | Crosby et al., 2016b | N/A |
| pJK09; sak deletion vector (pJB38 sak::tetM), CmR / AmpR | This Paper | N/A |
| pKS80; L. lactis expression vector, ErmR | O’Brien et al., 2002 | N/A |
| pKS80::clfA; vector for expression of ClfA in L. lactis, ErmR | O’Brien et al., 2002 | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| FIJI - ImageJ | Schindelin et al., 2012 | https://imagej.net/software/fiji/ |
| CFX Manager, v. 3.1 | Bio-Rad | https://www.bio-rad.com/en-us/sku/1845000-cfx-manager-software?ID=1845000 |
| Imaris, v. 9.5.1 | Oxford Instruments | https://imaris.oxinst.com/versions/9-5 |
| Prism, v.7 | GraphPad Software | https://www.graphpad.com/scientific-software/prism |
|
| ||
| Other | ||
|
| ||
| Angiogenesis μ-slide, tissue culture treated | Ibidi | Cat# 81506 |
| 8 well μ-slide, tissue culture treated | Ibidi | Cat# 80826 |
| CFX96 Touch Real-Time PCR System | Bio-Rad | N/A |
| BZ-X710 microscope | Keyence | N/A |
| Eclipse TE2000-E microscope | Nikon | N/A |
| Leica TCS SP8 2-photon microscope | Leica Microsystems | N/A |
| InSight DeepSee laser | Spectra-Physics | N/A |
Mice
Male and female C57BL/6J mice were purchased from the Jackson Laboratories and were housed in groups of two to five in SPF ABSL-2 animal facility of University of Colorado Anschutz Medical Campus. Male and female CatchupIVM-red mice (Hasenberg et al., 2015) were bred in the CCMG facility at the University of Calgary Cumming School of Medicine, and were housed in groups of two to five. All mice were provided with nesting material for enrichment. Mice were 8–10 week (C57BL/6J mice) or 7–8 week (CatchupIVM-red) old when they were used for experiments, and were randomly assigned to experimental groups. At the experiments’ endpoints, mice were euthanized according to local guidelines. Animal experiments were approved by the University of Colorado Institutional Animal Care and Use Committee (protocol 00486) and by the University of Calgary Animal Care Committee (protocol AC19–0138).
Human blood collection
Human heparin-anticoagulated whole blood was collected from anonymous adult volunteers of both sexes and used directly for experiments, or centrifuged at 2000 g for 20 min to obtain plasma. Blood collection was approved by the Colorado Multiple Institutional Review Board (protocol 17–1926).
Mouse dermal fibroblasts
Dermal fibroblasts were isolated from combined ear skin of three 8-week old female C57BL/6J mice and were cultivated in RPMI medium with fetal calf serum, asparagine, glutamine, 2-mercaptoethanol, and penicillin-streptomycin supplementation as described previously (Khan and Gasser, 2016).
METHOD DETAILS
Construction of bacteria mutants
The Δagr::tetM, ΔarlRS::tetM, chs:: ϕNΣ, lukA:: ϕNΣ, and sraP::ϕNΣ (ϕNΣ transposons from Nebraska Transposon Library(Fey et al., 2013)) mutation cassettes were transduced between S. aureus strains with phage 80α or 11, as described previously (Novick, 1991). The lukS:: ϕNΣspc mutant was created by exchanging Erm resistance cassette in the corresponding ϕNΣ Nebraska Transposon Library mutant for Spc resistance, and transducing it into the WT strain (Bose et al., 2013). The Δsak::tetM mutant was created using pJB38 deletion plasmid as described before (Kwiecinski et al., 2019), with regions flanking the sak amplified using primer pairs JK41/JK42 and JK43/JK44 (Table S1), and the constructed cassette was afterward transduced with phage 11.
Mouse skin infection
Previously described murine skin infection model was used (Grundstad et al., 2019; Kwiecinski et al., 2014). S. aureus from mid-log growth phase in TSB was washed with phosphate-buffered saline (PBS), and resuspended for infection in a sterile saline. Abdomens of mice were shaved with a microtome blade, wiped with alcohol pads, and 50 μl of bacteria suspension, containing either 1 × 108 CFU (for necrotic lesion scoring) or 1–2 × 106 CFU (for histology and skin CFU count), were injected subcutaneously through an insulin syringe. Developing lesions were photographed daily, and lesion area was measured with the FIJI software. At pre-determined days mice were euthanized, and the infected skin area was either excised, fixed with a phosphate-buffered 4% formaldehyde (4% PBF), embedded in paraffin, sliced, and stained with hematoxylin and eosin or with a modified tissue gram stain (Becerra et al., 2016), or an 8 mm diameter punch biopsy of the infected area was taken, homogenized, and used for skin CFU counts. The skin histopathology slides were assessed for the abscess area (measured with the FIJI) and the presence of staphylococcal abscess communities by an investigator unaware of the experimental groups.
In vitro staphylococcal abscess community model
A model of S. aureus growing in a 3-dimensional collagen/fibrin(ogen) gel (Guggenberger et al., 2012) was used to model behavior of the staphylococcal abscess community in the skin matrix. Mid-log phase bacteria were suspended in 1.7 mg/ml rat type I collagen solution in RPMI, pH 7.4, at 1 × 105 CFU/ml, and 10 μl of the solution was allowed to solidify for 45 min in wells of an “angiogenesis μ-slide” chamber at 37°C, 5% CO2. Afterward, gel was overlaid with 50 μl of RPMI containing 3 mg/ml human fibrinogen and 5% human plasma, incubated for 16 h at 37°C, 5% CO2, and communities growing inside the gel matrix were afterward imaged with an Eclipse TE2000-E microscope or BZ-X710 microscope. Same procedure was followed for L. lactis, except that 30°C was used for incubation, and gels were stained with 5 μM Syto9 dye to help bacteria visualization.
Neutrophil isolation
Peripheral blood polymorphonuclear leukocytes (PMNs) were isolated from blood of healthy adult human volunteers using Polymorphprep density gradient (Oh et al., 2008), resulting in approximately 95% pure preparation (assessed with Wright-Giemsa stain), and were suspended in RPMI with 2% human serum albumin (RPMI/HSA) for subsequent assays.
Neutrophil challenge of in vitro abscess model
After 16 h growth of S. aureus gel abscess models, the medium above the gels was aspirated, wells washed with PBS, and filled with 50 μl of neutrophils in RPMI/HSA at 3 × 105 PMNs/well. Before addition, neutrophils were stained with carboxyfluorescein diacetate succinimidyl ester (CFDA-SE). After 3 h incubation at 37°C, 5% CO2, propidium iodide was added in order to stain lysed cells and extracellular DNA, and the wells were imaged with BZ-X710 microscope.
Blood survival
To measure S. aureus survival in whole human blood, 50 μl of mid log-phase bacteria suspension in PBS containing 2.5 × 106 CFU was added to 450 μl of human whole blood, and incubated on rotating platform at 37°C for 1 h. Afterward, blood was mixed with 500 μl of PBS with 0.5% saponin, 200 U/ml streptokinase, 100 μg/ml trypsin, 2 U/ml DNase I to lyse cells and break bacterial clumps, surviving CFU counted, and expressed as % of original inoculum (Thomer et al., 2016). As survival of MRSA WT varied from 10% to 70%, depending on a blood donor, surviving % of mutant strains was normalized to WT survival in each donor.
Co-culture with neutrophils
Neutrophils in 100 μl RPMI/HSA were added at 3 × 105 cells /well to 96 well cell culture plates precoated for 1 h at 37°C with 50% human serum in PBS, and were allowed to settle for 15 min at a room temperature. Afterward, 100 μl of RPMI/HSA with 3 × 105 CFU of mid-log phase S. aureus was added to wells (MOI = 1), and to synchronize neutrophil response, the plate was centrifuged for 7 min at 500 g, 4°C. The plate was subsequently incubated at 37°C, 5% CO2, and at the predetermined time points 22 μl of 1% saponin were added per well to lyse all neutrophils, and the viable S. aureus CFU in the well was counted (Lu et al., 2014).
qPCR experiments
For quantitative PCR (qPCR) analysis, MRSA strains were grown in RPMI to a mid-log phase, and their RNA was isolated and transcribed to cDNA as described before (Kwiecinski et al., 2019). qPCR was performed by amplifying 20 ng of cDNA in 20 μl total reaction volume with iTaq Universal SYBR Green Supermix in CFX96 Touch Real-Time PCR System, under the following conditions: 3 min at 95°C, 40 cycles of 15 s at 95°C and 30 s at 55°C, followed by a dissociation curve. “No template” and “no reverse transcription” controls were performed in parallel. Primers for qPCR are listed in Table S1, and primer pairs efficiencies were 85% (ebh), 93% (sraP), 100% (nuc), 86% (lukS), 92% (lukF), 91% (lukA), 93% (lukB), 93% (chs), 91% (scin), and 88% (gyrB). Data were analyzed and Cq determined with CFX manager. Expression was normalized to that of gyrB, and values represent three biological replicates.
Nuclease activity
Supernatants from 16–18h S. aureus cultures in RPMI were used to quantify nuclease activity using the previously described Förster resonance energy transfer (FRET) assay (Kavanaugh et al., 2019). To be in the linear range of the assay, supernatants were diluted 100 × with distilled water. Nuclease activity was expressed as the initial rate of the DNA cleavage reaction (Vinit).
NETs degradation
To visualize degradation of neutrophil extracellular traps (NETs) by supernatants from 16–18h S. aureus cultures in RPMI, a previously described method was used (Schilcher et al., 2014). Neutrophils in RPMI/HSA were seeded into “μ-slide 8 well” coverslip chambers at 2 × 105 cells per 1 cm2, and stimulated with 25 nM phorbol 12-myristate 13-acetate (PMA) for 90 min at 37°C, 5% CO2, to induce NET formation. Afterward, culture supernatants diluted 20 × in RPMI were added to the chambers and incubated for 30 min at 37°C, 5% CO2, to allow degradation of NETs. Chambers were fixed with 4% PBF for 15 min, DNA was stained with 20 μM propidium iodide, and slides were imaged with the BZ-X710 microscope. To quantify amount of remaining NETs, in another set of experiments fixation with PBF was omitted in order for propidium iodide to stain exclusively the extracellular DNA in NETs and the cells undergoing lysis or NET secretion, and stained % of total area of random fields of view was measured with the FIJI software.
Killing of neutrophils
To measure killing of neutrophils by supernatants from 16–18h S. aureus cultures in RPMI, a previously described assay was used (Dumont et al., 2011). Neutrophils were seeded at 1 × 105 cells per well into 96-well plate in 90 μl of RPMI/HSA, and 10 μl of bacterial supernatants were added (final concentration of 10%). After 3h incubation at 37°C, 5% CO2, the plates were centrifuged at 250g, 10 min, and resulting supernatants were used to measure lactate dehydrogenase (LDH) leakage from damaged cells as the marker of neutrophil lysis with an LDH Cytotoxicity Detection Kit. The % neutrophil lysis was calculated using neutrophils incubated with 10% of RPMI as “0% lysis” control, and incubated with 0.2% Triton X-100 as “100% lysis” control.
Chemotaxis inhibition
To measure inhibition of neutrophil chemotaxis by supernatants from 16–18 h S. aureus cultures in RPMI, an under-agarose chemotaxis method was used (Hii et al., 2004). Two 2 mm diameter wells were punched 5 mm apart in 0.5% agarose/RPMI gel, and filled with 5 μl of 1 × 107 neutrophils in RPMI/HSA with 5% of bacterial supernatants (first well) or with 5 μl of 5 × 10−7 M N-Formyl-Met-Leu-Phe (fMLP) chemoattractant peptide (second well). After 90 min at 37°C, 5% CO2, images were taken with the BZ-X710 microscope, and the distance traveled by neutrophils under the agarose from the border of their well toward the well with the fMLP was measured. The % inhibition of chemotaxis was calculated in comparison to neutrophils mixed with 5% RPMI instead of supernatant (positive chemotaxis control). The supernatant concentration used (5%, 90 min incubation) was sub-lytic, confirmed by aspiration of neutrophils remaining in the well after the experiment, and staining them with trypan blue dye for viability measurement, consistently showing above 90% viability.
S. aureus susceptibility to α-defensins and oxygen radicals
Agar radial diffusion assays were used to measure susceptibility of the S. aureus from mid-log TSB culture. Agar overlay technique was used to detect zones of S. aureus growth inhibition caused by human α-defensin neutrophil peptide 1 (HNP-1) (Lehrer et al., 1991). Standard EUCAST disk diffusion susceptibility testing method (version 8.0) was used to detect susceptibility to ROS, except that instead of using antimicrobial disks, a 5 mm dimeter whole was punched in agar plates and filled with 10 μl of 10% hydrogen peroxide, or 20 μl of sodium hypochlorite solution at concentration equal to 2.5% active chlorine.
S. aureus survival after phagocytosis by neutrophils
A previously described method to measure intraphagosomal killing of S. aureus by neutrophils in suspension was adapted for adherent neutrophils (Pang et al., 2010). Human neutrophils in Hank’s Balanced Salt Solution with calcium and magnesium, 10% human serum, and 1% human serum albumin (HBSS+++) were placed in 48-well cell culture plate at 250 μl containing 1 × 105 cells per well, centrifuged at 300 g for 3min, and allowed to adhere to well bottom for 30 min at 37°C. S. aureus from mid-log phase was opsonized by incubation in HBSS+++ for 15 min at 37°C, and was added to wells with neutrophils at MOI = 10 in 250 μl volume. The plates were centrifuged at 500 g for 5 min to put bacteria in a direct contact with the neutrophils, and were incubated at 37°C for 15 min to allow for phagocytosis. Afterward, wells were extensively washed to remove non-phagocyted S. aureus, filled with 250 μl fresh HBSS+++, and incubated at 37°C to allow for the killing of the ingested bacteria. After 1 h, the medium was aspirated from the wells, the neutrophils were lysed by filling the wells with 250 μl of 1% saponin, and viable CFUs were counted. The same lysis and counting procedure was performed on parallel wells immediately before the 1 h incubation to determine the baseline 100% viable CFU.
S. aureus adhesion to fibroblasts
Mouse dermal fibroblasts were grown to confluency in “μ-slide 8 well” coverslip chambers and washed with PBS. Mid-exponential phase S. aureus strains carrying sGFP-expressing plasmid were washed with PBS, resuspended in unsupplemented RPMI, and added to fibroblasts at MOI = 20. After 1h incubation at 37°C, 5% CO2, medium was aspirated, wells washed with PBS, adhered fluorescent bacteria in 5 random sites per each chamber were visualized with a BZ-X710 microscope, and % of area with adhered bacteria was measured with the FIJI software.
Intravital microscopy
Resonant-scanning multiphoton microscopy was used to image the skin of CatchupIVM-red mice, in which neutrophils are tagged with a tdTomato red fluorescent protein (Hasenberg et al., 2015), and which was infected by subcutaneous injection of 5 × 106 CFU of S. aureus strains expressing sGFP into a back flank. Mice were anaesthetized with xylazine and ketamine and a jugular catheter was inserted to maintain anesthesia as previously described (Yipp et al., 2012). Superfusion buffer (HBSS with no calcium, magnesium, nor phenol red) was then perfused across the exteriorized skin tissue to keep the skin moist at a flow rate set to 0.05.
The infected dermal skin tissue was imaged with a Leica SP8 2-photon microscope, equipped with 25 × 0.95 NA water objective lens, two InSight DeepSee pulsed infrared lasers (fixed 1040 nm and tunable 680–1300 nm) and a high speed 8 kHz resonant scanner. Laser excitation at 940 nm was used to excite tdTomato and GFP with external detectors (HyD-RLD2 BP 585/40 for tdTomato, HyD-RLD3 BP 525/50 for GFP) and second harmonic generation (external detector HyD-RLD4 BP 450/70) to visualize skin collagen. Laser power, detector settings and acquisition settings were maintained throughout each experiment.
Intravital image analysis
A 3D tile scan (4 × 4 fields of view; each field 350 × 350 × 100 μm3) was first collected to get an overview of the infection region. From there, 3 fields of view (350 × 350 × 50 μm3) were selected within the infection region to select the time-lapse positions. Images were collected every 30 s for 20 minutes to capture dynamic neutrophil behavior. The first 10 minutes of each video was analyzed using Imaris version 9.5.1 (Oxford Instruments). Pre-processing of videos included the MATLAB extension “normalize time points” to exclude voxels less than 1, and a manual stabilization to minimize translational drift. From there, neutrophil spots were detected with automated thresholding and tracks were detected with Brownian motion. The neutrophil X,Y,Z- position for each time point was exported and track displacement and velocity was analyzed in R.
Neutrophil spots from the 3D tile scan were detected using automated thresholding and default spot settings. S. aureus surface volume was detected using manual thresholding and specific threshold values was noted for each mouse. A masked channel was applied to the S. aureus surface and the “intensity max Ch-x(S. aureus mask)” filter was applied to quantify S. aureus-positive neutrophils (a measure of neutrophils interacting with S. aureus). Bacterial discovery was analyzed by creating a total neutrophil surface, applying a mask to the neutrophil channel and filtering the S. aureus surface as “intensity max Ch-y(neutrophil mask)” (a measure of infiltration of S. aureus layer by neutrophils).
QUANTIFICATION AND STATISTICAL ANALYSIS
For each experiment, the number of replicates (N) and manner of data presentation (definition of center and precision of measures) is provided in figure legends. For all assays, data were pooled from at least two independent experiments. Differences between S. aureus strains were analyzed by ANOVA with a Dunnett’s multiple comparisons post-test (for comparison of mutant groups to WT strain) or Sidak’s multiple comparison post-test (for comparison of multiple groups with each other); by Kruskal-Wallis test with a Dunn’s multiple comparisons post-test (for CFU); by chi-square test with Bonferroni correction (for presence of SAC in abscesses); or by unpaired t test (for assays with only 2 groups). Two-tailed p values were calculated, and p < 0.05 was considered significant. Prism software was used for statistical calculations. For non-quantitative microscopy images, two independent experiments were performed and representative images are shown.
Supplementary Material
Highlights.
S. aureus requires the ArlRS-MgrA regulatory cascade for virulence in skin infection
Inactivation of ArlRS or MgrA blocks S. aureus adhesion and abscess structuring
The ArlRS-MgrA cascade is essential for S. aureus immune evasion
ACKNOWLEDGMENTS
We thank Joan Geoghegan (Trinity College, University of Dublin) for providing L. lactis strains and plasmids; the UC Anschutz histology cores at the Gates Center for Regenerative Medicine and the Division of Pulmonary Sciences and Critical Care Medicine for help processing skin biopsy specimens; and Katrin Schilcher and Young-Saeng Cho for help with illustrations. A.R.H. was supported by merit award BX002711 from the Department of Veterans Affairs and NIH public health service grants AI141490 and AI153185. J.M.K. was supported by American Heart Association postdoctoral fellowship 17POST33670580. R.M.K. was supported by the Alberta Graduate Excellence Scholarship from the Government of Alberta. Intravital imaging was supported by a foundation grant from the Canadian Institutes of Health Research (FDN-143248).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109462.
REFERENCES
- Baba T, Takeuchi F, Kuroda M, Yuzawa H, Aoki K, Oguchi A, Nagai Y, Iwama N, Asano K, Naimi T, et al. (2002). Genome and virulence determinants of high virulence community-acquired MRSA. Lancet 359, 1819–1827. [DOI] [PubMed] [Google Scholar]
- Bai J, Zhu X, Zhao K, Yan Y, Xu T, Wang J, Zheng J, Huang W, Shi L, Shang Y, et al. (2019). The role of ArlRS in regulating oxacillin susceptibility in methicillin-resistant Staphylococcus aureus indicates it is a potential target for antimicrobial resistance breakers. Emerg. Microbes Infect 8, 503–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian D, Harper L, Shopsin B, and Torres VJ (2017). Staphylococcus aureus pathogenesis in diverse host environments. Pathog. Dis 75, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassetti M, Carnelutti A, and Righi E (2017). The role of methicillin-resistant Staphylococcus aureus in skin and soft tissue infections. Curr. Opin. Infect. Dis 30, 150–157. [DOI] [PubMed] [Google Scholar]
- Becerra SC, Roy DC, Sanchez CJ, Christy RJ, and Burmeister DM (2016). An optimized staining technique for the detection of Gram positive and Gram negative bacteria within tissue. BMC Res. Notes 9, 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benton BM, Zhang JP, Bond S, Pope C, Christian T, Lee L, Winter-berg KM, Schmid MB, and Buysse JM (2004). Large-scale identification of genes required for full virulence of Staphylococcus aureus. J. Bacteriol 186, 8478–8489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berends ET, Horswill AR, Haste NM, Monestier M, Nizet V, and von Köckritz-Blickwede M (2010). Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J. Innate Immun 2, 576–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boles BR, Thoendel M, Roth AJ, and Horswill AR (2010). Identification of genes involved in polysaccharide-independent Staphylococcus aureus biofilm formation. PLoS ONE 5, e10146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose JL, Fey PD, and Bayles KW (2013). Genetic tools to enhance the study of gene function and regulation in Staphylococcus aureus. Appl. Environ. Microbiol 79, 2218–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt SL, Putnam NE, Cassat JE, and Serezani CH (2018). Innate Immunity to Staphylococcus aureus: Evolving Paradigms in Soft Tissue and Invasive Infections. J. Immunol 200, 3871–3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PR, Bae T, Williams WA, Duguid EM, Rice PA, Schneewind O, and He C (2006). An oxidation-sensing mechanism is used by the global regulator MgrA in Staphylococcus aureus. Nat. Chem. Biol 2, 591–595. [DOI] [PubMed] [Google Scholar]
- Chen PR, Nishida S, Poor CB, Cheng A, Bae T, Kuechenmeister L, Dunman PM, Missiakas D, and He C (2009). A new oxidative sensing and regulation pathway mediated by the MgrA homologue SarZ in Staphylococcus aureus. Mol. Microbiol 71, 198–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng AG, Kim HK, Burts ML, Krausz T, Schneewind O, and Missiakas DM (2009). Genetic requirements for Staphylococcus aureus abscess formation and persistence in host tissues. FASEB J 23, 3393–3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng AG, DeDent AC, Schneewind O, and Missiakas D (2011). A play in four acts: Staphylococcus aureus abscess formation. Trends Microbiol 19, 225–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung GY, Wang R, Khan BA, Sturdevant DE, and Otto M (2011). Role of the accessory gene regulator agr in community-associated methicillin-resistant Staphylococcus aureus pathogenesis. Infect. Immun 79, 1927–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby HA, Kwiecinski J, and Horswill AR (2016a). Staphylococcus aureus Aggregation and Coagulation Mechanisms, and Their Function in Host-Pathogen Interactions. Adv. Appl. Microbiol 96, 1–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby HA, Schlievert PM, Merriman JA, King JM, Salgado-Pabón W, and Horswill AR (2016b). The Staphylococcus aureus Global Regulator MgrA Modulates Clumping and Virulence by Controlling Surface Protein Expression. PLoS Pathog 12, e1005604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby HA, Tiwari N, Kwiecinski JM, Xu Z, Dykstra A, Jenul C, Fuentes EJ, and Horswill AR (2020). The Staphylococcus aureus ArlRS two-component system regulates virulence factor expression through MgrA. Mol. Microbiol 113, 103–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haas CJ, Veldkamp KE, Peschel A, Weerkamp F, Van Wamel WJ, Heezius EC, Poppelier MJ, Van Kessel KP, and van Strijp JA (2004). Chemotaxis inhibitory protein of Staphylococcus aureus, a bacterial antiinflammatory agent. J. Exp. Med 199, 687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong NWM, van Kessel KPM, and van Strijp JAG (2019). Immune Evasion by Staphylococcus aureus. Microbiol. Spectr 7, GPP3–0061–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont AL, Nygaard TK, Watkins RL, Smith A, Kozhaya L, Kreiswirth BN, Shopsin B, Unutmaz D, Voyich JM, and Torres VJ (2011). Characterization of a new cytotoxin that contributes to Staphylococcus aureus pathogenesis. Mol. Microbiol 79, 814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favazzo LJ, Gill AL, Farnsworth CW, Mooney RA, and Gill SR (2019). The Response of nor and nos Contributes to Staphylococcus aureus Virulence and Metabolism. J. Bacteriol 201, e00107–e00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fey PD, Endres JL, Yajjala VK, Widhelm TJ, Boissy RJ, Bose JL, and Bayles KW (2013). A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. MBio 4, e00537–e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraunholz M, and Sinha B (2012). Intracellular Staphylococcus aureus: live-in and let die. Front. Cell. Infect. Microbiol 2, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundstad ML, Parlet CP, Kwiecinski JM, Kavanaugh JS, Crosby HA, Cho YS, Heilmann K, Diekema DJ, and Horswill AR (2019). Quorum Sensing, Virulence, and Antibiotic Resistance of USA100 Methicillin-Resistant Staphylococcus aureus Isolates. MSphere 4, e00553–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guggenberger C, Wolz C, Morrissey JA, and Heesemann J (2012). Two distinct coagulase-dependent barriers protect Staphylococcus aureus from neutrophils in a three dimensional in vitro infection model. PLoS Pathog 8, e1002434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RK, Alba J, Xiong YQ, Bayer AS, and Lee CY (2013). MgrA activates expression of capsule genes, but not the α-toxin gene in experimental Staphylococcus aureus endocarditis. J. Infect. Dis 208, 1841–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag AF, and Bagnoli F (2017). The Role of Two-Component Signal Transduction Systems in Staphylococcus aureus Virulence Regulation. Curr. Top. Microbiol. Immunol 409, 145–198. [DOI] [PubMed] [Google Scholar]
- Harper L, Balasubramanian D, Ohneck EA, Sause WE, Chapman J, Mejia-Sosa B, Lhakhang T, Heguy A, Tsirigos A, Ueberheide B, et al. (2018). Staphylococcus aureus Responds to the Central Metabolite Pyruvate To Regulate Virulence. MBio 9, e02272–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasenberg A, Hasenberg M, Männ L, Neumann F, Borkenstein L, Stecher M, Kraus A, Engel DR, Klingberg A, Seddigh P, et al. (2015). Catchup: a mouse model for imaging-based tracking and modulation of neutrophil granulocytes. Nat. Methods 12, 445–452. [DOI] [PubMed] [Google Scholar]
- Hii CS, Anson DS, Costabile M, Mukaro V, Dunning K, and Ferrante A (2004). Characterization of the MEK5-ERK5 module in human neutrophils and its relationship to ERK1/ERK2 in the chemotactic response. J. Biol. Chem 279, 49825–49834. [DOI] [PubMed] [Google Scholar]
- Jacobsson G, Dashti S, Wahlberg T, and Andersson R (2007). The epidemiology of and risk factors for invasive Staphylococcus aureus infections in western Sweden. Scand. J. Infect. Dis 39, 6–13. [DOI] [PubMed] [Google Scholar]
- Jenul C, and Horswill AR (2019). Regulation of Staphylococcus aureus Virulence. Microbiol. Spectr 7, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson IM, Lindholm C, Luong TT, Lee CY, and Tarkowski A (2008). mgrA regulates staphylococcal virulence important for induction and progression of septic arthritis and sepsis. Microbes Infect 10, 1229–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanaugh JS, Flack CE, Lister J, Ricker EB, Ibberson CB, Jenul C, Moormeier DE, Delmain EA, Bayles KW, and Horswill AR (2019). Identification of Extracellular DNA-Binding Proteins in the Biofilm Matrix. MBio 10, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M, and Gasser S (2016). Generating Primary Fibroblast Cultures from Mouse Ear and Tail Tissues. J. Vis. Exp (107), 53565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiedrowski MR, Kavanaugh JS, Malone CL, Mootz JM, Voyich JM, Smeltzer MS, Bayles KW, and Horswill AR (2011). Nuclease modulates biofilm formation in community-associated methicillin-resistant Staphylococcus aureus. PLoS ONE 6, e26714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi SD, Malachowa N, Whitney AR, Braughton KR, Gardner DJ, Long D, Bubeck Wardenburg J, Schneewind O, Otto M, and Deleo FR (2011). Comparative analysis of USA300 virulence determinants in a rabbit model of skin and soft tissue infection. J. Infect. Dis 204, 937–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi SD, Malachowa N, and DeLeo FR (2015). Pathogenesis of Staphylococcus aureus abscesses. Am. J. Pathol 185, 1518–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda M, Ohta T, Uchiyama I, Baba T, Yuzawa H, Kobayashi I, Cui L, Oguchi A, Aoki K, Nagai Y, et al. (2001). Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 357, 1225–1240. [DOI] [PubMed] [Google Scholar]
- Kwiecinski J, Jin T, and Josefsson E (2014). Surface proteins of Staphylococcus aureus play an important role in experimental skin infection. APMIS 122, 1240–1250. [DOI] [PubMed] [Google Scholar]
- Kwiecinski JM, Crosby HA, Valotteau C, Hippensteel JA, Nayak MK, Chauhan AK, Schmidt EP, Dufrêne YF, and Horswill AR (2019). Staphylococcus aureus adhesion in endovascular infections is controlled by the ArlRS-MgrA signaling cascade. PLoS Pathog 15, e1007800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey KA, Geoghegan JA, and McLoughlin RM (2016). The Role of Staphylococcus aureus Virulence Factors in Skin Infection and Their Potential as Vaccine Antigens. Pathogens 5, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum ML, Neumann C, Cook C, Chukwuma U, Ellis MW, Hospenthal DR, and Murray CK (2012). Epidemiology of Staphylococcus aureus blood and skin and soft tissue infections in the US military health system, 2005–2010. JAMA 308, 50–59. [DOI] [PubMed] [Google Scholar]
- Lehrer RI, Rosenman M, Harwig SS, Jackson R, and Eisenhauer P (1991). Ultrasensitive assays for endogenous antimicrobial polypeptides. J. Immunol. Methods 137, 167–173. [DOI] [PubMed] [Google Scholar]
- Lewis ML, and Surewaard BGJ (2018). Neutrophil evasion strategies by Streptococcus pneumoniae and Staphylococcus aureus. Cell Tissue Res 371, 489–503. [DOI] [PubMed] [Google Scholar]
- Li L, Wang G, Cheung A, Abdelhady W, Seidl K, and Xiong YQ (2019). MgrA Governs Adherence, Host Cell Interaction, and Virulence in a Murine Model of Bacteremia Due to Staphylococcus aureus. J. Infect. Dis 220, 1019–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CL, Chen ZJ, Wang F, Hou HY, Wang Y, Zhu XH, Jian C, Tian L, Yan SZ, Xu LQ, and Sun ZY (2014). The impact of mgrA on progression of Staphylococcus aureus sepsis. Microb. Pathog 71–72, 56–61. [DOI] [PubMed] [Google Scholar]
- Lu T, Porter AR, Kennedy AD, Kobayashi SD, and DeLeo FR (2014). Phagocytosis and killing of Staphylococcus aureus by human neutrophils. J. Innate Immun 6, 639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo M, Kato F, Oogai Y, Kawai T, Sugai M, and Komatsuzawa H (2010). Distinct two-component systems in methicillin-resistant Staphylococcus aureus can change the susceptibility to antimicrobial agents. J. Antimicrob. Chemother 65, 1536–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mölne L, Verdrengh M, and Tarkowski A (2000). Role of neutrophil leukocytes in cutaneous infection caused by Staphylococcus aureus. Infect. Immun 68, 6162–6167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery CP, Boyle-Vavra S, and Daum RS (2010). Importance of the global regulators Agr and SaeRS in the pathogenesis of CA-MRSA USA300 infection. PLoS ONE 5, e15177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery CP, Boyle-Vavra S, Roux A, Ebine K, Sonenshein AL, and Daum RS (2012). CodY deletion enhances in vivo virulence of community-associated methicillin-resistant Staphylococcus aureus clone USA300. Infect. Immun 80, 2382–2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick RP (1991). Genetic systems in staphylococci. Methods Enzymol 204, 587–636. [DOI] [PubMed] [Google Scholar]
- Nygaard TK, Pallister KB, Ruzevich P, Griffith S, Vuong C, and Voyich JM (2010). SaeR binds a consensus sequence within virulence gene promoters to advance USA300 pathogenesis. J. Infect. Dis 201, 241–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien L, Kerrigan SW, Kaw G, Hogan M, Penadés J, Litt D, Fitzgerald DJ, Foster TJ, and Cox D (2002). Multiple mechanisms for the activation of human platelet aggregation by Staphylococcus aureus: roles for the clumping factors ClfA and ClfB, the serine-aspartate repeat protein SdrE and protein A. Mol. Microbiol 44, 1033–1044. [DOI] [PubMed] [Google Scholar]
- Oh H, Siano B, and Diamond S (2008). Neutrophil isolation protocol. J. Vis. Exp (17), 745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang YY, Schwartz J, Thoendel M, Ackermann LW, Horswill AR, and Nauseef WM (2010). agr-Dependent interactions of Staphylococcus aureus USA300 with human polymorphonuclear neutrophils. J. Innate Immun 2, 546–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker D, Narechania A, Sebra R, Deikus G, Larussa S, Ryan C, Smith H, Prince A, Mathema B, Ratner AJ, et al. (2014). Genome Sequence of Bacterial Interference Strain Staphylococcus aureus 502A. Genome Announc 2, e00284–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Párraga Solórzano PK, Yao J, Rock CO, and Kehl-Fie TE (2019). Disruption of Glycolysis by Nutritional Immunity Activates a Two-Component System That Coordinates a Metabolic and Antihost Response by Staphylococcus aureus. MBio 10, e01321–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radin JN, Kelliher JL, Párraga Solórzano PK, and Kehl-Fie TE (2016). The Two-Component System ArlRS and Alterations in Metabolism Enable Staphylococcus aureus to Resist Calprotectin-Induced Manganese Starvation. PLoS Pathog 12, e1006040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray GT, Suaya JA, and Baxter R (2013). Microbiology of skin and soft tissue infections in the age of community-acquired methicillin-resistant Staphylococcus aureus. Diagn. Microbiol. Infect. Dis 76, 24–30. [DOI] [PubMed] [Google Scholar]
- Rooijakkers SH, Ruyken M, Roos A, Daha MR, Presanis JS, Sim RB, van Wamel WJ, van Kessel KP, and van Strijp JA (2005). Immune evasion by a staphylococcal complement inhibitor that acts on C3 convertases. Nat. Immunol 6, 920–927. [DOI] [PubMed] [Google Scholar]
- Schilcher K, Andreoni F, Uchiyama S, Ogawa T, Schuepbach RA, and Zinkernagel AS (2014). Increased neutrophil extracellular trap-mediated Staphylococcus aureus clearance through inhibition of nuclease activity by clindamycin and immunoglobulin. J. Infect. Dis 210, 473–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlievert PM, and Blomster DA (1983). Production of staphylococcal pyrogenic exotoxin type C: influence of physical and chemical factors. J. Infect. Dis 147, 236–242. [DOI] [PubMed] [Google Scholar]
- Scott BNV, Sarkar T, Kratofil RM, Kubes P, and Thanabalasuriar A (2019). Unraveling the host’s immune response to infection: Seeing is believing. J. Leukoc. Biol 106, 323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidl K, Leemann M, and Zinkernagel AS (2018). The ArlRS two-component system is a regulator of Staphylococcus aureus-induced endothelial cell damage. Eur. J. Clin. Microbiol. Infect. Dis 37, 289–292. [DOI] [PubMed] [Google Scholar]
- Seilie ES, and Bubeck Wardenburg J (2017). Staphylococcus aureus pore-forming toxins: The interface of pathogen and host complexity. Semin. Cell Dev. Biol 72, 101–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha B, François PP, Nüsse O, Foti M, Hartford OM, Vaudaux P, Foster TJ, Lew DP, Herrmann M, and Krause KH (1999). Fibronectin-binding protein acts as Staphylococcus aureus invasin via fibronectin bridging to integrin alpha5beta1. Cell. Microbiol 1, 101–117. [DOI] [PubMed] [Google Scholar]
- Spaan AN, van Strijp JAG, and Torres VJ (2017). Leukocidins: staphylococcal bi-component pore-forming toxins find their receptors. Nat. Rev. Microbiol 15, 435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreeramoju P, Porbandarwalla NS, Arango J, Latham K, Dent DL, Stewart RM, and Patterson JE (2011). Recurrent skin and soft tissue infections due to methicillin-resistant Staphylococcus aureus requiring operative debridement. Am. J. Surg 201, 216–220. [DOI] [PubMed] [Google Scholar]
- Stephan A, and Fabri M (2015). The NET, the trap and the pathogen: neutrophil extracellular traps in cutaneous immunity. Exp. Dermatol 24, 161–166. [DOI] [PubMed] [Google Scholar]
- Thammavongsa V, Missiakas DM, and Schneewind O (2013). Staphylococcus aureus degrades neutrophil extracellular traps to promote immune cell death. Science 342, 863–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomer L, Emolo C, Thammavongsa V, Kim HK, McAdow ME, Yu W, Kieffer M, Schneewind O, and Missiakas D (2016). Antibodies against a secreted product of Staphylococcus aureus trigger phagocytic killing. J. Exp. Med 213, 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng CW, Kyme PA, Arruda A, Ramanujan VK, Tawackoli W, and Liu GY (2012). Innate immune dysfunctions in aged mice facilitate the systemic dissemination of methicillin-resistant S. aureus. PLoS ONE 7, e41454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JN, Crosby HA, Spaulding AR, Salgado-Pabón W, Malone CL, Rosenthal CB, Schlievert PM, Boyd JM, and Horswill AR (2013). The Staphylococcus aureus ArlRS two-component system is a novel regulator of agglutination and pathogenesis. PLoS Pathog 9, e1003819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li X, Chen L, Yuan B, Liu T, Dong Q, Liu Y, and Yin H (2020). Interleukin-33 facilitates cutaneous defense against Staphylococcus aureus by promoting the development of neutrophil extracellular trap. Int. Immunopharmacol 81, 106256. [DOI] [PubMed] [Google Scholar]
- Wilson J, Guy R, Elgohari S, Sheridan E, Davies J, Lamagni T, and Pearson A (2011). Trends in sources of meticillin-resistant Staphylococcus aureus (MRSA) bacteraemia: data from the national mandatory surveillance of MRSA bacteraemia in England, 2006–2009. J. Hosp. Infect 79, 211–217. [DOI] [PubMed] [Google Scholar]
- Wörmann ME, Reichmann NT, Malone CL, Horswill AR, and Gründling A (2011). Proteolytic cleavage inactivates the Staphylococcus aureus lipoteichoic acid synthase. J. Bacteriol 193, 5279–5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, Pittman K, Asaduzzaman M, Wu K, Meijndert HC, et al. (2012). Infectioninduced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med 18, 1386–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper does not report original datasets.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.