

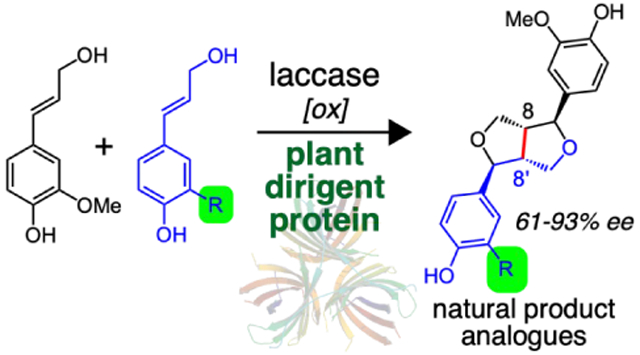
Abstract
Phenylpropanoids are a class of abundant building blocks found in plants derived from phenylalanine and tyrosine. Phenylpropanoid polymerization leads to the second most abundant biopolymer lignin, while stereo- and site-selective coupling generates an array of lignan natural products with potent biological activity, including the topoisomerase inhibitor and chemotherapeutic etoposide. A key step in etoposide biosynthesis involves a plant dirigent protein that promotes selective dimerization of coniferyl alcohol, a common phenylpropanoid, to form (+)-pinoresinol, a critical C2 symmetric pathway intermediate. Despite the power of this coupling reaction for the elegant and rapid assembly of the etoposide scaffold, dirigent proteins have not been utilized to generate other complex lignan natural products. Here we demonstrate that dirigent proteins from Podophyllum hexandrum in combination with a laccase guide the heterocoupling of natural and synthetic coniferyl alcohol analogues for the enantioselective synthesis of pinoresinol analogs. This route for complexity generation is remarkably direct and efficient: three new bonds and four stereocenters are produced from two different achiral monomers in a single step. We anticipate our results will enable biocatalytic routes to difficult-to-access non-natural lignan analogues and etoposide derivatives. Furthermore, these dirigent-protein and laccase promoted reactions of coniferyl alcohol analogues represent new regio- and enantioselective oxidative heterocouplings for which no other chemical methods have been reported.
Graphical Abstract
INTRODUCTION
Ubiquitous phenylpropanoid monomers in plants (termed monolignols) are the starting point for the biosynthesis of two very different classes of molecules: polymeric lignin, the structural component of the lignified plant cell wall, and lignans, bioactive dimeric natural products that typically contain multiple chiral centers and aryl ethers (see Scheme 1A for common modes of coupling). While nonselective polymerization of monolignol radical species results in the synthesis of lignin,1 regio- and enantioselective coupling routes are thought to lead to diverse optically-active dimeric lignans that accumulate in the tissue of both edible and medicinal plants.2-4 One important example is etoposide, an anti-cancer drug that is semi-synthetically produced from a lignan natural product, (−)-podophyllotoxin, found in Podophyllum spp (Scheme 1A).
Scheme 1. Proposed enantioselective production of hetero-(+)-pinoresinol analogues for biosynthesis of etoposide analogues.

A. Laccase oxidizes coniferyl alcohol and generates CA radical species that undergo three unique modes of coupling.13, 18-19 (Note on the coupling product naming convention: coupling products of the same monomer combination are collectively referred by their assigned compound number, and specific coupling modes are denoted by lowercase letters: a, 8-O-4’; b, 8-8’; and c, 8-5’.) In presence of PhDIR, oxidized radical species of coniferyl alcohol form predominantly 8-8’ coupling products enantioselectively. The 8-8’ coupling product (+)-pinoresinol can be subsequently processed by enzymes involved in the etoposide aglycone (EA) biosynthetic pathway. The methoxy group of etoposide highlighted in green is prone to oxidation by human CYP3A4 and therefore is a target for modification using an engineered biosynthesis route.
B. For each coupling mode (8-8’ coupling mode shown here; see Figure S1 for all other possible products), there are three possible types of coupling products when a mixture of two monomers is used: one heterocoupling and two distinct homocoupling products (8-8’ coupling mode shown here). We propose that the use of coniferyl alcohol analogues for (+)-pinoresinol analogue synthesis will lead to etoposide variants with modifications at the E ring.
Etoposide and related drugs (etopophos and teniposide) act as reversible topoisomerase inhibitors by intercalating at the interface of the topoisomerase IIβ and DNA cleavage complex and stabilizing the double stranded break, thereby inducing apoptosis of cancer cells.5 However, one liability of etoposide is the propensity of the drug to undergo demethylation by human liver enzyme CYP3A4 to generate a quinone with an altered activity profile (Scheme 1A, highlighted in green).6-10 We hypothesized that an analogue of etoposide bearing E-ring substituents less prone to oxidation might limit unwanted hepatic metabolism.
Etoposide is currently manufactured using a semi-synthetic approach that requires sourcing a related lignan, (−)-podophyllotoxin, from the medicinal plant Podophyllum spp. Recently we established a complete biosynthetic pathway for 4’-demethyl (−)-epipodophyllotoxin, or etoposide aglycone (EA) through heterologous expression of previously known11-16 and newly identified17 Podophyllum hexaudrum (Ph) genes in Nicotiana benthamiana (Nb). Given the modular nature of EA biosynthesis, we considered that analogous couplings of alternative monolignol building blocks could enable efficient modification of the etoposide E ring for the formation of analogues.
The EA pathway involves convergent coupling of two coniferyl alcohol (CA) monomers for rapid complexity generation (route shown in Scheme 1A shows the native biosynthetic pathway that begins with dimerization of two CA monomers).13, 18-19 This key step involves oxidation of CA monomers to their corresponding radical species by a peroxidase or a laccase.19-20 Dirigent proteins (DIRs), described from Podophyllum and other plant species, then direct the resulting radicals towards a remarkable stereoselective C2 symmetric dimerization.13-21-22 DIRs are not known to affect the rate of monolignol oxidation, nor are they thought to impact the net rate of dimerization. However, DIRs do change the relative ratio of different coupling products. In the absence of DIRs, monolignol radicals undergo spontaneous and nonselective couplings. This mechanism is supported empirically such that stereoselectivity is observed only in the presence of both an oxidizing agent (e.g. enzymatic or inorganic) and a DIR.13, 23 In contrast, multiple racemic coupling products are formed if only an oxidizing agent is present (for examples of other coupling modes, see Scheme 1A).
Comparison of crystal structures of enantiocomplementary dirigent proteins (a (+)-pinoresinol-forming DIR from Pisum sativum [PsDRR206]24 and a (−)-pinoresinol-forming DIR from Arabidopsis thaliana [AtDIR6]25) suggests that the active sites exhibit contrasting conformations, likely serving as “molds” for the differential binding of two radical substrates for the si-si or re-re coupling, respectively. The ability of dirigent proteins to mediate a highly regio- and enantioselective coupling reaction to generate (+)-pinoresinol raises the intriguing possibility that these proteins could couple other monolignols, including those not typically found in Nature. To date, (+)-pinoresinol-forming DIRs have exhibited high substrate specificity towards CA,13 however their ability to guide heterocoupling reactions (beneficial for the desired etoposide analogue generation described here) has not been explored. Stereoselectivity determined at this PhDIR-mediated (+)-pinoresinol step that funnels the supply of CA into the EA pathway is crucial because several of the remaining pathway enzymes are known to be enantiospecific.15, 26 This proposed engineered route could yield valuable non-natural and heterocoupled lignan analogues that would enable specific modifications of the E ring of etoposide.
To determine the inherent substrate flexibility of DIRs and expand the utility of this direct complexity-generating reaction, we investigated whether DIRs could mediate stereoselective heterocoupling of various analogues of the natural substrate, coniferyl alcohol (Scheme 1B). Here, we show that PhDIR is capable of promoting the coupling of synthetic and natural monolignols to form non-natural heterocoupled lignans. The resulting non-natural heterocoupling products retain the necessary stereochemistry to be further processed by the rest of the EA pathway enzymes for etoposide analogue biosynthesis. We anticipate the complex lignan scaffolds we produced will have direct utility in a biosynthetic route to etoposide analogues that are less prone to E-ring metabolism and formation of the reactive etoposide quinone (Scheme 1B, highlighted in blue).
RESULTS AND DISCUSSION
CA analogue design and preparation.
In choosing substrate designs to test for dirigent protein mediated coupling, we considered previous structure-activity relationships that had been established for the parent drug etoposide. For example, it has been shown that replacement of both the 3’- and 5’-methoxy groups by hydrogens in the E ring results in only a moderately decreased topoisomerase inhibition, and replacement of only the 5’-methoxy group by a hydrogen results in comparable topoisomerase inhibition as the parent compound.27-28 In contrast, methylation of the 4’-hydroxy group resulted in the loss of drug activity, consistent with recent evidence from the ternary crystal structure of topoisomerase IIβ, DNA, and etoposide that suggests an important favorable interaction between the functional group and topoisomerase residue D479.5 These data, in addition to the crystal structure of the DNA cleavage complex, suggested that activity would not be compromised by replacement of the E ring methoxy substituents with functional groups at the 3’ and 5’ positions anticipated to prevent the unwanted metabolism. With this hypothesis in mind, analogues of 1 (3-11) were prepared, containing phenolic substituents with varying degrees of electronegativity (Figure 1). Compounds 3 and 4 are naturally-occurring monolignols produced by plants that differ by the number of methoxy substituents ortho to the phenol. Monomers 5, 6, and 7 were designed to explore significantly altered electronic states of the aromatic ring, relative to coniferyl alcohol, with the strongly electron-withdrawing trifluoromethoxy, fluorine, and bromine groups replacing the methoxy group, respectively. Additionally, the bromine group of 7 could serve as a synthetic handle for functionalization of the coupling product to diversify the product profile post-coupling. Compounds 8 and 9 replace the methoxy group with mildly electron-donating methyl groups. Both 10 and 11 retain the alkyl aryl ethers at the 3’ position. For 10, the electronic state of the phenolic ring is expected to be altered by the presence of the 5’-fluorine substitution compared to 1. Monomer 11 probes the effects of a bulkier alkoxy group substitution.
Figure 1. Relative rates of monolignol oxidation and coupling product formation catalyzed by TvLac.

Each monomer is present as an equimolar mixture with 1. 1, 3 and 4 are naturally-occurring monolignols; all others are synthetic. For substrate oxidation, data points indicate the mean of peak integrations for the corresponding monomer species (m/z of [M-H2O+H]+) detected by LC-MS; EIC integrations are normalized to the peak integration at the initial reaction time points per species. For coupling product formation, data points indicate the mean of peak integrations for the corresponding regioisomeric products detected by LC-MS (m/z of [M-H2O+H]+, a common ion to all regioisomeric products a-c); EIC integrations are normalized to the maximum product EIC integration observed over the entire time course; relative ratios of products are unchanged from the raw data. CA-CA (2) denotes the natural dimer, CA-X the heterocoupling product, and X-X the non-CA dimer, where X denotes the non-CA analogue. Error bars show standard deviations of data from triplicate reactions. Consumption of coupling products (e.g. 2 past ~5 min) is likely due to nonselective TvLac activity resulting in oligomerization
Oxidation rates of monolignol analogues.
Dirigent proteins are thought to harness reactive monolignol radicals and direct selective dimerization after an initial oxidation event. In these transformations, several oxidizing reagents, inorganic or enzymatic, have been used to catalyze the initial oxidation of coniferyl alcohol.13, 29 We anticipated that efficient heterocoupling between 1 and a different monolignol analogue, necessary for the E-ring modified etoposide analogue, would only occur if the oxidation rates of each were closely matched. To probe this, we first examined relative rates of oxidation of our panel of monomers in vitro, using a fungal laccase from Trametes versicolor (TvLac) that is known to catalyze single electron oxidation of naturally-occurring monolignols (1, 3, and 4). Time-course of each substrate oxidation in the presence of 1 was investigated using liquid chromatography-mass spectrometry (LC-MS) to track substrate consumption during the reaction. For naturally-occurring monolignols, 4 exhibited the fastest oxidation rate with TvLac, followed by 1, and 3 with the slowest rate of oxidation (Figure 1, mixed substrates). As previously reported,29 the decreasing trend of oxidative rates with decreasing number of methoxy substituents attested to the importance of electron-donating nature of the methoxy group for single-electron oxidation mechanism of laccases. In line with these observations, compounds 5, 6, and 7, which lack the electron-donating aromatic substituents, exhibited noticeably slower oxidation rates in comparison to 1. Compound 8, with one methyl substituent, showed slower oxidation compared to 1, while compound 9 with two methyl groups showed a more closely matched oxidation trend as 1. More importantly, the rates of TvLac oxidation of 10 and 11 are sufficiently matched with that of 1, and both of these monomers would be particularly useful for generating E-ring modified etoposide variants. As anticipated, in the corresponding coupling reactions, appreciable levels of the desired heterocoupling products accumulate over the course of the assay (Figure 1, coupling products; see Figure S1 for enumeration of all possible coupling products). In contrast, slower-to-oxidize electron-deficient monolignols (relative to 1) do not couple as efficiently with 1 to generate heterocoupling products. Taken together, a trend emerges where the relative rates of TvLac oxidation for the two monomers present in a mixture determine the identity of the major product formed. In this model, the collision of oxidized monolignols dictates identity of the coupling product, a process that is controlled by the relative concentration of these monolignol radical species. These data confirm that efficient formation of the desired heterocoupling products requires a close match in monomer oxidation rates.
Expression and extraction of functional PhDIR.
To test if dirigent proteins could influence the regio- and enantioselectivity of these heterocoupling reactions, we first needed sufficient supply of PhDIR for in vitro assays. PhDIR was expressed in N. benthamiana leaves using Agrobacterium-mediated transient expression as described previously.17 While DIRs have previously been successfully expressed in heterologous hosts including Pichia pastoris and plant cell cultures,30-32 for plant leaf expression, the apoplastic fluid wash method (APW) has been employed as a secretion and extraction method for heterologous protein expression in N. benthamiana, without the need for further processing of the extracted proteins.25, 33-34 Since many annotated dirigent proteins, including PhDIR, are computationally predicted (by SignalP) to contain N-terminal signal peptides that direct the proteins to the apoplast, the extracellular space in plants,35-36 we applied a similar apoplastic extraction strategy to the N. benthamiana leaves expressing PhDIR without adding an extrinsic signal peptide and carried out activity assays of the protein without further purification. To control for different environmental factors such as salt concentration and presence of native N. benthamiana laccases and peroxidases, the APW protocol was performed in parallel using plants transformed with an empty vector (EV), and the resulting extracts were used as negative controls. To validate dirigent protein expression, 1 was subjected to oxidation by TvLac in the presence of plant extract from leaves expressing PhDIR. As expected based on previously characterized activity of a (+)-pinoresinol-forming DIR,13 the regioisomeric profile nearly inverted compared to that in the absence of PhDIR, with the (+)-pinoresinol (2b) regioisomeric content increasing from 33% (EV control) to 89% (PhDIR extract) (Figure S2a and Table 1; reaction scheme shown in Scheme 1A).
Table 1.
Reaction yields of 8-5' (c) and 8-8' (b) coupling products and enantiomeric excess of (+)-pinoresinol analogues (b)
| monomers | coupling | oxidant | plant extracta | 8-5’ (μM)b | [%]c | 8-8’ (μM)b | [%]c | 8-8' regioisomeric content (%)d |
% eee |
|---|---|---|---|---|---|---|---|---|---|
| 1 | CA-CA (2) | TvLac | EV | 250 ± 47 | [50] | 122 ± 26 | [24] | 33 | 2 |
| PhDIR | 57 ± 10 | [11] | 446 ± 29 | [89] | 89 | 88 | |||
| 1 + 8 | CA-MCA (17) | TvLac | EV | 86 ± 11 | [17] | 33 ± 5 | [7] | 28 | 2 |
| PhDIR | 55 ± 2 | [11] | 83 ± 3 | [17] | 60 | 91 | |||
| MCA-MCA (26) | TvLac | EV | 62 ± 14 | [12] | 15 ± 5 | [3] | 19 | 0 | |
| PhDIR | 41 ± 3 | [8] | 11 ± 2 | [2] | 21 | 31 | |||
| 1 + 10 | CA-FMCA (19) | TvLac | EV | 136 ± 19 | [27] | 24 ± 5 | [5] | 15 | 1 |
| PhDIR | 10 ± 2 | [2] | 145 ± 4 | [29] | 94 | 94 | |||
| FMCA-FMCA (28) | TvLac | EV | N/Af | 49 ± 8 | [10] | N/Af | −2 | ||
| PhDIR | N/Af | 129 ± 3 | [26] | N/Af | 82 | ||||
| 1 + 11 | CA-ECA (20) | TvLac | EV | 182 ± 12 | [36] | <10g | [<2] | <5 | 0 |
| PhDIR | 54 ± 11 | [11] | 88 ± 16 | [18] | 62 | 89 | |||
| ECA-ECA (29) | TvLac | EV | 168 ± 11 | [34] | <10g | [<2] | <6 | −1 | |
| PhDIR | 147 ± 23 | [29] | 20 ± 10 | [4] | 12 | 47 |
plant extracts obtained with APW from leaves expressing empty vector (EV) or PhDIR.
Reaction yield was estimated from a standard curve using EIC peak integration as analyzed by LC-MS on C18 chromatography. For 17 and 26, standard curves of 2b and 2c were used to estimate yields. Error values are standard deviations of triplicates.
100% theoretical yield corresponds to 500 μM product generated from coupling of 500 μM each substrate (1mM total substrate concentration).
8-8' regioisomeric content was calculated by the following formula: [8-8', b]/([8-8', b]+[8-5', c])*100%.
ee: enantiomeric excess of (+)-pinoresinol or analogue over (−)-pinoresinol or analogue, calculated using the EIC peak integration ratios on chiral chromatography.
Not applicable; 8-5’ coupling product was not observed with the fluorine substituent.
Below limit of linearity of 10 μM.
Altered regioselective product distributions mediated by PhDIR.
With functional PhDIR in hand, we next tested its capacity to direct the outcome of oxidative heterocoupling of CA and each of the alternative monomers shown in Figure 1. In our initial analysis of the reaction outcomes, we observed that the addition of PhDIR to reactions catalyzed by TvLac did not alter the ratio of all homo- and heterocoupling products compared to TvLac alone (see Figure 2, unfilled bars in the graphs indicating all products from homocoupling, e.g. 2 and 21-29, or heterocoupling, e.g. 12-20). This observation is in accordance with previous literature findings that dirigent proteins do not possess catalytic activity; accordingly, the rate of formation of the three coupling product types is dictated only by the TvLac oxidation step.
Figure 2. End-point coupling product distributions in the presence and absence of PhDIR.

Couplings of equimolar (a) 1 and 10, (b) 1 and 11, (c) 1 and 6 and (d) 1 and 8. Total content of the three coupling product types for each reaction was estimated by summing up peak integrations of the corresponding ion species (m/z of [M-H2O+H]+) detected by LC-MS, and each coupling product composition (type and coupling mode) was normalized to the total integrations. Values reflect only relative product concentrations to compare reactions. Colors indicate reactions carried out with empty vector control (black, on the left) or PhDIR (blue, on the right) extracts. Filled, slash-patterned columns indicate levels of 8-8’ coupling products. Bar heights indicate the mean of triplicates, and error bars show standard deviations.
We next quantified the impact of the PhDIR addition on the distribution of regioisomers formed for each coupling type. This analysis revealed that the addition of PhDIR significantly influences the regioselectivity of the heterocoupling reaction, shifting the major product to the 8-8’ regioisomer for heterocoupling of all monolignol analogues with 1 (Figures 2 and S2, filled, slash-patterned columns for products 12-20; and Table 1). More specifically, the desired 8-8’ heterocoupling product 19b comprised 94% of the quantified CA-FMCA heterocoupling products 19b-c (6.2-fold increase compared to the negative control, i.e. reactions with TvLac and EV extract, at 15%). Similarly, in reactions with PhDIR, 20b constitutes 62% of the quantified CA-ECA heterocoupling products 20b-c (~12-fold increase from <5% in control reaction). In addition, couplings of 1 with 3-9 each resulted in a similar trend of regioselectivity increase in heterocoupling reaction (Figures 2c-d and S2b-f for products 12-18), revealing remarkable flexibility in the ability of PhDIR to couple 1 to both natural and non-natural monolignols. These data show promise that the E ring modification of etoposide could be accomplished through a biosynthetic route from production of (+)-pinoresinol analogues. Moreover, in the context of plant metabolism, it is notable that 1, 3, and 4 are common and abundant metabolites found throughout the plant kingdom. Our in vitro results raise the possibility that naturally-occurring optically-active heterocoupled lignans, e.g. (+)-demethoxypinoresinol (CA-pCA 8-8’ heterocoupling product, 12b) from Gladiolus segetum37 and (+)-medioresinol (CA-SA 8-8’ heterocoupling product, 13b) from Eucommia ulmoides,38 originate with pinoresinol-like couplings catalyzed by an oxidase and mediated by dirigent proteins. However, the biosynthetic origins of these molecules have not been described to date.
Unlike the high regioselectivity observed for PhDIR-mediated heterocoupling reaction, homocoupling of CA analogues was not as selective. Regioselectivity observed for homocouplings of 10 and 11 only moderately increased compared to the striking heterocoupling regioselectivity increase (e.g. ~two-fold increase for 29b vs. ~12-fold increase for 20b). In the coupling reaction of 1 and 6, the addition of PhDIR did not increase the apparent percent composition of 8-8’ homocoupled product FCA-FCA, 24b (Figure S6). More generally, we did not observe any increase in regioselectivity in the formation of non-natural dimers derived from 3-9 (Figures S3-S9). In line with these results, dirigent proteins have previously been shown to not impact the oxidative dimerization of 3 and 4.13 It is possible that the larger increase in regioselectivity observed in heterocoupling reaction compared to non-CA dimerization (when it occurs) can be explained by substrate specificity in the binding pocket of PhDIR. Although speculative, PhDIR binding pocket is thought to take two units of monolignol radicals to facilitate coupling; one hypothesis is that the pocket preferentially binds an initial CA monolignol but is tolerant of CA analogues as the second monomer.
Asymmetric heterocoupling guided by PhDIR.
Encouraged by the increased regioselectivity observed in the PhDIR-mediated heterocoupling of CA with all monolignol analogues,we next investigated enantiomeric ratios of the heterocoupled 8-8’ products formed in our assays. In the dimerization of 1, the native substrate of PhDIR, the major product was confirmed to be (+)-pinoresinol with an enantiomeric excess (ee) >85% compared to <5% ee in control experiments without PhDIR (Figure 3 and Table 1). Notably, for the heterocoupling products formed at the highest levels, e.g. 19b and 20b, we determined that the 8-8’ heterocoupling products formed selectively in the presence of PhDIR were also produced in high enantioselectivity with 94% ee and 89% ee, respectively. In addition, heterocoupling products formed (albeit at lower levels) in the coupling reactions of 1 with each of 3-9 also proceeded with high enantioselectivity (71-93% ee) in the presence of PhDIR (Table 2 and Figures S3-S9). These results reveal the remarkable flexibility of dirigent proteins for guiding the enantioselective formation not only of C2 symmetric dimers, but also for 8-8’ heterocoupling products bearing four contiguous stereocenters.
Figure 3. PhDIR-mediated regio- and stereoselective heterocoupling of coniferyl alcohol and selected analogues.

EICs as analyzed by LC-MS with C18 and chiral chromatography show separation of regioisomers and enantiomers, respectively, of end-point TvLac reaction products in the absence and presence of PhDIR extracts (black and blue lines, respectively) from an equimolar substrate mixture. Authentic standards, purchased [(+)-2b] or purified from preparatory-scale reactions with and without PhDIR (all other standards), are shown in grey. (For 17 and 20, the c/c’ suffix denotes the 5-8’/8-5’ regioisomeric coupling products.) Control reactions were supplemented with an equal volume of plant extracts transformed with an empty vector (EV). C18 traces are to scale, and chiral traces are normalized per reaction. Chromatographic peaks corresponding to the (+)-pinoresinol analogues (structures shown above) are denoted with red dotted lines, and their regioisomers (C18) and enantiomers (chiral) with black dotted lines. Relative stereochemistry assignments are supported by the identical 1H NMR spectra of purified racemic 8-8’ coupling products isolated from the TvLac-only control reactions and enantioenriched 8-8’ coupling products from TvLac-PhDIR reactions (Figure S11). Regioisomeric products are distinguishable based on MS/MS fragmentation pattern (Figure S12).
Table 2.
Selective heterocoupling mediated by PhDIR
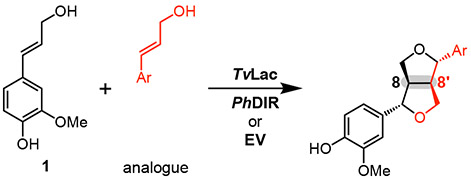




Relative to the 8-8’ heterocoupling products, we observed lower regio- and enantioselectivity for the formation of the corresponding non-natural 8-8’ homocoupling products in the presence of PhDIR (Table 1 and Figure S10). Of these products, the highest ee was obtained for 28b (82% ee), suggesting that PhDIR is capable of accepting the monolignols that closely resemble 1 in structure (e.g. 10). In line with this hypothesis, monomer 11, bearing a bulky ethoxy substituent, was converted to its 8-8’ dimer (29b) with only modest selectivity in the presence of PhDIR (47% ee), and homocoupling of 8, with a methyl group substituent, showed only a modest increase in enantioselectivity in the presence of PhDIR (31% ee for 26b); in these reactions, the 8-5’ regioisomer was still the major product. Moreover, in the coupling of 1 with 3-7 and 9, no enantioselectivity increase was observed for the homocouplings of the monolignol analogues (Figures S3-S9), further suggesting that the promiscuity of PhDIR is specific to the asymmetric heterocoupling mechanism.
Optimization of oxidation reaction for heterocoupling.
For the formation of some of the desired pinoresinol analogs (e.g. 15b and 17b), yield was limited by the slow oxidation of the monolignol analogue relative to 1. To address this, different reaction parameters were considered. In an effort to better couple the oxidation rates of monolignol substrates, we compared the activities of various oxidants: TvLac, horseradish peroxidase (HRP), and ammonium persulfate (APS). When we subjected an equimolar mixture of 1 and 6 to a free couplingreaction in the presence of each of these oxidants, we observed that the relative oxidation rate differences observed between 1 and 6 were similar regardless of the oxidant used (Figures 4a), where 6 was much slower to oxidize compared to 1. We also examined the effect of varying pH on the reaction. Because the pKa of the phenols vary depending on the substituent identity, we hypothesized that the pH of TvLac oxidation reactions could impact the relative rate of monomer oxidations. While TvLac activity was comparable at either pH 4 and the enzyme’s optimal pH of 5.5, relative oxidation rates did not differ at different pH conditions (Figure S13).
Figure 4. Optimization of heterocoupling product formation.

(a) relative oxidation rates and product formation in a coupling reaction of 1 and 6, with TvLac, HRP, and APS as oxidant. For data normalization, see Figure 1 caption. (b) coupling product formation in a TvLac oxidation of 0.5 mM 1 and 0, 0.5, 1.5, or 4.5 mM 6 (1:0, 1:1, 1:3, and 1:9 ratios, respectively). Data points indicate the mean of EIC peak integration (m/z of [M-H2O+H]+) normalized to the maximum product level per coupling product type. For (c) and (d), end-point product distributions of (c) 1 and 6 at a molar ratio of 1:9 (500 μM and 4.5 mM, respectively) and (d) 1 and 8 at a molar ratio of 1:3 (500 μM and 1.5 mM, respectively). See Figure 2 caption for data normalization.
As an alternative approach to improve heterocoupling product yields, we tested whether superstoichiometric amounts of 6 compared to 1 would result in higher accumulation of heterocoupled products 15 (and homocoupling products 24) (Figure 4b, c and S14). While CA homocoupling products (2) remained as the major product, this strategy was effective for increasing the formation of 15 from the coupling of 1 and 6. Altered stoichiometry was even more beneficial in the coupling of 1 and the methyl analogue 8 (Figure 4d), and enabled the product isolation described below. However, these yield improvements are maximal at a 1:3 ratio of 1 and 8—at higher ratios, enantioselectivity begins to drop when DIR is included in the reaction (Figure S15). Additionally, increasing the amount of PhDIR in the assay improved regioselectivity while the absolute yield of 17b remained constant.
Preparatory-scale biosynthesis of synthetic lignan analogues using PhDIR plant extracts.
Careful optimization of in vitro TvLac-PhDIR reactions allowed isolation and characterization of desired non-natural (+)-pinoresinol analogues (Table 3). Time-courses of TvLac-EV and TvLac-PhDIR reactions were taken to gain more insight into this coupled enzyme-protein reaction prior to product isolation and to avoid further apparent TvLac-catalyzed polymerization of the desired coupling products (Figure S16). In assays containing PhDIR, accumulation of the desired products was highest at a point coinciding with full consumption of monolignol substrates. These reaction conditions were employed for preparatory-scale in vitro coupling reactions of 1 with 10 or 11. For coupling of 1 and 8, a molar ratio of 1:3 (8 in excess) was used, while the amount of PhDIR extract was kept consistent as other coupling reactions. The 18% isolated yield (29% reaction yield, Table 1) of the CA-FMCA 8-8’ heterocoupling product 19b (Table 3) exceeds the yields of racemic (±)-pinoresinols (natural coupling product; at most 20%) obtained from coupling reactions promoted by either laccase alone or a synthetic oxidant, as previously reported in literature.35, 39 This is an exciting proof of concept for the robustness and flexibility of using plant chemistry for the direct enantioselective synthesis of natural product analogues not readily obtained through chemical methods.
Table 3.
Isolated yields of the (+)-pinoresinol analogues
| product | isolated yield (mg) |
PhDIR extract used (ml) |
yield (%)a |
ee (%)b |
|---|---|---|---|---|
| 8-8’ CA-MCA, 17b | 0.2 | 3 | 4 | 73 |
| 8-8’ CA-FMCA, 19b | 1.2 | 3.5 | 18 | 93 |
| 8-8’ CA-ECA, 20b | 0.7 | 4 | 9 | 93 |
| 8-8’ FMCA-FMCA, 28b | 1.2 | 3.5 | 19 | 82 |
| 8-8’ ECA-ECA, 29b | 0.5 | 2.9 | 9 | 61 |
100% theoretical yield corresponds to 500 μM product generated from 500 μM limiting monomer (1) concentration (1:1 molar ratio of 1 and 10 or 11, and 1:3 molar ratio of 1 and 8).
ee: enantiomeric excess of (+)-pinoresinol analogue over (−)-pinoresinol analogue, calculated using the EIC peak integration ratios on chiral chromatography.
CONCLUSIONS
Taken together, our results show that plant-derived dirigent protein can be used to promote the regio- and stereo-selective coupling of non-natural monolignols and CA to make functionalized variants of (+)-pinoresinol, a precursor to clinically used chemotherapeutics. The transformation involves coupling of achiral precursors and formation of three new bonds and four stereocenters in a single step. Importantly, there is no analogous synthetic method for this direct coupling and complexity-generating reaction. We anticipate that the expanded substrate versatility of DIRs described here, combined with future protein engineering and optimization efforts to overcome the limitations of the initial oxidation step, will enable the production of etoposide analogues with altered substituents that still function as topoisomerase inhibitors but are resistant to degradation by human liver cytochrome P450s.
While in vitro biochemical characterization of dirigent proteins has begun to shed light on their role in selective coupling of monolignols, our observation that heterocoupling products can be formed suggests the potential for previously unrealized activities of dirigent proteins in plants, including those characterized to date that mediate (+)-pinoresinol or (−)-pinoresinol-forming reactions.13, 30, 35, 40-41 In addition, a related protein from cotton, the (+)-gossypol-forming dirigent protein, promotes stereoselective aryl-aryl bond formation.34, 42 Notably, low concentrations of both (+)-gossypol and gossypol-6-methyl ether (which can be viewed as a heterocoupling product of hemigossypol and hemigossypol-6-methyl ether) have been found in the native plants, suggesting the possibility of other dirigent-protein-mediated heterocoupling reactions occurring in Nature.43
The typical plant genome, e.g. Arabidopsis thaliana, contains on the order of 20-50 putative DIR homologs, the vast majority of which have not been characterized and it is unclear whether they are associated with natural product biosynthesis.44-45 However, the diversity of known optically-active lignans found in plants suggest additional members of this class of proteins could also promote alternative stereoselective coupling reactions.46-47 In our study, we demonstrate an expanded substrate versatility of DIRs promoting stereoselective heterocoupling of natural and non-natural monolignol analogues. Furthermore, we showcase synthesis and isolation of optically-active coupling products derived from a broad substrate scope using crude plant extracts containing functional DIRs. These results show promise for combining conventional synthetic chemistry and biosynthesis engineering techniques to access synthetic analogues of complex natural products that can be optimized for use in the clinic.
EXPERIMENTAL
Transient expression of P. hexandrum genes by agro-infiltration.
Agrobacterium tumafaciens (GV3101, gentamicinR; agrobacteria or agro) transformants with P. hexandrum genes in pEAQ-HT (kanamycinR) vectors were prepared as described previously.17 Agrobacteria (gentR and kanR) grown on LB plates supplemented with gentamicin and kanamycin were resuspended in LB media and centrifuged at 8,000 x g for 5 min. Supernatant was discarded, and the cell pellets were resuspended in 500 μl of 10 mM MES buffer at pH 5.6 with 10 mM MgCl2 and 150 μM acetosyringone for 1-2 hr induction at room temperature. The induced cell suspension was further diluted with induction media to the desired inoculum level measured by optical density measurement at 600 nm. The agrobacterial suspension was infiltrated on the underside of the N. benthamiana leaves with a needleless 1-ml syringe. Plants were 5-6 weeks old at the time of infiltration, grown under a 16-hr light cycle. Biological replicates consisted of leaves from different N. benthamiana plants from the same batch, unless otherwise noted.
Apoplastic fluid wash protocol (APW).
Plant extracts were obtained from N. benthamiana leaves by APW as described previously.25, 48 Briefly, PhDIR-harboring or empty pEAQ-HT vector was transiently expressed in N. benthamiana by agro-infiltration (OD600nm = 0.1-0.3) for 5 days. Leaves were vacuum-infiltrated with 0.1 M MES and 0.3 M NaCl at pH 5.5 through three cycles of pulling vacuum to 150 torr and slow release to atmospheric pressure. The infiltrated leaves were layered on top of a piece of parafilm and wrapped around a pipette tip and placed inside a syringe in place of a plunger. The syringe assembly was placed in a conical tube and was centrifuged at 1500 x g for 15 min at 4°C. The extracts collected at the bottom of the conical tubes were transferred to clean tubes and centrifuged at 14,000 x g for 10 min at 4°C to clear undesired cellular debris. The supernatants were ultrafiltrated (Amicon Ultra-0.5ml 10-kD) at 14,000 x g for 10 min at 4°C, and the retentate was aliquoted, flash-frozen in liquid nitrogen and stored at −80°C until use at 5-fold dilution.
Time-course of TvLac oxidation on LC-MS.
Enzyme assays contained a final concentration of 1 mM equimolar mixture (1% MeOH in water) and 0.1 mU/μl TvLac in 1.5 mM MES buffer at pH 5.5. When TvLac was substituted with HRP or APS (Figure 4a), 5 pg/μl HRP and 10 mM H2O2, or 1 mM APS was used. When the molar ratio of the mixed substrates was varied (Figures 4b-d, S14 and S15), the amount of the limiting monomer (often 1) was kept constant at 500 μM with the final total substrate concentration ranging 0.5-5 mM. (e.g. For a molar ratio of 1:3, the final concentrations of the limiting and excess monomers were 500 μM and 1.5 mM, respectively, with the final total substrate concentration of 2 mM.) For the varied pH study (Figure S13), citrate buffer was used for pH 2 and 4, HEPES buffer at pH 7, and Tris buffer at pH 8.5. (Because there is no chromatography in this analysis, to minimize salt contamination in the mass spectrometer, it was important to use organic buffer reagents and ammonium hydroxide or sulfuric acid for adjusting pH.) For standards (Figure S17), bovine serum albumin was supplemented instead of TvLac at 152 ng/μl Reactions were initiated by addition of 2 mM substrate stock solutions (equi-volume dilution to a final concentration of 1 mM). Substrates were added to the reaction at ~1 min prior to the first injection of each sample. Real-time reaction samples were analyzed on the LC-MS on the positive ion mode through direct injection. Three samples were analyzed within the same worklist at a time, and reaction replicates were randomized over different worklists. Time points were recorded every 1.5-1.6 min, which accounts for the time of data collection as well as sample injection performed by the instrument.
Time-course of in-vitro TvLac-EV vs. TvLac-PhDIR assays on LC-MS.
Enzyme assays contained a final concentration of 1 mM equimolar substrate mixture, 1% MeOH, 0.333 mU/μl TvLac, and 20% v/v PhDIR or EV plant extract (transformed with agrobacteria at OD600nm = 0.3) in 20 mM MES buffer at pH 5.5. Reactions were initiated by addition of substrate stock solutions at 1.875 mM (1.875% DMSO in water). Substrates were added to the reaction at 1 min prior to the first injection of each sample. Reaction replicates were analyzed on the LC-MS on the positive ion mode at the same time. Time points were recorded every 1.5-1.6 min, which accounts for the time of data collection as well as sample injection performed by the instrument.
In-vitro LC-MS end-point analysis with C18 and chiral chromatography.
For assays involving PhDIR apoplastic plant extract, the protocols by Gasper et al. were followed with modifications.25 Enzyme assays were prepared by diluting the plant extract of leaves expressing either PhDIR or EV (transformed with agrobacteria at OD600nm = 0.1) five-fold in 0.1 M MES buffer containing 0.3 M NaCl and 0.333 mU/μl TvLac at pH 5.5. Substrate stock solutions were prepared from 100 mM solutions in DMSO for 1 and 8-11, and in MeOH for 1 and 3-7. (In our hands, using methanol stock solutions resulted in increased production of coupling products derived from 3-7 (CA-X and X-X) compared to when DMSO stock solutions were used.) Reactions were initiated by addition of 1.875 mM equimolar substrate solutions in 1.875% DMSO or MeOH for a final total substrate concentration of 1 mM at 30 °C. After 20 min for assays containing 4 and 40 min for the rest, the coupling reactions were quenched by addition of an equal volume of acetonitrile. The reactions were centrifuged at 15,000 rpm for 10 min, and the supernatants were filtered to be analyzed on the LC-MS on the positive ion mode.
Supplementary Material
ACKNOWLEDGMENT
We would like to thank Warren Lau for help and guidance in the initial development of this project, Mark Smith (Medicinal Chemistry Knowledge Center, ChEM-H) for help with designing medicinally relevant monolignol analogues, Russ Li for preparation of p-coumaryl alcohol, Curt Fischer and Yuqin Dai (Metabolic Knowledge Center, ChEM-H) for helpful discussions and 6545 qToF usage, Stephen Lynch (Stanford NMR facility director) for consultation on NMR techniques, Kevin Smith for help with NMR analysis, Alex Engel for critical reading of the manuscript, and Kevin Smith, Ryan Nett, Ricardo De La Peña and Bailey Schultz for helpful discussions. This work was supported by the National Science Foundation Graduate Research Fellowship under Grant No. DGE-1656518, NIH R01GM121527, and a AAAS Marion Milligan Mason Award for Women in the Chemical Sciences.
ABBREVIATIONS
- LC-MS
liquid chromatography-mass spectrometry
- EIC
extracted ion chromatogram
- HPLC
high-performance liquid chromatography
- Lac
laccase
- EV
empty vector (control)
- DIR
dirigent protein
- CYP
cytochrome P450
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
General methods and materials, synthetic procedures, supplementary figures, supplementary table, NMR spectra, and supplementary references (PDF)
The authors declare no competing financial interests.
REFERENCES
- 1.Boerjan W; Ralph J; Baucher M, Lignin Biosynthesis. Annual Review of Plant Biology 2003, 54 (1), 519–546. [DOI] [PubMed] [Google Scholar]
- 2.Davin LB; Lewis NG, Dirigent Proteins and Dirigent Sites Explain the Mystery of Specificity of Radical Precursor Coupling in Lignan and Lignin Biosynthesis. Plant Physiology 2000, 123 (2), 453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saleem M; Kim HJ; Ali MS; Lee YS, An Update on Bioactive Plant Lignans. Natural Product Reports 2005, 22 (6), 696–716. [DOI] [PubMed] [Google Scholar]
- 4.Adlercreutz H, Lignans and Human Health. Critical Reviews in Clinical Laboratory Sciences 2007, 44 (5–6), 483–525. [DOI] [PubMed] [Google Scholar]
- 5.Wu C-C; Li T-K; Farh L; Lin L-Y; Lin T-S; Yu Y-J; Yen T-J; Chiang C-W; Chan N-L, Structural Basis of Type II Topoisomerase Inhibition by the Anticancer Drug Etoposide. Science 2011, 333 (6041). 459–462. [DOI] [PubMed] [Google Scholar]
- 6.Jacob DA; Mercer SL; Osheroff N; Deweese JE, Etoposide Quinone Is a Redox-Dependent Topoisomerase II Poison. Biochemistry 2011, 50 (25), 5660–5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith NA; Byl JAW; Mercer SL; Deweese JE; Osheroff N, Etoposide Quinone Is a Covalent Poison of Human Topoisomerase IIβ. Biochemistry 2014, 53 (19), 3229–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ratain MJ; Kaminer LS; Bitran JD; Larson RA; Le Beau MM; Skosey C; Purl S; Hoffman PC; Wade J; Vardiman JW, Acute Nonlymphocytic Leukemia Following Etoposide and Cisplatin Combination Chemotherapy for Advanced Non-Small-Cell Carcinoma of the Lung. Blood 1987, 70 (5), 1412. [PubMed] [Google Scholar]
- 9.Pui C-H; Ribeiro RC; Hancock ML; Rivera GK; Evans WE; Raimondi SC; Head DR; Behm FG; Mahmoud MH; Sandlund JT; Crist WM, Acute Myeloid Leukemia in Children Treated with Epipodophyllotoxins for Acute Lymphoblastic Leukemia. New England Journal of Medicine 1991, 325 (24), 1682–1687. [DOI] [PubMed] [Google Scholar]
- 10.Relling MV; Nemec J; Schuetz EG; Schuetz JD; Gonzalez FJ; Korzekwa KR, O-Demethylation of Epipodophyllotoxins is Catalyzed by Human Cytochrome P450 3A4. Molecular Pharmacology 1994, 45 (2), 352. [PubMed] [Google Scholar]
- 11.Chu A; Dinkova A; Davin LB; Bedgar DL; Lewis NG, Stereospecificity of (+)-Pinoresinol and (+)-Lariciresinol Reductases from Forsythia intermedia. Journal of Biological Chemistry 1993, 268 (36), 27026–27033. [PubMed] [Google Scholar]
- 12.Dinkova-Kostova AT; Gang DR; Davin LB; Bedgar DL; Chu A; Lewis NG, (+)-Pinoresinol/(+)-Lariciresinol Reductase from Forsythia intermedia: Protein Purification, cDNA Cloning, Heterologous Expression and Comparison to Isoflavone Reductase. Journal of Biological Chemistry 1996, 271 (46), 29473–29482. [DOI] [PubMed] [Google Scholar]
- 13.Davin LB; Wang H-B; Crowell AL; Bedgar DL; Martin DM; Sarkanen S; Lewis NG, Stereoselective Bimolecular Phenoxy Radical Coupling by an Auxiliary (Dirigent) Protein Without an Active Center. Science 1997, 275 (5298), 362–367. [DOI] [PubMed] [Google Scholar]
- 14.Xia Z-Q; Costa MA; Proctor J; Davin LB; Lewis NG, Dirigent-Mediated Podophyllotoxin Biosynthesis in Linum flavum and Podophyllum peltatum. Phytochemistry 2000, 55 (6), 537–549. [DOI] [PubMed] [Google Scholar]
- 15.Xia Z-Q; Costa MA; Pélissier HC; Davin LB; Lewis NG, Secoisolariciresinol Dehydrogenase Purification, Cloning, and Functional Expression: Implications for Human Health Protection. Journal of Biological Chemistry 2001, 276 (16), 12614–12623. [DOI] [PubMed] [Google Scholar]
- 16.Marques JV; Kim K-W; Lee C; Costa MA; May GD; Crow JA; Davin LB; Lewis NG, Next Generation Sequencing in Predicting Gene Function in Podophyllotoxin Biosynthesis. Journal of Biological Chemistry 2013, 288 (1), 466–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lau W; Sattely ES, Six Enzymes from Mayapple that Complete the Biosynthetic Pathway to the Etoposide Aglycone. Science 2015, 349 (6253), 1224–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Freudenberg K, Biosynthesis and Constitution of Lignin. Nature 1959, 183 (4669), 1152–1155. [DOI] [PubMed] [Google Scholar]
- 19.Freudenberg K, Lignin: Its Constitution and Formation from p-Hydroxycinnamyl Alcohols. Science 1965, 148 (3670), 595–600. [DOI] [PubMed] [Google Scholar]
- 20.Hapiot P; Pinson J; Neta P; Francesch C; Mhamdi F; Rolando C; Schneider S, Mechanism of Oxidative Coupling of Coniferyl Alcohol. Phytochemistry 1994, 36 (4), 1013–1020. [Google Scholar]
- 21.Davin LB; Lewis NG, Dirigent Phenoxy Radical Coupling: Advances and Challenges. Current Opinion in Biotechnology 2005, 16 (4), 398–406. [DOI] [PubMed] [Google Scholar]
- 22.Pickel B; Schaller A, Dirigent proteins: Molecular Characteristics and Potential Biotechnological Applications. Applied Microbiology and Biotechnology 2013, 97 (19), 8427–8438. [DOI] [PubMed] [Google Scholar]
- 23.Halls SC; Davin LB; Kramer DM; Lewis NG, Kinetic Study of Coniferyl Alcohol Radical Binding to the (+)-Pinoresinol Forming Dirigent Protein. Biochemistry 2004, 43 (9), 2587–2595. [DOI] [PubMed] [Google Scholar]
- 24.Kim K-W; Smith CA; Daily MD; Cort JR; Davin LB; Lewis NG, Trimeric Structure of (+)-Pinoresinol Forming Dirigent Protein at 1.95 Å Resolution with Three Isolated Active Sites. Journal of Biological Chemistry 2014, 290 (3), 1308–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gasper R; Effenberger I; Kolesinski P; Terlecka B; Hofmann E; Schaller A, Dirigent Protein Mode of Action Revealed by the Crystal Structure of AtDIR6. Plant Physiology 2016, 172 (4), 2165–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Katayama T; Davin LB; Chu A; Lewis NG, Novel Benzylic Ether Reductions in Lignan Biogenesis in Forsythia intermedia. Phytochemistry 1993, 33 (3), 581–591. [Google Scholar]
- 27.Saulnier MG; Vyas DM; Langley DR; Doyle TW; Rose WC; Crosswell AR; Long BH, E-ring Desoxy Analogs of Etoposide. Journal of Medicinal Chemistry 1989, 32 (7), 1418–1420. [DOI] [PubMed] [Google Scholar]
- 28.Long BH; Casazza A-M, Structure-Activity Relationships of VP-16 Analogues. Cancer Chemotherapy and Pharmacology 1994, 34 (1), S26–S31. [DOI] [PubMed] [Google Scholar]
- 29.Wallace G; Fry SC, Action of Diverse Peroxidases and Laccases on Six Cell Wall-Related Phenolic Compounds. Phytochemistry 1999, 52 (5), 769–773. [Google Scholar]
- 30.Kim K-W; Moinuddin SGA; Atwell KM; Costa MA; Davin LB; Lewis NG, Opposite Stereoselectivities of Dirigent Proteins in Arabidopsis and Schizandra Species. Journal of Biological Chemistry 2012, 287 (41), 33957–33972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kazenwadel C; Klebensberger J; Richter S; Pfannstiel J; Gerken U; Pickel B; Schaller A; Hauer B, Optimized expression of the Dirigent Protein AtDIR6 in Pichia pastoris and Impact of Glycosylation on Protein Structure and Function. Applied Microbiology and Biotechnology 2013, 97 (16), 7215–7227. [DOI] [PubMed] [Google Scholar]
- 32.Effenberger I; Harport M; Pfannstiel J; Klaiber I; Schaller A, Expression in Pichia pastoris and Characterization of Two Novel Dirigent Proteins for Atropselective Formation of Gossypol. Applied Microbiology and Biotechnology 2017, 101 (5), 2021–2032. [DOI] [PubMed] [Google Scholar]
- 33.Pen J; Molendijk L; Quax WJ; Sijmons PC; van Ooyen AJJ; van den Elzen PJM; Rietveld K; Hoekema A, Production of Active Bacillus licheniformis Alpha-Amylase in Tobacco and its Application in Starch Liquefaction. Bio/Technology 1992, 10, 292. [DOI] [PubMed] [Google Scholar]
- 34.Effenberger I; Zhang B; Li L; Wang Q; Liu Y; Klaiber I; Pfannstiel J; Wang Q; Schaller A, Dirigent Proteins from Cotton (Gossypium sp.) for the Atropselective Synthesis of Gossypol. Angewandte Chemie International Edition 2015, 54 (49), 14660–14663. [DOI] [PubMed] [Google Scholar]
- 35.Pickel B; Constantin M-A; Pfannstiel J; Conrad J; Beifuss U; Schaller A, An Enantiocomplementary Dirigent Protein for the Enantioselective Laccase-Catalyzed Oxidative Coupling of Phenols. Angewandte Chemie International Edition 2010, 49 (1), 202–204. [DOI] [PubMed] [Google Scholar]
- 36.Almagro Armenteros JJ; Tsirigos KD; Sønderby CK; Petersen TN; Winther O; Brunak S; von Heijne G; Nielsen H, SignalP 5.0 Improves Signal Peptide Predictions Using Deep Neural Networks. Nature Biotechnology 2019, 37 (4), 420–423. [DOI] [PubMed] [Google Scholar]
- 37.Mohamed KM, Chemical Constituents of Gladiolus segetum Ker-gawl. Bull Pharm Sci 2005, 28 (1), 71–8. [Google Scholar]
- 38.Deyama T; Ikawa T; Kitagawa S; Nishibe S, The Constituents of Eucommia ulmoides OLIV. V. Isolation of Dihydroxydehydrodiconiferyl Alcohol Isomers and Phenolic Compounds. Chemical and Pharmaceutical Bulletin 1987, 35 (5), 1785–1789. [Google Scholar]
- 39.Lancefield CS; Westwood NJ, The Synthesis and Analysis of Advanced Lignin Model Polymers. Green Chemistry 2015, 17 (11), 4980–4990. [Google Scholar]
- 40.Kim MK; Jeon J-H; Fujita M; Davin LB; Lewis NG, The Western Red Cedar (Thuja plicata) 8–8 ′ DIRIGENT Family Displays Diverse Expression Patterns and Conserved Monolignol Coupling Specificity. Plant Molecular Biology 2002, 49 (2), 199–214. [DOI] [PubMed] [Google Scholar]
- 41.Seneviratne HK; Dalisay DS; Kim K-W; Moinuddin SGA; Yang H; Hartshorn CM; Davin LB; Lewis NG, Non-Host Disease Resistance Response in Pea (Pisum sativum) Pods: Biochemical Function of DRR206 and Phytoalexin Pathway Localization. Phytochemistry 2015, 113, 140–148. [DOI] [PubMed] [Google Scholar]
- 42.Liu J; Stipanovic RD; Bell AA; Puckhaber LS; Magill CW, Stereoselective Coupling of Hemigossypol to Form (+)-Gossypol in Moco Cotton is Mediated by a Dirigent Protein. Phytochemistry 2008, 69 (18), 3038–3042. [DOI] [PubMed] [Google Scholar]
- 43.Stipanovic RD; Puckhaber LS; Liu J; Bell AA, Total and Percent Atropisomers of Gossypol and Gossypol-6-methyl Ether in Seeds from Pima Cottons and Accessions of Gossypium barbadense L. Journal of Agricultural and Food Chemistry 2009, 57 (2), 566–571. [DOI] [PubMed] [Google Scholar]
- 44.Ralph SG; Jancsik S; Bohlmann J, Dirigent Proteins in Conifer Defense II: Extended Gene Discovery, Phylogeny, and Constitutive and Stress-Induced Gene Expression in Spruce (Picea spp.). Phytochemistry 2007, 68 (14), 1975–1991. [DOI] [PubMed] [Google Scholar]
- 45.Li Q; Chen J; Xiao Y; Di P; Zhang L; Chen W, The Dirigent Multigene Family in Isatis indigotica: Gene Discovery and Differential Transcript Abundance. BMC Genomics 2014, 15 (1), 388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Umezawa T, Diversity in Lignan Biosynthesis. Phytochemistry Reviews 2003, 2 (3), 371–390. [Google Scholar]
- 47.Davin LB; Lewis NG, An Historical Perspective on Lignan Biosynthesis: Monolignol, Allylphenol and Hydroxycinnamic Acid Coupling and Downstream Metabolism. Phytochemistry Reviews 2003, 2 (3), 257. [Google Scholar]
- 48.O'Leary BM; Rico A; McCraw S; Fones HN; Preston GM, The Infiltration-Centrifugation Technique for Extraction of Apoplastic Fluid from Plant Leaves Using Phaseolus vulgaris as an Example. JoVE 2014, (94), e52113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.